
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Objective
This study explored the molecular mechanisms by which dexmedetomidine (Dex) alleviates cisplatin (CP)-induced acute kidney injury (AKI) in rats.
Methods
CP-induced AKI models were established, and Dex was intraperitoneally injected at different concentrations into rats in the model groups. Subsequently, rats were assigned to the control, CP, CP + Dex 10 μg/kg, and CP + Dex 25 μg/kg groups. After weighing the kidneys of the rats, the kidney arterial resistive index was calculated, and CP-induced AKI was evaluated. In addition, four serum biochemical indices were measured: histopathological damage in rat kidneys was detected; levels of inflammatory factors, interleukin (IL)-1β, IL-18, IL-6, and tumor necrosis factor alpha, in kidney tissue homogenate of rats were assessed through enzyme-linked immunosorbent assay (ELISA); and levels of NLRP-3, caspase-1, cleaved caspase-1, gasdermin D (GSDMD), and GSDMD-N in kidney tissues of rats were determined via western blotting.
Results
Dex treatment reduced nephromegaly and serum clinical marker upregulation caused by CP-induced AKI. In addition, hematoxylin and eosin staining revealed that Dex treatment relieved CP-induced kidney tissue injury in AKI rats. ELISA analyses demonstrated that Dex treatment reduced the upregulated levels of proinflammatory cytokines in the kidney tissue of AKI rats induced by CP, thereby alleviating kidney tissue injury. Western blotting indicated that Dex alleviated CP-induced AKI by inhibiting pyroptosis mediated by NLRP-3 and caspase-1.
Conclusion
Dex protected rats from CP-induced AKI, and the mechanism may be related to NLRP-3/Caspase-1-mediated pyroptosis.
Introduction
Acute kidney injury (AKI) is associated with sudden loss of renal function. AKI is a common clinical disease among hospitalized patients and is considered a heterogeneous syndrome that affects the mortality and prognosis of patients [Citation1]. In addition, AKI causes acute kidney disease and is a global condition. In low- and middle-income countries, infection and hypovolemic shock are the leading causes of AKI, whereas in high-income countries, AKI mainly occurs in hospitalized elderly patients and is associated with sepsis, medication, and invasive procedures [Citation2]. Cisplatin (CP) is a therapeutic agent that is widely used for the treatment of solid organ cancer, although its side effects include nephrotoxicity, and the incidence of AKI in patients receiving CP treatment is 30% [Citation3]. These patients must suspend CP treatment because no treatment has yet been approved for AKI.
Recent studies have claimed that CP induces renal tubular cell injury through various signaling pathways and factors [Citation4]. Epigenetic changes play a role in CP-induced AKI and chronic kidney disease [Citation5–7]. However, the molecular mechanism underlying CP-induced AKI has not been fully elucidated. The mechanism of CP-induced AKI may be related to cellular stress responses, including DNA damage, mitochondrial pathology, oxidative stress, endoplasmic reticulum stress, autophagy, cell cycle arrest, senescence, apoptosis, programed necrosis, and inflammation.
Dexmedetomidine (Dex), a highly selective α2-adrenoceptor agonist, is beneficial in the prevention of various kidney diseases, including AKI. Recent studies have revealed the protective effect of Dex on kidney injury [Citation8]. Dex also inhibits the release of proinflammatory cytokines, weakens apoptosis, and reduces the production of reactive oxygen species (ROS) [Citation9–11]. However, the protective effect of Dex on acute stress-induced kidney injury is unclear. Moreover, several studies have discovered that Dex can alleviate septic heart injury by inhibiting ferroptosis [Citation12] and protect nerve cells from oxidative damage by maintaining iron homeostasis [Citation13]. Dex can also improve kidney function and reduce myocardial ischemia–reperfusion injury [Citation14]. However, the molecular mechanism by which Dex affects CP-induced AKI has not yet been fully elucidated.
Dex can inhibit brain injury by inhibiting the activation of the inflammasome (NLRP-3) [Citation15]. Furthermore, Dex plays a neuroprotective role by reducing oxidative stress by inhibiting NLRP-3-mediated neuroinflammation [Citation16]. The NLRP-3 inflammasome is one of the largest and most representative inflammasomes known to date and contains NLRP-3, an apoptosis-associated speck-like protein containing a CARD (ASC), and pro-casp-1 [Citation17]. Pyroptosis is a form of cell death mediated by gasdermin (GSDM) family proteins such as gasdermin D (GSDMD) and GSDME and is characterized by the activation of NLRP-3, formation of cell membrane pores, and release of interleukin (IL)-1β and IL-18 [Citation18]. The GSDMD N-terminal domain (GSDMD-N) is the final executor of pyroptosis [Citation19, Citation20] and is cleaved by active caspase-1 through classical and nonclassical inflammasome signaling pathways. GSDMD-N, generated by the cleavage of GSDMD, quickly inserts into the plasma membrane to form active pores and induces cytolysis. In addition, the vitamin D/vitamin D receptor can attenuate CP-induced AKI by downregulating the NLRP-3/caspase-1/GSDMD pyroptosis pathway [Citation21], and pentoxifylline and thiamin can ameliorate rhabdomyolysis-induced AKI in rats by suppressing pyroptosis mediated by TLR4/NF-κB and NLRP-3/caspase-1/GSDMD [Citation22]. Therefore, we hypothesized that Dex protects rats from CP-induced AKI by inhibiting NLRP-3/caspase-1-mediated pyroptosis.
