
Abstract
Acute kidney injury (AKI) is associated with a high mortality rate. Pathologically, renal ischemia/reperfusion injury (RIRI) is one of the primary causes of AKI, and hypoxia-inducible factor (HIF)-1α may play a defensive role in RIRI. This study assessed the role of hypoxia-inducible factor 1α (HIF-1α)-mediated mitophagy in protection against RIRI in vitro and in vivo. The human tubular cell line HK-2 was used to assess hypoxia/reoxygenation (H/R)-induced mitophagy through different in vitro assays, including western blotting, immunofluorescence staining, terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL), and reactive oxygen species (ROS) measurement. Additionally, a rat RIRI model was established for evaluation by renal histopathology, renal Doppler ultrasound, and transmission electron microscopy to confirm the in vitro data. The selective HIF-1α inhibitor LW6 reduced H/R-induced mitophagy but increased H/R-induced apoptosis and ROS production. Moreover, H/R treatment enhanced expression of the FUN14 domain-containing 1 (FUNDC1) protein. Additionally, FUNDC1 overexpression reversed the effects of LW6 on the altered expression of light chain 3 (LC3) BII and voltage-dependent anion channels as well as blocked the effects of HIF-1α inhibition in cells. Pretreatment of the rat RIRI model with roxadustat, a novel oral HIF-1α inhibitor, led to decreased renal injury and apoptosis in vivo. In conclusion, the HIF-1α/FUNDC1 signaling pathway mediates H/R-promoted renal tubular cell mitophagy, whereas inhibition of this signaling pathway protects cells from mitophagy, thus aggravating apoptosis, and ROS production. Accordingly, roxadustat may protect against RIRI-related AKI.
Introduction
Acute kidney injury (AKI), also known as acute renal failure, is defined by a sudden reduction in renal functions within seven days, as evidenced clinically by an increased serum creatinine level and decreased urine production [Citation1]. Pathologically, renal ischemia/reperfusion injury (RIRI) contributes to the occurrence of AKI [Citation2]. The rapid loss of renal function contributes to a high mortality rate, and among patients who survive, AKI is a risk factor for the development of chronic kidney disease (CKD) [Citation3]. Histologically, renal tubule cells are vulnerable to damage during RIRI, including in the forms of tissue hypoxia and mitochondrial dysfunction [Citation4]. To date, effective treatment options for AKI are still lacking. Hence, further investigation into the risk factors and pathogenesis of AKI may support the discovery of the molecular mechanisms underlying AKI and thus, the identification of novel approaches for managing AKI.
Any causes of a sudden reduction in the glomerular filtration rate or renal function as well as urinary tract obstruction collectively result in kidney damage and RIRI, thus contributing to AKI development [Citation5]. Increased expression of hypoxia-inducible factor (HIF)-1 has been reported to be a hallmark of RIRI [Citation6]. The HIF-1 protein contains two subunits, i.e., hypoxia-inducible factor 1α (HIF-1α) (an oxygen-inducible subunit) and HIF-1β (a normally and always expressed subunit in the cells) [Citation7]. In AKI, HIF-1α is thought to function to protect against RIRI by regulating the expression of HIF-1α-targeting genes, apoptosis-related genes, and macrophage infiltration-related genes as well as the expression of vascular cell adhesion molecule 1 (VCAM1) [Citation8,Citation9]. Activation of endothelial HIF-1 signaling by inhibiting prolyl hydroxylase domain-containing protein 2 expression was demonstrated to defend against RIRI by suppressing both the expression of proinflammation-related genes and inflammatory cell recruitment into the kidney [Citation10]. However, to date, the underlying pathogenesis of AKI and the protective role of HIF-1α against AKI remain to be determined.
To this end, it is well documented that maintaining normal mitochondrial functions is critical for supplying cell energy and survival. In contrast, any damage to the mitochondria can promote cellular production of reactive oxygen species (ROS) and cell death factors, which induce cell apoptosis [Citation11]. Mitophagy, the autophagy-related degradation of damaged, incompetent, or stressed mitochondria, helps to maintain cell health (homeostasis) to avoid the potential accumulation of dysfunctional mitochondria in cells [Citation12,Citation13]. Previous studies have demonstrated that mitophagy is a significant and important defense mechanism against RIRI in AKI [Citation12,Citation14]. In addition, a previous study showed that the newly discovered FUN14 domain-containing 1 (FUNDC1) protein in the outer mitochondrial membrane facilitates the receptor-dependent mitophagy in response to cell hypoxia [Citation15]. FUNDC1 directly interacts with light chain 3 (LC3), an autophagy-promoting protein, to induce mitophagy [Citation16]. Thus, FUNDC1 is vital for mitophagy, and aberrant FUNDC1 expression, phosphorylation, and regulation are associated with the development, progression, and prognosis of various human diseases [Citation17–23]. A previous study showed that silencing fundc1 expression caused deficiencies in cardiomyocyte mitophagy, a decrease in the protective effect of hypoxia adaptation against acute myocardial infarction, and an increase in cardiomyocyte apoptosis in vivo [Citation17]. Deficiencies in FUNDC1 expression and activity have been shown to impair the mitochondrial value and worsen dietary-related obesity and metabolic syndrome [Citation24]. Moreover, hypoxia/ischemia has been demonstrated to induce FUNDC1-related mitophagy [Citation15].
In the current study, we assessed the effects of inhibition of HIF-1α-induced mitophagy as well as FUNDC1 expression and activity in vitro and in vivo to elucidate their protective roles against RIRI-induced cell and tissue damage. Our results provide insight for a unique future strategy of targeting HIF-1α and FUNDC1 signaling for the therapeutic management of AKI.
Materials and methods
Cell line and in vitro H/R model
The human proximal tubular cell line HK-2 was originally obtained from Fuheng Biology (Shanghai, China) and maintained in Dulbecco’s modified Eagle medium/F-12 (DMEM/F12; Gibco, Waltham, MA; catalog number: 812209) containing 10% fetal bovine serum (FBS; Gibco, Waltham, MA; catalog number: ST30-3302) in a humidified incubator supplied with 95% air and 5% CO2 at 37 °C. For establishment of the in vitro hypoxia/reoxygenation (H/R) model, HK-2 cells were grown in DMEM/F12 with a low level of glucose (5.5 mM glucose) in a humidified incubator containing 1% O2 and 5% CO2 at 37 °C for 24 h. Thereafter, the cell medium was replaced with the normal complete culture medium, and HK-2 cells were moved to a humidified incubator supplied with 21% O2 and 5% CO2 at 37 °C for up to 48 h before use in subsequent experiments.
Drug treatment and gene transfection
The selective HIF-1α inhibitor LW6 (Selleck, Houston, TX; catalog number: S844101) was dissolved in dimethyl sulfoxide (DMSO) to prepare a stock solution of 10 mM LW6. The control and H/R cells were treated with or without LW6 at a dose of 20 µM.
