
Abstract
Chronic kidney disease (CKD) with high morbidity and mortality all over the world is characterized by decreased kidney function, a condition which can result from numerous risk factors, including diabetes, hypertension and obesity. Despite significant advances in our understanding of the pathogenesis of CKD, there are still no treatments that can effectively combat CKD, which underscores the urgent need for further study into the pathological mechanisms underlying this condition. In this regard, animal models of CKD are indispensable. This article reviews a widely used animal model of CKD, which is induced by adenine. While a physiologic dose of adenine is beneficial in terms of biological activity, a high dose of adenine is known to induce renal disease in the organism. Following a brief description of the procedure for disease induction by adenine, major mechanisms of adenine-induced CKD are then reviewed, including inflammation, oxidative stress, programmed cell death, metabolic disorders, and fibrillation. Finally, the application and future perspective of this adenine-induced CKD model as a platform for testing the efficacy of a variety of therapeutic approaches is also discussed. Given the simplicity and reproducibility of this animal model, it remains a valuable tool for studying the pathological mechanisms of CKD and identifying therapeutic targets to fight CKD.
1. Introduction
Chronic kidney disease (CKD) is a progressive disease with no cure and high morbidity and mortality [Citation1–3]. The causes of CKD are complex, including common and well-researched factors such as diabetes, hypertension, obesity and chronic inflammation [Citation4,Citation5]. The pathological features of CKD are characterized by fibrosis, which is a repair cascade initiated by several different stimuli and pathogenic factors, leading to excessive accumulation of matrix connective tissue components [Citation6]. However, at present, there is no accepted systematic strategy for CKD standardized screening and treatment, stressing a continual need to elucidate the underlying pathological mechanisms of CKD. In this regard, animal models of CKD have been invaluable in that utilization of these animal models not only enhances our understanding of the pathogenesis of kidney disease, but also provides excellent platforms for disease intervention in which the efficacy of testing compounds or pharmacological agents can be quantitatively assessed [Citation7–11].
Numerous animal models have been employed to elucidate the pathological mechanisms of CKD [Citation8,Citation12–14]. CKD animal models induced by 5/6 nephrectomy [Citation15–17], diabetic nephropathy [Citation18–23], Hypertension-induced renal injury [Citation4,Citation24], and primary glomerular nephropathy are widely utilized [Citation25–27]. These models have also been used to test the therapeutic effect of various drugs or compound [Citation28–33]. However, this review will specifically focus on the highly prevalent adenine-induced rodent model of CKD, which is implemented in both mice and rats [Citation34–37]. presents a comparative analysis of the adenine model and other CKD-induced animal models.
Table 1. Comparison of animal models of CKD induced by a variety of approaches.
Among all the CKD animal models induced by various approaches outlined in , the adenine-induced model provides certain advantages that are lacking in other models. Firstly, as a nucleobase (a purine derivative), adenine is environmentally nontoxic, presenting no hazard in routine laboratory handling. Secondly, in contrast to modalities such as 5/6 nephrectomy for modeling CKD, adenine induction is administered as a simple gavage or dietary feeding, which is non-surgical, noninvasive, and animal-friendly. Third, the method of adenine induction allows the production of mild, moderate and severe chronic kidney models with a stable disease course and a high success rate, which vary with the adenine dosage and duration of administration [Citation38, Citation79]. Fourth, the adenine model is capable of effectively simulating the progression of renal disease in both male and female test animals, and has provided key insights into sexual dimorphism, reflecting the clinical reality of CKD that affects both genders [Citation80–84]. Undeniably, the adenine-induced CKD model has its own disadvantages. One disadvantage is that the impact of adenine on reproductive and other organ systems remains speculative, as well as the fact that specific biomarkers of adenine-induced CKD have not been identified. Furthermore, while adenine-induced CKD primarily affects renal tubules, the intricate molecular and biochemical mechanisms leading to nephron injury by adenine have yet to be elucidated.
2. Adenine metabolism and renal implications
Adenine, a fundamental purine nucleobase, plays a crucial role in a variety of biochemical and physiological processes within cells. These processes including DNA and RNA synthesis, the generation of cellular energy through ATP and its related compounds, the modulation of extracellular signaling via ATP, ADP, and adenosine, the formation of the second messenger cAMP, and serving as vital cofactors for numerous enzymes, such as nicotinamide adenine dinucleotide (NAD) and flavin adenine dinucleotide (FAD) [Citation85]. Adenine is a common dietary component present in foods like meats, fish, beer and legumes [Citation86, Citation87], and while it is endogenously generated in the body, intake from these dietary sources also contributes to its overall pool [Citation88]. Within the gastrointestinal tract, physiological levels of adenine are absorbed and then metabolized by adenine phosphoribosyl transferase (APRT) to yield AMP, a precursor to a variety of adenine derivatives including adenosine, inosine 5′-monophosphate, hypoxanthine, and xanthine [Citation89]. Ultimately, xanthine is further metabolized into uric acid, which is excreted by the kidney [Citation90–94]. Additionally, adenine undergoes metabolism through a distinct path at physiological doses, in a reaction catalyzed by xanthine oxidase (XO), which leads to the formation of 2,8-dihydroxyadenine (2,8-DHA) eventually excreted in urine () [Citation95,Citation96]. APRT deficiency is an uncommon autosomal recessive disorder of purine metabolism that can lead to kidney stones and CKD because the enzymatic activity of APRT is crucial for the normal recycling of adenine. [Citation97–99].
Figure 1. Adenine metabolism pathways. APRT: adenine phosphoribosyl transferase; PRPP: 5-phosphoribosyl-1-pyrophosphate; AMP: adenosine monophosphate; IMP: inosine monophosphate; AMPD1: adenosine monophosphate deaminase 1; ADA: adenosine deaminase; PNP: purine nucleoside phosphorylase; XO: xanthine oxidase; PRPS1: phosphoribosyl pyrophosphate synthetase 1; PRPPs: phosphate diphosphokinase; ADSL: adenylosuccinate lyase; ATIC: 5-aminoimidazole-4-carboxamide ribonucleotide formyltransferase/IMP cyclohydrolase; ADSS: Adenylosuccinate synthase 2; ADP: adenosine diphosphate; ATP: adenosine triphosphate; AdeR: adenine receptors.
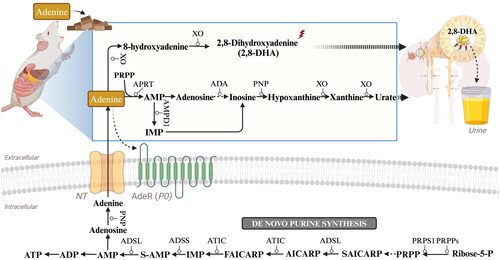
There is growing body of evidence that adenine metabolism is important for both renal function and disease [Citation100–102]. Acting beyond its traditional intracellular roles, adenine can also function as an extracellular signaling molecule, engaging with receptors widely expressed across various organs, the kidney included [Citation103,Citation104]. Intriguingly, an orphan G protein-coupled membrane receptor originally cloned from dorsal root ganglia has been subsequently shown to be a receptor selective for adenine [Citation92,Citation105,Citation106]. Although the highest expression of this adenine receptor (AdeR) has been found in the dorsal root ganglion, significant mRNA levels of the receptor have also been found in other organs such as the kidney [Citation105]. The AdeR is expressed in the vasculature in the cortical region, including glomerular arterioles, and throughout the collecting duct system [Citation107–109]. AdeR binds adenine with high affinity and potentially reduces cellular cAMP levels [Citation41]. Adenine itself can act as an extracellular signaling molecule, but dietary adenine is metabolized in the intestines before it contributes to various biological processes () [Citation110,Citation111]. Currently, the only known pathway for endogenous adenine production in mammals so far is the intracellular polyamine synthesis pathway, where adenine is produced as a byproduct [Citation104]. Numerous concentrative nucleoside transporters (CNTs) and equilibrative nucleoside transporters (ENTs) possess the ability to facilitate the transportation of nucleobases, specifically adenine [Citation112]. While ENTs facilitate diffusion, CNTs function as active transporters that rely solely on Na+ ions [Citation113]. Both ENT1 and ENT2 have been observed to participate in nucleobase transport within cells derived from healthy human kidney (renal proximal tubule cells) as well as renal cancers [Citation114, Citation115]. Nevertheless, the ENT1 transporter is unlikely to play a significant role in renal adenine transportation due to its low affinity for adenine [Citation116,Citation117]. The two nucleoside transporters exhibiting the highest expression levels in the kidney are CNT1 and ENT3, therefore potentially being involved in adenine transport [Citation118–120]. Initially described as a sodium-independent lysosomal transporter with an optimal pH range of 5.5 to 6.5, ENT3 has recently been detected on the mitochondrial membranes and the cell surface in select human cell lines derived from various tissues. These findings indicate that ENT3 is localized differently within subcellular compartments across various cell types [Citation118,Citation119,Citation121]. Given its abundance in the kidney, this transporter may indeed play a vital role in renal function. In summary, endogenous adenine is transported from cytosol to the extracellular space via transporter proteins, before it functions as a signaling molecule through the activation of the G-protein-coupled AdeR [Citation108,Citation122].
3. Adenine-induced renal dysfunction
Although physiological level doses of adenine play an important role, high doses of adenine can be used to induce kidney disease in animals [Citation85]. A search in the Google Scholar database indicates that the first report of a renal problem caused by adenine was published in 1902, and described 2,8-DHA crystals were deposited in the kidneys following adenine administration [Citation123]. This focused subsequent interest of toxicologists on the kidney and its function [Citation124]. The rat model of adenine-induced CKD was first proposed by Yokozawa in 1986, in which adenine feeding or gavage caused tubular obstruction and massive loss of renal units, leading to azotemia, toxin accumulation, and disorders of electrolyte and amino acid metabolism, resulting in renal failure [Citation34,Citation90]. Now, the procedures of adenine-induced CKD in mice and rats are well established and widely used. As shown in , feeding mice and rats with diets containing 0.075–0.75% adenine or intraperitoneally injecting them with an adenine solution for several weeks resulted in the development of CKD in both species. In general, the recommended induction dosage for rats is 0.75% w/w, while for mice, it is 0.20% w/w. Rats are more tolerant to adenine, so the dosage can typically be set at 0.75%. Conversely, mice are more sensitive or less tolerant to adenine intake, so the dosage for mice is generally controlled at 0.20%. Additionally, taste enhancers such as casein may need to be added to the adenine solution for mice [Citation44]. It is, however, crucial to note that elevating the dosage beyond these established benchmarks can culminate in renal toxicity or acute kidney failure. Furthermore, higher doses may also have detrimental effects on other organs, thereby deviating from the intended modeling of CKD and potentially leading to the construction of different disease models such as acute kidney failure or reproductive dysfunction. It is also important to consider the ethical implications and scientific validity of using very high doses, as it can result in a significant number of animal deaths within a short period. On the other hand, reducing the induction dosage below the recommended levels may lead to a more gradual and persistent decline in kidney function. However, this could result in excessively long experimental periods or the absence of the phenotypic expression of CKD.
Figure 2. General experimental scheme of adenine-induced CKD. Adenine is usually given at a dose of 0.75% w/w for rats or 0.20% w/w for mice, and is mixed into normal diets for ad libitum consumption (note: it is advisable to add a certain level of casein to the diets for mice). The progression of CKD may be studied for up to, or beyond, 4 weeks following the start of adenine feed administration.
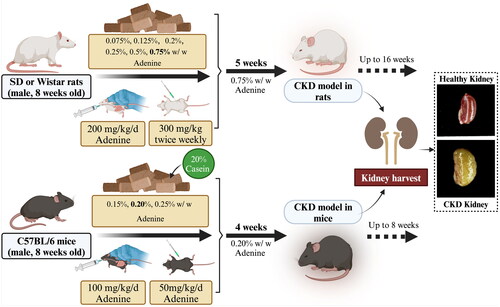
Symptoms of adenine-induced CKD via the different modes of administration are very similar but each has its own advantages and disadvantages [Citation125]. Gavage, for example, allows precise control of the dose of adenine given to the animal; however, due to the invasive nature of this method, it may cause stress to the animal, which can have confounding effects on the development of CKD. In contrast, the addition of adenine to food provides a less invasive method of continuous administration, thereby reducing the stress to which the animal is subjected. Of course, differences in food intake between individual animals can lead to dose inconsistencies, but standardized measures can be used to improve the reliability and reproducibility of study results. Intraperitoneal (IP) administration, although not common in adenine-induced CKD studies, can be applied in studies about CKD-related gastrointestinal dysfunction [Citation126]. However, IP administration may not accurately mimic the natural pathway of human exposure to adenine, and the injection process itself may introduce confounding factors, such as inflammation or local tissue damage, that can affect disease progression. Therefore, considering all factors, CKD is now frequently induced in rodents via feeding administration of adenine to model the disease in preclinical experimental studies.
