
Abstract
Liver failure and cirrhosis are characterized by abnormal hemostasis with aberrant platelet activation. In particular, the consequences of cholestatic liver disease and molecular mechanisms, including the role of bile acids leading to impaired platelet responses, are not well understood. Here, we demonstrate that bile acids inhibit human and murine platelet activation, adhesion and spreading, leading to reduced thrombus formation under flow conditions. We identified the G-protein coupled receptor TGR5 in platelets and provide support for its role as mediator of bile acid-induced impairment of platelet activation. In the liver, TGR5 couples to Gαs proteins, activates the adenylate cyclase to induce a transient cAMP rise and stimulates the MAPK signaling pathway to regulate cholangiocyte proliferation, hepatocyte survival and inflammation. In this report, we demonstrate that the genetic deficiency of TGR5 in mice led to enhanced platelet activation and thrombus formation, suggesting that TGR5 plays an important role in hemostasis. Mechanistically, platelet inhibition is achieved by TGR5 mediated PKA activation and modulation of AKT and ERK1/2 phosphorylation. Thus, this report provides evidence for the ability of TGR5 ligands to reduce platelet activation and identifies TGR5 agonism as a new target for the prevention of cardiovascular diseases.
Plain Language Summary
What is the context?
Liver failure or cirrhosis are related to impaired hemostasis and a role of bile acids in impaired platelet responses is known but only less understood.
Platelets express the bile acid receptor FXR. Ligand binding to the FXR on platelets causes a shift in platelet reactivity and is atheroprotective suggesting that the FXR is a potential target for the prevention of atherothrombotic diseases.
What is new?
Treatment of murine and human blood with bile acids in low molecular quantity led to reduced platelet activation, adhesion and thrombus formation.
The bile acid receptor TGR5 was identified on human and murine platelets.
TGR5 plays an important role in hemostasis because TGR5 deficient mice showed elevated platelet reactivity and enhanced thrombus formation.
Loss of TGR5 led to enhanced PKA activation and modulated the phosphorylation of MAPK such as AKT and ERK1/2.
What is the impact?
Impairment of platelet activation by bile acids is mediated by TGR5 via the protein kinase A signaling pathway.
Our findings provide evidence for the modulation of TGR5 activation as a potential new target of both, anti-platelet therapy in cardiovascular diseases and the restoration of hemostasis upon liver injury.
Introduction
Platelet activation and thrombus formation are crucial to achieve hemostasis after vascular damage. However, dysregulated platelet activation can lead to acute thrombotic complications leading to vessel occlusion at the sides of atherosclerotic plaque rupture, known as pathophysiological mechanism that is causative for myocardial infarction or stroke.Citation1,Citation2 Thus, the hemostatic system is susceptible to a balance of pro- and anti-hemostatic processes to avoid both, bleeding and thrombotic complications. Liver failure and cirrhosis are characterized by abnormal hemostasis because many proteins involved in hemostasis are produced in the liver and a reduced capacity to clear hemostatic proteins has been described for patients with liver disease.Citation3,Citation4 Thrombocytopenia decreased platelet production and a reduced half-life of platelets are causative for defective hemostasis.Citation5–7 In addition, defective platelet activation and aggregation are described upon liver failure. However, the consequences of cholestatic liver disease and the molecular mechanisms leading to impaired platelet responses are not well understood. It was shown that bile acids inhibit platelet aggregation in vitroCitation8,Citation9 and in vivo.Citation10 A very elegant model is the surgical ligation of the common bile duct known as the bile duct ligation model (BDL), where obstructive cholestatic injury in rodents is induced to allow the analysis of molecular and cellular processes as consequences of inappropriate bile flow.Citation11 In BDL mice, elevated nitric oxide (NO) and prostacyclin levels