
Abstract
Hydrogen sulfide (H2S), an endogenous gaseous signaling transmitter, has gained recognition for its physiological effects. In this review, we aim to summarize and discuss existing studies about the roles of H2S in renal functions and renal disease as well as the underlying mechanisms. H2S is mainly produced by four pathways, and the kidneys are major H2S–producing organs. Previous studies have shown that H2S can impact multiple signaling pathways via sulfhydration. In renal physiology, H2S promotes kidney excretion, regulates renin release and increases ATP production as a sensor for oxygen. H2S is also involved in the development of kidney disease. H2S has been implicated in renal ischemia/reperfusion and cisplatin–and sepsis–induced kidney disease. In chronic kidney diseases, especially diabetic nephropathy, hypertensive nephropathy and obstructive kidney disease, H2S attenuates disease progression by regulating oxidative stress, inflammation and the renin–angiotensin–aldosterone system. Despite accumulating evidence from experimental studies suggesting the potential roles of H2S donors in the treatment of kidney disease, these results need further clinical translation. Therefore, expanding the understanding of H2S can not only promote our further understanding of renal physiology but also lay a foundation for transforming H2S into a target for specific kidney diseases.
1. Introduction
Hydrogen sulfide (H2S) is a toxic, colorless gas with an odor of rotten eggs. It exists in nature and can be found in natural gas, volcanic emissions and petroleum [Citation1]. In 1989, Warenycia and Goodwin [Citation2] first demonstrated that the human body contains H2S, which mainly exists in the brain, and indicated that the brainstem is more sensitive to exogenous H2S than other parts of the brain. The physiological function of H2S has only recently been gradually recognized. High concentrations of H2S may lead to complete inhibition of cell respiration, mitochondrial membrane potential depolarization and superoxide generation [Citation3]. Low levels of H2S can regulate homeostatic mechanisms such as blood pressure (BP) control and apoptosis and participate in pathological mechanisms including oxidative stress (OS) and inflammation [Citation4,Citation5]. In the kidneys, H2S is actively involved in renal regulation, and H2S production disorders are involved in the onset and development of many kidney diseases [Citation6]. Although exogenous H2S has been shown to play key roles in alleviating various animal models of kidney damage, its specific molecular mechanism is unknown.
In this review, we first describe H2S generation and functions. Next, we introduce the role of H2S in renal physiology. Furthermore, we discuss H2S as a related factor in the occurrence and progression of renal disease and reveal some mechanisms. Finally, we summarize the application of H2S donors and inhibitors in preclinical work and logically evaluate the therapeutic potential of H2S in kidney diseases.
2. H2S generation and functions
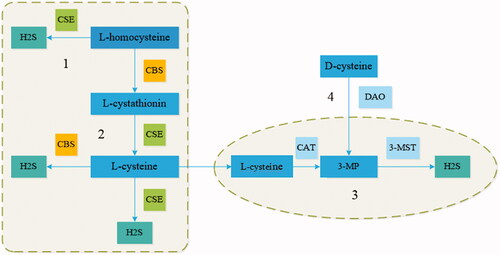
Although originally viewed as only a toxic gas, H2S is now recognized as a gaseous signaling molecule that is in some ways similar to nitric oxide (NO) and carbon monoxide (CO) [Citation7]. Unlike NO and CO, H2S is acidic, which allows it to dissolve in water. In addition, because H2S is highly lipophilic, it can spread freely to the cell membranes of all cell types [Citation8]. The enzymes responsible for the generation of endogenous H2S include cystathionine–β–synthase (CBS), cystathionine–γ–lyase (CSE), and mitochondrial 3–mercaptopyruvate sulfurtransferase (3–MST) [Citation9]. CBS and CSE both produce endogenous H2S in the cytosol, while 3–MST produces endogenous H2S in mitochondria [Citation4,Citation10]. Endogenous H2S is produced in four main ways. In the first mechanism, L–homocysteine and serine produce L–cystathionine under the action of CBS; the L–cystathionine is then changed into L–cysteine by CSE. Finally, H2S is formed in a process mediated by CBS and CSE in the cytoplasm [Citation6]. In the second mechanism CSE reacts with L–homocysteine to produce H2S, α–ketobutyrate and L–homolanthionine [Citation4]. In the third mechanism, cysteine aminotransferase converts L–cysteine to 3–mercaptopyruvate (3–MP), which is then utilized by 3–MST for the production of H2S in mitochondria [Citation11]. In the final mechanism, D–amino acid oxidase mediates the transformation of D–cysteine to 3–MP, and H2S is subsequently produced under the action of 3–MST. It is worth noting that 3–MP needs to be imported to mitochondria for the next step. In the kidneys, the main substrate for H2S production is D–cysteine, and H2S from D–cysteine is much more abundant than that from L–cysteine [Citation12] ().
Figure 1. Endogenous synthesis of H2S by four pathways. (A) CSE catalyzes the reaction of L–homocysteine to induce the production of H2S. (B) CBS reacts with L–homocysteine, increasing the generation of L–cystathionine, which is then converted into L–cysteine via CSE, which further produces H2S. (C) L–cysteine is converted into 3MP by CAT, and 3–MST catalyzes the reaction of 3MP to induce H2S generation in mitochondria. (D) DAO reacts with D–cysteine to generate 3MP, which then enters mitochondria and serves as a substrate for the production of H2S. CBS (cystathionine β–synthase); CSE (cystathionine γ–lyase); CAT (cysteine aminotransferase); 3MP (3–mercaptopyruvate); DAO (D–amino acid oxidase); 3–MST (3–mercaptopyruvate sulfurtransferase).
How does H2S perform biological functions? Recent studies have provided answers. H2S can regulate different signaling pathways that affect cell metabolism. H2S is involved in signal transmission through signaling pathways via sulfhydration, during which it reacts with cysteine residues of various target proteins to form persulfide bonds. The reactivity of sulfhydration is determined by the acid dissociation constants of cysteine residues [Citation13]. Mustafa et al. [Citation14] found that approximately 10–25% of liver proteins can be activated by S–sulfhydration, such as actin, tubulin, and glyceraldehyde–3–phosphate dehydrogenase. S–sulfhydration is essential for the functions of liver proteins; for example, it enhances glyceraldehyde–3–phosphate dehydrogenase activity and actin polymerization. H2S is an endothelium–derived hyperpolarizing factor that can lead to hyperpolarization and vasodilation of vascular endothelial and smooth muscle cells. This vasodilation is mainly achieved via activation of the ATP–sensitive, intermediate and small conductance potassium channels, and the most critical step for channel activation is S–sulfhydration [Citation15]. H2S participates in inflammatory reactions as a messenger molecule, and the downstream effects of sulfhydration affect nuclear factor κB (NF–κB). NF–κB plays a key role in the inflammatory response in cells. Nil Kantha et al. [Citation5] found that tumor necrosis factor–α (TNF–α) can stimulate the transcription of CSE to generate H2S. H2S sulfhydrates Cys38 of p65, enhancing its binding to the coactivator ribosomal protein S3, thereby regulating the nuclear functions of NF–κB. In CSE–deficient mice, p65 cannot be sulfhydrated, resulting in decreased NF–κB target gene activity. The protein tyrosine phosphatase–1B is located on the cytoplasmic face of the endoplasmic reticulum (ER) and has been implicated in ER stress signaling. H2S–induced sulfhydration of protein tyrosine phosphatase–1B participates in the ER stress response [Citation16]. P66Shc is an upstream activator of mitochondrial redox signaling. In response to OS, p66Shc is activated through protein kinase C–bII–mediated phosphorylation at Ser36. Xie et al. [Citation17] found that H2S downregulates the phosphorylation of p66Shc through the sulfhydration of Cys59 residue, thus reducing mitochondrial production of reactive oxygen species (ROS) and achieving antioxidant effects. Nuclear factor–erythroid 2–related factor 2 (Nrf2) is a master regulator of the antioxidant response. Normally, Nrf2 is ubiquitinated and rapidly degraded by the proteasome under the action of Kelch–like ECH–associated protein 1 (Keap1). Sodium sulfide (NaHS) has been reported to S–sulfhydrate Keap1 at Cys151 and promote Nrf2 nuclear translocation [Citation18].
3. H2S production in the kidneys and role in kidney physiology
3.1. H2S production in the kidneys
Some studies have found that the three enzymes that produce endogenous H2S are highly expressed in certain tissues, such as the kidneys [Citation19]. CBS, CSE and 3–MST can be detected in renal proximal tubules. CSE is also mainly expressed in renal glomeruli, interstitia, and interlobular arteries [Citation19,Citation20]. Under normal conditions, the CSE protein is expressed at the highest level in the kidneys, reaching levels 20 times those of CBS. In the kidney tissues of Sprague–Dawley rats, all three H2S–producing enzymes are present, and CSE mRNA is expressed more abundantly than 3MST and CBS mRNA [Citation4]. Therefore, CSE plays a leading role in the production of H2S [Citation21,Citation22]. CBS and CSE synergistically produce H2S, and these two enzymes can jointly increase the production of endogenous H2S in the kidneys [Citation23].
Three traditional H2S–synthesizing pathways (involving CSE, CBS, and 3–MST coupled with cysteine aminotransferase) have been identified in the kidneys, as described in the “H2S Generation and Functions” section. Therefore, we focused on a fourth H2S generation pathway, namely, the DAO/3–MST pathway [Citation24]. In this pathway, D–cysteine is transformed into 3–MP by peroxisome–located DAO. Due to metabolite exchanges between peroxisomes and mitochondria, 3–MP is imported into mitochondria and catalyzed into H2S by 3–MST [Citation24] (). Shibuya et al. showed that kidney lysate can produce 60 times more H2S when D–cysteine is used as a substrate than when L–cysteine is used [Citation12]. The discovery of the unique DAO/3–MST pathway in the kidneys and brain may imply a significant role of 3–MST–mediated H2S generation in these organs. This possibility is worth exploring further.
3.2. H2S in kidney physiology
3.2.1. Effect of H2S on renal excretory function
H2S plays an important role in renal excretion. Xia et al. found that both CBS and CSE can produce H2S in the kidney and that when either enzyme is inhibited, the expression of the other increases to compensate. They also found that in anesthetized Sprague–Dawley rats, infusion of NaHS in the renal artery can increase renal blood flow and the glomerular filtration rate (GFR). Because of the increase in the filtration rate, those authors speculated that the role of H2S in vasodilating blood vessels was greater in preglomerular arterioles than in postglomerular arterioles. H2S can also inhibit the Na–K–2Cl cotransporter in the ascending limb of the loop of Henle and the Na–K ATPase enzyme, potentially increasing the excretion of sodium and potassium from urine. Therefore, H2S participates in both vascular and tubular actions in the kidneys [Citation23].
