
Abstract
Juvenile myelomonocytic leukemia (JMML) is an aggressive pediatric leukemia with few effective treatments and poor outcomes even after stem cell transplantation, the only current curative treatment. We developed a JMML patient-derived xenograft (PDX) mouse model and demonstrated the in vivo therapeutic efficacy and confirmed the target of trametinib, a RAS-RAF-MEK-ERK pathway inhibitor, in this model. A PDX model was created through transplantation of patient JMML cells into mice, up to the second generation, and successful engraftment was confirmed using flow cytometry. JMML PDX mice were treated with trametinib versus vehicle control, with a median survival of 194 days in the treatment group versus 124 days in the control group (p = 0.02). Trametinib’s target as a RAS pathway inhibitor was verified by showing inhibition of ERK phosphorylation using immunoblot assays. In conclusion, trametinib monotherapy significantly prolongs survival in our JMML PDX model by inhibiting the RAS pathway. Our model can be effectively used for assessment of novel targeted treatments, including potential combination therapies, to improve JMML outcomes.
Introduction
Juvenile myelomonocytic leukemia (JMML) is a rare, aggressive leukemia commonly affecting children under the age of four, leading to symptoms of monocytic and granulocytic cell overproduction, hepatosplenomegaly, lymphadenopathy, and anemia.Citation1 The only current curative therapy for JMML is hematopoietic stem cell transplantation; however, the median 5-year survival rate after hematopoietic stem cell transplantation is only 52–64%, with relapse occurring at nearly a 50% rate within 1 year.Citation2,Citation3 With such poor outcomes and high treatment-related morbidity and mortality, the need for new and timely effective treatments is paramount. To develop novel therapies for JMML, an accurate disease model is necessary. To date, while there are reports of successful JMML patient-derived xenograft (PDX) models,Citation4–9 as well as RAS inhibitor assessment in genetically engineeredmouse modelsof JMML,Citation10–13 there are no current reports of in vivo RAS inhibitor assessment in a JMML PDX model. 90% of JMML cases involve constitutive activation of the RAS/MAPK signaling pathway due to fusions or upstream mutations, with mutations in NRAS, KRAS, NF1, PTPN11 and/or CBL.Citation5,Citation14,Citation15 This offers a rationale for RAS pathway-targeting therapeutics to treat JMML. Trametinib, a MEK inhibitor which inhibits ERK phosphorylation, was selected for our PDX modelbased on the patient in this case harboring RAS pathway mutations in PTPN11 and NF1().Citation16–18 In addition, a downstream RAS pathway targeting agent such as trametinib was preferentially chosen due to the potential of upstream inhibition being bypassed through escape pathways—a known mechanism of signaling pathway resistance.Citation19,Citation20 Trametinib monotherapy has been tested in JMML patients in trials and case reports with very small sample sizes, and with limited efficacy; obtaining sufficient efficacy data in patient trials is difficult due to disease rarity.Citation10,Citation21,Citation22 Thus, the significance of our study evaluating trametinib in a successfully created JMML PDX animal model is to verify its retained target in a preclinical model with known mutations and pave the way for future targeted combination therapy regimens. The ability to study mutation-specific treatments based on genetic profiles, in a model that retains the therapy’s target, is critical considering the absence of effective therapies for JMML. In this study, we report the successful creation of a JMML mouse xenograft model from primary patient cells. Furthermore, we demonstrate the therapeutic efficacy of a RAS pathway inhibitor, trametinib, inthis JMML PDX model. Trametinib’s target is confirmed in our model through immunoblot analysis. These findings establish a reliable disease model for the future evaluation of targeted treatment approaches, combination therapies, or large-scale drug screening.