induced the phosphorylation of the platelet inhibitor vasodilator-stimulated phosphoprotein (VASP) that together with elevated bile acid plasma levels led to prolonged platelet activation defects and impaired hemostasis in these mice.Citation12,Citation13 Primary bile acids are produced in the liver and are secreted as glycine and taurine conjugate into the intestine. Bile acids undergo several modifications induced by enteric anaerobic bacteria to produce secondary bile acids in the gut. They have been established as important signaling molecules involved in liver regeneration, tissue remodeling, energy balance, aging, immune homeostasis and intestinal function.Citation14 The most studied bile acid receptors are the farnesoid X receptor (FXR) and the Takeda G-protein-coupled receptor 5 (TGR5). A role for the FXR has already been demonstrated by Moraes and colleagues.Citation15 However, the presence of TGR5 in platelets and its role in platelet activation has not been described to date. The G-protein coupled receptor (GPCR) TGR5 is a member of the rhodopsin-like subfamily of GPCRs and is expressed in almost all tissues or cell types.Citation14 TGR5 is activated by conjugated and unconjugated bile acids, whereas taurine-conjugated bile acids are more potent activators. A role for TGR5 has been described in the liver and in metabolic diseases, such as diabetes and atherosclerosis.Citation14,Citation16–18 In this report, we demonstrate that bile acids strongly affect platelet activation, adhesion and thrombus formation. We identified TGR5 as a mediator of impaired platelet activation that has not been described in platelets so far. Genetic deficiency of TGR5 led to enhanced platelet activation and thrombus formation, suggesting that TGR5 plays an important role in hemostasis at physiological bile acid plasma levels.
Material and methods
Chemicals and antibodies
For platelet isolation, apyrase (Grade II from potato, #A7646, Sigma-Aldrich) and prostacyclin (#P5515, Calbiochem) were used. Platelets were activated with collagen-related peptide (CRP; Richard Farndale, University of Cambridge, Cambridge, UK), adenosine diphosphate (ADP; #A2754, Sigma-Aldrich), the thromboxane A2 analog U46619 (U46; #1932, Tocris), PAR4 peptide (PAR4; St. Louis, Missouri, MO, USA), thrombin (Thr; #10602400001, Roche Diagnostics, Germany), or convulxin (CVX, ALX-350-100-C050, ENZO Life Sciences). For immunoblotting antibodies targeting phosphorylated protein kinase (PK)A Substrate (RRXS*/T*, #9624, Cell Signaling, 1:1000), total Akt (#9272S, Cell Signaling, 1:1000), and phosphorylated Akt Ser473 (#9271S, Cell Signaling, 1:1000), total ERK1/2 (#4695, Cell Signaling, 1:1000) and phosphorylated ERK1/2 (#4370, Cell Signaling, 1:1000), total VASP (#3112, Cell Signaling, 1:1000) and phosphorylated VASP(#3111, Cell Signaling, 1:500), α-Tubulin (#2144, Cell Signaling, 1:2000), glyceraldehyde 3-phosphate dehydrogenase (GAPDH, #2118S, Cell Signaling, 1:2000) and horseradish peroxidase (HRP)-linked anti-rabbit secondary antibodies (#NA9340, GE Healthcare, 1:2500) were used. For immunoblotting targeting human TGR5, an anti-TGR5 antiserum (M39) was raised against amino acids 298–318 of human TGR5 (NP_733800) and characterized using TGR5-YFP transfected HEK293 cells and human gallbladder tissue as previously described.Citation19 Glyceraldehyde 3-phosphate dehydrogenase (GAPDH, # Sc -32233, Santa Cruz Biotechnology, 1:10000) and horseradish peroxidase-conjugated anti-mouse secondary antibody (#P0447, DAKO, 1:5000) served as loading control.
For flow cytometric analysis fluorophore conjugated antibodies labeling P-selectin (murine: #D200, Emfret Analytics; human: #555524, BD Biosciences), active integrin αIIbβ3 (murine: #D200; Emfret Analytics; human: #340507, BD Biosciences), GPIbα (#M040-2 Emfret Analytics), GPVI (#M011-1, Emfret Analytics), integrin α5 (#M080-1, Emfret Analytics) and integrin β3 (#M031-1, Emfret Analytics) were used. For experiments with bile acids, human and murine platelets were treated with the bile acids TCDC (taurochenodeoxycholic acid; #580221, Sigma-Aldrich) or TLC (taurolithocholic acid; # 903922, Sigma-Aldrich). As TLC is dissolved in DMSO, DMSO treatment served as control for TLC treated samples.
