
Summary
Platelets are the major cellular contributor to arterial thrombosis. However, activated platelets form two distinct subpopulations during thrombosis. Pro-aggregatory platelets aggregate to form the main body of the thrombus. In contrast, procoagulant platelets expose phosphatidylserine on their outer surface and promote thrombin generation. This apparently all-or-nothing segregation into subpopulations indicates that, during activation, platelets commit to becoming procoagulant or pro-aggregatory. Although the signaling pathways that control this commitment are not understood, distinct cytosolic and mitochondrial Ca2+ signals in different subpopulations are likely to be central. In this review, we discuss how these Ca2+ signals control procoagulant platelet formation and whether this process can be targeted pharmacologically to prevent arterial thrombosis.
Introduction – a Procoagulant Platelet Subpopulation in Arterial Thrombosis
Platelets play a critical role in arterial thrombosis, a major cause of myocardial infarction, stroke, and ultimately death. However, current anti-platelet therapies carry increased bleeding risk, have substantial inter-individual variation, and may have lower and reduced effectiveness in some high-risk groups such as patients with diabetes [Citation1,Citation2]. These problems, and the continuing incidence of arterial thrombosis despite current anti-platelet strategies, indicate that new targets for thrombosis prevention are required.
During thrombosis, platelets adhere to exposed matrix proteins such as collagen fibers. These platelets activate and release soluble platelet activators that recruit further platelets[Citation3]. Two distinct sub-populations of platelets form [Citation4,Citation5]. Pro-aggregatory platelets have active integrin αIIbβ3, which allows them to bind fibrinogen and aggregate with other pro-aggregatory platelets to form the body of the thrombus. Current anti-platelet therapies reduce platelet aggregation, either by blocking the signals that lead to αIIbβ3 activation, or by directly blocking αIIbβ3[Citation3]. In contrast, procoagulant platelets expose phosphatidylserine (PS), an efficient surface for assembly of coagulation complexes, generating a burst of thrombin that is responsible for producing an occlusive thrombus [Citation6,Citation7]. Procoagulant platelets also release PS-exposing extracellular vesicles (EVs), often called microparticles, that further promote coagulation [Citation8,Citation9]. Both pro-aggregatory and procoagulant platelets are essential to arterial thrombus growth[Citation10]. Therefore, procoagulant platelets may be an alternative target to thrombosis prevention.
PS exposure is commonly monitored using annexin V, which binds to PS with high affinity in a Ca2+-dependent manner. Following stimulation with physiological activators, platelets segregate into two subpopulations, either with high annexin V binding or with binding no higher than unstimulated platelets [Citation4,Citation11–13]. The extent of high annexin V binding is similar to that seen with high concentrations of Ca2+ ionophores, which induce annexin V binding in all platelets. It is possible that a distinction based on annexin V obscures a more complex picture. Lactadherin can bind membranes with lower PS density in their outer leaflet than can annexin V [Citation14,Citation15]. In one study, lactadherin bound all activated platelets under conditions where annexin V bound only a small subpopulation[Citation15], suggesting that all activated platelets expose a low level of PS, but that only a subpopulation expose a high level of PS. This high level appears functionally relevant, as coagulation factors, FXa, FVa, FVIII, prothrombin (FII) and FIXa bind specifically to the high annexin V-binding subpopulation [Citation4,Citation16–19]. These annexin V-binding, procoagulant platelets also have depolarized mitochondrial membrane potential (∆Ψm) and inactive integrin αIIbβ3, whereas annexin V-negative activated platelets have normal ∆Ψm and have active integrin αIIbβ3 [Citation4,Citation12,Citation20,Citation21], underlining the distinction between procoagulant platelets and pro-aggregatory platelets.
This apparently all-or-nothing segregation into subpopulations indicates that during activation, platelets commit to becoming procoagulant or pro-aggregatory. Although the signaling pathways that control this commitment are not understood, distinct cytosolic Ca2+ signals in different subpopulations may play an important role. Cytosolic Ca2+ concentration ([Ca2+]cyt) is a regulator of many platelet actions during thrombosis including PS exposure and release of PS-exposing EVs. Ca2+ activates the phospholipid scramblase, TMEM16F (also known as Anoctamin 6 [Ano6]), and inactivates an aminophospholipid flippase [Citation22,Citation23]. This results in net movement of PS to the outer leaflet of the plasma membrane where coagulation factors can bind. PS-exposing EV release requires PS exposure and activation of calpain, a Ca2+-dependent protease [Citation24,Citation25], complemented by other, less well-defined pathways [Citation9,Citation26]. Cytosolic Ca2+ signaling is therefore central to procoagulant platelet formation. Although platelet granule secretion, αIIbβ3 activation and clot retraction also depend on cytosolic Ca2+ signaling, differences in the amplitude or temporal pattern of [Ca2+]cyt signals between individual platelets may commit them to form different subpopulations.
Mitochondrial Ca2+ signaling also has a central role in procoagulant platelet formation. Opening of the mitochondrial permeability transition pore (PTP) is an important event in procoagulant platelets and is regulated by mitochondrial Ca2+ concentration ([Ca2+]mito). Although less is known about mitochondrial Ca2+ signaling, [Ca2+]mito signals are also likely to be heterogenous in activated platelets.
In this review, we discuss the interplay between platelet cytosolic and mitochondrial Ca2+ signals, highlighting how this might lead to the all-or-nothing commitment to becoming procoagulant, and consider the implications for therapeutic targeting of procoagulant platelets.
Cytosolic Ca2+ Signaling
Most platelet activators trigger a rapid increase in [Ca2+]cyt from resting levels <100 nM to up to 1–2 μM. Collagen fibers cluster GPVI, triggering a cascade of protein tyrosine phosphorylation to activate phospholipase Cγ2 [Citation27]. ADP and thromboxane A2, released by stimulated platelets, act via Gαq-coupled P2Y12 and TP receptors, respectively, to activate PLCβ. Thrombin acts via Gαq-coupled PAR1 and PAR4, also activating PLCβ. The phospholipases generate inositol 1,4,5 trisphosphate (IP3) and diacylglycerol (DAG) from phosphoinositide 4,5 bisphosphate (PIP2). IP3 opens IP3R receptors in the dense tubular system, releasing Ca2+ stored there into the cytoplasm[Citation3].
This initial increase in [Ca2+]cyt is amplified and sustained by Ca2+ entry across the plasma membrane. Ca2+ store depletion is detected by STIM1, which activates the channel ORAI1 in the plasma membrane. This process is called ‘store-operated Ca2+ entry’ (SOCE) and is a major mechanism for Ca2+ entry in platelets [Citation28–31]. Procoagulant platelet formation is inhibited in Orai1−/- murine platelets or human platelets treated with ORAI1 inhibitors [Citation28,Citation29,Citation32–35]. The nonselective cation channel, TRPC6, also contributes, probably activated by DAG. Na+ entry through TRPC6 drives further Ca2+ entry via reverse-mode Na+-Ca2+ exchange (rNCX)[Citation33]. The contribution of TRPC6 is less prominent than that of ORAI1, however, and may be restricted to providing additional Ca2+ entry when platelets are coincidentally stimulated by thrombin and collagen-like agonists [Citation32,Citation33]. P2X1, a ligand-gated ion channel activated by extracellular ATP, is a further route for Ca2+ entry[Citation36].