This study investigated whether Dex protects rats from CP-induced AKI by inhibiting NLRP-3/caspase-1-mediated pyroptosis, thereby providing a new theoretical basis for the treatment of CP-induced AKI.
Materials and methods
Construction of the rat animal models
Forty male Sprague–Dawley rats (aged 2–3 months, 190–220 g) were purchased from the experimental animal center of Guangdong Medical Experimental Animal Center and housed in a specific pathogen-free environment. In vivo research was approved by the Animal Ethics Committee of the Guangdong Medical Experimental Animal Center (D202310-7), and animal experiments were performed in strict accordance with relevant ethical standards. The pain and suffering caused by the experiments were minimized, and rats were housed and protected in a standardized manner. Anesthesia and analgesia were administered to the animals strictly following the relevant procedures. Male rats were divided into four groups, with 10 rats per group [Citation1]: Control group: the same volume of dimethylformamide (DMF) solution as the experimental group was administered to rats through a single intraperitoneal injection. After 1 h, normal saline was injected intraperitoneally. This was performed once a day for three days [Citation2]; CP group: rats were injected with CP (dissolved in DMF, 7 mg/kg, S1166, Selleck, MA, USA) intraperitoneally on the first day, and after 1 h, they were injected with normal saline once a day for 3 days [Citation3, Citation23]; CP + Dex 10 μg/kg group: rats were injected with CP (dissolved in DMF, 7 mg/kg) intraperitoneally on the first day, and 1 h later, Dex (dissolved in normal saline, 10 μg/kg, 145108-58-3, Sigma, MO, USA) was injected intraperitoneally, and the injection of Dex was performed once a day for three consecutive days [Citation4, Citation24]; CP + Dex 25 μg/kg group: a single intraperitoneal injection of CP (7 mg/kg) was administered to rats on the first day, and after 1 h, Dex (25 μg/kg) was injected intraperitoneally once a day for three consecutive days [Citation25]. Rats were euthanized 96 h after CP injection, and kidney tissue and peripheral blood were collected.
Determination of the kidney arterial resistive index
Rat kidneys from the control, CP, CP + Dex 10 μg/kg, and CP + Dex 25 μg/kg groups were collected and weighed. The renal arterial resistive index (RI) was then calculated as follows:
Detection of serum clinical markers of kidney tissue injury
Peripheral blood samples were collected and centrifuged (3000 rpm for 10 min) to separate serum. The levels of blood urea nitrogen (BUN; BC1535, Solarbio, Beijing, China), creatinine (Cr; MAK080, MO, USA), creatine kinase (CK; E-El-R1322c, Houston, USA), and kidney injury molecule 1 (Kim-1; E-EL-R3019, Houston, USA) in rat serum in the control, CP, CP + Dex 10 μg/kg, and CP + Dex 25 μg/kg groups were detected.
Hematoxylin and eosin (H&E) staining
Collected kidney tissue samples were washed with normal saline, soaked and fixed in paraformaldehyde overnight, gradient dehydrated, embedded in paraffin, and sectioned. After dewaxing, the sections were stained with H&E (H8070, Solarbio). Subsequently, kidney tissues in the control, CP, CP + Dex 10 μg/kg, and CP + Dex 25 μg/kg groups were observed under an optical microscope, and renal tubule injury scored at the same time according to Altındağ et al. [Citation23].
Enzyme-linked immunosorbent assay (ELISA)
ELISA was performed to detect the levels of IL-1β, IL-18, IL-6, and tumor necrosis factor alpha (TNF-α) in the kidney tissues of rats in the control, CP, CP + Dex 10 μg/kg, and CP + Dex 25 μg/kg groups. The collected kidney tissues were supplemented with phosphate-buffered saline containing 1% Triton X-100 and Roche EDTA-free protease inhibitor, placed on ice, and minced with high-pressure scissors. Minced kidney tissues were homogenized using a tissue homogenizer, and the kidney tissue homogenate was ultrasonicated using an ultrasonic apparatus at an intensity of 45% at 4 °C for 2 min. Treated kidney tissue samples were centrifuged at 12,000 rpm at 4 °C, and the supernatant was collected. Subsequently, the levels of IL-1β (SEKR-0002, Solarbio), IL-18 (SEKR-00054, Solarbio), IL-6 (SEKR-0005, Solarbio), and TNF-α (SEKR-0009, Solarbio) in rat kidney tissues were assessed according to the kit instructions. The absorbance of the supernatant of each tissue sample was read using a Thermo Multiskan Ascent Microplate Reader, and the concentration of the analyte was calculated using ProcartaPlex Analyst 1.0 software.