For gene transfection, a plasmid carrying FUNDC1 cDNA was purchased from GeneCopoeia (Rockville, MD, catalog number: 20922-1). Specifically, the human FUNDC1 cDNA (access number: NM-173794) was cloned using the polymerase chain reaction and inserted into the CMV-MCS-3FLAG-SV40-neomycin lentiviral vector between the Agel and EcoRI sites. Prior to transfection, HK-2 cells were plated into six-well plates at a density of 1 × 106 cells/well and cultured in complete DMEM/F12 overnight. The medium was then replaced with serum-free and antibiotic-free medium, and the cells were cultured for 1 h before transient transfection with 4 µg of the FUNDC1-overexpression plasmid for 6 h using polyethyleneimine (Proteintech, Wuhan, China, catalog number: PR40001) under normal culture conditions.
Western blotting
Total cellular protein was obtained from renal cortex tissues or HK-2 cells using radioimmunoprecipitation assay buffer (Solarbio, Beijing, China, catalog number: R0010-100). After quantitation, the protein extracts were separated by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred onto the polyvinylidene difluoride membranes (Millipore, Billerica, MA; catalog number: ISEQ00010). For western blotting, the membranes were first incubated in 5% skim milk in phosphate-buffered saline (PBS) at room temperature for 1 h before incubation in primary antibody solution at 4 °C overnight. The following primary antibodies were used: anti-HIF-1α (cat. no. #ab179483; Abcam, Cambridge, UK), anti-FUNDC1 (cat. no. #ab224722; Abcam, Cambridge, UK), anti-Light chain 3B (LC3B) (cat. no. #ab48394; Abcam, Cambridge, UK), and anti-cleaved caspase-3 (cat. no. #ab13847; Abcam, Cambridge, UK), all diluted in PBS at 1:1000; anti-voltage-dependent anion channel (VDAC; cat. no. #AP10866; Proteintech, Wuhan, China) diluted at 1:2000; and anti-β-actin (cat. no. #bs-0061R; Bioss, Beijing, China) diluted at 1:10,000. After incubation in the primary antibody solution, the membranes were incubated in a solution containing the secondary antibody, Licor goat anti-rabbit conjugated with IRDye 800 CW (cat. no. #926-32211; Licor Biosciences, Lincoln, NE). Then, the intensity of protein expression bands was quantified using the Odyssey Clx imaging system (Licor Biosciences, Lincoln, NE). The housekeeping protein β-actin was used as a loading control. The relative densities of all bands were quantified using ImageJ software (version 1.8.0; National Institutes of Health, Bethesda, MD).
Immunofluorescence staining
After the specified treatments, HK-2 cells were washed with PBS, fixed in 4% fresh paraformaldehyde solution for 10 min, then treated with 0.01% Triton X-100 solution for 15 min, and finally incubated in 5% bovine serum albumin at room temperature for 1 h. Next, the cell samples were incubated overnight at 4 °C in primary antibody solution. The primary antibodies used were: anti-HIF-1α (cat. no. #66730-1-Ig; Proteintech, Wuhan, China), anti-FUNDC1 (cat. no. #ab224722; Abcam, Cambridge, UK), and anti-LC3B (cat. no. #83506; Cell Signaling Technology, Danvers, MA), all diluted in PBS at 1:100. After incubation in primary antibody solution, the samples were washed thrice in PBS and then incubated with an anti-Alexa Fluor 488- or Alexa Fluor 594-coupled secondary antibody at room temperature for 1 h. Thereafter, the samples were incubated in 4,6-diamidino-2-phenylindole (DAPI) solution and sealed by a coverslip using a fluorescence-mounting medium (Zsbio, Beijing, China, catalog number: ZLI-9557). The cells were reviewed and scored under a fluorescence microscope (STEMI 305, ZEISS, Oberkochen, Germany).
To visualize mitochondria or lysosomes, HK-2 cells were first cultured in 15-mm dishes and treated with 500 nM MitoTracker Red or 50 nM LyoTracker Green (cat. no. #C1049B and C1047S, respectively; Beyotime, Shanghai, China), following the manufacturer’s protocols. Next, the HK-2 cells were viewed and scored under a two-photon laser confocal microscope (LSM880, Carl Zeiss, Oberkochen, Germany).
For double-labeling renal tissue immunofluorescence, paraffin-embedded renal tissue sections (4 µm in thickness) were dewaxed in dimethylbenzene and rehydrated in a series of ethanol solutions in water (100–50%). The sections were then subjected to antigen retrieval using ethylenediaminetetraacetic acid and incubated in primary antibody solution containing anti-FUNDC1, anti-LC3B, anti-VDAC, or anti-lysosome-associated membrane protein 1 (LAMP1) (all diluted 1:100 in PBS) at 4 °C overnight. After incubation in secondary antibody solution, images of three randomly selected visual fields were photographed under a fluorescence microscope (ZEISS, Oberkochen, Germany), and staining was quantified.
Hoechst and TUNEL staining
Changes in the cell apoptosis rate were assessed by terminal deoxynucleotidyl transferase-mediated dUTP nick end labeling (TUNEL) assay. Briefly, the cells were seeded in six-well plates, treated, and stained with Hoechst 33258 (Beyotime, China, catalog number: C1017) stain, as described in a previous study [Citation25]. The stained cells were viewed and scored under a fluorescence microscope (IX53, Olympus, Tokyo, Japan).
For evaluation of apoptosis in renal tissue samples, paraffin-embedded kidney sections (4-µm thick) were dewaxed in dimethylbenzene, rehydrated in a series of ethanol solutions (100–50%), stained with a TUNEL kit (Roche, Basel, Switzerland, catalog number: 11684817910) following the manufacturer’s protocol, and counterstained with DAPI. The stained cells were viewed and scored under a fluorescence microscope (Olympus, Tokyo, Japan).
Measurement of the intracellular ROS
The level of ROS production in HK-2 cells was measured with the dichloro-dihydrofluorescein diacetate (DCFH-DA) reagent [Citation26]. Briefly, after treatment, HK-2 cells were treated with 10 µM DCFH-DA (Solarbio, Beijing, China, catalog number: BC0170) at room temperature for 20 min, and thereafter, images were taken using an inverted fluorescence microscope (Olympus, Tokyo, Japan) for quantitation.