Ca2+ is removed from the cytosol by transporters and exchangers. SERCA2b and 3 sequester Ca2+ into membrane-bound stores. PMCA4b pumps Ca2+ across the plasma membrane. NCX removes Ca2+ in exchange for extracellular Na+, though whether NCX acts in forward-mode to remove Ca2+, or reverse-mode to increase [Ca2+]cyt, depends on the cytosolic Na+ concentration[Citation37]. Ca2+ is also taken into mitochondria, which will be considered in more detail below.
The interplay between Ca2+ release, Ca2+ entry, and Ca2+ removal produces a wide variety of cytosolic Ca2+ signals, including individual transient spikes, irregular trains of spikes (‘ragged spiking’), and sustained increases in [Ca2+]cyt[Citation38]. Several factors may contribute to the differences between individual platelets. The platelets themselves may be different, with variation in the copy number of receptors and key Ca2+ signaling proteins (e.g. IP3Rs); the extent of dense tubular system will affect how much stored Ca2+ is available for release; and variation in platelet volume may affect the volume of cytosol and hence [Ca2+]cyt dynamics[Citation39]. Each of these factors could also differ with platelet age. In addition to platelet-intrinsic factors, different platelets could be exposed to different local concentrations of platelet activators depending, for example, on their proximity to other activated platelets and activators released from them. The heterogeneity of platelet Ca2+ signaling is likely to be a complex interplay of many small differences. However, many platelet Ca2+ signaling experiments measure the average [Ca2+]cyt in whole platelet populations, such as in cuvettes or microplate wells. This approach, although convenient, blurs the dynamics of such signals in individual platelets and the differences between them. Insights from studies of Ca2+ signaling in individual platelets (e.g. Refs [Citation38,Citation40,Citation41]) show that there remains a lot to learn about the different patterns of Ca2+ signaling and how they are decoded into functional responses.
Mitochondrial Ca2+ Signaling and the Permeability Transition Pore (PTP)
Mitochondrial matrix Ca2+ concentration ([Ca2+]mito) is a key regulator of mitochondrial oxygen consumption and ATP synthesis in many cells, and also regulates procoagulant platelet formation[Citation42]. To enter the matrix, Ca2+ must cross the outer and inner mitochondrial membranes (OMM and IMM, respectively). Ca2+ readily crosses the OMM through voltage-dependent anion-selective channels (VDAC; despite the name, Ca2+ permeates open and closed states of these channels)[Citation43]. Ca2+ movement across the IMM is more tightly regulated. The dominant pathway is via the mitochondrial Ca2+ uniporter (MCU) complex. The MCU complex consists of the pore-forming unit, MCU itself (CCDC109a), regulators in the IMM such as MCUb (CCDC109b) and EMRE/SMDT1, the latter coupled to the MICU1/MICU2 heterodimer in the intramembrane space [Citation44,Citation45]. The MCU complex is inward-rectifying (toward the mitochondrial matrix), highly Ca2+ selective, and with a very large capacity to transport Ca2+. The mitochondrial membrane potential (∆Ψm, negative on the matrix side) provides the driving force for Ca2+ entry. Despite this driving force, mitochondrial Ca2+ entry is limited in cells with low [Ca2+]cyt due to inhibition by MICU2. When [Ca2+]cyt increases, MICU2 is inhibited and MICU1 is activated, leading to Ca2+ entry and an increase in [Ca2+]mito[Citation46]. Ca2+ leaves mitochondrial matrix via a 2 H+/Ca2+ antiporter (mHCX) and a Na+/Ca2+ exchange (mNCX). Together, these pathways generate dynamic increases in [Ca2+]mito in response to increased [Ca2+]cyt[Citation47].
Mitochondrial Ca2+ uptake is a key step in procoagulant platelet formation. Platelets from Mcu−/- mice, or human platelets treated with the MCU inhibitor, Ru360, generate fewer procoagulant platelets in response to physiological stimuli without effect on integrin activation[Citation48]. Notably, however, increasing [Ca2+]cyt with a Ca2+ ionophore bypasses the requirement for MCU[Citation48].
A large rise in [Ca2+]mito triggers the permeability transition pore (PTP), a large conductance pore of the IMM resulting in depolarization of ∆Ψm and movement of ions and metabolites up to 1.5 kDa[Citation49]. Cyclophilin D (CypD) controls the sensitivity of PTP to [Ca2+]mito[Citation50]. In the absence of CypD, the threshold [Ca2+]mito for PTP opening is much higher. CypD-deficient mouse platelets (Ppif−/-) generate significantly fewer procoagulant platelets than wild-type in response to physiological agonists[Citation12]. Cyclosporine A (CsA), which inhibits CypD, also inhibits the formation of procoagulant platelets[Citation34]. As with MCU, CypD does not appear necessary for PS exposure in response to high concentrations of Ca2+ ionophores, suggesting that mitochondrial Ca2+ entry and PTP opening can be bypassed by sufficient high [Ca2+]cyt. However, in response to physiological stimulation, it is clear the PS exposure and procoagulant platelet formation requires mitochondrial Ca2+ entry and PTP opening.
How Does PTP Regulate Procoagulant Platelet Formation?
Although mitochondrial Ca2+ entry is likely to occur in all activated platelets, PTP opening only occurs in platelets that become procoagulant. Panteleev and colleagues proposed an elegant model in which PTP opening is the step that commits platelets to becoming procoagulant [Citation40,Citation51]. In this model, physiological activators trigger an increase in [Ca2+]cyt through Ca2+ release and Ca2+ entry. Some of this Ca2+ is taken into mitochondria via MCU leading to an increase in [Ca2+]mito. However, since cytosolic Ca2+ signaling varies between individual platelets, the increase in [Ca2+]mito will also vary between individual platelets. In some platelets, [Ca2+]mito will be above the threshold for PTP opening. In these platelets, PTP opening commits the platelet to become procoagulant. They lose ∆Ψm (because of PTP opening), expose PS, and inactivate integrin αIIbβ3. In contrast, activated platelets without PTP opening maintain αIIbβ3 activation and are pro-aggregatory. In this model, the Ca2+ threshold for PTP opening is therefore the commitment point that converts heterogenous Ca2+ signaling into an all-or-nothing response.
How does PTP opening commit a platelet to becoming procoagulant? Specifically, how is this mitochondrial event communicated to TMEM16F in the plasma membrane? We envisage two possibilities: either PTP opening changes cytosolic Ca2+ signaling in such a way that it can now activate TMEM16F, or PTP opening releases an additional signal that regulates TMEM16F in addition to cytosolic Ca2+ (or, conceivably, removes an inhibitory factor).