3.2.2. H2S as an oxygen sensor
H2S may act as an oxygen (O2) sensor to restore O2 balance, a phenomenon that has been confirmed in various O2–sensing tissues, such as the carotid body, adrenal medulla and other chemoreceptive tissues, as well as in smooth muscle in systemic and respiratory vessels and airways [Citation25]. H2S metabolism is highly dependent on the concentration of O2 [Citation25]. Under physiological conditions, pO2 is reduced in the renal medulla, and oxidation of H2S is negatively correlated with pO2 in mitochondria, so the activity of H2S in the medulla is likely higher than that in the renal cortex [Citation4]. H2S, which accumulates in increased amounts in the renal medulla under hypoxic conditions, may restore O2 supply by increasing medullary blood flow [Citation26]. Moreover, studies have shown that under conditions of sufficient oxygen, the levels of CBS and CSE in mitochondria are low. Once hypoxia occurs, the concentrations of CBS and CSE increase, which increases the production of H2S [Citation27,Citation28]. H2S served as an electron donor and increases ATP production [Citation27]. Hypoxia is the most important risk factor for the pathogenesis and progression of many renal diseases. Endogenous H2S deficiency can further contribute to compromised medullary oxygenation and aggravate the occurrence and development of kidney disease [Citation26]. However, the specific mechanism remains unclear and needs further study.
3.2.3. H2S modulates renin release
H2S attenuates pathological signaling of the renin–angiotensin–aldosterone system (RAAS) to preserve kidney function. The RAAS is a humoral regulatory system composed of hormones and corresponding enzymes that regulates the excretion of water and sodium. The release of renin from juxtaglomerular cells determines the onset and development of renovascular hypertension, a procedure adjusted by intracellular 3′–5′–cyclic adenosine monophosphate (cAMP). H2S has been reported to downregulate cAMP by inhibiting adenylate cyclase activity, thereby regulating renin release and controlling BP [Citation29,Citation30]. In primary cultures of renin–rich kidney cells, NaHS significantly reduces the levels of intracellular cAMP and reduces renin activity. In a Dahl rat model of high–salt–induced hypertension, treatment with H2S has been found to inhibit RAAS system activation in the kidneys and to regulate BP [Citation31]. H2S also regulates BP via angiotensin–converting enzyme, which belongs to the RAAS system. In human endothelial cells, H2S can directly interfere with zinc in the active center of angiotensin–converting enzyme [Citation32].
In brief, H2S plays a key role in renal physiology; however, further study is required to establish the specific mechanisms involved.
4. Role of H2S in acute kidney injury
H2S plays diverse roles in the onset and development of kidney disease. Acute kidney injury (AKI) is the clinical syndrome caused by a rapid decline in renal function due to a number of causes, which can occur in patients without kidney disease or on the basis of original kidney disease. The main features of this type of injury are a rapid decrease in GFR, an increase in serum creatinine, oliguria and even anuria. Approximately 20% of hospitalized patients experience AKI, although among critically ill patients, the percentage can reach 50%. AKI is divided into different types according to etiology and stage, and the prognoses and treatments differ among the types. However, the main complications include volume overload, uremia complications and electrolyte disorders. AKI causes 2 million deaths a year, placing huge burdens on society and the medical system [Citation33,Citation34].
4.1. Role of H2S in renal ischemia/reperfusion injury
The damage to microvascular and parenchymal organs caused by ischemia reperfusion injury (IRI) mainly occurs via ROS, as has been proven in many organs. The kidneys are the main target organs of this type of injury, which results in the clinical syndrome of AKI. The production of ROS can lead not only to cell necrosis and apoptosis but also to lipid peroxidation and ATP depletion [Citation35,Citation36]. Recent studies have shown that H2S is strongly associated with renal IRI. Han et al. [Citation37] found that the expression and activity of CSE and CBS in the kidneys were decreased and that the levels of H2S were also decreased in mice subjected to bilateral renal ischemia for 30 min. NaHS treatment restored renal function and accelerated the return of tubular morphology to normal. Furthermore, NaHS treatment improved renal function by reducing OS. Eelke et al. [Citation38] revealed that CSE participates in the improvement of renal ischemia, most likely via production of H2S to mitigate OS. Azizi et al. [Citation39] found that H2S can protect against ischemia/reperfusion–induced AKI by reducing OS.
H2S may exert its renal IRI–improving effects through several mechanisms. Ischemia has profound effects on the renal endothelium, resulting in microvascular dysregulation and continued ischemia and further injury [Citation40]. In the first mechanism, H2S dilates blood vessels in the smooth muscle tissue of the kidneys, by activating K+ ATP channels, thereby increasing renal blood flow, which is helpful for the recovery of renal tubules [Citation15]. Notably, it has been confirmed in previous studies that H2S can reduce the renal burden in mice with angiotensin (Ang) II–induced hypertension, while IRI can activate the RAAS system [Citation41]. Thus, we speculate that in the second mechanism, H2S inhibits the RAAS system and protects kidney functions. In the third mechanism, H2S protects mitochondrial function. Treatment with H2S donors can induce reversible hypometabolism. The proposed mechanism involves reductions in mitochondrial activity via reversible binding to cytochrome c oxidase. Under hypoxic conditions, the integrity and function of mitochondria are impaired, but this impairment is alleviated after administration of H2S. Therefore, H2S reduces the use of O2, protects tissues from hypoxia and shock, and protects organs from IRI [Citation42,Citation43]. In the fourth mechanism, homocysteine can be reduced to cysteine in cells for glutathione synthesis. H2S can enhance the transport of homocysteine, increase the production of glutathione, inhibit the activity of nicotinamide adenine dinucleotide phosphate (NADPH) oxidase, play a role in scavenging free radicals and increase the activity of antioxidant enzymes [Citation1,Citation44]. In addition, A39, as a mitochondria–targeting H2S donor, has been reported to be related to the reduction of cellular OS and to exert dose–dependent protective effects against renal epithelial cell injury in vitro and IRI in vivo [Citation45].
4.2. Role of H2S in drug–induced acute kidney injury
Medications are important means of disease prevention and treatment but can also cause AKI. There are many kinds of nephrotoxic drugs that can be used in combination with single or multiple drugs. The pathogenesis of drug–induced AKI varies but mainly involves acute tubular/tubulointerstitial injury. High–risk factors include drug use and patient factors [Citation46].
Cisplatin (Cisp) is a chemotherapeutic agent that is widely used for solid tumors [Citation47]. It has been reported that approximately 25%–30% of patients treated with Cisp develop forms of nephrotoxicity, such as AKI. The most common damage caused by Cisp is DNA injury, which can also activate the apoptotic pathway and cause damage to other organelles through OS and inflammation. The most severe damage occurs to the ER and mitochondria [Citation48]. Cisp is mainly excreted by the kidneys and accumulates in renal proximal tubular cells, leading to subsequent tubular cell death and AKI [Citation49]. Mitochondrial dysregulation is central to tubular injury. Some research has shown that Cisp accumulates in the mitochondria of renal proximal tubular cells and impairs mitochondrial redox balance, finally leading to mitochondrial dysfunction [Citation50,Citation51]. Recent studies have reported that in mice with Cisp–induced AKI mice, damaged mitochondria not only compromise cellular energy metabolism but also induce mitochondrial fragmentation and ROS overproduction, which determined the degree of tubular epithelial cell injury and death [Citation52]. Previous studies have suggested that H2S alleviates Cisp–induced AKI and contrast–induced AKI [Citation53]. However, the underlying mechanism remains largely unclear. Yuan et al. [Citation54] found that H2S increases Sirtuin 3 (SIRT3) expression and enhances deacetylase activity by sulfhydrating SIRT3 at two CXXC zinc finger motifs. SIRT3 mainly exists in mitochondria and is a NAD+–dependent deacetylase. As one of the major mitochondrial deacetylases, SIRT3 improves mitochondrial bioenergetics and inhibits mitochondrial dysfunction by mediating deacetylation of its target proteins [Citation55]. Therefore, it has been concluded that H2S alleviates AKI in Cisp–induced mice [Citation54]. Recent reports have demonstrated that low concentrations of H2S stimulate mitochondrial oxidative phosphorylation and inhibit mitochondrial oxidant production [Citation56]. Akram et al. [Citation57] also found that H2S prevents the progression of Cisp nephrotoxicity in rats, possibly through its antioxidant properties.
However, recent studies have found that H2S can exacerbate Cisp–induced renal damage. Francescato et al. [Citation58] used DL–propargylglycine (PAG) to inhibit endogenous H2S production in Wistar rats and found that inflammation was reduced and kidney damage was alleviated. Liu et al. found that the slow–releasing H2S donor GYY4137 aggravated Cisp–induced renal injury, which was associated with inflammation, OS and apoptosis. Notably, the dose of GYY4143 they used was rather low (21 mg/kg), which may have led to insufficient or even a lack of production of H2S [Citation59]. One study has also revealed that in the context of doxorubicin–induced renal damage, PAG exerts its protective effect by decreasing H2S production and reducing OS and inflammation in the kidneys [Citation60].
H2S plays an important role in drug–induced AKI, but previous studies have yielded quite different results. The differences may have been related to the different H2S concentrations used in the experiments, which needs further study and discussion.
4.3. Role of H2S in sepsis–associated acute kidney injury
The probability of sepsis in critical patients with AKI is approximately 40%–50%. When sepsis and AKI occur simultaneously, a condition known as sepsis–associated AKI (SA–AKI), the mortality can be as high as 70% [Citation61]. Kidney hypoperfusion is the major contributor to SA–AKI. The pathogenesis of SA–AKI is complicated and includes renal macrocirculatory and microcirculatory disturbances, surges in inflammatory markers and OS and coagulation cascade activation [Citation62].
Lipopolysaccharide (LPS), TNF–α and interleukin (IL)–1β, which are typical inflammatory cytokines that function via Toll–like receptor 4 signaling pathways, have been found to participate in OS production in an AKI mouse model. Excessive OS production leads to dysfunction of tubular epithelial cells. NaHS improves renal function and attenuates kidney histopathological changes, LPS–induced inflammation and OS. Chen et al. [Citation63] found that plasma H2S levels are reduced in rabbits with SA–AKI and that exogenous H2S can suppress NF–κB and TNF–α activity and increase IL–10 content, thus delaying kidney damage. Caitlyn et al. [Citation64] also suggested that NF–κB transactivation can induce inflammation and cell hyperpermeability and that the CSE/H2S system can block NF–κB transactivation.
