
Abstract
Objective
Lipoprotein glomerulopathy (LPG) is a rare disorder characterized by the development of glomerular lipoprotein thrombosis. LPG exhibits familial aggregation, with mutations in the apolipoprotein E (APOE) gene identified as the leading cause of this disease. This study aimed to investigate APOE gene mutations and the clinicopathological features in eleven LPG patients.
Methods
Clinicopathological and follow-up data were obtained by extracting DNA, followed by APOE coding region sequencing analysis. This study analyzed clinical and pathological manifestations, gene mutations, treatment and prognosis.
Results
The mean age of the eleven patients was 33.82 years. Among them, five had a positive family history for LPG, ten presented with proteinuria, four exhibited nephrotic syndrome, and six presented with microscopic hematuria. Dyslipidemia was identified in ten patients. In all renal specimens, there was evident dilation of glomerular capillary lumens containing lipoprotein thrombi, and positive oil red O staining was observed in frozen sections of all samples. APOE gene testing revealed that one patient had no mutations, while the remaining ten patients exhibited mutations in the APOE gene, with three patients presenting with multiple mutations simultaneously. Following the confirmation of LPG diagnosis, treatment with angiotensin-converting enzyme inhibitor (ACEI)/angiotensin receptor blocker (ARB) was initiated, and the disease progressed slowly.
Conclusion
LPG is histologically characterized by lamellated lipoprotein thrombi in glomeruli, and kidney biopsy is essential for diagnosis. Mutations in the APOE gene are the leading cause of LPG. This study revealed clinicopathological characteristics and APOE gene mutations in patients with LPG, which helps us better understand the disease.
1. Introduction
Lipoprotein glomerulopathy (LPG), first reported approximately 30 years ago, is a glomerular disease characterized by markedly dilated glomerular capillary loops containing lipoprotein thrombi. In 1987, Saito et al. first reported this disease [Citation1]. Since then, an increasing number of cases have been identified globally, particularly in Asian countries such as China and Japan. The onset of this disease ranges from 4 to 69 years, with an average age of 31.5 years. It exhibits a higher prevalence in males than in females. Since the first reported case of lipoprotein nephropathy in China in 1997, over 200 cases of LPG have been reported, including approximately 60 cases in Nanjing (reported by the General Military Hospital) [Citation2, Citation3], 46 cases [Citation4–6] in Sichuan, 24 cases [Citation7–9] in Beijing (including 11 cases reported in our study), 10 cases [Citation10–12] in Guangdong, and 8 cases [Citation13–15] in Henan, along with sporadic cases in other regions.
Lipoprotein nephropathy is an autosomal recessive hereditary nephropathy. Apolipoprotein E (ApoE), a glycoprotein consisting of 299 amino acids with a relative molecular mass of 34 kDa, can bind to LDL receptors or LDL-related proteins, therefore participating in lipid transport and metabolism. ApoE facilitates the cellular uptake of triacylglycerol-rich lipoproteins and is essential for blood lipid metabolism. Currently, 20 gene mutations in APOE have been reported worldwide, and APOE Maebashi and APOE Kyoto are more prevalent in the Chinese population [Citation4, Citation5]. Currently, the pathogenesis of LPG remains unclear, but abnormal ApoE protein and genetic variations are suspected to be involved. There are few Chinese studies focusing on the pathogenesis and APOE gene mutations associated with LPG [Citation16–18].
In this study, a total of 11 LPG patients diagnosed over the past 18 years at the Department of Nephrology of Peking University First Hospital were enrolled. The study summarized and analyzed the clinical manifestations, pathological characteristics, gene mutations, treatment, and prognosis of LPG within this patient group.
2. Materials and methods
2.1. Patients
Clinical and pathological data of 11 patients with LPG treated at the Department of Nephrology of Peking University First Hospital from January 2001 to December 2018 were collected and subjected to followed up.
2.2. Methods
Routine examinations, including routine blood tests, urine tests, 24 h urinary protein (24h UTP) quantification, liver and kidney function tests, blood lipid profile tests and kidney ultrasound, were conducted on the 11 patients with LPG.
Kidney biopsies were performed, and the specimens were routinely embedded in paraffin and sliced with a thickness of 2-3 µm for observation under an optical microscope using multiple staining methods, such as Hematoxylin-Eosin (HE), Periodic Acid-Schiff (PAS), Masson, periodic acid-silver metheramine (PASM), PASM + Masson and Oil Red O staining. Notably, Oil Red O staining is a commonly used experimental technique for detecting lipid content in cells or tissues. Firstly, Red O stain solution was applied to the slides, which were subsequently incubated for 5–10 min. After dehydration in graded ethanol, the slides were immersed in xylene and covered with transparent coverslips. Finally, the tissue sections were observed and imaged under microscope.
Immunofluorescence staining was performed on frozen sections to observe the characteristics and extent of various immunoglobulins, complement deposition, ApoE and ApoB in the kidney. Glutaraldehyde-fixed specimens were examined with electron microscopy. The diagnosis of LPG was confirmed based on morphological changes. Genomic DNA was extracted from 5 µl blood samples of the 11 patients with LPG. PCR was performed to amplify APOE exons 1, 2 and 3, while touchdown PCR was used for amplification of exon 4. The sequencing results were analyzed using Chormas version 2.6.5 (Technelysium, Australia).