Obydennyy and colleagues demonstrated that procoagulant platelet formation was accompanied by a change in cytosolic Ca2+ signaling[Citation40]. Thrombin triggered a rapid train of spikes in [Ca2+]cyt that varied in frequency and amplitude between individual platelets. However, a transition to high, sustained [Ca2+]cyt during procoagulant platelet formation coincided with loss of ∆Ψm and PS exposure. The extent of this high, sustained [Ca2+]cyt is difficult to estimate, however, as [Ca2+]cyt was mostly measured using Fura Red in these experiments. Fura Red’s fluorescence decreases with Ca2+ binding. The high sustained [Ca2+]cyt levels that coincide with PTP opening resulted in very low fluorescence. PTP inhibition with CsA reduced the percentage of platelets that became procoagulant but did not affect the thrombin-induced spikes in [Ca2+]cyt. Similarly, Arachiche et al. also found that Fura Red fluorescence was lower in PS-exposing platelets, and this was also reversed by CsA[Citation52]. These observations suggest that PTP opening causes a further increase in [Ca2+]cyt, or transition from spiking to sustained cytosolic Ca2+ signaling.
In contrast, although Jobe and colleagues found that [Ca2+]cyt and [Ca2+]mito were higher in procoagulant platelets, they found no difference in cytosolic or mitochondrial Ca2+ signaling in CypD-deficient platelets[Citation53]. Similarly, pharmacological inhibition of mitochondrial Ca2+ uptake reduced [Ca2+]mito, ∆Ψm loss and PS exposure, but had no effect on [Ca2+]cyt. In this study, [Ca2+]cyt was measured with Fluo-4, a commonly-used probe with high affinity for Ca2+. From these results it appears that PTP opening is downstream of cytosolic and mitochondrial Ca2+ signaling, but that PTP opening does not further affect [Ca2+]cyt, in clear contrast to the conclusions of the studies described above. However, whilst we can readily repeat these results[Citation34], we propose an alternative explanation that reconciles these differing observations.
A ‘Supramaximal’ Ca2+ Signal in Procoagulant Platelets
Recently, we proposed that [Ca2+]cyt in procoagulant platelets is much higher than previously appreciated, and much higher than in pro-aggregatory (non-coagulant) platelets[Citation34]. We termed this very high [Ca2+]cyt signal a supramaximal Ca2+ signal, that is, much higher than normally considered maximal. Our observations are based on the use of fluorescent Ca2+ probes with different affinities for Ca2+. The Kd (dissociation constant) of Fluo-4 in vitro is approximately 390 nM. Although the Kd will be affected by the intracellular environment, the high affinity of Fluo-4 means that it is likely to be saturated by Ca2+ in the low micromolar range. In contrast, Fluo-5N has a very low affinity for Ca2+ (Kd = 90 μM in vitro). This low affinity means that changes [Ca2+]cyt in the nanomolar and low micromolar range will have little effect on Ca2+ binding to Fluo-5N, whereas [Ca2+]cyt of tens of micromolar will increase Ca2+ binding and give a fluorescent signal. The fluorescence of platelets loaded with either Fluo-4 or Fluo-5N showed very different patterns in procoagulant and non-coagulant platelets. The Fluo-4 fluorescence in stimulated platelets was much higher than in unstimulated platelets and was similar in procoagulant and non-coagulant platelets. In contrast, in Fluo-5N fluorescence of non-coagulant platelets was similar to unstimulated platelets whereas pro-coagulant platelets had brighter fluorescence. Our interpretation is that [Ca2+]cyt in procoagulant platelets is very high compared to non-coagulant platelets. In both subpopulations, [Ca2+]cyt is high enough to saturate Fluo-4, which is why there is no apparent difference in Fluo-4 fluorescence between the two subpopulations. This would normally lead to the conclusion that [Ca2+]cyt is similar in the two subpopulations. However, although [Ca2+]cyt is not high enough in non-coagulant platelets to give a signal with Fluo-5N, [Ca2+]cyt is much higher in procoagulant platelets because it is high enough to give a signal with Fluo-5N. The low affinity of Fluo-5N means that [Ca2+]cyt must be very high indeed. We estimated [Ca2+]cyt as approximately 160 μM.34 Although this fairly crude estimate needs further refining, it shows that [Ca2+]cyt in procoagulant platelets may be much higher than previously thought.
Very high [Ca2+]cyt signals have been reported in other cells. For example, [Ca2+]cyt was estimated as greater than 100 μM using low-affinity dyes in ATP-depleted renal proximal tubular cells [Citation54] and MDCK cells[Citation55]. Similarly, in excitotoxic neurons, [Ca2+]cyt was estimated as >5 µM by a low-affinity dye but only 0.3–0.4 µM with high-affinity fura-2[Citation56]. Interestingly, these examples are all of cells undergoing cell death. Procoagulant platelets are also necrotic[Citation57], although we have shown that the supramaximal [Ca2+]cyt signal in these procoagulant platelets is not simply due to loss of plasma membrane integrity[Citation34].
Supramaximal Ca2+ signals provide a means to activate effectors with low Ca2+ affinity, including TMEM16F and calpain. The half-maximal Ca2+ sensitivity of TMEM16F or phospholipid scramblase activity has been estimated at 10–80 μM in various cells, including platelets, lymphocytes and red blood cells [Citation58–62]. Similarly, calpain, which is needed for release of PS-exposing EVs[Citation24], also requires micromolar [Ca2+]cyt for activation [Citation63]. While the Ca2+ sensitivity may be altered in cells, with the Ca2+ sensitivity of TMEM16F increased by PIP2 and PIP3, for example[Citation64], supramaximal Ca2+ signaling provides a route to specifically activate these low-affinity effectors in procoagulant platelets. However, this does not exclude the possibility that there are other signals that are also released from mitochondria by PTP opening that also regulate PS exposure.
A Model for Commitment to Becoming Procoagulant
Our model for procoagulant platelet commitment involves three stages of Ca2+ signaling (). It is based on the model proposed by Panteleev and colleagues [Citation51] but with the addition of supramaximal Ca2+ signaling. First, platelet activators such as thrombin and collagen trigger an increase in [Ca2+]cyt. This occurs through Ca2+ release from intracellular stores and Ca2+ entry from outside (step 1 in ). These cytosolic Ca2+ signals are sufficient to trigger pro-aggregatory platelet activation in all platelets. However, these signals also vary between platelets, in part due to differences in the expression level of key receptors and Ca2+ signaling proteins. Second, some of the cytosolic Ca2+ is taken into mitochondria via MCU, which leads to mitochondrial Ca2+ signals that vary between platelets. In some platelets, [Ca2+]mito increases beyond a threshold such that PTP opening is triggered (2). PTP opening is the commitment to becoming procoagulant, the point of no return. The threshold for PTP opening is reduced by CypD, so when CypD is absent or inhibited, fewer platelets reach this threshold. Third, PTP opening triggers a supramaximal Ca2+ signal that activates TMEM16F and leads to PS exposure (3). In contrast, supramaximal Ca2+ signaling is not triggered in platelets that do not undergo PTP opening, and these platelets do not become procoagulant but remain pro-aggregatory. A heterogenous initial cytosolic Ca2+ is therefore converted into an all-or-nothing response.
