
Abstract
Collagen, the most thrombogenic constituent of blood vessel walls, activates platelets through glycoprotein VI (GPVI). In suspension, following platelet activation by collagen, GPVI is cleaved by A Disintegrin And Metalloproteinase (ADAM)10 and ADAM17. In this study, we use single-molecule localization microscopy and a 2-level DBSCAN-based clustering tool to show that GPVI remains clustered along immobilized collagen fibers for at least 3 hours in the absence of significant shedding. Tyrosine phosphorylation of spleen tyrosine kinase (Syk) and Linker of Activated T cells (LAT), and elevation of intracellular Ca2+, are sustained over this period. Syk, but not Src kinase-dependent signaling is required to maintain clustering of the collagen integrin α2β1, whilst neither is required for GPVI. We propose that clustering of GPVI on immobilized collagen protects GPVI from shedding in order to maintain sustained Src and Syk-kinases dependent signaling, activation of integrin α2β1, and continued adhesion.
Introduction
Platelets are small anucleate blood cells that play important roles in many physiological and pathological processes including hemostasis, thrombosis, inflammation, and cancer [Citation1]. Collagen, one of the major components of the extracellular matrix (ECM), is a potent activator of platelets acting through its interaction with the immunoglobulin receptor glycoprotein VI (GPVI). GPVI is considered to be a promising anti-thrombotic drug target due to its restricted expression pattern in platelets and megakaryocytes and relatively minor role in hemostasis [Citation2]. More recently GPVI has been shown to be involved in maintaining vascular integrity at sites of inflammation [Citation3].
GPVI is an ~65 kDa transmembrane protein found at the platelet surface in association with the Fc receptor gamma chain (FcRɣ). Ligand engagement of GPVI triggers Src family kinase-dependent phosphorylation of the conserved tyrosines in the immunoreceptor tyrosine-based activation motif (ITAM) in the associated FcRγ chain and binding of the spleen tyrosine kinase (Syk) via its tandem SH2 domains [Citation4–6]. Activation of Syk, by Src-dependent phosphorylation and autophosphorylation, initiates formation of a Linker of Activated T cells (LAT)-based signalosome that culminates in the activation of phospholipase Cγ2 (PLCγ2), elevation of intracellular Ca2+, integrin activation (including the collagen-binding integrin α2β1) and platelet activation [Citation7]. The GPVI-FcRγ complex is primarily a signaling rather than an adhesive receptor complex, with activation of integrins required for stable platelet adhesion under flow [Citation8].
It has recently been demonstrated that GPVI is also a receptor for fibrin [Citation9–12] and fibrinogen [Citation10,Citation13], with ligand binding also resulting in platelet activation. Different ligands induce different degrees of higher order clustering of GPVI thereby regulating signal strength [Citation14–17]. In suspension, activation of platelets by collagen and other ligands leads to shedding of GPVI through the action of A Disintegrin And Metalloproteinase (ADAM)10 and ADAM17 [Citation18], thereby limiting activation. Paradoxically, this could potentially lead in vivo to the unwanted embolization, or breaking away, of single platelets and platelet aggregates which could lead to further complications. To date, shedding of GPVI in platelets adhered to and spreading on an immobilized collagen substrate, which would represent the first steps of thrombus formation or adhesion of platelets to the ECM in a blood vessel with compromised integrity, has not been investigated.
In the present study, we have used single-molecule localization microscopy (SMLM) to monitor clustering of GPVI on platelets adhering and spreading on immobilized fibrillar collagen and correlated this with biochemical data and other complementary imaging techniques to assess platelet activation and GPVI shedding. We show that, in contrast to studies in solution, GPVI is not shed from platelets activated by immobilized collagen and that signaling is sustained, with Syk, LAT and Ca2+ mobilization remaining active over several hours of platelet spreading. GPVI clustering is not reversed by inhibition of Src or Syk kinase activation, in contrast to integrin α2β1, which requires sustained Syk but not Src-dependent signaling, highlighting the different nature of the two collagen receptors. We further show that the metalloproteinase ADAM10 does not co-localize with the GPVI clustered at collagen fibrils, suggesting exclusion of the sheddase from the clusters. Our findings confirm the hypothesis that sustained GPVI signaling is further regulated by higher-order clustering, acting to protect GPVI from shedding, essential for prolonged platelet activation, including integrin activation which is required to maintain platelet spreading. This is likely to be important for thrombus stability and the prevention of bleeding associated with disrupted vascular integrity driven by inflammation.
Materials and Methods
Reagents and Antibodies
GPVI and the GPVI-cytoplasmic tail were detected using 1G5 Fab and affinity-purified anti-GPVI cytoplasmic tail IgG [Citation19], respectively. The other antibodies used in the study were purchased from commercial suppliers. Monoclonal antibodies: anti-CD49b (integrin α2β1, abD Serotec, Oxford, UK); anti-ADAM10 [clone 11G2] (Abcam, Cambridge UK); anti-phosphotyrosine (clone 4G10). Polyclonal antibodies: anti-LAT IgG (Millipore Merck, Abingdon, UK); anti-Syk IgG (#sc1077) and anti-PLCγ2 IgG (Q-20):sc-407 (Santa Cruz Biotechnology, Dallas, USA); anti-phospho LAT Tyr200 (Abcam, Cambridge UK); anti-phospho Syk Tyr525/526, anti-phospho PLCγ2 Tyr1217 and AlexaFluor 647-conjugated anti-phospho-Syk Tyr525/526 (Cell Signaling Technology, Hitchin, UK). All the fluorescent secondary antibodies, phalloidin-Alexa 488 and Oregon green 488 BAPTA-1-AM Ca2+ dye used for imaging, were purchased from Thermo Fisher Scientific (Waltham, MA, USA). The HRP- and fluorescence-conjugated secondary antibodies, used for immunoblotting, were obtained from Amersham Biosciences (GE Healthcare, Bucks, UK) and antibodies-online.com GmbH (Aachen, Germany), respectively. Horm collagen was obtained from Nycomed Pharma GmbH (Munich, Germany). 10% neutral buffered Formalin solution and Hydromount solution were purchased from Sigma (Poole, UK) and National Diagnostics (Atlanta, USA), respectively. The broad spectrum metalloproteinases inhibitor GM6001 and N-ethylmaleimide (NEM) were obtained from Millipore Merck (Abingdon, UK). The ADAM10 inhibitor GI254023 was purchased from Scientific Laboratory Supplies (Nottingham, UK). The Src-family kinases inhibitor PP2 and Syk inhibitor PRT-060318 were purchased from TOCRIS (Abingdon, UK) and Caltag Medsystems (Buckingham, UK), respectively. The inhibitors indomethacin and apyrase were obtained from Sigma (Poole, UK).
Human Platelet Preparation
Human washed platelets were prepared from blood samples donated by healthy, consenting volunteers under the following license: ERN_11-0175 ‘The regulation of activation of platelets’ (University of Birmingham). Blood was drawn via venipuncture into the anticoagulant sodium citrate and then acid/citrate/dextrose (ACD) added to 10% (v:v). Blood was centrifuged at 200 × g for 20 min. Platelet-rich plasma (PRP) was collected and centrifuged at 1,000 × g for 10 min in the presence of 0.1 μg ml−1 prostacyclin. Plasma was removed and the platelet pellet was resuspended in modified Tyrode’s buffer (129 mM NaCl, 0.34 mM Na2HPO4, 2.9 mM KCl, 12 mM NaHCO3, 20 mM HEPES, 5 mM glucose, 1 mM MgCl2; pH 7.3) containing ACD and 0.1 μg ml−1 prostacyclin before centrifugation at 1,000 × g for 10 min. The washed platelet pellet was resuspended in modified Tyrode’s buffer, left to rest for 30 min at room temperature (RT) and the platelet count adjusted to the desired concentration.
