
Abstract
Dexamethasone (Dex) is a type of glucocorticoid drug. Long term use can induce growth plate chondrocytes (GPCs) apoptosis, impair differentiation, and inhibit cell proliferation and bone growth. It has been reported that Krüppel-like factor 2 (KLF2) inhibits osteoblast damage induced by Dex, but the role in Dex-induced GPCs remains unclear. Dex was used to construct a model of growth plate injury in vitro. CCK-8 and TUNEL kits were used to determine cell viability and apoptosis. A model of growth plate injury was established by intraperitoneal injection of Dex. Immunohistochemistry was used to investigate the expression of KLF2 in rats. The results showed that KLF2 expression of rat tibial GPCs was down-regulated after Dex stimulation. Overexpression of KLF2 promoted cell viability and cell cycle, while inhibited apoptosis of growth plate Dex-induced chondrocytes. Moreover, KLF2 inhibited Runx2-mediated PI3K/AKT and ERK signalling pathways. And PI3K/AKT and ERK signalling pathways, which were involved in the regulation of KLF2 on GPCs. Further studies showed that KLF2 alleviated growth plate injury in vivo. In conclusion, our study found that KLF2 promoted proliferation and inhibited apoptosis of Dex-induced GPCs by targeting the Runx2-mediated PI3K/AKT and ERK signalling pathways.
1. Introduction
Epiphyseal injury is a unique bone disease in children [Citation1]. Once this damage, it will affect the growth of children and the normal growth and development of the children’s bones [Citation2]. Epiphyseal plate is a component of cartilage, which is closely related to the proliferation and apoptosis of chondrocytes [Citation3]. Dexamethasone (Dex) is a kind of glucocorticoid (GCs) with anti-inflammatory, detoxification and anti-allergy effects [Citation4]. Glucocorticoids have immune suppression, anti-shock, antipyretic and detoxification effects, and are widely used in paediatric clinics for asthma, chemotherapy, anti-shock, antipyretic and the treatment of other diseases treatment [Citation5–7]. However, long-term use of Dex can induce growth plate chondrocytes (GPCs) apoptosis, impair differentiation, and inhibit cell proliferation and bone growth [Citation7, Citation8].
Krueppel-like factor 2 (KLF2) is a transcription factor that binds zinc finger structure to DNA [Citation9]. KLF2 is expressed in MC3T3-E1 cells and primary osteoblasts. Interestingly, the expression of KLF2 increases during osteoblast differentiation. Overexpression of KLF2 in MC3T3-E1 cells promotes the expression of Alp, Osx and Ocn, and promotesd the expression of Runx2 [Citation10]. These results indicate that KLF2 is a new regulator of osteoblast differentiation, suggesting that KLF2 may be a new target for the treatment of bone diseases.
Runt-related transcription factor 2 (Runx2) is a transcription factor that is essential for osteoblast differentiation and chondrocyte maturation [Citation11]. Runx2 regulates the expression of Ihh, Col10α1, Spp1, Ibsp, MMP13 and VEGF-A in growth plate [Citation12]. Studies have found that Runx2 plays an important role in bone metastasis of breast and prostate cancer [Citation13]. In addition, Runx2 is one of the genes leading to osteoarthritis (OA) [Citation14]. Studies have found that inhibition of the PI3K/AKT and ERK signalling pathway can significantly inhibit the apoptosis of chondrocytes and inhibit the progress of osteoarthritis [Citation15]. In addition, Runx2 can play a carcinogenic role in oesophageal cancer by activating the PI3K/AKT and ERK pathways, indicating that Runx2 has a regulatory role in PI3K/AKT and ERK pathways [Citation16].
This study was based on Dex-treated rat tibial GPCs, and analysed the expression of KLF2 in Dex-treated rat tibial GPCs. Furthermore, through the up-regulation of KLF2 expression, the role of KLF2 in the proliferation cycle and apoptosis of rat tibial GPCs treated with Dex was explored. In addition, a rat Dex model was constructed to analyse the effects of Dex-treated rat body weight, tibia length, nose-tail length, and growth plate thickness after KLF2 overexpression. The purpose of this study was to clarify the mechanism of KLF2-mediated GPCs apoptosis and provide a potential treatment for Dex-induced growth retardation in children.