In contrast, the role of H2S in sepsis is not clear. Plasma H2S concentrations have been found to be high in patients with septic shock. In a mouse model of LPS–induced inflammation, injection of LPS has been found to increase the gene expression of CSE in the kidneys. PAG administration before LPS injection reduces the content of H2S and the infiltration of leukocytes in the kidneys. After NaHS injection, the levels of plasma TNF–α increase significantly [Citation65]. Nevertheless, further research is needed to elucidate the specific role of H2S in SA–AKI. The role of H2S in AKI is summarized in [Citation37–39,Citation54,Citation57,Citation65–69].
Table 1. Previous studies on the effects of H2S in acute kidney injury.
5. Involvement of H2S in chronic kidney disease
Chronic kidney disease (CKD) is defined as the occurrence of renal structural alteration and dysfunction with multiple causes over a course of more than three months. Characteristics of CKD include abnormal blood or urine test results, pathological damage to the structure and function of the kidneys, and an unexplained decrease in GFR, such as a decrease to a level less than 60 mL/min. Diabetes mellitus (DM) and hypertension are common causes of CKD. CKD cannot be cured; even if the external pathogenic factors have been removed, CKD tends to deteriorate internally, and glomerular and renal tubular damage continue to be aggravated, such that CKD eventually progresses to end–stage renal failure. In this context, the renal structural changes in patients lead to OS, and excessive OS can activate redox–sensitive proinflammatory transcription factors and signal transduction pathways so that the system triggers an inflammatory response, resulting in a sustained exacerbation of renal injury [Citation70]. In recent years, the progression of CKD has been able to be delayed with strict control of BP and blood glucose, suppression of proteinuria and suppression of kidney damage. Complications of CKD include volume and electrolyte abnormalities, anemia, mineral and bone abnormalities, endocrine abnormalities, cardiovascular and cerebrovascular diseases and uremia [Citation71].
CKD can be divided into five stages according to GFR, and chronic renal failure (CRF) corresponds to stages 3–5. Among patients with CRF and end–stage renal failure, the morbidity and mortality of cardiovascular diseases are increased significantly [Citation72]; therefore, compared to patients with other chronic diseases, patients with CRF tend to require longer and more frequent hospitalizations [Citation73]. CRF has become a major health problem worldwide, and its pathological foundations are renal tubular cell loss and fibrosis. Inflammation, OS and excessive autophagy are the main causes of incurable disease or continuous progression [Citation74].
In both patients and animal models, plasma levels of H2S are lower in subjects with CRF than in normal subjects [Citation75]. The enzyme that produces H2S in the residual kidneys is downregulated, and H2S production is also significantly reduced, which is associated with increased inflammation and OS [Citation76]. Mohammad et al. [Citation76] found that 5/6–nephrectomized (5/6–Nx) rats exhibited marked OS, inflammation, GSH synthase declines and NADPH oxidase upregulation. Treatment with NaHS reduced BP and increased creatinine clearance. NaHS has also been shown to reduce malondialdehyde levels and increase superoxide dismutase activity [Citation77].
Apoptosis is an intrinsic cell–suicide program that is critical for the normal development and maintenance of tissue homeostasis in multicellular organisms [Citation78]. Many researchers believe that high levels of ROS can lead to imbalances in redox reactions, inducing apoptosis or cell necrosis [Citation79]. The proteins of the Bcl–2 family are involved in apoptotic signaling pathways and include proapoptotic and antiapoptotic members [Citation79]. In the renal tissues of CRF rats, apoptosis is significantly increased, as indicated by evidence including upregulation of the expression of the proapoptotic factor Bax, and downregulation of the expression of the antiapoptotic factor Bcl–2 [Citation80].
One study has shown that H2S can effectively inhibit cleaved caspase–3 activity and thus reduce the extent of renal apoptosis [Citation77]. Therefore, we predict that a reduction in cleaved caspase–3 activity may be a key step for H2S to play an antiapoptotic role, but a large number of experiments are still needed to verify this hypothesis [Citation81]. Beclin, LC3A/B, and mTOR are characteristic markers of autophagy, and a recent study found that the expression of these markers was unequivocally increased in 5/6–Nx rats. After administration of NaHS, these markers returned to normal levels. The study also suggested that the interplay between H2S and NO contributes to renal functions. Chronic renal injury inhibited endothelial NOS (eNOS) expression while inducing inducible NO synthase (iNOS) expression, but treatment with H2S reversed these changes [Citation74].
In rats with adenine–induced CRF, administration of H2S not only reduces the concentrations of blood urea nitrogen and Scr but also reduces the extent of anemia. Furthermore, H2S can inhibit apoptosis and inflammation in these rats through the ROS/mitogen–activated protein kinase (MAPK) and NF–κB signaling pathways [Citation82].
H2S ameliorates CRF by inhibiting OS, inflammation, and autophagy and interacting with NO. However, the specific mechanism still needs to be proven.
5.1. Role of H2S in diabetic nephropathy
DM is a widespread disease that seriously threatens human health. DM can affect important organs and poses huge medical burdens on families and society [Citation83]. Diabetic nephropathy (DN) is one of the most severe microvascular complications of DM. The principal pathological features of DN are glomerular sclerosis and interstitial fibrosis [Citation84], and the mechanism is thought to involve induction of OS [Citation85], accumulation of advanced glycosylation end–products (AGEs) [Citation86], activation of inflammatory and fibrosis signaling pathways [Citation87], abnormal renal hemodynamics and abnormal activation of the RAAS, secondary to chronic hyperglycemia [Citation88].
The current evidence suggests that H2S deficiency contributes to the development of DN. Plasma H2S levels are lower in patients with type 2 DM (T2DM) than in normal individuals, and the same results have been found in diabetic rats [Citation89]. High levels of urinary sulfate are related to high plasma H2S concentrations, and high urinary sulfate can reduce the risk of renal disease progression in T2DM patients [Citation90].
In addition, the plasma H2S content in DM patients undergoing dialysis is lower than that in the normal population, and in chronic hemodialysis patients, a lower level of H2S is associated with a higher propensity for uremic atherosclerosis [Citation91]. Recent studies have shown that H2S supplementation can slow the development of DN. Treatment with NaHS ameliorates kidney lesions in T2DM by increasing glucose uptake in myotubes and adipocytes [Citation92]. Supplementation with H2S or an endogenous precursor of H2S (L–cysteine) in culture medium prevents the release of proinflammatory cytokines such as IL–8 and monocyte chemotactic protein–1 in high–glucose–treated human U937 monocytes [Citation89]. We summarize the role of H2S in DN in [Citation93–100].
Table 2. Previous studies on the effects of H2S in diabetic nephropathy.
5.1.1. H2S alleviates diabetic nephropathy by reducing oxidative stress
OS develops due to an imbalance between the levels of ROS in cells and the antioxidant system [Citation101]. The increase in blood glucose levels leads to activation of the protein kinase C pathway in kidney cells. Protein kinase C activates NADPH II in mitochondria and then increases OS, which leads to expansion of the glomerular mesangial region, thickening of the basement membrane, disorder of endothelial cell function and proteinuria [Citation102]. ROS production also results in accumulation of extracellular matrix, activation of fibrosis factors, and podocyte injury [Citation103]. Glucose can form AGEs with various proteins under nonenzymatic conditions, and AGEs can induce the OS response and increase protein kinase C activity, thus inducing increased production of inflammatory chemokines [Citation104].
Overall, high glucose induces OS in kidney cells, and H2S can participate in this process. Marwa et al. [Citation105] found that kidney damage caused by hyperglycemia through OS can be antagonized by NaHS. H2S can protect kidney functions in T1DM rats in a manner related to suppression of OS via increased activity of superoxide dismutase [Citation106].
Under physiological conditions, Keap1 ubiquitinates and degrades Nrf2. However, under conditions of antioxidation, shear stress, or electrophilic agent exposure, Nrf2 dissociates from Keap1 and translocates to the nucleus, where it promotes the expression of downstream antioxidant genes [Citation107]. Animal experiments have shown that activation of the renal Nrf2 signaling pathway alleviates OS injury and proteinuria and increases extracellular matrix and basement membrane thickening [Citation108]. In high–glucose–treated endothelial cells, H2S induces Nrf2 dissociation from Keap1 by increasing S–sulfhydration of Keap1, increasing Nrf2 translocation to the nucleus and inducing downstream antioxidant gene expression, thereby reducing OS and adhesion molecule levels [Citation109]. Previous studies have shown that H2S activates Nrf2 and increases the protein expression of heme oxygenase–1, thus improving renal resistance to OS in DM rats [Citation110].
Recent studies have suggested that high glucose can induce NADPH oxidase 4 (NOX4) expression and extracellular matrix synthesis by downregulating adenosine monophosphate kinase–activated protein kinase (AMPK) in kidney cells [Citation111]. NOX4 is a member of the group of NADPH oxidases of the NOX family and is a main source of ROS in the kidneys [Citation112]. H2S recruits iNOS to generate NO in order to inhibit high–glucose–induced NOX4 expression, OS, and matrix protein accumulation [Citation111].
High glucose recruits mammalian target of rapamycin complex 1 to exacerbate hypertrophy and increase protein synthesis in renal epithelial and mesangial cells. In DN, phosphatidylinositol 3 kinase/protein kinase B/mammalian target of rapamycin complex 1 signaling is activated due to the suppression of AMPK activity [Citation6,Citation113]. Treatment with NaHS reverses the decrease in AMPK phosphorylation caused by high glucose, which reduces renal damage [Citation114].
Autophagy consists of a number of complex steps, primarily formation of phagocytic vesicles, synthesis of autophagosomes, formation of autophagolysosomes after fusion of autophagosomes and lysosomes and gradual degradation of autophagolysosomes. Autophagy can scavenge waste within cells to achieve cellular homeostasis and organelle renewal [Citation115]. In DM patients, the concentration of ROS increases as hyperglycemia persists, which can lead to excessive autophagy or disrupted autophagy, which can damage endothelial cells [Citation116]. In endothelial cells, high–glucose–/palmitate–induced mitochondrial damage is associated with OS. Mitochondrial damage reduces ATP production and causes persistent overactivation of the AMPK signaling pathway, thereby leading to excessive autophagy and further damaging endothelial cells. H2S treatment can inhibit OS and attenuate excessive autophagy [Citation117].
In recent years, an increasing number of studies have found that H2S can alleviate OS and inhibit excessive autophagy caused by OS, which may provide new avenues for therapeutic treatment of DN.