Renal disease occurs at non-physiologically high concentrations of adenine due to the fact that exogenously administered adenine is not only metabolized to AMP by APRT, but also becomes a substrate for XO, which is converted to 8-hydroxyadenine, and subsequently to the highly insoluble 2,8-DHA, and then the 2,8-DHA forms intratubular precipitates that lead to chronic tubulointerstitial injury, tissue remodeling of crystalline interstitial inclusions, inflammatory and fibrotic processes, renal insufficiency, and metabolic abnormalities similar to the symptoms of CKD in humans [Citation16,Citation34,Citation95,Citation127]. Certainly, the etiology of adenine-induced chronic renal failure is different from that of human chronic renal failure, especially because of the low incidence of crystalline nephropathy as a causative factor in human disease[Citation128]. Despite the different etiology, the adenine model has been exemplary in elucidating the mechanisms of extrarenal comorbidities associated with CKD. Therefore, this method of animal model establishment is commonly used to study the pathological mechanisms of CKD [Citation11,Citation44,Citation79,Citation129,Citation130]. Moreover, it is possible to study the progression of different disease stages in CKD by controlling for the progression after induction periods [Citation131–133]. Additionally, as adenine is administered by gavage or mixed in feed, the process of kidney disease induction is simple and straightforward and as it circumvents the need for surgical intervention. Importantly, adenine-induced renal disease can recapitulate the clinical symptoms of human kidney disease and demonstrates a high degree of reproducibility in experimental settings [Citation39].
With respect to the adenine-induced CKD, the adenine-induced model may have some advantages over other CKD models, including the UUO model, the 5/6 nephrectomy model, the folic acid toxicity model, and the cisplatin model. Combined with those described in the historical literature and practiced in the authors’ laboratories, the major advantages of the adenine-induced model are that it does not require surgical construction, it can be administered by diet to reduce animal suffering and human labor, it can be controlled for CKD disease stages [Citation79,Citation134,Citation135]. Furthermore, the optimization of the dietary ratios can reduce the animal mortality rate during the study, while taking into account the disease development in the animal model, thereby ensuring reproducibility issues [Citation44]. In addition, this model is particularly suitable for the study of CKD with metabolic disorders [Citation136–139]. Given that the mechanisms underlying CKD remain elusive, exploring and contrasting various CKD models could yield comprehensive insights into the pathophysiology. Nevertheless, in the adenine-induced CKD model, it is clear that inflammatory abnormalities, oxidative stress, fibrosis, renal metabolic disorders and ferroptosis are involved in CKD progression [Citation11,Citation140–143].
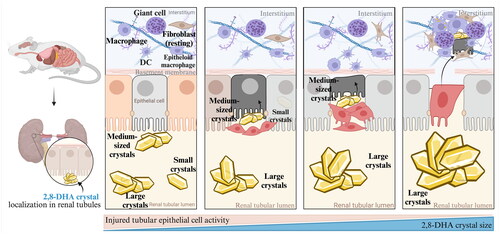
Tubular cells are highly susceptible to nephrotoxic injuries, in particular, the metabolically active and energy-intensive proximal tubular epithelial cells (PTECs) [Citation144]. Notably, crystal deposition and tubular damage in the animal model of adenine-induced CKD are mostly located in the proximal tubular compartment () [Citation135,Citation145]. The accumulation of crystal deposits within renal tubular epithelium leads to mechanical obstruction, interstitial inflammation, and programmed cell death [Citation146]. Notably, a mouse model with CKD induced by an adenine-rich diet exhibits the distinctive characteristics of human CKD patients with renal filtration failure and cardiovascular symptoms [Citation79,Citation147,Citation148]. In addition, the formation of 2,8-DHA crystals in the kidney was found in both the adenine-induced CKD mouse model and human patients with APRT deficiency [Citation149–151]. Thus, the adenine-induced CKD model, which is a laboratory animal model, basically summarizes many of the clinical manifestations of patients with clinical CKD [Citation144]. As is inherent in all animal models of human disease, any model of kidney disease in animals can only serve as a substitute and will never be identical to human kidney disease.
Figure 3. Process of 2,8-DHA crystal granuloma formation. Supersaturation of 2,8-DHA in the urine leads to the formation of 2,8-DHA crystal nuclei. These nuclei adhere to renal tubular cells, creating a nidus for further crystal growth and leading to the formation of crystal plugs in the tubular lumen, causing obstruction of the tubules. Very small crystal plugs were internalized and likely degraded by renal tubular cells, while medium-sized crystal plugs trigger dedifferentiation and proliferative growth of renal tubular cells, eventually resulting in the translocation of plugs into the interstitium. Within the interstitial compartment, renal tubular cells cooperate with macrophages and other cells in the formation of interstitial crystal granulomas that isolate the crystal plug from the renal parenchyma and initiate crystal degradation.
4. Major mechanisms of adenine-induced CKD
When given non-physiological doses of adenine via the diet, adenine is metabolized to form 2,8-DHA, which rapidly accumulates crystal deposits in the renal tubules and leads to recurrent episodes of renal injury that leads to CKD transformation, characterized by a loss of renal function (increased proteinuria and blood urea nitrogen, plasma uric acid, and plasma creatinine) and tubular injury (increased number of dilated tubules, increased cellular debris in the tubular lumen and increased fibrosis) [Citation84,Citation145]. This sequence of events is similar to other animal models of induced CKD, and little is known about the in vivo pathogenesis of the CKD by excessive adenine and 2,8-DHA crystal accumulation. With the increase in the number of days of adenine dietary intake, the model animals showed gradual wasting, chills and cold limbs, significantly enlarged kidneys, grayish-white kidneys with granular surfaces, and elevated serum creatinine (SCr) and blood urea nitrogen (BUN), and other biochemical indexes of CKD [Citation34, Citation79]. In this article, the main mechanisms involved in adenine-induced CKD are summarized.
4.1. Inflammations
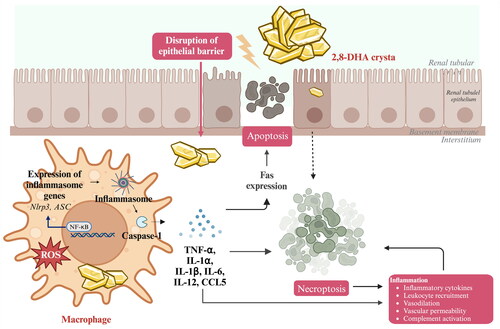
Renal inflammation is a protective response that is induced following kidney injury, which seeks to eliminate the cause of injury and establish tissue repair [Citation152]. Studies have shown that 2,8-DHA crystals can induce renal tubular cell injury and promote an inflammatory phenotype. This involves the activation of toll-like receptors (TLRs) on renal mononuclear phagocytes, the expression of high-mobility group protein B1 (HMGB1), and the activation of nuclear factor-kappa B (NF-κB) in B cells [Citation135]. If left unresolved, persistent generation of 2,8-DHA crystals can lead to a series of biochemical pathways involving immune system cells and intrinsic renal cells. In the context of chronic inflammation, DHA crystal-induced renal damage is characterized by sustained inflammation with increased expression of the chemokine Ccl5 and monocyte infiltration, mainly composed of F4/80+ macrophages and resident renal dendritic cells, as well as ErHr3+ monocytes and monocyte-derived macrophages representing a subset of F4/80+ cells [Citation153–155]. Macrophages accumulate around damaged cortical and medullary tubules and often form interstitial granulomas around the crystals [Citation156]. In this process, tubular cells in cooperation with macrophages overgrow and transfer crystals into the interstitium, restoring the patency of the tubular lumen; this was followed by degradation of interstitial crystals as a result of granulomatous inflammation [Citation135, Citation157]. If renal tubular injury and persistent inflammation are not resolved, the intrinsic cells of the kidney exhibit increased reactivity to the inflammatory and fibrotic processes in the environment of chronic inflammation. Changes in their gene expression profile enable them to produce and release more pro-fibrotic cytokines, driving fibroblast differentiation and progressive transformation toward myofibroblasts. This process is associated with oxidative stress and cell death () [Citation152, Citation158].
Figure 4. Processes of the primary mechanism of adenine-induced renal inflammation.
4.2. Oxidative stress
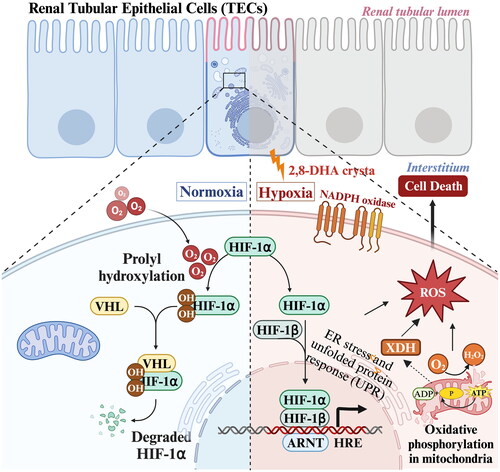
Oxidative stress is already found in early stages of renal disease and increases with declining kidney function [Citation159–162]. Numerous studies demonstrate renal oxidative stress in the adenine-induced CKD model [Citation163–167]. The occlusion of renal tubules by 2,8-DHA crystals not only affects the tubules themselves but also imposes a pressure load on the peritubular capillaries, resulting in subsequent renal hypoxia [Citation131, Citation134, Citation168]. Hypoxia accelerates degradation of cellular ATP and increases the production of hypoxanthine, which activates renal XO, subsequently leading to the production of oxidative stress [Citation169]. For instance, in adenine-induced mouse model, Keiichi Ohata et al. found that the levels of HIF-1α mRNA and XO activity in the kidneys of the adenine diet group were significantly higher than those of the normal diet group, but the levels of catalase mRNA were significantly lower than those of the normal diet group; thus, higher levels of oxidative stress promote the progression of renal tubulointerstitial injury in the adenine model [Citation165]. Furthermore, in their research on an adenine-induced CKD model, Ali, Badreldin H. et al. found that the concentrations of glutathione and superoxide dismutase, markers of oxidative stress, were significantly increased in an adenine-induced CKD model. Meanwhile, reactive oxygen species directly impaired mitochondrial function, DNA synthesis, triggered genomic instability and induced cell death [Citation164]. These findings suggest that adenine is closely related to oxidative stress ().
Figure 5. Processes of the major mechanism of adenine-induced renal oxidative stress.
4.3. Programmed cell death
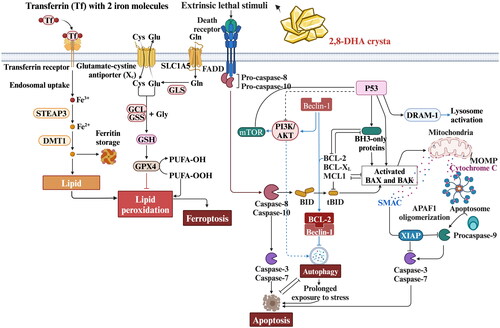
The pathophysiology of renal diseases is significantly connected with programmed renal cell death, including apoptosis, necroptosis, ferroptosis, pyroptosis, and autophagy, as is now recognized [Citation170–176]. In rodent models of adenine-induced nephropathy, where crystal deposition and tubular injury are predominantly observed in the proximal tubular compartment, Muhammad Ali Khan et al. confirmed the pathobiological process of ferroptosis within these structures. Their research employed an experimental rat model of adenine-induced nephropathy and extended to adenine-exposed human primary proximal tubular epithelial cells (PTEC). Notably, in rats subjected to an adenine-rich diet, substantial elevations in iron accumulation and lipid peroxidation, specifically 4-hydroxynonenal (4-HNE), were discovered within the tubules—hallmarks signaling ferroptosis. Furthermore, in human primary PTEC cultures challenged with adenine, substantial rise in mitochondrial superoxide production, mitochondrial depolarization, DNA damage, and incidence of necrotic cell death were observed, when compared to untreated PTEC. Detailed molecular studies into adenine-challenged PTEC confirmed these findings, highlighting a significant reduction in the lipid-repairing enzyme, glutathione peroxidase 4 (GPX4), and a corresponding increase in 4-HNE. Such changes further validate the occurrence of ferroptotic cell death in the context of excessive adenine. Furthermore, Ferrostatin-1 (a potent inhibitor of ferroptosis) treatment attenuated PTEC death in the adenine-induced CKD model by targeting the aforementioned ferroptosis pathway through the inhibition of lipid peroxidation, which further demonstrated that ferroptosis plays a more important role in the adenine-induced CKD model [Citation143, Citation177]. Notably, other types of programmed cell death, such as cellular apoptosis and autophagy, have also been reported in adenine-induced CKD () [Citation38, Citation178–183].