Platelet Spreading and Staining
For confocal and direct stochastic optical reconstruction microscopy (dSTORM) imaging, 13 mm #1.5 glass coverslips (VWK, UK) and 35 mm #1.5 (0.17 mm) uncoated glass-bottomed dishes (MatTek Corporation, Ashland, MA, USA) were coated with 10 μg ml−1 Horm collagen diluted in manufacturer-supplied diluent, and left overnight at 4°C, before being blocked in 5 mg ml−1 BSA for 1 hour at RT. Washed and rested platelets were diluted to 2 × 107 cells ml−1 in modified Tyrode’s buffer and allowed to spread on the collagen-coated surface for the required times at 37°C. When required, washed platelets were labeled with 2 μg ml−1 1G5-Fab against intact GPVI for 10 min at 37°C prior to spreading. Where stated, platelets were also incubated with specific inhibitors before and/or after spreading on collagen. Adhered cells were PBS-washed, fixed for 10 min with 10% neutral buffered Formalin solution, permeabilized for 5 min with 0.1% (v:v) Triton X-100 in PBS, blocked with 1% (w/v) BSA + 2% (v:v) goat serum in PBS and then labeled with antibodies or phalloidin for 1 h at RT as required. For confocal and epifluorescence imaging, samples were mounted on glass slides using Hydromount solution and stored at RT. Samples for dSTORM were stored in PBS at RT until imaged.
Confocal Imaging
Spread platelets mounted on glass slides were imaged using a Leica TCS SP2 confocal microscope and the 63 × 1.4NA objective. For each condition, 5 z-stacks (step size 0.25 µm) were imaged at random positions on the coverslip at a resolution of 1024 × 1024 pixels and a zoom of 4. Phalloidin-Alexa 488-labeled platelets were imaged with a 488 argon laser and Alexa-647 labeled platelets with a 647 HeNe laser. Dual-color labeled platelets were imaged simultaneously. Single-plane reflection images were also taken to monitor the distribution of the collagen fibrils. The images were then processed using ImageJ v1.48 (NIH, Bethesda, USA).
Colocalisation Analyses
Qualitative and quantitative colocalisation analyses of GPVI and phospho-proteins were performed using Fiji v1.52 [Citation20]. For the qualitative colocalisation analysis, the maximum intensity projections for each channel were thresholded using the ImageJ Default method (a variation of the IsoData algorithm) and multiplied together to obtain colocalisation masks showing only the colocalising pixels. For the quantitative colocalisation analysis, regions of interest were drawn around single platelets or groups of platelets randomly chosen within the field of view (FOV) and the Plugin Coloc2 was used to measure the degree of colocalisation through the Pearson’s correlation coefficient.
Epifluorescence Imaging
Dual-color imaging of Alexa 488 and 647-labeled platelets spread on collagen was performed using a Zeiss Axio Observer 7 Epifluorescent microscope equipped with a 63 × 1.4NA oil immersion lens, Colibri 7 LED light source, Zeiss Filter sets 38 and 50 for GFP/FITC and Cy5/647, respectively, and Hammamatsu ORCA Flash 4 LT sCMOS camera for image acquisition. DIC images were also taken (intensity = 7.0 V and exposure time = 100 ms) to visualize the collagen distribution. For each condition, five random FOVs were imaged (intensity = 20% and 25% and exposure time: 200 ms and 100 ms for 488 and 647, respectively) and then analyzed in Fiji v1.52 [Citation20].
Platelet Spreading Analysis
The confocal and epifluorescence images of platelets spread on collagen were assessed for platelet count and surface area using specialized semi-automated workflow [Citation21] built on the open-source KNIME software [Citation22] and Ilastik [Citation23]. A pixel classifier was used to construct a binary cell segmentation and the center of each single platelet was then set manually to facilitate the separation of touching cells. The coordinates of the cell center positions were then used to generate the final segmentation by employing a watershed-based transformation algorithm. Only objects with a size > 1 μm2 were included in the final per cell measurements including area and circularity.
dSTORM Imaging
Super-resolution imaging of GPVI receptors was performed using a Nikon N-STORM system in TIRF and dSTORM mode with a 100 × 1.49NA TIRF objective lens. The microscope system includes a Ti-E stand with Perfect Focus, an Agilent Ultra High Power Dual Output Laser bed with 170-mW 647-nm and 20-mW 405-nm lasers for the fluorophore excitation and an Andor IXON Ultra 897 EMCCD camera for the image capture. DIC and TIRF images were acquired on random FOVs containing both platelets and collagen fibers. An oxidizing and reducing buffer (100 mM MEA, 50 μg ml−1 glucose oxidase and 1 μg ml−1 catalase diluted in PBS, pH 7.5 [Citation24]) was used to allow the photoswitching performance. For single color (Alexa647) imaging of labeled GPVI, the N-STORM emission cube was used, combined with the gradual increase of the 405 laser power (5% every 30 sec) to reactivate the fluorophore switching. Simultaneous dual-color (Alexa647 and Alexa488) imaging was carried out using the Nikon Quad Cube. For each FOV, 20,000 frames were acquired in Nikon NIS Elements v4.5 software using an exposure time of 20 ms, gain 300 and conversion gain 3. To generate the final super-resolved images, the ThunderSTORM plugin for Fiji was implemented with the application of the Gaussian PSF model and maximum likelihood fitting [Citation25]. Drift correction and photon intensity filtering (>1000 photons) were applied within ThunderSTORM to post-process the reconstructed images. To minimize the ‘multiblinking’ artifacts, detections within 75 nm of another detection, either in the same or subsequent frames, were merged. The normalized Gaussian method was then used to allow visualization of the reconstructed dSTORM images which contain the spatial coordinates for each detected fluorescent blink. The image datasets were then exported as text files and analyzed for clustering in KNIME software [Citation22].
Two-level Cluster Analysis of dSTORM Data
Two-level cluster analysis was performed by first segmenting large clusters, corresponding to collagen fibers (Level I), and then segmenting denser clusters within the large clusters using different algorithm parameters (Level II). This analysis was implemented using the R library RSMLM [Citation26] and KNIME [Citation22] (workflow available upon request). Both segmentation levels were calculated using density-based spatial clustering of applications with noise (DBSCAN) [Citation27]. The radii of the local neighborhood were set to 75 nm and 30 nm for Level I and II, respectively, and the minimum number of reachable detections was set to either 3 or 10 for the different levels. Clusters with less than either 100 (Level I) or 10 (Level II) detections were removed. The union of circular regions (radius = 30 nm) positioned around each point in a cluster was used to measure the cluster area applying a grid of 5 nm pixels and image-based dilation. The analysis was performed on images using the whole FOV and the quantitative data relative to all clusters were outputted to an Excel spreadsheet.
Measurements of Ca2+ Mobilization
For Ca2+ mobilization analysis, washed platelets, diluted to 2 × 108 cells ml−1 in modified Tyrode’s buffer, were incubated for 45 min at 37°C with 1 μM Oregon green-488 BAPTA-1-AM and centrifuged at 1000 × g for 10 min in the presence of 2.8 μM PGI2 and ACD. The platelet pellet was resuspended in the same volume of modified Tyrode’s buffer and rested for at least 30 min before diluting to 2 × 107 platelets ml−1 for spreading. Rested platelets were then plated on collagen-coated MatTek dishes, prepared as described above, to adhere and spread. Live-cell imaging of Ca2+ mobilization was tracked using the Zeiss Axio Observer 7 Epifluorescent microscope as detailed above. Where indicated, the Src inhibitor PP2 (20 µM) and the Syk inhibitor PRT-060318 (10 µM) or the vehicle (DMSO) were added to the platelets after 45 min of spreading. Images were then acquired every 1 sec for 2 min using Zen Pro v2.3 software. Once processed in Fiji v1.52 [Citation20], the percentage of spiking platelets, number of spikes per platelet, amplitude, and peak duration were measured using MATLAB (Mathworks, Inc., Natick, MA) within one representative FOV (~150 cells) for each condition. Signals were baseline-corrected by application of a linear top-hat filter. A threshold of 20 fluorescence units was set for identification of spikes and the duration of a spike was defined as the time for fluorescence to reduce to below this threshold.