Figure 1. Ca2+ signaling in procoagulant platelets
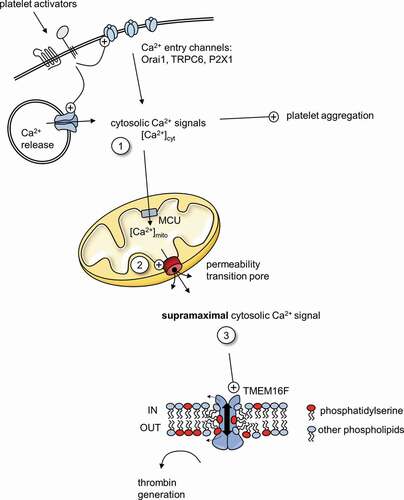
Our proposed model alters how we might interpret the effect of genetic or pharmacological inhibition on Ca2+ signaling in procoagulant platelets. CsA inhibits supramaximal Ca2+ signaling measured by low affinity Fluo-5 N, without appearing to affect cytosolic Ca2+ signaling measured by high-affinity Fluo-4 [Citation34,Citation53]. We predict that a similar difference will be seen with any pharmacological or genetic approach that blocks mitochondrial Ca2+ or PTP opening (steps 2 and 3) but does not affect the initial cytosolic Ca2+ signal (step 1). For example, we have seen a similar inhibition of supramaximal Ca2+ signaling, with little apparent effect on Fluo-4-measured signals, when mitochondrial Ca2+ uptake was inhibited. In contrast, inhibition of Orai1 did inhibit the Fluo-4-measured signals, indicating that Orai1 is involved in the initial cytosolic Ca2+ signal[Citation34]. We and others have extensively used high-affinity Ca2+ dyes (such as Fluo-4, Fura-2 or similar analogues) to investigate Ca2+ signaling during procoagulant platelet formation. In the future, such studies should also include analysis of mitochondrial Ca2+ and supramaximal Ca2+ signals. Analyzing the variation in pattern of cytosolic Ca2+ signaling between individual platelets would also be very informative.
Another source of variation between platelets is in their number of mitochondria. Although we found no significant difference in staining with a fluorescent mitochondrial dye between procoagulant and pro-aggregatory platelets[Citation34], Obydenni and colleagues found that the number of mitochondria in a platelet was a key regulator of procoagulant platelet formation. In this elegant study, platelets with only 1–4 mitochondria were considerably more likely to become procoagulant than platelets with 5 or more mitochondria[Citation65]. This may contribute to the increased procoagulant platelet formation in Wiskott-Aldrich syndrome (WAS), as a greater proportion of platelets had three mitochondria or fewer, although additional mechanisms are also likely to be involved[Citation65]. Fewer mitochondria could mean that more cytosolic Ca2+ is taken up by each mitochondrion, increasing the likelihood that [Ca2+]mito reaches threshold for PTP opening. Differences in mitochondrial number between healthy platelets could result from small differences in packaging mitochondria into megakaryocyte proplatelets, or release of mitochondria into the circulation[Citation66].
How the supramaximal Ca2+ signal is generated is not yet clear. It could be a direct consequence of PTP opening, with Ca2+ that has accumulated in the mitochondrial matrix being rapidly released back into the cytosol[Citation50]. PTP opening appears to coincide with a decrease in [Ca2+]mito (although this could also be explained by release of the mitochondrial Ca2+ dye, Rhod-2, from the mitochondria)[Citation40]. Alternatively, lysis of intracellular organelles may release stored Ca2+. Necrosis often involves lysis of intracellular lysosomes, for example[Citation67]. In addition, plasma membrane pores or channels could be involved. Pore-forming executioner proteins are implicated in cell death and could be a route for Ca2+ entry, including MLKL in necroptosis[Citation68], gasdermin D in pyroptosis [Citation69,Citation70] and gasdermin E in secondary necrosis[Citation71]. Clarifying the mechanisms that generate the supramaximal Ca2+ could identify new anti-thrombotic targets.
Inhibiting Procoagulant Platelet Formation to Prevent Arterial Thrombosis
Selective inhibition of procoagulant platelet formation, without affecting platelet aggregation, might be a therapeutic approach to prevent arterial thrombosis. In our model, the first step uses the same molecular machinery in procoagulant and pro-aggregatory subpopulations, such as the receptors (e.g., PARs, GPVI) and Ca2+ release and inhibition of this step is unlikely to lead to selectivity. There may be some selectivity achieved by inhibiting Ca2+ entry channels, such as ORAI1 or TRPC6. Ca2+ entry is required to amplify cytosolic Ca2+ signaling to a sufficient level to lead to PTP opening, whereas aggregation can take place in the absence of extracellular Ca2+. Although procoagulant platelet formation is reduced in Orai1−/- mouse platelets and with ORAI1 inhibitors [Citation29,Citation32,Citation34,Citation35], there was limited inhibition of platelet aggregation, particularly at higher concentrations of platelet activators [Citation28,Citation72]. However, thrombus formation under arterial shear in vitro was reduced, even under non-coagulating conditions, suggesting that platelet aggregation is inhibited. Moreover, loss of ORAI1 in T cells results in severe combined immunodeficiency (SCID)[Citation73], limiting the potential use of ORAI1 blockers as anti-thrombotics.
Ca2+ uptake into mitochondria via MCU is another potential target. Mcu−/- platelets have reduced procoagulant platelet formation with no apparent effect on aggregation[Citation48]. Mcu−/- mice are viable and appear grossly normal[Citation74], suggesting that targeting MCU may be relatively safe. Moreover, dysregulation of mitochondrial Ca2+ and subsequent mPTP opening also underlies cardiac ischemia-reperfusion injury, making MCU a potential target to limit myocardial injury following arterial thrombosis.
Similarly, PTP inhibition is a potential target. CsA reduces procoagulant platelet formation [Citation12,Citation34]. CsA inhibits CypD and so reduces the Ca2+ sensitivity of PTP opening. However, CsA also has other targets, including cyclophilin A, leading to immunosuppression through inhibition of calcineurin, a Ca2+-dependent phosphatase. More selective PTP inhibitors might prevent procoagulant platelet formation without immunosuppression.
PTP opening leads to the supramaximal Ca2+ signal that we have described. As noted above, better understanding of the mechanism of the supramaximal signal may identify new anti-thrombotic targets, but we are not yet at that stage. At present, the more promising avenues for inhibiting Ca2+ signaling in procoagulant platelet formation appear to be to inhibit mitochondrial Ca2+ uptake or PTP opening.
Alternatively, platelet procoagulant activity could be directly blocked by inhibiting TMEM16F. Loss of TMEM16F causes Scott Syndrome, a moderate, very rare bleeding disorder[Citation75]. Several Tmem16f−/- mouse strains have been reported with divergent effects on bleeding time, from no difference to severe bleeding[Citation75]. Inhibition of TMEM16F might therefore be associated with bleeding. However, there are very few reported TMEM16F inhibitors and these are of little use to test whether acute inhibition of TMEM16F increases bleeding risk. R5421 was described as an inhibitor of platelet scramblase activity[Citation76], and though it may directly inhibit TMEM16F with low potency, it has off-target effects on cytosolic Ca2+ signaling and on αIIbβ3 activation[Citation77]. Similarly, although epigallocatechin gallate (EGCG), and related polyphenols, are also reported TMEM16F inhibitors [Citation78,Citation79], (though this action has been disputed [Citation80]), these polyphenols inhibit platelet aggregation through a variety of mechanisms [Citation81–86]. Understanding how these molecules affect platelet procoagulant activity could lead to more selective TMEM16F inhibitors in the future.