Figure 6. Processes of the major mechanism of adenine-induced renal programmed cell death.
4.4. Metabolic disorders
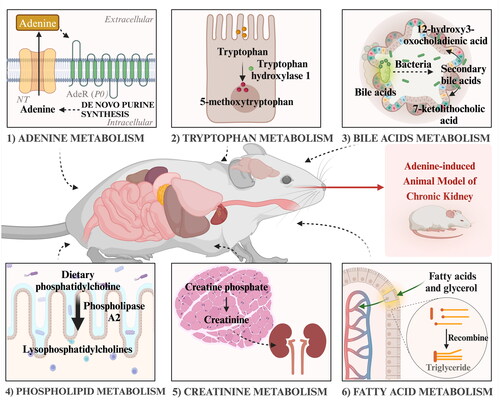
Multiple metabolic pathways, including those involving adenine, tryptophan, bile acids, phospholipids, creatinine, and fatty acid metabolism, have also been reported to be dysregulated in the adenine-induced CKD model [Citation137–139, Citation163, Citation184–187]. In a study exploring biomarkers of early stages of CKD, Toshihiro Kobayashi et al. applied metabolomics to blood samples collected from adenine-induced CKD rats for 28 days, and analyzed them to find that indolyl sulfate, phenyl sulfate, p-cresol sulfate, and phenylacetylglycine were increased with the progression of renal hypoplasia, suggesting that tryptophan and glycocholic acid may reflect the development of CKD [Citation138]. Ying-Yong Zhao et al. identified 12 endogenous metabolites in the urine of CKD rats, and the changes in these metabolites suggest that disturbances of amino acid metabolism, phospholipid metabolism, and energy metabolism are associated with adenine-induced CKD rats [Citation185]. It was also found that indoxyl sulfate and p-cresyl sulfate were significantly increased in adenine-induced CKD rats and stimulate progressive tubulointerstitial fibrosis by increasing the expression of transforming growth factor-β1 (TGF-β1) [Citation187]. Similarly, in an animal model of adenine induction and in patients with early-stage CKD, it was found that reduced levels of tryptophan metabolite 5-MTP and its regulatory enzyme, TPH-1, causes renal injury by activating NF-κB and inhibiting Nrf2 pathways, and this dysfunction leads to accumulation of renal extracellular matrix and progression of renal fibrosis [Citation188, Citation189]. Tomoko Kimura et al. showed that the main cause of anemia in CKD is a lack of adequate erythropoietin (EPO), but other factors may also be involved, including dysregulation of iron metabolism, using an adenine-induced mouse model of CKD [Citation40]. Overall, these studies suggest that dysregulation of various metabolic pathways, including those involving adenine, tryptophan, bile acids, phospholipid, creatinine, and fatty acid metabolism, is associated with the development and progression of adenine-induced CKD (). Moreover, the findings regarding the role of specific metabolites in promoting renal injury and fibrosis through activation of pro-fibrotic pathways highlight the potential therapeutic targets for the treatment of CKD.
Figure 7. Processes of the major mechanism of adenine-induced renal metabolic disorders.
4.5. Fibroblast/myofibroblast activation
When renal tubular epithelial cells (RTECs) are persistently injured, such as with sustained 2,8-DHA accumulation, RTECs produce a large number of growth factors and cytokines, including TGF-β, PDGF, hedgehog, and Wnt ligands [Citation190–193]. These cytokines secreted by injured RTECs can accelerate the regeneration of injured RTECs and promote the differentiation of fibroblasts into myofibroblasts [Citation194]. Activated myofibroblasts undergo rapid proliferation and produce a large amounts of ECM components, such as fibronectin and collagen, which contribute to the progression of renal tubulointerstitial fibrosis [Citation195]. For instance, numerous studies have demonstrated that adenine significantly upregulated the expression of type IV collagen, fibronectin, and overall extracellular matrix in the kidneys of mice with adenine-induced kidney injury, resulting in disruption of TGF-β/Smad signaling and epithelial mesenchymal transition (EMT) in RTECs [Citation166, Citation196–200]. During this process, the activation of TGF-β1 stimulates ERK, p38 MAPK, and JNK pathways, mediating the progression of renal fibrosis [Citation166, Citation196, Citation201]. Additionally, fibroblast-derived αvβ1 integrin, a critical regulator of renal fibrosis, is the only αv integrin on renal fibroblasts capable of binding to and activating TGF-β. Inhibiting αvβ1 integrin in an adenine-induced model has been shown to the activation of the TGF-β complex and ameliorate renal failure and fibrosis [Citation202]. Furthermore adenine was found to upregulate Wnt1 protein expression in the kidneys of CKD rats, along with renal β-catenin protein expression. This was accompanied by an upregulation of downstream proteins, including Snail1, Twist, MMP-7, PAI-1, and FSP1, suggesting that the overactivity of the Wnt/β-catenin signaling pathway promotes myofibroblast differentiation and renal injury through RAS activation [Citation203]. Therefore, adenine-induced fibroblast/myofibroblast activation communicates is associated with multiple signaling pathways, forming a feedback loop that exacerbates the progression of renal fibrosis by leading to exacerbation of EMT (). Overall and mechanistically, it should be noted that adenine-diet animal model primarily damages the kidneys, with the main damage occurring in the proximal tubules. Importantly, this model can develop CKD-related complication that mimic those seen in humans, such as vascular calcification, mineral and bone disorders, muscle loss, and others [Citation79, Citation204–208]. As outlined in the literature above, in experimental animals with adenine-induced CKD, inflammation and oxidative stress contribute to the repair of renal tubular damage while simultaneously increasing the expression of pro-fibrotic cytokines and collagen deposition to aid in the recovery of kidney damage. However, the persistent production of 2,8-DHA crystals can lead to renal tubule epithelial cell death via multiple pathways, including apoptosis, necroptosis, ferroptosis, and others. This process involves multifactorial upstream signal transduction abnormalities, including TGF-β/Smad and Wnt/β-catenin signaling disruptions, among a series of interrelated factors. Therefore, the precise mechanism underlying adenine-induced CKD still require thorough investigation.
Figure 8. Processes of the major mechanism of adenine-induced renal fibroblast/myofibroblast activation.

5. Research applications of the adenine-induced CKD model
Beyond shedding light on the pathophysiological underpinnings of renal diseases, the adenine-induced animal model of kidney disease serves a pivotal role in evaluating the efficacy of various therapeutic interventions. These interventions include not only pharmacological agents but also chemical and natural compounds. As illustrated in , a curated selection of studies employing this model emphasizes the diverse array of experimental therapies tested against CKD triggered by adenine. It should be noted that all compounds listed are in the preclinical stage, as their beneficial effects on CKD trials have involved experimental animals.
Table 2. Adenine-induced animal models for testing therapeutic interventions in CKD.
It is worth noting that animal models of kidney disease, regardless of the inducing agents or triggers used, commonly exhibit exacerbated oxidative damage, which is a shared mechanism leading to renal inflammation and fibrosis [Citation237]. Given these findings, the natural products with antioxidant properties outlined in may offer therapeutic potential for managing adenine-induced kidney injury. However, it must be acknowledged that determining the most effective natural products may be challenging due to differences in testing conditions and experimental designs among different laboratories. For the prevention and treatment of CKD, the use of adenine-induced animal models to evaluate drug effectiveness is well-established, which will contribute to the development of novel medications and drive advancements in the field of nephrology.
6. Miscellaneous
Adenine-induced animal models are valuable experimental tools for elucidating the mechanisms underlying CKD and for studying its common complications. For instance, vascular calcification and bone pathology are frequent complications in CKD progression, scenarios often observed in clinical CKD patients [Citation238–241]. Most of the CKD animal models do not report cardiovascular-related complications. However, 0.75% adenine diet for 6 weeks showed most of the vascular calcification changes expected in CKD animal models [Citation242–244]. Additionally, Neven et al. demonstrated bone and mineral disorders for the first time in the adenine-model of induced CKD [Citation245], and subsequently, several studies have utilized the adenine-induced CKD model to assess CKD-related osteoporosis and mineral bone disease [Citation42, Citation43, Citation246, Citation247]. Indeed, abnormal mineral metabolism in patients with clinical nephropathy, which leads to secondary hyperparathyroidism, has been replicated in the adenine-induced CKD model [Citation44, Citation223, Citation246]. It’s also worth noting that male CKD patients may develop gonadal abnormalities leading to impotence, decreased libido, reduced testicular size, impaired spermatogenesis, and gynecomastia [Citation248–250]. Yuichi Adachi et al. first reported that adenine-induced CKD reduces testicular function by inhibiting 17β-hydroxysteroid oxidoreductase, which is crucial for converting androstenedione to testosterone [Citation251], and they reported that the hormonal status in male rats with adenine-induced CKD changes over time post-induction [Citation252]. In conclusion, adenine diet animal models not only present a stable progression of CKD but also model multiple comorbidities, effectively mimicking the complexities of human CKD progression. Consequently, they are indispensable for the assessment of disease mechanisms and therapeutic strategies in preclinical CKD studies.
7. Conclusion and future perspectives
The adenine-induced CKD animal model holds great promise for future CKD research. This model can facilitate drug screening and efficacy evaluation, paving the way to the discovery of innovative therapeutic targets and medications. Furthermore, it serves as a valuable tool for investigating the underlying pathological mechanisms of CKD, facilitating a comprehensive understanding of molecular, cellular, and tissue-level changes. Future research may involve integrating omics technologies, such as transcriptomics, proteomics and metabolomics to harness this preclinical animal model for revealing the complex pathogenesis and pathophysiological alterations of CKD. Additionally, the model offers a prime opportunity to be leveraged for the development of innovative therapeutic strategies (e.g. for addressing APRT deficiency), including cellular therapies and gene editing technologies, aiming to ultimately reverse or cure the disease. Finally, interdisciplinary collaborations can further enhance research efforts, exploring the interconnections between common complications of CKD and other fields such as cardiovascular, reproductive and orthopedic medicine. These perspectives demonstrate the vast potential and broad application prospects of adenine-induced CKD animal models in advancing our understanding and treatment of CKD.
Authors’ contributions
Yang Q.: conceptualization, project administration, and writing; Su S.Y., and Luo N.: data curation; Cao G.: Funding acquisition.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Yan M-T, Chao C-T, Lin S-H. Chronic kidney disease: strategies to retard progression. IJMS. 2021;22(18):1. doi: 10.3390/ijms221810084.
- Kalantar-Zadeh K, Jafar TH, Nitsch D, et al. Chronic kidney disease. Lancet. 2021;398(10302):786–22. doi: 10.1016/S0140-6736(21)00519-5.
- Ruiz-Ortega M, Rayego-Mateos S, Lamas S, et al. Targeting the progression of chronic kidney disease. Nat Rev Nephrol. 2020;16(5):269–288. doi: 10.1038/s41581-019-0248-y.
- Ku E, Lee BJ, Wei J, et al. Hypertension in CKD: core curriculum 2019. Am J Kidney Dis. 2019;74(1):120–131. doi: 10.1053/j.ajkd.2018.12.044.
- Webster AC, Nagler EV, Morton RL, et al. Chronic kidney disease. Lancet. 2017;389(10075):1238–1252. doi: 10.1016/S0140-6736(16)32064-5.
