
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Objectives
Calcium oxalate (CaOx) crystal deposition in acute kidney injury (AKI) patients is under recognized but impacts renal outcomes. This study investigates its determinants and effects.
Methods
We studied 814 AKI patients with native kidney biopsies from 2011 to 2020, identifying CaOx crystal deposition severity (mild: <5, moderate: 5–10, severe: >10 crystals per section). We assessed factors like urinary oxalate, citrate, urate, electrolytes, pH, tubular calcification index, and SLC26A6 expression, comparing them with creatinine-matched AKI controls without oxalosis. We analyzed how these factors relate to CaOx severity and their impact on renal recovery (eGFR < 15 mL/min/1.73 m2 at 3-month follow-up).
Results
CaOx crystal deposition was found in 3.9% of the AKI cohort (32 cases), with 72% due to nephrotoxic medication-induced tubulointerstitial nephritis. Diuretic use, higher urinary oxalate-to-citrate ratio induced by hypocitraturia, and tubular calcification index were significant contributors to moderate and/or severe CaOx deposition. Poor baseline renal function, low urinary chloride, high uric acid and urea nitrogen, tubular SLC26A6 overexpression, and glomerular sclerosis were also associated with moderate-to-severe CaOx deposition. Kidney recovery was delayed, with 43.8%, 31.2%, and 18.8% of patients having eGFR < 15 mL/min/1.73 m2 at 4, 12, and 24-week post-injury. Poor outcomes were linked to high urinary α1-microglobulin-to-creatinine (α1-MG/C) ratios and active tubular injury scores. Univariate analysis showed a strong link between this ratio and poor renal outcomes, independent of oxalosis severity.
Conclusions
In AKI, CaOx deposition is common despite declining GFR. Factors worsening tubular injury, not just oxalate-to-citrate ratios, are key to understanding impaired renal recovery.
Introduction
The deposition of calcium-containing crystals in the kidney pathology of patients with acute kidney injury (AKI) has been occasionally observed; however, it has not gained adequate attention. Renal oxalosis, also known as oxalate nephropathy (OxN), is a prevalent type of crystalline nephropathy that is typically characterized by kidney injury induced by an abundance of calcium oxalate (CaOx). These crystals damage the kidney tubular epithelial cells, trigger interstitial inflammation and fibrosis, and ultimately result in disease progression with poor kidney outcomes [Citation1–3]. In addition to crystals, the pathogenic effects of urinary soluble oxalate on the kidney have been increasing recognized, especially in patients with chronic kidney disease (CKD). In non-transplanted kidneys, secondary OxN is primarily caused by enterogenic hyperoxaluria resulting from non-genetic conditions, such as fat malabsorption [Citation4,Citation5]. However, the occurrence of CaOx deposition in AKI pathology due to nonenteric risk factors has been sporadically reported and may have been overlooked [Citation6], despite decreased excretion of urinary oxalate due to a decline in glomerular filtration rate.
Hyperoxaluria greatly contributes to the formation of intraluminal CaOx crystals [Citation7]. The excretion of urinary oxalate is primarily regulated by the serum oxalate levels through glomerular filtration, with a small portion secreted by tubular transporters, specifically the solute carrier 26 family 6 (SLC26A6) and eliminated through the gut [Citation8]. However, a cohort study conducted by Mayo Clinic revealed that 23.5% of non-transplanted kidneys with OxN had no identifiable traditional risk factors [Citation6], whereas our case series reported a prevalence of 74.9% [Citation2]. Further, other urinary constituents may contribute to intraluminal crystallization and the internalization of crystals. The level of citraturia, for example, has a significant impact on the formation of either calcium oxalate monohydrate (COM) or calcium oxalate dihydrate crystals. COM formed in the proximal tubule lumen is associated with increased inflammation and cell damage [Citation9]. Nonetheless, tubular crystal casts and plugs rarely obstruct enough nephrons to explain AKI, which prompts the question of whether inflammation is the main factor driving crystal-induced AKI. Previous studies on toxic and post-ischemic AKI have revealed that kidney injuries are largely driven by an unspecific innate immunity response [Citation10–12], rather than merely dependent on the type of crystal involved.
This study aimed to characterize the differences of determinants including urinary parameters, urinary pH, tubular calcification, and tubular SLC26A6 expression between AKI patients with CaOx deposition and serum creatinine-matched AKI controls without oxalosis, and further analyze their effect on oxalosis and potential contribution to adverse long-term kidney outcomes.