5.1.2. Inflammation and H2S in diabetic nephropathy
An abnormal immune system and chronic inflammation play important roles in the pathological progression of DN [Citation102]. NF–κB is the central link and common pathway regulating the transcription of many inflammatory factors and the key factor in endothelial inflammatory injury [Citation118]. When the NF–κB pathway of kidney cells is activated, the cells secrete adhesion factors such as intercellular adhesion molecule–1 and vascular cell adhesion protein–1 as well as chemokines such as monocyte chemotactic protein–1 and IL–1. These adhesion molecules and chemokines attract monocytes, macrophages, and T lymphocytes into the kidneys, thereby activating TNF–α signaling and leading to increased renal pathology and fibrosis [Citation119,Citation120]. In rats with streptozotocin (STZ)–induced DM, NaHS exerts anti–inflammatory actions by inhibiting NF–κB signaling in glomerular mesangial cells [Citation110].
MMP–9, a zinc–dependent endopeptidase, is an inflammatory cytokine that leads to extracellular matrix degradation and renal vascular remodeling [Citation121]. Kundu et al. [Citation122] showed that MMP–9 reduces the expression of CBS and CSE and decreases the content of H2S in DM mice and that treatment with H2S reverses MMP–9–induced DM kidney remodeling.
5.1.3. H2S improves diabetic nephropathy by attenuating RAAS activity
The classic RAAS includes several steps. Briefly, the kidneys secrete renin, which activates angiotensinogen produced from the liver to produce Ang I. Ang I is then converted into Ang II in the pulmonary circulation. Ang II, the main effector of the RAAS, acts on Ang II type I receptors, causing the smooth muscle of arterioles to contract and stimulating the secretion of aldosterone into the spheroid of the adrenal cortex [Citation123]. Hyperglycemia activates the renal RAAS, increases glomerular hydrostatic pressure, and causes proteinuria, glomerulosclerosis and interstitial fibrosis [Citation124]. Blockade of the RAAS has been shown to delay the progression of DN [Citation125]. As previously mentioned, renin is secreted by juxtaglomerular cells, and H2S can regulate renin release by downregulating cAMP. In a murine model of transverse aortic constriction–induced heart failure, treatment with H2S reduced renal RAAS pathological activation and protected the heart, kidney and blood vessels [Citation126]. In DN, H2S may play a preventive role by regulating renin release and activity [Citation22].
H2S can also inhibit the RAAS system by regulating other links. Compared with normal–glucose–stimulated cells, high–glucose–stimulated cells exhibit significant upregulation of angiotensinogen, angiotensin–converting enzyme and Ang II type I receptor mRNA levels, but this upregulation can be reversed by treatment with H2S [Citation127]. One study has found that the protein expression levels of angiotensin–converting enzyme, Ang II type I receptor receptors and Ang II are significantly upregulated in DN. These proteins are downregulated after NaHS treatment [Citation110].
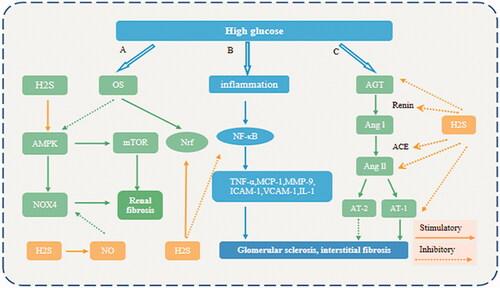
Therefore, H2S can attenuate OS and the inflammatory response and inhibit pathological activation of the RAAS. In the early stage of DN, H2S can ameliorate glomerular basement membrane thickening and mesangial matrix hyperplasia. In the late stage, H2S can delay renal fibrosis and ameliorate renal remodeling. H2S is thus involved in the different stages of DN ().
Figure 2. Mechanisms underlying the protective effect of H2S in DN. (A) H2S stimulates NO formation, which reduces NOX4 levels; H2S activates AMPK, thereby suppressing mTOR signaling; and H2S activates the Nrf2 pathway. (B) H2S acts as an anti–inflammatory factor by blocking NF–κB signaling in the renal system. (C) Renin converts AGT into Ang I. Under the action of ACE, Ang I is further converted to Ang II, and Ang II binds to AT–1, which exacerbates DN. The activation of RAAS in DN is ameliorated by H2S treatment via inhibition of AGT, renin, ACE, Ang II and AT–1 receptors. OS (Oxidative stress); AMPK (AMP–activated protein kinase); NO (nitric oxide); mTOR (mechanistic target of rapamycin); Nrf (Nuclear factor–erythroid 2–related factor 2); NOX4 (NADPH oxidase 4). TNF–α (tumor necrosis factor α); IL–1β (interleukin–1β); VCAM–1 (vascular cell adhesion molecule–1); ICAM–1(intercellular adhesion molecule–1); MCP–1 (monocyte chemotactic protein–1); MMP–9 (matrix metalloproteinase–9). AGT (Angiotensinogen); ACE (angiotensin converting enzyme); AngI (angiotensin I); AngII (angiotensin II); AT–1 (Ang II type I receptor1).
5.2. Role of H2S in hypertensive nephropathy
Hypertensive nephropathy is the second–leading cause of end–stage renal disease after DM [Citation128]. Most hypertensive patients develop mild–to–moderate hypertensive nephrosclerosis, with only relatively few patients entering end–stage renal disease. Nevertheless, the occurrence of end–stage renal disease dramatically increases when BP values are uncontrolled for a long time or when kidney disease is a preexisting condition [Citation129,Citation130]. Poor control of long–term hypertension aggravates kidney damage. Kidney damage further increases BP levels. Thus, long–term hypertension and kidney damage form a vicious cycle, resulting in multiple organ damage [Citation131].
Although the specific mechanism is unclear, studies have revealed that mice lacking CSE show significant hypertension and reduced endothelium–dependent vasorelaxation [Citation132]. In rats, BP can also be increased via inhibition of H2S–producing enzymes (CBS and CSE) [Citation133]. As mentioned above, H2S increases the estimated GFR (eGFR) and the urinary excretion of sodium. In addition, H2S controls BP by directly regulating vascular tension. H2S is acknowledged as an endothelium–derived hyperpolarizing factor that acts by sulfhydrating cysteine residues of K–ATP channels [Citation15]. In addition, H2S can activate the cAMP/protein kinase A signaling pathway, release calcium, and regulate vasorelaxation [Citation134].
PPARs are a class of ligand–activated nuclear transcription factors that belong to the receptor superfamily. Three subtypes of PPARs, PPAR–α, PPAR–β/δ, and PPAR–γ, have been found. Each PPAR subtype can alleviate metabolic abnormalities under the action of agonists; however, their mechanisms of action are different. For example, activation of PPAR–β/δ can significantly improve BP by increasing NO and serve as a new therapeutic target for hypertension [Citation135,Citation136]. Recent studies have shown that the role of H2S in regulating BP might be related to PPARβ/δ activation. H2S is thought to work with NO in synergy to regulate vascular tone. The specific molecular mechanism may involve H2S–mediated upregulation of PPAR–δ expression, increases in protein kinase B or AMPK phosphorylation, and enhancement of eNOS phosphorylation with a consequent increase in NO production [Citation137,Citation138]. In rats, the use of NOS inhibitors, such as Nx–nitro–l–arginine methyl ester, can cause hypertension, which can be reversed by treatment with NaHS [Citation139]. However, in aortic rings of rats, low concentrations of NaHS (10–100 μM) can downregulate NO production and consequently induce vasoconstriction. In contrast, administration of high doses of NaHS has been reported to directly relax aortic rings [Citation140]. These findings suggest a role of H2S/NO crosstalk in BP regulation. The antihypertensive effect of H2S is also dose dependent, but the specific mechanism needs further study [Citation6].
Ang II can contract whole arterioles. In addition, it can promote secretion of aldosterone in the adrenal cortex. Aldosterone acts on the renal tubules and increases blood volume. Furthermore, Ang II induces OS by binding angiotensin receptor 1 to mediate excessive ROS accumulation, leading to endothelial damage. In the kidneys, Ang II exacerbates target organ inflammation by increasing superoxide formation and chemokine release [Citation141,Citation142]. In addition to H2S inhibiting renin release, another possible mechanism is that exogenous H2S inhibits the binding of Ang II to the Ang II type I receptor, reduces Ang II–mediated signaling and inhibits pathological progression in mice. In mice with Ang II–induced hypertension, H2S can also inhibit ROS production in blood vessels and remove ROS, improving antioxidant capacity [Citation143].
In summary, H2S plays indispensable roles in regulating BP, reducing proteinuria, delaying renal dysfunction and structural deterioration by inhibiting Ang II and increasing NO production [Citation143]. Thus, H2S may evolve into a unique target for the treatment of hypertensive nephropathy.
5.3. Role of H2S in obstructive kidney disease
Obstructive nephropathy is a common urological disease that includes kidney stones, polycystic kidney disease, and renal artery stenosis. Ureteral obstruction accelerates renal fibrosis, which is associated with the onset and development of obstructive nephropathy [Citation144]. Renal fibrosis is also a key pathological feature in CKD [Citation145]. In a unilateral ureteral obstruction (UUO) mouse model, the activity levels of H2S and H2S production enzymes, including CBS and CSE, have been confirmed to decrease significantly. After administration of NaHS, H2S levels increase and renal fibrosis is alleviated, and these changes are accompanied by an increase in H2S concentration [Citation146]. Kidney damage caused by UUO is closely related to inflammation and renal fibrosis. Previous studies have shown that H2S participates in this pathological state [Citation147]. Supplementation with exogenous H2S reduces UUO–induced kidney injury. Furthermore, recent research has shown that in a rat model of obstructive nephropathy, GYY4137 ameliorates inflammatory damage and tubulointerstitial fibrosis [Citation148,Citation149], as does NaHS [Citation150,Citation151]. We summarize the role of H2S in obstructive kidney disease in [Citation145–152].
Table 3. Previous studies on the effects of H2S in obstructive kidney disease.
5.3.1. H2S alleviates obstructive kidney disease by reducing oxidative stress and inflammation
Inflammation, OS, vascular tension and intracellular signaling pathways are involved in the progression of renal fibrosis. The induction of fibrosis is exacerbated by oxidative stress, as ROS have been reported to increase the expression of Ang II and TGF–β1 [Citation153,Citation154]. H2S can increase the activity of antioxidant enzymes such as glutathione and superoxide dismutase to resist OS. Nrf2 is a transcription factor that regulates the adaptive response to OS. H2S has been shown to induce the gene expression of antioxidant enzymes and the activation of Nrf2 [Citation107,Citation146]. Excessive autophagy induced by an increase in OS can lead to vascular endothelial dysfunction. Treatment with NaHS significantly ameliorates kidney damage in UUO mice, possibly by inhibiting the ROS–AMPK pathway [Citation152].