Normality of the data was evaluated by kurtosis and skewness (both the absolute values were <3). For normally distributed data, differences of quantitative parameters between groups were analyzed using the t-test and means ± standards deviation (SD) were presented as descriptive statistics. For non-normally distributed data, differences of quantitative parameters between groups were analyzed using the non-parametric test and median and interquartile range (IQR) were presented as descriptive statistics. Differences were considered to be significant if p < 0.05. Data analysis was performed with SPSS version 22.0 (SPSS, Chicago, IL, USA).
Our research was in compliance with the Declaration of Helsinki and approved by the clinical research ethics committee of the Peking University First Hospital (No.2014[749]).
3. Results
3.1. Demographic and general data
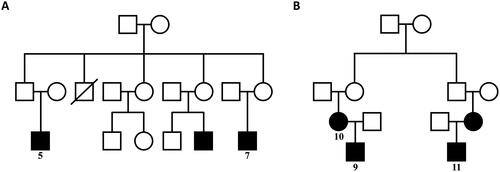
The study included eleven patients from multiple regions in China, namely Beijing (2 cases), Yinchuan (2 cases), Xinjiang (1 case), Henan (2 cases), Shanxi (1 case), Sichuan (1 case), Hubei (1 case), and Gansu (1 case). Among these patients, five patients (Cases 5, 7, 9, 10 and 11) had a clear family history of LPG. Notably, two patients from Yinchuan (Cases 9 and 10) were identified as mother and son, and one patient from Xinjiang (Case 11), whose mother was also an LPG patient, shared a familial connection with the patient from Yinchuan (Case 10), who was a cousin. Additionally, two patients (Cases 5 and 7) from Cangzhou were identified as cousins (). The remaining six patients had no significant family history.
Figure 1. Patient pedigree diagram: The black squares and circles with corresponding numbers represent 5 patients of LPG (Case 5,7,9,10,11) who had clear family histories diagnosed in our hospital. Two patients from Yinchuan (Case 9,10) were mother and son. One patient from Xinjiang (Case 11), whose mother was also an LPG patient, and the patient from Yinchuan (Case 10) were cousins. Two patients (Case 5 and 7) from Cangzhou were cousins. The two black squares without numbers represent patients of LPG diagnosed in other hospitals. The square with oblique line represents a person who has passed away.
Clinical features such as edema and/or proteinuria were observed in these patients. The ages of the patients ranged from 13 to 53 years old, with a mean age of 33.82 ± 13.91 years old. Among the 11 patients with LPG, 9 patients were male, and 2 were female. The average follow-up time was 78.4 months. Cases 4 and 8 were lost to follow-up. Four patients had hypertension, and Case 8 had malignant hypertension with a blood pressure as high as 210/160 mmHg. The average systolic and diastolic blood pressures of the 11 patients were 143.09 ± 32.88 mmHg and 90.55 ± 26.08 mmHg, respectively. With the exception for Case 6, who exhibited negative urine protein, the remaining patients presented with moderate to severe proteinuria. Four patients were diagnosed with nephrotic syndrome, six patients experienced hematuria. Five patients displayed hyperuricemia, and four patients demonstrated elevated blood creatinine, placing them at CKD stage 2-4 ().
Table 1. General data and prognosis of the patients.
3.2. Laboratory examination
Case 6 exhibited negative for urine protein, while the remaining 10 patients presented with moderate to massive proteinuria. The 24-h urinary protein (24h UTP) quantity of the patients ranged from 0.04-14.0 [median (IQR) 3.02 (8.09)] g, with 4 cases diagnosed with nephrotic syndrome. Six patients developed microscopic hematuria. The hemoglobin levels in the 11 patients with LPG ranged from 75-150 (mean ± SD 118.82 ± 24.05) g/L. Mild anemia was observed in six patients with hemoglobin levels ranging from 94-123 g/L, while Case 1 demonstrated moderate anemia with a hemoglobin level of 75 g/L, attributed to renal anemia due to CKD stage 3. Blood creatinine levels ranged from 52.2-275.0 [median (IQR) 91.0 (66.0)] μmol/L, with 4 patients classified as CKD stage 2-4 at diagnosis. Blood uric acid levels ranged from 213 to 643 (mean ± SD 421.73 ± 112.84) µmol/L, and 5 patients exhibited hyperuricemia. Except for Case 5, the other 10 patients showed abnormal blood lipid profiles, including increased levels of triglycerides (TG), total cholesterol (TC) and low-density lipoprotein (LDL-C). High-density lipoprotein (HDL-C) levels were within the normal reference range (1.40-1.56 mmol/L), except for one patient with a decreased HDL-C level. All patients exhibited varying degrees of hypoproteinemia, with albumin (ALB) levels ranging from 22-39.2 (mean ± SD 30.11 ± 5.79) g/L. Case 9, a heavy alcohol consumer, had elevated glutamyl-transpeptidase (GGT) levels, while the remaining 10 patients had normal liver function. Serum immunological examinations revealed that case 3 was ANA-positive, while all the other patients tested negative for ANA, anti-dsDNA antibody, antineutrophil cytoplasmic antibody (ANCA), anti-glomerular basement membrane (GBM) antibodies and complement 3 (C3)/complement 4 (C4). Kidney ultrasound examinations for the 11 patients showed that 6 LPG patients had enlarged kidney volumes, nine patients had diffuse kidney lesions (e.g. blurred internal structures, enhanced echogenicity of renal parenchyma and unclear boundary between cortex and medulla), and the remaining two patients’ kidneys were roughly normal. The results of the major laboratory examinations are shown in .