Western blotting
RIPA lysis buffer (P0013B, Beyotime Biotechnology, Shanghai, China) containing 1% protease inhibitor and phosphorylase inhibitor was used to lyse the rat kidney tissue cells. Briefly, tissue samples were mixed with lysis solution and ground to obtain the protein supernatant. Protein concentration was determined using a BCA protein quantitative kit (A53226, Thermo Fisher Scientific, Rockford, IL, USA). Subsequently, protein loading buffer (5× loading buffer, P1040, Solarbio) was added to the protein samples, which were then heated at 99 °C for 10 min. Next, the protein was separated by sodium dodecyl sulfate- polyacrylamide gel electrophoresis, and the separated protein was transferred to a polyvinylidene fluoride membrane, which was blocked with 5% bovine serum albumin at ambient temperature for 1 h. Corresponding primary antibodies, including NLRP-3 (1:1000, ab263899, Abcam, Cambridge, UK), caspase-1 (1:1000, ab238972, Abcam), cleaved caspase-1 (1:1000, ab238972, Abcam), GSDMD (1:1000, ab219800, Abcam), and GSDMD-N (1:1000, ab263899, Abcam), were incubated with the membrane at 4 °C overnight. The following day, the membrane was washed with TBST, and the secondary antibody solution (1:5000, AP160P, AP187P, Sigma, MO, USA) was added for 1 h of incubation at ambient temperature. The membrane was then washed with TBST. Next, immunoreactive bands were developed (WBAVDCH01, Sigma) and imaged (Amersham Imager 600, USA). ImageJ was used for the gray analysis.
Statistical analysis
SPSS 21.0 (IBM SPSS Statistics, Chicago, IL, USA) was used for statistical analysis. Data are expressed as the mean ± standard deviation. One-way analysis of variance and Bonferroni t-test for multiple comparisons were used to compare the measurement data among multiple groups. Significance was set at p < 0.05.
Results
Dex treatment improves AKI and reduces the generation of related kidney injury markers induced by cisplatin in rats
As previously reported, CP can induce AKI and Dex treatment can improve CP-induced AKI [Citation8]. However, the regulation of CP-induced AKI by Dex treatment in rats remains unclear. In this study, the weight of kidneys from rats in the CP group was significantly higher (p < 0.0001) than that in the control group because of CP-induced AKI. Nevertheless, relative to the CP group, the kidney weight in the CP + Dex 10 and 25 μg/kg groups significantly decreased with Dex treatment (p = 0.0026 and p < 0.0001, respectively). Moreover, kidney weight in the CP + Dex 25 μg/kg group decreased more significantly than that in the CP + Dex 10 μg/kg group (p = 0.0087) (F [3, 36] = 31.40) (). The RI of rats in the CP group (p < 0.0001) significantly increased compared with that of the control group rats, whereas the RI of rats in the CP + Dex 10 and 25 μg/kg groups significantly decreased significantly with Dex treatment relative to that of rats in the CP group (p = 0.0091 and p < 0.0001, respectively); furthermore, this decrease was more pronounced in the CP + Dex 25 μg/kg group than in the CP + Dex 10 μg/kg group (p = 0.0099) (F [3, 36] = 32.31) (). Thus, kidney weight and RI analyses in rats indicated that Dex treatment reduced nephromegaly in CP-induced AKI.
Figure 1. Impact of dexmedetomidine on kidney weight and related kidney injury markers in rats induced by cisplatin. An acute kidney injury model induced by cisplatin (CP) in rats was simulated by intraperitoneal injection of CP with dexmedetomidine (Dex) at different concentrations (10 and 25 μg/kg). A, Rat kidney weight (F = 31.40); B, kidney arterial resistive index (RI) of rats, RI = weight of both kidneys/body weight × 100% (F = 32.31). (C–F) Serum clinical markers of kidney injury in rats, including blood urea nitrogen (F = 143.4) (C), creatinine (Cr) (F = 107.9) (D), creatine kinase (CK) (F = 233.0) (E), and kidney injury molecule 1 (Kim-1) (F = 254.5) (F). Data are expressed as mean ± standard deviation (n = 10). **p < 0.01 vs. Control group; ## p < 0.01 vs. CP group; & p < 0.01 vs. CP + Dex 10 μg/kg.
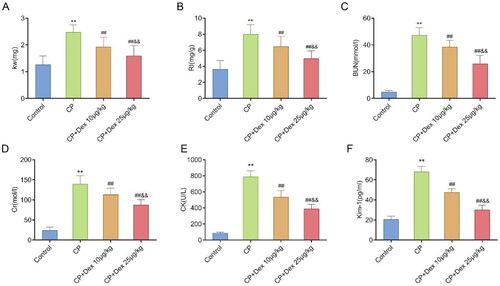
AKI is associated with the loss of kidney function. The clinical signs of kidney tissue injury include increased serum levels of Cr, BUN, CK, and Kim-1, which can be used to evaluate CP-induced nephrotoxicity [Citation1]. Herein, compared with those in the control group, the serum levels of BUN (p < 0.0001) (F [3, 36] = 143.4), Cr (p < 0.0001) (F [3, 36] = 107.9), CK (p < 0.0001) (F [3, 36] = 233.0), and Kim-1 (p < 0.0001) (F [3, 36] = 254.5) in rats in the CP group were significantly increased, whereas the serum levels of these clinical markers of kidney tissue injury in the CP + Dex 10 and 25 μg/kg groups were significantly decreased (p < 0.001). In addition, relative to the CP + Dex 10 μg/kg group, the RI of rats in the CP + Dex 25 μg/kg group decreased more significantly (p < 0.001), indicating that Dex treatment reduced the production of clinical markers related to CP-induced AKI (). These results suggest that Dex could improve nephromegaly and kidney function injury in rats with CP.