2. Materials and methods
2.1. Animal model
A total of 30 two-week-old male Sprague Dawley (SD) rats with a body weight of (80 ± 2) g were purchased from Experimental Animal Centre of the Shandong University and raised at the Experimental Animal Centre. Rats were housed at 25 ± 1 °C with a12‑h light/dark cycles, 50 ± 5% humidity, and free access to food and water. The rats were randomly divided into three groups, each with 10 rats. Control group: every day at 8:00 am, the control group received an intraperitoneal injection of 0.9% NaCl (200 μg/100 g); Dex (Sanjian, Tianjin, China) group: the Dex treatment group received intraperitoneal injection of Dex (200 μg/100 g); and Dex + KLF2 group: the Dex + KLF2 group not only received an intraperitoneal injection of Dex (200 μg/100 g), but also received an injection of a KLF2 overexpression lentiviral vector (GenePharma Shanghai, China, 1 × 109 PFU) into the joint. Ten days later, the rats were anaesthetised with 10% chloral hydrate. Under anaesthesia, the rats were placed in the abdomen position and the length of the nose and tail was measured. This was repeated three times and took the average value was used. This study was supported by the Ethics Committee of the Xi’an Children’s Hospital (20220080).
2.2. Culture of rat tibial growth plate chondrocytes
After the rats were culled, the proximal tibia was aseptically removed on a clean bench, the growth plate cartilage was separated from the osteochondral junction, the muscle and periosteum outside the tibia were carefully removed, and the cartilage pieces were cut into 1 mm3 pieces. After two step enzymatic digestion steps (0.25% trypsin EDTA digested for 30 min, and 0.2% collagenase digested for 3 h), the cells were collected and counted. Then, the trypan blue test was used to determine the cell viability, and the chondrocytes were seeded in a 25 cm2 culture flask at a density of 5 × 105 cells/bottle, and cultured in an incubator at 37 °C, 5% CO2, and 95% saturated humidity. Then the rat tibial GPCs were treated with Dex, and the treatment time and concentration determined in a previous study [Citation17].
2.3. Cell transfection
When the cell fusion reached 50%–80%, the constructed overexpression lentivirus, pcDNA3.1-KLF2, pcDNA3.1-ERK and their negative control vectors (Sanjian, Tianjin, China) were transfected into the chondrocytes.
2.4. CCK-8 assay
The transfected chondrocytes were evenly spread in a 96-well plate with 1 × 104 cells per well, and cultured in an incubator at 37 °C and 5% CO2. After adding different concentrations of Dex, 10 μL of CCK-8 solution (Dojindo Laboratories, Kumamoto, Japan) was added to each well at 0, 24, 48 and 72 h, respectively. Then the plates were placed in the incubator at 37 °C, 5% CO2 for 2 h. The absorbance of each group was detected at 450 nm on a plate reader.
2.5. Apoptosis assay
We determined the apoptosis of cells using the flow cytometry. Cells were seeded in a 6-well plate and treated with Dex for 48 h for analysing flow cytometry. Then, the adherent and floating cells were harvested, washed these with cooled PBS, and stained them with FITC-conjugated Annexin V/PI (Sanjian, Tianjin, China) according to the kit’s instruction. After this, we detected double-stained cells using flow cytometry (BD Bioscience, Franklin Lakes, NJ, USA). The data were analysed and processed by Cell Quest software.
2.6. Immunohistochemistry
The tibia sections were dewaxed and rehydrated, then incubated overnight with a primary KLF2 antibody (Abcam, USA) at 4 °C, followed by HRP-conjugated anti-rabbit IgG at room temperature for 1 h. Diaminobenzidine (DAB, Sangong, China) was used for colour development. The sections were also counterstained with haematoxylin.
2.7. Quantitative real-time PCR
We used a TRIzol reagent (Takara, Dalian, Shenyang, China) to isolate total RNA samples from cells and tissues in accordance with the instructions. We used the reverse transcription kit (Takara) for cDNA synthesis. With cDNA as a template, we conducted qRT-PCR using SYBR Green (Takara) in an ABI 6500 under the condition of 95 °C for 90 s and 40 cycles of 95 °C for 15 s, 58 °C for 21 s, and 72 °C for 24 s. The primers used are tabulated primers of genes in . With β-actin as the internal control, we calculated the relative gene expression using the 2-ΔΔCt method.
Table 1. The primer sequence of RT-PCR.