Protein Phosphorylation and Receptor Shedding Assay
Washed platelets were diluted to 5 × 108 cells ml−1 in modified Tyrode’s buffer supplemented with 2 mM CaCl2 and allowed to spread on a collagen-coated surface (10 μg ml−1) for specific time periods at 37°C. Non-adherent platelets were removed and lysed in 2x lysis buffer (300 mM NaCl, 20 mM Tris, 2 mM EGTA, 2 mM EDTA, and 2% NP-40 detergent, pH 7.4 supplemented with protease inhibitors: 2 mM Na3VO4, 2 mM 4-(2-Aminoethyl) benzenesulfonyl fluoride hydrochloride (AEBSF), 10 μg ml−1 leupeptin, 10 μg ml−1 aprotinin and 1 μg ml−1 pepstatin). Adherent and spread platelets were PBS-washed and then lysed in 1x lysis buffer. The protein concentration of both lysates was measured using a Bio-Rad Protein Assay, diluted to the same concentration in lysis buffer and resuspended in appropriate volumes of 5x reducing sodium dodecyl sulfate (SDS) sample buffer (10 mg ml−1 SDS, 25% (v:v) 2-mercaptoethanol, 50% (v:v) glycerol, 25% (v:v) Stacking buffer (0.5 M Tris HCl, pH 6.8), containing a trace amount of Brilliant Blue) and stored at −20°C.
Western Blot
Lysate samples were denatured at 95°C for 10 min and centrifuged at 18000 × g for 5 min at 4°C to pellet insoluble material. Pre-cast polyacrylamide gels, Bolt 4–12% Bis-Tris Plus (Invitrogen) were used to run samples. Resolved proteins were transferred to PVDF membrane (Trans-Blot Turbo RTA transfer kit, LF PVDF from Bio-Rad). After transfer, the membrane was blocked for 1 h in 5% (w:v) BSA buffer and then incubated with specific primary antibodies overnight at 4°C. Membrane washing (3 x 10 min) to remove unbound primary antibodies was executed in TBST (50 mM Tris-Cl pH 7.5, 150 mM NaCl, 0.05% (v:v) Tween 20) before incubating with fluorophore- or HRP-conjugated secondary antibodies (1:10,000) for 1 h at RT. To remove unbound secondary antibodies, washing (3 x 10 min) was repeated. For chemiluminescence-based detection of the proteins, the membrane was then incubated with the Pierce ECL Western Blotting Substrate (Thermo Fisher Scientific). Both chemiluminescent and fluorescent antibody binding were visualized using the Odyssey Fc System (LI-COR). Quantification of the intensity of the bands was performed using Image Studio v5.2. The percentage of cleaved GPVI was calculated using the band intensities of full-length GPVI (~62kDa) and GPVI tail remnant (~10kDa) as follows: % shed GPVI = (remnant GPVI/(full-length GPVI + remnant GPVI)) * 100.
Statistical Analysis
Results are expressed as means ± standard error of the mean (SEM). Data were analyzed using GraphPad Prism 8 software (GraphPad Software, Inc., La Jolla, CA, USA). Significant differences were determined using unpaired two-tailed Student’s t-test or one-way Analysis of variance (ANOVA) with Tukey’s posthoc test for multiple comparisons. The significance was set at p ≤ 0.05.
Results
GPVI Signaling Is Sustained over Time in Platelets Adhering to Immobilized Collagen
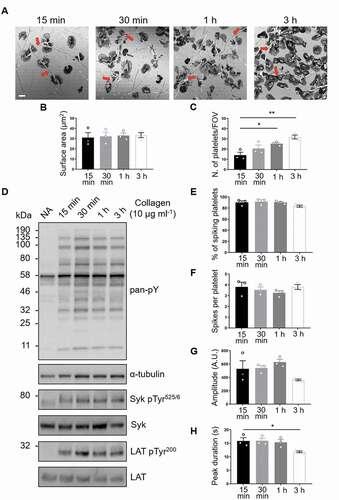
Studies in vitro using platelets and cell lines activated with GPVI receptor agonists in suspension have demonstrated that fibrillar collagen triggers slow but sustained ITAM signaling [Citation28] and metalloproteolytic shedding of GPVI [Citation18]. This raises the question of whether this is also the case on an immobilized surface, where shedding might lead to ablation of adhesion to collagen, through loss of signaling, and embolization of platelets. Reflection images show that human platelets remain spread on immobilized collagen for at least 3 h (). As expected, there was a steady increase in the number of platelets coming into contact with the collagen fibers over time, but there was no change in mean platelet surface area (, c). To assess platelet signaling in these cells, whole-cell tyrosine phosphorylation and specific phosphorylation sites on two key GPVI signaling mediators, Syk, and LAT, were measured by western blot (). Total phospho-tyrosine levels, detected by the antibody clone 4G10, were elevated in adherent platelets relative to cells in suspension (non-adhered, NA) and remained increased over time (). Site-specific phosphorylation of both Syk Tyr 525/6 and LAT Tyr200, residues associated with their activation, was maintained, with no significant change in signal over 3 h ( and Supp. , b). An additional step of GPVI signaling transduction involves Ca2+ movement from intracellular compartments upon activation of PLCγ2. Therefore, we monitored the Ca2+ spiking in platelets labeled with the Ca2+ dye Oregon green 488 BAPTA-1-AM that had been spread on immobilized collagen for up to 3 h (Supp. movie 1). Single-cell Ca2+ dynamics was analyzed using MATLAB. The results show that the percentage and frequency of spiking platelets spread on collagen for 3 h remained constant (, f), although the amplitude and peak duration slightly declined (, h). Overall, immobilized collagen supports platelet spreading via GPVI signaling for at least 3 h.
Figure 1. GPVI signaling remains constant in platelets spread on fibrous collagen for at least 3 h. (A) Confocal reflection images of washed human platelets spread on Horm collagen for the indicated times. Quantification of spread platelet surface area (B) and platelet number per field of view (C) for the specified time points from 5 FOVs from three independent experiments. Arrows indicate collagen fibers. Scale bar: 5 μm. (D) Western blot analysis of platelets spread for the indicated time points or non-adhered platelets (NA) probed for total phosphotyrosine (4G10), phosphorylated Syk (Tyr525/6; pSyk) and LAT (Tyr200; pLAT). The antibodies against α-tubulin, pan-Syk and pan-LAT were used as loading controls for total phosphotyrosines, phospho-Syk and phospho-LAT, respectively. Quantification of live-cell Ca2+ imaging of platelets spread for the indicated time points showing percentage of platelets exhibiting Ca2+ spiking (E), number of spikes per platelet in the 2 min imaging period (F), spike amplitude (G), spike duration (H). Data taken from three representative FOVs for each time point (~150 platelets in each FOV) from three independent experiments. All data are expressed as mean ± SEM. Significance was calculated using one-way ANOVA with Tukey’s multiple comparisons test (* p < .05)
Sustained Signaling Colocalises with GPVI on Collagen in Spread Platelets
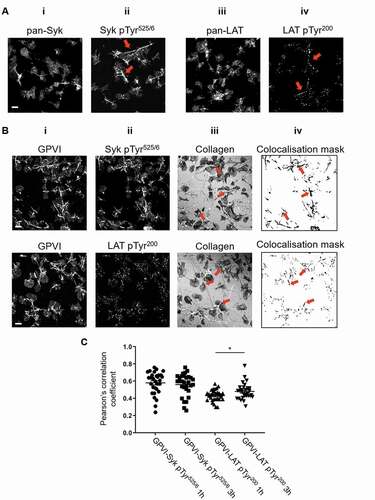
Western blot analysis quantifies platelet-signaling events but does not provide spatial information. To investigate the location of signaling events, platelets spread on immobilized collagen for 1 h were labeled for either pan-Syk, phospho-Syk Tyr525/6, pan-LAT or phospho-LAT Tyr200 and imaged by confocal microscopy (). Fig. 2Ai, iii illustrate that total (pan) Syk and LAT are homogeneously distributed over the platelet surface. However, both pSyk and pLAT were enriched along collagen fibrils, with a lower density of phospho-proteins detected in areas where there is no visible collagen (Fig. 2Aii, iv). Dual-color confocal imaging of GPVI and phosphorylated proteins was carried out to assess their colocalisation (). GPVI, labeled with 1G5-Fab, was highly enriched along collagen fibers (Fig. 2Bi). A combination of qualitative (colocalisation mask, showing pixels which contain both GPVI and phospho-proteins, Fig. 2Biv) and quantitative (Pearson’s correlation coefficient, ) analysis demonstrated significant colocalisation between the phospho-proteins and GPVI along collagen. This colocalisation was sustained for at least 3 h, with the pLAT/GPVI colocalisation slightly, but significantly, increased at the later time point.