Dex treatment reduces pathological damage to kidney tissue in CP-induced AKI rats
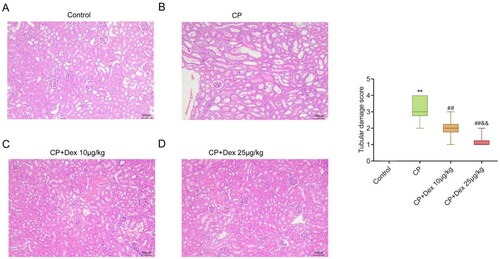
H&E staining was performed to investigate the effect of Dex treatment on CP-induced pathological damage to kidney tissue in AKI rats. Rats in the control group exhibited normal kidney tissue morphology and structure, intact basement membranes of glomerular capillaries and tubular epithelium, and a clear tubular structure (). Rats in the CP group showed severe tubular damage, swelling, and degeneration of tubular epithelial cells, and substantial necrosis of the proximal tubules compared with those in the control group (). In addition, different degrees of kidney tissue injury were observed in the CP + Dex 10 and CP + Dex 25 μg/kg groups. Compared with the CP group, Dex treatment not only improved kidney tissue injury in rats but also reduced the swelling and necrosis of tubular epithelial cells (p < 0.001). Conversely, kidney tissue injury in rats in the CP + Dex 25 μg/kg group was significantly improved compared with that in the CP + Dex 10 μg/kg group (p = 0.0029) (F [3, 36] = 65.67) (). Collectively, Dex treatment improved the pathological damage to kidney tissue in CP-induced AKI rats.
Figure 2. Effect of dexmedetomidine on the histopathology of kidney tissue in rats treated with cisplatin. Pathological changes in the kidney tissue of acute kidney injury rats were detected using hematoxylin and eosin staining. A, Representative pathology images of kidneys in the control (A), CP (B), CP + Dex 10 μg/kg (C), and CP + Dex 25 μg/kg groups (F = 65.67). ** p < 0.01 vs. Control group; ## p < 0.01 vs. CP group; & p < 0.01 vs. CP + Dex 10 μg/kg.
Dex treatment improves the levels of proinflammatory cytokines in kidney tissue of AKI rats induced by CP
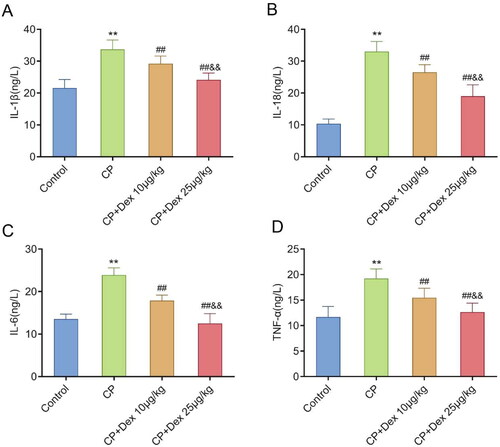
AKI promotes the production of proinflammatory cytokines, including IL-1β, IL-18, IL-6, and TNF-α [Citation26]. Therefore, the levels of these cytokines in rat kidney tissues were measured in this study. ELISA results showed that the levels of IL-1β (p < 0.0001) (F [3, 36] = 44.50), IL-18 (p < 0.0001) (F [3, 36] = 44.50), IL-6 (p < 0.0001) (F [3, 36] = 95.76), and TNF-α (p < 0.0001) (F [3, 36] = 31.36) in the CP group were much higher than those in the control group. However, compared with those in the CP group, the levels of these cytokines in the CP + Dex 10 and 25 μg/kg groups were markedly decreased (p < 0.001) and were markedly lower in the CP + Dex 25 μg/kg group than those in the CP + Dex 10 μg/kg group (p < 0.001). Overall, Dex treatment reduced the production of inflammatory factors in the kidney tissues of CP-induced AKI rats, and this effect increased with the increase in Dex concentration (). Thus, Dex could improve the level of proinflammatory cytokines in the kidney tissue of rats with CP-induced AKI, thereby alleviating kidney tissue injury.