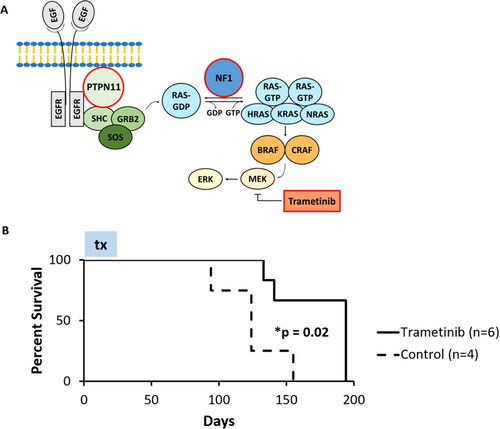
Figure 1. Trametinib, a MEK inhibitor, prolongs survival time in a JMML PDX mouse model compared to control. (A) A simplified schematic of the RAS pathway demonstrating the mutations identified in the patient in this case (PTPN11 and NF1, highlighted with red borders) and the mechanism of action of trametinib, a MEK inhibitor (highlighted with red borders) which acts by downregulation of ERK phosphorylation. (B) Kaplan-Meier survival curve for JMML PDX mice treated with trametinib versus vehicle control. The duration of trametinib treatment in the experiment (28 days) is denoted by the box labeled ‘tx’. The trametinib treatment group showed a statistically significant increase in median survival compared to control.
Materials and methods
Case
The patient was a 4-year-old previously healthy male who presented with fevers, fatigue, weight loss, splenomegaly, and facial petechiae for 3 weeks. Initial laboratory investigations showed leukocytosis, anemia, and thrombocytopenia. Bone marrow (BM) biopsy and flow cytometry showed 6.5% abnormal myeloblasts and 20% monocytes with no monotypic B cell population. Karyotype analysis showed 47,XY,+22[13]/46,XY[7] and genetic testing indicated PTPN11 (c.179G > T, 47%), ASXL1 (c.1900_1922del23, 18% and c.1918_1939del21, 3%), and NF1(c.2382_2385delTCCA, 8% and c.908T > C, 8%) mutations, confirming the diagnosis of JMML. The patient was initially treated per the Children’s Oncology Group AAML1421 protocol with cytarabineand fludarabine, achieving remission. Two haploidentical allogeneic peripheral blood stem cell transplants (PBSCT) were attempted, both resulting in acute graft rejection. A third PBSCT was successfully completed. Despite multiple post-transplant complications, the patient has remained in remission from his leukemia. A statement of written informed consent was obtained from the patient’s parents for this case.
PDX creation
Leukemia cells were collected from the patient through BM aspiration at diagnosis using the UC Davis Internal Review Board approved protocol. Mononuclear cells were isolated using standardized Ficoll-Paque protocol (Sigma-Aldrich), stored frozen in dimethylsulfoxide (DMSO), and thawed immediately prior to transplantation. Female mice were used based on availability. The JMML PDX mouse model, over two generations, was created by bilateral intratibial injection of 0.5–5 million leukemia cells per mouse into healthy 6- to 12-week-old NOD/SCID/IL2Rg−/− (NSG) mice, without conditioning, using our institutionally approved animal care protocol. Leukemia cells were transplanted into four female mice (1st generation). Leukemia cells were harvested from the spleens of primary mice and serially transplanted into 12 female recipient mice (2nd generation). Mice were monitored biweekly and euthanized when they showed signs of leukemia in accordance with Institutional Animal Care and Use Committee (IACUC) policy on Humane Endpoints. Leukemia cells harvested from the BM and spleen were confirmed to be human leukocytes by flow cytometry using anti-human HLA-ABC antibodies (Biolegend & BD Biosciences).
Trametinib efficacy study
Ten male mice were injected with five million patient JMML cells per mouse and randomly enrolled into one of two groups. Group 1: vehicle control with 1% DMSO dissolved in a solution of 0.5% hydroxypropyl methylcellulose (HPMC, Sigma-Aldrich) in Tween 20 (Sigma-Aldrich) by daily oral gavage (n = 4). Group 2: trametinib (MedKoo Biosciences) 1 mg/kg by daily oral gavage (n = 6). Treatment started on day 4 for 28 days, based on the previously published regimens in acute lymphoblastic leukemia (ALL) models.Citation23,Citation24 The dose of trametinib was modified to 0.5 mg/kg daily at week 2 and 0.5 mg/kg every other day at week 3 due to intolerance of the high viscosity of the trametinib solution as evidenced by signs of aspiration. The dosage was then increased to 1 mg/kg every other day at week 4 until 28 total days of administration as the mice tolerated the treatment. The control group was administered the vehicle following the same schedule as the treatment group. Statistical significance for survival time was determined by the log-rank test. Analyses used Prism 8.3 software (GraphPad). Leukemia development was confirmed by flow cytometry using an anti-HLA-ABC antibody (Biolegend & BD Biosciences).