Study approval
All animal experiments were conducted according to the Declaration of Helsinki and approved by the Ethics Committee of the State Ministry of Agriculture, Nutrition and Forestry State of North Rhine-Westphalia, Germany (Reference number: AZ 81-02.05.40.21.041).
Experiments using human tissues and blood were reviewed and approved by the Ethics Committee of the Heinrich-Heine-University. All healthy volunteers provided their written informed consent to participate in this study according to the Ethics Committee of the University Clinic of Duesseldorf, Germany (2018-140-kFogU, study number: 2018064710). This study was conducted according to the Declaration of Helsinki and the International Council for Harmonization Guidelines on Good Clinical Practice.
Animals
Gene-targeted mice lacking TGR5 (TGR5-/-) and the corresponding wild-type littermates were bred from breeder pairs and genotyped by PCR. Experiments were performed with male and female mice aged 2–4 months. Mice were maintained in an environmentally controlled room at 22 ± 1°C with a 12 h day-night cycle. Mice were housed in Macrolon cages type III with ad libitum access to food (standard chow diet) and water.
Human sample preparation
Whole blood: Fresh citrate-anticoagulated blood (BD-Vacutainer®; Becton, Dickinson and Company; #367714) was collected from healthy volunteers and centrifuged at 200 g for 10 min at room temperature (RT). After centrifugation, the platelet-rich plasma (PRP) was collected. For platelet isolation, PBS was added (pH 6.5; supplemented with 2.5 U/mL apyrase and 1 μM PGI2) in a volumetric ratio of 1:1 to PRP, followed by centrifugation at 1000 g for 6 min. The platelet pellet was resuspended in human Tyrode´s buffer (pH 7.4, 140 mM NaCl2, 2.8 mM KCl, 12 mM NaHCO3, 0.5 mM Na2HPO4, 5.5 mM Glucose). The platelet number was measured using a hematology analyzer (Sysmex - KX21N) and adjusted for the following experiments.
Gallbladder tissue: Gallbladder tissue was obtained from patients undergoing cholecystectomy for chronic gallstone disease, as part of a resection of a pancreatic or gastric cancer, or as part of a hepatectomy for metastasis resection as described before.Citation19 Tissue was obtained immediately after cholecystectomy and directly washed with ice-cold phosphate-buffered saline (PBS) and divided as follows. Part of the tissue was shock frozen in liquid nitrogen and stored at −80°C until used for RNA isolation and cryosectioning. HEK293 cells underwent co-transfection, with a construct containing a cAMP-sensitive reporter gene, rat TGR5-YFP, and a Renilla expression vector using LipofectAMINE (Invitrogen) as described earlier.Citation19
Mouse platelet preparation
Mice were given a regulated dose of isoflurane (2%) and oxygen (1 L/min) to induce anesthesia. The blood was drawn from the retrobulbar venous plexus using glass capillaries coated in sodium heparin. The isolation of murine platelets was done as previously mentioned.Citation20 Blood was swiftly drawn into a 300 mL heparin solution (20 U/mL in PBS). The samples of whole blood were centrifuged at 250 g for 5 min at room temperature. Centrifugation at 50 g for 6 min at room temperature was used to extract platelet-rich plasma (PRP). The PRP was centrifuged twice at 650 g for 5 min at room temperature in murine Tyrode’s buffer (136 mM NaCl2, 0.4 mM Na2HPO4, 2.7 mM KCl, 12 mM NaHCO3, 0.1% glucose, 0.35% bovine serum albumin (BSA), pH 7.4), containing apyrase (0.02 U/mL) and prostacyclin (0.5 μM). The separated platelets were resuspended in Tyrode’s buffer with 1 mM CaCl2 added for use in experiments. Cell counts were determined using a hematology analyzer (Sysmex - KX21N).
