
Abstract
Unlike Fc receptors for switched immunoglobulin (Ig) isotypes, Fc receptor for IgM (FcµR) is selectively expressed by lymphocytes. The ablation of the FcµR gene in mice impairs B cell tolerance as evidenced by concomitant production of autoantibodies of IgM and IgG isotypes. In this essay, we reiterate the autoimmune phenotypes observed in mutant mice, ie IgM homeostasis, dysregulated humoral immune responses including autoantibodies, and Mott cell formation. We also propose the potential phenotypes in individuals with FCMR deficiency and the model for FcµR-mediated regulation of self-reactive B cells.
Introduction
The existence of FcµR has been suggested for decades, but its molecular identification has defied until 2009, when the authentic FcµR cDNA was isolated from human B-lineage cDNA libraries by the functional expression cloning strategy [Citation1]. Unlike FcRs for switched Ig isotypes (eg FcγRs, FcεRs, FcαR, Fcα/µR), FcµR is selectively expressed by lymphocytes: B, T and NK cells in humans and only B cells in mice [Citation1–10], although there are several reports describing the possible expression of FcµR by other cell types in both mice and humans [Citation11–15]. FcµR is a type I sialoglycoprotein with a Mr of ∼60 kDa and has a unique structure consisting of a single N-terminal Ig-like domain and membrane proximal stalk region in the ectodomain, a transmembrane (TM) segment carrying a charged His residue, and a long cytoplasmic tail containing conserved three Tyr and five Ser residues. The Ig-like domain is responsible for its exclusive binding to IgM with a high affinity of ∼10 nM and has a unique first complementary determining region (CDR1) distinct from that of two promiscuous IgM- and IgA-binding receptors, polymeric Ig receptor (pIgR) and Fcα/μR [Citation1]. Recent crystallographic and cryo-electron microscopic (cryo-EM) analyses have identified critical amino acid (aa) residues of FcµR binding to the fourth constant domain of IgM (Cµ4) and also unravel the intricate mechanisms for the interaction of FcµR with secreted pentameric IgM and membrane-bound monomeric IgM molecules [Citation16,Citation17]. The existence of His residue in the FcµR TM suggests a possible requirement for another membrane protein with a negatively charged aa residue in its TM segment to neutralise the positive His residue, but the identification of such adaptor protein remains to be elucidated. The conserved Tyr and Ser residues in the cytoplasmic tail of FcµR undergo phosphorylation upon ligation with IgM ligands [Citation1]. More recently, the phosphorylated C-terminal Tyr residue (DY385INV) of FcµR has been found to promote cellular activation by triggering signaling events that are analogous to those described for the Ig tail tyrosine motif (ITT), a signal amplifier of memory-type B cell receptors (BCRs) of IgG and IgE isotypes with Ca2+ mobilization and recruitment of growth factor receptor bound protein 2 (Grb2) after cross-linkage of receptor in vitro [Citation18,Citation19].
Since the cellular distribution of FcµR is different between humans (B, T, and NK cells) and mice (only B cells), the real in vivo functions of FcµR in humans await until identification of the pathophysiological manifestations in FCMR-deficient individuals. The current potential functions of FcµR are thus mainly implicated from studies of Fcmr-deficient (KO) mice. Although some conflicting views exist among five different strains of Fcmr KO mice, in this short essay we will focus on the common findings observed in most strains of the mutant mice along with our models of FcµR functions related to autoimmunity.
Lessons from murine FcµR deficiency
Homeostasis of serum IgM
FcµR is involved in IgM homeostasis as suggested by the following observations. Pre-immune (or natural) serum IgM (and IgG3) levels are increased by twofold in Fcmr KO mice compared with their wild type (WT) mice [Citation3], and this IgM increase is correlated with the number of its null mutation alleles (ie Fcmr−/− > Fcmr+/- > Fcmr+/+) [Citation4]. Since the affinity of human FcµR for pentameric IgM is ∼10 nM, which is much (∼100-fold) lower than the concentration of plasma IgM (∼1 µM), we initially* thought that FcµR on murine B cells in vivo must always be occupied with plasma IgM, and the IgM-bound FcµR should be internalized, rapidly retrieved from early endosomes and returned to the plasma membrane, thereby maintaining the plasma IgM concentration to a constant level, as do neonatal IgG FcR (FcRn) on endothelial cells and FcαR on blood phagocytes [Citation20–22]. [*Later, we learned that the IgM ligand binding activity between human and mouse FcµRs was distinct (see below).] However, the half-life of exogenously administered IgM was comparable in both Fcmr KO and WT mice, hence ruling out the above receptor recycling possibility [Citation3]. The increased serum level of natural IgM in Fcmr KO mice seems to be the consequence of more production of IgM by either enhanced differentiation from IgM B cells to plasma cells or an increased number of IgM secreting plasma cells. In supporting this, CD5+ B-1 (B-1a) B cells in the spleen [Citation3] and CD138+ plasmablats/plasma cells in the spleen and bone marrow [Citation7] are increased in the mutant mice. Notably, it has been shown that serum natural IgM levels in mice raised under germ-free conditions are similar to those in mice held under conventional or specific pathogen-free housing conditions [Citation23–25]. The natural IgM might be the consequence of exposure to self-antigens associated with cell corpses and B-1a B cells are a major source of such natural IgM antibodies [Citation26,Citation27], consistent with the findings of increases in splenic B-1a B cells in the spleen and serum levels of natural IgM in Fcmr KO mice. Among IgM-binding receptors [ie FcµR on B cells, pIgR on mucosal epithelial cells [Citation28], Fcα/μR on follicular dendritic cells and other non-hematopoietic cells [Citation29], FcµR is the sole receptor involved in serum IgM homeostasis [Citation30,Citation31].