Table 2. Laboratory examination results.
3.3. Pathological manifestations
Characteristic lesions were identified in 11 patients with LPG under a light microscope, revealing markedly enlarged glomerular capillary lumens containing a large amount of lightly stained reticular substances. Capillaries exhibited balloon-like changes or capillary hemangioma-like expansion (). PAS staining resulted in a light pink color, Masson and PASM staining were light blue, and the oil red O staining demonstrated a strongly positive reaction (). The mesangial cells and matrix exhibited mild to moderate proliferation (). In Cases 1, 5, 7, 8 and 11, mesangial proliferative lesions and mesangial insertion were evident, accompanied by irregular thickening of the basement membrane and segmental double-track formation (). Case 2 presented with segmental sclerosis and balloon adhesion. Renal tubular lesions included vacuolization, granular degeneration and focal atrophy of epithelial cells (). Case 1 exhibited focal compensated hypertrophy with protein tubular casts within the lumen, while Case 2 demonstrated focal brush border loss. Renal interstitial lesions featured focal lymphocytic and mononuclear cell infiltration with (Cases 1, 2, 4, 5, 6, 7, 8, 9 and 11) or without (Cases 3 and 10) fibrosis. Small arterial walls exhibited hypertrophy in all patients; specifically, fibrous intimal hyperplasia and sclerosis were observed in Cases 1 and 2, luminal stenosis were observed in Cases 4 and 8, and vitreous changes were observed in Case 6. Above all, in kidney specimens of LPG, light microscopy revealed lesions in the glomerular, tubular, interstitial and vascular compartments.
Figure 2. Light microscopy: Eosin staining showed dilated capillary loops exhibiting an eosinophilic lipoprotein thrombus inside the capillary lumens.
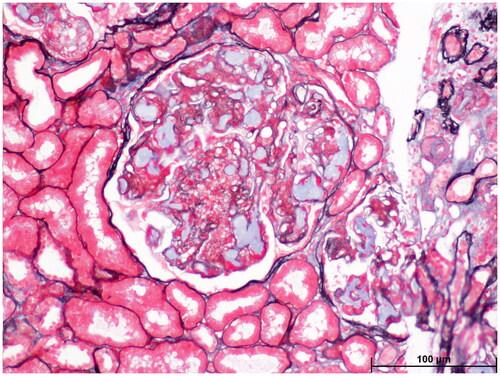
Figure 3. Light microscopy: Oil red O staining showed large amounts of red droplets in the thrombus-like substances in the renal specimen.
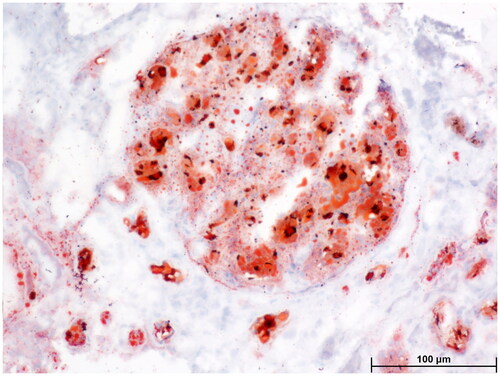
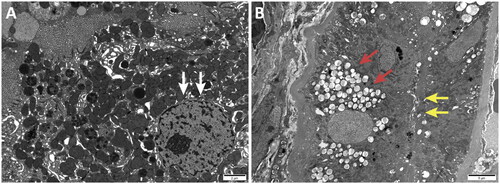
Figure 4. Electron microscopy: Proliferative lesions of mesangial matrix (4 A) and mesangial cells (4B) were observed in the renal specimen.
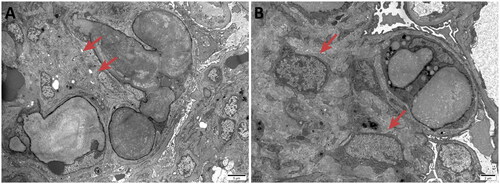
Figure 5. Light microscopy: Proliferation of mesangial cells and matrix was observed in the renal specimen.
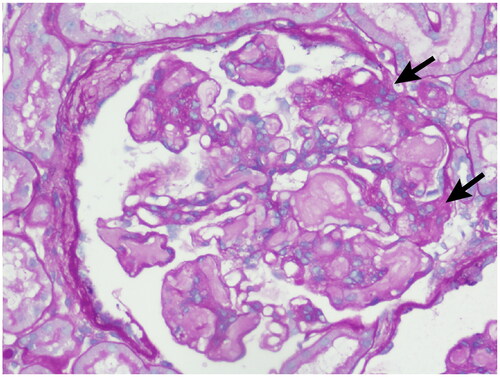
Figure 6. Electron microscopy: Irregular thickening of the basement membrane and segmental double-track formation was observed in the renal specimen.
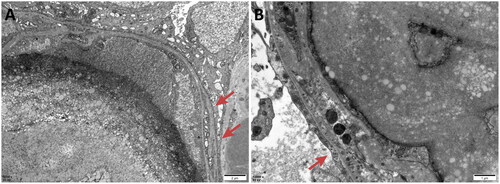
Figure 7. Electron microscopy: Renal tubular lesions including vacuolization (red arrows), granular degeneration (white arrows) and focal atrophy (yellow arrows) of epithelial cells were observed in the renal specimen.
