
Abstract
Introduction
Renal involvement of primary biliary cholangitis (PBC) usually presents as distal renal tubular acidosis. Proximal tubular (PT) dysfunctions in PBC were rarely reported with unclear clinicopathological characteristics and renal prognosis.
Methods
We identified 11 cases of PBC with PT dysfunctions (PBC-PT). Their medical document, kidney pathology, and follow-up data were retrospectively reviewed and analyzed.
Results
The 11 PBC-PT patients were mainly middle-aged (57.8 ± 5.2 years) females (81.8%). Most of them were asymptomatic PBC (7, 63.6%) with a high prevalence of elevated serum immunoglobulin M (IgM, 81.8%) and G (IgG, 54.5%) levels. In the kidney, they had a mean estimated glomerular filtration rate (eGFR) level of 46.54 ± 23.03 ml/min/1.73m2, and 81.8% of them had eGFR below 60 ml/min/1.73m2. They showed different degrees of PT dysfunctions, including hyperuricosuria, hypouricemia, normoglycemic glycosuria, generalized aminoaciduria, hyperphosphaturia, and hypophosphatemia. Their kidney pathology showed tubulointerstitial nephritis with lymphoplasmacytic infiltrates, brush border defects, and proximal tubulitis. After glucocorticoids treatment, the PT dysfunctions manifesting as hypophosphatemia, hypouricemia, and renal glycosuria all recovered, and the eGFR levels were improved from 43.24 ± 19.60 ml/min/1.73m2 to 55.02 ± 21.14 ml/min/1.73m2 (p = 0.028), accompanied by significant improvements of serum IgM levels (from 5.97 ± 4.55 g/L to 2.09 ± 1.48 g/L, p = 0.019).
Conclusions
The PT dysfunctions were rare in PBC patients, and glucocorticoids treatment could benefit the improvements of eGFR and tubular functions.
1. Introduction
As a women’s susceptibility disease, primary biliary cholangitis (PBC) is an autoimmune disease characterized by elevations of alkaline phosphatase (ALP) or γ-glutamyltransferase (GGT), with a high prevalence of anti-mitochondrial antibodies (AMAs) [Citation1–3]. Being a chronic non-suppurative small bile duct cholangitis, more than half of the PBC patients coexist with other autoimmune diseases, such as primary Sjӧgren’s syndrome (pSS) [Citation4,Citation5]. Kidney involvements in PBC are mainly presented as tubulointerstitial nephritis (TIN) with the most clinical manifestation of distal renal tubular acidosis (RTA) [Citation6,Citation7] and infrequently presented as glomerular diseases such as membranous nephropathy and microscopic polyangiitis [Citation8,Citation9]. Proximal tubular (PT) dysfunctions, namely renal Fanconi syndrome (FS), have also been rarely reported in PBC, and they are characterized as some or all of the following clinical features: generalized aminoaciduria, renal glycosuria, hyperphosphaturia, hyperuricosuria, proximal RTA, and low molecular weight proteinuria. The mechanisms and renal prognosis of PT dysfunctions in PBC (PBC-PT) remain unclear. In this retrospective study, we reported a single-center cohort of 11 PBC-PT patients, focused on their clinicopathologic features and treatment responses.
2. Methods
2.1. Patients
We retrospectively reviewed the clinical and pathological data of 11 PBC patients with PT dysfunctions, who were admitted to Peking Union Medical College Hospital between February 2003 and July 2021. The diagnosis of PBC [Citation2, Citation3], required two of the three following criteria: (i) biochemical evidence of chronic cholestasis with an elevation of serum ALP (>100 U/L) or GGT (> 45 U/L) levels; (ii) presence of AMAs, or other PBC-specific autoantibodies including anti-sp100 or anti-gp210 antibodies if AMAs were negative; (iii) liver biopsy demonstrating nonsuppurative cholangitis and destruction of interlobular bile ducts. For the accompanying autoimmune diseases, the diagnosis of pSS was made according to the revised version of the American-European Consensus Group [Citation10]. The diagnosis of PT dysfunctions required at least one of the following manifestations [Citation11–13]: (i) generalized aminoaciduria; (ii) Normoglycemic glycosuria, (iii) hyperphosphaturia with or without hypophosphatemia; (iv) hyperuricosuria, or hypouricemia in condition of unavailable urine uric acid level which should exclude other factors leading to hypouricemia such as drugs, malnutrition, intractable diarrhea, malignant tumor, Wilson’s disease, syndrome of inappropriate secretion of antidiuretic hormone, and pregnancy [Citation14]; and (v) proximal RTA. The exclusion criteria were tubular dysfunctions induced by drugs or toxins, multiple myeloma, light chain proteinuria, and amyloidosis [Citation12,Citation13]. For patients with more than three kinds of the above PT dysfunctions or with a coexistence of two PT dysfunctions and PT lesions in kidney pathology, the diagnosis of renal FS was made [Citation11–13].