Autoantibodies
In addition to the increase in pre-immune serum IgM levels, Fcmr-KO mice have also high titers of serum autoantibodies of both IgM and IgG isotypes against dsDNA, chromatin or other cellular components as compared with WT mice. FcµR thus plays an important regulatory role in the development of self-reactive B cells [Citation3–5,Citation7,Citation8]. In mice unable to secrete IgM due to the ablation of secretory exon (µs), they cannot control infections, because of inefficient induction of protective IgG antibody responses and also suffer from the autoimmune pathology, paradoxically associated with IgG autoantibodies, possibly because of impaired clearance of autoantigen-containing apoptotic cells [Citation27,Citation32,Citation33]. Given these precedents, we have hypothesized that the introduction of Fcmr null mutation onto autoimmune-prone mice may affect their autoimmune pathological outcomes depending on the balance of protective IgM vs pathologic IgG autoantibodies. We have chosen a Fas-deficient C57BL/6 mouse strain (B6.MRL Faslpr; hereafter B6/lpr for simplicity), which has a milder form of lupus-like disease compared with the original MRL Faslpr mice [Citation34–36]. FcµR(+) and FcµR(−) B6/lpr mice are thus generated by crossing Fcmr−/− B6 mice with autoimmune B6/lpr mice. FcµR(−) B6/lpr mice are found to have a rapid increase in serum autoantibody levels of IgM and, to a lesser extent, IgG isotypes against dsDNA at a relatively early age as compared with FcµR(+) B6/lpr mice, suggesting that FcµR deficiency affects the kinetics and production of autoantibodies [Citation37]. However, FcµR deficiency has no significant impacts on the autoimmune pathology associated with B6/lpr mice, including glomerulonephritis, the glomerular deposits of IgM, IgG, IgA, and C3, the renal function (as determined by blood urea nitrogen levels) and the mouse survival [Citation37]. This is contrasted with the inhibitory FcγRIIb; Fcgr2b-deficient B6/lpr mice develop severe systemic autoimmune diseases and die within 30–40 weeks of age [Citation38–40]. Results from subsequent studies on the contribution of chromosome 1 region around the Fcgr2b, particularly the major lupus susceptibility locus Sle16, to the severity of autoimmunity-associated pathology in comparing between 129 ES- and B6 ES-derived Fcgr2b−/− mice, have suggested that Fcgr2b is a modifier of autoimmune susceptibility [Citation41–44]. The finding of the B6-derived, but not 129-derived, Sle16 locus in our FcµR(−) B6/lpr mice may thus account for no impacts on the development of severe lupus-like autoimmune disease [Citation37].
Another intriguing finding comes from comparative assessments of serum autoantibody titers against Smith antigen (Sm) or ribonuclear protein (RNP) among four different groups of mice: FcµR(+) vs FcµR(−) B6 and FcµR(+) vs FcµR(−) B6/lpr mice [Citation37]. At 10-wk of age, IgM anti-Sm/RNP autoantibody titers are highest in FcµR(−) B6/lpr mice in the order of FcµR(−) B6/lpr ≫ FcµR(+) B6/lpr, FcµR(−) B6 > FcµR(+) B6, but later (at 20-wk of age) are elevated in three groups of mice in the order of FcµR(−) B6/lpr, FcµR(−) B6 > FcµR(+) B6/lpr ≫ FcµR(+) B6. By contrast, IgG anti-Sm/RNP autoantibody titers are elevated in the only B6/lpr mouse strain irrespective of the absence or presence of FcµR. Thus, FcµR deficiency alone clearly contributes to an increase of IgM anti-Sm/RNP autoantibodies and its early increase appears to result from the synergy of both deficiencies of Fcmr and Fas genes.
Marginal zone (MZ) B cells, innate-like B cells, are considered as a major cell type producing ant-Sm/RNP autoantibodies [Citation45–47]. Notably, the number of MZ B cells in FcµR(−) B6/lpr mice is markedly reduced as compared with that in FcµR(+) B6/lpr mice, the results being opposite to their serum levels of IgM anti-Sm/RNP autoantibodies [Citation37]. The marked reduction of MZ B cells may result from their rapid differentiation into plasma cells in the absence of FcµR [Citation3,Citation37]. Alternatively, FcµR-deficient MZ B cells may undergo cell death due to a lack of survival signals through FcµR upon cross-linkage with BCR [Citation48], as shown by cross-talk downstream of FcµR and BCR signaling via the non-canonical NFκB pathway [Citation49]. In µs KO mice, the number of MZ B cells is increased by 3-fold and this increase can be normalized by passive administration of natural polyclonal IgM [Citation50].
Collectively, FcµR deficiency affects the kinetics and production of autoantibodies against dsDNA or chromatins, but have no impact on the severity of lupus nephritis associated with B6/lpr mice. The latter finding is in agreement with the Sle16 locus of B6 origin in our mutant mice. FcµR is also involved in the regulation of the production of autoantibodies against the Sm/RNP antigen by MZ B cells.
Mott cells
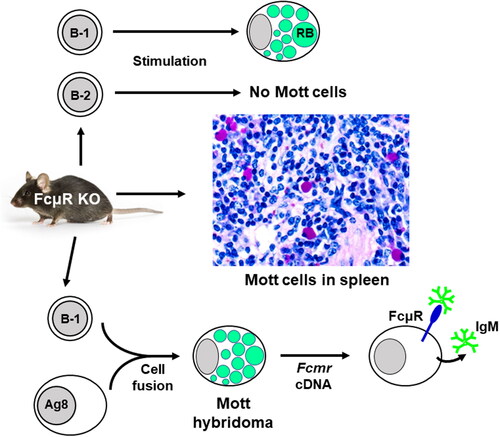
Mott cells, also called morular or grape cells, are bizarre plasma cells containing Ig-inclusion bodies termed Russell bodies within the cisterna of dilated rough ER and represent a cellular response to the accumulation of abundant non-degradable Igs that fail to exit from the ER, like “constipated” plasma cells [Citation51–53]. Mott cells are rarely detected in normal tissues but are frequently observed in various pathological conditions including autoimmune disorders, B-cell neoplasms, and chronic infections [Citation51,Citation53–56]. Remarkably, significant increases in Mott cells in spleen and lymph node tissues as defined by strong staining with a periodic acid-Schiff (PAS) reagent are demonstrated in three different strains of Fcmr KO mice as compared with their control mice [Citation37,Citation57]. Furthermore, splenic B cells from the mutant mice clearly generate more Mott cells than those from their WT mice, when stimulated ex vivo with lipopolysaccharide (LPS) alone or a B-1 B cell activation cocktail (LPS/dextran-anti-IgD/IL-4/IL-5), but not with a B-2 B cell activation cocktail (anti-CD40/dextran-anti-IgD/IL-4/IL-5) (). A single IgMλ+ Mott hybridoma clone developed from splenic B-1a B cells of mutant mice has a nearly identical VH or a completely identical Vλ gene to the corresponding germline gene segments, along with the addition of several nucleotides at the VH/DH and DH/JH junctions, consistent with the characteristics of B-1a B cell-derived natural IgM. Transduction of FcµR cDNA into this FcµR-negative IgMλ Mott hybridoma results in a reduction of cells containing IgM-inclusion bodies with concomitant enhancement in IgM secretion and binding of the secreted IgM to FcµR expressed on the Mott transductant [Citation57]. Thus, these findings indicate a regulatory role of FcµR in the formation of Mott cells and IgM-inclusion bodies.