UUO causes inflammation in the kidneys. Studies have found that exogenous H2S inhibits NF–κB activation, leads to a reduced inflammatory response and controls the production of proinflammatory cytokines such as IL–1β and TNF–α, which are also found in H9C2 cardiac cells [Citation146,Citation155]. The characteristic signaling mechanisms of renal fibrosis involve the TGF–β1–Smad and MAPK pathways. H2S weakens Smad3 phosphorylation and blocks MAPK kinase activation. In addition, administration of NaHS reduces the expression of proliferation–related genes, such as proliferating cell nuclear antigen and c–Myc, to inhibit fibroblast proliferation and the phenotypic transition to myofibroblasts [Citation147].
5.3.2. H2S and macrophages in obstructive kidney disease
Zhou et al. [Citation145] found that in obstructed kidneys of UUO mice, reduced H2S production is related to increased macrophage infiltration. Macrophages are immune cells located in tissues that are derived from monocytes and have broad functions. Macrophages preferentially induce iNOS or arginase and are classified as M1 or M2 macrophages [Citation156]. Activated M1 macrophages cause tissue damage and inflammation by increasing the levels of proinflammatory cytokines, NO and ROS. However, M2 macrophages promote tissue repair and collagen production and inhibit immune activity. In previous studies, M1 and M2 macrophages have been shown to be involved in the pathogenesis of renal fibrosis. During renal fibrosis, the NF–κB pathway is activated, which can activate and modulate M1 macrophages. The interferon regulatory factor/signal transducer and activator of transcription (IRF/STAT, through STAT6) signaling pathway, activated by IL–4 and IL–13, can transform macrophages to the M2 phenotype [Citation157–159]. H2S can significantly reduce M1 and M2 macrophage infiltration by inhibiting NF–κB and IL–4/STAT6. In addition, NF–κB and IL–4/STAT6 pathway activation in UUO–induced mice is accompanied by activation of the NLRP3 inflammasome. H2S donors suppress NLRP3 inflammasome activation, which may contribute to the anti–inflammatory and antifibrotic effects of H2S [Citation145].
On the basis of the positive role of H2S in obstructive kidney disease, H2S may be a novel potential therapy against renal injury caused by urinary obstruction [Citation160].
6. Methodology of preclinical work
6.1. H2S donors
The most common class of H2S donors employed in biological studies is the class of sulfide salts, which includes sodium hydrosulfide (NaHS) and sodium sulfide (Na2S). However, these salts do not release H2S; rather, they dissociate to yield H2S in an instantaneous and pH–dependent manner. At physiological pH, approximately 85% of the sulfide delivered by the salts will be in the dissociated hydrosulfide form (HS−), and 15% will be the dissolved gas H2S [Citation161].
Sulfide salts can provide direct, instantaneous access to the biologically relevant forms of sulfide (H2S and HS−), in contrast to endogenous sources, which slowly and steadily produce H2S [Citation161,Citation162]. Sulfide saltshydrolyze immediately upon dissolution in water, establishing equilibrium among H2S, HS−, and S2– species. Once this equilibrium is established, H2S volatilizes rapidly, and the air oxidation of HS− is catalyzed by trace metals in water, lowering the total concentration of sulfur species [Citation163].
In the aortic rings of rats, low concentrations of NaHS (10–100 μM) can induce vasoconstriction. However, high doses of NaHS can directly relax aortic rings. It has been suggested that high concentrations of H2S may exert a rapid “knockdown” effect, perhaps because high concentrations of H2S cause transient inhibition of complex IV, resulting in inhibition of mitochondrial respiration [Citation161]. In previous studies, NaHS treatment (100 µmol kg−1 day−1) for 4 weeks reversed diabetes–induced vascular dysfunction in mouse aortas [Citation100] and ameliorated the cardiovascular changes induced by obesity [Citation164]. However, it is worth noting that the NaHS was injected intraperitoneally, which may have led to a high initial circulating concentration of H2S followed by a decline and would not have provided 24–hour “coverage” for H2S delivery in vivo [Citation161]. In addition, the hygroscopicity of sulfide salts introduced error during calculation of the exact H2S concentration. Sulfide salts also emit a pungent odor, which is a real problem when considering the use of these compounds for pharmaceutical and human therapeutic applications.
Although NaHS has been successfully used in a variety of studies, unformulated sulfide salts are not the best choices for pharmaceutical development due to their short half–life, rapid and uncontrolled release, and unpleasant smell. H2S donation can be achieved through slow–releasing H2S donors, such as GYY4137, which have been used in hundreds of studies in vitro and in vivo. Recent studies have also found many donors with regulated H2S release profiles, including oxidant–triggered donors, pH–dependent donors, esterase–activated donors, and organelle–targeted compounds [Citation161].
As the field continues to develop, we expect that H2S–related compounds will find their way into clinical trials. Garlic, which can produce the gasotransmitter H2S, has been used for T2DM patients and has been reported to significantly lower glycemia and lipid metabolism with concomitant amelioration of redox metabolism [Citation165]. H2S–releasing nonsteroidal anti–inflammatory drugs represent examples of new anti–inflammatory drugs. Coupling of either naproxen or diclofenac to an H2S–releasing moiety has been reported to enhance gastrointestinal and cardiovascular safety [Citation166]. ATB–346 has been used in clinical trials and shows milder adverse effects than other drugs [Citation167]. The novel H2S prodrug (SG1002) increases H2S levels and attenuates increases in type B natriuretic peptide in patients with heart failure and is safe and well tolerated at all doses (200, 400, and 800 mg twice daily for 7 days) [Citation168]. Novel H2S–releasing agents warrant further research in larger clinical studies. However, the benefit–risk ratio and usage should be taken into account at an individual patient level, especially during assessment of underlying conditions that may increase the risk of adverse events.
6.2. Pharmacological inhibitors of H2S
For inhibition of H2S synthesis, there are now several small molecule compounds targeting each of the three H2S–producing enzymes CBS, CSE, and 3–MST. The most commonly used inhibitors of CSE are PGG and b–cyano–L–alanine (BCA) [Citation169]. PGG and BCA are often used at very high levels (1–10 mM) in cell–based assays, suggesting that they poorly cross the cell membrane [Citation170]. Unlike that of BCA, the action of PGG is irreversible. In addition to PAG and BCA, another glycine analog and natural toxin, aminoethoxyvinylglycine, has been found to block CSE and is more potent than PAG [Citation171]. All three inhibitors suffer from the same selectivity drawbacks, as they inhibit additional pyridoxal 59–phosphate–dependent enzymes [Citation161].
Although PGG is the drug of choice to pharmacologically inhibit CSE, the limitations associated with its lack of selectivity remain. AVG has not been used in cellular or in vivo assays thus far [Citation161]. Therefore, it is necessary to investigate more approaches.
7. Conclusions
In this review, we have comprehensively discussed the roles of H2S in renal diseases, especially DN, and explored the related molecular mechanisms. DN is one of the most serious complications of DM, and numerous drug classes are available for treatment, but their efficacy remains limited. H2S can regulate autophagy, apoptosis, OS, and inflammation through multiple signaling pathways, providing new targets for treatment. However, compared to sodium–glucose cotransporter 2 inhibitors, which constitute a new class of blood glucose–lowering medications that block renal glucose reabsorption and have protective effects on the kidneys, H2S may have mild efficacy in lowering glucose [Citation172]. In addition, the role of H2S in AKI, such as sepsis–associated AKI and drug–induced AKI, is unclear. Research on such topics will advance and critically broaden our understanding of the therapeutic potential of H2S in kidney disease.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Rodrigues C, Percival SS. Immunomodulatory effects of glutathione, garlic derivatives, and hydrogen sulfide. Nutrients. 2019;11(2):295.
- Warenycia MW, Goodwin LR, Benishin CG, et al. Acute hydrogen sulfide poisoning. Demonstration of selective uptake of sulfide by the brainstem by measurement of brain sulfide levels. Biochem Pharmacol. 1989;38(6):973–981.
- Wedmann R, Bertlein S, Macinkovic I, et al. Working with "H2S": facts and apparent artifacts. Nitric Oxide. 2014;41:85–96.
- Koning AM, Frenay AR, Leuvenink HG, et al. Hydrogen sulfide in renal physiology, disease and transplantation–the smell of renal protection. Nitric Oxide. 2015;46:37–49.
- Sen N, Paul BD, Gadalla MM, et al. Hydrogen sulfide-linked sulfhydration of NF-κB mediates its antiapoptotic actions. Mol Cell. 2012;45(1):13–24.
- Feliers D, Lee HJ, Kasinath BS. Hydrogen sulfide in renal physiology and disease. Antioxid Redox Signal. 2016;25(13):720–731.
- Wen YD, Wang H, Zhu YZ. The drug developments of hydrogen sulfide on cardiovascular disease. Oxid Med Cell Longev. 2018;2018:4010395.
- Mathai JC, Missner A, Kügler P, et al. No facilitator required for membrane transport of hydrogen sulfide. Proc Natl Acad Sci USA. 2009;106(39):16633–16638.
- Yetik-Anacak G, Sevin G, Ozzayım O, et al. Hydrogen sulfide: a novel mechanism for the vascular protection by resveratrol under oxidative stress in mouse aorta. Vascul Pharmacol. 2016;87:76–82.
- Ngowi EE, Sarfraz M, Afzal A, et al. Roles of hydrogen sulfide donors in common kidney diseases. Front Pharmacol. 2020;11:564281.
- Módis K, Coletta C, Erdélyi K, et al. Intramitochondrial hydrogen sulfide production by 3-mercaptopyruvate sulfurtransferase maintains mitochondrial electron flow and supports cellular bioenergetics. Faseb J. 2013;27(2):601–611.
- Shibuya N, Koike S, Tanaka M, et al. A novel pathway for the production of hydrogen sulfide from D-cysteine in mammalian cells. Nat Commun. 2013;4:1366.
- Paul BD, Snyder SH. H2S signalling through protein sulfhydration and beyond. Nat Rev Mol Cell Biol. 2012;13(8):499–507.
- Mustafa AK, Gadalla MM, Sen N, et al. H2S signals through protein S-sulfhydration. Sci Signal. 2009;2(96):ra72.
- Mustafa AK, Sikka G, Gazi SK, et al. Hydrogen sulfide as endothelium-derived hyperpolarizing factor sulfhydrates potassium channels. Circ Res. 2011;109(11):1259–1268.
- Krishnan N, Fu C, Pappin DJ, et al. H2S-Induced sulfhydration of the phosphatase PTP1B and its role in the endoplasmic reticulum stress response. Sci Signal. 2011;4(203):ra86.