Trametinib target confirmation
JMML cells were harvested from the BM of PDX mice and stored frozen in 10% DMSO and 90% FBS in liquid nitrogen. Three untreated, control JMML samples with high HLA positivity (77–87%) were used for ex vivo trametinib target confirmation. Upon thawing, the cells were cultured in Human Plasma-like Medium (HPLM) supplemented with IL-3 (10 ng/mL), FLT-3 ligand (10 ng/mL), TPO (10 ng/mL), and SCF (25 ng/mL) (all Thermo Fisher Scientific),Citation25 at 1.0 × 106 cells per well with or without trametinib at 200 nM. At 1 h and 24 h after treatment, the cells were washed with PBS and total protein was extracted using RIPA Buffer (Thermo Fisher Scientific) supplemented with complete protease inhibitor (Sigma Aldrich) and Halt Phosphatase Inhibitor Cocktail (Thermo Fisher Scientific). 15 μg of protein were resolved using SDS-PAGE (12.5%), blotted onto a nitrocellulose membrane, and then blocked in PBS with 0.05% (vol/vol) Tween 20 (T-PBS) containing 1% (wt/vol) bovine serum albumin (BSA). The blots were incubated overnight at 4 °C with primary antibodies phospho-p44/42 MAPK (Erk1/2) (Thr202/Tyr204) and p44/42 MAPK (Erk1/2) (Cell Signaling Technologies). Both antibodies were diluted at 1:1000 in T-PBS containing 1% (wt/vol) BSA. Secondary anti-rabbit HRP-linked (Cell Signaling Technologies) and anti-mouse HRP-linked (Abcam)antibodies were used at 1:2,000 and 1:10,000 dilution, respectively. Blots were washed, then incubated with SuperSignal™ West Femto Maximum Sensitivity Substrate (Thermo Fisher Scientific) for imaging. Bands were visualized using ChemiDoc MP Imaging System (Bio-Rad). The averaged protein expression was normalized to the actin expression (Santa Cruz Biotechnology, Inc).
Results
A JMML PDX model, with serial transplantation to the second generation, was successfully created. Leukemia symptoms, including decreased activity, rough coat, and splenomegaly, developed in 3 of 4 primary mice at 16 weeks on average. Necropsy showed leukemia cell- infiltrated BM and spleen. Harvested cells were confirmed to be HLA-ABC positive by flow cytometry.Citation5,Citation26,Citation27 Serial transplantation of primary mouse spleen cells into secondary mice resulted in development of leukemia symptoms in 12 of 12 secondary mice at 21 weeks on average. Harvested BM cells from both primary and secondary mice were positive for HLA-ABC ranging from 2.8% to 95.3% (Supplemental Table 1). Successful engraftment is defined as 0.5% or more human HLA-ABC in murine BM, based on convention for xenograft models.Citation4,Citation28–30
To determine the in vivo therapeutic efficacy of trametinib, 10 new mice transplanted with patient JMML cells were treated with trametinib (n = 6) and compared against the control (n = 4). Trametinib treatment significantly prolonged survival time in the treated mice with a median survival of 194 days (95% confidence interval: 133–194 days) in the treatment group versus 124 days (95% confidence interval: 94–155) in the control group (p = 0.02, ). Leukemia was confirmed in the harvested cells upon euthanization by HLA-ABC positivity by flow cytometry, which ranged from 41.8-92.1% (Supplemental Table 2).