Flow cytometry
Heparinized murine whole blood was washed three times with Tyrode’s buffer by centrifugation at 650 g for 5 min. After washing the whole blood, samples were diluted in Tyrode’s buffer containing 1 mM CaCl2. Citrate-anticoagulated human blood samples were diluted in a 1:10 ratio with human Tyrode’s buffer. For platelet activation analysis, samples were stimulated with indicated agonists and specifically labeled with antibodies against P-selectin and active integrin αIIbβ3 in a ratio of 1:10 for 15 min at 37°C. Reaction was stopped by adding 300 µL of PBS to all samples. To analyze glycoprotein (GP) expression, washed whole blood was labeled for GPIbα, GPVI, integrin α5, or integrin β3 in a ratio of 1:10 for 15 min at RT. For quantification, MFI (mean fluorescence intensity) values of the platelet specific FSC/SSC population was analyzed using a FACSCalibur flow cytometer (BD Biosciences).
Adhesion and spreading
Cover slips were coated with 100 µg/mL human fibrinogen (#F3879, Sigma-Aldrich) at a defined area (10 × 10 mm) and incubated at 4°C overnight. After blocking with 1% BSA for 60 min, isolated platelets (8 × 104) resuspended in Tyrode’s buffer (with CaCl2 (0.1 M)) were allowed to adhere on the cover slips for indicated time points. The cover slips were rinsed with PBS, adherent platelets were fixed with phosphate-buffered formaldehyde (Roti®-Histofix 4%, Carl Roth) and covered using Fluoromount™ (# F4680, Sigma Aldrich). For recording of differential interference contrast images, a Axio Observer.D1 microscope (Carl Zeiss Microscopy) with 40× magnification (objective LD Plan-Neofluar 40 × 0.6 Korr. Ph2 M27) was used. The adherent cells were counted with ImageJ software. For the analysis of the spreading a 100× magnification (objective Plan Apochromat 100×/1.40 Oil DIC M27) was recorded. Platelet spreading was analyzed using ZEN 2012 (blue edition, Carl Zeiss Software).
Immunofluorescence staining
For immunofluorescence detection of TGR5 expression in murine and human platelets, platelets were allowed to adhere to a fibrinogen matrix (see above). Following PFA fixation, platelets were stained with an anti TGR5 antibody (human and murine: #PA5-23182, Thermo Fisher Scientific, 1:50), rhodamine-phalloidin staining was used to visualize actin filaments ((#R415, invitrogen) at 4°C overnight. Secondary antibody labeling followed the next day after rinsing the samples with PBS with respective secondary antibodies (#A-32733, invitrogen). Samples were mounted and recorded as described above.
Flow chamber
Cover slips (24 × 60 mm) were blocked with 1% BSA and coated with 200 µg/ml fibrillary type I collagen (Nycomed). A collagen-coated surface was then perfused with heparinized murine or citrate-anticoagulated human whole blood at shear rates of 450 and 1000s−1, and platelet adhesion and aggregate formation were assessed from five different microscopic areas using Image J software (LD Plan-Neofluar x 40, Axio Observer D1; Carl Zeiss). For fluorescent detection of platelet aggregate formation, murine platelets were labeled with fluorescently labeled anti-GPIb antibody (#x488; Emfret Analytics) and human platelets with mepacrine (#Q3251, Sigma) 10 min prior assessing blood to flow chamber. Thrombus formation/height was determined as the mean integrated fluorescence intensity and to analyze surface coverage, total area covered by fluorescent positive signal was analyzed per image using ImageJ software.
Western blot
Detection of antibody binding was determined in platelets lysates that had been incubated with indicated agonists. Each sample had a total number of 20 × 106 platelets, which were sedimented by centrifugation. Lysates were prepared, separated on SDS-polyacrylamide gel and transferred onto nitrocellulose blotting membranes. Subsequently, the membrane was blocked using 5% powdered skim milk in TBST (TBS with 0.1% Tween 20) and probed with respective primary antibody. Antibody against α-Tubulin or GAPDH served as loading control. The antibody incubation was performed following the manufacturer´s manual. For visualizing protein bands Immobilon Western Chemiluminescent HRP substrate solution (BioRad), the Vilber Fusion-FX6-EDGE V.070 system, and for quantification ImageJ was used.
cAMP ELISA
Levels of cAMP were determined using Mouse/Rat cAMP Parameter Assay Kit (#KGE012B, R&DSystems) following the manufacture’s protocol. Briefly, the murine platelets were isolated as described above and activated with indicated agonists for 5 min. Reaction was stopped by addition of platelet inhibitors (apyrase and prostacyclin) and platelets were sedimented to collect supernatant and prepare platelet lysates to analyze cAMP content.