- Panizo S, Martínez-Arias L, Alonso-Montes C, et al. Fibrosis in chronic kidney disease: pathogenesis and consequences. IJMS. 2021;22(1):408. doi: 10.3390/ijms22010408.
- Djudjaj S, Boor P. Cellular and molecular mechanisms of kidney fibrosis. Mol Aspects Med. 2019;65:16–36. doi: 10.1016/j.mam.2018.06.002.
- Becker GJ, Hewitson TD. Animal models of chronic kidney disease: useful but not perfect. Nephrol Dial Transplant. 2013;28(10):2432–2438. doi: 10.1093/ndt/gft071.
- Noone D, Licht C. Chronic kidney disease: a new look at pathogenetic mechanisms and treatment options. Pediatr Nephrol. 2014;29(5):779–792. doi: 10.1007/s00467-013-2436-5.
- Musgrove J, Wolf M. Regulation and effects of FGF23 in chronic kidney disease. Annu Rev Physiol. 2020;82(1):365–390. doi: 10.1146/annurev-physiol-021119-034650.
- Kumakura S, Sato E, Sekimoto A, et al. Nicotinamide attenuates the progression of renal failure in a mouse model of adenine-induced chronic kidney disease. Toxins (Basel). 2021;13(1):50. doi: 10.3390/toxins13010050.
- Bao Y-W, Yuan Y, Chen J-H, et al. Kidney disease models: tools to identify mechanisms and potential therapeutic targets. Zool Res. 2018;39(2):72–86. doi: 10.24272/j.issn.2095-8137.2017.055.
- Fu Y, Tang C, Cai J, et al. Rodent models of AKI-CKD transition. Am J Physiol Renal Physiol. 2018;315(4):F1098–F1106. doi: 10.1152/ajprenal.00199.2018.
- Orlando LA, Belasco EJ, Patel UD, et al. The chronic kidney disease model: a general purpose model of disease progression and treatment. BMC Med Inform Decis Mak. 2011;11(1):41. doi: 10.1186/1472-6947-11-41.
- Singh S, Grabner A, Yanucil C, et al. Fibroblast growth factor 23 directly targets hepatocytes to promote inflammation in chronic kidney disease. Kidney Int. 2016;90(5):985–996. doi: 10.1016/j.kint.2016.05.019.
- Vaziri ND, Wong J, Pahl M, et al. Chronic kidney disease alters intestinal microbial flora. Kidney Int. 2013;83(2):308–315. doi: 10.1038/ki.2012.345.
- Watanabe H, Miyamoto Y, Honda D, et al. p-Cresyl sulfate causes renal tubular cell damage by inducing oxidative stress by activation of NADPH oxidase. Kidney Int. 2013;83(4):582–592. doi: 10.1038/ki.2012.448.
- Alicic RZ, Rooney MT, Tuttle KR. Diabetic kidney disease. Clin J Am Soc Nephrol. 2017;12(12):2032–2045. doi: 10.2215/CJN.11491116.
- Wada J, Makino H. Inflammation and the pathogenesis of diabetic nephropathy. Clin Sci (Lond). 2013;124(3):139–152. doi: 10.1042/CS20120198.
- Reidy K, Kang HM, Hostetter T, et al. Molecular mechanisms of diabetic kidney disease. J Clin Invest. 2014;124(6):2333–2340. doi: 10.1172/JCI72271.
- Putta S, Lanting L, Sun G, et al. Inhibiting microRNA-192 ameliorates renal fibrosis in diabetic nephropathy. J Am Soc Nephrol. 2012;23(3):458–469. doi: 10.1681/ASN.2011050485.
- Thomas MC, Brownlee M, Susztak K, et al. Diabetic kidney disease. Nat Rev Dis Primers. 2015;1(1):15018. doi: 10.1038/nrdp.2015.18.
- Tuttle KR, Bakris GL, Bilous RW, et al. Diabetic kidney disease: a report from an ADA consensus conference. Am J Kidney Dis. 2014;64(4):510–533. doi: 10.1053/j.ajkd.2014.08.001.
- Udani S, Lazich I, Bakris GL. Epidemiology of hypertensive kidney disease. Nat Rev Nephrol. 2011;7(1):11–21. doi: 10.1038/nrneph.2010.154.
- Bertani T, Rocchi G, Sacchi G, et al. Adriamycin-induced glomerulosclerosis in the rat. Am J Kidney Dis. 1986;7(1):12–19. doi: 10.1016/s0272-6386(86)80051-8.
- Arif E, Solanki AK, Nihalani D. Adriamycin susceptibility among C57BL/6 substrains. Kidney Int. 2016;89(3):721–723. doi: 10.1016/j.kint.2015.10.019.
- Xiong T, Attar M, Gnirck A-C, et al. Interleukin-9 protects from early podocyte injury and progressive glomerulosclerosis in adriamycin-induced nephropathy. Kidney Int. 2020;98(3):615–629. doi: 10.1016/j.kint.2020.04.036.
- Breyer MD, Susztak K. The next generation of therapeutics for chronic kidney disease. Nat Rev Drug Discov. 2016;15(8):568–588. doi: 10.1038/nrd.2016.67.
- Chen D-Q, Hu H-H, Wang Y-N, et al. Natural products for the prevention and treatment of kidney disease. Phytomedicine. 2018;50:50–60. doi: 10.1016/j.phymed.2018.09.182.
- Jivishov E, Nahar L, Sarker SD. Nephroprotective natural products. In: Annual reports in medicinal chemistry. Elsevier; 2020. p. 251–271. doi: 10.1016/bs.armc.2020.02.003.
- Rahman M, Akter S, Dorotea D, et al. Renoprotective potentials of small molecule natural products targeting mitochondrial dysfunction. Front Pharmacol. 2022;13:925993. doi: 10.3389/fphar.2022.925993.
- Uddin MJ, Kim EH, Hannan M, et al. Pharmacotherapy against oxidative stress in chronic kidney disease: promising small molecule natural products targeting Nrf2-HO-1 signaling. Antioxidants. 2021;10(2):258. doi: 10.3390/antiox10020258.
- Zhou Z, Qiao Y, Zhao Y, et al. Natural products: potential drugs for the treatment of renal fibrosis. Chin Med. 2022;17(1):98. doi: 10.1186/s13020-022-00646-z.
- Yokozawa T, Zheng PD, Oura H, et al. Animal model of adenine-induced chronic renal failure in rats. Nephron. 1986;44(3):230–234. doi: 10.1159/000183992.
- Liu X, Deng R, Chen Y, et al. Jian-Pi-Yi-Shen formula improves adenine-induced chronic kidney disease via regulating tryptophan metabolism and aryl hydrocarbon receptor signaling. Front Pharmacol. 2022;13:922707. doi: 10.3389/fphar.2022.922707.
- Li X, Li W, Zhang Z, et al. SIRT6 overexpression retards renal interstitial fibrosis through targeting HIPK2 in chronic kidney disease. Front Pharmacol. 2022;13:1007168. doi: 10.3389/fphar.2022.1007168.
- Li L, Li A, Gan L, et al. Roxadustat improves renal osteodystrophy by dual regulation of bone remodeling. Endocrine. 2022;79(1):180–189. doi: 10.1007/s12020-022-03199-1.
- Diwan V, Mistry A, Gobe G, et al. Adenine-induced chronic kidney and cardiovascular damage in rats. J Pharmacol Toxicol Methods. 2013;68(2):197–207. doi: 10.1016/j.vascn.2013.05.006.
- Kim K, Anderson EM, Thome T, et al. Skeletal myopathy in CKD: a comparison of adenine-induced nephropathy and 5/6 nephrectomy models in mice. Am J Physiol Renal Physiol. 2021;321(1):F106–F119. doi: 10.1152/ajprenal.00117.2021.
- Kimura T, Kuragano T, Yamamoto K, et al. Deregulated iron metabolism in bone marrow from adenine-induced mouse model of chronic kidney disease. Int J Hematol. 2019;109(1):59–69. doi: 10.1007/s12185-018-2531-2.
- Kobuchi S, Ayajiki K. Impairment of endothelial function in adenine-induced renal injury in rats. J. Pharmacol. Sci. 2016;130:s246.
- Mohamed FF, Amadeu de Oliveira F, Kinoshita Y, et al. Dentoalveolar alterations in an adenine-induced chronic kidney disease mouse model. J Bone Miner Res. 2023;38(8):1192–1207. doi: 10.1002/jbmr.4829.
- Saito H, Miyakoshi N, Kasukawa Y, et al. Analysis of bone in adenine-induced chronic kidney disease model rats. Osteoporos Sarcopenia. 2021;7(4):121–126. doi: 10.1016/j.afos.2021.11.001.
- Jia T, Olauson H, Lindberg K, et al. A novel model of adenine-induced tubulointerstitial nephropathy in mice. BMC Nephrol. 2013;14(1):116. doi: 10.1186/1471-2369-14-116.
- Mazumder MK, Giri A, Kumar S, et al. A highly reproducible mice model of chronic kidney disease: evidences of behavioural abnormalities and blood-brain barrier disruption. Life Sci. 2016;161:27–36. doi: 10.1016/j.lfs.2016.07.020.
- Aparicio-Trejo OE, Avila-Rojas SH, Tapia E, et al. Chronic impairment of mitochondrial bioenergetics and β-oxidation promotes experimental AKI-to-CKD transition induced by folic acid. Free Radic Biol Med. 2020;154:18–32. doi: 10.1016/j.freeradbiomed.2020.04.016.
- Perales-Quintana MM, Saucedo AL, Lucio-Gutiérrez JR, et al. Metabolomic and biochemical characterization of a new model of the transition of acute kidney injury to chronic kidney disease induced by folic acid. Peer J. 2019;7:e7113. doi: 10.7717/peerj.7113.
- Zhou Y, Luo Z, Liao C, et al. MHC class II in renal tubules plays an essential role in renal fibrosis. Cell Mol Immunol. 2021;18(11):2530–2540. doi: 10.1038/s41423-021-00763-z.
- Luan J, Fu J, Jiao C, et al. IL-18 deficiency ameliorates the progression from AKI to CKD. Cell Death Dis. 2022;13(11):957. doi: 10.1038/s41419-022-05394-4.
- Katagiri D, Hamasaki Y, Doi K, et al. Interstitial renal fibrosis due to multiple cisplatin treatments is ameliorated by semicarbazide-sensitive amine oxidase inhibition. Kidney Int. 2016;89(2):374–385. doi: 10.1038/ki.2015.327.
- Torres R, Velazquez H, Chang JJ, et al. Three-dimensional morphology by multiphoton microscopy with clearing in a model of cisplatin-induced CKD. J Am Soc Nephrol. 2016;27(4):1102–1112. doi: 10.1681/ASN.2015010079.
- Sharp CN, Doll MA, Megyesi J, et al. Subclinical kidney injury induced by repeated cisplatin administration results in progressive chronic kidney disease. Am J Physiol Renal Physiol. 2018;315(1):F161–F172. doi: 10.1152/ajprenal.00636.2017.
- Sears SM, Siskind LJ. Potential therapeutic targets for cisplatin-induced kidney injury: lessons from other models of AKI and fibrosis. J Am Soc Nephrol. 2021;32(7):1559–1567. doi: 10.1681/ASN.2020101455.
- Xu Z, Li W, Han J, et al. Angiotensin II induces kidney inflammatory injury and fibrosis through binding to myeloid differentiation protein-2 (MD2). Sci Rep. 2017;7(1):44911. doi: 10.1038/srep44911.
- Yang Q, Hu J, Yang Y, et al. Sirt6 deficiency aggravates angiotensin II-induced cholesterol accumulation and injury in podocytes. Theranostics. 2020;10(16):7465–7479. doi: 10.7150/thno.45003.
- Su M, Li W, Yuan Y, et al. Epididymal white adipose tissue promotes angiotensin II-induced cardiac fibrosis in an exosome-dependent manner. Transl Res. 2022;248:51–67. doi: 10.1016/j.trsl.2022.05.004.
- Matsushita Y, Ogawa D, Wada J, et al. Activation of peroxisome proliferator–activated receptor δ inhibits streptozotocin-induced diabetic nephropathy through anti-inflammatory mechanisms in mice. Diabetes. 2011;60(3):960–968. doi: 10.2337/db10-1361.