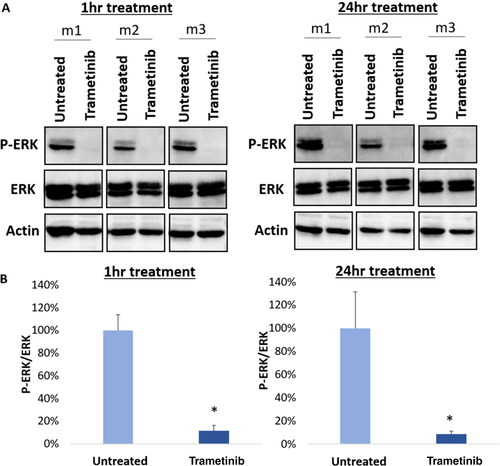
Furthermore, trametinib’s target confirmation was determined using harvested JMML cells from untreated PDX mice. Immunoblot analysis () showed decreased expression of p-ERK relative to total ERK in the ex vivo trametinib-treated JMML cells compared to untreated control, both 1 h and 24 h after treatment. The relative expression of p-ERK compared to total ERK, expressed as a percentage of control, was only 11% (p < 0.001) after 1 h of treatment and 9% (p = 0.007) after 24 h of treatment (). This verifies trametinib’s mechanism of action–specifically, MEK inhibition–through downregulation of ERK phosphorylationCitation16–18
Figure 2. Trametinib downregulates p-ERK in JMML PDX cells. (A) Immunoblotting was performed in cells after 1 h (left, n = 3) and 24 h (right, n = 3) of treatment with trametinib at 200 nM. The control group was untreated cells. A representative blot is depicted. (B) Digital quantification of the bands is shown. Trametinib significantly decreases p-ERK expression relative to total ERK compared to untreated cells (1 h p < 0.001, n = 3; 24 h p = 0.007, n = 3), confirming its mechanism of MEK inhibition. The student’s t-test was used and the error bars depict standard deviation.
Discussion
In this study, we developed a JMML PDX model and demonstrated that our model can be used for preclinical drug testing, such as our in vivo assessment of RAS inhibitor efficacy. Previously published JMML PDX modelsCitation4–8 have demonstrated successful PDX creation through serial transplantation and genomic analysis, and existing therapeutic assessments include azacitadine versus cytarabine or the anti-CD47 antibody magrolimab.Citation8,Citation9 In the azacitadine study, there were reduced CD34+ progenitor cells within the leukemia population in the azacitadine group compared to the cytarabine group. In the magrolimab study, magrolimab is reported to result in depletion of human JMML cells in the spleen and liver. In this study, we demonstrated the in vivo efficacy of a RAS inhibitor in our JMML PDX model. Given the absence of any curative regimens for JMML besides stem cell transplantation, as well as the inefficacy of generalized cytotoxic chemotherapy, the ability to trial mutation-specific therapies becomes exceptionally important. Moreover, our ex vivo JMML culture can pave the way to the discovery of new effective JMML therapies, as it offers a larger scale method to screen drugs, and combination therapies, for their efficacy in JMML. These drugs can then be further assessed in in vivo PDX models.
A key limitation in this study is that the PDX model is based on a single patient case, due to the rarity of JMML. A larger sample of cases could allow our findings to be more externally generalizable. However, RAS pathway mutations are known to be canonical in JMML and the RAS-associated PTPN11 and NF1 mutations harbored in this case are among the most ubiquitous genes altered in 90–95% of all JMML patients,Citation14,Citation31 making our findings potentially broadly applicable. In addition, while we only report a monotherapy regimen using trametinib in this study, there is also potential for other targeted mono-or combination therapies to be effective. For example, the patient in this case also harbors an ASXL1 mutation, which has been shown to induce hematopoiesis through Akt/mTOR activation;Citation32 in follow up studies, combination therapy with a mTOR inhibitor such as rapamycin could prove to be efficacious. Our PDX model can be used to develop new targeted combination therapies such as these, as well as conduct high volume screening of new drugs or predict responses to new treatments prior to use in patients. It can also be used to study drug resistance or pro-survival alterations in the leukemia microenvironment. These are particularly critical investigations in JMML, which displays significant treatment resistance and refractoriness to known therapies.
Author contributions
All authors had full access to the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. AL, HK, MI, AP, JC, and NS contributed to the conception and design of the study. AL, HK, and MI conducted mouse experiments and mechanistic studies. HK and YL performed the statistical analysis. AL wrote the first draft of the manuscript. HK wrote sections of the manuscript. NS provided overall supervision and funding acquisition. All authors contributed to manuscript revision, read, and approved the submitted version.