Animals and RIRI model
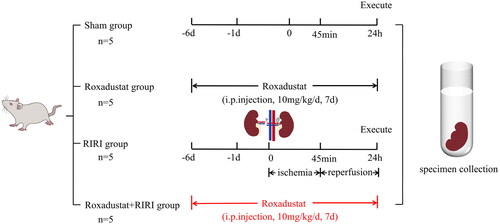
The experimental protocol for this study was previously approved by the Ethics Committee of Lanzhou University Second Hospital (ID #D2022-045) and followed the Chinese Council on Animal Care guidelines. Additionally, this study was reported following the ARRIVE guidelines. Briefly, male Wistar rats aged 10 weeks (weighing 205 ± 15 g) were obtained from the Lanzhou Veterinary Research Institute, Chinese Academy of Agricultural Sciences (Lanzhou, China). The animals were housed in standard laboratory cages with free access to food and water. As shown in , we randomly divided them into sham, roxadustat treatment, RIRI model, and roxadustat + RIRI groups (n = 5 in each group). Roxadustat (cat. no. #S1007; Selleck Chemicals, Houston, TX) was dissolved in DMSO to obtain the stock solution and then added into PBS to prepare a working solution of 1 mg/mL. The rats in the roxadustat and roxadustat + RIRI treatment groups received intraperitoneal (i.p.) injections of roxadustat (10 mg/kg/day) daily for seven days before renal ischemia/reperfusion. The RIRI model of rats was established as described previously [Citation27] by tightening of left renal pedicles for 45 min with an atraumatic vascular clamp and removing the right kidney during the ischemic period. The clips were removed, and the left kidney was watched for 5 min to observe the occurrence of reperfusion. The sham group of rats was subjected to the same laparotomy procedure as the model animals without the RIRI procedure. After 24 h of reperfusion, the animals were sacrificed by intravenous injection of an overdose of pentobarbital (Shanghai Pharmaceutical New Asia Pharmaceutical Co., Ltd., Shanghai, China, catalog number: H31020502). A blood sample was withdrawn from the abdominal aorta, and the left kidney was resected for immediate use in experiments or immediately frozen in liquid nitrogen and stored at −80 °C for use in later experiments.
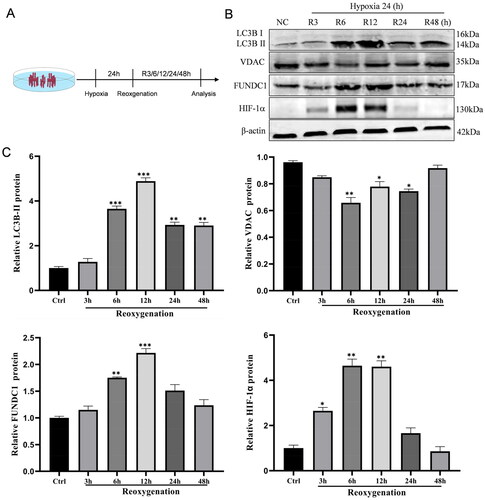
Figure 1. Hypoxia/reoxygenation (H/R) induction of mitophagy in HK-2 cell. (A) Illustration of the in vitro H/R procedure. HK-2 cells were grown for 24 h in hypoxic conditions and then returned to reoxygenation conditions for the indicated periods (3, 6, 12, 24, and 48 h), while cells growing under normal conditions were used as a control. (B, C) Western blots. The H/R and control cells were subjected to protein extraction and western blot analysis of the expression levels of HIF-1α, FUNDC1, VDAC, and LC3B proteins. The data are quantified as mean ± SEM. *p < .05, **p < .01, and ***p < .001 vs. control.
Renal histopathology
The serum level of creatinine was detected with spectrophotometry in a clinical lab. The tissue morphology was assessed in 4-µm-thick paraffin-embedded renal tissue sections after staining with the PAS stain or hematoxylin and eosin (HE). The tubular injury was scored according to the percentage of damaged tubules, as follows: 0, no damage; 1, <25% damaged tubules; 2, 25–49% damaged tubules; 3, 50–75% damaged tubules; and 4, >75% damaged tubules, according to the method reported in a previous study [Citation21]. Two pathologists reviewed and scored the stained tissue sections under a light microscope (Olympus, Tokyo, Japan) using two randomly selected microscopic fields (magnification, ×200) in a double-blinded fashion.
Renal Doppler ultrasonography
At the end of 24 h of reperfusion, renal Doppler ultrasonography using an M-mode ultrasound system with a 22-MHz linear transducer (EPIQ7, Philips, Amsterdam, The Netherlands) was performed to evaluate the degree of kidney injury in rats after exposure to 2% isoflurane. The investigator was blinded to the animal assignment groups. The ultrasonography examination included measurement of the peak-systolic velocity (PSV), end-diastolic velocity (EDV), and renal resistive index (RRI). The PSV and EDV were calculated as cm/s, while the RRI was calculated as (PSV − EDV)/PSV.
Transmission electron microscopy (TEM)
After resection from the experimental rats, the kidneys were cut into 1 mm3 pieces and incubated in 3% glutaraldehyde/PBS solution (Sigma, St. Louis, MO) at 4 °C overnight. On the next day, the samples were washed in PBS thrice, further incubated in 1% (w/v) osmium tetroxide (Sigma, St. Louis, MO) at room temperature for 2 h, and then dehydrated in a series of ethanol solutions (70–100%). The dehydrated tissue samples were embedded in propylene oxide and Epon (Sigma, St. Louis, MO), and solidified at 60 °C for 48 h in 100% epoxy resin. After that, ultrathin sections were cut and stained using uranyl acetate and lead citrate. The stained sections were viewed by TEM (HT7800, HITACHI, Tokyo, Japan).
Statistical analysis
The data are expressed as the mean ± standard error of the mean (SEM) and were statistically analyzed using Prism 8.2.1 (GraphPad Software, San Diego, CA, www.graphpad.com). Student’s t-test was performed to detect statistical differences between two groups, and one-way analysis of variance analysis was used to detect statistical differences among multiple groups. A value of p < .05 indicated statistical significance.
Results
Effect of H/R on regulation of HIF-1α and mitophagy-related proteins
We first assessed the effect of H/R on the regulation of HIF-1α and mitophagy-related proteins in the human tubular cell line HK-2 (). After H/R, the HIF-1α level was significantly increased in HK-2 cells and peaked at 6 h and 12 h after reoxygenation (). HIF-1α expression gradually decreased and reached the initial level at 48 h after reoxygenation. Moreover, the expression of FUNDC1 and LC3BII proteins followed a trend similar to that of HIF-1α, first increasing and then decreasing after H/R. However, the level of the mitochondrial VDAC protein showed a decreasing trend followed by an increasing trend, with the lowest level observed at 6 h after reoxygenation and a return to the initial level at 48 h after reoxygenation.
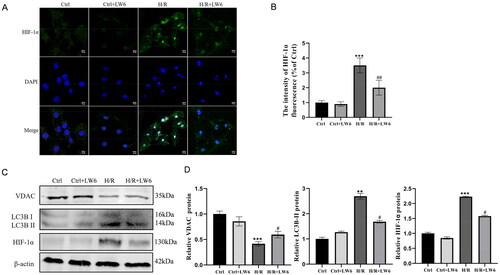
Figure 2. LW6 reduction of hypoxia/reoxygenation (H/R)-induced mitophagy in HK-2 cells. (A) Immunofluorescence. The H/R and control cells were treated with 20 µM LW6 for 12 h and then subjected to immunofluorescence staining. The data showed HIF-1α accumulation in the H/R-treated cells, while LW6 effectively inhibited the H/R-induced HIF-1α expression. Scale bar = 10 µm. (B) Quantified data from the assay as mean ± SEM. (C, D) Western blots. The H/R and control cells were treated with 20 µM LW6 for 12 h and then subjected to protein extraction and western blot analysis of the expression levels of HIF-1α, FUNDC1, VDAC, and LC3B proteins. **p < .01 and ***p < .001 vs. control; #p < .05 and ##p < .01 vs. H/R group.