2.2. Data collection
We systemically reviewed the patients’ medical documents and collected their demographic data, clinical symptoms such as jaundice and pruritus, history of drug usage (especially drugs which could lead to tubular injuries like non-steroidal anti-inflammatory drugs, antiviral drugs, herbs, and proton pump inhibitors), physical examinations, laboratory tests, therapy regimens, and follow-up information. Laboratory tests included routine tests, renal tubular function evaluations and immunological assays. The routine tests were complete blood count, urinalysis, 24-h urine protein, liver and renal function, serum protein electrophoresis, serum and urine immunofixation electrophoresis. The renal tubular function evaluations were arterial blood gas analysis, blood and 24-h urine electrolytes, and urine amino acid detection. The immunological assays were serum immunoglobulins (IgG, IgA, IgM), complements, rheumatoid factor, antinuclear antibodies, and AMAs. Renal pathology was reevaluated by two experienced renal pathologists independently.
2.3. Evaluations of renal tubular functions
Hyperuricosuria was defined by one of the following standards: (i) increased urinary fractional excretion of uric acid (FEUA = [urine uric acid × plasma creatinine]/[plasma uric acid × urine creatinine] × 100%, > 15%) [Citation12]; (ii) increased 24-h uric acid excretion (≥ 750 mg/d for women, ≥800 mg/d for men) in the condition of hypouricemia [Citation15]. Hyperphosphaturia and RTA diagnosis were made according to the criteria we concluded in our previous work as follows [Citation13]. Hyperphosphaturia was defined by one of the three following standards: (i) increased urinary fractional excretion of phosphate (FEPO4 = [urine phosphate × plasma creatinine]/[plasma phosphate × urine creatinine] × 100%, > 20%) [Citation12]; (ii) increased 24 h phosphorus excretion (> 100 mg/d) in the condition of hypophosphatemia [Citation15]; (iii) decreased renal tubular reabsorption of phosphate (TmP/GFR < 0.77 mmol/L) [Citation11,Citation16]. Hyperchloremic metabolic acidosis was diagnosed when serum pH was < 7.35 and serum bicarbonate levels were < 24 mmol/L with normal anion gap. If a patient with hyperchloremic metabolic acidosis simultaneously had urine pH > 5.5, positive urine anion gap and hypokalemia, distal RTA was considered. The RTA type was further validated by sodium bicarbonate infusion test: sodium bicarbonate infusion was given till the serum bicarbonate reached 24 mmol/L and fractional excretion of bicarbonate (FEHCO3) was measured by the formula [urine bicarbonate × plasma creatinine]/[plasma bicarbonate × urine creatinine] × 100%. When FEHCO3 was > 15%, proximal RTA was diagnosed. When FEHCO3 was < 5%, distal RTA was confirmed [Citation15,Citation17,Citation18].
2.4. Statistical analysis
Continuous and categorical variables were shown as mean ± standard deviation and percentages, respectively. We evaluated the improvements of renal function and serum IgM levels after treatment by paired t-tests. All the statistical analyses were finished by the Statistical Package for the Social Sciences (SPSS, version 20.0, SPSS Inc., Chicago, IL, USA). p < 0.05 was defined as statistically significant.
3. Results
3.1. Demographic features
From February 2003 to July 2021, 11 patients were diagnosed as PBC with PT dysfunctions in all the 935 PBC inpatients in Peking Union Medical College Hospital. None of them had received any suspicious drugs leading to tubular injuries, which helped us rule out the possibility of drug-induced tubular dysfunctions. Among the 11 PBC-PT patients, 9 (81.8%) were female corresponding to a female: male ratio of 4.5:1. Their mean age at diagnosis was 57.8 ± 5.2 years, and the median disease duration was 36 months (range 2–163 months) (). Among them, seven cases (63.6%) had the diagnosis of PT dysfunctions before the diagnosis of PBC with a median interval of 38 ± 30 months (range 3–84 months). Furthermore, two of them had liver dysfunctions lagged far behind PT dysfunction onset, namely nephropathy-onset PBC, as elevations of serum ALP or GGT levels emerged until 40 and 60 months after PT dysfunction onset, respectively. None of our PBC-PT patients had a family history of PBC, other autoimmune diseases, or renal tubular dysfunctions.
Table 1. Summary of the clinical characteristics of the 11 PBC-PT patients.