Methods
Patients
We enrolled patients who underwent native kidney biopsies with CaOx crystals deposition identified in native kidney biopsies from a cohort of 814 AKI patients who underwent kidney biopsies at the Renal Pathology Center of Peking University First Hospital from January 2011 to December 2020. AKI diagnosis followed the Kidney Disease Improving Global Outcomes criteria [Citation13], and CaOx crystals were confirmed as transparent high-birefringence crystals under polarized light microscopy. Control patients with AKI but without oxalosis were selected, matched for serum creatinine levels. Patients with primary hyperoxaluria as determined by genomic sequencing and family history would not be included. Patients with AKI due to severe glomerulonephritis (GN) were excluded from prognosis analysis (). Severe GN was defined as glomerular disease with severe activity, as determined by the ‘glomerular active index’, and was considered the primary contributor to AKI.
Figure 1. Flow chart of the study.
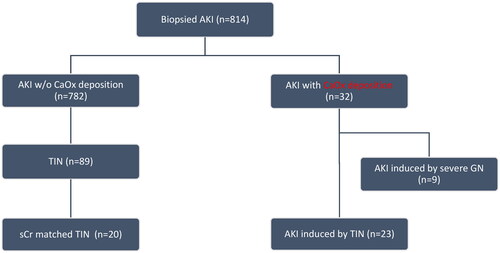
The study was approved by the Ethics Committee of Peking University First Hospital, and written informed consent was obtained from all participants (2021Y042). illustrates the study’s flow chart. The study follows the STROBE guidelines (Supplementary file ‘STROBE checklist’).
Evaluation of Clinical and laboratory parameters
Clinical and laboratory parameters were assessed. Demographic information and medical history, including hypertension, diabetes mellitus, CKD, nephrolithiasis, alcohol, and smoking habits, were extracted from the electronic medical system. Traditional factors associated with hyperoxaluria, such as diarrhea, malabsorption, excessive consumption of oxalate-rich foods, and vitamin C supplements, were recorded. Furthermore, data on other factors that could impact pre-renal glomerular filtration rate were collected. These factors include the use of renin–angiotensin–aldosterone system inhibitors (RAASi), diuretics, and non-steroidal anti-inflammatory drugs (NSAIDs), as well as the presence of hypotension, hypoalbuminemia, blood urea nitrogen (BUN)-to-creatinine ratio (BUN/Cr) >10, and filtration excretion of sodium (FeNa) <1%.
Upon admission, routine biochemical tests were performed, and the daily urine output was meticulously documented. The evaluation of kidney function was based on the measurement of serum creatinine levels and estimated glomerular filtration rate (eGFR) using the CKD-EPI equation [Citation14]. Other parameters including brain natriuretic peptide (BNP), albumin, erythrocyte sedimentation rate (ESR), and C-reactive protein (CRP) levels were measured. For urine analysis, morning spot-urine parameters were assessed. This included the examination of urine pH, identification of sediments, such as crystals, casts, pyuria, and hematuria. Additionally, the albumin-to-creatinine ratio, glucose levels, N-acetyl-β-D-glucosaminidase (NAG), and α1-microglobulin (α1-MG) were analyzed. To gather comprehensive data on urate and electrolyte levels, 24-h urine collections were conducted. The supernatants of spot-urine samples were utilized to measure the concentration ratios of urinary oxalate and citrate to creatinine (uOx/C, uCi/C) through the implementation of Thermo Fisher IC-MS workflow and an oxalate-citrate assay kit (Colorimetric, ab196990; Abcam, Cambridge, MA). Derivative parameters to further characterize the relationship between urine oxalate, citrate and calcium were calculated as follows: concentration ratio of urinary oxalate to citrate (uOx/uCi), concentration ratio of urinary calcium to citrate (uCa/uCi), uCi/C multiply by uCa/C (uCi/C x uCa/C).
Evaluation of renal pathology
Kidney biopsy specimens underwent a sequential process of staining with hematoxylin and eosin and Masson stain, followed by examination under immunofluorescence, light, and electron microscopy. Polarized light microscopy and electron microscopy were utilized to quantify and localize the CaOx crystals. The severity of crystal deposition was graded based on the number of crystals per section: mild (<5 crystals), moderate (5–10 crystals), or severe (>10 crystals). To detect and measure tubular calcification, the von Kossa staining method was employed, using the von Kossa kit from Jiemei Gene in Shanghai, China. This allowed for the determination of the ‘tubular calcification index’.