RT-PCR
Isolation of RNA from murine platelets was performed with a minimum of 1 billion platelets for each biological replicate by pooling platelet from at least three mice. Subsequently, the platelets were lysed using 500 µL TRI Reagent® (#T9424, Sigma-Aldrich; 5 min, RT). Chloroform (100 µL) was added, samples were vortexed, incubated (3 min, RT) and centrifuged (12.000× g, 4°C). The upper phase was collected, mixed with 250 µL isopropanol and incubated (10 min, RT) following a centrifugation (12.000× g, 4°C). Afterward the pellet was washed twice with 75% ethanol following centrifugation (7.500 g, 5 min). Finally, the pellet was dried (10 min, RT) and dissolved in 40 µL RNAse-free water. RNA concentration was measured with an Eppendorf BioPhotometer® D30 using 2 µL sample in an Eppendorf µCuvette® G1.0. RNA quality was checked with the 260/230 nm and 260/280 nm ratio. The RNA was diluted to 0.1 µg/ml in ddH2O, pretreated with DNaseI (Roche, Germany) for DNA degradation and cDNA was generated with the InPromII Reverse transcription System (Promega; #A3800) following the manufacturer’s protocol. PCR amplification was performed with the following oligonucleotide primers: murine TGR5 fwd 5′-GAGCGTCGCCCACCACTAGG-3′; murine TGR5 rev 5′-CGCTGATCACCCAGCCCCATG-3′; murine GAPDH for 5′-GGTGAAGGCGGTGTGAACG-3′; murine GAPDH rev 5′-CTCGCTCCTGGAAGATGGTG-3.’ After PCR reaction, samples were separated using gel agarose electrophoresis following standard procedure.
Quantification of serum bile acid levels
Bile acid analysis was performed as described earlier.Citation20 Bile acids and their glycine and taurine derivatives were analyzed by UPLC-MS/MS. The system consisted of an Acquity UPLC-H Class (Waters, UK) coupled to a Xevo-TQS tandem mass spectrometer (Waters, UK) which is equipped with an ESI source in the negative ion mode. Data were collected in the multiple reaction monitoring (MRM) mode.
Statistical analysis
GraphPad Prism 8 (version 8.4.3) was used to do the statistical analysis, and the data are provided as arithmetic means with SEM (Standard Error of Mean). The Sidak’s multiple comparison post-hoc test, a two-way or one-way ANOVA, or a (un)paired Student's t-test was used to analyze statistical differences. Asterisks (***p .001, **p .01; *p .05) are used to denote significant differences.
Results
Bile acids modulate human platelet activation
The effects of different bile acids on human platelet activation were analyzed by flow cytometry. First, platelets (platelet-rich plasma, PRP) were treated with different bile acids for 1 h and 6 h and P-selectin exposure at the platelet membrane (marker for degranulation) and active integrin αIIbβ3 (fibrinogen receptor) were measured following platelet stimulation with CRP because defective platelet activation in BDL mice was mainly due to GPVI activation.Citation12 In addition, the platelets were stimulated with ADP and PAR4 peptide to activate the G-protein coupled receptors. As shown in , no major alterations in P-selectin exposure have been detected in bile acid treated platelets compared to controls. In contrast, reduced integrin activation was detected in the presence of the hydrophilic bile acids taurocholic acid (TC) and taurochenodeoxycholic acid (TCDC) and in the presence of glycochenodeoxycholic acid (GCDC) following ADP stimulation for 1 h, while the incubation with TC and TCDC following CRP stimulation of platelets led to reduced integrin activation after 6 h (, Supplemental Figure S1A,B).