Figure 2. Phosphorylated Syk and LAT colocalize with GPVI along collagen fibers. (A) Confocal microscopy imaging of human washed platelets spread on collagen (10 µg ml−1) for 1 h, labeled for (Ai) Pan-Syk, (Aii) pSyk Tyr525/6, (Aiii) Pan-LAT, (Aiv) pLAT Tyr200. The enrichment of pSyk Tyr525/6 and pLAT Tyr200 along collagen fibers is indicated by red arrows (Aii, iv). (B) Dual-color confocal imaging of platelets, pre-incubated for Pan-GPVI using 1G5-Fab (Bi), spread on collagen for 1 h and post-labeled for pSyk Tyr525/6 (Bii, first panel) or pLAT Tyr200 (Bii, second panel). The position of collagen fibers is shown in the confocal reflection image (Biii). The qualitative colocalisation mask in (Biv) shows the colocalisation of GPVI and the respective phosphoprotein with enrichment at the collagen fibers (arrows). (C) Quantification of GPVI and phosphoprotein colocalisation using Pearson’s correlation coefficient for platelets spreading for 1 h and 3 h. Scatter plot represents mean ± SEM of n = 30 platelets taken from three independent experiments. Significance was measured using unpaired two-tailed t-test, * p < .05. Scale bar: 5 μm
GPVI Clustering and Nanoclustering on Collagen Fibers Is Sustained over Time
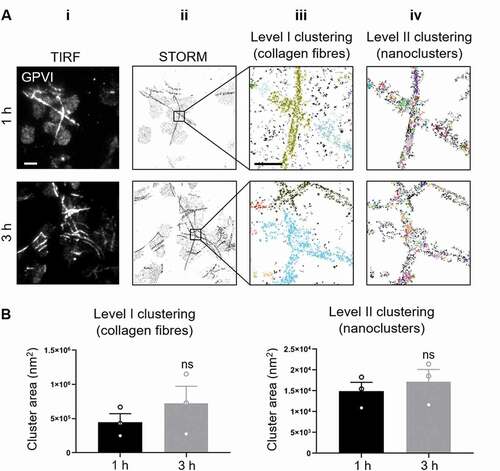
In order to correlate GPVI clustering with the platelet signaling events detected using biochemical methods, we used the SMLM technique dSTORM (characterized by a resolution of 20–40 nm) [Citation29]. dSTORM permitted the nanoscale spatial organization of GPVI receptors in platelets that have been spread on collagen fibers for 1 h and 3 h to be examined and quantified relatively between conditions. The diffraction-limited TIRF images (Fig. 3Ai) show the same alignment of GPVI receptors along collagen fibers as that observed in confocal imaging (Fig. 2Bi). dSTORM allowed us to super-resolve the receptor distribution on fibrous collagen (Fig. 3Aii). A customized two-level cluster analysis workflow based on DBSCAN [Citation27] was implemented to first identify and isolate large clusters of GPVI along collagen fibers (Level I; Fig. 3Aiii) and then take those clusters and subdivide them into nanoclusters (Level II; Fig. 3Aiv). This approach allowed us to assess only GPVI clusters associated with the visible collagen fibers, to identify any differences between the platelets spread for different durations. The algorithm clusters the localized data points by grouping detections with high local density. Different clusters are represented by different colors (Fig. 3Aiii, iv). Quantitative analysis of the clusters showed no significant difference in cluster area between 1 h and 3 h of spreading at either Level I or Level II clustering (). Cluster density and number of detections per cluster also remained constant over time (Supp. ,). This suggests that GPVI-collagen interactions had stabilized within 1 h of platelet spreading and remained constant regardless of the time.
Figure 3. dSTORM imaging and two-level DBSCAN cluster analysis of GPVI in platelets spread on collagen for 1 h and 3 h
GPVI Shedding Is Minimal in Platelets Spreading on Collagen
In human platelets in suspension, GPVI is cleaved upon treatment with different agonists, predominantly by ADAM10 [Citation18]. However, whether GPVI is cleaved in platelets that have been spread on an immobilized ligand is unknown. Using an antibody raised against the cytosolic tail of GPVI, the degree of GPVI shedding in spread platelets was assessed. Intact GPVI has a molecular weight of ~65 kDa, but when the extracellular domain is shed, a 10-kDa remnant fragment containing the cytosolic tail remains bound to the platelet. Using western blot we found that in platelets that did not contact collagen (non-adhered, NA, ) no GPVI remnant fragment was detected. In platelets spread on collagen, only a minimal amount of GPVI shedding was detected, with ~15% loss of intact GPVI after 3 h (, b). As a positive control, the thiol-modifying reagent N-ethylmalemide (NEM), which triggers activation of metalloproteinases, was added to spread platelets for 30 min before cell lysis. NEM induced ~100% of GPVI shedding (, (). We carried out two-color immunolocalization of the intracellular tail of GPVI (using GPVI-tail antibody) with the extracellular domain (using 1G5-Fab) to obtain spatial information on the intact receptor (). Epifluorescence imaging reveals a strong overlap of two receptor domains , iii), which was particularly prevalent along the collagen fibers, as highlighted in the colocalisation mask (iv). This indicates that this pool of GPVI has not been shed from the platelets in the 3 h timeframe, in accordance with the biochemical data.
Figure 4. GPVI is not shed in platelets spread on fibrous collagen
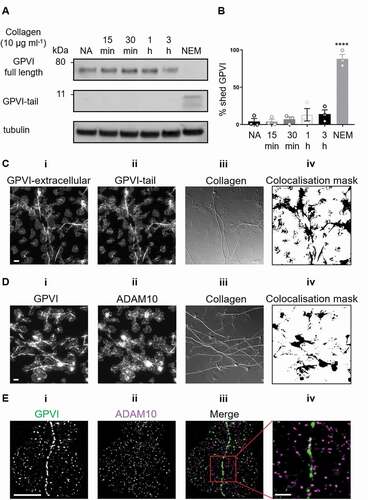
To further investigate whether GPVI shedding occurred in platelets spread on collagen we evaluated the location of the GPVI sheddase ADAM10 in relation to GPVI oligomers using two-color epifluorescence microscopy () and dSTORM (). The data demonstrate differential distribution of the two proteins, with no visible enrichment of ADAM10 at collagen fibers as was seen for GPVI (Fig. 4Di, iii, iv). Dual-color dSTORM imaging enabled us to assess the nanoscale organization of GPVI (Fig. 4Ei) and ADAM10 (Fig. 4Eii) at the single molecule level. ADAM10 molecules rarely colocalized with the tightly packed GPVI clusters (Fig. 4Eiii, iv), suggesting that the metalloproteinase was not present in GPVI clusters, thus preventing shedding and allowing signaling to continue.
Effect of Metalloproteinases Inhibition and Activation on GPVI Cluster Formation
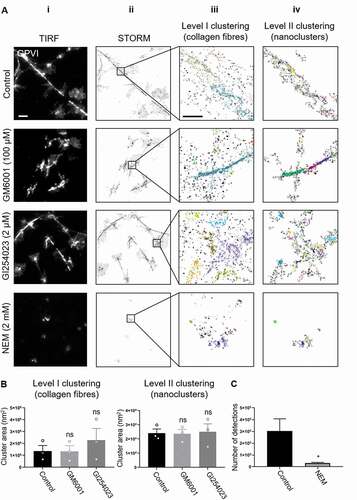
To investigate whether GPVI remains intact and clustered at the cell surface, dSTORM was carried out on platelets that had been spread in the presence of the broad-spectrum metalloproteinase inhibitor GM6001 or the ADAM10-specific inhibitor GI254023, at concentrations previously shown to inhibit GPVI metalloproteolysis [Citation18,Citation30,Citation31]. If GPVI shedding did occur in spread platelets we would expect to see an increase in the measured cluster parameters when the metalloproteinase inhibitors were present. To confirm that shedding was possible, spread platelets were post-treated with NEM. Both metalloproteinase inhibitors had no effect on platelet spreading whereas NEM significantly impaired the ability of platelets to remain spread on collagen, showing a significant reduction in platelet surface area in comparison to controls (Supp. ,,). In platelets treated with either of the metalloproteinase inhibitors, discrete GPVI clusters were detected (). These clusters were not significantly different from those formed in control platelets in terms of area (), density (Supp. ) or number of detections per cluster (Supp. ) at both clustering levels. In contrast, very few clusters were detected in NEM-treated platelets () and there was loss of 90% of the GPVI-related single-molecule detections () making the 2-level cluster analysis unfeasible. This again supports the hypothesis that GPVI shedding does not occur in platelets spread on collagen.