Finally, procoagulant platelets release PS-exposing EVs, which further promote coagulation. Indeed, EVs contribute approximately 5% of the volume of an arterial thrombi[Citation87]. Their mechanism of release is poorly understood but requires PS exposure and calpain activity [Citation88,Citation89]. Intriguingly, PS-exposing EV release can be inhibited by 2-aminoethoxydiphenylborate (2-APB), a commonly used Ca2+ channel modulator, and a related molecule, 3-(diphenylphosphino)-1-propylamine (DP3A). This inhibition was not due to inhibition of PS exposure or calpain activity, indicating that there are as yet unidentified components to the EV release machinery that can be targeted[Citation9].
Conclusion
The mechanisms that commit a platelet to becoming procoagulant are still incompletely understood. A complex interplay between cytosolic and mitochondrial Ca2+ signals is required, leading to permeability transition pore opening and an overload of cytosolic Ca2+. In this, procoagulant platelet formation resembles a rapid form of regulated necrosis [Citation57,Citation67,Citation90]. Like necrotic cells, procoagulant platelets may have pro-inflammatory and well as pro-coagulant effects[Citation91]. Inhibiting the formation and functions of procoagulant platelets is a potential therapeutic approach but requires us to further decipher the Ca2+ signaling code that commits a platelet to becoming procoagulant.
Acknowledgements
The authors were supported by British Heart Foundation project grants PG/17/45/33071 and PG/20/12/34982.
Disclosure statement
The authors have no financial or scientific conflicts of interest related to this work.
Additional information
Funding
References
- Huber K, Bates ER, Valgimigli M, Wallentin L, Kristensen SD, Anderson JL, Lopez Sendon JL, Tubaro M, Granger CB, Bode C, et al. Antiplatelet and anticoagulation agents in acute coronary syndromes: what is the current status and what does the future hold? Am Heart J 2014;1685:611–621. DOI:https://doi.org/10.1016/j.ahj.2014.06.014.
- Capranzano P, Capodanno D. Dual antiplatelet therapy in patients with diabetes mellitus: special considerations. Expert Rev Cardiovasc Ther 2013;113:307–317. DOI:https://doi.org/10.1586/erc.13.3.
- Jackson SP. Arterial thrombosis-insidious, unpredictable and deadly. Nat Med 2011;1711:1423–1436. DOI:https://doi.org/10.1038/nm.2515.
- Munnix ICA, Kuijpers MJE, Auger J, Thomassen CMLGD, Panizzi P, van Zandvoort MAM, Rosing J, Bock PE, Watson SP, Heemskerk JWM, Munnix ICAA, Kuijpers MJEE, Auger J, Thomassen CMLGDLGD, Panizzi P, van Zandvoort MAMM, Rosing J, Bock PE, Watson SP, Heemskerk JWMM. Segregation of platelet aggregatory and procoagulant microdomains in thrombus formation - regulation by transient integrin activation. Arterioscler Thromb Vasc Biol 2007;2711:2484–2490. DOI:https://doi.org/10.1161/ATVBAHA.107.151100.
- Heemskerk JWM, Mattheij NJA, Cosemans JMEM. Platelet-based coagulation: different populations, different functions. J Thromb Haemost 2013;111:2–16. DOI:https://doi.org/10.1111/jth.12045.
- Monroe DM, Hoffman M, Roberts HR. Platelets and Thrombin Generation. Arterioscler Thromb Vasc Biol 2002;229:1381–1389. DOI:https://doi.org/10.1161/01.ATV.0000031340.68494.34.
- Monroe DM, Hoffman M. What does it take to make the perfect clot? Arterioscler Thromb Vasc Biol 2006;261:41–48. DOI:https://doi.org/10.1161/01.ATV.0000193624.28251.83.
- Varon D, Shai E. Platelets and their microparticles as key players in pathophysiological responses. J Thromb Haemost 2015;13:S40–S46. DOI:https://doi.org/10.1111/jth.12976.
- Wei H, Davies JE, Harper MT. 2-Aminoethoxydiphenylborate (2-APB) inhibits release of phosphatidylserine-exposing extracellular vesicles from platelets. Cell Death Discov 2020;61:10. DOI:https://doi.org/10.1038/s41420-020-0244-9.
- Kuijpers MJE, Munnix ICA, Cosemans JMEM, Van Vlijmen BJ, Reutelingsperger CPM, Egbrink MGAO, Heemskerk JWM. Key role of platelet procoagulant activity in tissue factor- and collagen-dependent thrombus formation in arterioles and venules in vivo - Differential sensitivity to thrombin inhibition. Microcirculation 2008;154:269–282. DOI:https://doi.org/10.1080/10739680701653517.
- Dachary-Prigent J, Freyssinet J-M, Pasquet J-M, Carron J-C, Nurden AT Annexin V as a probe of aminophospholipid exposure and platelet membrane vesiculation: a flow cytometry study showing a role for free sulfhydryl groups.
- Jobe SM, Wilson KM, Leo L, Raimondi A, Molkentin JD, Lentz SR, Di Paola J. Critical role for the mitochondrial permeability transition pore and cyclophilin D in platelet activation and thrombosis. Blood 2008;1113:1257–1265. DOI:https://doi.org/10.1182/blood-2007-05-092684.
- Harper MT, Poole AW. PKC inhibition markedly enhances Ca2+ signaling and phosphatidylserine exposure downstream of protease-activated receptor-1 but not protease-activated receptor-4 in human platelets. J Thromb Haemost 2011;98:1599–1607. DOI:https://doi.org/10.1111/j.1538-7836.2011.04393.x.
- Dasgupta SK, Guchhait P, Thiagarajan P. Lactadherin binding and phosphatidylserine expression on cell surface-comparison with annexin A5. Transl Res 2006;1481:19–25. DOI:https://doi.org/10.1016/j.lab.2006.03.006.
- SHI J, Pipe SW, Rasmussen JT, CW HEEGAARD, Gilbert GE. Lactadherin blocks thrombosis and hemostasis in vivo: correlation with platelet phosphatidylserine exposure. J Thromb Haemost 2008;67:1167–1174. DOI:https://doi.org/10.1111/j.1538-7836.2008.03010.x.
- London FS, Marcinkiewicz M, Walsh PN. PAR-1-stimulated factor IXa binding to a small platelet subpopulation requires a pronounced and sustained increase of cytoplasmic calcium. Biochemistry 2006;4523:7289–7298. DOI:https://doi.org/10.1021/bi060294m.
- London FS, Marcinkiewicz M, Walsh PN. A subpopulation of platelets responds to Thrombin- or SFLLRN-stimulation with binding sites for Factor IXa. J Biol Chem 2004;27919:19854–19859. DOI:https://doi.org/10.1074/jbc.M310624200.
- Panteleev MA, Ananyeva NM, Greco NJ, Ataullakhanov FI, Saenko EL. Two subpopulations of thrombin-activated platelets differ in their binding of the components of the intrinsic factor X-activating complex. J Thromb Haemost 2005;311:2545–2553. DOI:https://doi.org/10.1111/j.1538-7836.2005.01616.x.