All kidney specimens from the 11 patients were subjected to electron microscopy, which revealed consistent LPG characteristics. Glomerular capillary lumens exhibited significant dilation containing several particles of different sizes and electron densities (). Vacuoles of varying sizes within the lumen were arranged in clusters or layers (). Mesangial cells and the matrix within the glomerulus showed light to moderate proliferation with segmental mesangial insertion. The basement membrane exhibited segment thickening along with widening of the internal loose layer (Cases 1, 2, 4, 5, 7 and 8), consistent with LPG along with thrombotic microangiopathy (TMA)-like changes. Extensive fusion of epithelial foot processes was observed. Kidney tubular epithelial vacuolation degeneration, elevated lysosomes and focal atrophy were observed. Lymphocytic and monocytic infiltration along with collagen fiber hyperplasia was observed in the renal interstitium in Cases 2 and 4. Case 3 had massive electron dense deposits under the epithelium, extensive fusion of epithelial foot processes and no obvious tubular or renal stromal lesions, which was consistent with stage I-II membranous nephropathy (MN) combined with LPG. Case 1 had LPG combined with mesangial proliferative IgA nephropathy. Case 6 had LPG combined with IgA nephropathy (M0E0S0T0). In Case 9, small electron transparent vacuolar structures were observed within the glomerular capillary lumen. Irregular thickening of the GBM was observed. The basement membrane displayed diffuse layering with visible holes and lacerations in the epithelial foot process, which was consistent with lipoprotein glomerulopathy, without ruling out Alport syndrome ().
Figure 8. Electron microscopy: Dilated glomerular capillary lumens were filled with a large number of particles of different sizes and electron densities.
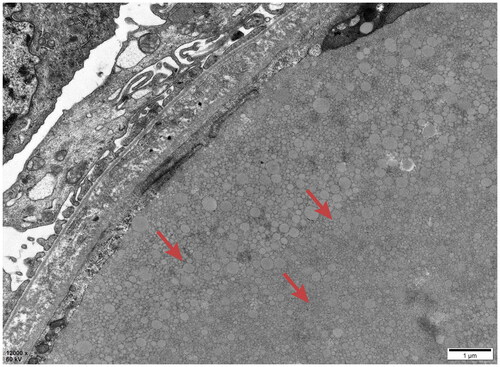
Figure 9. Electron microscopy: Vacuoles were observed in the glomerular capillary lumen.
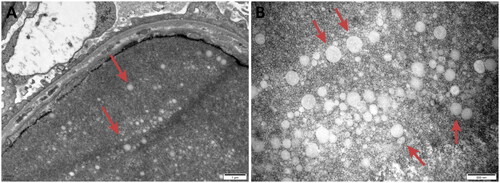
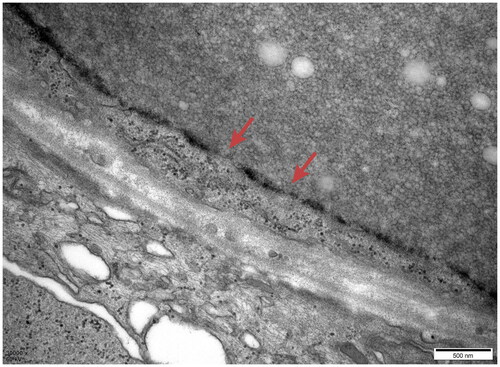
Figure 10. Electron microscopy: The basement membrane showed insect eroding and tear-like changes.
Routine immunofluorescence staining, including immunoglobulin (Ig) G, IgA, IgM, C3, C1q, FRA, and Alb staining, showed no characteristic changes. Nine of the 11 patients tested positive for IgM, five for C3, and all were negative for C1q and Alb. Among them, Case 3 was strongly positive for IgG (IgG1 + −++, IgG4 ++ − +++), Case 1 showed IgA +++, and Patient 6 showed IgA + − ++ ().
Table 3. Immunofluorescence staining.
3.4. APOE genetic test results
Gene testing for APOE exons 1, 2, 3 and 4 was conducted for the 11 patients with LPG. The results of the sequencing analysis of the coding region of the APOE gene in patients are shown in . No mutations were identified in exons 1 or 2 among any of the 11 patients. Mutations in the APOE gene were detected in exons 3 and 4. Three patients (Cases 1, 4 and 8) had an APOE KYOTO (R43C) mutation in exon 3; in particular, one of the three patients (Case 1) also had an APOE3 (c130R) mutation in exon 4. Six patients (Cases 2, 5, 7, 9, 10 and 11) had the APOE Maebashi (p.160_162delRKL) mutation in exon 4. Among them, one patient (Case 11) also had an APOE3 (c130R) mutation, while another patient (Case 2) had three mutations, namely APOE Maebashi (p.160_162delRKL), APOE2 (p. R176C) and APOE3 (c130R) simultaneously. In addition, an APOE2 (p. R176C) mutation was detected in Case 3. No mutation was found in Case 6. The APOE3 (c130R) mutation, which was not previously reported in LPG cases, did not occur independently in the patients with LPG in this study; instead, it coexisted with other known APOE gene mutations associated with LPG. The APOE3 (c130R) mutation may not be a pathogenetic gene for LPG. The sequencing results indicated the occurrence of two or more coexisting APOE gene mutations in patients with LPG, which has rarely been reported in the previous literature.
Table 4. APOE gene mutations.