- Kostic S, Hauke T, Ghahramani N, et al. Expression pattern of apoptosis-inducing factor in the kidneys of streptozotocin-induced diabetic rats. Acta Histochem. 2020;122(8):151655. doi: 10.1016/j.acthis.2020.151655.
- Li YJ, Chen X, Kwan TK, et al. Dietary fiber protects against diabetic nephropathy through short-chain fatty acid–mediated activation of G protein–coupled receptors GPR43 and GPR109A. J Am Soc Nephrol. 2020;31(6):1267–1281. doi: 10.1681/ASN.2019101029.
- Choi SY, Lim SW, Salimi S, et al. Tonicity-responsive enhancer-binding protein mediates hyperglycemia-induced inflammation and vascular and renal injury. J Am Soc Nephrol. 2018;29(2):492–504. doi: 10.1681/ASN.2017070718.
- Bing P, Maode L, Li F, et al. Expression of renal transforming growth factor-β and its receptors in a rat model of chronic cyclosporine-induced nephropathy. Transplant Proc. 2006;38(7):2176–2179. doi: 10.1016/j.transproceed.2006.07.015.
- Du H, Le G, Hou L, et al. Nontoxic concentration of ochratoxin a aggravates renal fibrosis induced by adriamycin/cyclosporine a nephropathy via TGF-β1/SMAD2/3. J Agric Food Chem. 2022;70(43):14005–14014. doi: 10.1021/acs.jafc.2c03577.
- Chen A, Sheu L-F, Ho Y-S, et al. Experimental focal segmental glomerulosclerosis in mice. Nephron. 1998;78(4):440–452. doi: 10.1159/000044974.
- Bucciarelli E, Binazzi R, Santori P, et al. Nephrotic syndrome in rats due to adriamycin chlorhydrate. Lav Ist Anat Istol Patol Univ Studi Perugia. 1976;36:53–69.
- Cao Q, Lu J, Li Q, et al. CD103+ dendritic cells elicit CD8+ T cell responses to accelerate kidney injury in adriamycin nephropathy. J Am Soc Nephrol. 2016;27(5):1344–1360. doi: 10.1681/ASN.2015030229.
- Yang B, Xie Y, Guo M, et al. Nephrotoxicity and Chinese herbal medicine. Clin J Am Soc Nephrol. 2018;13(10):1605–1611. doi: 10.2215/CJN.11571017.
- Furusho T, Sohara E, Mandai S, et al. Renal TNFα activates the WNK phosphorylation Cascade and contributes to salt-sensitive hypertension in chronic kidney disease. Kidney Int. 2020;97(4):713–727. doi: 10.1016/j.kint.2019.11.021.
- Heidarian E, Jafari-Dehkordi E, Valipour P, et al. Nephroprotective and anti-inflammatory effects of Pistacia atlantica leaf hydroethanolic extract against gentamicin-induced nephrotoxicity in rats. J Diet Suppl. 2017;14(5):489–502. doi: 10.1080/19390211.2016.1267062.
- Rizwan F, Yesmine S, Banu SG, et al. Renoprotective effects of stevia (stevia rebaudiana bertoni), amlodipine, valsartan, and losartan in gentamycin-induced nephrotoxicity in the rat model: biochemical, hematological and histological approaches. Toxicol Rep. 2019;6:683–691. doi: 10.1016/j.toxrep.2019.07.003.
- Ryu SH, Kim JW, Yoon D, et al. Serum and urine toxicometabolomics following gentamicin-induced nephrotoxicity in male Sprague-Dawley rats. J Toxicol Environ Health A. 2018;81(11):408–420. doi: 10.1080/15287394.2018.1451180.
- S L, X J, Y L, et al. Sodium/calcium overload and Sirt1/Nrf2/OH-1 pathway are critical events in mercuric chloride-induced nephrotoxicity. Chemosphere. 2019;234:579–588. doi: 10.1016/j.chemosphere.2019.06.095.
- Joshi D, Srivastav SK, Belemkar S, et al. Zingiber officinale and 6-gingerol alleviate liver and kidney dysfunctions and oxidative stress induced by mercuric chloride in male rats: a protective approach. Biomed Pharmacother. 2017;91:645–655. doi: 10.1016/j.biopha.2017.04.108.
- Tao Y-y, Wang Q-L, Yuan J-L, et al. Effects of vitamin E on mercuric chloride-induced renal interstitial fibrosis in rats and the antioxidative mechanism. Zhong Xi Yi Jie He Xue Bao. 2011;9(2):201–208. doi: 10.3736/jcim20110214.
- Nørregaard R, Mutsaers HAM, Frøkiær J, et al. Obstructive nephropathy and molecular pathophysiology of renal interstitial fibrosis. Physiol Rev. 2023;103(4):2827–2872. doi: 10.1152/physrev.00027.2022.
- Sárközy M, Watzinger S, Kovács ZZA, et al. Neuregulin-1β improves uremic cardiomyopathy and renal dysfunction in rats. JACC Basic Transl Sci. 2023;8(9):1160–1176. doi: 10.1016/j.jacbts.2023.03.003.
- Chen Y, Mao C, Gu R, et al. Nidogen-2 is a novel endogenous ligand of LGR4 to inhibit vascular calcification. Circ Res. 2022;131(12):1037–1054. doi: 10.1161/CIRCRESAHA.122.321614.
- Chou Y-H, Pan S-Y, Shao Y-H, et al. Methylation in pericytes after acute injury promotes chronic kidney disease. J Clin Invest. 2020;130(9):4845–4857. doi: 10.1172/JCI135773.
- Walker JA, Richards S, Belghasem ME, et al. Temporal and tissue-specific activation of aryl hydrocarbon receptor in discrete mouse models of kidney disease. Kidney Int. 2020;97(3):538–550. doi: 10.1016/j.kint.2019.09.029.
- Diwan V, Brown L, Gobe GC. Adenine-induced chronic kidney disease in rats. Nephrology (Carlton). 2018;23(1):5–11. doi: 10.1111/nep.13180.
- Makhloufi C, Nicolas F, McKay N, et al. Female AhR knockout mice develop a minor renal insufficiency in an adenine-diet model of chronic kidney disease. Int J Mol Sci. 2020;21(7):2483. doi: 10.3390/ijms21072483.
- Melsom T, Norvik JV, Enoksen IT, et al. Sex differences in age-related loss of kidney function. J Am Soc Nephrol. 2022;33(10):1891–1902. doi: 10.1681/ASN.2022030323.
- Merz CNB, Dember LM, Ingelfinger JR, et al. Sex and the kidneys: current understanding and research opportunities. Nat Rev Nephrol. 2019;15(12):776–783. doi: 10.1038/s41581-019-0208-6.
- Neugarten J, Golestaneh L. Influence of sex on the progression of chronic kidney disease. Mayo Clin Proc. 2019;94(7):1339–1356. doi: 10.1016/j.mayocp.2018.12.024.
- Diwan V, Small D, Kauter K, et al. Gender differences in adenine-induced chronic kidney disease and cardiovascular complications in rats. Am J Physiol Renal Physiol. 2014;307(11):F1169–F1178. doi: 10.1152/ajprenal.00676.2013.
- Lutwak-Mann C. Adenine derivatives and their biological functions. Biol Rev. 1939;14(4):399–419. doi: 10.1111/j.1469-185X.1939.tb00850.x.
- Wu B, Roseland JM, Haytowitz DB, et al. Availability and quality of published data on the purine content of foods, alcoholic beverages, and dietary supplements. J Food Compos Anal. 2019;84:103281. doi: 10.1016/j.jfca.2019.103281.
- Hou C, Xiao G, Amakye WK, et al. Guidelines for purine extraction and determination in foods. Food Frontiers. 2021;2(4):557–573. doi: 10.1002/fft2.100.
- Kaneko K, Aoyagi Y, Fukuuchi T, et al. Total purine and purine base content of common foodstuffs for facilitating nutritional therapy for gout and hyperuricemia. Biol Pharm Bull. 2014;37(5):709–721. doi: 10.1248/bpb.b13-00967.
- Salati LM, Gross CJ, Henderson LM, et al. Absorption and metabolism of adenine, adenosine-5’-monophosphate, adenosine and hypoxanthine by the isolated vascularly perfused rat small intestine. J Nutr. 1984;114(4):753–760. doi: 10.1093/jn/114.4.753.
- Yokozawa T, Oura H, Okada T. Metabolic effects of dietary purine in rats. J Nutr Sci Vitaminol (Tokyo). 1982;28(5):519–526. doi: 10.3177/jnsv.28.519.
- Shuvy M, Nyska A, Beeri R, et al. Histopathology and apoptosis in an animal model of reversible renal injury. Exp Toxicol Pathol. 2011;63(4):303–306. doi: 10.1016/j.etp.2010.02.002.
- Gorzalka S, Vittori S, Volpini R, et al. Evidence for the functional expression and pharmacological characterization of adenine receptors in native cells and tissues. Mol Pharmacol. 2005;67(3):955–964. doi: 10.1124/mol.104.006601.
- Coade SB, Pearson JD. Metabolism of adenine nucleotides in human blood. Circ Res. 1989;65(3):531–537. doi: 10.1161/01.RES.65.3.531.
- Choi HK, Mount DB, Reginato AM. Pathogenesis of gout. Ann Intern Med. 2005;143(7):499. doi: 10.7326/0003-4819-143-7-200510040-00009.
- Wyngaarden JB, Dunn JT. 8-Hydroxyadenine as the intermediate in the oxidation of adenine to 2,8-dihydroxyadenine by xanthine oxidase. Arch Biochem Biophys. 1957;70(1):150–156. doi: 10.1016/0003-9861(57)90088-7.
- George J, Struthers AD. Role of urate, xanthine oxidase and the effects of allopurinol in vascular oxidative stress. Vasc Health Risk Manag. 2009;5(1):265–272. doi: 10.2147/VHRM.S4265.
- Runolfsdottir HL, Palsson R, Agustsdottir IM, et al. Kidney disease in adenine phosphoribosyltransferase deficiency. Am J Kidney Dis. 2016;67(3):431–438. doi: 10.1053/j.ajkd.2015.10.023.
- Stockelman MG, Lorenz JN, Smith FN, et al. Chronic renal failure in a mouse model of human adenine phosphoribosyltransferase deficiency. Am J Physiol. 1998;275(1):F154–F163. doi: 10.1152/ajprenal.1998.275.1.F154.
- Van Acker KJ, Simmonds HA, Potter C, et al. Complete deficiency of adenine phosphoribosyltransferase. Report of a family. N Engl J Med. 1977;297(3):127–132. doi: 10.1056/NEJM197707212970302.
- Sharaf El Din UAA, Salem MM, Abdulazim DO. Uric acid in the pathogenesis of metabolic, renal, and cardiovascular diseases: a review. J Adv Res. 2017;8(5):537–548. doi: 10.1016/j.jare.2016.11.004.
- Katsyuba E, Romani M, Hofer D, et al. NAD + homeostasis in health and disease. Nat Metab. 2020;2(1):9–31. doi: 10.1038/s42255-019-0161-5.
- Burnstock G. Purinergic signalling: therapeutic developments. Front Pharmacol. 2017;8:661. doi: 10.3389/fphar.2017.00661.
- Fukuda T, Majumder K, Zhang H, et al. Adenine has an anti-inflammatory effect through the activation of adenine receptor signaling in mouse macrophage. J Funct Foods. 2017;28:235–239. doi: 10.1016/j.jff.2016.11.013.
- Thimm D, Schiedel AC, Peti-Peterdi J, et al. The nucleobase adenine as a signalling molecule in the kidney. Acta Physiol (Oxf). 2015;213(4):808–818. doi: 10.1111/apha.12452.
- Bender E, Buist A, Jurzak M, et al. Characterization of an orphan G protein-coupled receptor localized in the dorsal root ganglia reveals adenine as a signaling molecule. Proc Natl Acad Sci USA. 2002;99(13):8573–8578. doi: 10.1073/pnas.122016499.
- Knospe M, Müller CE, Rosa P, et al. The rat adenine receptor: pharmacological characterization and mutagenesis studies to investigate its putative ligand binding site. Purinergic Signal. 2013;9(3):367–381. doi: 10.1007/s11302-013-9355-6.