To evaluate the extent of tubular injury, tissue inflammation, or fibrotic changes, two indices were created to provide a standardized measurement of the extent of these different renal conditions: an activity index and a chronicity index. These indices, presented in Supplementary Table 1, were developed using a modified Banff working classification as a basis [Citation15]. Briefly, activity index was composed of acute tubular injury score (ATI), acute interstitial injury score (ATI), and acute tubulointerstitial injury score (ATI, calculated as: AT + AI); chronicity index contained chronic tubular injury score (CT), chronic interstitial injury score (CI) and chronic tubulointerstitial score (CTI, calculated as: CT + CI). The glomerular active index was used to evaluate the presence of mesangial injury, endocapillary hypercellularity, segmental sclerosis, and crescents. These findings were then categorized into three groups: no glomerulonephropathy (GN), mild-to-moderate activity, or severe activity. On the other hand, the glomerular chronic index was calculated based on the percentage of globally sclerosed glomeruli. The scores ranged from 0 (sclerosed glomeruli <5), 1 (sclerosed glomeruli 5–10%), 2 (sclerosed glomeruli 11–20%), to 3 (sclerosed glomeruli >20%). This index provided an indication of the degree of glomerular sclerosis present. If a specific GN such as diabetic nodular glomerulosclerosis or focal segmental glomerulosclerosis (FSGS) was identified, the score defaulted to 3 [Citation16]. The pathological diagnosis and semi-quantitative scores were validated by two experienced pathologists.
The expression of tubular SLC26A6 was analyzed using antibodies against SLC26A6 (Santa Cruz, CA) implemented in immunoperoxidase staining on paraffin sections of the kidney biopsy. The findings were quantified as the percentage of positive renal tubules out of the total tubules (%) under a 400× microscope. The expression levels were then categorized as mild (<15%), moderate (15–20%), or severe (>20%).
Treatment, follow-up, and outcomes
Clinical data and follow-up outcomes were retrieved from medical records and conducted via telephone interviews. The correlations of determinants on severity of CaOx deposits and their impact of kidney outcomes were analyzed based on the most recent follow-up within a 6-month period. Patients with their 3-month eGFR <15 mL/min/1.73 m2 or kidney failure with replacement therapy (abbreviated as ‘KFRT subgroup’) were recognized as indicating poor long-term outcomes.
Statistical analyses
Normally distributed data are presented as means ± SD, whereas non-normally distributed data are presented as medians (Q25, Q75). Categorical data were summarized as counts and percentages. To compare the clinicopathological and laboratory characteristics of AKI patients with CaOx crystal deposition to the selected controls, the Student’s t-test, Mann–Whitney U test, Kruskal–Wallis test, or Wilcoxon signed-rank test were employed according to appropriateness. The correlation analysis, coupled with univariate regression, was performed using the backward stepwise elimination method to identify risk factors that influenced oxalosis severity and poor kidney outcomes. Regression analyses were reported as odds ratios (ORs) with 95% confidence intervals (CIs). A significance level of p value <0.05 was set. All statistical analyses were conducted using Statistical Software for Social Sciences version 26.0 (SPSS Inc., Chicago, IL).
Results
Clinical features
The prevalence of CaOx crystal deposition was 3.9% of the whole AKI cohort (32 cases). The baseline clinical features of these patients are presented in (32 vs. 20 cases). The average age of these patients was 52.8 ± 13.8 years, and there was a higher proportion of males, with a male-to-female ratio of 5.4:1. Most AKI patients with CaOx deposition were linked to tubulointerstitial nephritis caused by nephrotoxic medications, with 63% of these cases being classified as AKI stage 3. The average daily urine output was 1769 ± 628 mL. Nine patients (28%) had traditional predisposing factors for secondary hyperoxaluria. Further, 28 patients (88%) exhibited pre-renal conditions related to renal hypoperfusion (significantly higher than that in the control group, 28 vs. 12 patients, p = 0.04): the use of RAASi in 10 patients (31%), NSAIDs in 6 patients (19%), and diuretics in 14 patients (43.8%). Thirteen patients (41%) reported a history of alcohol abuse, and four (12.5%) had alcohol exposure prior to the onset of the disease. However, all patients denied exposure to toxic alcohol. AKI patients with CaOx deposition tended to have higher rate of comorbidities, including diabetes mellitus (53% vs. 20%, p = 0.02), hypertension (69% vs. 15%, p = < 0.001), alcohol abuse (41% vs. 5%, p = 0.008), and infection (44% vs. 15%, p = 0.04) ().
Table 1. Baseline features of patients with acute kidney injury (AKI) and CaOx deposition.