Figure 1. Bile acids modulate human platelet activation. Human platelet-rich plasma (PRP) was incubated with indicated bile acids for 1 h and 6 h. (A) integrin αIIbβ3 activation and (B) P-selectin externalization after agonist stimulation was measured in flow cytometric analysis to detect platelet activation state. Mean fluorescence intensity (MFI) in the platelet specific FSC/SSC gate was recorded (n = 4). Isolated human platelets were preincubated for 1 h with hydrophilic bile acids (C+D) or hydrophobic bile acids (E+F) and analyzed for their activation pattern (n = 6). Statistical significance was determined by two-way ANOVA with Tukey’s multiple comparisons test. *p < .05, **p < .01. Data shown as mean + SEM. CRP = collagen-related peptide, PAR4 = PAR4 activating peptide. For each bile acid, a concentration of 50 µM was used.
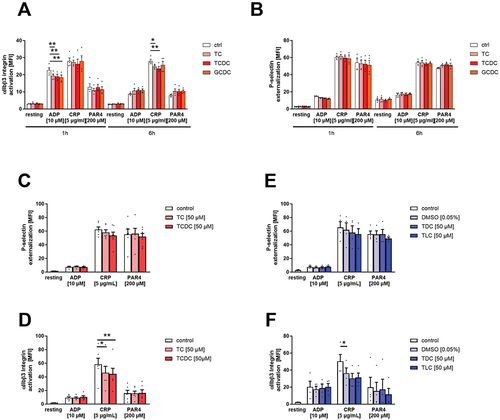
Next, we analyzed platelet activation in response to ADP, CRP and PAR4 peptide to activate different signaling pathways in platelets in the absence and presence of indicated bile acids using our standard protocol (for detail see the material and method section). Again, no differences in P-selectin exposure were measured, while integrin activation was reduced when platelets were activated in the presence of TC and TCDC (, Supplemental Figure S1C,D). To analyze the impact of hydrophobic bile acids on platelet activation, platelets were activated with indicated agonists in the presence and absence (control with and without DMSO) of taurodeoxycholic acid (TDC) and taurolithocholic acid (TLC). In contrast to the results obtained with hydrophilic bile acids, we detected unaltered platelet activation (, Supplemental Figure S1E,F).
Impact of bile acids on human platelet adhesion and thrombus formation
To analyze the impact of bile acids on platelet adhesion, we allowed platelets to adhere to immobilized fibrinogen for different time points (5, 20 and 60 min). As shown in , reduced platelet adhesion has been detected after 20 and 60 min when platelets were treated with the hydrophilic bile acids TC and TCDC (). In addition to platelet adhesion, we determined the formation of filopodia, lamellipodia and fully spread morphology of platelets to analyze the impact of bile acids on the reorganization of the platelet cytoskeleton. No differences were measured neither at early (5 min) nor at later time points (20 and 60 min) of platelet spreading on immobilized fibrinogen ().
Figure 2. Bile acids inhibit human platelet adhesion and thrombus formation. Human platelets were isolated and 1 h after preincubation with hydrophilic (A-D) or hydrophobic (E-H) bile acids, isolated platelets were allowed to adhere on a fibrinogen matrix. (H+K) total adhesion (A+E) and spreading (C+G) was recorded after 60 min (n = 6). All images show platelet adhesion and spreading after 60 min. Human whole blood was preincubated with hydrophilic (I-K) or hydrophobic (M-L) bile acids and thrombus formation on a collagen matrix under arterial shear rate (1700−1) was analyzed with representative images (1700s−1; n = 3–5). Surface coverage and number of adherent cells was determined using Fiji software. Relative thrombus volume was determined as mean fluorescence intensity of fluorescently labeled platelets present in different thrombi. Spreaded platelets were classified in 4 different stages and counted. Statistical significance was determined by two-way ANOVA with Tukey’s multiple comparisons test. *p < .05, **p < .01, ***p < .001. Data shown as mean + SEM. CRP = collagen-related peptide, PAR4 = PAR4 activating peptide.