Figure 5. Effect of metalloproteinase inhibitors and NEM on GPVI clustering
Secondary Mediators are Not Required for GPVI Clustering
Release of secondary mediators, thromboxane A2 (TxA2) and adenosine diphosphate (ADP), enhance integrin activation on adherent platelets and aid thrombus development through the recruitment and activation of additional platelets [Citation32]. We evaluated whether secondary mediators had any effect on platelet spreading on collagen and GPVI clustering along the collagen fibers. Pre-incubation of platelets with 10 μM indomethacin and 2 U/ml apyrase, to inhibit TxA2 and ADP, did not prevent platelet adhesion and spreading (Supp. , , ). In addition, the ability of GPVI to form clusters on collagen was not perturbed by the abolishment of secondary mediator activity (Supp. , , , ).
Figure 6. Effect of Src-family and Syk inhibitors on platelet spreading, GPVI signaling and clustering in response to fibrous collagen
Active Src-family and Syk Kinases are Not Required to Maintain GPVI Clustering on Collagen
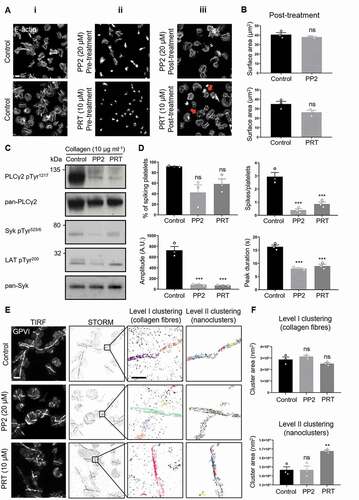
Our results show that ITAM signaling is sustained in spread platelets. In order to investigate the action of Src and Syk on the clustering of GPVI, we treated platelets with selective inhibitors of these kinases. In accordance with previous results [Citation17], pre-incubation of platelets with either the Src-family kinase inhibitor PP2 or Syk kinase inhibitor PRT-060318 (PRT) significantly reduced the extent of platelet spreading on collagen (ii and Supp. ). PRT had a more marked effect with a complete abolishment of lamellipodia formation. In order to investigate whether Src-family and Syk kinases are required to maintain GPVI clustering and activation in spread platelets, we post-treated fully spread platelets for 15 min with each of the inhibitors or the vehicle control. We found that sustained spreading did not require Src-family kinase signaling as demonstrated by the lack of an effect of PP2 on platelet surface area (Fig. 6Aiii, B). However, PRT-treatment slightly reduced platelet surface area and had a visible effect on the actin cytoskeleton, with loss of stress fibers and appearance of actin nodules (Fig. 6Aiii, B). Both inhibitors impaired GPVI downstream signaling; western blot analysis showed a decrease in pPLCγ2, pSyk, and pLAT levels ( and Supp. Fig. 8A, B, C), and Ca2+ imaging showed a significant reduction in the number of spiking platelets, with those spiking doing so at a much lower number, amplitude, and duration than controls (, Supp. movie 2). These results confirmed that Src and Syk kinases are required to maintain collagen-induced GPVI-mediated platelet activation. Finally, we evaluated the potential feedback exerted by Src and Syk kinases on GPVI receptor clustering. dSTORM imaging demonstrated that GPVI was still clustered along collagen fibers with Src-family or Syk kinases inhibition (). Indeed, GPVI binding to collagen was not affected by the inhibition of either kinase activity as shown by the Level I clustering analysis ( and Supp. Fig. 9A). Syk inhibition perturbed the nanoscale receptor organization within the larger collagen-associated clusters, with clusters of increased area compared to control samples (). However, the density and number of detections per cluster remained unchanged (Supp. Fig. 9B).
Figure 7. Syk but not Src-family inhibitor impairs integrin α2β1 clustering on collagen in spread platelets
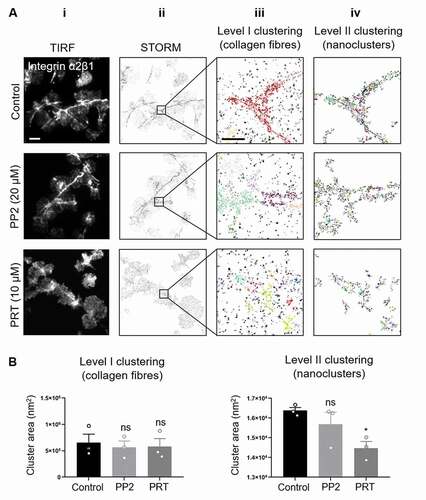
Inhibition of Syk but Not Src Kinase Activity Impaired Clustering of Integrin α2β1 along Collagen Fibers
To further address the effect of Src and Syk inhibition on collagen-induced platelet spreading and signaling, we investigated the distribution of the other collagen receptor integrin α2β1 using dSTORM and 2-level cluster analysis. Consistent with previous work [Citation17], in spread platelets integrin α2β1 also accumulated along collagen fibers ( first row). Post-spreading treatment with the Src kinase inhibitor PP2 had no effect on integrin clusters ( second row; 7B and Supp Fig 10A, B). However, whilst post-spreading treatment with the Syk inhibitor PRT did not have any effect on cluster density or number of detections per cluster (Supp. Fig 10A, B), it resulted in a visible redistribution of α2β1 clusters (, third row). Large integrin α2β1 clusters were still detected (Level I analysis, Fig. 7Aiii, iv third row) but these were no longer closely associated with the collagen, and the nanoclusters detected in the Level II analysis were significantly smaller (), suggesting a compromised interaction of the integrin with its ligand with Syk inhibition. These results suggest that, in contrast to GPVI, active Syk kinase is required for the maintenance of integrin α2β1 clustering along collagen fibers in spread platelets.
Discussion
This study has used advanced microscopy, image analysis and biochemical techniques to demonstrate that GPVI-mediated signaling is sustained in platelets adhering and spreading on an immobilized collagen surface. Our data show that GPVI clusters along the collagen fibers, leading to an enrichment of the receptor and sustained activation of downstream signaling molecules such as pSyk and pLAT and continued Ca2+ spiking in platelets spread for up to 3 h. Our imaging data show little overlap of GPVI with its sheddase ADAM10, which is consistent with no significant shedding of GPVI in collagen-adhered platelets. Furthermore, once bound to collagen, GPVI did not require positive feedback from Src-family and Syk kinases to maintain the clusters. This is in contrast to integrin α2β1 which requires continued Syk, but not Src, activity to remain bound to collagen fibers.
The platelet receptor GPVI has been identified as a promising antithrombotic target due to its restricted expression on platelets and megakaryocytes and the fact that loss of GPVI protects against thrombosis whilst having negligible effects on primary hemostasis [Citation33]. It is therefore important to understand the basic biology behind GPVI signaling and platelet activation in order to develop novel approaches to modulate GPVI receptor function. This is also important as more recent work has shown that GPVI plays a role in maintaining vascular integrity in inflammation, so this will need to be taken into account when assessing new anti-GPVI therapies [Citation3].