Upon admission, laboratory tests revealed a mean BUN/Cr ratio of 12.4 ± 5.8. Median serum albumin levels were measured at 36.1 (24.5, 41.2) g/L, and median BNP levels were at 256.5 (42.5, 448.8) pg/mL. Crystalluria of CaOx monohydrate crystals was observed in the urine sediments of 2 patients (6.3%) (), whereas sterile leukocyturia was present in 17 patients (53%). Renal glycosuria, elevated urinary levels of NAG, and α1-MG were frequently observed. The urine pH was measured at 6.0 (5.0, 6.5). Among AKI patients with CaOx deposition, 72% (23 cases) were attributed to tubulointerstitial nephritis based on clinical evaluation. When comparing to the selected non-oxalosis AKI control induced by tubulointerstitial nephritis, these patients demonstrated no significant difference in the daily urine electrolyte excretion but had the higher level of urinary oxalate (median, 0.2875 mmol/L, p < 0.001), an increased oxalate-to-citrate ratio (0.7110 ± 0.3972, p = 0.005) ().
Figure 2. CaOx crystal cast and intrarenal depositions. Immunohistochemistry staining showed the expression of SLC26A6 transporter in (A and B) urinary sediments of a cast containing CaOx crystals. (C and D) Intraluminal and intracellular CaOx crystals (original magnification, ×5000). (E) Tubular expression of SLC26A6 transporter in a patient with severe oxalosis and (F) another patient with minimal change disease as the control (original magnification, ×200). (G) CaOx crystals under polarized microscopy (original magnification, ×200). (H) von Kossa staining showed calcium-containing crystals.
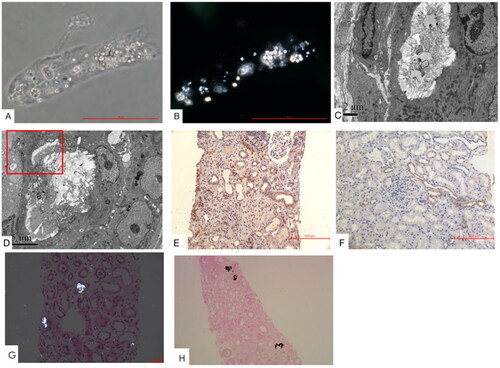
Table 2. Comparison of clinical features among AKI patients with oxalosis and non-oxalosis.
Pathological characteristics
Renal pathological findings predominantly included degenerative changes in tubular injury, characterized by diffuse interstitial edema, mild interstitial inflammation, and varying degrees of interstitial fibrosis. Half of the patients displayed tubular necrosis, whereas a few had a naked basement membrane. Only one patient presented with tubulitis (Supplementary Table 2). Intraluminal localization of CaOx crystals was evident in all patients, with intracellular localization observed in five patients (16%) (). In the cohort, severe, moderate, and mild CaOx crystal deposition was present in 40.6%, 18.8%, and 40.6% of the cases, respectively. Calcium phosphate crystals were easily identifiable (). There existed a close relationship between the severity of tubular calcification and CaOx deposition (R = 0.550, p = 0.007). When compared to the selected control, AKI patients with CaOx deposition exhibited higher active tubular injury scores (score 3.5 vs. 2.0, p = 0.01), tubular calcification index (11 vs. 0, p < 0.001), and tubular expression of SLC26A6 (0.18 ± 0.05 vs. 0.14 ± 0.04, p = 0.009) ( and ). Subgroup analysis of patients with non-to-moderate vs. severe CaOx deposition (Supplementary Table 3) and non-to-mild versus moderate-to-severe CaOx deposition (Supplementary Table 4) consistently revealed similar findings.
Although a considerable proportion of patients exhibited concurrent GN (including diabetic kidney disease, IgA nephropathy, membranous nephropathy, lupus nephritis, and minimal change disease), only 28% of these AKI patients were attributed to severe GN. Notably, all of these patients exhibited mild CaOx.