2.8. Western blot
We lysed cells and tissue samples with a RIPA lysis buffer containing protease inhibitors (Sigma). The protein suspension was then collected and quantified using a BCA protein Assay Kit (Pierce). Following this, an equal quantity of protein was boiled with loading buffer for 10 min, separated with 10% SDS-PAGE, and transferred onto PVDF membranes (Takara, Dalian, China). Then, we used 5% skim milk to block the membrane at room temperature for 0.5 h, followed by primary antibody incubation at 4 °C overnight. After washing with a Tris-buffered saline and polysorbate buffer, the membrane was incubated at room temperature with the appropriate secondary antibody for 1 h. Then, the bands on membranes were visualised using the ChemiDoc XRS System (Bio-Rad, Hercules, CA, USA). We utilised primary antibodies in this study as follows: KLF2 (1:1000), Ki67 (1:1000), c-caspase-3 (1:1000), Bax (1:1000), Bcl-2 (1:1000), Runx2 (1:1000), PI3K (1:1000), ERK (1:1000), AKT (1:1000), and GAPDH (1:5000).
2.9. Statistical analysis
We used GraphPad Prism 7.0 (GraphPad Software, San Diego, CA, USA) for statistical analysis in this study. All experiments were conducted in triplicate, and the data are presented as mean ± standard deviation (SD). Comparisons between groups were made using Student’s t-test, and multiple comparisons were made using a one-way analysis of variance, followed by Turkey’s test. For all comparisons, p < 0.05 was considered statistically significant.
3. Results
3.1. Different concentrations of Dex inhibit the expression of KLF2 in GPCs
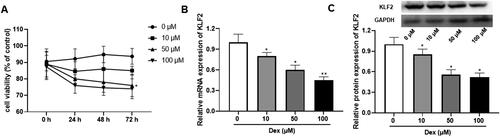
We firstly treated GPCs with different concentrations of Dex (0, 10, 50, 100 μM) to construct the growth plate injury model. According to the results, Dex significantly inhibited the cell viability of GPCs in a dose-dependent manner (). Compared with the control group, different concentrations of Dex inhibited the expression of KLF2 at different degrees in a dose-dependent manner, including mRNA and protein (). In conclusion, Dex significantly inhibited the cell viability, and the expression of KLF2 in GPCs.
Figure 1. Dex at different concentrations inhibited the expression of KLF2 in GPCs. The isolated GPCs were treated with dexamethasone (Dex, 0, 10, 50, 100 μM) for 72 h. (A) The cell viability of rat GPCs treated with different doses of Dex. (B) The mRNA expression of KLF2 in chondrocytes after treated with different doses of Dex. (C) The protein expression of KLF2 in chondrocytes after treated with different doses of Dex. “*” means compared with 0 μM group p < 0.05. GAPDH was used as an invariant internal control for calculating protein-fold changes.
3.2. Overexpression of KLF2 promotes proliferation of Dex-induced GPCs
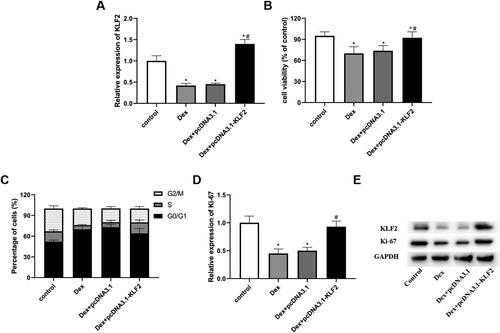
To explore the role of KLF2 in chondrocytes, we overexpressed KLF2 using an overexpression vector. The results showed that Dex decreased the expression of KLF2, while pcDNA3.1-KLF2 significantly increased the expression of KLF2 (). As presented in , pcDNA3.1-KLF2 significantly promoted the cell viability compared with the pcDNA3.1 and Dex group (). Then, the data in showed that Dex reduced the proportion of cells in S phase, while KLF2 overexpression increased S-phase cells, resulting in fewer G0/G1-phase cells (). Further study revealed that Dex significantly promoted the expression of Ki-67 compared with pcDNA3.1 and Dex group (). These results indicated that KLF2 promotes the proliferation of Dex-induced GPCs.