HIF-1α mediates H/R-facilitated renal tubular cell mitophagy
Based on the observed changes in HIF-1α expression after H/R, we assessed the role of HIF-1α in H/R-induced mitophagy in vitro after 6 h of reoxygenation. We treated HK-2 cells with the selective HIF-1α inhibitor LW6 and found that LW6 could significantly block H/R-induced HIF-1α expression (). Moreover, western blot analysis showed that LW6 effectively inhibited expression of the H/R-induced autophagy marker LC3BII but upregulated the mitochondrial protein VDAC ().
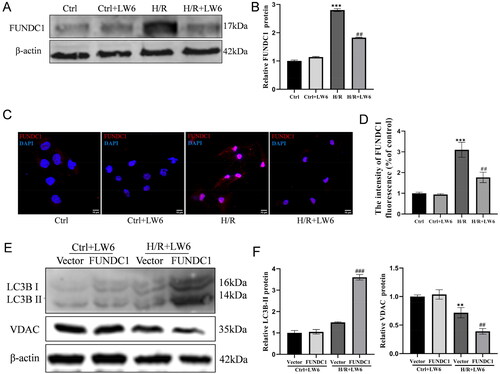
Figure 3. FUNDC1 reversal of the decrease in hypoxia/reoxygenation (H/R)-induced mitophagy in HK-2 cells. (A, B) Western blots. The H/R and control cells were treated with 20 µM LW6 for 12 h and then subjected to protein extraction and western blot analysis. The data showed the inhibitory effect of HIF-1α inhibition by LW6 on H/R-induced FUNDC1 expression. (C) Immunofluorescence. The H/R and control cells were treated with 20 µM LW6 for 12 h and then subjected to immunofluorescence staining. The data showed changes in FUNDC1 expression in H/R-treated cells with or without LW6 treatment. Scale bar = 10 µm. (D) Quantified immunofluorescence data. (E, F) Western blots. The H/R and control cells were grown, treated with 20 µM LW6 for 12 h, transfected with a plasmid carrying FUNDC1 cDNA for FUNDC1 overexpression, and then subjected to protein extraction and western blot analysis. The data showed that FUNDC1 overexpression reversed the changes in LC3B and VDAC expression in H/R-treated cells. Data are quantified as mean ± SEM. ***p < .001 vs. control; ##p < .01 vs. H/R group in B and D. **p < .05 vs. vector group without H/R; ##p < .01 and ###p < .001 vs. vector group with H/R in F.
FUNDC1 is a downstream regulator of HIF-1α in mitophagy in renal tubular cells
As FUNDC1 mediates ischemia/hypoxia-induced mitophagy [Citation15], we assessed FUNDC1 expression in this context. We found that the level of FUNDC1 protein was upregulated in H/R-treated HK-2 cells, whereas such increased expression was dramatically blocked by treatment with the selective HIF-1α inhibitor LW6 (). In addition, induced overexpression of FUNDC1 reversed the effect of LW6 on LC3BII and VDAC levels ().
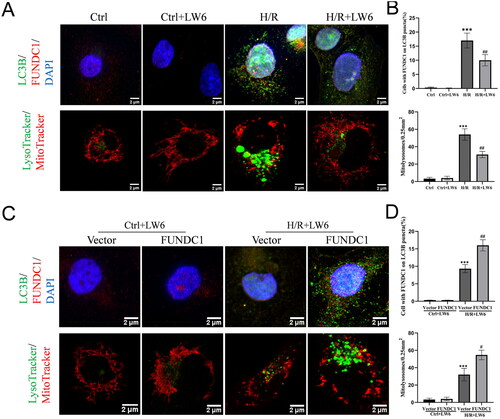
Figure 4. HIF-1α/FUNDC1 signaling mediates mitolysosome formation in HK-2 cells. (A, B) Immunofluorescence. Hypoxia/reoxygenation (H/R) and control cells were treated with 20 µM LW6 for 12 h and then subjected to double labeling of LC3B and FUNDC1, as well as MitoTracker and LyoTracker. The data showed the inhibitory effect of LW6 on the formation of H/R-induced mitolysosomes. (C, D) Immunofluorescence. The H/R and control cells were transfected with the FUNDC1 cDNA plasmid, and double-labeling of LC3B and FUNDC1, as well as MitoTracker and LyoTracker labeling were performed. The data showed that FUNDC1 overexpression reversed the inhibitory effect of LW6 on H/R-induced mitolysosome formation. Scale bar = 2 µm. Data are quantified as mean ± SEM. ***p < .001 vs. control; ##p < .01 vs. H/R group in B. ***p < .001 vs. vector group without H/R; #p < .05 and ##p < .01 vs. vector group with H/R in D.
HIF-1α/FUNDC1 signaling mediates the H/R-induced increase in mitolysosome formation
Next, we simultaneously observed the locations LC3B and FUNDC1 expression in HK-2 cells using LysoTracker Green and MitoTracker Red, respectively (). We found dramatic mitophagy induction and colocalization of LC3B and FUNDC1 proteins in the mitochondria and lysosomes. However, such cell activities were blocked after treatment with the selective HIF-1α inhibitor LW6 (). In contrast, FUNDC1 overexpression in HK-2 cells reversed the LW6-mediated inhibition of H/R-induced mitophagy ().
Figure 5. Effect of HIF-1α/FUNDC1 signaling in attenuating H/R-induced apoptosis and ROS production in vitro. (A, B) Hoechst staining. The H/R and control cells were treated with 20 µM LW6 for 12 h and subjected to Hoechst staining. The data showed the inhibitory effect of LW6 on H/R-induced tubular apoptosis. Scale bar = 10 µm. Arrows indicate apoptotic cells. (C, D) Western blots. The H/R and control cells were treated with 20 µM LW6 for 12 h and subjected to western blot analysis of cleaved caspase-3 expression. The data showed the effect of HIF-1α inhibition on the increase in H/R-induced cleaved caspase-3. (E, F) Immunofluorescence. The H/R and control cells were treated with 20 µM LW6 for 12 h and subjected to immunofluorescence staining. The data showed the effect of HIF-1α inhibition on H/R-induced ROS production. Scale bar = 6 µm. (G, H) Hoechst staining. The H/R and control cells were grown and transfected with a plasmid carrying FUNDC1 cDNA for FUNDC1 overexpression for 6 h and subjected to Hoechst staining. The data showed the effect of FUNDC1 overexpression on H/R-induced apoptosis. Scale bar = 10 µm. (I, J) Western blots. The H/R and control cells were grown and transfected with a plasmid carrying FUNDC1 cDNA for FUNDC1 overexpression for 6 h and subjected to western blotting. The data showed the effect of FUNDC1 overexpression on the increase in H/R-induced cleaved caspase-3. (K, L) Immunofluorescence. The H/R and control cells were transfected with the FUNDC1 cDNA plasmid for 6 h and subjected to immunofluorescence staining. The data showed the effect of FUNDC1 overexpression on H/R-induced ROS production. Scale bar = 6 µm. **p < .01 and ***p < .001 vs. control; #p < .05 and ##p < .01 vs. H/R group in B, D, and F. ***p < .001 vs. vector group without H/R; #p < .05 and ##p < .01 vs. vector group with H/R in H, J, and L.