Factors contributing to severity of oxalosis deposition and renal outcomes
Univariable regression analysis revealed significant associations between moderate-to-severe CaOx deposition and the following factors: peak serum creatinine ( mol/L) (OR = 1.002; 95% CI, 1.000–1.004), nadir eGFR (mL/min/1.73 m2) (OR = 0.896; 95% CI, 0.810–0.991), use of diuretics (OR = 7.700; 95% CI, 2.063–28.742), 24-h urine chloride (mol/24h) (OR = 0.773; 95% CI, 0.633–0.994), 24-h uric acid (mol/24h) (OR = 1.006; 95% CI, 1.001–1.012), 24-h urea nitrogen (mol/24 h) (OR = 1.198; 95% CI, 1.025–1.400), uOx/uCi ratio (mmol/mmol, per 0.01 increase) (OR = 1.023; 95% CI, 1.004–1.042), tubular expression of SLC26A6 (OR = 1.401; 95% CI, 1.102–1.780), tubular calcification index (OR = 1.320; 95% CI, 1.105–1.575), and glomerular global sclerosis (OR = 2.026; 95% CI, 1.233–3.330) (). Furthermore, use of diuretics (OR = 3.889; 95% CI, 1.038–14.566), urinary citrate levels (mol/L) (OR = 0.996; 95% CI, 0.992–1.000), uCi/C ratio (mmol/mmol, per 0.01 increase) (OR = 0.905, 95% CI, 0.822–0.996), uOx/uCi ratio (mmol/mmol, per 0.01 increase) (OR = 1.026; 95% CI, 1.006–1.047), and tubular calcification index (OR = 1.065; 95% CI, 1.002–1.032) were found to be associated with severe CaOx deposition ().
Table 3. Univariable regression analysis of risk factor to oxalosis.
All AKI patients with CaOx deposition received supportive therapy, and three of them were given potassium citrate supplements. However, renal recovery was delayed, as 43.8%, 31.2%, and 18.8% of AKI patients with CaOx deposition had an eGFR < 15 mL/min/1.73 m2 at the 4, 12, and 24-week follow-up post-injury, respectively. Serum creatinine was much higher in CaOx patients at 4-week (243.41[156.00–425.65] vs. 153.00 [122.00–193.88] mol/L, p = 0.02), while the gap was narrowed down at 12-week (161.00 [112.00–271.50] vs. 120.50 [105.75–145.50]
mol/L, p = 0.09) and 24-week (134.00 [93.06–183.530] vs. 121.41 [106.00–161.68]
mol/L, p = 0.77) (Supplementary Figure 1). When comparing long-term outcomes, it was found that patients in the KFRT subgroup had a high prevalence of severe GN, reaching 80% (Supplementary Table 5), no difference of CaOx deposition severity found within subgroups having various renal outcomes. In the 23 cases of patients with CaOx deposition but without severe GN, higher levels of tubular injury, as indicated by the urinary α1-microglobulin-to-creatinine (α1-MG/C) ratio and the pathologic active tubular injury score, were significantly associated with poor renal outcomes (eGFR < 15 mL/min/1.73 m2 at the 3-month follow-up) compared to others (). Instead of severity of CaOx deposition, univariate regression analysis confirmed a strong association between urinary α1-MG/C ratio and an unfavorable kidney outcome (OR = 1.007) .
Table 4. Comparison between AKI patients having CaOx deposition (having no severe glomerulonephropathy) with a 3-month eGFR <15 and ≥ 15 mL/min/1.73 m2.
Table 5. Univariate regression analysis of poor long-term kidney outcome (eGFR < 15 mL/min/1.73 m2 or on KFRT vs. eGFR ≥ 15 mL/min/1.73 m2 [reference] at 3 months) in patients having no glomerulonephritis.
Discussion
Our study offers crucial clinical and pathological insights into patients with AKI who exhibit intrarenal CaOx deposition, a condition observed in 3.9% of our AKI patient cohort. These patients characterized by significant CaOx deposition, not only have compromised renal function but also present with unique features: an elevated oxalate-to-citrate ratio associated with hypocitraturia and increased tubular expression of the SLC26A6 transporter; a higher tubular calcification index; and a status of intrarenal ischemia indicated by reduced urinary excretion of chloride and citrate, along with a high prevalence of glomerular global sclerosis and frequent use of diuretics. However, the relatively low incidence of crystalluria in these patients indicates that the clinical diagnosis of CaOx deposition is often underappreciated. Oxalosis-enabling conditions in AKI patients suggest the involvement of underlying chronic kidney injury related to comorbidities (diabetes, hypertension, etc.) or renal ischemic lesions (glomerulosclerosis), which might explain the delayed remission in these patients. Rather than severity of oxalosis, our findings suggest that patients with more pronounced tubular injury experience worse kidney outcomes. We propose that tubular injury is a consequence of a multifactorial response to CaOx crystals, exacerbated by intrarenal ischemia, which impairs the renal tubules’ capacity to prevent crystallization. These observations merit further investigation [Citation2,Citation7].