Figure 1. Enhanced Mott cell formation in FcµR-deficient mice. Middle: Mott cells in splenic and lymph node (not shown) tissues from three different strains of Fcmr KO mice as defined by their strong purple staining with PAS reagent are significantly increased as compared with their WT control mice. Bottom: Splenic CD5+B220+ B-1a B cells (106 cells) enriched from Fcmr KO and WT control mice are activated with LPS before fusing with Ag8.653 cells (Ig non-producing plasmacytoma). One IgMλ+ Mott hybridoma clone containing Russel bodies (green) is generated from the total 16 Fcmr-deficient hybridomas, whereas none from the total 6 WT hybridomas. When FcµR cDNA is transduced into the Mott hybridoma, the number of cells containing IgM-inclusion bodies is reduced, the secretion of pentameric IgM (green) in culture media is increased, and secreted IgM binds FcµR (blue) expressed on the plasma membrane. None of these changes are observed when the empty vector as a control is transduced into the Mott hybridoma (not shown). Top: Splenic B cells from Fcmr KO and WT mice are stimulated for 4 d with LPS alone, B-1 (LPS/dextran-anti-IgD/IL-4/IL-5) or B-2 cocktail (anti-CD40/dextran-anti-IgD/IL-4/IL-5). Mott cells are clearly generated in the mutant B cell cultures than the WT control B cell cultures when stimulated with LPS alone or the B-1 stimulation cocktail, but not with the B-2 stimulation cocktail.
As to the mechanisms of the FcµR-mediated regulation of Mott cell formation, we have initially proposed the following model based on the precedents of the propensity to produce autoantibodies in Fcmr KO mice [Citation3–5,Citation7,Citation8] as well as of the predominant interaction of FcμR with IgM ligands in cis (over in trans) on the same cell surface [Citation1,Citation9,Citation58]. A given B-cell expresses an IgM BCR with self-reactivity to an intracellular membrane component but may not interact with the corresponding antigen because of its low affinity. When the cell receives a certain signal to switch from the μm to μs exon usage (eg via a Toll-like receptor), along with the synthesis of the J chain, during the translocation from the ER to the Golgi, the resultant pentameric IgM is accumulated inside the vesicles, where it binds its cognate membrane antigen via the Fab regions and to FcμR via its Fc portions. This cis engagement of self-antigen/secreted pentameric IgM/FcμR within the vesicles may prevent further development of such autoreactive B cells, thereby contributing to peripheral tolerance. IgM-inclusion bodies in Mott cells are byproducts in the absence of FcμR [Citation9,Citation37,Citation57]. Since the identification of intracellular membrane antigen recognized by the aforementioned Mott hybridoma IgMλ has defied [Citation57], we have not yet formally validated this model. Another possibility is that FcµR may help release of membrane vesicles carrying cognate membrane antigen/pentameric IgM autoantibody/FcµR to extracellular space as exosomes. There is a precedent in a transgenic mouse model that certain autoreactive B-cell hybridomas accumulate IgM in the Golgi, like Mott hybridomas, due to the formation of immune complexes between IgM and glycosaminoglycan and release large (up to 2 µm in diameter) spherical IgM complexes, termed spherons, into media [Citation59]. Whatever the molecular bases are, Mott cell formation is linked to the FcµR deficiency.
Many different factors have also been implicated in the formation of Mott cells. These include: (i) structural alterations of Ig heavy chains (eg their truncation at Cµ1 domain) to prevent their appropriate processing, (ii) impairment of Ig light chains to prevent Ig heavy chain aggregation as shown in their deficient mice, (iii) inability to degrade or to export Ig, leading to its aggregation, and (iv) deficiency of Src homology 2 domain-containing protein tyrosine phosphatase SHP1/PTP6 (motheaten mice or B cell-specific Ptp6 KO mice) [Citation54,Citation60–64]. By analysis of NZB/W F1 x NZW backcross mice, the locus contributing to Mott cell formation, called Mott-1, is mapped to a satellite marker locus between Mit48 and Mit70 on chromosome 4 of NZB mice, which is in close proximity to the locus for IgM hypergammaglobulinemia Imh-1 [Citation65]. Thus, multiple genes or loci, including Fcmr on chromosome 1, are apparently involved in the formation of Mott cells and Ig-inclusion bodies.
Dysregulated humoral immune responses
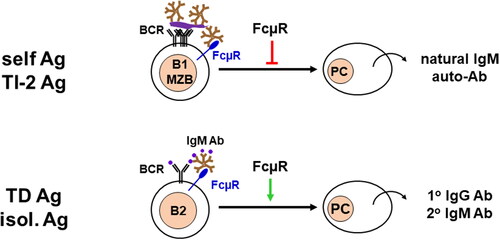
Although there are some discrepancies in antibody responses to T cell-independent (TI) or -dependent (TD) antigens among different strains of Fcmr KO mice that may be due to differences in mouse ages, antigen doses and forms, administration routes, kinetics, targeting exons, breeding environments including commensal bacteria, and others [Citation3–5,Citation7]. However, the mutant mice generally exhibit enhanced TI type 2 immune responses for multiple cross-linkage of BCRs but impaired TD responses for limited cross-linkage of BCRs, particularly at suboptimal doses of antigens [Citation3]. Given the findings that similar selective enhancement of TI-2 immune responses is also observed in mice deficient for IgM secretion (µs KO) or components of the BCR complex (eg Cd19 KO or Cd81 KO) [Citation33,Citation66,Citation67], FcµR seems to regulate B cell responses to TI-2 and TD antigens by interacting differently with BCR complexes on the plasma membrane (). FcµR inhibits immune responses when multiple IgM BCRs are cross-linked with repetitive epitopes on particulate antigens or cell corpses, whereas FcµR stimulate immune responses when a few BCRs are ligated with limited epitopes on isolated proteins.
Figure 2. Dysregulated antibody responses. FcµR plays a distinct regulatory role in immune responses depending on the forms of antigens. For cell corpse containing self-antigens, particulate bacteria-associated polysaccharides, and proteins, or TI type2 antigens, FcµR inhibits their antibody reposes as the consequence of multiple BCR cross-linkages (top). On the other hand, for isolated proteins or TD antigens, FcµR enhances the antibody responses as the result of a limited number of BCR cross-linkages.