Figure 3. Impact of dexmedetomidine treatment on the levels of proinflammatory cytokines in kidney tissues of acute kidney injury rats induced by cisplatin. Levels of the proinflammatory cytokines interleukin (IL)-1β, IL-18, IL-6, and tumor necrosis factor alpha (TNF-α) in the kidney tissues of rats were detected using enzyme-linked immunosorbent assay. A, IL-1β (F = 44.50); B, IL-18 (F = 125.2); C, IL-6 (F = 95.76); D, TNF-α (F = 31.36). Data are expressed as mean ± standard deviation (n = 10). ** p < 0.01 vs. Control group; ## p < 0.01 vs. CP group; & p < 0.01 vs. CP + Dex 10 μg/kg.
Dex improves AKI in rats induced by CP by inhibiting NLRP-3/caspase-1-mediated pyroptosis
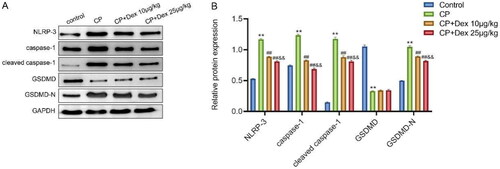
Previous studies have shown that Dex treatment can improve CP-induced AKI in rats. In addition, CP can induce human tubular epithelial cell pyroptosis [Citation27], whereas Dex can inhibit the activation of NLRP-3 induced by HIR by triggering SIRT3-mediated mitochondrial autophagy [Citation15]. After the activation of NLRP-3, pro-caspase-1 is converted to active caspase-1, which converts IL-1β and IL-18 precursors into mature IL-1β and IL-18. The subsequent formation of GSDMD-N and GSDMD-C from the cleavage of GSDMD by active caspase-1 causes cell pyroptosis. A higher degree of pyroptosis caused by GSDMD-N induces swelling of the cell, which then ruptures, causing the release of proinflammatory mediators in a proinflammatory form of programed cell death known as pyroptosis [Citation28, Citation29]. Therefore, we explored whether Dex inhibited NLRP-3/caspase-1-mediated pyroptosis in CP-induced AKI rats. Specifically, western blotting was performed to examine the expression levels of NLRP-3, caspase-1, cleaved caspase-1, GSDMD, and GSDMD-N in the kidney tissues of rats in the control, CP, and CP + Dex 10 and 25 μg/kg groups. As shown in , compared with those of the control group, the levels of NLRP-3 (p < 0.0001), caspase-1 (p < 0.0001), cleaved caspase-1 (p < 0.0001), and GSDMD-N (p < 0.0001) in the kidney tissue of rats in the CP group increased significantly, whereas the level of GSDMD decreased notably (p < 0.0001). Nevertheless, compared with the CP group, the levels of NLRP-3, caspase-1, cleaved caspase-1, and GSDMD-N in the kidney tissues of rats in the CP + Dex 10 and 25 μg/kg groups were significantly decreased (p < 0.01), whereas the level of GSDMD was not significantly different. In addition, the levels of NLRP-3, caspase-1, cleaved caspase-1, and GSDMD-N in the kidney tissues of rats in the CP + Dex 25 μg/kg group were significantly lower than those in the CP + Dex 10 μg/kg group (p < 0.01). These results revealed that Dex inhibited pyroptosis mediated by NLRP-3/caspase-1 in rats with CP, proving that Dex may alleviates CP-induced AKI by inhibiting NLRP-3/caspase-1-mediated pyroptosis.
Figure 4. Influence of dexmedetomidine on the levels of pyroptosis-related proteins in kidney tissues of rats induced by cisplatin. A, Western blotting assessment of the expression levels of NLRP-3, caspase-1, cleaved caspase-1, gasdermin D (GSDMD), and GSDMD-N proteins in the kidney tissues of rats. B, Gray value analysis of immunoreactive bands in each group. Data are expressed as mean ± standard deviation (n = 10) (Alpha = 0.05). ** p < 0.01 vs. Control group; ## p < 0.01 vs. CP group; & p < 0.01 vs. CP + Dex 10 μg/kg.
Discussion
CP is widely used as an antitumor drug to treat solid tumors such as testicular, ovarian, colorectal, lung, and head and neck cancers. Clinically, CP is one of the main causes of AKI in patients with cancer [Citation30]. As a common severe syndrome, AKI is characterized by tubular injury and sudden loss of kidney function within a few hours and is a global health problem with high morbidity and mortality [Citation1]. Because the exact molecular mechanism of CP-induced AKI remains unclear, strategies for preventing and treating CP-induced AKI are limited. Previous studies have focused on the apoptosis and necrosis of tubular cells induced by CP, but specific inhibitors of apoptosis and necrosis have failed to completely prevent or cure CP-induced AKI [Citation31]. Despite its wide range of therapeutic effects, CP also induces nephrotoxicity, which limits its clinical application. Hence, minimizing CP-induced nephrotoxicity is key to optimizing the clinical results of CP [Citation32, Citation33]. In this study, we established a CP animal model and found that CP-induced AKI in rats, increased the weight of rat kidneys, and upregulated the levels of kidney tissue injury markers in the serum and of proinflammatory factors in kidney tissues. Pathological examination showed that the tubules were severely damaged, the epithelial cells of the tubules were swollen and degenerated, and substantial necrosis had occurred in the proximal tubules. Therefore, CP has severe nephrotoxic effects.