It is known that the surface expression and distribution of GPVI is tightly regulated by a variety of stimulatory and inhibitory mechanisms to coordinate platelet responses and prevent improper activation. Indeed, GPVI spatial organization and cluster formation at the plasma membrane play a central role in regulating signaling [Citation17,Citation34]. Platelet GPVI levels at the plasma membrane can be regulated by endocytosis [Citation35,Citation36] or by receptor shedding [Citation37], the latter being the subject of significant research [Citation38]. The extracellular region of GPVI can be cleaved by members of the ADAM family of sheddases [Citation31,Citation39]. ADAM10 is the major GPVI sheddase on human platelets, with ADAM17 and potentially other metalloproteinases also causing shedding of mouse GPVI [Citation39]. To date, GPVI shedding has been shown to be induced in platelet suspensions by a number of physiological and non-physiological stimuli, including ligands of GPVI and other platelet ITAM receptors [Citation40]. GPVI shedding induced by ligand engagement is thought to require receptor clustering, dissociation of calmodulin from the cytoplasmic tail and activation of the intracellular signaling cascade [Citation18]. Recent work using an ADAM10-specific substrate, which becomes fluorescent when cleaved, has shown that ADAM10 is constitutively active at the surface of resting platelets although GPVI is not constitutively shed [Citation30]. Activation of platelets with GPVI ligands such as collagen-related peptide or the purified venom protein convulxin do not alter the level of ADAM10 activity but do induce GPVI shedding, whereas exposure to pathophysiological fluid shear stress or NEM increase ADAM10 activity and cause GPVI shedding. Taken together, these observations suggest another level of control, which could include altered ADAM10 or GPVI localization within the membrane.
This study has investigated shedding in platelets adhered to an immobilized substrate, which more closely mimics platelets adherence to collagen in vivo. We have shown that GPVI is enriched along collagen fibers and remains clustered for at least 3 h. We hypothesized that the ability of GPVI to cluster along collagen would allow signaling complexes to be formed and maintained and may safeguard GPVI from shedding by marginalization of the sheddase ADAM10 due to steric hindrance. A similar situation is seen with the T-cell immunological synapse where the phosphatases CD148 [Citation41] and CD45 [Citation42] are marginalized from clusters of the ITAM-containing T-cell receptor, to allow signaling to be prolonged. The data support our hypothesis, with minimal GPVI shedding detected and high degrees of clustered GPVI colocalising with pSyk and pLAT but very little ADAM10 and GPVI colocalisation observed. The enriched degree of colocalisation of the intra- and extracellular domains of GPVI in the dual-color imaging combined with no evidence of remnant GPVI fragment by western blot, indicate that the receptor in contact with collagen remains intact in spread platelets. Nevertheless, GPVI shedding could be induced in spread platelets by treatment with the thiol-modifying agent NEM, commonly used to induce metalloproteinase activity. How this shedding is achieved is unclear given the distinct distributions of the two proteins. However, NEM is not specific for ADAM10 and is membrane-permeable. NEM-treated spread platelets themselves start to detach from the collagen-coated surface. It is possible that NEM treatment could result in rearrangement of membrane proteins, enabling GPVI cleavage. Furthermore, ADAM17, as well as other platelet metalloproteinases, are known to share substrate specificity with ADAM10 and would also be activated by NEM treatment so could play a role.
Upon collagen adhesion, activated platelets release soluble agonists from the storage organelles such as ADP and TxA2 which act as pro-thrombotic triggers through amplification of intracellular signaling cascades, ultimately leading to enhanced integrin activation and platelet aggregation [Citation43]. Despite these roles in platelet responses, the discharge of secondary mediators is not needed to potentiate collagen adhesion and GPVI cluster formation, as these were not altered when platelets spread in the presence of indomethacin and apyrase. These results suggest that under these experimental conditions, secondary mediators do not function as a positive feedback loop for the initial stimulus on collagen, in contrast to their fundamental role in supporting platelet aggregation during thrombus growth. This is different to the related platelet hemITAM receptor CLEC-2, where secondary mediators play a vital role in its signaling [Citation44].
Similar to GPVI, CLEC-2 also forms clusters upon interaction with its endogenous ligand podoplanin and the clustering of CLEC-2 requires active Src and Syk kinases [Citation45]. Since GPVI receptor cluster formation seems to maintain ITAM signaling over time, signaling may contribute to the ability of GPVI to maintain large clusters through positive feedback. Pre-incubation of platelets with the Src inhibitor PP2 or the Syk inhibitor PRT prevented complete spreading of platelets on collagen as previously shown [Citation17] and therefore prevented detailed analysis of GPVI clustering. However, the effect of positive feedback of these kinases on cluster formation could be tested by addition of the inhibitor to platelets that had already undergone spreading. Treatment of spread platelets with both the inhibitors had very little effect on numbers and extent of GPVI clusters that had already formed on the collagen, with only a slight increase in the GPVI nanoclusters seen when Syk was inhibited. However, the inhibitors reduced pSyk, pLAT and pPLCɣ2 and had an almost immediate effect on Ca2+ signaling. This demonstrates that Src-family and Syk kinases signaling are fundamental for sustaining full GPVI-mediated platelet activation. Syk, but not Src, inhibition resulted in a disruption of the actin cytoskeleton, with loss of stress fibers and a slight reduction in platelet spreading. Previous work has shown that GPVI dimer formation is dependent on the actin cytoskeleton [Citation17] so the actin depolymerization caused by Syk inhibition may disrupt GPVI dimers and could explain the appearance of slightly larger GPVI nanoclusters at the platelet surface. Syk has a well-established role in cytoskeleton organization upon immunoreceptor stimulation in several cell types, including platelets [Citation46]. Work in B-cells has also shown a differential dependence upon Src and Syk family kinases in B-cell receptor (BCR) signaling. Under conditions of BCR clustering by multivalent ligands, Syk is sufficient to induce signaling. However, with monomeric ligands where no clustering is initiated, both Syk and Src family kinases are required for BCR activation [Citation47]. In spread platelets, actin disruption caused by inhibiting Syk may be mediated through the action of Syk on the Vav-family of Rho/Rac GTPase guanine nucleotide exchange factors (GEFs), which have been shown to require Syk-mediated tyrosine phosphorylation to become activated [Citation48]. Vav1 and Vav3 play redundant roles in platelet activation by collagen and loss of these proteins prevents PLCɣ2 phosphorylation and full platelet spreading [Citation49]. Together, these results suggest that, whereas sustained collagen-induced platelet activation requires intact Src and Syk activity, prolonged GPVI cluster formation does not.
In platelets, there is a second receptor for collagen, integrin α2β1, which has previously been shown to also cluster along collagen fibers [Citation17]. In contrast to GPVI, our current and previous [Citation26] results show that Syk, but not Src, kinase activity is required for maintaining integrin α2β1 clustering along collagen. Unlike GPVI, integrins fluctuate between an ‘on’ and ‘off’ state and only the conversion into an active conformation allows high-affinity ligand binding [Citation50,Citation51]. The F-actin cytoskeleton plays an important role in the up-regulation of integrin activity [Citation52] through the engagement of different adaptor proteins such as kindlin, vinculin and talin which couple integrins to the F-actin [Citation53]. Indeed, the importance of this interaction has been demonstrated by platelet-specific talin1 knockout mice, where loss of talin from platelets caused a massive reduction in β1 integrin-mediated platelet adhesion under flow conditions, with no stable adhesion or microthrombi formed [Citation54]. Our results suggest that the actin disruption caused by Syk-inhibition causes inactivation of the integrin and its subsequent dissociation from collagen. In contrast, as GPVI clusters are largely unaffected by PRT treatment it implies that, once bound to collagen, GPVI remains bound and does not have such a dependence on the actin cytoskeleton. As integrin α2β1 is the main adhesive receptor, dissociation from collagen would have a negative impact on platelet adhesion, especially under flow conditions where GPVI has been shown to be the main signaling receptor and integrin α2β1 plays a role in stable adhesion [Citation55].
Taken together our results support a model in which GPVI clustering induced by immobilized collagen triggers the assembly of signaling complexes at the platelet surface via the engagement of downstream signaling proteins, increasing the avidity for ligand-binding and strengthening the signal. We show that GPVI clustering is maintained over time and helps to prevent negative regulation mediated by the sheddase ADAM10. The sustained, primarily Syk-mediated, signaling is required to prolong calcium signals, maintain actin cytoskeletal arrangements and integrin α2β1 engagement with collagen to ensure that platelets remain properly spread and adhered to the substrate. Recent work by Ahmed et al [Citation56] have also demonstrated a role for GPVI signaling in maintaining a stable thrombus. They show this is through the binding of GPVI to fibrin(ogen), highlighting why protecting GPVI from shedding is important. Thus, GPVI clustering at collagen is important for both the initiation and maintenance of a stable thrombus, as well as the prevention of bleeding associated with disrupted vascular integrity driven by inflammation, where GPVI plays a critical role [Citation3]. Due to the relatively minor role of GPVI in hemostasis, this receptor has been identified as a promising anti-thrombotic target. The selective inhibition of receptor clustering induced by collagen provides a new avenue for the development of novel therapeutic approaches to improve the functional outcomes of a variety of thrombotic and inflammatory conditions.