Potential clues for identification of individuals with FCMR deficiency
FCMR deficiency in humans has not yet been identified but, if it exists, the clinical manifestations may be much more complex and profound than those in Fcmr KO mice, because additional cell types (ie T and NK lymphocytes) express FcµR in humans. Notwithstanding, some aspects seen in the mutant mice may also be preserved in the individuals affected with FCMR deficiency. They may include: (i) no (or markedly diminished) cell-surface expression of FcµR by lymphocytes (B, T, and NK cells), (ii) elevated serum levels of IgM, but not IgG and IgA, like hyper-IgM syndrome [Citation68,Citation69], (iii) autoimmune manifestations such as increased serum autoantibody titers of IgM and IgG isotypes, (iv) dysregulated humoral immune responses to certain microorganisms depending on antigenic epitopes (isolated vs repetitive) [Citation70], (v) generation of Mott cells in pokeweed mitogen-activated blood mononuclear cells ex vivo [Citation71], and (vi) no apparent abnormalities in complete blood counts with differential and in T and B lymphocyte populations. Certain abnormalities in cellular immunity in FCMR-deficient individuals against viral infection are also predicted depending on the function of FcµR on T and NK cells. There are several findings of FcµRs on non-B cells in humans. FcµR on T and NK cells are strongly down-modulated upon IL-2 stimulation in vitro, consistent with the findings that the cell surface FcµR levels on freshly prepared effector memory T cells are much lower than naive T cells [Citation72,Citation73]. The binding of IgM to FcµR on NK cells initiates intracellular signals but does not mediate NK cell cytotoxicity [Citation72]. FcµR-mediated IgM uptake by T cells enhances their surface expression of TCR and co-stimulatory molecules, thereby facilitating T cell activation particularly when antigen concentrations are low [Citation74]. Because of limited findings of FcµR on T and NK cells in humans, it is too difficult to speculate the potential abnormalities of these cell types in FCMR-deficient individuals. Given the fact that FcµR is expressed by Treg cells in humans [Citation73], it is conceivable that FCMR deficiency may affect not only systemic but also organ-specific autoimmune disorders.
Furthermore, seven aa residues of human FcµR interacting with IgM ligands are now identified by crystallographic and cryo-EM analyses, ie Arg45 in the CDR1, Thr57, and Thr60 in the C’ strand, Phe67 and Lys69 in the CDR2, Thr110, and Asp111 in the CDR3 [Citation16,Citation17]. (The numbers indicate the aa position from the first Met residue.) The two residues of Phe67 and Thr110 turn out to be immediately adjacent to the Asn66 and Asn109 residues previously identified by our site-directed mutagenesis based on the difference in IgM binding between human (constitutive binding) and mouse (transient binding) FcµRs [Citation75,Citation76]. The above seven aa residues are found to interact with five aa residues in the Cµ4 domain of the human IgM molecule at positions 465-468 (Asn-Leu-Arg-Glu) and position 562 (Glu) in the α helices [Citation16,Citation17]. (The numbers in IgM indicate the aa position from the first N-terminal aa residue of the Cµ1 domain.) It remains to be elucidated if a homozygous missense mutation of each of these aa residues affects the interaction of FcµR with IgM ligands, although it was the case for the identification of Asp66 and Asn109 of FcµR by site-directed mutagenesis [Citation75,Citation76]. The importance of the homozygous state is suggested by the genotype analysis of FcγRIIa that loss of the His131 allele in both chromosomes is a high heritable risk factor for lupus nephritis in African American [Citation77].
A potential model for FcµR-mediated regulation of self-reactive B cells
Two-thirds of newly generated immature B cells in the bone marrow express a self-reactive IgM BCR [Citation78], and the majority of such autoreactive immature B cells die probably as a consequence of this autoantigen recognition [Citation79]. This is consistent with the earlier findings that neonatal injection of anti-IgM antibodies induces agammaglobulinemia in mice and chickens [Citation80]. The immune system develops several regulatory mechanisms to eliminate or inactivate the self-reactivity of BCR in the bone marrow, called “central” tolerance. If immature B cells express IgM with high avidity for self-antigens, they are aborted by apoptosis (clonal deletion). Alternatively, they change the antigen specificity of IgM by another or several attempt(s) of Ig light chain gene rearrangement (receptor editing). If immature B cells express IgM with low avidity for self-antigens, they down-modulate the cell surface density and signaling of IgM BCR to become functional unresponsiveness (anergy). If immature B cells are not reactive with self-antigens in the bone marrow microenvironment, they are ignored by this repertoire-selecting process and exit the bone marrow to enter the peripheral mature B cell pools [Citation79,Citation81–83]. A number of genes and proteins in mice and humans have been implicated to be involved in these processes of central B cell tolerance (please see Table 1 in ref. [Citation82]), including phosphoinositide 3-kinase (Pi3k) [Citation84–86], phosphatase and tension homolog (Pten) [Citation87–89], and recombination activating gene 1 (RAG1) [Citation90]. Several additional genes are subsequently identified to affect B cell tolerance, including chemokine CXC receptor 4 (CXCR4) [Citation91], lysosomal protein transmembrane 5 (Laptm5) [Citation92], and interferon α receptor (Ifnar) [Citation93].
As to the potential mechanisms for the contribution of FcµR to central B cell tolerance, we have proposed the following model [Citation94]. In immature B cells in the bone marrow, self-reactive IgM BCR may predominantly interact with FcµR containing ITT motifs in two ways, based on the findings of the predominant binding of FcµR with IgM in cis on the same cell surface than in trans between neighboring cells [Citation9,Citation58] and of the opsonization activity of natural IgM. In one way, self-antigens (eg cell corpse) opsonised by polyreactive natural IgM are able to bind both IgM BCR via its self-reactivity and FcµR via the Fc portion of natural IgM on the surface of immature B cells (IgM BCR/self-antigen/natural IgM/FcµR). In another way, according to the predicted interaction between IgM BCR and FcµR based on the recent structural analyses [Citation16,Citation17], IgM on immature B cells binds a free form of self-antigens (without IgM opsonization) and the resultant cross-linked IgM BCRs facilitate to bind FcµR via the Cµ4 domain of IgM BCR (self-antigen/IgM BCR/FcµR). In either case, upon recognition of self-antigens, immature B cells are able to receive both signals from the BCR complex (Igα/ß and CD19) and FcµR. In immature B cells, PI3K kinase activity is balanced by its antagonist PTEN phosphatase, and the co-engagement of BCR and FcµR up-regulate PTEN activity to convert phosphatidylinositol 3,4,5-triphosphate (PIP3) to phosphatidylinositol 4,5-biphosphate (PIP2), prevents the phosphorylation of AKT Ser/Thr kinase, thereby allowing the transcription factor of forkhead box translocation factor O1 (FOXO1) to translocate into nucleus, where FOXO1 upregulates RAG1/RAG2 expression, leading to receptor editing or clonal deletion. The signal-amplifying ITT motif of FcµR is likely to affect the signal strength and biological outcome of the self-reactive IgM BCR following cross-linkage of the two receptors. In fact, the situation is structurally reminiscent to that of IgG or IgE memory-type BCR, which have the ITT motif in their cytoplasmic tails [Citation18]. The ubiquitously expressed adaptor protein Grb2 integrates signals as the proximal ITT signal effector from the outside of cells to intracellular signaling pathways via its central SH2 and the two flanking SH3 domains and is known to play a pivotal role in the negative or/and positive selection(s) in the thymus [Citation95,Citation96]. Further studies are required to unravel the intricate signal crosstalk between IgM BCR and FcµR and whether it may also contribute to central B cell tolerance.