Figure 2. Overexpression of KLF2 promotes proliferation of Dex-induced GPCs. GPCs were transfected with pcDNA3.1-KLF2 or control pcDNA3.1, and then treated with Dex (50 μM) for 24 h. (A) The protein expression of KLF2 was detected by Western blot. (B) Cell viability of chondrocytes was detected by CCK-8 assay. (C) Cell cycle analysis of chondrocytes treated with Dex was quantified by PI staining and fluorescence flow cytometry. (D–E) The protein expression of Ki-67 was detected by Western blot. “*” means compared with control group p < 0.05, and “#” means compared with Dex + pcDNA3.1 group p < 0.05. GAPDH was used as an invariant internal control for calculating protein-fold changes.
3.3. Overexpression of KLF2 inhibits Dex-induced apoptosis of GPCs
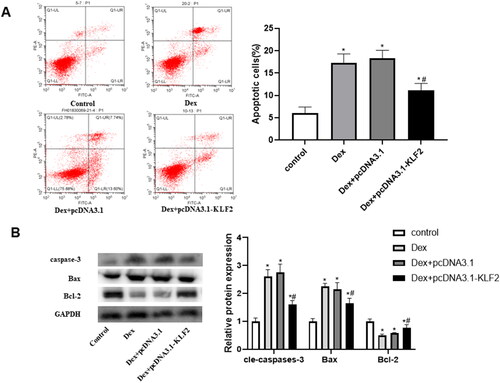
Our research further explored the role of KLF2 in apoptosis of GPCs. As presented in , Dex induced the apoptosis of GPCs, as well as increased the expression of Bax and c-caspase-3, while decreased the expression of Bcl-2. On the contrary, KLF2 overexpression significantly reversed these effects, decreased the expression of Bax and c-caspase-3, and increased the expression of Bcl-2 (). These results indicated that KLF2 inhibited the apoptosis of Dex-induced GPCs.
Figure 3. Overexpression of KLF2 inhibits Dex-induced apoptosis of GPCs. GPCs were transfected with pcDNA3.1-KLF2 or control pcDNA3.1, and then treated with Dex (50 μM) for 24 h. (A) The apoptosis rate of Dex-induced chondrocytes was measured by fluorescence flow cytometry. (B) The protein expression of c-caspase-3, Bax and Bcl-2 was detected by Western blot. “*” means compared with control group p < 0.05, and “#” means compared with Dex + pcDNA3.1 group p < 0.05. GAPDH was used as an invariant internal control for calculating protein-fold changes.
3.4. Overexpression of KLF2 inhibits Runx2-mediated activation of the PI3K/AKT and ERK signalling pathways
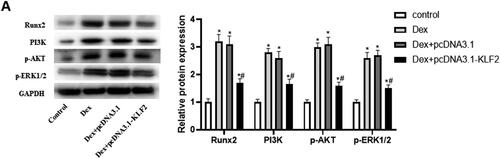
It has been reported that PI3K/AKT and ERK signalling are involved in the regulation of growth plate injury [Citation18]. Our research further explored the role of KLF2 in the Runx2-mediated PI3K/AKT and ERK signalling pathways. As expected, Dex significantly elevated the protein expression of Runx2, PI3K, p-AKT and p-ERK1/2 compared with control, and KLF2 overexpression significantly decreased their expression in Dex-induced GPCs (). The result indicated that KLF2 inhibits the activation of Runx2-mediated PI3K/AKT and ERK signalling pathways.
Figure 4. Overexpression of KLF2 inhibits Runx2 mediated activation of PI3K/AKT and ERK signalling pathways. GPCs were transfected with pcDNA3.1-KLF2 or control pcDNA3.1, and then treated with Dex (50 μM) for 24 h. (A) The protein expression of Runx2, PI3K, AKT and ERK was detected by Western blot. (B) Quantification of protein expression of Runx2, PI3K, AKT and ERK. “*” means compared with control group p < 0.05, and “#” means compared with Dex + pcDNA3.1 group p < 0.05. GAPDH was used as an invariant internal control for calculating protein-fold changes.