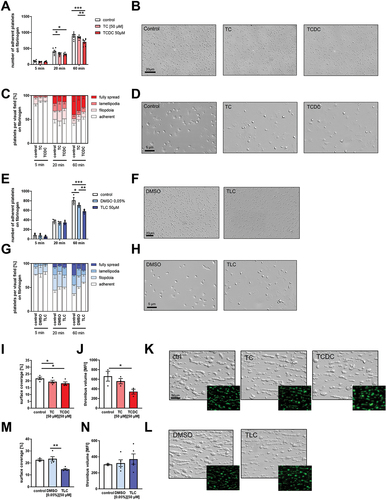
We next analyzed the influence of the hydrophobic bile acid TLC on platelet adhesion and the reorganization of the cytoskeleton. Again, reduced platelet adhesion has been detected. However, this was only true when we allowed platelets to adhere to fibrinogen for 60 min, while no differences were detected after 5 and 20 min (). Again, no major alterations were measured when we analyzed the formation of filopodia and lamellipodia in the presence of TLC ().
To investigate the role of bile acids and their impact on platelet activation and aggregation under more physiological conditions, we performed flow chamber experiments and analyzed the formation of three-dimensional thrombi on collagen under flow conditions (shear rate 1700 sec−1). As shown in , reduced surface coverage of collagen-adherent platelet aggregates was detected when we incubated whole blood with TC, TCDC or TLC () compared to controls. Furthermore, reduced thrombus volume was detected in the presence of TCDC, suggesting that reduced platelet adhesion and moderate platelet activation defects under static conditions led to impaired thrombus formation under flow.
Impact of bile acids on murine platelet activation and adhesion
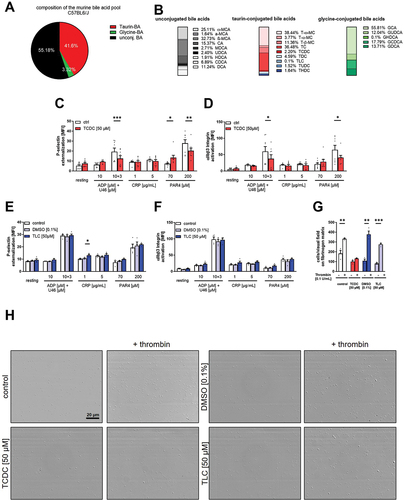
To analyze the impact of bile acids on murine platelet activation and adhesion, we first determined the composition of the murine bile acid pool using C57/BL6/J mice (). We detected 55.18% unconjugated, 41.6% taurine-conjugated and only 3.32% glycine-conjugated bile acids. A detailed analysis of different bile acids is shown in . Based on the results in and the fact that taurine conjugated bile acids are more potent activators for TGR5, we investigated the impact of the hydrophilic bile acid TCDC and the hydrophobic bile acid TLC on murine platelet activation.Citation21 The treatment of murine platelets with TCDC led to a significant reduction in platelet activation as shown by reduced P-selectin exposure and decreased integrin αIIbβ3 activation following platelet activation with ADP and the thromboxane analog U46619, and PAR4-peptide (). However, the activation of platelets with CRP did not lead to any alterations in the presence of TCDC. In contrast, we only observed slightly elevated P-selectin exposure when murine platelets were treated with TLC and low doses of CRP (). However, murine platelet adhesion on fibrinogen was reduced after treatment of platelets with either TCDC or TLC, respectively. The reduction in platelet adhesion was more pronounced when platelets were stimulated with low doses of thrombin, which is necessary to allow full spreading of murine platelets ().
Figure 3. Effect of taurine conjugated bile acids on murine platelets. Serum samples of C57Bl6/J mice were analyzed regarding their specific bile acid profile (n = 5). (A) Total variation of conjugation state of the murine bile acid pool and (B) differentiated appearance of each bile acid in percent. Murine whole blood was preincubated for 1 h with TCDC (C+D, n = 8) or TLC with respective DMSO control (E+F, n = 4). Integrin αIIbβ3 activation after agonist stimulation was measured in flow cytometric analysis. Mean fluorescence intensity (MFI) in the platelet specific FSC/SSC gate was recorded. 1 h after preincubation with TCDC, TLC or DMSO control, isolated platelets were allowed to adhere on a fibrinogen matrix for 60 min and total adhesion was recorded (n = 3). Surface coverage and number of adherent cells was determined using Fiji software. Statistical significance was determined by student’s unpaired t-test. *p < .05, **p < .01, *p < .001. Data shown as mean + SEM. CRP = collagen-related peptide, PAR4 = PAR4 activating peptide, U46 (U46619) = thromboxane A2 analogue.