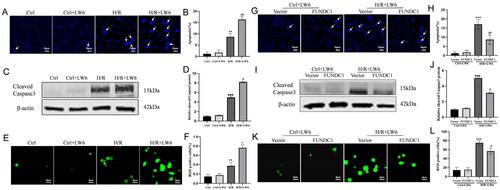
HIF-1α/FUNDC1 signaling acts to reduce H/R-induced apoptosis and ROS production in vitro
We further evaluated the function of HIF-1α/FUNDC1 signaling to protect against H/R-increased cell apoptosis and ROS production by assessing changes in H/R-increased HK-2 cell apoptosis and ROS levels. As shown in , H/R dramatically induced apoptosis among HK-2 cells, which was further enhanced after treatment with the selective HIF-1α inhibitor LW6. Consistently, the level of the cleaved caspase 3 protein (a marker of apoptosis) was dramatically increased in H/R-treated HK-2 cells and further increased after LW6 treatment (). Similarly, H/R dramatically enhanced ROS production in HK-2 cells, and this increase was exacerbated by treatment with LW6 (). Furthermore, overexpression of FUNDC1 protein suppressed the stimulatory effects of LW6 on cell apoptosis, cleaved caspase-3 expression, and ROS production ().
Figure 6. Roxadustat activates HIF-1α/FUNDC1 signaling in a rat renal I/R model. (A) Serum creatinine levels. Four groups of rats were prepared (as detailed in ‘Materials and methods’ section), and serum samples were collected for measurement of the creatinine level. Data are quantified as mean ± SEM. (B, C) Periodic acid-Schiff (PAS) staining. Renal tissues were resected from the rats and subjected to tissue processing and PAS staining. Data are quantified as mean ± SEM. Scale bar = 100 µm (D, E) Renal Doppler ultrasound. The four groups of rats were prepared and subjected to renal Doppler ultrasound. The graphs show the quantified data. (F, G) Western blots. The renal cortex tissues were resected from the rats and subjected to protein extraction and western blot analysis. Data are quantified as mean ± SEM. (H, I) Immunofluorescence. Resected tissues were double-labeled with anti-LC3B and anti-FUNDC1, anti-LC3B and anti-VDAC, or anti-VDAC and anti-LAMP1. Scale bar = 20 µm. The graphs show the quantified data. (J) Transmission electron microscopy (TEM) for evaluation of mitochondrial morphology in renal tubular epithelial cells. White arrows indicate normal mitochondria; yellow arrows indicate damaged mitochondria; and red arrows indicate the mitophagosome. Scale bar = 500 nm. (K, M) TdT-mediated dUTP-nick-end labeling (TUNEL) assay. Renal cortex tissues were resected and subjected to tissue processing and TUNEL assay. The graph in M depicts the quantified data. Scale bar = 100 µm. (L) Western blots. Renal cortex tissues were resected and subjected to protein extraction and western blot analysis. Data are quantified as mean ± SEM. (N) The graph in N depicts the quantified data. *p < .05, **p < .01, and ***p < .001 vs. sham group. #p < .05, ##p < .01, and ###p < .001 vs. RIRI group.
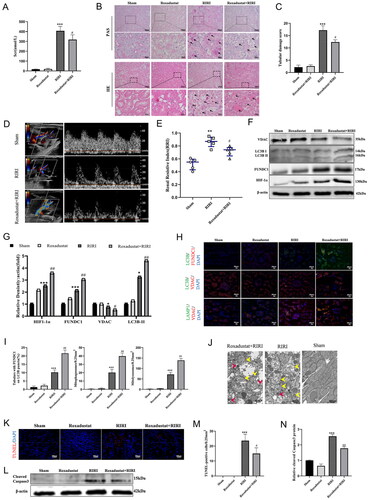
Roxadustat activates HIF-1α/FUNDC1-facilitated mitophagy and attenuates apoptosis in vivo
An animal RIRI model was established to confirm the in vitro results observed in this study. In our in vivo model, the HIF polyhydroxylase inhibitor roxadustat significantly reduced the serum creatinine level of the RIRI rats (p = .03; ). The levels of the tubular epithelial vacuolar degeneration, tubular necrosis, and cast formation also were decreased in the roxadustat-treated RIRI rats compared with the corresponding levels in the untreated RIRI rats (). Hematoxylin and eosin and periodic acid-Schiff (PAS) staining further confirmed these findings (). Additionally, the RRI of the roxadustat-treated RIRI rats was less than that of the untreated RIRI rats (0.71 ± 0.07 vs. 0.86 ± 0.08, p = .4), as shown in . Western blot analysis also demonstrated that roxadustat pretreatment upregulated the levels of HIF-1α, FUNDC1, and LC3BII but downregulated the level of VDAC in the renal cortex of the RIRI animals (). In addition, LC3B co-localized intracellularly with FUNDC1 and VDAC proteins, while VDAC and LAMP1 proteins also co-localized intracellularly (). The TEM evaluation also showed that roxadustat treatment alleviated the RIRI-induced histology changes in the mitochondria (mitochondrial swelling and loss of mitochondrial cristae) of the tubular epithelia and that the level of mitophagosomes was increased in the roxadustat-treated RIRI rats (). However, roxadustat treatment also decreased the levels of cleaved caspase-3 and TUNEL-positive cells in the renal tubular tissues (), indicating that roxadustat treatment alleviates RIRI-induced apoptosis.
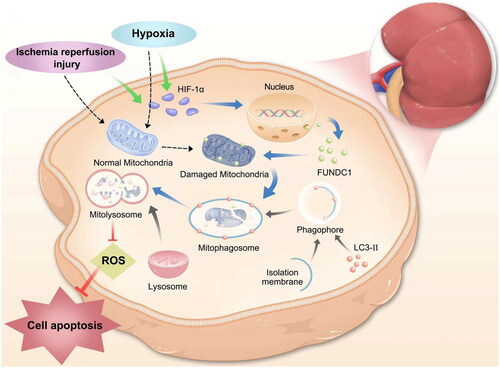
Figure 7. Illustration of HIF-1α/FUNDC1 signaling in vitro. The graph illustrates the signal transduction pathways of ischemia/reperfusion injury or hypoxia-induced HIF-1α expression. HIF-1α/FUNDC1 signaling mediates mitophagy in renal tubular cells. Inhibition of HIF-1α/FUNDC1 signaling decreases cellular ROS production and apoptosis, thereby maintaining cell viability.