It remains unclear how FcµR is involved in the selection of self-reactive B-1 B cells in the fetal liver (FL) and why splenic B-1a B cells are increased in Fcmr KO mice. During ontogeny, hematopoietic stem and progenitor cells undergo a functional switch from FL to adult bone marrow (BM) sites as hallmarked by reducing their self-renewal activity and becoming quiescent. Fetal-specific RNA-binding protein Lin28b is identified as a post-transcriptional molecular switch and the Lin28b and Let-7 microRNA are differentially expressed in FL and adult BM [Citation97,Citation98]. Developing B cells are positively and negatively selected by self-antigens, and the Lin28b and Let-7 axis is also involved in fetal and adult B lymphopoieses. Ectopic provision of Lin28b at adult pro-B cells can redirect them to fetal B-1 B cell development. Conversely, expression of Let-7 in fetal pro-B cells can switch to the conventional B-2 B cell development [Citation97,Citation98]. Two transcription factors, Arid3a (or Bright) and Bhlhe41, are found to be key regulators for promoting fetal B lymphopoiesis through the Lin28b-Let-7 axis [Citation99,Citation100]. The Arid3a and Bhlhe41 mRNAs contain multiple Let-7 target motifs, supporting the involvements of these molecules (Lin28b-Let-7-Arid3/Bhlhe41) in fetal lymphopoiesis. Intact BCR signaling is required for the generation of B-1 B cells from the Lin28b-induced BM-derived progenitors [Citation99]. Although the surface expression of FcµR by FL B cells has never been assessed, if expressed, it will be curious to know how FcµR interacts with BCR on FL B cells under possibly different ranges of self-antigens and lower concentrations of natural IgM compared with BM microenvironments. (According to the heat map data [Citation99], Fcmr (or Faim3) transcripts of pro-B cells in FL are significantly less as compared with those in adult BM.)
As to the increase in splenic B-1a B cells observed in Fcmr KO mice, altered expressions of many genes, especially involved in BCR signaling, are found to affect the B-1 B cell compartment. For example, weaken BCR signaling causes the lack of or reduced number of B-1 B cells, including the ablation of Cd19, complement receptor 2 (Cr2; Cd21), Bruton tyrosine kinase (Btk), Pi3k p85α subunit, guanine nucleotide exchange factor (Vav), and phosphatidylinositol-specific phospholipase Cγ2 (Plcg2) [Citation66,Citation101–108]. Conversely, strengthen BCR signaling causes an increase in B-1 B cells such as over-expression of CD19, ablation of inhibitory receptors with ITIM [eg Cd22 and paired Ig-like receptor B (Pirb)], and Ptp6/Shp1 [Citation54,Citation64,Citation109–111]. Notably, several other genes also affect the B-1 B cell development. For example, B cell-specific deletion of Ser/Thr kinase Tak1 (Map3k7) impairs the B-1 B cell development possibly due to increased susceptibility to cell death, but enhances the MZ B cell development resulted from increased NF-κB2 activity [Citation112]. ER-associated Kelch-like protein 14 (Klhl14) promotes the development of B-1a B cells but inhibits that of B-1b B cells [Citation113]. Transcription factor, the nuclear factor of activated T cells calcineurin-dependent 1 (Nfatc1), is essential for the B-1a B cell development in both the peritoneal cavity and spleen [Citation114]. Paralogous zinc finger transcription factors, early growth response 1 and 2 (EGR1/2), are induced by chronic activation of BCR with self-antigens through Ca2+- and calcineurin-activated NFAT transcription factor and regulate the developments of B-1 B cells and CD21low age-associated B cells as determined by B cell-specific deletion of Egr1/2, suggesting the EGR1/2 as a tolerance checkpoint [Citation115]. Collectively, further studies are required to determine how BCR and FcµR signals crosstalk with the aforementioned potential signaling pathways to regulate the development of self-reactive B cells and to contribute to B cell tolerance.
Acknowledgements
We thank Dr. Paolo Casali for providing this opportunity, Dr. Jürgen Wienands for valuable comments, Mr. Hilmar Fünning for literature support, and Mr. Hilmar Frank for informatics support. Some data described in this essay were indebted to the collaboration with Drs. Yusuke Suzuki, Hiroshi Ohno, Yoshiki Kubagawa, Christopher Skopnik, Uwe Klemn, Kyeong-Hee Lee, Nicole Baumgarth, Nikles Engels, and Jürgen Wienands.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Kubagawa H, Oka S, Kubagawa Y, et al. Identity of the elusive IgM Fc receptor (FcμR) in humans. J Exp Med. 2009;206(12):1–9.
- Shima H, Takatsu H, Fukuda S, et al. Identification of TOSO/FAIM3 as an Fc receptor for IgM. Int Immunol. 2010;22(3):149–156.
- Honjo K, Kubagawa Y, Jones DM, et al. Altered Ig levels and antibody responses in mice deficient for the Fc receptor for IgM (FcµR). Proc Natl Acad Sci U S A. 2012;109(39):15882–15887.
- Ouchida R, Mori H, Hase K, et al. Critical role of the IgM Fc receptor in IgM homeostasis, B-cell survival, and humoral immune responses. Proc Natl Acad Sci U S A. 2012;109(40):E2699–E2706.
- Choi SC, Wang H, Tian L, et al. Mouse IgM Fc receptor, FCMR, promotes B cell development and modulates antigen-driven immune responses. J Immunol. 2013;190(3):987–996.
- Wang H, Coligan JE, Morse HC. Emerging functions of natural IgM and its Fc receptor FCMR in immune homeostasis. Front Immunol. 2016;7:99.
- Nguyen TTT, Kläsener K, Zürn C, et al. The IgM receptor FcµR limits tonic BCR signaling by regulating expression of the IgM BCR. Nat Immunol. 2017;18(3):321–333.
- Yu J, Duong VHH, Westphal K, et al. Surface receptor Toso controls B cell-mediated regulation of T cell immunity. J Clin Invest. 2018;128(5):1820–1836.
- Kubagawa H, Honjo K, Ohkura N, et al. Functional roles of the IgM Fc receptor in the immune system. Front Immunol. 2019;10:945.
- Liu J, Wang Y, Xiong E, et al. Role of the IgM Fc receptor in immunity and tolerance. Front Immunol. 2019;10:529.
- Nguyen XH, Lang PA, Lang KS, et al. Toso regulates the balance between apoptotic and nonapoptotic death receptor signaling by facilitating RIP1 ubiquitination. Blood. 2011;118(3):598–608.
- Lang KS, Lang PA, Meryk A, et al. Involvement of Toso in activation of monocytes, macrophages, and granulocytes. Proc Natl Acad Sci U S A. 2013;110(7):2593–2598.