Supplemental Material
Download MS Excel (10.4 KB)Disclosure statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Additional information
Funding
References
- Gupta AK, Meena JP, Chopra A, Tanwar P, Seth R. Juvenile myelomonocytic leukemia-a comprehensive review and recent advances in management. Am J Blood Res. 2021;11(1):1–21.
- Dvorak CC, Loh ML. Juvenile myelomonocytic leukemia: molecular pathogenesis informs current approaches to therapy and hematopoietic cell transplantation. Front Pediatr. 2014;2:25. doi:10.3389/fped.2014.00025.
- Locatelli F, Niemeyer CM. How I treat juvenile myelomonocytic leukemia. Blood. 2015;125(7):1083–1090. doi:10.1182/blood-2014-08-550483.
- Krombholz CF, Aumann K, Kollek M, et al. Long-term serial xenotransplantation of juvenile myelomonocytic leukemia recapitulates human disease in Rag2-/- c-/- mice. Haematologica. 2016;101(5):597–606. doi:10.3324/haematol.2015.138545.
- Yoshimi A, Balasis ME, Vedder A, et al. Robust patient-derived xenografts of MDS/MPN overlap syndromes capture the unique characteristics of CMML and JMML. Blood. 2017;130(4):397–407. doi:10.1182/blood-2017-01-763219.
- Caye A, Rouault-Pierre K, Strullu M, et al. Despite mutation acquisition in hematopoietic stem cells, JMML-propagating cells are not always restricted to this compartment. Leukemia. 2019;34(7):1973–1668. doi:10.1038/s41375-019-0662-y.
- Louka E, Povinelli B, Rodriguez-Meira A, et al. Heterogeneous disease-propagating stem cells in juvenile myelomonocytic leukemia. J Exp Med. 2021;218(2):e20180853.
- Krombholz CF, Gallego-Villar L, Sahoo SS, et al. Azacitidine is effective for targeting leukemia-initiating cells in juvenile myelomonocytic leukemia. Leukemia. 2019;33(7):1805–1810. doi:10.1038/s41375-018-0343-2.
- Wang J, Racjak J, Koleci N, et al. Inhibition of the “don’t eat me signal” CD47 to prevent relapse in juvenile myelomonocytic leukemia. Klin Padiatr. 2023;235(03):0060.
- Pasupuleti SK, Chao K, Ramdas B, et al. Potential clinical use of azacitidine and MEK inhibitor combination therapy in PTPN11-mutated juvenile myelomonocytic leukemia. Mol Ther. 2023;31(4):986–1001. doi:10.1016/j.ymthe.2023.01.030.
- Kong G, Wunderlich M, Yang D, et al. Combined MEK and JAK inhibition abrogates murine myeloproliferative neoplasm. J Clin Invest. 2014;124(6):2762–2773. doi:10.1172/JCI74182.
- Chang T, Krisman K, Theobald EH, et al. Sustained MEK inhibition abrogates myeloproliferative disease in Nf1 mutant mice. J Clin Invest. 2013;123(1):335–339. doi:10.1172/JCI63193.
- Lyubynska N, Gorman MF, Lauchle JO, et al. A MEK inhibitor abrogates myeloproliferative disease in Kras mutant mice. Sci Transl Med. 2011;3(76):76ra27. doi:10.1126/scitranslmed.3001069.
- Niemeyer CM. JMML genomics and decisions. Hematology Am Soc Hematol Educ Program. 2018;2018(1):307–312. doi:10.1182/asheducation-2018.1.307.
- Stieglitz E, Taylor-Weiner AN, Chang TY, et al. The genomic landscape of juvenile myelomonocytic leukemia. Nat Genet. 2015;47(11):1326–1333. doi:10.1038/ng.3400.
- Long GV, Fung C, Menzies AM, et al. Increased MAPK reactivation in early resistance to dabrafenib/trametinib combination therapy of BRAF-mutant metastatic melanoma. Nat Commun. 2014;5(1):5694. doi:10.1038/ncomms6694.