Hyperoxaluria has long been recognized as a critical factor in the development of secondary oxalosis. Unlike in CKD, where SLC26A6-mediated enhancement of enteric oxalate secretion occurs, AKI patients are unable to effectively compensate for oxalate regulation in the intestines, resulting in an increased oxalate burden [Citation17]. The upregulation of tubular SLC26A6 expression in response to a significant decrease in GFR can result in an increased presence of oxalate in the tubular fluid, specifically reaching the distal tubules where calcium crystallization primarily takes place. However, our study has unveiled an interesting finding that patients with intrarenal CaOx deposition exhibited lower urinary oxalate excretion compared to those without crystal. It follows that the presence of inhibitors such as citrate and uromodulin that prevent crystal formation in the urine, plays an essential role in the genesis of oxalosis. As a result, massive CaOx crystals can form. The higher ratio of oxalate-to-citrate observed in this study indicated the contribution of urinary citrate to inhibition of CaOx deposition. Citrate plays an essential role in preventing the formation of CaOx crystal by binding to calcium and oxalate, forming soluble complexes that are excreted in the urine. Citrate is synthesized in the mitochondria and is crucial for energy production [Citation18]. It is produced by the liver, filtered by the kidneys, and affected by factors, such as bone metabolism, kidney clearance, metabolic acidosis, and hypokalemia [Citation19–20]. Medications and certain conditions can further decrease urinary citrate levels. Avid reabsorption of citrate in the tubules may indicate an energy imbalance in AKI patients [Citation18]. Restoring plasma citrate by reducing its excretion in urine may have positive effects on energy utilization. In patients with gastrointestinal malabsorption, the lack of daily urinary citrate excretion contributes to the development of oxalosis, along with secondary hyperoxaluria [Citation21,Citation22]. The use of angiotensin-converting enzyme inhibitors can cause hypocitraturia, unrelated to acidosis or hypokalemia [Citation23]. Studies have shown that enalapril reduces urinary citrate excretion and increases renal cortical ATP citrate lyase activity. The activation of the proximal tubular Na/H antiporter by angiotensin II and subsequent changes in intracellular pH may affect urinary citrate excretion and renal citrate metabolism [Citation24]. Despite the increased activity of the succinate transporter NaDC-1, urinary citrate levels remain low [Citation25]. Moreover, citrate attachment to crystal surfaces enhances the protective effect of other inhibitors, including the Tamm–Horsfall protein [Citation26].
From the study result, the calcium-containing crystal was more evident than the CaOx crystal, and the tubular calcification index on pathology was significantly elevated in AKI patients with CaOx deposition. We believe that hypocitraturia weakens the defenses against the formation of both COM crystal and calcium phosphate crystals, leading to increased cytotoxicity and inflammation [Citation27,Citation28]. Administration of potassium citrate increases urinary citrate levels, and alleviates renal pathological changes possibly through the expression of NaDC-1, ultimately reducing the occurrence of nephrolithiasis [Citation29]. Furthermore, the activation of the SLC26A6 transporter not only functions as a 1Cl−/1oxalate transporter but also contributes to calcium phosphate precipitation by acting as an ion exchanger of 1Cl−/2HCO3− [Citation30]. This leads to increased HCO3− excretion and urine alkalization, which in turn predisposes to phosphate crystallization. The buildup of phosphate and calcium in the tubular fluid can be attributed to decreased urine flow caused by transient dehydration during glomerular hypoperfusion. This condition is commonly induced by the combined use of diuretics, RAASi, and NSAIDs, particularly in the presence of severe hypoalbuminemia. Moreover, the increased reabsorption of salt and water in both the proximal tubule and descending limb of Henle’s loop further reduces phosphorus reabsorption due to the limited permeability of calcium and phosphate in this loop segment. The subsequent formation of calcium phosphate crystals can directly damage tubular epithelial cells. Oxidative stress from reduced extracellular fluid exacerbates CaOx crystal formation in hyperoxaluric rat kidneys [Citation31]. Damaged tubular cells linked to pre-renal glomerular ischemia and expressing surface-bound hyaluronan and osteopontin, bind crystals, leading to adhesion and activation of NLRP3 inflammasomes, thereby worsening AKI [Citation12].
Our study had some limitations. The sample size of our study was too small to better stratify the severity of CaOx deposition and to conduct multivariate regression to further analyze the relationships of important influencing factors (including crystal severity, higher urinary α1-MG/C ratio, and active tubular injury scores) and kidney outcomes in patients with AKI. We were unable to examine plasma oxalate and citrate owing to the retrospective nature of this study. Additionally, based on the nature of AKI that inhibitors against crystals formation are dramatically reduced, the influences of urinary level and function of uromodulin should be considered. Lastly, we used creatinine as the only matching criterion between patients with CaOx deposition and the control group since the sample size was small, which might lead to selection bias due to other confounding factors like underlying causes of AKI, age, and comorbidities. A more comprehensive balancing of the potential confounding factors would be achieved in further cohort studies with larger sample size.