- Rochereau N, Michaud E, Waeckel L, et al. Essential role of TOSO/FAIM3 in intestinal IgM reverse transcytosis. Cell Rep. 2021;37(7):110006.
- Anania JC, Westin A, Adler J, et al. A novel image analysis approach reveals a role for complement receptors 1 and 2 in follicular dendritic cell organization in germinal centers. Front Immunol. 2021;12:655753.
- Hopp CS, Sekar P, Diouf A, et al. Plasmodium falciparum-specific IgM B cells dominate in children, expand with malaria, and produce functional IgM. J Exp Med. 2021;218(4):e20200901.
- Li Y, Shen H, Zhang R, et al. Immunoglobulin M perception by FcµR. Nature. 2023;615(7954):907–912.
- Chen Q, Menon RP, Masino L, et al. Structural basis for Fc receptor recognition of immunoglobulin M. Nat Struct Mol Biol. 2023;30(7):1033–1039.
- Engels N, König LM, Heemann C, et al. Recruitment of the cytoplasmic adaptor Grb2 to surface IgG and IgE provides antigen receptor-intrinsic costimulation to class-switched B cells. Nat Immunol. 2009;10(9):1018–1025.
- Kubagawa H, Clark C, Skopnik CM, et al. Physiological and pathophysiological roles of IgM Fc receptor (FcµR) isoforms. Int J Mol Sci. 2023;24(6):5728.
- Mellman I, Warren G. The road taken: past and future foundations of membrane traffic. Cell. 2000;100(1):99–112.
- Ghetie V, Ward ES. Multiple roles for the major histocompatibility complex class I- related receptor FcRn. Annu Rev Immunol. 2000;18(1):739–766.
- Stewart WW, Kerr MA. The binding of monomeric IgA to myeloid FcαR: evidence for receptor re-cycling and determination of its affinity. Adv Exp Med Biol. 1995;371A:655–658.
- Hashimoto K, Handa H, Umehara K, et al. Germfree mice reared on an “antigen-free” diet. Lab Anim Sci. 1978;28(1):38–45.
- Haury M, Sundblad A, Grandien A, et al. The repertoire of serum IgM in normal mice is largely independent of external antigenic contact. Eur J Immunol. 1997;27(6):1557–1563.
- Thurnheer MC, Zuercher AW, Cebra JJ, et al. B1 cells contribute to serum IgM, but not to intestinal IgA, production in gnotobiotic Ig allotype chimeric mice. J Immunol. 2003;170(9):4564–4571.
- Kim SJ, Gershov D, Ma X, et al. I-PLA(2) activation during apoptosis promotes the exposure of membrane lysophosphatidylcholine leading to binding by natural immunoglobulin M antibodies and complement activation. J Exp Med. 2002;196(5):655–665.
- Ehrenstein MR, Notley CA. The importance of natural IgM: scavenger, protector and regulator. Nat Rev Immunol. 2010;10(11):778–786.
- Kaetzel CS. The polymeric immunoglobulin receptor: bridging innate and adaptive immune responses at mucosal surfaces. Immunol Rev. 2005;206(1):83–99.
- Kikuno K, Kang DW, Tahara K, et al. Unusual biochemical features and follicular dendritic cell expression of human Fcα/µ receptor. Eur J Immunol. 2007;37(12):3540–3550.
- Shimada S, Kawaguchi-Miyashita M, Kushiro A, et al. Generation of polymeric immunoglobulin receptor-deficient mouse with marked reduction of secretory IgA. J Immunol. 1999;163(10):5367–5373.
- Honda S, Kurita N, Miyamoto A, et al. Enhanced humoral immune responses against T-independent antigens in fcα/µR-deficient mice. Proc Natl Acad Sci U S A. 2009;106(27):11230–11235.
- Boes M, Prodeus AP, Schmidt T, et al. A critical role of natural immunoglobulin M in immediate defense against systemic bacterial infection. J Exp Med. 1998;188(12):2381–2386.
- Ehrenstein MR, O’Keefe TL, Davies SL, et al. Targeted gene disruption reveals a role for natural secretory IgM in the maturation of the primary immune response. Proc Natl Acad Sci U S A. 1998;95(17):10089–10093.
- Cohen PL, Eisenberg RA. Lpr and gld: single gene models of systemic autoimmunity and lymphoproliferative disease. Annu Rev Immunol. 1991;9(1):243–269.
- Suda T, Nagata S. Why do defects in the Fas-Fas ligand system cause autoimmunity? J Allergy Clin Immunol. 1997;100(6 Pt 2):S97–S101.
- Izui S, Kelley VE, Masuda K, et al. Induction of various autoantibodies by mutant gene lpr in several strains of mice. J Immunol. 1984;133(6):3010–3014.
- Honjo H, Kubagawa Y, Suzuki Y, et al. Enhanced autoantibody and Mott cell formation in FcµR-decient autoimmune mice. Int Immunol. 2014;26(12):659–672.
- Bolland S, Ravetch JV. Spontaneous autoimmune disease in FcγRIIB-deficient mice results from strain-specific epistasis. Immunity. 2000;13(2):277–285.
- Yajima K, Nakamura A, Sugahara A, et al. FcγRIIB deficiency with Fas mutation is sufficient for the development of systemic autoimmune disease. Eur J Immunol. 2003;33(4):1020–1029.
- McGaha TL, Karlsson MC, Ravetch JV. FcγRIIB deficiency leads to autoimmunity and a defective response to apoptosis in mrl-MpJ mice. J Immunol. 2008;180(8):5670–5679.
- Carlucci F, Cortes-Hernandez J, Fossati-Jimack L, et al. Genetic dissection of spontaneous autoimmunity driven by 129-derived chromosome 1 loci when expressed on C57BL/6 mice. J Immunol. 2007;178(4):2352–2360.
- Fossati-Jimack L, Cortes-Hernandez J, Norsworthy PJ, et al. Regulation of B cell tolerance by 129-derived chromosome 1 loci in C57BL/6 mice. Arthritis Rheum. 2008;58(7):2131–2141.
- Boross P, Arandhara VL, Martin-Ramirez J, et al. The inhibiting Fc receptor for IgG, FcγRIIB, is a modifier of autoimmune susceptibility. J Immunol. 2011;187(3):1304–1313.
- Sato-Hayashizaki A, Ohtsuji M, Lin Q, et al. Presumptive role of 129 strain-derived Sle16 locus in rheumatoid arthritis in a new mouse model with fcγ receptor type IIb-deficient C57BL/6 genetic background. Arthritis Rheum. 2011;63(10):2930–2938.
- Qian Y, Conway KL, Lu X, et al. Autoreactive MZ and B-1 B-cell activation by faslpr is coincident with an increased frequency of apoptotic lymphocytes and a defect in macrophage clearance. Blood. 2006;108(3):974–982.