Dex is a highly selective α2-adrenoceptor agonist and its receptors are widely distributed in the proximal and distal tubules [Citation34]. Dex can alleviate lipopolysaccharide (LPS)-induced kidney dysfunction and histological damage induced by LPS [Citation6]. Moreover, Dex weakened endoplasmic reticulum stress-induced apoptosis through the α2AR/PI3K/AKT signaling pathway, thereby alleviating CP-induced AKI. The protective mechanism of Dex may be related to the inhibition of apoptosis via the α2AR/PI3K/AKT signaling pathway [Citation25]. Herein, the in vivo study showed that Dex treatment significantly reduced the kidney weight and RI of rats induced by CP and significantly inhibited the levels of BUN, Cr, CK, and Kim-1 in serum. The pathological results revealed that Dex treatment markedly reduced CP-induced renal tubular injury and necrosis. Moreover, ELISA detection of IL-1β, IL-18, IL-6, and TNF-α showed that Dex treatment reduced the production of proinflammatory factors in kidney tissues of rats with CP-induced AKI, and the improvement effect of Dex was concentration dependent. Thus, this study confirmed the protective effects of Dex on CP-induced AKI.
To investigate the protective mechanism of Dex against CP-induced AKI, we determined that the protective effects of Dex on oxidative stress, apoptosis, and pyroptosis in the brain and peripheral tissues have been revealed in studies using various in vitro and in vivo models [Citation35]. For example, Dex can activate the FoxO3a/ARC axis by downregulating miR-29b, thereby inhibiting pyroptosis following myocardial ischemia–reperfusion injury [Citation35]. Chemotherapeutic drugs, such as CP or doxorubicin, can induce pyroptosis in human renal tubular epithelial cells through the ROS/JNK/Caspase-3/GSDME signaling pathway. Treatments targeting GSDME can effectively reduce nephrotoxicity induced by chemotherapeutic drugs [Citation27]. However, further research is needed to determine whether Dex improves AKI by mediating pyroptosis.
Pyroptosis is a form of programed cell death. Specifically, the innate immune receptor NOD-like receptor assembles into the intracellular NLRP-3 inflammasome protein complex upon sensing plasma membrane perturbations induced by specific microbial products or sterile danger signals. NLRP-3 and caspase-1 then form macromolecular structures. Furthermore, caspase-1 is activated when recruited to the inflammasome and subsequently cleaves inactive pro-IL-18β and pro-IL-1 into biologically active IL-18β and IL-2, respectively. Moreover, the cleavage of GSDMD produces N-terminal fragments that can oligomerize to form pores in the plasma membrane. IL-18β and IL-4 are secreted through these pores. However, excessive pore formation is harmful to the integrity of the plasma membrane and causes pyroptosis [Citation36–38]. In this study, the protein levels of NLRP-3, caspase-1, cleaved caspase-1, and GSDMD-N were significantly increased in rat kidney tissue after CP induction. However, Dex decreased the levels of NLRP-3, caspase-1, cleaved caspase-1, and GSDMD-N in rat kidney tissues, thereby reducing pyroptosis and kidney injury. Moreover, the inhibitory effect of Dex on pyroptosis increased with increasing Dex concentration. Collectively, these results suggest that the protective effect of Dex against CP-induced AKI may be related to the inhibition of NLRP-3/caspase-1-mediated pyroptosis.
Notably, although the AKI rat model induced by CP was constructed for in vivo assessment in this study, in vitro cell experiments at the level of kidney tissue cells were not performed for further verification. Furthermore, because of the lack of kidney tissue-specific NLRP-3 or caspase-1 to knockdown in rats, it is uncertain whether Dex inhibits only the NLRP-3/caspase-1 pathway in the kidney.
Overall, in an AKI rat model induced by CP, treatment with Dex improved CP-induced AKI in a gradient-dependent manner. Dex treatment inhibited nephromegaly, increased serum markers of kidney tissue injury, and alleviated the pathological damage in kidney tissues caused by CP-induced AKI in a concentration dependent manner. In addition, Dex treatment improved the upregulation of proinflammatory cytokines in the kidney tissue of rats with CP, thereby alleviating kidney tissue injury. Dex may alleviated CP-induced AKI by inhibiting pyroptosis mediated by NLRP-3/caspase-1. Therefore, this study provides a theoretical basis for exploring the potential diagnosis and treatment targets of CP-induced AKI and demonstrates the potential value of Dex in protecting against CP-induced AKI.
Conclusion
Dex administration protected rats from CP-induced AKI by inhibiting NLRP-3/caspase-1-mediated pyroptosis. Nevertheless, a number of issues remain to be addressed by applying Dex to alleviate CP-induced AKI by inhibiting NLRP-3/caspase-1-mediated pyroptosis, such as the mode of administration, concentration of administered drug, and clinical safety.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Datasets used in this article are available from the corresponding author on reasonable request.