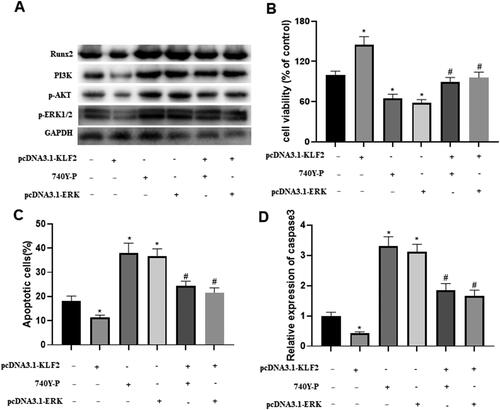
3.5. Runx2-mediated PI3K/AKT and ERK signalling pathways are involved in the regulation of GPCs by KLF2
Next, we explored the underlying mechanism by which KLF2 inhibited the progression of growth plate injury. The western blot results indicated that KLF2 decreased the protein levels of Runx2, PI3K, AKT and ERK, while a PI3K agonist (740Y-P) or pcDNA3.1-ERK significantly reversed the effect of KLF2 (). Interestingly, KLF2 significantly increased the cell viability of GPCs, and 740Y-P or pcDNA3.1-ERK hindered the cell viability (). As for apoptosis, KLF2 suppressed the apoptosis in GPCs, but 740Y-P or pcDNA3.1-ERK significantly increased the apoptosis of GPCs (). Meanwhile, KLF2 suppressed the expression of c-caspase-3, while 740Y-P or pcDNA3.1-ERK significantly promoted the expression of c-caspase-3 (). These results suggest that Runx2-mediated PI3K/AKT and ERK signalling pathways are involved in the mechanism by which KLF2 affects Dex-induced GPCs.
Figure 5. Runx2-mediated PI3K/AKT and ERK signalling pathways are involved in KLF2 regulation of GPCs. After GPCs were transfected with pcDNA3.1-KLF2 or PI3K agonist (740Y-P) or pcDNA3.1-ERK, GPCS were treated with Dex (50 μM) for 24 h. (A) The protein expression of Runx2, PI3K, AKT and ERK was detected by Western blot. (B) Cell viability of chondrocytes was detected by CCK-8 assay. (C) The apoptosis rate of Dex-induced chondrocytes was measured by fluorescence flow cytometry. (D) The protein expression of c-caspase-3 was detected by Western blot. “*” means compared with control group p < 0.05, and “#” means compared with pcDNA3.1-KLF2 group p < 0.05. GAPDH was used as an invariant internal control for calculating protein-fold changes.
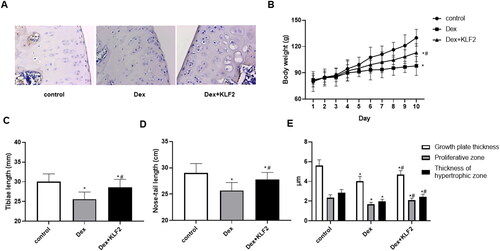
3.6. Overexpression of KLF2 alleviates growth retardation in rats
To further verify the role of KLF2 in Dex-induced chondrocytes, we established a rat growth plate injury model. As shown in , Dex treatment significantly decreased the expression of KLF2, and after KLF2 overexpression lentivirus infection, the expression of KLF2 was up-regulated in rat growth plate sections. Moreover, Dex treatment permanently reduced the rat body weight as well as length of tibiae and nose-tail, while KLF2 inhibited these decreases (). Interestingly, KLF2 significantly increased the thickness of the growth plate, proliferation zone and hypertrophy zone induced by Dex ().
Figure 6. Overexpression of KLF2 alleviates growth retardation in rats. SD rats were injected with Dex intraperitoneally and KLF2 overexpression vector in the joint, and were divided into three groups: control, Dex, Dex + KLF2. (A) Immunohistochemistry was used to analyse the expression of KLF2 in rat tibiae growth plate. (B-D) Quantification of the rat body weight, length of tibiae and nose-tail. (E) Quantification of the thickness of growth plate, proliferative zone and hypertrophic zone. “*” means compared with control group p < 0.05, and “#” means compared with Dex group p < 0.05.
4. Discussion
Dex is a synthetic GC that has been widely used to treat inflammatory and autoimmune diseases. However, clinical evidence shows that the prolonged treatment with Dex causes growth retardation in treated children by triggering chondrocyte apoptosis within the growth plate [Citation19, Citation20]. In this study, we found that KLF2 expression was increased in chondrocytes of the tibial growth plate in Dex-treated rats. In addition, KLF2 promoted the cell activity of GPCs induced by Dex. KLF2 promoted the proliferation of GPCs and alleviated Dex-induced apoptosis in GPCs through the Runx2-mediated PI3K/AKT and ERK signalling pathways.