- Clarke SH. Anti-Sm B cell tolerance and tolerance loss in systemic lupus erythematosus. Immunol Res. 2008;41(3):203–216.
- Kishi Y, Higuchi T, Phoon S, et al. Apoptotic marginal zone deletion of anti-Sm/ribonucleoprotein B cells. Proc Natl Acad Sci U S A. 2012;109(20):7811–7816.
- Liu J, Zhu H, Qian J, et al. Fcµ receptor promotes the survival and activation of marginal zone B cells and protects mice against bacterial sepsis. Front Immunol. 2018;9:160.
- Ouchida R, Lu Q, Liu J, et al. FcµR interacts and cooperates with the B cell receptor to promote B cell survival. J Immunol. 2015;194(7):3096–3101.
- Baker N, Ehrenstein MR. Cutting edge: selection of B lymphocyte subsets is regulated by natural IgM. J Immunol. 2002;169(12):6686–6690.
- Bangle R. A morphologic and histochemical study of cytoplasmic Russell bodies in cancer cells. Am J Pathol. 1963;43(3):437–448.
- Alanen A, Pira U, Lassila O, et al. Mott cells are plasma cells defective in immunoglobulin secretion. Eur J Immunol. 1985;15(3):235–242.
- Bain BJ. Russell bodies and mott cells. Am J Hematol. 2009;84(8):516–516.
- Shultz LD, Coman DR, Lyons BL, et al. Development of plasmacytoid cells with russell bodies in autoimmune “viable motheaten” mice. Am J Pathol. 1987;127(1):38–50.
- Maldonado JE, Brown AL, Bayrd ED, et al. Cytoplasmic and intranuclear electron-dense bodies in the myeloma cell. Arch Pathol. 1966;81(6):484–500.
- Pizzolitto S, Camilot D, DeMaglio G, et al. Russell body gastritis: expanding the spectrum of Helicobacter pylori - related diseases? Pathol Res Pract. 2007;203(6):457–460.
- Mahmoudi Aliabadi P, Al-Qaisi K, Jani PK, et al. Enhanced mott cell formation linked with IgM Fc receptor (FcµR) deficiency. Eur J Immunol. 2023;53(7):e2250315.
- Honjo K, Kubagawa Y, Kearney JF, et al. Unique ligand-binding property of the human IgM Fc receptor. J Immunol. 2015;194(4):1975–1982.
- Khan SN, Cox JV, Nishimoto SK, et al. Intra-Golgi formation of IgM-glycosaminoglycan complexes promotes Ig deposition. J Immunol. 2011;187(6):3198–3207.
- Kopito RR, Sitia R. Aggresomes and Russell bodies. Symptoms of cellular indigestion? EMBO Rep. 2000;1(3):225–231.
- Decourt C, Galea HR, Sirac C, et al. Immunologic basis for the rare occurrence of true nonsecretory plasma cell dyscrasias. J Leukoc Biol. 2004;76(3):528–536.
- Mattioli L, Anelli T, Fagioli C, et al. ER storage diseases: a role for ERGIC-53 in controlling the formation and shape of Russell bodies. J Cell Sci. 2006;119(Pt 12):2532–2541.
- Corcos D, Osborn MJ, Matheson LS, et al. Immunoglobulin aggregation leading to Russell body formation is prevented by the antibody light chain. Blood. 2010;115(2):282–288.
- Pao LI, Lam KP, Henderson JM, et al. B cell-specific deletion of protein-tyrosine phosphatase Shp1 promotes B-1a cell development and causes systemic autoimmunity. Immunity. 2007;27(1):35–48.
- Jiang Y, Hirose S, Hamano Y, et al. Mapping of a gene for the increased susceptibility of B1 cells to Mott cell formation in murine autoimmune disease. J Immunol. 1997;158(2):992–997.
- Sato S, Steeber DA, Tedder TF. The CD19 signal transduction molecule is a response regulator of B-lymphocyte differentiation. Proc Natl Acad Sci U S A. 1995;92(25):11558–11562.
- Tsitsikov EN, Gutierrez-Ramos JC, Geha RS. Impaired CD19 expression and signaling, enhanced antibody response to type II T independent antigen and reduction of B-1 cells in CD81-deficient mice. Proc Natl Acad Sci U S A. 1997;94(20):10844–10849.
- Yazdani R, Fekrvand S, Shahkarami S, et al. The hyper IgM syndromes: epidemiology, pathogenesis, clinical manifestations, diagnosis and management. Clin Immunol. 2019;198:19–30.
- Barbouche MR, Chen Q, Carbone M, et al. Comprehensive review of autoantibodies in patients with hyper-IgM syndrome. Cell Mol Immunol. 2018;15(6):610–617.
- Chattopadhyay G, Chen Q, Colino J, et al. Intact bacteria inhibit the induction of humoral immune responses to bacterial-derived and heterologous soluble T cell-dependent antigens. J Immunol. 2009;182(4):2011–2019.
- Posnett DN, Mouradian J, Mangraviti DJ, et al. Mott cells in a patient with a lymphoproliferative disorder. Differentiation of a clone of B lymphocytes into mott cells. Am J Med. 1984;77(1):125–130.
- Murakami Y, Narayanan S, Su S, et al. Toso, a functional IgM receptor, is regulated by IL-2 in T and NK cells. J Immunol. 2012;189(2):587–597.
- Kubagawa H, Oka S, Kubagawa Y, et al. The long elusive IgM Fc receptor, FcµR. J Clin Immunol. 2014;34(1):S35–S45.
- Meryk A, Pangrazzi L, Hagen M, et al. Fcµ receptor as a costimulatory molecule for T cells. Cell Rep. 2019;26(10):2681–2691 e5.
- Skopnik CM, Al-Qaisi K, Calvert RA, et al. Identification of amino acid residues in human IgM Fc receptor (FcµR) critical for IgM binding. Front Immunol. 2020;11:618327.
- Kubagawa H, Skopnik CM, Al-Qaisi K, et al. Differences between human and mouse IgM Fc receptor (FcµR). Int J Mol Sci. 2021;22(13):7024.
- Salmon JE, Millard S, Schachter LA, et al. Fc gamma RIIA alleles are heritable risk factors for lupus nephritis in African Americans. J Clin Invest. 1996;97(5):1348–1354.
- Wardemann H, Yurasov S, Schaefer A, et al. Predominant autoantibody production by early human B cell precursors. Science. 2003;301(5638):1374–1377.
- Melchers F. Checkpoints that control B cell development. J Clin Invest. 2015;125(6):2203–2210.
- Lawton AR, Cooper MD. Modification of B lymphocyte differentiation by anti-immunoglobulins. Contemp Top Immunobiol. 1974;3:193–225.
- Healy JI, Goodnow CC. Positive versus negative signaling by lymphocyte antigen receptors. Annu Rev Immunol. 1998;16(1):645–670.