In conclusion, CaOx deposition is relatively frequent even as the glomerular filtration rate decreases in AKI. This delays and impairs renal function restoration. Beyond the increased urinary oxalate-to-citrate ratio, instead of CaOx deposition, it is crucial to consider factors that exacerbate active tubular injury, as these hinder the recovery of renal function. Understanding the mechanisms can help clinicians develop proactive interventions to minimize negative effects of CaOx formation in this population.
Authors’ contributions
WY, TZ, XC, SW, YW, and TS participated in data collection and clinical follow-up. WY, TZ, and TS made the draft, and TZ and TS revised the draft. SW made the pathological diagnosis. TS designed and revised the manuscript. All authors read and approved the final manuscript.
Supplemental Material
Download PDF (274.2 KB)Acknowledgements
The authors thank the Editage (https://www.editage.com/) for the help of English editing.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
All data relevant to the study are included in the article or uploaded as supplementary information.
Additional information
Funding
References
- Fanger H, Esparza A. Crystals of calcium oxalate in kidneys in uremia. Am J Clin Pathol. 1964;41(6):1–12. doi: 10.1093/ajcp/41.6.597.
- Zhou J, Yu X, Su T, et al. Critically ill, tubular injury, delayed early recovery: characteristics of acute kidney disease with renal oxalosis. Ren Fail. 2021;43(1):425–432. doi: 10.1080/0886022X.2021.1885443.
- Buysschaert B, Aydin S, Morelle J, et al. Etiologies, clinical features, and outcome of oxalate nephropathy. Kidney Int Rep. 2020;5(9):1503–1509. doi: 10.1016/j.ekir.2020.06.021.
- Demoulin N, Aydin S, Gillion V, et al. Pathophysiology and management of hyperoxaluria and oxalate nephropathy: a review. Am J Kidney Dis. 2022;79(5):717–727. doi: 10.1053/j.ajkd.2021.07.018.
- Rosenstock JL, Joab TMJ, DeVita MV, et al. Oxalate nephropathy: a review. Clin Kidney J. 2021;15(2):194–204. doi: 10.1093/ckj/sfab145.
- Reddy S, Bolen E, Abdelmalek M, et al. Clinical outcomes and histological patterns in oxalate nephropathy due to enteric and nonenteric risk factors. Am J Nephrol. 2021;52(12):961–968. doi: 10.1159/000520286.
- Schepers MSJ, Van Ballegooijen ES, Bangma CH, et al. Oxalate is toxic to renal tubular cells only at supraphysiologic concentrations. Kidney Int. 2005;68(4):1660–1669. doi: 10.1111/j.1523-1755.2005.00576.x.
- Jiang H, Pokhrel G, Chen Y, et al. High expression of SLC26A6 in the kidney may contribute to renal calcification via an SLC26A6-dependent mechanism. PeerJ. 2018;6:e5192. doi: 10.7717/peerj.5192.
- Sun XY, Gan QZ, Ouyang JM. Calcium oxalate toxicity in renal epithelial cells: the mediation of crystal size on cell death mode. Cell Death Discov. 2015;1(1):15055. doi: 10.1038/cddiscovery.2015.55.
- Mankan AK, Kubarenko A, Hornung V. Immunology in clinic review series; focus on autoinflammatory diseases: inflammasomes: mechanisms of activation. Clin Exp Immunol. 2012;167(3):369–381. doi: 10.1111/j.1365-2249.2011.04534.x.
- Mulay SR, Evan A, Anders HJ. Molecular mechanisms of crystal-related kidney inflammation and injury. Implications for cholesterol embolism, crystalline nephropathies and kidney stone disease. Nephrol Dial Transplant. 2014;29(3):507–514. doi: 10.1093/ndt/gft248.
- Mulay SR, Kulkarni OP, Rupanagudi KV, et al. Calcium oxalate crystals induce renal inflammation by NLRP3-mediated IL-1β secretion. J Clin Invest. 2013;123(1):236–246. doi: 10.1172/JCI63679.
- Khwaja A. KDIGO clinical practice guidelines for acute kidney injury. Nephron Clin Pract. 2012;120(4):c179–84. doi: 10.1159/000339789.