- Xie ZZ, Shi MM, Xie L, et al. Sulfhydration of p66Shc at cysteine59 mediates the antioxidant effect of hydrogen sulfide. Antioxid Redox Signal. 2014;21(18):2531–2542.
- Yang G, Zhao K, Ju Y, et al. Hydrogen sulfide protects against cellular senescence via S-sulfhydration of Keap1 and activation of Nrf2. Antioxid Redox Signal. 2013;18(15):1906–1919.
- Kamoun P. Endogenous production of hydrogen sulfide in mammals. Amino Acids. 2004;26(3):243–254.
- Zhang S, Pan C, Zhou F, et al. Hydrogen sulfide as a potential therapeutic target in fibrosis. Oxid Med Cell Longev. 2015;2015:593407.
- Kabil O, Vitvitsky V, Xie P, et al. The quantitative significance of the transsulfuration enzymes for H2S production in murine tissues. Antioxid Redox Signal. 2011;15(2):363–372.
- Sun HJ, Wu ZY, Cao L, et al. Hydrogen sulfide: recent progression and perspectives for the treatment of diabetic nephropathy. Molecules. 2019;24(15):2857.
- Xia M, Chen L, Muh RW, et al. Production and actions of hydrogen sulfide, a novel gaseous bioactive substance, in the kidneys. J Pharmacol Exp Ther. 2009;329(3):1056–1062.
- Cao X, Bian JS. The role of hydrogen sulfide in renal system. Front Pharmacol. 2016;7:385.
- Olson KR. Hydrogen sulfide as an oxygen sensor. Antioxid Redox Signal. 2015;22(5):377–397.
- Bełtowski J. Hypoxia in the renal medulla: implications for hydrogen sulfide signaling. J Pharmacol Exp Ther. 2010;334(2):358–363.
- Teng H, Wu B, Zhao K, et al. Oxygen-sensitive mitochondrial accumulation of cystathionine β-synthase mediated by Lon protease. Proc Natl Acad Sci USA. 2013;110(31):12679–12684.
- Fu M, Zhang W, Wu L, et al. Hydrogen sulfide (H2S) metabolism in mitochondria and its regulatory role in energy production. Proc Natl Acad Sci USA. 2012;109(8):2943–2948.
- Lu M, Liu YH, Goh HS, et al. Hydrogen sulfide inhibits plasma renin activity. J Am Soc Nephrol. 2010;21(6):993–1002.
- Liu YH, Lu M, Xie ZZ, et al. Hydrogen sulfide prevents heart failure development via inhibition of renin release from mast cells in isoproterenol-treated rats. Antioxid Redox Signal. 2014;20(5):759–769.
- Huang P, Chen S, Wang Y, et al. Down-regulated CBS/H2S pathway is involved in high-salt-induced hypertension in Dahl rats. Nitric Oxide. 2015;46:192–203.
- Laggner H, Hermann M, Esterbauer H, et al. The novel gaseous vasorelaxant hydrogen sulfide inhibits angiotensin-converting enzyme activity of endothelial cells. J Hypertens. 2007;25(10):2100–2104.
- Levey AS, James MT. Acute kidney injury. Ann Intern Med. 2017;167(9):ITC66–ITC80.
- Farrar A. Acute kidney injury. Nurs Clin North Am. 2018;53(4):499–510.
- Banaei S. Novel role of microRNAs in renal ischemia reperfusion injury. Ren Fail. 2015;37(7):1073–1079.
- Casey TM, Arthur PG, Bogoyevitch MA. Necrotic death without mitochondrial dysfunction-delayed death of cardiac myocytes following oxidative stress. Biochim Biophys Acta. 2007;1773(3):342–351.
- Han SJ, Kim JI, Park JW, et al. Hydrogen sulfide accelerates the recovery of kidney tubules after renal ischemia/reperfusion injury. Nephrol Dial Transplant. 2015;30(9):1497–1506.
- Bos EM, Wang R, Snijder PM, et al. Cystathionine γ-lyase protects against renal ischemia/reperfusion by modulating oxidative stress. J Am Soc Nephrol. 2013;24(5):759–770.
- Azizi F, Seifi B, Kadkhodaee M, et al. Administration of hydrogen sulfide protects ischemia reperfusion-induced acute kidney injury by reducing the oxidative stress. Ir J Med Sci. 2016;185(3):649–654.
- Verma SK, Molitoris BA. Renal endothelial injury and microvascular dysfunction in acute kidney injury. Semin Nephrol. 2015;35(1):96–107.
- Snijder PM, Frenay AR, Koning AM, et al. Sodium thiosulfate attenuates angiotensin II-induced hypertension, proteinuria and renal damage. Nitric Oxide. 2014;42:87–98.
- Bos EM, Leuvenink HG, Snijder PM, et al. Hydrogen sulfide-induced hypometabolism prevents renal ischemia/reperfusion injury. J Am Soc Nephrol. 2009;20(9):1901–1905.
- Elrod JW, Calvert JW, Morrison J, et al. Hydrogen sulfide attenuates myocardial ischemia-reperfusion injury by preservation of mitochondrial function. Proc Natl Acad Sci USA. 2007;104(39):15560–15565.
- Parsanathan R, Jain SK. Hydrogen sulfide increases glutathione biosynthesis, and glucose uptake and utilisation in C(2)C(12) mouse myotubes. Free Radic Res. 2018;52(2):288–303.
- Ahmad A, Olah G, Szczesny B, et al. AP39, a mitochondrially targeted hydrogen sulfide donor, exerts protective effects in renal epithelial cells subjected to oxidative stress in vitro and in acute renal injury in vivo. Shock. 2016;45(1):88–97.
- Perazella MA. Drug-induced acute kidney injury: diverse mechanisms of tubular injury. Curr Opin Crit Care. 2019;25(6):550–557.
- Holditch SJ, Brown CN, Lombardi AM, et al. Recent advances in models, mechanisms, biomarkers, and interventions in Cisplatin-Induced acute kidney injury. Int J Mol Sci. 2019;20(12):3011.
- Manohar S, Leung N. Cisplatin nephrotoxicity: a review of the literature. J Nephrol. 2018;31(1):15–25.
- Ciarimboli G. Membrane transporters as mediators of cisplatin side-effects. Anticancer Res. 2014;34(1):547–550.
- Tsushida K, Tanabe K, Masuda K, et al. Estrogen-related receptor α is essential for maintaining mitochondrial integrity in cisplatin-induced acute kidney injury. Biochem Biophys Res Commun. 2018;498(4):918–924.
- Bajwa A, Rosin DL, Chroscicki P, et al. Sphingosine 1-phosphate receptor-1 enhances mitochondrial function and reduces cisplatin-induced tubule injury. J Am Soc Nephrol. 2015;26(4):908–925.
- Liu Z, Li H, Su J, et al. Numb depletion promotes Drp1-Mediated mitochondrial fission and exacerbates mitochondrial fragmentation and dysfunction in acute kidney injury. Antioxid Redox Signal. 2019;30(15):1797–1816.
- Yan L, Jiaqiong L, Yue G, et al. Atorvastatin protects against contrast-induced acute kidney injury via upregulation of endogenous hydrogen sulfide. Ren Fail. 2020;42(1):270–281.
- Yuan Y, Zhu L, Li L, et al. S-Sulfhydration of SIRT3 by hydrogen sulfide attenuates mitochondrial dysfunction in Cisplatin-induced acute kidney injury. Antioxid Redox Signal. 2019;31(17):1302–1319.
- Wang T, Cao Y, Zheng Q, et al. SENP1-Sirt3 signaling controls mitochondrial protein acetylation and metabolism. Mol Cell. 2019;75(4):823–834 e5.
- Gerő D, Torregrossa R, Perry A, et al. The novel mitochondria-targeted hydrogen sulfide (H(2)S) donors AP123 and AP39 protect against hyperglycemic injury in microvascular endothelial cells in vitro. Pharmacol Res. 2016;113(Pt A):186–198.
- Ahangarpour A, Abdollahzade Fard A, Gharibnaseri MK, et al. Hydrogen sulfide ameliorates the kidney dysfunction and damage in cisplatin-induced nephrotoxicity in rat. Vet Res Forum. 2014;5(2):121–127.
- Della Coletta Francescato H, Cunha FQ, Costa RS, et al. Inhibition of hydrogen sulphide formation reduces cisplatin-induced renal damage. Nephrol Dial Transplant. 2011;26(2):479–488.
- Liu M, Jia Z, Sun Y, et al. A H 2 S donor GYY4137 exacerbates Cisplatin-Induced nephrotoxicity in mice. Mediators Inflamm. 2016;2016:8145785.
- Francescato HD, Marin EC, Cunha Fde Q, et al. Role of endogenous hydrogen sulfide on renal damage induced by adriamycin injection. Arch Toxicol. 2011;85(12):1597–1606.
- Hoste EA, Bagshaw SM, Bellomo R, et al. Epidemiology of acute kidney injury in critically ill patients: the multinational AKI-EPI study. Intensive Care Med. 2015;41(8):1411–1423.
- Shum HP, Yan WW, Chan TM. Recent knowledge on the pathophysiology of septic acute kidney injury: a narrative review. J Crit Care. 2016;31(1):82–89.
- Chen X, Xu W, Wang Y, et al. Hydrogen sulfide reduces kidney injury due to urinary-derived sepsis by inhibiting NF-κB expression, decreasing TNF-α levels and increasing IL-10 levels. Exp Ther Med. 2014;8(2):464–470.
- Bourque C, Zhang Y, Fu M, et al. H(2)S protects lipopolysaccharide-induced inflammation by blocking NFκB transactivation in endothelial cells. Toxicol Appl Pharmacol. 2018;338:20–29.
- Li L, Bhatia M, Zhu YZ, et al. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 2005;19(9):1196–1198.
- Cao X, Xiong S, Zhou Y, et al. Renal protective effect of hydrogen sulfide in cisplatin-induced nephrotoxicity. Antioxid Redox Signal. 2018;29(5):455–470.
- Sun HJ, Leng B, Wu ZY, et al. Polysulfide and hydrogen sulfide ameliorate Cisplatin-Induced nephrotoxicity and renal inflammation through persulfidating STAT3 and IKKβ. Int J Mol Sci. 2020;21(20):7805.
- Chen Y, Jin S, Teng X, et al. Hydrogen sulfide attenuates LPS-Induced acute kidney injury by inhibiting inflammation and oxidative stress. Oxid Med Cell Longev. 2018;2018:6717212.