- Nemazee D. Mechanisms of central tolerance for B cells. Nat Rev Immunol. 2017;17(5):281–294.
- Nemazee D, Weigert M. Revising B cell receptors. J Exp Med. 2000;191(11):1813–1817.
- Llorian M, Stamataki Z, Hill S, et al. The PI3K p110delta is required for down-regulation of RAG expression in immature B cells. J Immunol. 2007;178(4):1981–1985.
- Verkoczy L, Duong B, Skog P, et al. Basal B cell receptor-directed phosphatidylinositol 3-kinase signaling turns off RAGs and promotes B cell-positive selection. J Immunol. 2007;178(10):6332–6341.
- Srinivasan L, Sasaki Y, Calado DP, et al. PI3 kinase signals BCR-dependent mature B cell survival. Cell. 2009;139(3):573–586.
- Cheng S, Hsia CY, Feng B, et al. BCR-mediated apoptosis associated with negative selection of immature B cells is selectively dependent on Pten. Cell Res. 2009;19(2):196–207.
- Benhamou D, Labi V, Novak R, et al. A c-Myc/miR17-92/Pten axis controls PI3K-mediated positive and negative selection in B cell development and reconstitutes CD19 deficiency. Cell Rep. 2016;16(2):419–431.
- Gonzalez-Martin A, Adams BD, Lai M, et al. The microRNA miR-148a functions as a critical regulator of B cell tolerance and autoimmunity. Nat Immunol. 2016;17(4):433–440.
- Walter JE, Rucci F, Patrizi L, et al. Expansion of immunoglobulin-secreting cells and defects in B cell tolerance in rag-dependent immunodeficiency. J Exp Med. 2010;207(7):1541–1554.
- Alves da Costa T, Peterson JN, Lang J, et al. Central human B cell tolerance manifests with a distinctive cell phenotype and is enforced via CXCR4 signaling in hu-mice. Proc Natl Acad Sci USA. 2021;118(16):e2021570118.
- Wang Y, Liu J, Akatsu C, et al. LAPTM5 mediates immature B cell apoptosis and B cell tolerance by regulating the WWP2-PTEN-AKT pathway. Proc Natl Acad Sci U S A. 2022;119(36):e2205629119.
- Ferri DM, Nassar C, Manion KP, et al. Elevated levels of interferon-α act directly on B cells to breach multiple tolerance mechanisms promoting autoantibody production. Arthritis Rheumatol. 2023;75(9):1542–1555.
- Jani PK, Kubagawa H, Melchers F. A rheostat sets B-cell receptor repertoire selection to distinguish self from non-self. Curr Opin Immunol. 2020;67:42–49.
- Lowenstein EJ, Daly RJ, Batzer AG, et al. The SH2 and SH3 domain-containing protein GRB2 links receptor tyrosine kinases to ras signaling. Cell. 1992;70(3):431–442.
- Jang IK, Zhang J, Chiang YJ, et al. Grb2 functions at the top of the T-cell antigen receptor-induced tyrosine kinase Cascade to control thymic selection. Proc Natl Acad Sci U S A. 2010;107(23):10620–10625.
- Yuan J, Nguyen CK, Liu X, et al. Lin28b reprograms adult bone marrow hematopoietic progenitors to mediate fetal-like lymphopoiesis. Science. 2012;335(6073):1195–1200.
- Copley MR, Babovic S, Benz C, et al. The Lin28b-let-7-Hmga2 axis determines the higher self-renewal potential of fetal haematopoietic stem cells. Nat Cell Biol. 2013;15(8):916–925.
- Zhou Y, Li YS, Bandi SR, et al. Lin28b promotes fetal B lymphopoiesis through the transcription factor Arid3a. J Exp Med. 2015;212(4):569–580.
- Xu X, Deobagkar-Lele M, Bull KR, et al. An ontogenetic switch drives the positive and negative selection of B cells. Proc Natl Acad Sci U S A. 2020;117(7):3718–3727.
- Rickert RC, Rajewsky K, Roes J. Impairment of T-cell-dependent B-cell responses and B-1 cell development in CD19-deficient mice. Nature. 1995;376(6538):352–355.
- Ahearn JM, Fischer MB, Croix D, et al. Disruption of the Cr2 locus results in a reduction in B-1a cells and in an impaired B cell response to T-dependent antigen. Immunity. 1996;4(3):251–262.
- Khan WN, Alt FW, Gerstein RM, et al. Defective B cell development and function in btk-deficient mice. Immunity. 1995;3(3):283–299.
- Fruman DA, Satterthwaite AB, Witte ON. Xid-like phenotypes: a B cell signalosome takes shape. Immunity. 2000;13(1):1–3.
- Suzuki H, Terauchi Y, Fujiwara M, et al. Xid-like immunodeficiency in mice with disruption of the p85alpha subunit of phosphoinositide 3-kinase. Science. 1999;283(5400):390–392.
- Tarakhovsky A, Turner M, Schaal S, et al. Defective antigen receptor-mediated proliferation of B and T cells in the absence of Vav. Nature. 1995;374(6521):467–470.
- Zhang R, Alt FW, Davidson L, et al. Defective signalling through the T- and B-cell antigen receptors in lymphoid cells lacking the Vav proto-oncogene. Nature. 1995;374(6521):470–473.
- Wang D, Feng J, Wen R, et al. Phospholipase Cγ2 is essential in the functions of B cell and several Fc receptors. Immunity. 2000;13(1):25–35.
- O’Keefe TL, Williams GT, Davies SL, et al. Hyperresponsive B cells in CD22-deficient mice. Science. 1996;274(5288):798–801.
- Nakamura A, Kobayashi E, Takai T. Exacerbated graft-versus-host disease in pirb−/− mice. Nat Immunol. 2004;5(6):623–629.
- Inaoki M, Sato S, Weintraub BC, et al. CD19-regulated signaling thresholds control peripheral tolerance and autoantibody production in B lymphocytes. J Exp Med. 1997;186(11):1923–1931.
- Shinohara H, Kurosaki T. Negative role of TAK1 in marginal zone B-cell development incidental to NF-κB noncanonical pathway activation. Immunol Cell Biol. 2016;94(9):821–829.
- Li S, Liu J, Min Q, et al. Kelch-like protein 14 promotes B-1a but suppresses B-1b cell development. Int Immunol. 2018;30(7):311–318.
- Berland R, Wortis HH. Normal B-1a cell development requires B cell-intrinsic NFATc1 activity. Proc Natl Acad Sci U S A. 2003;100(23):13459–13464.
- Masle-Farquhar E, Peters TJ, Miosge LA, et al. Uncontrolled CD21low age-associated and B1 B cell accumulation caused by failure of an EGR2/3 tolerance checkpoint. Cell Rep. 2022;38(3):110259.