- Berny MA, Munnix ICA, Auger JM, Schols SEM, Cosemans JMEM, Panizzi P, Bock PE, Watson SP, McCarty OJT, Heemskerk JWM. Spatial distribution of factor Xa, Thrombin, and Fibrin(ogen) on Thrombi at Venous Shear Kleinschnitz C, editor. PLoS ONE 2010;54:e10415. DOI:https://doi.org/10.1371/journal.pone.0010415.
- Mattheij NJA, Gilio K, van Kruchten R, Jobe SM, Wieschhaus AJ, Chishti AH, Collins P, Heemskerk JWM, Cosemans JMEM. Dual mechanism of integrin alpha(IIb)beta(3) closure in procoagulant platelets. J Biol Chem 2013;28819:13325–13336. DOI:https://doi.org/10.1074/jbc.M112.428359.
- Choo H-J, Kholmukhamedov A, Zhou C, Jobe S. Inner mitochondrial membrane disruption links apoptotic and agonist-initiated phosphatidylserine externalization in platelets. Arterioscler Thromb Vasc Biol 2017;378:1503–1512. DOI:https://doi.org/10.1161/ATVBAHA.117.309473.
- Baig AA, Haining EJ, Geuss E, Beck S, Swieringa F, Wanitchakool P, Schuhmann MK, Stegner D, Kunzelmann K, Kleinschnitz C, et al. TMEM16F-mediated platelet membrane phospholipid scrambling is critical for hemostasis and thrombosis but not thromboinflammation in mice—brief report. Arterioscler Thromb Vasc Biol 2016;3611:2152–2157. DOI:https://doi.org/10.1161/ATVBAHA.116.307727.
- Bevers EM, Comfurius P, Zwaal RFA. Platelet procoagulant activity: physiological significance and mechanisms of exposure. Blood Rev 1991;53:146–154. DOI:https://doi.org/10.1016/0268-960X(91)90031-7.
- Fox JEB, Austin CD, Reynolds CC, Steffen PK. Evidence that agonist-induced activation of calpain causes the shedding of procoagulant-containing microvesicles from the membrane of aggregating platelets. J Biol Chem 1991;26620:13289–13295. DOI:https://doi.org/10.1016/S0021-9258(18)98837-X.
- Wei H, Harper MT. ABT-737 triggers caspase-dependent inhibition of platelet procoagulant extracellular vesicle release during apoptosis and secondary necrosis in vitro. Thromb Haemost 2019;19:1665–1674.
- Morel O, Jesel L, Freyssinet JM, Toti F. Cellular mechanisms underlying the formation of circulating microparticles. Arterioscler Thromb Vasc Biol 2011;311:15–26. DOI:https://doi.org/10.1161/ATVBAHA.109.200956.
- Watson SP, Auger JM, McCarty OJT, Pearce AC. GPVI and integrin αIIbβ3 signaling in platelets. J Thromb Haemost 2005;38:1752–1762. DOI:https://doi.org/10.1111/j.1538-7836.2005.01429.x.
- Braun A, Varga-Szabo D, Kleinschnitz C, Pleines I, Bender M, Austinat M, Boesl M, Stoll G, Nieswandt B. Orai1 (CRACM1) is the platelet SOC channel and essential for pathological thrombus formation. Blood 2009;1139:2056–2063. DOI:https://doi.org/10.1182/blood-2008-07-171611.
- Gilio K, van Kruchten R, Braun A, Berna-Erro A, Feijge MAH, Stegner D, van der Meijden PEJ, Kuijpers MJE, Varga-Szabo D, Heemskerk JWM, et al. Roles of platelet STIM1 and orai1 in glycoprotein VI- and Thrombin-dependent procoagulant activity and thrombus formation. J Biol Chem 2010;28531:23629–23638. DOI:https://doi.org/10.1074/jbc.M110.108696.
- Gamage TH, Gunnes G, Lee RH, Louch WE, Holmgren A, Bruton JD, Lengle E, Kolstad TRS, Revold T, Amundsen SS, et al. STIM1 R304W causes muscle degeneration and impaired platelet activation in mice. Cell Calcium 2018;76:87–100. DOI:https://doi.org/10.1016/j.ceca.2018.10.001.
- Varga-Szabo D, Braun A, Kleinschnitz C, Bender M, Pleines I, Pham M, Renné T, Stoll G, Nieswandt B. The calcium sensor STIM1 is an essential mediator of arterial thrombosis and ischemic brain infarction. J Exp Med 2008;2057:1583–1591. DOI:https://doi.org/10.1084/jem.20080302.
- Chen W, Thielmann I, Gupta S, Subramanian H, Stegner D, van Kruchten R, Dietrich A, Gambaryan S, Heemskerk JWM, Hermanns HM, et al. Orai1-induced store-operated Ca 2+ entry enhances phospholipase activity and modulates canonical transient receptor potential channel 6 function in murine platelets. J Thromb Haemost 2014;124:528–539. DOI:https://doi.org/10.1111/jth.12525.
- Harper MT, Londono JEC, Quick K, Londono JC, Flockerzi V, Philipp SE, Birnbaumer L, Freichel M, Poole AW. Transient receptor potential channels function as a coincidence signal detector mediating phosphatidylserine exposure. Sci Signal 2013;6281:281. DOI:https://doi.org/10.1126/scisignal.2003701.
- Abbasian N, Millington-Burgess SL, Chabra S, Malcor JD, Harper MT. Supramaximal calcium signaling triggers procoagulant platelet formation. Blood Adv 2020;41:154–164. DOI:https://doi.org/10.1182/bloodadvances.2019000182.
- Harper MT, Poole AW. Store-operated calcium entry and non-capacitative calcium entry have distinct roles in thrombin-induced calcium signalling in human platelets. Cell Calcium 2011;504:351–358. DOI:https://doi.org/10.1016/j.ceca.2011.06.005.
- Fung CYE, Jones S, Ntrakwah A, Naseem KM, Farndale RW, Mahaut-Smith MP, Platelet C. 2+ responses coupled to glycoprotein VI and Toll-like receptors persist in the presence of endothelial-derived inhibitors: roles for secondary activation of P2X1 receptors and release from intracellular Ca 2+ stores. Blood 2012;11915:3613–3621. DOI:https://doi.org/10.1182/blood-2011-10-386052.
- Aliotta A, Bertaggia Calderara D, Zermatten MG, Alberio L. Sodium–calcium exchanger reverse mode sustains dichotomous ion fluxes required for procoagulant COAT platelet formation. Thromb Haemost 2020.
- Heemskerk JWM, Willems GM, Rook MB, Sage SO. Ragged spiking of free calcium in ADP-stimulated human platelets: regulation of puff-like calcium signals in vitro and ex vivo. J Physiol 2001;5353:625–635. DOI:https://doi.org/10.1111/j.1469-7793.2001.00625.x.
- Purvis JE, Chatterjee MS, Brass LF, Diamond SL. Amolecular signaling model of platelet phosphoinositide and calcium regulation during homeostasis and P2Y1 activation. Blood 2008;11210:4069–4079. DOI:https://doi.org/10.1182/blood-2008-05-157883.
- Obydennyy S, Sveshnikova AN, Ataullakhanov FI, Panteleev MA. Dynamics of calcium spiking, mitochondrial collapse and phosphatidylserine exposure in single platelets during activation. J Thromb Haemost 2015;13:649.