3.2. Hepatic and systemic manifestations
The major hepatic manifestations in our PBC-PT patients were elevations of serum GGT (90.9%), ALP (81.8%), aspartate aminotransferase (AST) (72.7%), alanine aminotransferase (ALT) (63.6%), and total bilirubin levels (45.5%), with fewer symptoms of jaundice (18.2%) and pruritus (9.1%). According to the natural course of PBC, seven patients were in the asymptomatic stage, one in symptomatic stage and three patients in the decompensated stage with portal hypertension. Only one patient (patient 6) received liver biopsy and showed typical features of PBC as chronic non-suppurative cholangitis with a prominent reduction in biliary ducts and periportal inflammatory infiltrations. It also showed fibrous septa and scattered nodules complying with the histological criterion of stage 3-4 in PBC [Citation19]. Other organ involvements mainly included interstitial lung disease (21.9%) and cytopenia of autoimmune origin (9.4%). The most common immunological abnormalities were positive anti-mitochondrial antibody M2 subtype (AMA-M2) (100.0%), anti-nuclear antibody (100%), rheumatoid factor (85.7%), and elevated serum IgM (81.8%) and IgG (54.5%) levels. The accompanying pSS happened in 63.6% of our patients ( and ).
Table 2. Detailed clinicopathological features and renal prognosis of the 11 PBC-PT patients.
3.3. Renal and tubular manifestations
The median serum creatinine (Scr) level of our PBC-PT patients was 118 μmol/L (range 66 ∼ 396 μmol/L) with a mean estimated glomerular filtration rate (eGFR) of 46.54 ± 23.03 mL/min/1.73m2. Most of them (81.8%) had eGFR below 60 mL/min/1.73m2. Proteinuria (> 0.5 g/d) was detected in 45.5% of them, with a median urine protein level of 0.50 g/d (range 0.10–1.81 g/d). They showed different degrees of PT dysfunctions, including hypouricemia (seven cases, 63.6%), normoglycemic glycosuria (seven cases, 63.6%), generalized aminoaciduria [five (62.5%) of the eight cases who had urine amino acid test], and hypophosphatemia (six cases, 54.5%). Only five patients had evaluations of urine phosphorus and uric acid excretion levels, and 3 (60.0%) and 4 (80.0%) of them were confirmed to have hyperphosphaturia and hyperuricosuria, respectively. The PBC-PT patients also presented with some other tubular dysfunctions including renal potassium loss with hypokalemia (10 cases, 90.9%), RTA (eight cases, 72.7%), and renal-origin metabolic alkalosis (2 cases, 18.2%). All eight patients with RTA fit the characteristics of distal RTA with urine pH > 5.5, positive urine anion gap and hypokalemia. None of them carried out sodium bicarbonate infusion tests to confirm if there was a coexistence of proximal RTA. Accumulatively, five patients with PT dysfunctions further met the diagnosis criteria of renal FS with more than three kinds of PT dysfunctions or with coexistence of two PT dysfunctions and PT lesions in kidney pathology ( and ).
3.4. Kidney pathology
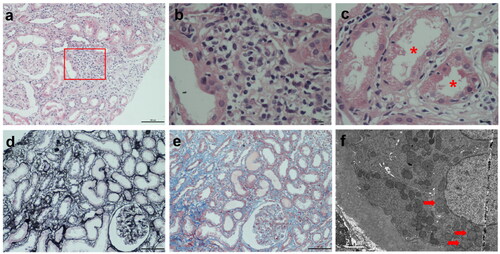
In total, five PBC-PT patients received kidney biopsies (). They all had TIN as the primary lesion, presenting as minor or mild glomerular changes and focal tubulointerstitial inflammatory infiltrates with major components of lymphocytes and plasma cells (). Most of them also showed PT injuries including defective brush border, proximal tubulitis characterized as inflammatory cell infiltration in the PT epithelium () and proximal tubule atrophy (), as well as different degrees of interstitial fibrosis (). Immunofluorescence staining of immunoglobulins (IgG, IgA, IgM), complements (C3, C4, C1q) and kappa- and lambda-chains were all negative in the glomeruli and tubules. Mitochondrial changes as swollen mitochondria with destroyed or disappeared cristae in the PT epithelium were indicated by electron microscopy ().
Figure 1. PBC-PT patients presented with tubulointerstitial nephritis in kidney biopsy. (a) Focal interstitial inflammatory infiltrates with major components of lymphocytes and plasma cells were shown by HE staining (×100). (b) Enlarged boxed area in (a) (×400). (c) Proximal tubulitis characterized as inflammatory cell infiltration in proximal tubular epithelium (asterisks) was revealed by HE staining (×400). (d) Focal tubular atrophy was shown by periodic acid-silver metheramine staining (×100). (e) Focal interstitial fibrosis was indicated by masson trichrome staining (×100). (f) Swollen mitochondria with destroyed or disappeared cristae (arrows) in proximal tubular epithelium were shown by transmission electron microscopy (×5000). Abbreviations: PBC-PT, primary biliary cholangitis with proximal tubular dysfunctions; HE, hematoxylin and eosin staining.