- Li T, Zhao J, Miao S, et al. Protective effect of H(2)S on LPS‑induced AKI by promoting autophagy. Mol Med Rep. 2022;25(3)
- Akchurin OM, Kaskel F. Update on inflammation in chronic kidney disease. Blood Purif. 2015;39(1–3):84–92.
- Girndt M. Diagnosis and treatment of chronic kidney disease. Internist. 2017;58(3):243–256.
- Padmanabhan A, Gohil S, Gadgil NM, et al. Chronic renal failure: an autopsy study. Saudi J Kidney Dis Transpl. 2017;28(3):545–551.
- Lian Y, Xie L, Chen M, et al. Effects of an astragalus polysaccharide and rhein combination on apoptosis in rats with chronic renal failure. Evid Based Complement Alternat Med. 2014;2014:271862.
- Shirazi MK, Azarnezhad A, Abazari MF, et al. The role of nitric oxide signaling in renoprotective effects of hydrogen sulfide against chronic kidney disease in rats: involvement of oxidative stress, autophagy and apoptosis. J Cell Physiol. 2019;234(7):11411–11423.
- Perna AF, Lanza D, Sepe I, et al. Vasodilatation caused by endogenous hydrogen sulfide in chronic renal failure. G Ital Nefrol. 2013;30(2):gin/30.2.2.
- Aminzadeh MA, Vaziri ND. Downregulation of the renal and hepatic hydrogen sulfide (H2S)-producing enzymes and capacity in chronic kidney disease. Nephrol Dial Transplant. 2012;27(2):498–504.
- Askari H, Seifi B, Kadkhodaee M, et al. Protective effects of hydrogen sulfide on chronic kidney disease by reducing oxidative stress, inflammation and apoptosis. Excli J. 2018;17:14–23.
- Wu D, Si W, Wang M, et al. Hydrogen sulfide in cancer: friend or foe? Nitric Oxide. 2015;50:38–45.
- Circu ML, Aw TY. Reactive oxygen species, cellular redox systems, and apoptosis. Free Radic Biol Med. 2010;48(6):749–762.
- Tu Y, Sun W, Wan YG, et al. Dahuang Fuzi decoction ameliorates tubular epithelial apoptosis and renal damage via inhibiting TGF-β1-JNK signaling pathway activation in vivo. J Ethnopharmacol. 2014;156:115–124.
- Ford A, Al-Magableh M, Gaspari TA, et al. Chronic NaHS treatment is vasoprotective in High-Fat-Fed ApoE(–/–) mice. Int J Vasc Med. 2013;2013:915983.
- Wu D, Luo N, Wang L, et al. Hydrogen sulfide ameliorates chronic renal failure in rats by inhibiting apoptosis and inflammation through ROS/MAPK and NF-κB signaling pathways. Sci Rep. 2017;7(1):455.
- Migdalis I, Leslie D, Mavrogiannaki A, et al. Diabetes mellitus 2014. Int J Endocrinol. 2015;2015:845759.
- Stenvinkel P. Chronic kidney disease: a public health priority and harbinger of premature cardiovascular disease. J Intern Med. 2010;268(5):456–467.
- Stanton RC. Oxidative stress and diabetic kidney disease. Curr Diab Rep. 2011;11(4):330–336.
- Tesch GH, Lim AK. Recent insights into diabetic renal injury from the db/db mouse model of type 2 diabetic nephropathy. Am J Physiol Renal Physiol. 2011;300(2):F301–10.
- Lim AK, Tesch GH. Inflammation in diabetic nephropathy. Mediators Inflamm. 2012;2012:146154.
- Kamiyama M, Zsombok A, Kobori H. Urinary angiotensinogen as a novel early biomarker of intrarenal renin-angiotensin system activation in experimental type 1 diabetes. J Pharmacol Sci. 2012;119(4):314–323.
- Jain SK, Bull R, Rains JL, et al. Low levels of hydrogen sulfide in the blood of diabetes patients and streptozotocin-treated rats causes vascular inflammation? Antioxid Redox Signal. 2010;12(11):1333–1337.
- van den Born JC, Frenay AR, Bakker SJ, et al. High urinary sulfate concentration is associated with reduced risk of renal disease progression in type 2 diabetes. Nitric Oxide. 2016;55-56:18–24.
- Li H, Feng SJ, Zhang GZ, et al. Correlation of lower concentrations of hydrogen sulfide with atherosclerosis in chronic hemodialysis patients with diabetic nephropathy. Blood Purif. 2014;38(3–4):188–194.
- Xue R, Hao DD, Sun JP, et al. Hydrogen sulfide treatment promotes glucose uptake by increasing insulin receptor sensitivity and ameliorates kidney lesions in type 2 diabetes. Antioxid Redox Signal. 2013;19(1):5–23.
- Sun HJ, Xiong SP, Cao X, et al. Polysulfide-mediated sulfhydration of SIRT1 prevents diabetic nephropathy by suppressing phosphorylation and acetylation of p65 NF-κB and STAT3. Redox Biol. 2021;38:101813.
- Yu Y, Xiao L, Ren Z, et al. Glucose-induced decrease of cystathionine β-synthase mediates renal injuries. Faseb J. 2021;35(5):e21576.
- Hussain Lodhi A, Ahmad FU, Furwa K, et al. Role of oxidative stress and reduced endogenous hydrogen sulfide in diabetic nephropathy. Drug Des Devel Ther. 2021;15:1031–1043.
- Juin SK, Pushpakumar S, Sen U. GYY4137 regulates extracellular matrix turnover in the diabetic kidney by modulating retinoid X receptor signaling. Biomolecules. 2021;11(10):1477.
- Elbassuoni EA, Аziz NM, Habeeb WN. The role of activation of K(АTP) channels on hydrogen sulfide induced renoprotective effect on diabetic nephropathy. J Cell Physiol. 2020;235(6):5223–5228.
- Qian X, Li X, Ma F, et al. Novel hydrogen sulfide-releasing compound, S-propargyl-cysteine, prevents STZ-induced diabetic nephropathy. Biochem Biophys Res Commun. 2016;473(4):931–938.
- Ahmed HH, Taha FM, Omar HS, et al. Hydrogen sulfide modulates SIRT1 and suppresses oxidative stress in diabetic nephropathy. Mol Cell Biochem. 2019;457(1–2):1–9.
- Li Y, Li L, Zeng O, et al. H(2)S improves renal fibrosis in STZ-induced diabetic rats by ameliorating TGF-β1 expression. Ren Fail. 2017;39(1):265–272.
- Kumawat M, Sharma TK, Singh I, et al. Antioxidant enzymes and lipid peroxidation in type 2 diabetes mellitus patients with and without nephropathy. N Am J Med Sci. 2013;5(3):213–219.
- Mima A. Inflammation and oxidative stress in diabetic nephropathy: new insights on its inhibition as new therapeutic targets. J Diabetes Res. 2013;2013:248563.
- Eid AA, Gorin Y, Fagg BM, et al. Mechanisms of podocyte injury in diabetes: role of cytochrome P450 and NADPH oxidases. Diabetes. 2009;58(5):1201–1211.
- Zhang MH, Feng L, Zhu MM, et al. Antioxidative and anti-inflammatory activities of paeoniflorin and oxypaeoniflora on AGEs-induced mesangial cell damage. Planta Med. 2013;79(14):1319–1323.
- Safar MM, Abdelsalam RM. H2S donors attenuate diabetic nephropathy in rats: modulation of oxidant status and polyol pathway. Pharmacol Rep. 2015;67(1):17–23.
- Yang R, Liu XF, Ma SF, et al. Protective effect of hydrogen sulfide on kidneys of type 1 diabetic rats. Zhongguo Ying Yong Sheng Li Xue Za Zhi. 2016;32(2):181–184.
- Wei Y, Gong J, Thimmulappa RK, et al. Nrf2 acts cell-autonomously in endothelium to regulate tip cell formation and vascular branching. Proc Natl Acad Sci USA. 2013;110(41):E3910–8.
- Zheng H, Whitman SA, Wu W, et al. Therapeutic potential of Nrf2 activators in streptozotocin-induced diabetic nephropathy. Diabetes. 2011;60(11):3055–3066.
- Xie L, Gu Y, Wen M, et al. Hydrogen sulfide induces Keap1 S-sulfhydration and suppresses diabetes-accelerated atherosclerosis via Nrf2 activation. Diabetes. 2016;65(10):3171–3184.
- Zhou X, Feng Y, Zhan Z, et al. Hydrogen sulfide alleviates diabetic nephropathy in a streptozotocin-induced diabetic rat model. J Biol Chem. 2014;289(42):28827–28834.
- Lee HJ, Lee DY, Mariappan MM, et al. Hydrogen sulfide inhibits high glucose-induced NADPH oxidase 4 expression and matrix increase by recruiting inducible nitric oxide synthase in kidney proximal tubular epithelial cells. J Biol Chem. 2017;292(14):5665–5675.
- Gorin Y, Block K. Nox4 and diabetic nephropathy: with a friend like this, who needs enemies? Free Radic Biol Med. 2013;61:130–142.
- Lee MJ, Feliers D, Mariappan MM, et al. A role for AMP-activated protein kinase in diabetes-induced renal hypertrophy. Am J Physiol Renal Physiol. 2007;292(2):F617–27.
- Lee HJ, Mariappan MM, Feliers D, et al. Hydrogen sulfide inhibits high glucose-induced matrix protein synthesis by activating AMP-activated protein kinase in renal epithelial cells. J Biol Chem. 2012;287(7):4451–4461.
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell. 2008;132(1):27–42.
- Li L, Tan J, Miao Y, et al. ROS and autophagy: interactions and molecular regulatory mechanisms. Cell Mol Neurobiol. 2015;35(5):615–621.
- Liu J, Wu J, Sun A, et al. Hydrogen sulfide decreases high glucose/palmitate-induced autophagy in endothelial cells by the Nrf2-ROS-AMPK signaling pathway. Cell Biosci. 2016;6(1):33.
- Fernández-Sánchez A, Madrigal-Santillán E, Bautista M, et al. Inflammation, oxidative stress, and obesity. Int J Mol Sci. 2011;12(5):3117–3132.
- Wada J, Makino H. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci (Lond). 2013;124(3):139–152.
- Wang W, Sun W, Cheng Y, et al. Role of sirtuin-1 in diabetic nephropathy. J Mol Med. 2019;97(3):291–309.
- Naduk-Kik J, Hrabec E. The role of matrix metalloproteinases in the pathogenesis of diabetes mellitus and progression of diabetes retinopathy. Postepy Hig Med Dosw. 2008;62:442–450.