- Bye AP, Gibbins JM, Mahaut-Smith MP. Ca2+ waves coordinate purinergic receptor-evoked integrin activation and polarization. Sci Signal 2020;13:615. DOI:https://doi.org/10.1126/scisignal.aav7354.
- Glancy B, Balaban RS. Role of mitochondrial Ca 2+ in the regulation of cellular energetics. Biochemistry 2012;5114:2959–2973. DOI:https://doi.org/10.1021/bi2018909.
- Shoshan-Barmatz V, De S. Mitochondrial VDAC, the Na+/Ca2+ exchanger, and the Ca2+ uniporter in Ca2+ dynamics and signaling. Advances in experimental medicine and biology Vol. 981. Springer New York LLC; 2017. 323–347.
- Belosludtsev KN, Dubinin MV, Belosludtseva NV, Mironova GD. Mitochondrial Ca2+ transport: mechanisms, molecular structures, and role in cells. Biochemistry (Moscow) 2019;846:593–607. DOI:https://doi.org/10.1134/S0006297919060026.
- Nemani N, Shanmughapriya S, Madesh M. Molecular regulation of MCU: implications in physiology and disease. Cell Calcium 2018;74:86–93. DOI:https://doi.org/10.1016/j.ceca.2018.06.006.
- Patron M, Checchetto V, Raffaello A, Teardo E, VecellioReane D, Mantoan M, Granatiero V, Szabò I, DeStefani D, Rizzuto R. MICU1 and MICU2 finely tune the mitochondrial Ca2+ uniporter by exerting opposite effects on MCU activity. Mol Cell 2014;535:726–737. DOI:https://doi.org/10.1016/j.molcel.2014.01.013.
- Mammucari C, Gherardi G, Rizzuto R. Structure, activity regulation, and role of the mitochondrial calcium uniporter in health and disease. Front Oncol 2017;7. DOI:https://doi.org/10.3389/fonc.2017.00139.
- Kholmukhamedov A, Janecke R, Choo H-J, Jobe SM. The mitochondrial calcium uniporter regulates procoagulant platelet formation. J Thromb Haemost 2018;1611:2315–2321. DOI:https://doi.org/10.1111/jth.14284.
- Brenner C, Moulin M. Physiological Roles of the Permeability Transition Pore. Circ Res 2012;1119:1237–1247. DOI:https://doi.org/10.1161/CIRCRESAHA.112.265942.
- Bernardi P, von Stockum S. The permeability transition pore as a Ca2+ release channel: new answers to an old question. Cell Calcium 2012;521:22–27. DOI:https://doi.org/10.1016/j.ceca.2012.03.004.
- Sveshnikova AN, Ataullakhanov FI, Panteleev MA. Compartmentalized calcium signaling triggers subpopulation formation upon platelet activation through PAR1. Mol Biosyst 2015;114:1052–1060. DOI:https://doi.org/10.1039/C4MB00667D.
- Arachiche A, Kerbiriou-Nabias D, Garcin I, Letellier T, Dachary-Prigent J. Rapid procoagulant phosphatidylserine exposure relies on high cytosolic calcium rather than on mitochondrial depolarization. Arterioscler Thromb Vasc Biol 2009;2911:1883–1889. DOI:https://doi.org/10.1161/ATVBAHA.109.190926.
- Choo H-J, Saafir TB, Mkumba L, Wagner MB, Jobe SM. Mitochondrial calcium and reactive oxygen species regulate agonist-initiated platelet phosphatidylserine exposure. Arterioscler Thromb Vasc Biol 2012;3212:2946–2955. 2946-+. DOI:https://doi.org/10.1161/ATVBAHA.112.300433.
- Weinberg JM, Davis JA, Venkatachalam MA. Cytosolic-free calcium increases to greater than 100 micromolar in ATP-depleted proximal tubules. J Clin Invest 1997;1003:713–722. DOI:https://doi.org/10.1172/JCI119584.
- Dong Z, Saikumar P, Griess GA, Weinberg JM, Venkatachalam MA. Intracellular Ca2+ thresholds that determine survival or death of energy-deprived cells. Am J Pathol 1998;1521:231–240.
- Hyrc K, Handran SD, Rothman SM, Goldberg MP. Ionized intracellular calcium concentration predicts excitotoxic neuronal death: observations with low-affinity fluorescent calcium indicators. The Journal of Neuroscience 1997;1717:6669–6677. DOI:https://doi.org/10.1523/JNEUROSCI.17-17-06669.1997.
- Hua VM, Abeynaike L, Glaros E, Campbell H, Pasalic L, Hogg PJ, Chen VMY, Vu Minh H, Abeynaike L, Glaros E, et al. Necrotic platelets provide a procoagulant surface during thrombosis. Blood 2015;12626:2852–2862. DOI:https://doi.org/10.1182/blood-2015-08-663005.
- Yang H, Kim A, David T, Palmer D, Jin T, Tien J, Huang F, Cheng T, Coughlin SR, Jan YN, et al. TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell 2012;1511:111–122. DOI:https://doi.org/10.1016/j.cell.2012.07.036.
- Basse F, Stout JG, Sims PJ, Wiedmer T. Isolation of an erythrocyte membrane protein that mediates Ca2+-dependent transbilayer movement of phospholipid. J Biol Chem 1996;27129:17205–17210. DOI:https://doi.org/10.1074/jbc.271.29.17205.
- Kojima H, Newtonnash D, Weiss HJ, Zhao J, Sims PJ, Wiedmer T. PRODUCTION AND CHARACTERIZATION OF TRANSFORMED B-LYMPHOCYTES EXPRESSING THE MEMBRANE DEFECT OF SCOTT SYNDROME. J Clin Invest 1994;946:2237–2244. DOI:https://doi.org/10.1172/JCI117586.
- Verhoven B, Schlegel RA, Williamson P. RAPID LOSS AND RESTORATION OF LIPID ASYMMETRY BY DIFFERENT PATHWAYS IN RESEALED ERYTHROCYTE-GHOSTS. Biochim Biophys Acta 1992;11041:15–23. DOI:https://doi.org/10.1016/0005-2736(92)90126-7.
- Malvezzi M, Chalat M, Janjusevic R, Picollo A, Terashima H, Menon AK, Accardi A. Ca2+-dependent phospholipid scrambling by a reconstituted TMEM16 ion channel. Nat Commun 2013;4. DOI:https://doi.org/10.1038/ncomms3367.
- Moldoveanu T, Hosfield CM, Lim D, Elce JS, Jia Z, Davies PL. A Ca2+ switch aligns the active site of calpain. Cell 2002;1085:649–660. DOI:https://doi.org/10.1016/S0092-8674(02)00659-1.
- Ye W, Han TW, Nassar LM, Zubia M, Jan YN, Jan LY. Phosphatidylinositol-(4, 5)-bisphosphate regulates calcium gating of small-conductance cation channel TMEM16F. Proc Natl Acad Sci U S A 2018;1157:E1667–E1674. DOI:https://doi.org/10.1073/pnas.1718728115.