Previous studies have shown that KLF2 is expressed and plays a regulatory role in the lung, T cells, T-reg cells, endothelial cells, monocytes and other related tissues and cells, and is related to a variety of inflammatory and pathological states [Citation21, Citation22]. In addition, KLF2 is expressed in osteoblasts and primary osteoblasts [Citation23]. Interestingly, KLF2 expression is increased in osteoblasts during osteoblast differentiation [Citation24]. Importantly, we found that KLF2 can physically interact with Runx2. KLF2 promotes osteoblast differentiation by regulating Runx2 and the physical interaction with Runx2. Based on these results, this study found that KLF2 is a new regulatory factor involved in osteoblast differentiation, suggesting that KLF2 may be a new target for the treatment of bone diseases [Citation10]. In this study, we found that KLF2 expression was down-regulated in the Dex-treated rat GPCs, and played a protective role in the proliferation reduction and apoptosis of chondrocytes. Furthermore, our in vivo data suggested that overexpression of KLF2alleviated Dex-induced growth retardation in rats by inhibiting GPCs.
To explore the underlying molecular mechanisms of KLF2 in Dex-induced chondrocytes, we studied the regulator that bind to KLF2. Consistent with previous data [Citation10], our results also show that KLF2 significantly promoted the expression of Runx2 in GPCs. Runx2 is the key to osteoblast differentiation and chondrocyte maturation [Citation25]. During the differentiation of osteoblasts, Runx2 was weakly expressed in undifferentiated mesenchymal cells, up-regulated in pre-osteoblasts, highly expresses in immature osteoblasts, and down-regulated in mature osteoblasts [Citation26]. Runx2 enhances the proliferation of osteoblast progenitor cells by directly regulating FGFR2 and FGFR3 [Citation12]. Furthermore, it has been reported that Runx2 induces Sp7 and activates canonical Wnt signal to guide cells to differentiate into osteoblasts, thereby inhibiting chondrocyte differentiation [Citation11]. In the present study, our findings indicated that KLF2 affected Dex-treated rat GPCs functions through targeting Runx2.
It has been found that Runx2 can activate and thereby regulate the PI3K/AKT and ERK pathways [Citation16]. Previous studies have reported that PI3K/AKT and ERK MAPK pathways are involved in the regulation of FGF2 on human chondrocytes and play an important role in the occurrence and development of osteoarthritis [Citation27]. Our study found that activation of PI3K/AKT and ERK significantly inhibited the proliferation of GPCs and promoted cell apoptosis, suggesting that PI3K/AKT and ERK pathway may be the key way to regulate growth plate injury, which is basically consistent with previous research conclusions.
In this study, KLF2 promoted the cell activity of GPCs induced by Dex. KLF2 promoted the proliferation of GPCs and alleviated Dex-induced apoptosis of GPCs through Runx2-mediated PI3K/AKT and ERK signalling pathways. In addition, overexpression of KLF2 in vivo can alleviate the symptoms of poor growth in rats. This study has clarified the role and mechanism of KLF2 and provides a possible treatment for children with growth retardation.
Authors’ contributions
Xiaowei Wang was involved in study design, implementation of the experiments and data analysis. Yulong Ma and Tao Peng were involved in data analysis and interpretation, and drafting of the manuscript. Xudong Yao and Chaonan Sun were involved in data analysis and revision of the manuscript.
Data availability statement
The datasets used and/or analysed during the current study are available from the corresponding author on reasonable request.
Disclosure statement
The authors declare that they have no competing interests.
Funding
The author(s) reported there is no funding associated with the work featured in this article.
References
- Schwab SA. Epiphyseal injuries in the growing athlete. Can Med Assoc J. 1977;117(6):626–630.
- Launay F. Sports-related overuse injuries in children. Orthop Traumatol Surg Res. 2015;101(1 Suppl):S139–S47.
- Chauhan DK, Dhillon MS, Tripathy SK. Neuro-epiphyseal injury around the ankle: a case report. Foot (Edinb). 2009;19(2):133–136.
- Sinner B. Perioperative dexamethasone. Anaesthesist. 2019;68(10):676–682.
- Harrington J, Holmyard D, Silverman E, et al. Bone histomorphometric changes in children with rheumatic disorders on chronic glucocorticoids. Pediatr Rheumatol Online J. 2016;14(1):58.
- Deng J, Chalhoub NE, Sherwin CM, et al. Glucocorticoids pharmacology and their application in the treatment of childhood-onset systemic lupus erythematosus. Semin Arthritis Rheum. 2019;49(2):251–259.