3.5. Treatment and prognosis
Besides the basic prescription of ursodeoxycholic acid (UDCA) (100.0%) and renal supplementary treatments such as potassium and phosphorus supplements in all the 11 PBC-PT patients, eight (72.7%) of them were treated with glucocorticoids (GCs) upon diagnosis of renal injuries. The median initial dose of GCs equivalent to prednisone was 40 mg/d (range 15–55 mg/d) and six (75.0%) of them received doses of 0.8–1.0 mg/kg/d. Four cases also received immunosuppressants in addition to GCs, including cyclophosphamide (n = 3) and azathioprine (n = 1) ().
The median follow-up time was 27 months (range 0–216 months). One patient died of liver failure in the 9th year. None of them developed the end-stage renal disease (ESRD) during follow-up. Their average eGFR levels remained stable at around 55 mL/min/1.73m2 (). For the patients with GCs treatment, their serum IgM levels significantly decreased from 5.97 ± 4.55 g/L to 2.09 ± 1.48 g/L (p = 0.019, ) and the serum IgG levels decreased from 20.10 ± 6.86 g/L to 12.90 ± 4.51 g/L (p = 0.240). Their eGFR levels were significantly improved from 43.24 ± 19.60 mL/min/1.73m2 to 55.02 ± 21.14 mL/min/1.73m2 (p = 0.028, ), and the Scr levels decreased from 145 ± 44 μmol/L to 115 ± 31 μmol/L (p = 0.052, ). There were five PBC-PT patients who had reevaluations of PT functions in the follow-up. Among them, three cases (Patients 1, 3 and 11) with GCs treatment showed the recovery of hypophosphatemia, hypouricemia, and glycosuria from the first month of GCs treatment and remained stable with no relapses. The other two cases (patients 2 and 4) with only UDCA and renal supplementary treatments showed stable PT functions ( and ).
Figure 2. Long-term renal prognosis and eGFR improvements after GCs treatment in PBC-PT patients. Patients who had follow-up time less than three months (n = 2) and insufficient eGFR follow-up data (n = 1) were excluded. (a) Evolution of eGFR in the whole population of PBC-PT patients (n = 8 at baseline). (b–d) After receiving GCs treatment, PBC-PT patients (n = 6) showed improvements in serum IgM (p = 0.019), eGFR (p = 0.028) and Scr (p = 0.052) during follow-up. Abbreviations: IgM, immunoglobulin M; eGFR, estimated glomerular filtration rate; GCs, glucocorticoids; PBC-PT, primary biliary cholangitis with proximal tubular dysfunctions; mo, months; Scr, serum creatinine.
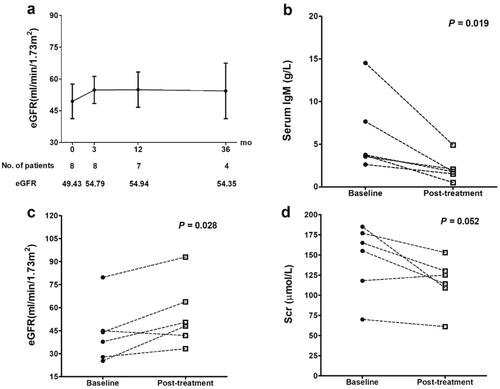
Table 3. Improvements of PT functions after GCs treatment in PBC-PT patients.
There had been 24 PBC-PT patients reported in the literature [Citation20–32]. Among the 16 cases who received GCs treatment, 10 (62.5%) PBC-PT patients showed eGFR improvements, five had stable eGFR and only one case had progressed renal insufficiency. For the other four cases without GCs treatment, only one patient had follow-up data and showed progressed renal insufficiency. (Table S1 in the Supplement)
4. Discussion
Renal involvement of distal RTA in PBC was well known, but PT dysfunctions in PBC were rarely identified with only about 20 cases reported in the literature [Citation20,Citation24,Citation27,Citation29]. We reported 11 cases of PBC-PT with detailed clinicopathological records and follow-up data, to our limited knowledge, which is the largest sample in a single center so far. These gradually accumulated PBC-PT cases remind us PBC-PT may need more attention. If PBC patients encounter hypokalemia, hypophosphatemia, hypouricemia or normoglycemic glycosuria, PT dysfunctions should be considered and nephrologists’ consulting would be necessary. It was noteworthy that more than half of our PBC-PT patients were diagnosed as PBC after the diagnosis of PT dysfunctions. Furthermore, two of them presented as nephropathy-onset PBC, with elevations of serum ALP or GGT levels until 40 months and 60 months after PT dysfunctions onset, respectively. Therefore, we recommend all the patients with PT dysfunctions for unknown reasons should regularly receive screening for PBC including both liver enzymes and AMA-M2 antibody measurements.