Discussion
The results of the present study demonstrate that HIF-1α/FUNDC1 signaling can mediate H/R-induced mitophagy in kidney tubular cells, and inhibiting this signaling pathway can protect cells and tissues from mitophagy, apoptosis, and ROS production. This finding suggests that roxadustat should be further assessed as a novel potential approach for the future clinical control of RIRI-related AKI. Indeed, previous studies reported that HIF-1α can protect the kidney against RIRI [Citation9,Citation28]. Additional studies also showed that direct injection of recombinant HIF-1α protein had a protective effect in an ischemic AKI rat model [Citation29], while HIF-1α knockout in kidney tubular cells significantly aggravated I/R injury in a model animal [Citation30]. Our findings are consistent with these studies in demonstrating that HIF-1α can play a protective effect against AKI in renal tubular cells. This conclusion is supported by our findings that LW6 inhibition of HIF-1α activity enhanced apoptosis and ROS in H/R-induced HK-2 cells and stabilization of HIF-1α protein using roxadustat alleviated RIRI in the rat model of RIRI. In addition, growing evidence suggests that HIF-1α promotes mitophagy by upregulating mitophagy-related proteins [Citation31]. In cells, mitophagy can be activated via two distinct pathways, i.e., the receptor-dependent and ubiquitin-dependent pathways. Various outer mitochondrial membrane proteins, like BCL2/adenovirus E1B 19-kDa protein-interacting protein 3 (BNIP3) and FUNDC1, can directly bind to LC3B, while ubiquitin-dependent mitophagy occurs via the PTEN-induced kinase 1 (PINK1) and parkin-related pathway. One study implicated HIF-1α in PINK1/parkin-mediated mitophagy [Citation32]. Moreover, a recent study revealed that HIF-1α can regulate PINK1 and mitophagy in hepatocellular carcinoma cells [Citation33]. In this regard, our current study demonstrates that H/R-induced HIF-1α expression mediates the H/R-induced mitophagy in renal tubular cells. Additionally, we also revealed that intervention of H/R-upregulated FUNDC1 by using the selective HIF-1α inhibitor LW6 concomitantly blocked the induced expression of other mitophagy-related proteins in HK-2 cells. In contrast, overexpression of FUNDC1 antagonized the reduction of LW6-mediated mitophagy-related proteins. These findings suggest that H/R-induced HIF-1α expression and mitophagy might occur via induction of FUNDC1 expression in HK-2 cells, consistent with the findings of a previous study [Citation34].
Mitophagy, a fundamental process removes damaged mitochondria through the autophagosomes and lysosomes to maintain cellular and mitochondrial homeostasis [Citation35]. However, aberrant mitophagy can lead to loss of function and structural integrity among renal tubular epithelial cells [Citation36]. In our current study, TEM analysis revealed mitochondrial swelling and mitochondrial cristae loss in the renal tubular epithelia in a rat AKI model. Moreover, our current data also demonstrated the role of HIF-1α/FUNDC1 signaling in mediating H/R-induced mitophagy and reducing ROS production. ROS are generated and produced by the mitochondria through the electron transport chain, which induces oxidative phosphorylation or downstream chain reactions [Citation11], but injured and stressed mitochondria can produce and release ROS. In contrast, because mitophagy may prevent ROS production and release from cells, the removal of injured mitochondria via mitophagy is a key protective effect of ischemic preconditioning in the kidney [Citation12,Citation37]. Furthermore, FUNDC1-mediated mitophagy has been proven to attenuate oxidative stress-induced mitochondrial dysfunction and apoptosis in a cell septic cardiomyopathy model [Citation21]. In addition, empagliflozin, an antidiabetes drug used in the clinic, has been reported to diminish the effects of cardiac microvascular ischemia/reperfusion through FUNDC1-induced mitophagy, neutralized ROS, and suppression of apoptosis [Citation38]. Our current data further support these findings by showing that HIF-1α/FUNDC1 signaling in H/R-induced mitophagy blocks the increase in ROS. However, the association of mitophagy with ROS production requires further exploration. A previous study demonstrated that ROS are able to upregulate and downregulate HIF-1α expression under both normoxic and hypoxic conditions, respectively, indicating the role of ROS in mitophagy [Citation7]. A close relationship between ROS and autophagy has also been reported, in which oxidative stress leads to increased autophagy and autophagy leads to reduced ROS levels [Citation39]. Imbalance among mitophagy, ROS, and mitochondrial injury also was suggested to promote aging [Citation40]. Also, FUNDC1-mediated mitophagy was shown to have a protective effect in ischemia/reperfusion-induced damage to the intestine and brain [Citation18]. Another study showed that NOD-like receptor X1/FUNDC1 signaling also regulates mitophagy, reduces the increase in injured mitochondria and apoptosis, and alleviates intestinal ischemia/reperfusion [Citation19]. Moreover, HIF-1α/FUNDC1-related mitophagy plays a protective role in the retina against anti-vascular endothelial growth factor therapy-induced hypoxia [Citation33]. The results of our current study support these previously reported findings [Citation18,Citation19,Citation33]. Furthermore, FUNDC1-induced mitophagy was found to be protective or destructive in cardiac ischemia injury; e.g., several molecules, including mammalian STE20-like kinase 1, NR4A1, and Ripk3, aggravate cardiac ischemia injury through suppression of FUNDC1-mediated mitophagy [Citation41–43]. Lost peroxisome proliferator-activated receptor-γ expression also leads to FUNDC1-related mitophagy in ischemia/reperfusion-increased cardiac damages, which in turn further induces mitochondrial respiratory activities and ATP production, resulting in myocardial dysfunction [Citation44]. Additionally, electroacupuncture preconditioning was able to reduce myocardial ischemia/reperfusion damage by inhibiting mTORC1–ULK1–FUNDC1 pathway-regulated mitophagy [Citation20]. These data indicate that FUNDC1-regulated mitophagy may have different effects in different tissues.
Roxadustat has been successfully used to treat renal anemia in CKD and dialysis patients [Citation45,Citation46]. Intraperitoneal injection of Roxadustat into animals also showed a protective effect in cisplatin/contrast-induced acute kidney damage and apoptosis via HIF-1α upregulation [Citation21,Citation47]. In another study, roxadustat treatment blocked the AKI-to-CKD transition by maintaining vascular regeneration and antioxidative activity [Citation48]. Indeed, the results of our current study demonstrated that RIRI induced HIF-1α protein expression and that roxadustat treatment further increased HIF-1α protein in vivo, further supporting the conclusions of previous studies [Citation21,Citation47–49]. Nevertheless, our current study does have some limitations. For example, HK-2 cells lack the morphological characteristics of primary renal tubular epithelial cells in vivo, R0 samples should be added as positive control, and no HIF-1α agonists were used for further validation in vitro experiments. Moreover, this study did not analyze functional mitochondrial damage, such as changes in mitochondrial membrane potential. Furthermore, only one dose of roxadustat (10 mg/kg) was tested in our in vivo experiment, and thus, these results need to be further verified.
Conclusions
The current study demonstrated the effect of HIF-1α/FUNDC1 signaling in mediating H/R-induced mitophagy via inhibition of tubular cell apoptosis and ROS generation in vitro and in vivo (). Our findings indicate the potential of Roxadustat as a useful therapeutic agent for future clinical control of RIRI-AKI.