We compared the laboratory test results (e.g., 24-h total urine protein, albumin, hemoglobin, serum creatinine, uric acid, triglyceride and low-density lipoprotein levels) between groups with different APOE gene mutations (i.e., patients with the APOE Maebashi mutation vs. those without the APOE Maebashi mutation), no statistically significant differences were found (p > 0.05). Compared with those in patients with the APOE Maebashi mutation, the serum creatinine levels were higher in patients without this mutation (mean ± SD 144.60 ± 78.94 μmol/L vs mean ± SD 87.23 ± 32.42 μmol/L), although the difference was not statistically significant (p = 0.136). Clinical features such as edema were observed in all these patients. No differences in morphological changes under microscope were observed between the groups with different APOE gene mutations.
3.5. Treatment and prognosis
Among the 11 patients with LPG, 6 individuals (Cases 4, 6, 7, 8, 9 and 11) were previously treated with glucocorticoids and immunosuppressants but exhibited poor responses. Upon confirmation of the diagnosis of LPG through repeated kidney biopsies, a reduction in steroids and immunosuppressive agents was initiated. Instead, patients were prescribed ACEI/ARB and atorvastatin/fenofibrate for the management of urinary protein and blood lipids. Case 3 discontinued medication upon achieving complete remission (proteinuria under 0.3 g/day with stable serum creatinine [Citation4]), with 24h UTP levels decreasing from 1.36 g to 0.01 g. Case 2 achieved partial remission, with a transaminase increase attributed to adverse reactions to lipid-lowering drugs after one month of treatment; these levels returned to normal after discontinuing fenofibrate. The 24h UTP levels in Case 2 decreased from 9.45 g to 1.85 g. Case 7 achieved partial remission and remained stable throughout the 89-month follow-up, with no progression observed in serum creatinine levels (73 μmol/L). Cases 6, 9, 10 and 11 were followed up for 33, 141, 179 and 212 months, respectively. Their kidney function transitioned from normal to CKD stage 2 to 5, and their eGFR levels were 55.2, 41.1, 60.7 and 11.7 mL/min/1.73 m2, respectively. Case 1 was already in CKD stage 4 at diagnosis and had an eGFR level of 25.7 mL/min/1.73m2. Case 5 did not achieve remission (). The overall trend of disease progression in the 11 patients was gradual.
Table 5. Laboratory examination results before and after treatment.
4. Discussion
LPG is a renal disorder characterized by lipid deposition in the glomeruli and tubular epithelial cells [Citation1]. Previous studies have indicated that patients with LPG typically demonstrate moderate to severe proteinuria, with occasional presentation of nephrotic syndrome [Citation19]. Hematuria, hypertension and even kidney failure, may present in some patients, with a family history of LPG [Citation20]. Our findings were consistent with previous reports.
One patient demonstrated normal levels of lipids, consistent with previous reports highlighting cases of LPG with normal lipids [Citation21]. Ten patients had dyslipidemia; among them, nine patients had elevated TG levels, six patients had elevated total cholesterol levels, and seven patients had elevated LDL-C. Saito et al. [Citation21] reported that both plasma triglyceride levels and total cholesterol levels were significantly increased in LPG patients. Plasma triglyceride levels appeared to be significantly elevated compared to total cholesterol levels, their further analysis revealed that mainly very low-density lipoprotein (VLDL) and intermediate-density lipoprotein (IDL) were increased. However, neither VLDL nor IDL was measured in the 11 patients with LPG in this study. Seven patients had anemia, among whom one had stage 3 CKD, which could be attributed to renal anemia. The remaining six patients exhibited mild anemia, potentially related to the effect of lipids on bone marrow hematopoietic function [Citation22, Citation23]. Distinctive histological changes provide important evidence for diagnosing lipoprotein nephropathy.
The pathological characteristics include a significantly dilated glomerular capillary lumen filled with lightly stained light reticular substances, mesangial dissolution, and mild to moderate mesangial proliferation. This proliferation can create a double-track sign by the insertion of the mesangial matrix into the glomerular basement membrane. In the renal tubular interstitium, tubular atrophy, interstitial lymphocyte and monocyte infiltration and hyperplasia of fibroconnective tissue were observed. Therefore, the pathology might lead to misdiagnosis of membranoproliferative glomerulonephritis (MPGN), focal segmental glomerulosclerosis (FSGS), proliferative sclerosing nephritis, or even sclerosing nephritis. Such misdiagnoses make it difficult to identify the primary lesion of lipoprotein nephropathy [Citation24]. Among our 11 patients, Cases 4, 6 and 7 were previously misdiagnosed with MPGN, which was further diagnosed as LPG through repeated kidney biopsy. Previous reports from China have also identified misdiagnoses of segmental proliferation with FSGS and mild MPGN (IgM type) [Citation25]. These patients received treatments with steroids and immunosuppressants (cyclophosphamide, cyclosporine, rituximab, etc.) but had a poor prognosis. Liu et al. [Citation20] reported two patients misdiagnosed with lupus nephritis and MPGN before being confirmed as LPG. These studies, including our results, suggested that more attention should be given to changes under light microscopy to avoid misdiagnosis.