It was interesting that there were some clues reminding that the PBC-PT patients might have dysfunctions of some other tubular segments beyond proximal tubules. One clue was that the eight patients of PBC-PT with RTA in our study all had distal RTA which suggested potential injuries of distal tubules or collecting ducts, although it remained unknown whether they had a coexistence of proximal RTA without sodium bicarbonate infusion tests. Another clue was that two of our PBC-PT cases had metabolic alkalosis with obvious renal loss of potassium, hypokalemia, hypocalciuria and normal blood pressure, which met the criteria of hypokalemic salt-loss tubulopathies including Gitelman syndrome and Bartter syndrome caused by dysfunctional transporters in distal tubules or thick ascending limbs [Citation33,Citation34]. Acquired hypokalemic salt-loss tubulopathies had been reported in pSS [Citation35–37] and systemic lupus erythematosus [Citation38] but not in PBC before. Further studies including chloride clearance test after hydrochlorothiazide or furosemide intake and genetic analysis could help in the differential diagnoses [Citation33].
The mechanism of renal tubular involvement in PBC is not yet fully understood. The potential hypotheses are a common genetic predisposition or cross-reacting local antigens between liver and kidney which may stimulate and attract autoantibodies or activated lymphocytes and plasma cells to kidney leading to TIN and tubular dysfunctions [Citation29,Citation39]. The cross-reactivity between hepatocyte membrane surface lipoproteins and Tamm-Horsfall glycoprotein produced by renal tubular cells were reported [Citation40,Citation41]. However, cross-reactivity between proximal tubules and hepatocytes, which may explain the PT injuries in PBC, remains unknown. Since 90–95% of PBC patients have AMAs [Citation1] and mitochondrial cytopathies can cause PT dysfunctions [Citation42,Citation43], it is reasonable to presume that AMAs and their immune complexes might lead to renal mitochondrial damages and thus cause PT dysfunctions. Lino et al. reported increased size of mitochondria with rarefaction of invaginations and accumulation of a granulous material in a PBC-PT patient [Citation24]. We observed mitochondrial swelling and mitochondrial cristae loss in the PT epithelium of PBC-PT patients. However, the causal relationship between these mitochondrial changes and PT injuries remained unknown and whether AMAs have a direct pathological role in PT injuries deserves further studies [Citation24,Citation44]. In addition, a high prevalence (81.8%) of elevated serum IgM levels in our PBC-PT patients was consistent with the previous case report [Citation30]. Mizoguchi et al. observed dominant IgM-positive cell infiltration in both the renal and liver pathology [Citation30], which indicated a potential link between IgM/IgM secreting cells and PT injuries in PBC.
More than half of our PBC-PT cases coexisted with pSS, which was similar to the previous reports as PBC had a high prevalence of 3.5–73% coexisted with pSS [Citation45,Citation46]. The two diseases have similar renal manifestations with distal renal tubular acidosis as the most common manifestation and occasional PT dysfunctions and glomerulitis. They may share some similar pathogenesis including similar genetic predisposition (HLA-DR2, HLA-DR3, STAT4, IRF5, and IL12A), unique epithelial apoptosis, and local infiltrations of B cells, T cytotoxic cells and T helper cells leading to altered expression of proinflammatory cytokines and chronic inflammation [Citation46,Citation47]. Although it is important to differentiate between PBC-associated and pSS-associated PT dysfunctions, there had been no reported methods to distinguish them as the mechanisms of renal involvements in PBC or pSS both remain unclear, and their differences in renal manifestations were largely unknown. More cases would be needed to compare their clinicopathological characteristics and further studies about their renal-specific mechanisms would provide potential differential points.
The good responses of eGFR and tubular functions to GCs treatment in our PBC-PT patients were consistent with previous studies. Komatsuda et al. reported 7 (77.8%) of nine PBC cases with biopsy proved TIN had eGFR improvements after GCs treatment (0.5–1 mg/kg/d) [Citation27]. Among the total 24 PBC-PT patients reported in the literature, 10 (62.5%) of the 16 cases who received GCs treatment showed eGFR improvements, and only one case had progressed renal insufficiency as Supplement Table S1 showed [Citation20–32]. Mizoguchi et al. tried to use methylprednisolone pulse therapy (500 mg/d) for three consecutive days followed by 10 mg/d of prednisolone in a recurrent PBC-PT case, and observed an improved eGFR without relapses [Citation30]. However, most previous reports ignored the treatment responses of tubular functions in PBC-PT patients. Only one PBC-PT case has been reported to show recoveries of tubular dysfunctions after three months of GCs treatment [Citation25]. In this study, we observed the recovery of hypophosphatemia, hypouricemia, and glycosuria in three PBC-PT cases from the first month of GCs treatment and remained stable without relapses. Therefore, a short course of GCs treatment was worth trying in PBC-PT patients. Whether long-term immunosuppressant treatment is beneficial needs further well-designed studies to prove.