Additional information
Funding
References
- Pickkers P, Darmon M, Hoste E, et al. Acute kidney injury in the critically ill: an updated review on pathophysiology and management. Intensive Care Med. 2021;47(8):1–9. doi:10.1007/s00134-021-06454-7.
- Kellum JA, Romagnani P, Ashuntantang G, et al. Acute kidney injury. Nat Rev Dis Primers. 2021;7(1):52. doi:10.1038/s41572-021-00284-z.
- Kim D-H, Choi H-I, Park JS, et al. Farnesoid X receptor protects against cisplatin-induced acute kidney injury by regulating the transcription of ferroptosis-related genes. Redox Biol. 2022;54:102382. doi:10.1016/j.redox.2022.102382.
- Volarevic V, Djokovic B, Jankovic MG, et al. Molecular mechanisms of cisplatin-induced nephrotoxicity: a balance on the knife edge between renoprotection and tumor toxicity. J Biomed Sci. 2019;26(1):25. doi:10.1186/s12929-019-0518-9.
- Wang Z, Wu J, Hu Z, et al. Dexmedetomidine alleviates Lipopolysaccharide-Induced acute kidney injury by inhibiting p75NTR-mediated oxidative stress and apoptosis. Oxid Med Cell Longev. 2020;2020:5454210–5454213. doi:10.1155/2020/5454210.
- Hao Y, Miao J, Liu W, et al. Formononetin protects against cisplatin‑induced acute kidney injury through activation of the PPARalpha/Nrf2/HO‑1/NQO1 pathway. Int J Mol Med. 2021;47(2):511–522. doi:10.3892/ijmm.2020.4805.
- Tang C, Livingston MJ, Safirstein R, et al. Cisplatin nephrotoxicity: new insights and therapeutic implications. Nat Rev Nephrol. 2023;19(1):53–72. doi:10.1038/s41581-022-00631-7.
- Chen Y, Feng X, Hu X, et al. Dexmedetomidine ameliorates acute stress-induced kidney injury by attenuating oxidative stress and apoptosis through inhibition of the ROS/JNK signaling pathway. Oxid Med Cell Longev. 2018;2018:4035310. doi:10.1155/2018/4035310.
- Chen Y, Luan L, Wang C, et al. Dexmedetomidine protects against lipopolysaccharide-induced early acute kidney injury by inhibiting the iNOS/NO signaling pathway in rats. Nitric Oxide. 2019;85:1–9. doi:10.1016/j.niox.2019.01.009.
- Si Y, Bao H, Han L, et al. Dexmedetomidine attenuation of renal ischaemia-reperfusion injury requires sirtuin 3 activation. Br J Anaesth. 2018;121(6):1260–1271. doi:10.1016/j.bja.2018.07.007.
- Qiu R, Yao W, Ji H, et al. Dexmedetomidine restores septic renal function via promoting inflammation resolution in a rat sepsis model. Life Sci. 2018;204:1–8. doi:10.1016/j.lfs.2018.05.001.
- Wang C, Yuan W, Hu A, et al. Dexmedetomidine alleviated sepsis‑induced myocardial ferroptosis and septic heart injury. Mol Med Rep. 2020;22(1):175–184. doi:10.3892/mmr.2020.11114.
- Cui Y, Zhang Y, Zhao X, et al. ACSL4 exacerbates ischemic stroke by promoting ferroptosis-induced brain injury and neuroinflammation. Brain Behav Immun. 2021;93:312–321. doi:10.1016/j.bbi.2021.01.003.
- Tao W-H, Shan X-S, Zhang J-X, et al. Dexmedetomidine attenuates Ferroptosis-Mediated renal ischemia/reperfusion injury and inflammation by inhibiting ACSL4 via alpha2-AR. Front Pharmacol. 2022;13:782466. doi:10.3389/fphar.2022.782466.
- Yu W, Lyu J, Jia L, et al. Dexmedetomidine ameliorates hippocampus injury and cognitive dysfunction induced by hepatic ischemia/reperfusion by activating SIRT3-Mediated mitophagy and inhibiting activation of the NLRP3 inflammasome in young rats. Oxid Med Cell Longev. 2020;2020:7385458–7385417. doi:10.1155/2020/7385458.
- Cho I, Koo BN, Kim SY, et al. Neuroprotective effect of dexmedetomidine against postoperative cognitive decline via NLRP3 inflammasome signaling pathway. Int J Mol Sci. 2022;23(15):8806.
- Sun L, Ma W, Gao W, et al. Propofol directly induces caspase-1-dependent macrophage pyroptosis through the NLRP3-ASC inflammasome. Cell Death Dis. 2019;10(8):542. doi:10.1038/s41419-019-1761-4.
- Li S, Sun Y, Song M, et al. NLRP3/caspase-1/GSDMD-mediated pyroptosis exerts a crucial role in astrocyte pathological injury in mouse model of depression. JCI Insight. 2021;6(23):e146852. doi:10.1172/jci.insight.146852.
- de Vasconcelos NM, Van Opdenbosch N, Van Gorp H, et al. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 2019;26(1):146–161. doi:10.1038/s41418-018-0106-7.