- Zaman F, Zhao Y, Celvin B, et al. Humanin is a novel regulator of hedgehog signaling and prevents glucocorticoid-induced bone growth impairment. Faseb J. 2019;33(4):4962–4974.
- Hoshino M, Kaneko K, Miyamoto Y, et al. 8-Nitro-cGMP promotes bone growth through expansion of growth plate cartilage. Free Radic Biol Med. 2017;110:63–71.
- Laha D, Deb M, Das H. KLF2 (kruppel-like factor 2 [lung]) regulates osteoclastogenesis by modulating autophagy. Autophagy. 2019;15(12):2063–2075.
- Hou Z, Wang Z, Tao Y, et al. KLF2 regulates osteoblast differentiation by targeting of Runx2. Lab Invest. 2019;99(2):271–280.
- Komori T. Runx2, an inducer of osteoblast and chondrocyte differentiation. Histochem Cell Biol. 2018;149(4):313–323.
- Komori T. Regulation of proliferation, differentiation and functions of osteoblasts by Runx2. Int J Mol Sci. 2019;20(7):1694–1702.
- Kim B, Kim H, Jung S, et al. A CTGF-RUNX2-RANKL axis in breast and prostate cancer cells promotes tumor progression in bone. J Bone Miner Res. 2020;35(1):155–166.
- Chen D, Kim DJ, Shen J, et al. Runx2 plays a Central role in osteoarthritis development. J Orthop Translat. 2020;23:132–139.
- Lin Z, Tian XY, Huang XX, et al. microRNA-186 inhibition of PI3K-AKT pathway via SPP1 inhibits chondrocyte apoptosis in mice with osteoarthritis. J Cell Physiol. 2019 May;234(5):6042–6053.
- Lu H, Jiang T, Ren K, et al. RUNX2 plays an oncogenic role in esophageal carcinoma by activating the PI3K/AKT and ERK signaling pathways. Cell Physiol Biochem. 2018;49(1):217–225.
- Liu X, She Y, Wu H, et al. Long non-coding RNA Gas5 regulates proliferation and apoptosis in HCS-2/8 cells and growth plate chondrocytes by controlling FGF1 expression via miR-21 regulation. J Biomed Sci. 2018;25(1):18.
- Scindia PD, Leeds MJ, Swaminathan MS. Iron homeostasis in healthy kidney and its role in acute kidney injury. Seminars in Nephrology. 2019;39(1):76–84.
- Su YW, Fan J, Fan CM, et al. Roles of apoptotic chondrocyte-derived CXCL12 in the enhanced chondroclast recruitment following methotrexate and/or dexamethasone treatment. J Cell Physiol. 2021;236(8):5966–5979.
- Huang Y, Cai GQ, Peng JP, et al. Glucocorticoids induce apoptosis and matrix metalloproteinase-13 expression in chondrocytes through the NOX4/ROS/p38 MAPK pathway. J Steroid Biochem Mol Biol. 2018;181:52–62.
- Jha P, Das H. KLF2 in regulation of NF-κB-Mediated immune cell function and inflammation. Int J Mol Sci. 2017;18(11):2383–2392.
- Turpaev KT. Transcription factor KLF2 and its role in the regulation of inflammatory processes. Biochemistry (Mosc). 2020;85(1):54–67.
- Maity J, Barthels D, Sarkar J, et al. Ferutinin induces osteoblast differentiation of DPSCs via induction of KLF2 and autophagy/mitophagy. Cell Death Dis. 2022;13(5):452.
- Kim I, Kim JH, Kim K, et al. The IRF2BP2-KLF2 axis regulates osteoclast and osteoblast differentiation. BMB Rep. 2019;52(7):469–474.
- Narayanan A, Srinaath N, Rohini M, et al. Regulation of Runx2 by MicroRNAs in osteoblast differentiation. Life Sci. 2019;232:116676.
- Rashid H, Chen H, Javed A. Runx2 is required for hypertrophic chondrocyte mediated degradation of cartilage matrix during endochondral ossification. Matrix Biol Plus. 2021;12:100088.
- Yu X, Qi Y, Zhao T, et al. NGF increases FGF2 expression and promotes endothelial cell migration and tube formation through PI3K/akt and ERK/MAPK pathways in human chondrocytes. Osteoarthritis Cartilage. 2019;27(3):526–534.