- Peti-Peterdi J, Kishore BK, Pluznick JL. Regulation of vascular and renal function by metabolite receptors. Annu. Rev. Physiol. 2016;78(1):391–414. doi: 10.1146/annurev-physiol-021115-105403.
- Thimm D, Knospe M, Abdelrahman A, et al. Characterization of new G protein-coupled adenine receptors in mouse and hamster. Purinergic Signal. 2013;9(3):415–426. doi: 10.1007/s11302-013-9360-9.
- Kishore BK, Zhang Y, Pop IL, et al. Cellular localization of P0 (adenine) receptor in rat kidney. Faseb J. 2012;26(S1):688.3–688.3. doi:10.1096/fasebj.26.1_supplement.688.3.
- Kamatani N, Carson DA. Dependence of adenine production upon polyamine synthesis in cultured human lymphoblasts. Biochim Biophys Acta. 1981;675(3–4):344–350. doi: 10.1016/0304-4165(81)90024-6.
- Caruso V, Zuccarini M, Di Iorio P, et al. Metabolic changes induced by purinergic signaling: role in food intake. Front Pharmacol. 2021;12:655989. doi: 10.3389/fphar.2021.655989.
- Wright NJ, Lee S-Y. Toward a molecular basis of cellular nucleoside transport in humans. Chem Rev. 2021;121(9):5336–5358. doi: 10.1021/acs.chemrev.0c00644.
- Inoue K. Molecular basis of nucleobase transport systems in mammals. Biol Pharm Bull. 2017;40(8):1130–1138. doi: 10.1248/bpb.b17-00374.
- Damaraju VL, Mowles D, Wilson M, et al. Comparative in vitro evaluation of transportability and toxicity of capecitabine and its metabolites in cells derived from normal human kidney and renal cancers. Biochem Cell Biol. 2013;91(6):419–427. doi: 10.1139/bcb-2013-0041.
- Hau RK, Wright SH, Cherrington NJ. Addressing the clinical importance of equilibrative nucleoside transporters in drug discovery and development. Clin Pharmacol Ther. 2023;114(4):780–794. doi: 10.1002/cpt.2984.
- SenGupta DJ, Lum PY, Lai Y, et al. A single glycine mutation in the equilibrative nucleoside transporter gene, hENT1, alters nucleoside transport activity and sensitivity to nitrobenzylthioinosine. Biochemistry. 2002;41(5):1512–1519. doi: 10.1021/bi015833w.
- Chen C, Xie T, Zhang Y, et al. Erythrocyte ENT1-AMPD3 axis is an essential purinergic hypoxia sensor and energy regulator combating CKD in a mouse model. J Am Soc Nephrol. 2023;34(10):1647–1671. doi: 10.1681/ASN.0000000000000195.
- Govindarajan R, Leung GPH, Zhou M, et al. Facilitated mitochondrial import of antiviral and anticancer nucleoside drugs by human equilibrative nucleoside transporter-3. Am J Physiol Gastrointest Liver Physiol. 2009;296(4):G910–922. doi: 10.1152/ajpgi.90672.2008.
- Lu H, Chen C, Klaassen C. Tissue distribution of concentrative and equilibrative nucleoside transporters in male and female rats and mice. Drug Metab Dispos. 2004;32(12):1455–1461. doi: 10.1124/dmd.104.001123.
- Pastor-Anglada M, Mata-Ventosa A, Pérez-Torras S. Inborn errors of nucleoside transporter (NT)-encoding genes (SLC28 and SLC29. Int J Mol Sci. 2022;23(15):8770. doi: 10.3390/ijms23158770.
- Cheng X, Klaassen CD. Tissue distribution, ontogeny, and hormonal regulation of xenobiotic transporters in mouse kidneys. Drug Metab Dispos. 2009;37(11):2178–2185. doi: 10.1124/dmd.109.027177.
- Kishore BK, Zhang Y, Gevorgyan H, et al. Cellular localization of adenine receptors in the rat kidney and their functional significance in the inner medullary collecting duct. Am J Physiol Renal Physiol. 2013;305(9):F1298–F1305. doi: 10.1152/ajprenal.00254.2013.
- Nicolaier A. Ueber die umwand lung des adenins im thierischen organismus, Z. Klin. Med. 1902;45:359.
- Warner WL. Toxicology and pharmacology of adenine in animals and man. Transfusion. 1977;17(4):326–332. doi: 10.1046/j.1537-2995.1977.17477216860.x.
- Al Za’abi M, Ali B, Yasin J, et al. Development of a new model for the induction of chronic kidney disease via intraperitoneal adenine administration, and the effect of treatment with gum acacia thereon. FASEB J. 2015;29(S1):938–933. doi: 10.1096/fasebj.29.1_supplement.938.3.
- Hayeeawaema F, Muangnil P, Jiangsakul J, et al. A novel model of adenine-induced chronic kidney disease-associated gastrointestinal dysfunction in mice: the gut-kidney axis. Saudi J Biol Sci. 2023;30(6):103660. doi: 10.1016/j.sjbs.2023.103660.
- Manfredi JP, Holmes EW. Purine salvage pathways in myocardium. Annu Rev Physiol. 1985;47(1):691–705. doi: 10.1146/annurev.ph.47.030185.003355.
- Herlitz LC, D’Agati VD, Markowitz GS. Crystalline nephropathies. Arch Pathol Lab Med. 2012;136(7):713–720. doi: 10.5858/arpa.2011-0565-RA.
- Mishima E, Ichijo M, Kawabe T, et al. Germ-free conditions modulate host purine metabolism, exacerbating adenine-induced kidney damage. Toxins (Basel). 2020;12(9):547. doi: 10.3390/toxins12090547.
- Motohashi H, Tahara Y, Whittaker DS, et al. The circadian clock is disrupted in mice with adenine-induced tubulointerstitial nephropathy. Kidney Int. 2020;97(4):728–740. doi: 10.1016/j.kint.2019.09.032.
- Ullah MM, Ow CPC, Hilliard Krause LM, et al. Renal oxygenation during the early stages of adenine-induced chronic kidney disease. Am J Physiol Renal Physiol. 2019;317(5):F1189–F1200. doi: 10.1152/ajprenal.00253.2019.
- Pruijm M, Phan O, Zanchi A. Metformin versus SGLT-2 inhibitors: how low can we go? Kidney Int. 2022;101(5):874–877. doi: 10.1016/j.kint.2022.02.012.
- Chen D-Q, Chen H, Chen L, et al. The link between phenotype and fatty acid metabolism in advanced chronic kidney disease. Nephrol Dial Transplant. 2017;32(7):1154–1166. doi: 10.1093/ndt/gfw415.
- Fong D, Ullah MM, Lal JG, et al. Renal cellular hypoxia in adenine-induced chronic kidney disease. Clin Exp Pharmacol Physiol. 2016;43(10):896–905. doi: 10.1111/1440-1681.12621.
- Klinkhammer BM, Djudjaj S, Kunter U, et al. Cellular and molecular mechanisms of kidney injury in 2,8-dihydroxyadenine nephropathy. J Am Soc Nephrol. 2020;31(4):799–816. doi: 10.1681/ASN.2019080827.
- Zhang Z-H, Chen H, Vaziri ND, et al. Metabolomic signatures of chronic kidney disease of diverse etiologies in the rats and humans. J Proteome Res. 2016;15(10):3802–3812. doi: 10.1021/acs.jproteome.6b00583.
- Miao H, Wu X, Wang Y, et al. 1-Hydroxypyrene mediates renal fibrosis through aryl hydrocarbon receptor signalling pathway. Br J Pharmacol. 2022;179(1):103–124. doi: 10.1111/bph.15705.
- Kobayashi T, Matsumura Y, Ozawa T, et al. Exploration of novel predictive markers in rat plasma of the early stages of chronic renal failure. Anal Bioanal Chem. 2014;406(5):1365–1376. doi: 10.1007/s00216-013-7472-2.
- Arinze NV, Yin W, Lotfollahzadeh S, et al. Tryptophan metabolites suppress the Wnt pathway and promote adverse limb events in chronic kidney disease. J Clin Invest. 2022;132(1):e142260. doi: 10.1172/JCI142260.
- Li Q-M, Chena H-R, Zha X-Q, et al. Renoprotective effect of Chinese chive polysaccharides in adenine-induced chronic renal failure. Int J Biol Macromol. 2018;106:988–993. doi: 10.1016/j.ijbiomac.2017.08.101.
- Zaaba NE, Al-Salam S, Beegam S, et al. Catalpol attenuates oxidative stress and inflammation via mechanisms involving sirtuin-1 activation and NF-κB inhibition in experimentally-induced chronic kidney disease. Nutrients. 2023;15(1):237. doi: 10.3390/nu15010237.
- Zhao Y-Y, Cheng X-L, Wei F, et al. Serum metabonomics study of adenine-induced chronic renal failure in rats by ultra performance liquid chromatography coupled with quadrupole time-of-flight mass spectrometry. Biomarkers. 2012;17(1):48–55. doi: 10.3109/1354750X.2011.637180.
- Khan MA, Nag P, Grivei A, et al. Adenine overload induces ferroptosis in human primary proximal tubular epithelial cells. Cell Death Dis. 2022;13(2):104. doi: 10.1038/s41419-022-04527-z.
- Chevalier RL. The proximal tubule is the primary target of injury and progression of kidney disease: role of the glomerulotubular junction. Am J Physiol Renal Physiol. 2016;311(1):F145–161. doi: 10.1152/ajprenal.00164.2016.
- Choi J, Choi M-S, Jeon J, et al. In vivo longitudinal 920 nm two-photon intravital kidney imaging of a dynamic 2,8-DHA crystal formation and tubular deterioration in the adenine-induced chronic kidney disease mouse model. Biomed Opt Express. 2023;14(4):1647–1658. doi: 10.1364/BOE.485187.
- Mulay SR, Shi C, Ma X, et al. Novel insights into crystal-induced kidney injury. Kidney Dis (Basel). 2018;4(2):49–57. doi: 10.1159/000487671.
- Fang C, Lau WL, Sun J, et al. Chronic kidney disease promotes cerebral microhemorrhage formation. J Neuroinflammation. 2023;20(1):51. doi: 10.1186/s12974-023-02703-2.
- Nguy L, Shubbar E, Jernås M, et al. Adenine-induced chronic renal failure in rats decreases aortic relaxation rate and alters expression of proteins involved in vascular smooth muscle calcium handling. Acta Physiol (Oxf). 2016;218(4):250–264. doi: 10.1111/apha.12724.
- Ali BH, Al-Salam S, Za’abi MA, et al. New model for adenine-induced chronic renal failure in mice, and the effect of gum acacia treatment thereon: comparison with rats. J Pharmacol Toxicol Methods. 2013;68(3):384–393. doi: 10.1016/j.vascn.2013.05.001.
- Rabe M, Schaefer F. Non-transgenic mouse models of kidney disease. Nephron. 2016;133(1):53–61. doi: 10.1159/000445171.
- Verma R, Niraimathi M, Prasad P, et al. Dihydroxyadenine crystal-induced nephropathy presenting with rapidly progressive renal failure. Kidney Res Clin Pract. 2018;37(3):287–291. doi: 10.23876/j.krcp.2018.37.3.287.
- Meng X-M, Nikolic-Paterson DJ, Lan HY. Inflammatory processes in renal fibrosis. Nat Rev Nephrol. 2014;10(9):493–503. doi: 10.1038/nrneph.2014.114.
- de Jong JP, Voerman JS, van der Sluijs-Gelling AJ, et al. A monoclonal antibody (ER-HR3) against murine macrophages. I. Ontogeny, distribution and enzyme histochemical characterization of ER-HR3-positive cells. Cell Tissue Res. 1994;275(3):567–576. doi: 10.1007/BF00318825.
- Rogers NM, Ferenbach DA, Isenberg JS, et al. Dendritic cells and macrophages in the kidney: a spectrum of good and evil. Nat Rev Nephrol. 2014;10(11):625–643. doi: 10.1038/nrneph.2014.170.
- Furuichi K, Kaneko S, Wada T. Chemokine/chemokine receptor-mediated inflammation regulates pathologic changes from acute kidney injury to chronic kidney disease. Clin Exp Nephrol. 2009;13(1):9–14. doi: 10.1007/s10157-008-0119-5.