- Yao F, Jin Z, Zheng Z, et al. HDAC11 promotes both NLRP3/caspase-1/GSDMD and caspase-3/GSDME pathways causing pyroptosis via ERG in vascular endothelial cells. Cell Death Discov. 2022;8(1):112. doi:10.1038/s41420-022-00906-9.
- Jiang S, Zhang H, Li X, et al. Vitamin D/VDR attenuate cisplatin-induced AKI by down-regulating NLRP3/caspase-1/GSDMD pyroptosis pathway. J Steroid Biochem Mol Biol. 2021;206:105789. doi:10.1016/j.jsbmb.2020.105789.
- Al-Kharashi L, Attia H, Alsaffi A, et al. Pentoxifylline and thiamine ameliorate rhabdomyolysis-induced acute kidney injury in rats via suppressing TLR4/NF-kappaB and NLRP-3/caspase-1/gasdermin mediated-pyroptosis. Toxicol Appl Pharmacol. 2023;461:116387. doi:10.1016/j.taap.2023.116387.
- Altındağ F, Ergen H. Sinapic acid alleviates cisplatin-induced acute kidney injury by mitigating oxidative stress and apoptosis. Environ Sci Pollut Res Int. 2023;30(5):12402–12411. doi:10.1007/s11356-022-22940-x.
- Lempiainen J, Finckenberg P, Mervaala EE, et al. Dexmedetomidine preconditioning ameliorates kidney ischemia-reperfusion injury. Pharmacol Res Perspect. 2014;2(3):e00045.
- Chai Y, Zhu K, Li C, et al. Dexmedetomidine alleviates cisplatin‑induced acute kidney injury by attenuating endoplasmic reticulum stress‑induced apoptosis via the alpha2AR/PI3K/AKT pathway. Mol Med Rep. 2020;21(3):1597–1605.
- Hu J, Gu W, Ma N, et al. Leonurine alleviates ferroptosis in cisplatin-induced acute kidney injury by activating the Nrf2 signalling pathway. Br J Pharmacol. 2022;179(15):3991–4009. doi:10.1111/bph.15834.
- Shen X, Wang H, Weng C, et al. Caspase 3/GSDME-dependent pyroptosis contributes to chemotherapy drug-induced nephrotoxicity. Cell Death Dis. 2021;12(2):186. doi:10.1038/s41419-021-03458-5.
- Yang J, Liu Z, Wang C, et al. Mechanism of gasdermin D recognition by inflammatory caspases and their inhibition by a gasdermin D-derived peptide inhibitor. Proc Natl Acad Sci U S A. 2018;115(26):6792–6797. doi:10.1073/pnas.1800562115.
- Shi J, Zhao Y, Wang K, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526(7575):660–665. doi:10.1038/nature15514.
- Yang Y, Adebali O, Wu G, et al. Cisplatin-DNA adduct repair of transcribed genes is controlled by two circadian programs in mouse tissues. Proc Natl Acad Sci U S A. 2018;115(21):E4777–E85.
- Linkermann A, Chen G, Dong G, et al. Regulated cell death in AKI. J Am Soc Nephrol. 2014;25(12):2689–2701. doi:10.1681/ASN.2014030262.
- Sami DH, Soliman AS, Khowailed AA, et al. 7-hydroxycoumarin modulates Nrf2/HO-1 and microRNA-34a/SIRT1 signaling and prevents cisplatin-induced oxidative stress, inflammation, and kidney injury in rats. Life Sci. 2022;310:121104. doi:10.1016/j.lfs.2022.121104.
- Ko J-W, Shin N-R, Jung T-Y, et al. Melatonin attenuates cisplatin-induced acute kidney injury in rats via induction of anti-aging protein, klotho. Food Chem Toxicol. 2019;129:201–210. doi:10.1016/j.fct.2019.04.049.
- Gu J, Sun P, Zhao H, et al. Dexmedetomidine provides renoprotection against ischemia-reperfusion injury in mice. Crit Care. 2011;15(3):R153. doi:10.1186/cc10283.
- Zhong Y, Li YP, Yin YQ, et al. Dexmedetomidine inhibits pyroptosis by down-regulating miR-29b in myocardial ischemia reperfusion injury in rats. Int Immunopharmacol. 2020;86:106768. doi:10.1016/j.intimp.2020.106768.
- Liu Y, Zhang T, Zhou Y, et al. Visualization of perforin/gasdermin/complement-formed pores in real cell membranes using atomic force microscopy. Cell Mol Immunol. 2019;16(6):611–620. doi:10.1038/s41423-018-0165-1.
- Huang Y, Xu W, Zhou R. NLRP3 inflammasome activation and cell death. Cell Mol Immunol. 2021;18(9):2114–2127. doi:10.1038/s41423-021-00740-6.
- Wang C, Yang T, Xiao J, et al. NLRP3 inflammasome activation triggers gasdermin D-independent inflammation. Sci Immunol. 2021;6(64):eabj3859. doi:10.1126/sciimmunol.abj3859.