- Zeiser R. Trametinib. Recent Results Cancer Res. 2014;201:241–248.
- Hashemzadeh S, Ramezani F, Rafii-Tabar H. Study of molecular mechanism of the interaction between MEK1/2 and trametinib with docking and molecular dynamic simulation. Interdiscip Sci. 2019;11(1):115–124. doi:10.1007/s12539-018-0305-4.
- Vitiello PP, Cardone C, Martini G, et al. Receptor tyrosine kinase-dependent PI3K activation is an escape mechanism to vertical suppression of the EGFR/RAS/MAPK pathway in KRAS-mutated human colorectal cancer cell lines. J Exp Clin Cancer Res. 2019;38(1):41. doi:10.1186/s13046-019-1035-0.
- Pomeroy EJ, Lee LA, Lee RDW, et al. Ras oncogene-independent activation of RALB signaling is a targetable mechanism of escape from NRAS(V12) oncogene addiction in acute myeloid leukemia. Oncogene. 2017;36(23):3263–3273. doi:10.1038/onc.2016.471.
- Stieglitz E, Loh ML, Meyer J, et al. MEK inhibition demonstrates activity in relapsed, refractory patients with juvenile myelomonocytic leukemia: results from COG study ADVL1521. Blood. 2021;138(Supplement 1):3679–3679. doi:10.1182/blood-2021-144850.
- Li J, Li J, Hu T, et al. Response to trametinib of two pediatric myeloid malignancies cases harboring RAS mutation and monosomy 7. Leuk Lymphoma. 2022;63(9):2238–2242. doi:10.1080/10428194.2022.2067998.
- Kerstjens M, Pinhancos SS, Castro PG, et al. Trametinib inhibits. Haematologica. 2018;103(4):e147–e150. doi:10.3324/haematol.2017.174060.
- Jones CL, Gearheart CM, Fosmire S, et al. MAPK signaling cascades mediate distinct glucocorticoid resistance mechanisms in pediatric leukemia. Blood. 2015;126(19):2202–2212. doi:10.1182/blood-2015-04-639138.
- Hofmans M, Lammens T, Depreter B, et al. Long non-coding RNAs as novel therapeutic targets in juvenile myelomonocytic leukemia. Sci Rep. 2021;11(1):2801. doi:10.1038/s41598-021-82509-5.
- Yan Y, Dong L, Chen C, et al. JMML tumor cells disrupt normal hematopoietic stem cells by imposing inflammatory stress through overproduction of IL-1β. Blood Adv. 2022;6(1):200–206. doi:10.1182/bloodadvances.2021005089.
- Nakamura Y, Ito M, Yamamoto T, et al. Engraftment of NOD/SCID/gammac(null) mice with multilineage neoplastic cells from patients with juvenile myelomonocytic leukaemia. Br J Haematol. 2005;130(1):51–57. doi:10.1111/j.1365-2141.2005.05578.x.
- Notta F, Mullighan CG, Wang JCY, et al. Evolution of human BCR-ABL1 lymphoblastic leukaemia-initiating cells. Nature. 2011;469(7330):362–367. doi:10.1038/nature09733.
- Lapidot T, Sirard C, Vormoor J, et al. A cell initiating human acute myeloid-leukemia after transplantation into scid mice. Nature. 1994;367(6464):645–648. doi:10.1038/367645a0.
- Díaz de la Guardia R, Velasco-Hernandez T, Gutiérrez-Agüera F, et al. Engraftment characterization of risk-stratified AML in NSGS mice. Blood Adv. 2021;5(23):4842–4854. doi:10.1182/bloodadvances.2020003958.
- Behnert A, Meyer J, Parsa J-Y, et al. Exploring the genetic and epigenetic origins of juvenile myelomonocytic leukemia using newborn screening samples. Leukemia. 2022;36(1):279–282. doi:10.1038/s41375-021-01331-0.
- Fujino T, Goyama S, Sugiura Y, et al. Mutant ASXL1 induces age-related expansion of phenotypic hematopoietic stem cells through activation of Akt/mTOR pathway. Nat Commun. 2021;12(1):1826. doi:10.1038/s41467-021-22053-y.