Kidney specimens from patients with LPG commonly exhibit no deposition of immunoglobulin, complement or fibrinogen according to fluorescence microscopy, while IgM deposition might be observed in some cases. Our study revealed that most of our patients had IgM deposition in the mesangial area and capillary walls. Additionally, half of the patients exhibited a small amount of C3 deposition, two patients exhibited IgA deposition in the mesangial area, and one patient exhibited IgG deposition in the capillary walls. In line with our results, several previous studies have shown the deposition of immunoglobulins and complements such as IgM, IgA, C1q and C3 in the mesangial area and capillary walls [Citation26–28]. However, the IgA deposits in Case 1 and Case 6, which appeared mild, may represent incidental IgA deposits, which are common in the Chinese population [Citation29]. Electron microscopy examination is helpful for the diagnosis of LPG. Under an electron microscope, one can observe significantly dilated glomerular capillary lumens filled with particles of varying sizes and electron densities, arranged in clustered or layered patterns.
Previous studies revealed that although the pathogenesis of LPG is closely related to APOE gene mutations, APOE gene mutations are not the only pathogenic factor. Previous studies have shown that some close relatives carrying APOE gene mutations do not develop this disease, suggesting the involvement of the local kidney microenvironment, defective macrophages, oxidative stress and environmental factors in the pathogenesis of LPG. One patient in our study had no APOE gene mutations, while the other 10 patients had mutations or abnormalities in the APOE gene, with three patients having two or more types of APOE gene mutations simultaneously. Worldwide, twenty genetic mutations in the APOE gene in patients with LPG have been reported, with studies in China contributing to the discovery of 10 different APOE gene mutations. Hu et al. [Citation6] reported that the APOE Kyoto mutation was detected in 35 patients and 28 asymptomatic relatives from 31 different families in the same county seat in southwestern China. Hu et al. [Citation5] also reported a case of a newly identified mutation, APOE Chengdu (L173P). Chen et al. [Citation9], Pan et al. and Luo et al. [Citation7, Citation12] reported 5 cases of APOE Maebashi mutation, 2 cases of APOE Kyoto mutation and 1 case of APOE Guangzhou mutation, respectively. One case of APOE Modena mutation has been discovered in Shenzhen [Citation10]. Fan et al. [Citation2] reported the two cases of APOE Kyoto mutation and APOE Tokyo mutation, respectively. However, Chen et al. [Citation3] did not find any APOE gene mutations through a full-length sequence analysis of the gene. The majority of LPG cases were reported in China and Japan globally. It is mainly distributed in southwestern and southeastern China and in central and north-central Japan. APOE Kyoto, APOE Guangzhou and Tokyo-Maebashi were reported in LPG cases of China. APOE Sendai (Arg145Pro) was mainly observed in north-central Japan, particularly in Yamagata and Miyagi [Citation30], whereas it has not been reported in China. APOE Kyoto (Arg25Cys) was reported as the most common mutation in LPG throughout the world, including in southwestern China, Japan, France and the USA. Several other APOE variants have recently been reported in Western countries, including the United States, Russia, Italy, Brazil, and France [Citation31–37].
Currently, no specific treatment is available for LPG. All patients included in this study received ACEIs/ARBs and symptomatic supportive treatment. Traditional treatment options for nephrotic syndrome, such as adrenal corticosteroids, cytotoxic drugs and anticoagulants, have demonstrated no clear curative effect [Citation38] and may even aggravate renal lesions. Reports have indicated that different types of lipid-lowering drugs and blood purification methods can alleviate LPG symptoms. Lipid-lowering therapy has been effective in relieving hyperlipidemia and proteinuria, maintaining renal function and improving the glomerular lesions observed through repeated kidney biopsy [Citation6, Citation39, Citation40]. Studies conducted in China have shown that fenofibrate alleviates proteinuria in patients with APOE Kyoto and APOE Tokyo gene mutations, therefore delaying the progression of renal function. Hu et al. [Citation6] reported on 35 patients with APOE Kyoto gene mutations, of whom 16 underwent fenofibrate treatment for more than 12 months. Six patients achieved complete remission (proteinuria lower than 0.3 g/d), and eight achieved partial remission. Fan et al. [Citation2] reported that compared with the control treatment, fenofibrate treatment resulted in significantly better proteinuria remission and renal survival rates during a follow-up of over 2 years. One patient with the APOE Kyoto mutation and two patients with the APOE Tokyo gene mutation received treatment of fenofibrate, resulting in urinary protein remission and stable renal function, while one patient with the APOE Kyoto mutation in the control group progressed to end-stage renal disease (ESRD). Various methods, such as Staphylococcus A protein immunoadsorption [Citation23, Citation41, Citation42], direct adsorption of whole blood lipoprotein (DALI) [Citation43], double plasma exchange[Citation2, Citation14, Citation44] and heparin-induced in vitro LDL precipitation isolation for LDL apheresis [Citation45] may also reduce proteinuria and improve the prognosis of patients with LPG. However, relapse is common upon discontinuation of treatment. Approximately 50% of patients with LPG gradually progress to ESRD, and the majority of patients in our follow-up exhibited slow progression. While kidney transplantation may improve the quality of life of ESRD patients, patients with postoperative transplanted kidneys may still experience LPG recurrence.
This study has several limitations. The study involved a single cohort, the number of patients was relatively small, and the follow-up time was not long enough. More patients and longer follow-up periods are needed to determine the overall prognosis of LPG in the Chinese population. Previous reports have identified misdiagnoses of other GN such as MPGN or FSGS, thus there may have been potential cases that were missed in this case series.
LPG is underrecognized. The diversity of renal tissue lesions and the coexistence of other renal diseases might be responsible for the lack of understanding of this disease. Kidney biopsy is of vital importance for the diagnosis of LPG. Performing Oil Red O staining on renal tissue was helpful for further confirming the diagnosis. APOE gene analysis is suggested for patients who may have LPG.