Figure 8. Illustration of the rat model of renal ischemia/reperfusion injury (RIRI). The rats were divided into the following groups: sham, roxadustat treatment, RIRI model, and roxadustat + RIRI (n = 5 per group). Rats in the roxadustat and roxadustat + RIRI treatment groups received intraperitoneal injections of roxadustat (10 mg/kg/day) for seven days before renal ischemia/reperfusion. After 24 h of reperfusion, the rats were euthanized. From each rat, a blood sample was collected from the abdominal aorta, and the left kidney was harvested.
Author contributions
WZ and LY conceived and designed the experiments; CG prepared the manuscript; YL, HW, TW, HBW, YW, HNW, and GC performed the experiments; SC, JM, and SF analyzed the data. All authors contributed to the manuscript revision and approved the submitted version.
Ethical approval
All experiments were approved by the Ethics Committee of Lanzhou University Second Hospital (ID #D2022-045) and performed according to the Chinese Council on Animal Care guidelines. Additionally, this study is reported following the ARRIVE guidelines.
Acknowledgements
We thank Medjaden Inc. for editing this manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
The datasets generated and analyzed during the current study are not publicly available, because none of the data types required uploading to a public repository. However, they are available from the corresponding author upon reasonable request.
Additional information
Funding
References
- Hashemi SS, Janfeshan S, Karimi Z. Acute lung injury induced by acute uremia and renal ischemic-reperfusion injury: the role of toll-like receptors 2 and 4, and oxidative stress. Iran J Basic Med Sci. 2022;25(5):1–14. doi: 10.22038/ijbms.2022.64025.14099.
- Ostermann M, Bellomo R, Burdmann EA, et al. Controversies in acute kidney injury: conclusions from a Kidney Disease: Improving Global Outcomes (KDIGO) Conference. Kidney Int. 2020;98(2):294–309. doi: 10.1016/j.kint.2020.04.020.
- Noble RA, Lucas BJ, Selby NM. Long-term outcomes in patients with acute kidney injury. Clin J Am Soc Nephrol. 2020;15(3):423–429. doi: 10.2215/cjn.10410919.
- Schödel J, Ratcliffe PJ. Mechanisms of hypoxia signalling: new implications for nephrology. Nat Rev Nephrol. 2019;15(10):641–659. doi: 10.1038/s41581-019-0182-z.
- de Ponte MC, Cardoso VG, Gonçalves GL, et al. Early type 1 diabetes aggravates renal ischemia/reperfusion-induced acute kidney injury. Sci Rep. 2021;11(1):19028. doi: 10.1038/s41598-021-97839-7.
- Zhang Z, Haimovich B, Kwon YS, et al. Unilateral partial nephrectomy with warm ischemia results in acute hypoxia inducible factor 1-alpha (HIF-1α) and Toll-like receptor 4 (TLR4) overexpression in a porcine model. PLOS One. 2016;11(5):e0154708. doi: 10.1371/journal.pone.0154708.
- Shu S, Wang Y, Zheng M, et al. Hypoxia and hypoxia-inducible factors in kidney injury and repair. Cells. 2019;8(3):207. doi: 10.3390/cells8030207.
- Bernhardt WM, Câmpean V, Kany S, et al. Preconditional activation of hypoxia-inducible factors ameliorates ischemic acute renal failure. J Am Soc Nephrol. 2006;17(7):1970–1978. doi: 10.1681/asn.2005121302.
- Xu ZH, Wang C, He YX, et al. Hypoxia-inducible factor protects against acute kidney injury via the Wnt/β-catenin signaling pathway. Am J Physiol Renal Physiol. 2022;322(6):F611–F624. doi: 10.1152/ajprenal.00023.2022.
- Rajendran G, Schonfeld MP, Tiwari R, et al. Inhibition of endothelial PHD2 suppresses post-ischemic kidney inflammation through hypoxia-inducible factor-1. J Am Soc Nephrol. 2020;31(3):501–516. doi: 10.1681/asn.2019050523.
- Li X, Fang P, Mai J, et al. Targeting mitochondrial reactive oxygen species as novel therapy for inflammatory diseases and cancers. J Hematol Oncol. 2013;6:19. doi: 10.1186/1756-8722-6-19.
- Livingston MJ, Wang J, Zhou J, et al. Clearance of damaged mitochondria via mitophagy is important to the protective effect of ischemic preconditioning in kidneys. Autophagy. 2019;15(12):2142–2162. doi: 10.1080/15548627.2019.1615822.
- Scott SV, Klionsky DJ. Delivery of proteins and organelles to the vacuole from the cytoplasm. Curr Opin Cell Biol. 1998;10(4):523–529. doi: 10.1016/s0955-0674(98)80068-9.
- Tang C, Han H, Yan M, et al. PINK1-PRKN/PARK2 pathway of mitophagy is activated to protect against renal ischemia–reperfusion injury. Autophagy. 2018;14(5):880–897. doi: 10.1080/15548627.2017.1405880.
- Liu L, Feng D, Chen G, et al. Mitochondrial outer-membrane protein FUNDC1 mediates hypoxia-induced mitophagy in mammalian cells. Nat Cell Biol. 2012;14(2):177–185. doi: 10.1038/ncb2422.
- Li X, Liu Q, Bao W, et al. Impact of blood pressure changes on myocardial work indices in hypertensive patients in a day. J Clin Hypertens. 2022;24(1):3–14. doi: 10.1111/jch.14379.
- Li Q, Liu Y, Huang Q, et al. Hypoxia acclimation protects against heart failure postacute myocardial infarction via Fundc1-mediated mitophagy. Oxid Med Cell Longev. 2022;2022:8192552. doi: 10.1155/2022/8192552.
- Cai Y, Yang E, Yao X, et al. FUNDC1-dependent mitophagy induced by tPA protects neurons against cerebral ischemia–reperfusion injury. Redox Biol. 2021;38:101792. doi: 10.1016/j.redox.2020.101792.
- Li S, Zhou Y, Gu X, et al. NLRX1/FUNDC1/NIPSNAP1-2 axis regulates mitophagy and alleviates intestinal ischaemia/reperfusion injury. Cell Prolif. 2021;54(3):e12986. doi: 10.1111/cpr.12986.
- Xiao Y, Chen W, Zhong Z, et al. Electroacupuncture preconditioning attenuates myocardial ischemia–reperfusion injury by inhibiting mitophagy mediated by the mTORC1–ULK1–FUNDC1 pathway. Biomed Pharmacother. 2020;127:110148. doi: 10.1016/j.biopha.2020.110148.
- Lin Q, Li S, Jiang N, et al. Inhibiting NLRP3 inflammasome attenuates apoptosis in contrast-induced acute kidney injury through the upregulation of HIF1A and BNIP3-mediated mitophagy. Autophagy. 2021;17(10):2975–2990. doi: 10.1080/15548627.2020.1848971.
- Wang J, Zhu P, Li R, et al. Fundc1-dependent mitophagy is obligatory to ischemic preconditioning-conferred renoprotection in ischemic AKI via suppression of Drp1-mediated mitochondrial fission. Redox Biol. 2020;30:101415. doi: 10.1016/j.redox.2019.101415.
- Zhang W. The mitophagy receptor FUN14 domain-containing 1 (FUNDC1): a promising biomarker and potential therapeutic target of human diseases. Genes Dis. 2021;8(5):640–654. doi: 10.1016/j.gendis.2020.08.011.