- Levey AS, Stevens LA, Schmid CH, et al. A new equation to estimate glomerular filtration rate. Ann Intern Med. 2009;150(9):604–612. doi: 10.7326/0003-4819-150-9-200905050-00006.
- Roufosse C, Simmonds N, Clahsen-van Groningen M, et al. A 2018 reference guide to the banff classification of renal allograft pathology. Transplantation. 2018;102(11):1795–1814. doi: 10.1097/TP.0000000000002366.
- Jia Y, Poor SMM, Dufault B, et al. Chronic kidney damage pathology score for systematic assessment of the non-neoplastic kidney tissue and prediction of post-operative renal function outcomes. Hum Pathol. 2022;124:76–84. doi: 10.1016/j.humpath.2022.03.003.
- Tiselius H-G, Lindbäck B, Fornander A-M, et al. Studies on the role of calcium phosphate in the process of calcium oxalate crystal formation. Urol Res. 2009;37(4):181–192. doi: 10.1007/s00240-009-0191-7.
- Martínez-Reyes I, Chandel NS. Mitochondrial TCA cycle metabolites control physiology and disease. Nat Commun. 2020;11(1):102. doi: 10.1038/s41467-019-13668-3.
- Costello LC, Franklin RB. Plasma citrate homeostasis: how it is regulated; and its physiological and clinical implications. An important, but neglected, relationship in medicine. HSOA J Hum Endocrinol. 2016;1(005):28286881.
- Aruga S, Wehrli S, Kaissling B, et al. A: chronic metabolic acidosis increases NaDC-1 mRNA and protein abundance in rat kidney. Kidney Int. 2000;58(1):206–215. doi: 10.1046/j.1523-1755.2000.00155.x.
- Rudman D, Dedonis JL, Fountain MT, et al. Hypocitraturia in patients with gastrointestinal malabsorption. N Engl J Med. 1980;303(12):657–661. doi: 10.1056/NEJM198009183031201.
- Tseng YS, Wu WB, Chen Y, et al. Small intestine resection increases oxalate and citrate transporter expression and calcium oxalate crystal formation in rat hyperoxaluric kidneys. Clin Sci (Lond). 2020;134(19):2565–2580. doi: 10.1042/CS20200973.
- Melnick JZ, Preisig PA, Haynes S, et al. Converting enzyme inhibition causes hypocitraturia independent of acidosis or hypokalemia. Kidney Int. 1998;54(5):1670–1674. doi: 10.1046/j.1523-1755.1998.00150.x.
- Geibel J, Giebisch G, Boron WF. Angiotensin II stimulates both Na(+)-H + exchange and Na+/HCO3- cotransport in the rabbit proximal tubule. Proc Natl Acad Sci USA. 1990;87(20):7917–7920. doi: 10.1073/pnas.87.20.7917.
- Ohana E, Shcheynikov N, Moe OW, et al. SLC26A6 and NaDC-1 transporters interact to regulate oxalate and citrate homeostasis. J Am Soc Nephrol. 2013;24(10):1617–1626. doi: 10.1681/ASN.2013010080.
- Hess B, Jordi S, Zipperle L, et al. Citrate determines calcium oxalate crystallization kinetics and crystal morphology—studies in the presence of tamm–horsfall protein of a healthy subject and a severely recurrent calcium stone former. Nephrol Dial Transplant. 2000;15(3):366–374. doi: 10.1093/ndt/15.3.366.
- Chutipongtanate S, Chaiyarit S, Thongboonkerd V. Citrate, not phosphate, can dissolve calcium oxalate monohydrate crystals and detach these crystals from renal tubular cells. Eur J Pharmacol. 2012;689(1–3):219–225. doi: 10.1016/j.ejphar.2012.06.012.
- He Y, Chen X, Yu Z, et al. Sodium dicarboxylate cotransporter-1 expression in renal tissues and its role in rat experimental nephrolithiasis. J Nephrol. 2004;17(1):34–42.
- Markowitz GS, Perazella MA. Acute phosphate nephropathy. Kidney Int. 2009;76(10):1027–1034. doi: 10.1038/ki.2009.308.
- Sindić A, Chang MH, Mount DB, et al. Renal physiology of SLC26 anion exchangers. Curr Opin Nephrol Hypertens. 2007;16(5):484–490. doi: 10.1097/MNH.0b013e3282e7d7d0.
- Huang HS, Ma MC. High sodium-induced oxidative stress and poor anticrystallization defense aggravate calcium oxalate crystal formation in rat hyperoxaluric kidneys. PLoS One. 2015;10(8):e0134764. doi: 10.1371/journal.pone.0134764.