Limitations
Our study had several limitations. Firstly, incomplete renal tubular function screening and monitoring might lead to missed PBC-PT cases and interfere with the assessment of tubular responses to UDCA or GCs treatment in some patients. Secondly, with limited renal biopsy samples, we could not do more research on renal pathology and the potential pathogenesis of PT injuries in PBC. Thirdly, with limited cases and unknown mechanisms of renal involvements in PBC or pSS, we could not distinguish between PBC-associated and pSS-associated PT dysfunctions despite a high prevalence of pSS coexistence in the PBC-PT patients.
Conclusions
We reported the largest single-center case series of PBC-PT and good responses of both eGFR and tubular functions to GCs treatment.
Ethical approval and consent to participate
The study was performed in accordance with the Helsinki Declaration and approved by the local Ethic Committee of Peking Union Medical College Hospital (JS-2582). Informed written consent has been obtained from our patients as specified in the ICMJE recommendations.
Author contributions
Xiaoxiao Shi: conceptualization, methodology, investigation, formal analysis, writing—original draft preparation. Tianchen Guo: investigation, validation, formal analysis, writing—original draft preparation. Yubing Wen, Wei Ye, and Wenling Ye: methodology, data curation. Ke Zheng, Yan Qin, and Xuemei Li: resources, writing—reviewing and editing, supervision. Fengchun Zhang: resources, writing—reviewing and editing. Limeng Chen: conceptualization, resources, data curation, writing—reviewing and editing, project administration, and funding acquisition.
Supplemental Material
Download PDF (587.6 KB)Acknowledgements
We acknowledge a conference abstract named “Xiaoxiao Shi, Tianchen Guo, Yubing Wen, Xuemei Li, Limeng Chen*. Renal Proximal Tubular Dysfunctions in Primary Biliary Cholangitis. Kidney Week, American society of nephrology, 2022” as this manuscript was derived from it by the same research team.
Disclosure statement
No conflicts of interest are relevant to this manuscript.
Data availability statement
The data underlying this article are available in the article and in its online supplementary material.
Additional information
Funding
References
- Kaplan MM, Gershwin ME. Primary biliary cirrhosis. N Engl J Med. 2005;353(12):1–10. doi: 10.1056/NEJMra043898.
- Lindor KD, Bowlus CL, Boyer J, et al. Primary biliary cholangitis: 2018 practice guidance from the American association for the study of liver diseases. Hepatology. 2019;69(1):394–419. doi: 10.1002/hep.30145.
- EASL clinical practice guidelines: the diagnosis and management of patients with primary biliary cholangitis. J Hepatol. 2017;67:145–172.
- Floreani A, Franceschet I, Cazzagon N, et al. Extrahepatic autoimmune conditions associated with primary biliary cirrhosis. Clin Rev Allergy Immunol. 2015;48(2-3):192–197. doi: 10.1007/s12016-014-8427-x.
- Chalifoux SL, Konyn PG, Choi G, et al. Extrahepatic manifestations of primary biliary cholangitis. Gut Liver. 2017;11(6):771–780. doi: 10.5009/gnl16365.
- Parés A, Rimola A, Bruguera M, et al. Renal tubular acidosis in primary biliary cirrhosis. Gastroenterology. 1981;80(4):681–686.
- Burnevich EZ, Lopatkina TN. System manifestations of primary biliary cirrhosis. Klin Med (Mosk). 2006;84(12):42–46.
- Zimmermann J, Harendza S, Noriega M, et al. Membranous nephropathy and primary biliary cholangitis: a case report and review of the literature. Clin Nephrol. 2021;96(1):36–45. doi: 10.5414/CN110363.
- Yamashita H, Suzuki A, Takahashi Y, et al. Anti-neutrophil cytoplasmic antibody (ANCA)-associated vasculitis associated with primary biliary cirrhosis: a case report and literature review. Intern Med. 2015;54(10):1303–1308. doi: 10.2169/internalmedicine.54.3678.
- Vitali C, Bombardieri S, Jonsson R, et al. Classification criteria for sjogren’s syndrome: a revised version of the European criteria proposed by the American-European consensus group. Ann Rheum Dis. 2002;61(6):554–558. doi: 10.1136/ard.61.6.554.
- Messiaen T, Deret S, Mougenot B, et al. Adult fanconi syndrome secondary to light chain gammopathy. Clinicopathologic heterogeneity and unusual features in 11 patients. Medicine (Baltimore). 2000;79(3):135–154. doi: 10.1097/00005792-200005000-00002.