Supplemental Material
Download PDF (223.6 KB)Acknowledgements
The authors thank Suxia Wang, Jin Xu, Xu Zhang and Yiyi Ma for their assistance in the evaluation of the pathology reports.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The data underlying this article are available in the article and in its online supplementary material.
Additional information
Funding
References
- Saito T, Sato H, Kudo K, et al. Lipoprotein glomerulopathy: glomerular lipoprotein thrombi in a patient with hyperlipoproteinemia. Am J Kidney Dis. 1989;13(2):1–12. doi: 10.1016/s0272-6386(89)80134-9.
- Fan WJ, Ge YC, Zhou Y, et al. Fenofibrate in the treatment of patients with lipoprotein nephropathy. Chin J Nephrol Dial Transplant. 2016;25:25–29.
- Chen S, Liu ZH, Zheng JM, et al. Genomic analysis of apolipoprotein E gene in patients with lipoprotein glomerulopathy. Chin J Nephrol Dial Transplant. 2005;14:218–223.
- Chen TY, Li GS, Zou YR, et al. Clinical characteristics and apolipoprotein E gene mutation in patients with lipoprotein glomerulopathy. Pract J Clin Med. 2013;10:57–59.
- Wu HY, Yang Y, Hu ZX. The novel apolipoprotein E mutation ApoE chengdu (c.518T > C, p.L173P) in a Chinese patient with lipoprotein glomerulopathy. J Atheroscler Thromb. 2018;25(8):733–740. doi: 10.5551/jat.41996.
- Hu ZX, Huang SM, Wu Y, et al. Hereditary features, treatment, and prognosis of the lipoprotein glomerulopathy in patients with the APOE Kyoto mutation. Kidney Int. 2014;85(2):416–424. doi: 10.1038/ki.2013.335.
- Pan YL, Chen YQ, Zhang H, et al. Screening for apolipoprotein E gene mutations in 4 patients with lipoprotein glomerulopathy. Chin J Nephrol. 2006;22:449–453.
- Zou GM, Zhuo L, Tan M, et al. Clinicopathologic features of lipoprotein glomerulopathy: observation of 6 cases. Natl Med J China. 2018;98:2910–2913.
- Chen YQ, Zhang H, Zhu SL, et al. A3-amino acid deletion of apolipoprotein E found in 3 Chinese lipoprotein glomerulopathy patients. Zhonghua Yi Xue Za Zhi. 2003;83(9):774–777.
- Ku M, Tao C, Zhou AA, et al. A novel apolipoprotein E mutation (p.Arg150Cys) in a Chinese patient with lipoprotein glomerulopathy. Chin Med J (Engl). 2019;132(2):237–239. doi: 10.1097/CM9.0000000000000050.
- Zhang B, Weng DS. Analysis of the relationship between the clinical manifestations and pathological characteristics of lipoprotein nephropathy. Chin J Integ Traditional Western Nephrol. 2005;6:463–465.
- Luo BX, Huang FX, Liu QC, et al. Apolipoprotein E guangzhou (arginine150 proline): a new variant in lipoprotein glomerulopathy. Chin J Nephrol. 2007;23:711–715.
- Ma S, Zhao ZZ, Wang XY, et al. Clinical features and pathological characteristics of lipoprotein glomerulopathy. J Med Forum. 2013;34:35–37.
- Chen J, Zhao XH, He HS, et al. Two cases of double filtration plasmapheresis treatment for familial hyperlipoproteinemia nephropathy. Pract J Med Pharm. 2012;10:930.
- Jia L, Zhang JJ, Shi PP, et al. One case report of lipoprotein glomerulopathy with literature review. J Clin Pediatr. 2016;2:120–124.
- Li Y, Chen J, Zou Y, et al. Lipoprotein glomerulopathy resulting from compound heterogeneous mutations of APOE gene: a case report. Medicine (Baltimore). 2022;101(5):e28718. doi: 10.1097/MD.0000000000028718.
- Wu H, Yang J, Liu YQ, et al. Lipoprotein glomerulopathy induced by ApoE Kyoto mutation in ApoE-deficient mice. J Transl Med. 2021;19(1):97. doi: 10.1186/s12967-021-02765-x.
- Han J, Pan Y, Chen Y, et al. Common apolipoprotein E gene mutations contribute to lipoprotein glomerulopathy in China. Nephron Clin Pract. 2010;114(4):c260–267. doi: 10.1159/000276578.
- Saito T, Oikawa S, Sato H, et al. Lipoprotein glomerulopathy: renal lipidosis induced by novel apolipoprotein E variants. Nephron. 1999;83(3):193–201. doi: 10.1159/000045511.
- Liu ZH. Lipoprotein nephropathy: new disease, new insights. Chin J Nephrol Dial Transplant. 2001;10:301–302.
- Saito T, Sato H, Oikawa S, et al. Lipoprotein glomerulopathy. Report of a normolipidemic case and review of the literature. Am J Nephrol. 1993;13(1):64–68. doi: 10.1159/000168591.
- Bansal P, Dahate P, Raghuwanshi S, et al. Current updates on role of lipids in hematopoiesis. Infect Disord Drug Targets. 2018;18(3):192–198. doi: 10.2174/1871526518666180405155015.
- Hermetet F, Buffière A, Aznague A, et al. High-fat diet disturbs lipid raft/TGF-beta signaling-mediated maintenance of hematopoietic stem cells in mouse bone marrow. Nat Commun. 2019;10(1):523. doi: 10.1038/s41467-018-08228-0.