- Wu H, Wang Y, Li W, et al. Deficiency of mitophagy receptor FUNDC1 impairs mitochondrial quality and aggravates dietary-induced obesity and metabolic syndrome. Autophagy. 2019;15(11):1882–1898. doi: 10.1080/15548627.2019.1596482.
- Cao Z, Wu Z, Duan T, et al. Curcumin ameliorates HO-induced injury through SIRT1–PERK–CHOP pathway in pancreatic beta cells. Acta Biochim Biophys Sin. 2022;54(3):370–377. doi: 10.3724/abbs.2022004.
- Liao W, Fu Z, Zou Y, et al. MicroRNA-140-5p attenuated oxidative stress in cisplatin induced acute kidney injury by activating Nrf2/ARE pathway through a Keap1-independent mechanism. Exp Cell Res. 2017;360(2):292–302. doi: 10.1016/j.yexcr.2017.09.019.
- Hosszu A, Antal Z, Lenart L, et al. σ1-receptor agonism protects against renal ischemia–reperfusion injury. J Am Soc Nephrol. 2017;28(1):152–165. doi: 10.1681/asn.2015070772.
- Wei Q, Sun H, Song S, et al. MicroRNA-668 represses MTP18 to preserve mitochondrial dynamics in ischemic acute kidney injury. J Clin Invest. 2018;128(12):5448–5464. doi: 10.1172/jci121859.
- Wang H, Liu N, Li R, et al. Nephropreventing effect of hypoxia-inducible factor 1α in a rat model of ischaemic/reperfusion acute kidney injury. Clin Exp Pharmacol Physiol. 2018;45(10):1076–1082. doi: 10.1111/1440-1681.12947.
- Fu ZJ, Wang ZY, Xu L, et al. HIF-1α-BNIP3-mediated mitophagy in tubular cells protects against renal ischemia/reperfusion injury. Redox Biol. 2020;36:101671. doi: 10.1016/j.redox.2020.101671.
- Li R, Zhou Y, Zhang S, et al. The natural (poly)phenols as modulators of microglia polarization via TLR4/NF-κB pathway exert anti-inflammatory activity in ischemic stroke. Eur J Pharmacol. 2022;914:174660. doi: 10.1016/j.ejphar.2021.174660.
- Yu L, Wang Y, Guo YH, et al. HIF-1α alleviates high-glucose-induced renal tubular cell injury by promoting parkin/PINK1-mediated mitophagy. Front Med. 2021;8:803874. doi: 10.3389/fmed.2021.803874.
- Zheng Y, Huang C, Lu L, et al. STOML2 potentiates metastasis of hepatocellular carcinoma by promoting PINK1-mediated mitophagy and regulates sensitivity to lenvatinib. J Hematol Oncol. 2021;14(1):16. doi: 10.1186/s13045-020-01029-3.
- Sun Y, Wen F, Yan C, et al. Mitophagy protects the retina against anti-vascular endothelial growth factor therapy-driven hypoxia via hypoxia-inducible factor-1α signaling. Front Cell Dev Biol. 2021;9:727822. doi: 10.3389/fcell.2021.727822.
- Zhou H, Zhu P, Wang J, et al. Pathogenesis of cardiac ischemia reperfusion injury is associated with CK2α-disturbed mitochondrial homeostasis via suppression of FUNDC1-related mitophagy. Cell Death Differ. 2018;25(6):1080–1093. doi: 10.1038/s41418-018-0086-7.
- Molitoris BA. Actin cytoskeleton in ischemic acute renal failure. Kidney Int. 2004;66(2):871–883. doi: 10.1111/j.1523-1755.2004.00818.x.
- Dan Dunn J, Alvarez LA, Zhang X, et al. Reactive oxygen species and mitochondria: a nexus of cellular homeostasis. Redox Biol. 2015;6:472–485. doi: 10.1016/j.redox.2015.09.005.
- Cai C, Guo Z, Chang X, et al. Empagliflozin attenuates cardiac microvascular ischemia/reperfusion through activating the AMPKα1/ULK1/FUNDC1/mitophagy pathway. Redox Biol. 2022;63:102738. doi: 10.1016/j.redox.2022.102288.
- Li L, Tan J, Miao Y, et al. ROS and autophagy: interactions and molecular regulatory mechanisms. Cell Mol Neurobiol. 2015;35(5):615–621. doi: 10.1007/s10571-015-0166-x.
- De Gaetano A, Gibellini L, Zanini G, et al. Mitophagy and oxidative stress: the role of aging. Antioxidants. 2021;10(5):794. doi: 10.3390/antiox10050794.
- Zhou H, Zhu P, Guo J, et al. Ripk3 induces mitochondrial apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury. Redox Biol. 2017;13:498–507. doi: 10.1016/j.redox.2017.07.007.
- Zhou H, Wang J, Zhu P, et al. NR4A1 aggravates the cardiac microvascular ischemia reperfusion injury through suppressing FUNDC1-mediated mitophagy and promoting Mff-required mitochondrial fission by CK2α. Basic Res Cardiol. 2018;113(4):23. doi: 10.1007/s00395-018-0682-1.
- Yu W, Xu M, Zhang T, et al. Mst1 promotes cardiac ischemia–reperfusion injury by inhibiting the ERK–CREB pathway and repressing FUNDC1-mediated mitophagy. J Physiol Sci. 2019;69(1):113–127. doi: 10.1007/s12576-018-0627-3.
- Zhou H, Li D, Zhu P, et al. Melatonin suppresses platelet activation and function against cardiac ischemia/reperfusion injury via PPARγ/FUNDC1/mitophagy pathways. J Pineal Res. 2017;63(4). doi: 10.1111/jpi.12438.
- Chen N, Hao C, Liu BC, et al. Roxadustat treatment for anemia in patients undergoing long-term dialysis. N Engl J Med. 2019;381(11):1011–1022. doi: 10.1056/NEJMoa1901713.
- Chen N, Hao C, Peng X, et al. Roxadustat for anemia in patients with kidney disease not receiving dialysis. N Engl J Med. 2019;381(11):1001–1010. doi: 10.1056/NEJMoa1813599.
- Yang Y, Yu X, Zhang Y, et al. Hypoxia-inducible factor prolyl hydroxylase inhibitor roxadustat (FG-4592) protects against cisplatin-induced acute kidney injury. Clin Sci. 2018;132(7):825–838. doi: 10.1042/cs20171625.
- Wu M, Chen W, Miao M, et al. Anti-anemia drug FG4592 retards the AKI-to-CKD transition by improving vascular regeneration and antioxidative capability. Clin Sci. 2021;135(14):1707–1726. doi: 10.1042/cs20210100.
- Regolisti G, Maggiore U, Cademartiri C, et al. Renal resistive index by transesophageal and transparietal echo-Doppler imaging for the prediction of acute kidney injury in patients undergoing major heart surgery. J Nephrol. 2017;30(2):243–253. doi: 10.1007/s40620-016-0289-2.