- Vignon M, Javaugue V, Alexander MP, et al. Current anti-myeloma therapies in renal manifestations of monoclonal light chain-associated Fanconi syndrome: a retrospective series of 49 patients. Leukemia. 2017;31(1):123–129. doi: 10.1038/leu.2016.195.
- Shi X, Chen Z, Wang J, et al. Primary sjӧgren’s syndrome with renal fanconi syndrome: good responses to treatment with glucocorticoids. Semin Arthritis Rheum. 2020;50(6):1326–1332. doi: 10.1016/j.semarthrit.2020.03.017.
- Nakayama A, Matsuo H, Ohtahara A, et al. Clinical practice guideline for renal hypouricemia (1st edition). Hum Cell. 2019;32(2):83–87. doi: 10.1007/s13577-019-00239-3.
- Gilbert SJ, Weiner DE, Gipson DS, et al. National kidney foundation’s primer on kidney diseases. 6th ed. New York: Elsevier; 2014.
- Payne RB. Renal tubular reabsorption of phosphate (TmP/GFR): indications and interpretation. Ann Clin Biochem. 1998;35 (Pt 2)(2):201–206. doi: 10.1177/000456329803500203.
- Ram R, Swarnalatha G, Dakshinamurty KV. Renal tubular acidosis in sjogren’s syndrome: a case series. Am J Nephrol. 2014;40(2):123–130. doi: 10.1159/000365199.
- Rodríguez Soriano J. Renal tubular acidosis: the clinical entity. J Am Soc Nephrol. 2002;13(8):2160–2170. doi: 10.1097/01.ASN.0000023430.92674.E5.
- Hirschfield GM, Dyson JK, Alexander GJM, et al. The british society of gastroenterology/UK-PBC primary biliary cholangitis treatment and management guidelines. Gut. 2018;67(9):1568–1594. doi: 10.1136/gutjnl-2017-315259.
- Izumi N, Hasumura Y, Takeuchi J. Hypouricemia and hyperuricosuria as expressions of renal tubular damage in primary biliary cirrhosis. Hepatology. 1983;3(5):719–723. doi: 10.1002/hep.1840030516.
- Kamouchi M, Tsuji H, Hirakata H, et al. Tubulointerstitial disorders in the kidney associated with primary biliary cirrhosis (PBC). Clin Nephrol. 1991;35(3):134–135.
- Kodama T, Imai H, Wakui H, et al. Tubulointerstitial nephritis with renal tubular acidosis and asymptomatic primary biliary cirrhosis accompanied by antibody to a 52-kDa mitochondrial protein alone. Clin Nephrol. 1996;45(6):401–405.
- Davidson BK, Haslock I. Osteomalacia secondary to renal tubular acidosis masquerading as primary biliary cirrhosis. Rheumatology (Oxford). 2000;39(12):1428–1429. doi: 10.1093/rheumatology/39.12.1428.
- Lino M, Binaut R, Noël LH, et al. Tubulointerstitial nephritis and fanconi syndrome in primary biliary cirrhosis. Am J Kidney Dis. 2005;46(3):e41-6–e46. doi: 10.1053/j.ajkd.2005.05.021.
- Terrier B, Fakhouri F, Berezne A, et al. Osteomalacia revealing celiac disease and primary biliary cirrhosis-related fanconi syndrome in a patient with systemic sclerosis. Clin Exp Rheumatol. 2008;26:467–470.
- Bando H, Hashimoto N, Hirota Y, et al. Severe hypophosphatemic osteomalacia with fanconi syndrome, renal tubular acidosis, vitamin D deficiency and primary biliary cirrhosis. Intern Med. 2009;48(5):353–358. doi: 10.2169/internalmedicine.48.1644.
- Komatsuda A, Wakui H, Ohtani H, et al. Tubulointerstitial nephritis and renal tubular acidosis of different types are rare but important complications of primary biliary cirrhosis. Nephrol Dial Transplant. 2010;25(11):3575–3579. doi: 10.1093/ndt/gfq232.
- Yamaguchi S, Maruyama T, Wakino S, et al. A case of severe osteomalacia caused by tubulointerstitial nephritis with Fanconi syndrome in asymptomotic primary biliary cirrhosis. BMC Nephrol. 2015;16(1):187. doi: 10.1186/s12882-015-0184-4.
- Takahashi N, Saeki T, Komatsuda A, et al. Tubulointerstitial nephritis with IgM-positive plasma cells. J Am Soc Nephrol. 2017;28(12):3688–3698. doi: 10.1681/ASN.2016101074.
- Mizoguchi S, Katayama K, Murata T, et al. IgM-positive tubulointerstitial nephritis associated with asymptomatic primary biliary cirrhosis. Kidney Int Rep. 2018;3(4):1004–1009. doi: 10.1016/j.ekir.2018.04.001.