- Obydennyi SI, Artemenko EO, Sveshnikova AN, Ignatova AA, Varlamova TV, Gambaryan S, Lomakina GY, Ugarova NN, Kireev II, Ataullakhanov FI, et al. Mechanisms of increased mitochondria-dependent necrosis in Wiskott-Aldrich syndrome platelets. Haematologica 2020;1054:1095–1106. DOI:https://doi.org/10.3324/haematol.2018.214460.
- Boudreau LH, Duchez AC, Cloutier N, Soulet D, Martin N, Bollinger J, Paré A, Rousseau M, Naika GS, Lévesque T, et al. Platelets release mitochondria serving as substrate for bactericidal group IIA-secreted phospholipase a to promote inflammation. Blood 2014;12414:2173–2183. DOI:https://doi.org/10.1182/blood-2014-05-573543.
- Vanden Berghe T, Linkermann A, Jouan-Lanhouet S, Walczak H, Vandenabeele P. Regulated necrosis: the expanding network of non-apoptotic cell death pathways. Nat Rev Mol Cell Biol 2014;152:134–146.
- Kolbrink B, Riebeling T, Kunzendorf U, Krautwald S. Plasma membrane pores drive inflammatory cell death. Front Cell Dev Biol 2020;8. DOI:https://doi.org/10.3389/fcell.2020.00817.
- Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, Lieberman J. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature 2016;5357610:153–158. DOI:https://doi.org/10.1038/nature18629.
- Sborgi L, Rühl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, Farady CJ, Müller DJ, Broz P, Hiller S. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. Embo J 2016;3516:1766–1778. DOI:https://doi.org/10.15252/embj.201694696.
- Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun 2017;8. DOI:https://doi.org/10.1038/ncomms14128.
- Bergmeier W, Masatsugu OH, McCarl CA, Roden RC, Bray PF, Feske S. R93W mutation in Orai1 causes impaired calcium influx in platelets. Blood 2009;1133:675–678. DOI:https://doi.org/10.1182/blood-2008-08-174516.
- Lacruz RS, Feske S. Diseases caused by mutations in ORAI1 and STIM1. Ann N Y Acad Sci 2015;13561:45–79. DOI:https://doi.org/10.1111/nyas.12938.
- Murphy E, Pan X, Nguyen T, Liu J, Holmström KM, Finkel T. Unresolved questions from the analysis of mice lacking MCU expression. Biochem Biophys Res Commun 2014;4494:384–385. DOI:https://doi.org/10.1016/j.bbrc.2014.04.144.
- Millington-Burgess SL, Harper MT. Gene of the issue: ANO6 and Scott Syndrome. Platelets 2019;317:964–967. DOI:https://doi.org/10.1080/09537104.2019.1693039.
- Dekkers DWC, Comfurius P, Vuist WMJ, Billheimer JT, Dicker I, Weiss HJ, Zwaal RFA, Bevers EM. Impaired Ca2+-induced tyrosine phosphorylation and defective lipid scrambling in erythrocytes from a patient with Scott syndrome: A study using an inhibitor for scramblase that mimics the defect in Scott syndrome. Blood 1998;916:2133–2138. DOI:https://doi.org/10.1182/blood.V91.6.2133.
- Millington-Burgess SL, Bonna AM, Rahman T, Harper MT. Ethaninidothioic acid (R5421) is not a selective inhibitor of platelet phospholipid scramblase activity. Br J Pharmacol 2020;17717:4007–4020. DOI:https://doi.org/10.1111/bph.15152.
- Suzuki T, Suzuki J, Nagata S. Functional Swapping between Transmembrane Proteins TMEM16A and TMEM16F. J Biol Chem 2014;28911:7438–7447. DOI:https://doi.org/10.1074/jbc.M113.542324.
- Watanabe R, Sakuragi T, Noji H, Nagata S. Single-molecule analysis of phospholipid scrambling by TMEM16F. Proc Natl Acad Sci U S A 2018;11512:3066–3071.
- Le T, Le SC, Zhang Y, Liang P, Yang H. Evidence that polyphenols do not inhibit the phospholipid scramblase TMEM16F. J Biol Chem 2020;29535:12537–12544. DOI:https://doi.org/10.1074/jbc.AC120.014872.
- Faggio C, Sureda A, Morabito S, Sanches-Silva A, Mocan A, Nabavi SF, Nabavi SM. Flavonoids and platelet aggregation: A brief review. Eur J Pharmacol 2017;807:91–101. DOI:https://doi.org/10.1016/j.ejphar.2017.04.009.
- Khan H, Jawad M, Kamal MA, Baldi A, Xiao J, Nabavi SM, Daglia M. Evidence and prospective of plant derived flavonoids as antiplatelet agents: strong candidates to be drugs of future. Food Chem Toxicol 2018;119:355–367. DOI:https://doi.org/10.1016/j.fct.2018.02.014.
- Sanchez M, Romero M, Gomez-Guzman M, Tamargo J, Perez-Vizcaino F, Duarte J. Cardiovascular effects of flavonoids. Curr Med Chem 2018;26: 6991-7034.
- Ed Nignpense B, Chinkwo KA, Blanchard CL, Santhakumar AB. Polyphenols: modulators of platelet function and platelet microparticle generation? Int J Mol Sci 2020;21:1.
- Wright B, Spencer JPE, Lovegrove JA, Gibbins JM. Insights into dietary flavonoids as molecular templates for the design of anti-platelet drugs. Cardiovasc Res 2013;971:13–22. DOI:https://doi.org/10.1093/cvr/cvs304.
- Lopez JJ, El Haouari M, Jardin I, Alonso N, Regodon S, Diez-Bello R, Redondo PC, Rosado JA. Flavonoids and Platelet-Derived Thrombotic Disorders. Curr Med Chem 2020;2639:7035–7047. DOI:https://doi.org/10.2174/0929867325666180417170218.
- Chernysh IN, Nagaswami C, Kosolapova S, Peshkova AD, Cuker A, Cines DB, Cambor CL, Litvinov RI, Weisel JW. The distinctive structure and composition of arterial and venous thrombi and pulmonary emboli. Sci Rep 2020;101:1. DOI:https://doi.org/10.1038/s41598-020-59526-x.
- Wei H, Malcor J, Harper M. Lipid rafts are essential for release of phosphatidylserine-exposing extracellular vesicles from platelets. Sci Rep 2018;81:9987. DOI:https://doi.org/10.1038/s41598-018-28363-4.
- Owens A, Mackman N. Microparticles in Hemostasis and Thrombosis. Circ Res 2011;108:1284–1297. DOI:https://doi.org/10.1161/CIRCRESAHA.110.233056.
- Jackson SP, Schoenwaelder SM. Procoagulant platelets: are they necrotic? Blood 2010;11612:2011–2018. DOI:https://doi.org/10.1182/blood-2010-01-261669.
- Ghasemzadeh M, Kaplan ZS, Alwis I, Schoenwaelder SM, Ashworth KJ, Westein E, Hosseini E, Salem HH, Slattery R, McColl SR, et al. The CXCR1/2 ligand NAP-2 promotes directed intravascular leukocyte migration through platelet thrombi. Blood 2013;12122:4555–4566. DOI:https://doi.org/10.1182/blood-2012-09-459636.