- Vervaet BA, Verhulst A, D’Haese PC, et al. Nephrocalcinosis: new insights into mechanisms and consequences. Nephrol Dial Transplant. 2009;24(7):2030–2035. doi: 10.1093/ndt/gfp115.
- Correa-Costa M, Braga TT, Felizardo RJF, et al. Macrophage trafficking as key mediator of adenine-induced kidney injury. Mediators Inflamm. 2014;2014:291024–291012. doi: 10.1155/2014/291024.
- Aminzadeh MA, Nicholas SB, Norris KC, et al. Role of impaired Nrf2 activation in the pathogenesis of oxidative stress and inflammation in chronic tubulo-interstitial nephropathy. Nephrol Dial Transplant. 2013;28(8):2038–2045. doi: 10.1093/ndt/gft022.
- Karamouzis I, Sarafidis PA, Karamouzis M, et al. Increase in oxidative stress but not in antioxidant capacity with advancing stages of chronic kidney disease. Am J Nephrol. 2008;28(3):397–404. doi: 10.1159/000112413.
- Srivastava A, Tomar B, Sharma D, et al. Mitochondrial dysfunction and oxidative stress: role in chronic kidney disease. Life Sci. 2023;319:121432. doi: 10.1016/j.lfs.2023.121432.
- Ebert T, Neytchev O, Witasp A, et al. Inflammation and oxidative stress in chronic kidney disease and dialysis patients. Antioxid Redox Signal. 2021;35(17):1426–1448. doi: 10.1089/ars.2020.8184.
- Dounousi E, Papavasiliou E, Makedou A, et al. Oxidative stress is progressively enhanced with advancing stages of CKD. Am J Kidney Dis. 2006;48(5):752–760. doi: 10.1053/j.ajkd.2006.08.015.
- Chen H, Cao G, Chen D-Q, et al. Metabolomics insights into activated redox signaling and lipid metabolism dysfunction in chronic kidney disease progression. Redox Biol. 2016;10:168–178. doi: 10.1016/j.redox.2016.09.014.
- Ali BH, Al-Husseni I, Beegam S, et al. Effect of gum Arabic on oxidative stress and inflammation in adenine–induced chronic renal failure in rats. PLoS One. 2013;8(2):e55242. doi: 10.1371/journal.pone.0055242.
- Ohata K, Kamijo-Ikemori A, Sugaya T, et al. Renoprotective effect of the xanthine oxidoreductase inhibitor topiroxostat under decreased angiotensin II type 1a receptor expression. Eur J Pharmacol. 2017;815:88–97. doi: 10.1016/j.ejphar.2017.09.005.
- Saleh MA, Awad AM, Ibrahim TM, et al. Small-dose sunitinib modulates p53, bcl-2, STAT3, and ERK1/2 pathways and protects against adenine-induced nephrotoxicity. Pharmaceuticals. 2020;13(11):397. doi: 10.3390/ph13110397.
- Ali BH, Al-Salam S, Al Suleimani Y, et al. Curcumin ameliorates kidney function and oxidative stress in experimental chronic kidney disease. Basic Clin Pharmacol Toxicol. 2018;122(1):65–73. doi: 10.1111/bcpt.12817.
- Kamijo-Ikemori A, Sugaya T, Hibi C, et al. Renoprotective effect of the xanthine oxidoreductase inhibitor topiroxostat on adenine-induced renal injury. Am J Physiol Renal Physiol. 2016;310(11):F1366–F1376. doi: 10.1152/ajprenal.00517.2015.
- Saugstad OD. Hypoxanthine as an indicator of hypoxia: its role in health and disease through free radical production. Pediatr Res. 1988;23(2):143–150. doi: 10.1203/00006450-198802000-00001.
- Wang Y, Cai J, Tang C, et al. Mitophagy in acute kidney injury and kidney repair. Cells. 2020;9(2):338. doi: 10.3390/cells9020338.
- Cuevas S, Pelegrín P. Pyroptosis and redox balance in kidney diseases. Antioxidants Redox Signal. 2021;35(1):40–60. doi: 10.1089/ars.2020.8243.
- Carney EF. Ferroptotic stress promotes the AKI to CKD transition. Nat Rev Nephrol. 2021;17(10):633–633. doi: 10.1038/s41581-021-00482-8.
- Belavgeni A, Meyer C, Stumpf J, et al. Ferroptosis and necroptosis in the kidney. Cell Chem Biol. 2020;27(4):448–462. doi: 10.1016/j.chembiol.2020.03.016.
- Ni L, Yuan C, Wu X. Targeting ferroptosis in acute kidney injury. Cell Death Dis. 2022;13(2):182. doi: 10.1038/s41419-022-04628-9.
- Maremonti F, Meyer C, Linkermann A. Mechanisms and models of kidney tubular necrosis and nephron loss. JASN. 2022;33(3):472–486. doi: 10.1681/ASN.2021101293.
- Tang C, Livingston MJ, Liu Z, et al. Autophagy in kidney homeostasis and disease. Nat Rev Nephrol. 2020;16(9):489–508. doi: 10.1038/s41581-020-0309-2.
- Dixon SJ, Lemberg KM, Lamprecht MR, et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149(5):1060–1072. doi: 10.1016/j.cell.2012.03.042.
- Song X, Pang H, Cui W, et al. Renoprotective effects of enzyme-hydrolyzed polysaccharides from auricularia polytricha on adenine-induced chronic kidney diseases in mice. Biomed Pharmaco. 2021;135:111004. doi: 10.1016/j.biopha.2020.111004.
- Wu D, Luo N, Wang L, et al. Hydrogen sulfide ameliorates chronic renal failure in rats by inhibiting apoptosis and inflammation through ROS/MAPK and NF-κB signaling pathways. Sci Rep. 2017;7(1):455. doi: 10.1038/s41598-017-00557-2.
- Awad AM, Saleh MA, Abu-Elsaad NM, et al. Erlotinib can halt adenine induced nephrotoxicity in mice through modulating ERK1/2, STAT3, p53 and apoptotic pathways. Sci Rep. 2020;10(1):11524. doi: 10.1038/s41598-020-68480-7.
- Tian F, Wang Z, He J, et al. 4-Octyl itaconate protects against renal fibrosis via inhibiting TGF-β/smad pathway, autophagy and reducing generation of reactive oxygen species. Eur J Pharmacol. 2020;873:172989. doi: 10.1016/j.ejphar.2020.172989.
- Yuan H, Zheng C, Zhu L, et al. Contribution of TFEB-mediated autophagy to tubulointerstitial fibrosis in mice with adenine-induced chronic kidney disease. Biomed Pharmaco. 2021;133:110949. doi: 10.1016/j.biopha.2020.110949.
- Khamis T, Alsemeh AE, Alanazi A, et al. Breast milk mesenchymal stem cells and/or derived exosomes mitigated adenine-induced nephropathy via modulating renal autophagy and fibrotic signaling pathways and their epigenetic regulations. Pharmaceutics. 2023;15(8):2149. doi: 10.3390/pharmaceutics15082149.
- Zhao Y-Y, Cheng X-L, Wei F, et al. Application of faecal metabonomics on an experimental model of tubulointerstitial fibrosis by ultra performance liquid chromatography/high-sensitivity mass spectrometry with MSE data collection technique. Biomarkers. 2012;17(8):721–729. doi: 10.3109/1354750X.2012.724450.
- Zhao Y-Y, Liu J, Cheng X-L, et al. Urinary metabonomics study on biochemical changes in an experimental model of chronic renal failure by adenine based on UPLC Q-TOF/MS. Clin Chim Acta. 2012;413(5–6):642–649. doi: 10.1016/j.cca.2011.12.014.
- Khattri RB, Thome T, Ryan TE. Tissue-specific 1H-NMR metabolomic profiling in mice with adenine-induced chronic kidney disease. Metabolites. 2021;11(1):45. doi: 10.3390/metabo11010045.
- Zhao Y-Y, Cheng X-L, Wei F, et al. Intrarenal metabolomic investigation of chronic kidney disease and its TGF-β1 mechanism in induced-adenine rats using UPLC Q-TOF/HSMS/MSE. J. Proteome Res. 2013;12(2):692–703. doi: 10.1021/pr3007792.
- Chen D-Q, Cao G, Chen H, et al. Identification of serum metabolites associating with chronic kidney disease progression and anti-fibrotic effect of 5-methoxytryptophan. Nat Commun. 2019;10(1):1476. doi: 10.1038/s41467-019-09329-0.
- Chen D-Q, Wu X-Q, Chen L, et al. Poricoic acid a as a modulator of TPH-1 expression inhibits renal fibrosis via modulating protein stability of β-catenin and β-catenin-mediated transcription. Therapeutic Adv Chronic Dis. 2020;11:204062232096264. doi: 10.1177/2040622320962648.
- Gewin L, Zent R, Pozzi A. Progression of chronic kidney disease: too much cellular talk causes damage. Kidney Int. 2017;91(3):552–560. doi: 10.1016/j.kint.2016.08.025.
- Succar L, Pianta TJ, Davidson T, et al. Subclinical chronic kidney disease modifies the diagnosis of experimental acute kidney injury. Kidney Int. 2017;92(3):680–692. doi: 10.1016/j.kint.2017.02.030.
- Perazella MA. Renal vulnerability to drug toxicity. Clinical J Am Soc Nephrol. 2009;4(7):1275–1283. doi: 10.2215/CJN.02050309.
- Edeling M, Ragi G, Huang S, et al. Developmental signalling pathways in renal fibrosis: the roles of notch, Wnt and hedgehog. Nat Rev Nephrol. 2016;12(7):426–439. doi: 10.1038/nrneph.2016.54.
- Wynn T. Cellular and molecular mechanisms of fibrosis. Journal Pathol. 2008;214(2):199–210. doi: 10.1002/path.2277.
- Duffield JS, Lupher M, Thannickal VJ, et al. Host responses in tissue repair and fibrosis. Annu Rev Pathol. 2013;8:241–276. doi: 10.1146/annurev-pathol-020712-163930.
- Yi H, Huang C, Shi Y, et al. Metformin attenuates renal fibrosis in a mouse model of adenine-induced renal injury through inhibiting TGF-β1 signaling pathways. Front Cell Dev Biol. 2021;9:603802. doi: 10.3389/fcell.2021.603802.
- Thakur R, Sharma A, Lingaraju MC, et al. Ameliorative effect of ursolic acid on renal fibrosis in adenine-induced chronic kidney disease in rats. Biomed Pharmaco. 2018;101:972–980. doi: 10.1016/j.biopha.2018.02.143.
- Zhang Z-H, Li M-H, Liu D, et al. Rhubarb protect against tubulointerstitial fibrosis by inhibiting TGF-β/smad pathway and improving abnormal metabolome in chronic kidney disease. Front Pharmacol. 2018;9:1029. doi: 10.3389/fphar.2018.01029.
- Cai H, Su S, Li Y, et al. Protective effects of salvia miltiorrhiza on adenine-induced chronic renal failure by regulating the metabolic profiling and modulating the NADPH oxidase/ROS/ERK and TGF-β/smad signaling pathways. J Ethnopharmacol. 2018;212:153–165. doi: 10.1016/j.jep.2017.09.021.
- Wang B, Jha JC, Hagiwara S, et al. Transforming growth factor-β1-mediated renal fibrosis is dependent on the regulation of transforming growth factor receptor 1 expression by let-7b. Kidney Int. 2014;85(2):352–361. doi: 10.1038/ki.2013.372.
- Meng X, Nikolic-Paterson DJ, Lan HY. TGF-β: the master regulator of fibrosis. Nat Rev Nephrol. 2016;12(6):325–338. doi: 10.1038/nrneph.2016.48.
- Chang Y, Lau WL, Jo H, et al. Pharmacologic blockade of αvβ1 integrin ameliorates renal failure and fibrosis in vivo. JASN. 2017;28(7):1998–2005. doi: 10.1681/ASN.2015050585.
- Wang Y-N, Liu H-J, Ren L-L, et al. Shenkang injection improves chronic kidney disease by inhibiting multiple renin-angiotensin system genes by blocking the Wnt/β-catenin signalling pathway. Front Pharmacol. 2022;13:964370. doi: 10.3389/fphar.2022.964370.