- Akagi R, Ishii A, Kaneko K, et al. A report of three cases of patients with tubulointerstitial nephritis with IgM-positive plasma cells, treatment, and serum-IgM as a sensitive marker for relapse. BMC Nephrol. 2023;24(1):201. doi: 10.1186/s12882-023-03253-8.
- Er C, Dyson J, Jones D, et al. Primary biliary cholangitis presenting with Fanconi syndrome: an important phenotype. BMJ Case Rep. 2022;15(8):e248461. doi: 10.1136/bcr-2021-248461.
- Blanchard A, Bockenhauer D, Bolignano D, et al. Gitelman syndrome: consensus and guidance from a kidney disease: improving global outcomes (KDIGO) controversies conference. Kidney Int. 2017;91(1):24–33. doi: 10.1016/j.kint.2016.09.046.
- Konrad M, Nijenhuis T, Ariceta G, et al. Diagnosis and management of bartter syndrome: executive summary of the consensus and recommendations from the European Rare Kidney Disease Reference Network Working Group for Tubular Disorders. Kidney Int. 2021;99(2):324–335. doi: 10.1016/j.kint.2020.10.035.
- Kim YK, Song HC, Kim WY, et al. Acquired gitelman syndrome in a patient with primary sjögren syndrome. Am J Kidney Dis. 2008;52(6):1163–1167. doi: 10.1053/j.ajkd.2008.07.025.
- Gu X, Su Z, Chen M, et al. Acquired gitelman syndrome in a primary sjögren syndrome patient with a SLC12A3 heterozygous mutation: a case report and literature review. Nephrology (Carlton). 2017;22(8):652–655. doi: 10.1111/nep.13045.
- Fraj A, El Euch M, Jaziri F, et al. Acquired bartter syndrome in primary sjögren syndrome. Saudi J Kidney Dis Transpl. 2020;31(5):1144–1147. doi: 10.4103/1319-2442.301185.
- Barathidasan GS, Krishnamurthy S, Karunakar P, et al. Systemic lupus erythematosus complicated by a gitelman-like syndrome in an 8-year-old girl. CEN Case Rep. 2020;9(2):129–132. doi: 10.1007/s13730-019-00440-1.
- Silveira MAD, Seguro AC, Gomes SA, et al. Distal renal tubular acidosis associated with autoimmune diseases: reports of 3 cases and review of mechanisms. Am J Case Rep. 2022;23:e933957. doi: 10.12659/AJCR.933957.
- Tsantoulas DC, McFarlane IG, Portmann B, et al. Cell-mediated immunity to human Tamm-Horsfall glycoprotein in autoimmune liver disease with renal tubular acidosis. Br Med J. 1974;4(5943):491–494. doi: 10.1136/bmj.4.5943.491.
- Sakaguchi K, Koide N, Kondow H, et al. Hepatocyte plasma membrane antigens. II. Characterization of liver-specific membrane lipoprotein (LP-1) and Tamm-Horsfall glycoprotein (THGP) like antigens (hepatic THGP) on the plasma membrane of chang liver cell. Gastroenterol Jpn. 1983;18(4):339–345. doi: 10.1007/BF02774951.
- Grünfeld JP, Niaudet P, Rötig A. Renal involvement in mitochondrial cytopathies. Nephrol Dial Transplant. 1996;11(5):760–761. doi: 10.1093/oxfordjournals.ndt.a027391.
- Cez A, Brocheriou I, Lescure F-X, et al. Decreased expression of megalin and cubilin and altered mitochondrial activity in tenofovir nephrotoxicity. Hum Pathol. 2018;73:89–101. doi: 10.1016/j.humpath.2017.12.018.
- Solís Herruzo JA, Solís Muñoz P, Muñoz Yagüe T. The pathogenesis of primary biliary cirrhosis. Revista Espanola de Enfermedades Digestivas: organo Oficial de la Sociedad Espanola de Patologia Digestiva. 2009;101:413–423.
- Deng X, Li J, Hou S, et al. Prevalence and impact of sjögren’s syndrome in primary biliary cholangitis: a systematic review and meta-analysis. Ann Hepatol. 2022;27(6):100746. doi: 10.1016/j.aohep.2022.100746.
- Sun Y, Zhang W, Li B, et al. The coexistence of sjögren’s syndrome and primary biliary cirrhosis: a comprehensive review. Clin Rev Allergy Immunol. 2015;48(2-3):301–315. doi: 10.1007/s12016-015-8471-1.
- Selmi C, Meroni PL, Gershwin ME. Primary biliary cirrhosis and sjögren’s syndrome: autoimmune epithelitis. J Autoimmun. 2012;39(1-2):34–42. doi: 10.1016/j.jaut.2011.11.005.