- Kundu S, Pushpakumar SB, Tyagi A, et al. Hydrogen sulfide deficiency and diabetic renal remodeling: role of matrix metalloproteinase-9. Am J Physiol Endocrinol Metab. 2013;304(12):E1365–78.
- Patel S, Rauf A, Khan H, et al. Renin-angiotensin-aldosterone (RAAS): the ubiquitous system for homeostasis and pathologies. Biomed Pharmacother. 2017;94:317–325.
- Thomas MC, Brownlee M, Susztak K, et al. Diabetic kidney disease. Nat Rev Dis Primers. 2015;1:15018.
- Bermejo S, García CO, Rodríguez E, et al. The renin-angiotensin-aldosterone system blockade in patients with advanced diabetic kidney disease. Nefrologia. 2018;38(2):197–206.
- Li Z, Organ CL, Kang J, et al. Hydrogen sulfide attenuates renin angiotensin and aldosterone pathological signaling to preserve kidney function and improve exercise tolerance in heart failure. JACC Basic Transl Sci. 2018;3(6):796–809.
- Xue H, Yuan P, Ni J, et al. H(2)S inhibits hyperglycemia-induced intrarenal renin-angiotensin system activation via attenuation of reactive oxygen species generation. PLoS One. 2013;8(9):e74366.
- Hart PD, Bakris GL. Hypertensive nephropathy: prevention and treatment recommendations. Expert Opin Pharmacother. 2010;11(16):2675–2686.
- Udani S, Lazich I, Bakris GL. Epidemiology of hypertensive kidney disease. Nat Rev Nephrol. 2011;7(1):11–21.
- Seccia TM, Caroccia B, Calò LA. Hypertensive nephropathy. Moving from classic to emerging pathogenetic mechanisms. J Hypertens. 2017;35(2):205–212.
- Bidani AK, Griffin KA. Pathophysiology of hypertensive renal damage: implications for therapy. Hypertension. 2004;44(5):595–601.
- Yang G, Wu L, Jiang B, et al. H2S as a physiologic vasorelaxant: hypertension in mice with deletion of cystathionine gamma-lyase. Science. 2008;322(5901):587–590.
- Roy A, Khan AH, Islam MT, et al. Interdependency of cystathione γ-lyase and cystathione β-synthase in hydrogen sulfide-induced blood pressure regulation in rats. Am J Hypertens. 2012;25(1):74–81.
- Zhao W, Wang R. H(2)S-induced vasorelaxation and underlying cellular and molecular mechanisms. Am J Physiol Heart Circ Physiol. 2002;283(2):H474–80.
- Gross B, Pawlak M, Lefebvre P, et al. PPARs in obesity-induced T2DM, dyslipidaemia and NAFLD. Nat Rev Endocrinol. 2017;13(1):36–49.
- Toral M, Romero M, Pérez-Vizcaíno F, et al. Antihypertensive effects of peroxisome proliferator-activated receptor-β/δ activation. Am J Physiol Heart Circ Physiol. 2017;312(2):H189–H200.
- Predmore BL, Julian D, Cardounel AJ. Hydrogen sulfide increases nitric oxide production from endothelial cells by an akt-dependent mechanism. Front Physiol. 2011;2:104.
- Xiao L, Dong JH, Teng X, et al. Hydrogen sulfide improves endothelial dysfunction in hypertension by activating peroxisome proliferator-activated receptor Delta/endothelial nitric oxide synthase signaling. J Hypertens. 2018;36(3):651–665.
- Ji W, Liu S, Dai J, et al. Hydrogen sulfide defends against the cardiovascular risk of Nw-nitro-L-argininemethyl ester-induced hypertension in rats via the nitric oxide/endothelial nitric oxide synthase pathway. Chin Med J. 2014;127(21):3751–3757.
- Ali MY, Ping CY, Mok YY, et al. Regulation of vascular nitric oxide in vitro and in vivo; a new role for endogenous hydrogen sulphide? Br J Pharmacol. 2006;149(6):625–634.
- Ruiz-Ortega M, Esteban V, Rupérez M, et al. Renal and vascular hypertension-induced inflammation: role of angiotensin II. Curr Opin Nephrol Hypertens. 2006;15(2):159–166.
- Liao TD, Yang XP, Liu YH, et al. Role of inflammation in the development of renal damage and dysfunction in angiotensin II-induced hypertension. Hypertension. 2008;52(2):256–263.
- Al-Magableh MR, Kemp-Harper BK, Hart JL. Hydrogen sulfide treatment reduces blood pressure and oxidative stress in angiotensin II-induced hypertensive mice. Hypertens Res. 2015;38(1):13–20.
- Stevens S. Obstructive kidney disease. Nurs Clin North Am. 2018;53(4):569–578.
- Zhou Y, Zhu X, Wang X, et al. H(2)S alleviates renal injury and fibrosis in response to unilateral ureteral obstruction by regulating macrophage infiltration via inhibition of NLRP3 signaling. Exp Cell Res. 2020;387(1):111779.
- Jung KJ, Jang HS, Kim JI, et al. Involvement of hydrogen sulfide and homocysteine transsulfuration pathway in the progression of kidney fibrosis after ureteral obstruction. Biochim Biophys Acta. 2013;1832(12):1989–1997.
- Song K, Wang F, Li Q, et al. Hydrogen sulfide inhibits the renal fibrosis of obstructive nephropathy. Kidney Int. 2014;85(6):1318–1329.
- Lin S, Visram F, Liu W, et al. GYY4137, a slow-releasing hydrogen sulfide donor, ameliorates renal damage associated with chronic obstructive uropathy. J Urol. 2016;196(6):1778–1787.
- Lin S, Lian D, Liu W, et al. Daily therapy with a slow-releasing H(2)S donor GYY4137 enables early functional recovery and ameliorates renal injury associated with urinary obstruction. Nitric Oxide. 2018;76:16–28.
- Jiang D, Zhang Y, Yang M, et al. Exogenous hydrogen sulfide prevents kidney damage following unilateral ureteral obstruction. Neurourol Urodyn. 2014;33(5):538–543.
- Dursun M, Otunctemur A, Ozbek E, et al. Protective effect of hydrogen sulfide on renal injury in the experimental unilateral ureteral obstruction. Int Braz j Urol. 2015;41(6):1185–1193.
- Chen Q, Yu S, Zhang K, et al. Exogenous H2S inhibits autophagy in unilateral ureteral obstruction mouse renal tubule cells by regulating the ROS-AMPK signaling pathway. Cell Physiol Biochem. 2018;49(6):2200–2213.
- Song K, Li Q, Yin X-Y, et al. Hydrogen sulfide: a therapeutic candidate for fibrotic disease? Oxid Med Cell Longev. 2015;2015:458720.
- Duni A, Liakopoulos V, Roumeliotis S, et al. Oxidative stress in the pathogenesis and evolution of chronic kidney disease: untangling Ariadne’s thread. Int J Mol Sci. 2019;20(15):3711.
- Xu W, Chen J, Lin J, et al. Exogenous H2S protects H9c2 cardiac cells against high glucose-induced injury and inflammation by inhibiting the activation of the NF-κB and IL-1β pathways. Int J Mol Med. 2015;35(1):177–186.
- Orecchioni M, Ghosheh Y, Pramod AB, et al. Macrophage polarization: different gene signatures in M1(LPS+) vs. Classically and M2(LPS-) vs. Front Immunol. 2019;10:1084.
- Wynn TA, Vannella KM. Macrophages in tissue repair, regeneration, and fibrosis. Immunity. 2016;44(3):450–462.
- Chakarov S, Lim HY, Tan L, et al. Two distinct interstitial macrophage populations coexist across tissues in specific subtissular niches. Science. 2019;363(6432):aau0964.
- Martinez FO, Helming L, Gordon S. Alternative activation of macrophages: an immunologic functional perspective. Annu Rev Immunol. 2009;27:451–483.
- Lin S, Juriasingani S, Sener A. Is hydrogen sulfide a potential novel therapy to prevent renal damage during ureteral obstruction? Nitric Oxide. 2018;73:15–21.
- Szabo C, Papapetropoulos A. International union of basic and clinical pharmacology. CII: pharmacological modulation of H(2)S levels: H(2)S donors and H(2)S biosynthesis inhibitors. Pharmacol Rev. 2017;69(4):497–564.
- Powell CR, Dillon KM, Matson JB. A review of hydrogen sulfide (H(2)S) donors: chemistry and potential therapeutic applications. Biochem Pharmacol. 2018;149:110–123.
- Hughes MN, Centelles MN, Moore KP. Making and working with hydrogen sulfide: the chemistry and generation of hydrogen sulfide in vitro and its measurement in vivo: a review. Free Radic Biol Med. 2009;47(10):1346–1353.
- Gomez CB, de la Cruz SH, Medina-Terol GJ, et al. Chronic administration of NaHS and L-cysteine restores cardiovascular changes induced by high-fat diet in rats. Eur J Pharmacol. 2019;863:172707.
- Melino S, Leo S, Toska Papajani V. Natural hydrogen sulfide donors from allium sp. as a nutraceutical approach in type 2 diabetes prevention and therapy. Nutrients. 2019;11(7):1581.
- Fiorucci S, Distrutti E. COXIBs, CINODs and H2S-releasing NSAIDs: current perspectives in the development of safer non steroidal anti-inflammatory drugs. Curr Med Chem. 2011;18(23):3494–3505.
- Wallace JL, Caliendo G, Santagada V, et al. Markedly reduced toxicity of a hydrogen sulphide-releasing derivative of naproxen (ATB-346). Br J Pharmacol. 2010;159(6):1236–1246.
- Polhemus DJ, Li Z, Pattillo CB, et al. A novel hydrogen sulfide prodrug, SG1002, promotes hydrogen sulfide and nitric oxide bioavailability in heart failure patients. Cardiovasc Ther. 2015;33(4):216–226.
- Wang Y, Qu R, Hu S, et al. Upregulation of cystathionine β-synthetase expression contributes to visceral hyperalgesia induced by heterotypic intermittent stress in rats. PLoS One. 2012;7(12):e53165.
- Krause NC, Kutsche HS, Santangelo F, et al. Hydrogen sulfide contributes to hypoxic inhibition of airway transepithelial sodium absorption. Am J Physiol Regul Integr Comp Physiol. 2016;311(3):R607–17.
- Asimakopoulou A, Panopoulos P, Chasapis CT, et al. Selectivity of commonly used pharmacological inhibitors for cystathionine β synthase (CBS) and cystathionine γ lyase (CSE). Br J Pharmacol. 2013;169(4):922–932.
- Nespoux J, Vallon V. SGLT2 inhibition and kidney protection. Clin Sci. 2018;132(12):1329–1339.