Author Contributions
C.P. did experimental work, analyzed the data, prepared figures and wrote the manuscript. J.A.P. implemented the cluster analysis and platelet spreading workflows. C.O’S. created the calcium analysis code and analyzed the data. R.K.A. and E.E.G. provided key reagents and expertise. S.P.W. provided supervision, devised experiments and interpreted the data. N.S.P. conceived the project, devised experiments, interpreted the data, prepared figures, and wrote the manuscript. All authors reviewed the manuscript.
Competing Interests
The authors declare no competing interests.
Supplemental Material
Download Microsoft Video (AVI) (6 MB)Supplemental Material
Download Microsoft Video (AVI) (9.6 MB)Supplemental Material
Download MS Word (1.7 MB)Acknowledgements
This work was funded by the British Heart Foundation, through the Chair award [CH0/03/003)] to Steve P. Watson, and the Centre of Membrane Proteins and Receptors (COMPARE), Universities of Birmingham and Nottingham, Midlands, UK. We thank Deirdre Kavanagh, the COMPARE microscope officer, for support with imaging and Mark Probert for preliminary imaging data. We also thank Steve Thomas, Abdullah Khan, Julie Rayes and Christopher Smith for helpful discussions.
Supplementary Material
Supplemental data for this article can be accessed on the publisher’s website.
Additional information
Funding
References
- Rayes J, Watson SP, Nieswandt B. Functional significance of the platelet immune receptors GPVI and CLEC-2. J Clin Invest 2019;129:12–23.
- Dütting S, Bender M, Nieswandt B. Platelet GPVI: a target for antithrombotic therapy?! Trends Pharmacol Sci 2012;33:583–590.
- Boulaftali Y, Mawhin M-A, Jandrot-Perrus M, Ho-Tin-Noé B. Glycoprotein VI in securing vascular integrity in inflamed vessels. Res Pract Thromb Haemost 2018;2:228–239.
- Ezumi Y, Shindoh K, Tsuji M, Takayama H. Physical and functional association of the Src family kinases Fyn and Lyn with the collagen receptor glycoprotein VI-Fc receptor γ chain complex on human platelets. J Exp Med 1998;188:267–276.
- Séverin S, Nash C, Mori J, Zhao Y, Abram C, Lowell C, Senis Y, Watson S. Distinct and overlapping functional roles of Src family kinases in mouse platelets. J Thromb Haemost 2012;10:1631–1645.
- Suzuki-Inoue K, Tulasne D, Shen Y, Bori-Sanz T, Inoue O, Jung SM, Moroi M, Andrews RK, Berndt MC, Watson SP. Association of Fyn and Lyn with the proline-rich domain of glycoprotein VI regulates intracellular signaling. J Biol Chem 2002;277:21561–21566.
- Watson S, Auger J, McCarty O, Pearce A. GPVI and integrin αIIbβ3 signaling in platelets. J Thromb Haemost 2005;3:1752–1762.
- Pugh N, Maddox BD, Bihan D, Taylor KA, Mahaut-Smith MP, Farndale RW. Differential integrin activity mediated by platelet collagen receptor engagement under flow conditions. Thromb Haemost 2017;117:1588–1600.
- Alshehri OM, Hughes CE, Montague S, Watson SK, Frampton J, Bender M, Watson SP. Fibrin activates GPVI in human and mouse platelets. Blood 2015;126:1601–1608.
- Induruwa I, Moroi M, Bonna A, Malcor JD, Howes JM, Warburton EA, Farndale RW, Jung SM. Platelet collagen receptor Glycoprotein VI‐dimer recognizes fibrinogen and fibrin through their D‐domains, contributing to platelet adhesion and activation during thrombus formation. J Thromb Haemost 2018;16:389–404.
- Mammadova-Bach E, Ollivier V, Loyau S, Schaff M, Dumont B, Favier R, Freyburger G, Latger-Cannard V, Nieswandt B, Gachet C, et al. Platelet glycoprotein VI binds to polymerized fibrin and promotes thrombin generation. Blood 2015;126:683.
- Onselaer M-B, Hardy AT, Wilson C, Sanchez X, Babar AK, Miller JL, Watson CN, Watson SK, Bonna A, Philippou H. Fibrin and D-dimer bind to monomeric GPVI. Blood Adv 2017;1:1495–1504.
- Mangin PH, Onselaer M-B, Receveur N, Le Lay N, Hardy AT, Wilson C, Sanchez X, Loyau S, Dupuis A, Babar AK. Immobilized fibrinogen activates human platelets through glycoprotein VI. Haematologica 2018;103:898–907.
- Haining EJ, Matthews AL, Noy PJ, Romanska HM, Harris HJ, Pike J, Morowski M, Gavin RL, Yang J, Milhiet P-E. Tetraspanin Tspan9 regulates platelet collagen receptor GPVI lateral diffusion and activation. Platelets 2017;28:629–642.
- Horii K, Brooks MT, Herr AB. Convulxin forms a dimer in solution and can bind eight copies of glycoprotein VI: implications for platelet activation. Biochemistry 2009;48:2907–2914.
- Jiang P, Loyau S, Tchitchinadze M, Ropers J, Jondeau G, Jandrot-Perrus M. Inhibition of glycoprotein VI clustering by collagen as a mechanism of inhibiting collagen-induced platelet responses: the example of losartan. PloS One 2015;10:e0128744.
- Poulter NS, Pollitt AY, Owen DM, Gardiner EE, Andrews RK, Shimizu H, Ishikawa D, Bihan D, Farndale RW, Moroi M, et al. Clustering of glycoprotein VI (GPVI) dimers upon adhesion to collagen as a mechanism to regulate GPVI signaling in platelets. JTH 2017;15:549–564.
- Gardiner EE, Arthur JF, Kahn ML, Berndt MC, Andrews RK. Regulation of platelet membrane levels of glycoprotein VI by a platelet-derived metalloproteinase. Blood 2004;104:3611–3617. Epub 2004/ 08/17.
- Al-Tamimi M, Mu FT, Arthur JF, Shen Y, Moroi M, Berndt MC, Andrews RK, Gardiner EE. Anti-glycoprotein VI monoclonal antibodies directly aggregate platelets independently of FcgammaRIIa and induce GPVI ectodomain shedding. Platelets 2009;20:75–82. Epub 2009/ 02/24.
- Schindelin J, Arganda-Carreras I, Frise E, Kaynig V, Longair M, Pietzsch T, Preibisch S, Rueden C, Saalfeld S, Schmid B. Fiji: an open-source platform for biological-image analysis. Nat Meth 2012;9:676.
- Khan AO, Maclachlan A, Lowe GC, Nicolson PLR, Al Ghaithi R, Thomas SG, Watson SP, Pike JA, Morgan NV. High-throughput platelet spreading analysis: a tool for the diagnosis of platelet- based bleeding disorders. Haematologica 2020;105(3):e124–e128.
- Berthold MR, Cebron N, Dill F, Gabriel TR, Kötter T, Meinl T, Ohl P, Thiel K, Wiswedel B. KNIME-the Konstanz information miner: version 2.0 and beyond. AcM SIGKDD Explorat Newslr 2009;11:26–31.
- Sommer C, Straehle C, Kӧthe U, Hamprecht FA. Ilastik: interactive learning and segmentation toolkit. In: 2011 IEEE international symposium on biomedical imaging: from nano to macro. 2011. p. 230–233.
- Metcalf DJ, Edwards R, Kumarswami N, Knight AE. Test samples for optimizing STORM super-resolution microscopy. J Vis Exp 2013;(79):50579.
- Ovesný M, Křížek P, Borkovec J, Švindrych Z, Hagen GM. ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics 2014;30:2389–2390.