- Massini G, Caldiroli L, Molinari P, et al. Nutritional strategies to prevent muscle loss and sarcopenia in chronic kidney disease: what do we currently know? Nutrients. 2023;15(14):3107. doi: 10.3390/nu15143107.
- Chillon J-M, Massy ZA, Stengel B. Neurological complications in chronic kidney disease patients, nephrol. Nephrol Dial. Transplant. 2016;31(10):1606–1614. doi: 10.1093/ndt/gfv315.
- Kelly DM, Ademi Z, Doehner W, et al. Chronic kidney disease and cerebrovascular disease. Stroke. 2021;52(7):e328–e346. doi: 10.1161/STROKEAHA.120.029680.
- Pellicori P, Kalra PR, Clark AL, et al. Chronic kidney disease (CKD) and CKD-ism in heart failure – what a mess!. European J Heart Fail. 2022;24(11):2196–2198. doi: 10.1002/ejhf.2696.
- Floege J, Drüeke TB. Mineral and bone disorder in chronic kidney disease: pioneering studies. Kidney Int. 2020;98(4):807–811. doi: 10.1016/j.kint.2020.07.002.
- Abdelrahman AM, Suleimani YA, Za’abi MA, et al. The renoprotective effect of the dipeptidyl peptidase-4 inhibitor sitagliptin on adenine-induced kidney disease in rats. Biomed Pharmacother. 2019;110:667–676. doi: 10.1016/j.biopha.2018.11.117.
- Ali BH, Al Za’abi M, Adham SA, et al. The effect of sildenafil on rats with adenine—induced chronic kidney disease. Biomed Pharmacother. 2018;108:391–402. doi: 10.1016/j.biopha.2018.09.061.
- Fang J, Guo Y, Yin W, et al. Neoxanthin alleviates the chronic renal failure-induced aging and fibrosis by regulating inflammatory process. Int Immunopharmacol. 2023;114:109429. doi: 10.1016/j.intimp.2022.109429.
- Horinouchi Y, Murashima Y, Yamada Y, et al. Pemafibrate inhibited renal dysfunction and fibrosis in a mouse model of adenine-induced chronic kidney disease. Life Sci. 2023;321:121590. doi: 10.1016/j.lfs.2023.121590.
- Hsu C-N, Hou C-Y, Chang-Chien G-P, et al. Sodium thiosulfate improves hypertension in rats with adenine-induced chronic kidney disease. Antioxidants. 2022;11(1):147. doi: 10.3390/antiox11010147.
- Subhash N, Sriram R, Kurian GA. Sodium thiosulfate protects brain in rat model of adenine induced vascular calcification. Neurochem Int. 2015;90:193–203. doi: 10.1016/j.neuint.2015.09.004.
- Hsu C-N, Yang H-W, Hou C-Y, et al. Melatonin prevents chronic kidney disease-induced hypertension in young rat treated with adenine: implications of gut microbiota-derived metabolites. Antioxidants. 2021;10(8):1211. doi: 10.3390/antiox10081211.
- Klaerner G, Shao J, Biyani K, et al. Mechanism of action of veverimer: a novel, orally administered, nonabsorbed, counterion-free, hydrochloric acid binder under development for the treatment of metabolic acidosis in chronic kidney disease. J Pharmacol Exp Ther. 2020;375(3):439–450. doi: 10.1124/jpet.120.000190.
- Matsui I, Hamano T, Inoue K, et al. L-lysine ameliorates vascular calcification in adenine-induced uremic rats. Nephrol Dial Transplant. 2013;28:102–102.
- Shimomura A, Matsui I, Hamano T, et al. Dietary L-lysine prevents arterial calcification in adenine-induced uremic rats. J Am Soc Nephrol. 2014;25(9):1954–1965. doi: 10.1681/ASN.2013090967.
- Ou Y, Hou W, Li S, et al. Sodium citrate inhibits endoplasmic reticulum stress in rats with adenine-induced chronic renal failure. Am J Nephrol. 2015;42(1):14–21. doi: 10.1159/000437235.
- Ou Y, Li S, Zhu X, et al. Citrate attenuates adenine-induced chronic renal failure in rats by modulating the Th17/treg cell balance. Inflammation. 2016;39(1):79–86. doi: 10.1007/s10753-015-0225-y.
- Patel P, Patel S, Chudasama P, et al. Roflumilast alleviates adenine-induced chronic kidney disease by regulating inflammatory biomarkers. Eur J Pharmacol. 2023;949:175731. doi: 10.1016/j.ejphar.2023.175731.
- Samaha MM, Nour OA, Sewilam HM, et al. Diacerein mitigates adenine-induced chronic kidney disease in rats: focus on TLR4/MYD88/TRAF6/NF-κB pathway. Life Sci. 2023;331:122080. doi: 10.1016/j.lfs.2023.122080.
- Sato H, Goto M, Nishimura G, et al. Upacicalcet, a positive allosteric modulator of the calcium-sensing receptor, prevents vascular calcification and bone disorder in a rat adenine-induced secondary hyperparathyroidism model. Bone. 2023;167:116613. doi: 10.1016/j.bone.2022.116613.
- Vaja V, Sun CC, Cappellini MD, et al. A hepcidin inhibitor mobilizes iron for incorporation into red blood cells in an adenine-induced chronic kidney disease model in rats. Blood. 2012;120(21):2082–2082. doi: 10.1182/blood.V120.21.2082.2082.
- Thibodeau J-F, Simard J-C, Holterman CE, et al. PBI-4050 via GPR40 activation improves adenine-induced kidney injury in mice. Clin Sci (Lond). 2019;133(14):1587–1602. doi: 10.1042/CS20190479.
- Tsuboi Y, Ohtomo S, Ichida Y, et al. EOS789, a novel pan-phosphate transporter inhibitor, is effective for the treatment of chronic kidney disease–mineral bone disorder. Kidney Int. 2020;98(2):343–354. doi: 10.1016/j.kint.2020.02.040.
- Wu M, Tang R-N, Liu H, et al. Cinacalcet attenuates the renal endothelial-to-mesenchymal transition in rats with adenine-induced renal failure. Am J Physiol Renal Physiol. 2014;306(1):F138–F146. doi: 10.1152/ajprenal.00375.2013.
- Zeng J, Huang H, Zhang Y, et al. Dapagliflozin alleviates renal fibrosis in a mouse model of adenine-induced renal injury by inhibiting TGF-β1/MAPK mediated mitochondrial damage. Front Pharmacol. 2023;14:1095487. doi: 10.3389/fphar.2023.1095487.
- Atteia HH, Alamri ES, Sirag N, et al. Soluble guanylate cyclase agonist, isoliquiritigenin attenuates renal damage and aortic calcification in a rat model of chronic kidney failure. Life Sci. 2023;317:121460. doi: 10.1016/j.lfs.2023.121460.
- Diwan V, Brown L, Gobe GC. The flavonoid rutin improves kidney and heart structure and function in an adenine-induced rat model of chronic kidney disease. J Funct Foods. 2017;33:85–93. doi: 10.1016/j.jff.2017.03.012.
- Li M, Wei Y, Cai M, et al. Perilla peptides delay the progression of kidney disease by improving kidney apoptotic injury and oxidative stress and maintaining intestinal barrier function. Food Biosci. 2021;43:101333. doi: 10.1016/j.fbio.2021.101333.
- Liu X, Gao L, Huang X, et al. Lipidomics reveals the potential mechanism of honokiol against adenine-induced chronic kidney disease. Front Pharmacol. 2022;13:1019629. doi: 10.3389/fphar.2022.1019629.
- Long M, Li Q-M, Fang Q, et al. Renoprotective effect of Laminaria japonica polysaccharide in adenine-induced chronic renal failure. Molecules. 2019;24(8):1491. doi: 10.3390/molecules24081491.
- Sharma A, Thakur R, Lingaraju MC, et al. Betulinic acid attenuates renal fibrosis in rat chronic kidney disease model. Biomed Pharmacother. 2017;89:796–804. doi: 10.1016/j.biopha.2017.01.181.
- Singh MP, Sharma C, Kang SC. Morin hydrate attenuates adenine-induced renal fibrosis via targeting cathepsin D signaling. Int Immunopharmacol. 2021;90:107234. doi: 10.1016/j.intimp.2020.107234.
- Zhou W, Chen M, Liu H, et al. Dihydroartemisinin suppresses renal fibrosis in mice by inhibiting DNA-methyltransferase 1 and increasing klotho. Acta Pharmacol Sin. 2022;43(10):2609–2623. doi: 10.1038/s41401-022-00898-3.
- Ratliff BB, Abdulmahdi W, Pawar R, et al. Oxidant mechanisms in renal injury and disease. Antioxid Redox Signal. 2016;25(3):119–146. doi: 10.1089/ars.2016.6665.
- Vervloet M, Cozzolino M. Vascular calcification in chronic kidney disease: different bricks in the wall? Kidney Int. 2017;91(4):808–817. doi: 10.1016/j.kint.2016.09.024.
- Hutcheson JD, Goettsch C. Cardiovascular calcification heterogeneity in chronic kidney disease. Circ Res. 2023;132(8):993–1012. doi: 10.1161/CIRCRESAHA.123.321760.
- Covic A, Vervloet M, Massy ZA, et al. Bone and mineral disorders in chronic kidney disease: implications for cardiovascular health and ageing in the general population. Lancet Diabetes Endocrinol. 2018;6(4):319–331. doi: 10.1016/S2213-8587(17)30310-8.
- Reiss AB, Miyawaki N, Moon J, et al. CKD, arterial calcification, atherosclerosis and bone health: inter-relationships and controversies. Atherosclerosis. 2018;278:49–59. doi: 10.1016/j.atherosclerosis.2018.08.046.
- Chang X, Cui L, Wang X, et al. Quercetin attenuates vascular calcification through suppressed oxidative stress in adenine-induced chronic renal failure rats. Biomed Res Int. 2017;2017:5716204. doi: 10.1155/2017/5716204.
- Yao Z, Xu Y, Ma W, et al. Magnesium citrate protects against vascular calcification in an adenine-induced chronic renal failure rat model. J Cardiovasc Pharmacol. 2018;72(6):270–276. doi: 10.1097/FJC.0000000000000590.
- Zhao M-M, Xu M-J, Cai Y, et al. Mitochondrial reactive oxygen species promote p65 nuclear translocation mediating high-phosphate-induced vascular calcification in vitro and in vivo. Kidney Int. 2011;79(10):1071–1079. doi: 10.1038/ki.2011.18.
- Neven E, Bashir-Dar R, Dams G, et al. Disturbances in bone largely predict aortic calcification in an alternative rat model developed to study both vascular and bone pathology in chronic kidney disease. J Bone Miner Res. 2015;30(12):2313–2324. doi: 10.1002/jbmr.2585.
- Metzger CE, Swallow EA, Stacy AJ, et al. Strain-specific alterations in the skeletal response to adenine-induced chronic kidney disease are associated with differences in parathyroid hormone levels. Bone. 2021;148:115963. doi: 10.1016/j.bone.2021.115963.
- Metzger CE, Swallow EA, Stacy AJ, et al. Adenine-induced chronic kidney disease induces a similar skeletal phenotype in male and female C57BL/6 mice with more severe deficits in cortical bone properties of male mice. PLoS One. 2021;16(4):e0250438. doi: 10.1371/journal.pone.0250438.
- Basson R, Rees P, Wang R, et al. Sexual function in chronic illness. J Sex Med. 2010;7(1 Pt 2):374–388. doi: 10.1111/j.1743-6109.2009.01621.x.
- Edey MM. Male sexual dysfunction and chronic kidney disease. Front Med (Lausanne). 2017;4:32. doi: 10.3389/fmed.2017.00032.
- Wang CJ, Cukor D, Johansen KL. Sexual dysfunction among patients with chronic kidney disease. Semin Nephrol. 2021;41(6):534–549. doi: 10.1016/j.semnephrol.2021.10.006.
- Adachi Y, Sasagawa I, Nakada T. Reproductive insufficiency in the male rat with adenine-induced chronic renal failure. Urol Int. 1993;51(4):228–230. doi: 10.1159/000282550.
- Adachi Y, Sasagawa I, Tateno T, et al. Influence of adenine-induced chronic renal failure on testicular function in the rat. Andrologia. 1998;30(2):115–118. doi: 10.1111/j.1439-0272.1998.tb01157.x.