- Ping H, Liu ZH, Gong RJ, et al. Clinical manifestations and pathological characteristics of lipoprotein glomerulopathy. Chin J Nephrol. 2002;18:403–407.
- Yang JR, He YN, Lin LR, et al. One case of immunoadsorption therapy for severe lipoprotein nephropathy. Chin J Blood Purifi. 2006;5:816.
- Kinomura M, Sugiyama H, Saito T, et al. A novel variant apolipoprotein E Okayama in a patient with lipoprotein glomerulopathy. Nephrol Dial Transplant. 2008;23(2):751–756. doi: 10.1093/ndt/gfm675.
- Boumendjel R, Papari M, Gonzalez M. A rare case of lipoprotein glomerulopathy in a white man: an emerging entity in Asia, rare in the white population. Arch Pathol Lab Med. 2010;134(2):279–282. doi: 10.5858/134.2.279.
- Li MS, Li Y, Liu Y, et al. An updated review and meta analysis of lipoprotein glomerulopathy. Front Med (Lausanne). 2022;9:905007. doi: 10.3389/fmed.2022.905007.
- Ji SM, Liu M, Chen JS, et al. The fate of glomerular mesangial IgA deposition in the donated kidney after allograft transplantation. Clin Transplant. 2004;18(5):536–540. doi: 10.1111/j.1399-0012.2004.00206.x.
- Toyota K, Hashimoto T, Ogino D, et al. A founder haplotype of APOE-Sendai mutation associated with lipoprotein glomerulopathy. J Hum Genet. 2013;58(5):254–258. doi: 10.1038/jhg.2013.8.
- Pêgas KL, Rohde R, Garcia CD, et al. Lipoprotein glomerulopathy: a case report of a rare disease in a Brazilian child. J Bras Nefrol. 2014;36(1):93–95. doi: 10.5935/0101-2800.20140015.
- Sam R, Wu H, Yue L, et al. Lipoprotein glomerulopathy: a new apolipoprotein E mutation with enhanced glomerular binding. Am J Kidney Dis. 2006;47(3):539–548. doi: 10.1053/j.ajkd.2005.12.031.
- Sipovskii VG, Klemina IK, Zverkov RV, et al. A case of diagnosing lipoprotein glomerulopathy in Russia. Arkh Patol. 2016;78(6):52–57. doi: 10.17116/patol201678652-57.
- Pasquariello A, Pasquariello G, Innocenti M, et al. Lipoprotein glomerulopathy: first report of 2 not consanguineous Italian men from the same town. J Nephrol. 2011;24(3):381–385. doi: 10.5301/JN.2011.7772.
- Meyrier A, Dairou F, Callard P, et al. Lipoprotein glomerulopathy: first case in a white European. Nephrol Dial Transplant. 1995;10(4):546–549. doi: 10.1093/ndt/10.4.546.
- Marinaki S, Kalaitzakis E, Kolovou K, et al. A case of lipoprotein glomerulopathy in a Greek caucasian male. Int Urol Nephrol. 2022;54(4):969–970. doi: 10.1007/s11255-021-02930-7.
- Ting JA, McRae SA, Schwartz D, et al. Lipoprotein glomerulopathy, first case report from Canada. Int J Nephrol Renovasc Dis. 2022;15:207–214. doi: 10.2147/IJNRD.S364890.
- Saito T, Matsunaga A, Oikawa S. Impact of lipoprotein glomerulopathy on the relationship between lipids and renal diseases. Am J Kidney Dis. 2006;47(2):199–211. doi: 10.1053/j.ajkd.2005.10.017.
- Usui R, Takahashi M, Nitta K, et al. Five-year follow-up of a case of lipoprotein glomerulopathy with APOE Kyoto mutation. CEN Case Rep. 2016;5(2):148–153. doi: 10.1007/s13730-016-0214-5.
- Arai T, Yamashita S, Yamane M, et al. Disappearance of intraglomerular lipoprotein thrombi and marked improvement of nephrotic syndrome by bezafibrate treatment in a patient with lipoprotein glomerulopathy. Atherosclerosis. 2003;169(2):293–299. doi: 10.1016/s0021-9150(03)00194-1.
- Zhan JF, Liu ZH, Li SJ, et al. Effect of immunoadsorption therapy in patients with lipoprotein glomerulopathy. Chin J Nephrol Dial Transplant. 2006;15:203–209.
- Xin Z, Liu ZH, Li SJ, et al. Successful treatment of patients with lipoprotein glomerulopathy by protein a immunoadsorption: a pilot study. Nephrol Dial Transplant. 2009;2:864–869.
- Academic committee of the people’s liberation army institute of nephrology. Whole blood lipoprotein adsorption therapy for lipoprotein nephropathy. Chin J Nephrol Dial Transplant. 2004;13:493–498.
- Li WC, Wang Y, Han ZW, et al. Apolipoprotein E mutation and double filtration plasmapheresis therapy on a new Chinese patient with lipoprotein glomerulopathy. Kidney Blood Press Res. 2014;39(4):330–339. doi: 10.1159/000355810.
- Russi G, Furci L, Leonelli M, et al. Lipoprotein glomerulopathy treated with LDL-apheresis (heparin-induced extracorporeal lipoprotein precipitation system): a case report. J Med Case Rep. 2009;3:9311.