- Pike JA, Khan AO, Pallini C, Thomas SG, Mund M, Ries J, Poulter NS, Styles IB. Topological data analysis quantifies biological nano-structure from single molecule localization microscopy. Bioinformatics 2020;36:1614–1621.
- Ester M, Kriegel H-P, Sander J, Xu X. A density-based algorithm for discovering clusters in large spatial databases with noise. In: KDD. vol. 96. Palo Alto, CA: Association for the Advancement of Artificial Intelligence (AAAI); 1996. p. 226–231.
- Tomlinson M, Calaminus S, Berlanga O, Auger J, Bori‐Sanz T, Meyaard L, Watson S. Collagen promotes sustained glycoprotein VI signaling in platelets and cell lines. J Thromb Haemost 2007;5:2274–2283.
- Nicovich PR, Owen DM, Gaus K. Turning single-molecule localization microscopy into a quantitative bioanalytical tool. Nat Protoc 2017;12:453.
- Facey A, Pinar I, Arthur JF, Qiao J, Jing J, Mado B, Carberry J, Andrews RK, Gardiner EE. A-disintegrin-and-metalloproteinase (ADAM) 10 activity on resting and activated platelets. Biochemistry 2016;55:1187–1194. Epub 2016/ 02/04.
- Gardiner EE, Karunakaran D, Shen Y, Arthur JF, Andrews RK, Berndt MC. Controlled shedding of platelet glycoprotein (GP) VI and GPIb–IX–V by ADAM family metalloproteinases. J Thromb Haemost 2007;5:1530–1537.
- Bye AP, Unsworth AJ, Gibbins JM. Platelet signaling: a complex interplay between inhibitory and activatory networks. J Thromb Haemost 2016;14:918–930.
- Andrews RK, Arthur JF, Gardiner EE. Targeting GPVI as a novel antithrombotic strategy. J Blood Med 2014;5:59–68. Epub 2014/ 06/06.
- Loyau S, Dumont B, Ollivier V, Boulaftali Y, Feldman L, Ajzenberg N, Jandrot-Perrus M. Platelet glycoprotein VI dimerization, an active process inducing receptor competence, is an indicator of platelet reactivity. Arterioscler Thromb Vasc Biol 2012;32:778–785.
- Takayama H, Hosaka Y, Nakayama K, Shirakawa K, Naitoh K, Matsusue T, Shinozaki M, Honda M, Yatagai Y, Kawahara T, et al. A novel antiplatelet antibody therapy that induces cAMP-dependent endocytosis of the GPVI/Fc receptor gamma-chain complex. J Clin Invest 2008;118:1785–1795. Epub 2008/ 04/03.
- Moroi M, Farndale RW, Jung SM. Activation-induced changes in platelet surface receptor expression and the contribution of the large-platelet subpopulation to activation. Res Pract Thromb Haemost 2020;4:285–297. Epub 2020/ 02/29.
- Stephens G, Yan Y, Jandrot-Perrus M, Villeval JL, Clemetson KJ, Phillips DR. Platelet activation induces metalloproteinase-dependent GP VI cleavage to down-regulate platelet reactivity to collagen. Blood 2005;105:186–191. Epub 2004/ 09/02.
- Bender M, Stegner D, Nieswandt B. Model systems for platelet receptor shedding. Platelets 2017;28:325–332. Epub 2016/ 06/24.
- Bender M, Hofmann S, Stegner D, Chalaris A, Bosl M, Braun A, Scheller J, Rose-John S, Nieswandt B. Differentially regulated GPVI ectodomain shedding by multiple platelet-expressed proteinases. Blood 2010;116:3347–3355. Epub 2010/ 07/21.
- Andrews RK, Gardiner EE. Basic mechanisms of platelet receptor shedding. Platelets 2017;28:319–324. Epub 2016/ 10/26.
- Lin J, Weiss A. The tyrosine phosphatase CD148 is excluded from the immunologic synapse and down-regulates prolonged T cell signaling. J Cell Biol 2003;162:673–682. Epub 2003/ 08/13.
- Furlan G, Minowa T, Hanagata N, Kataoka-Hamai C, Kaizuka Y. Phosphatase CD45 both positively and negatively regulates T cell receptor phosphorylation in reconstituted membrane protein clusters. J Biol Chem 2014;289:28514–28525. Epub 2014/ 08/17.
- Offermanns S. Activation of platelet function through G protein-coupled receptors. Circ Res 2006;99:1293–1304. Epub 2006/ 12/13.
- Pollitt AY, Grygielska B, Leblond B, Desire L, Eble JA, Watson SP. Phosphorylation of CLEC-2 is dependent on lipid rafts, actin polymerization, secondary mediators, and Rac. Blood 2010;115:2938–2946.
- Pollitt AY, Poulter NS, Gitz E, Navarro-Nunez L, Wang YJ, Hughes CE, Thomas SG, Nieswandt B, Douglas MR, Owen DM, et al. Syk and Src family kinases regulate C-type lectin receptor 2 (CLEC-2)-mediated clustering of podoplanin and platelet adhesion to lymphatic endothelial cells. J Biol Chem 2014;289:35695–35710.
- Mocsai A, Ruland J, Tybulewicz VL. The SYK tyrosine kinase: a crucial player in diverse biological functions. Nat Rev Immunol 2010;10:387–402. Epub 2010/ 05/15.
- Mukherjee S, Zhu J, Zikherman J, Parameswaran R, Kadlecek TA, Wang Q, Au-Yeung B, Ploegh H, Kuriyan J, Das J, et al. Monovalent and multivalent ligation of the B cell receptor exhibit differential dependence upon Syk and Src family kinases. Sci Signal 2013;6(256):ra1. Epub 2013/ 01/03.
- Deckert M, Tartare-Deckert S, Couture C, Mustelin T, Altman A. Functional and physical interactions of Syk family kinases with the Vav proto-oncogene product. Immunity 1996;5:591–604. Epub 1996/ 12/01.
- Pearce AC, Senis YA, Billadeau DD, Turner M, Watson SP, Vigorito E. Vav1 and vav3 have critical but redundant roles in mediating platelet activation by collagen. J Biol Chem 2004;279:53955–53962. Epub 2004/ 10/01.
- Hynes RO. Integrins: bidirectional, allosteric signaling machines. Cell 2002;110:673–687. Epub 2002/ 09/26.
- Spiess M, Hernandez-Varas P, Oddone A, Olofsson H, Blom H, Waithe D, Lock JG, Lakadamyali M, Stromblad S. Active and inactive beta1 integrins segregate into distinct nanoclusters in focal adhesions. J Cell Biol 2018;217:1929–1940. Epub 2018/ 04/11.
- Zhu J, Luo BH, Xiao T, Zhang C, Nishida N, Springer TA. Structure of a complete integrin ectodomain in a physiologic resting state and activation and deactivation by applied forces. Mol Cell 2008;32:849–861. Epub 2008/ 12/30.
- Horton ER, Byron A, Askari JA, Ng DHJ, Millon-Fremillon A, Robertson J, Koper EJ, Paul NR, Warwood S, Knight D, et al. Definition of a consensus integrin adhesome and its dynamics during adhesion complex assembly and disassembly. Nat Cell Biol 2015;17:1577–1587. Epub 2015/ 10/20.
- Petrich BG, Marchese P, Ruggeri ZM, Spiess S, Weichert RA, Ye F, Tiedt R, Skoda RC, Monkley SJ, Critchley DR, et al. Talin is required for integrin-mediated platelet function in hemostasis and thrombosis. J Exp Med 2007;204:3103–3111. Epub 2007/ 12/19.
- Pugh N, Simpson AM, Smethurst PA, de Groot PG, Raynal N, Farndale RW. Synergism between platelet collagen receptors defined using receptor-specific collagen-mimetic peptide substrata in flowing blood. Blood 2010;115:5069–5079. Epub 2010/ 03/31.
- Ahmed MU, Kaneva V, Loyau S, Nechipurenko D, Receveur N, Le Bris M, Janus-Bell E, Didelot M, Rauch A, Susen S, et al. Pharmacological Blockade of Glycoprotein VI Promotes Thrombus Disaggregation in the Absence of Thrombin. Arterioscler Thromb Vasc Biol 2020;40:2127–2142. Epub 2020/ 07/24.