The bile acid receptor TGR5 is expressed in human and murine platelets and mediates enhanced platelet adhesion on fibrinogen
TGR5 protein expression was analyzed in human and murine platelets. Immunoblot analysis of human cell lysates provided the first evidence for the presence of TGR5 in platelets (). The activation of platelets with CRP did not lead to any changes in the protein content of TGR5 in human platelets (). The human gall bladder tissue and TGR5 transfected HEK cells served as positive control. The presence of TGR5 protein was confirmed by platelet adhesion experiments showing TGR5 protein expression in human platelets. The actin cytoskeleton of the platelets was stained with phalloidin to visualize the morphology of the platelets ().
Figure 4. Platelets express the bile acid receptor TGR5. Human platelets lysates (40×106) were analyzed regarding their protein expression of TGR5 (A) with and without CRP stimulation. Lysates of human gallbladder tissue and TGR5 transfected HEK293 cells served as positive control. (B) Quantification of TGR5 expression in human platelets lysates (n = 3). (C) Human platelets were allowed to adhere on a fibrinogen matrix and were stained for TGR5 and Actin filaments (n = 3). (D) Analysis of cDNA isolated from murine platelets were analyzed regarding the expression of TGR5 mRNA (n = 3). (E) Murine platelets were allowed to adhere on a fibrinogen matrix and were stained for TGR5 and actin filaments (n = 3). (F) Murine platelets of wildtype and TGR5-/- mice were pre-incubated with TCDC, TLC or DMSO control for 1 h. After preincubation, platelets were allowed to adhere on a fibrinogen matrix for 60 min and total adhesion was recorded (n = 3–6). Surface coverage and number of adherent cells (E) was determined using Fiji software. Statistical significance was determined by two-way ANOVA with Tukey’s multiple comparisons test. *p < .05, *p < .001. Data shown as mean + SEM.
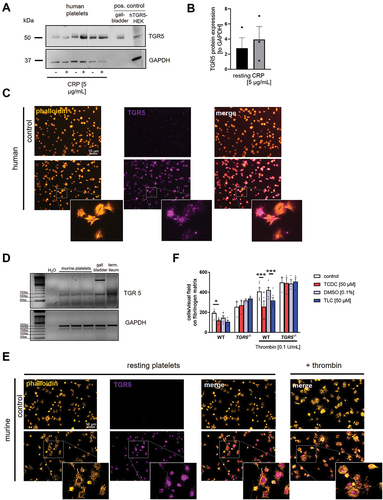
The presence of TGR5 in murine platelets was investigated by RT-PCR showing low expression of TGR in platelets and elevated levels of TGR5 in the gall bladder and the ileum that served as positive controls (). In addition, we performed platelet adhesion experiments and found TGR5 also expressed in mouse platelets. For visualization of the platelet morphology, we performed actin staining by phalloidin. Where indicated platelets were stimulated with thrombin to allow full platelet activation of adherent platelets ().
Next, we investigated the effects of the bile acids TCDC and TLC on platelet adhesion using platelets from wild type and TGR5 knockout mice to investigate if TGR5 plays any role in bile acid-induced alterations of platelet activation, adhesion and thrombus formation. Treatment of platelets with either TCDC or TLC led to a significant reduction of platelet adhesion to immobilized fibrinogen in the presence and in the absence of thrombin stimulation (, Supplemental Figure S2). However, bile acid induced reduction of platelet adhesion was completely abolished using TGR5 deficient platelets suggesting that TGR5 plays a crucial role in bile acid induced effects on platelet activation and adhesion.
Loss of TGR5 leads to increased platelet activation and thrombus formation
We first analyzed blood cell counts of TGR5 deficient mice and found slightly elevated platelet counts but unaltered platelet size (Mean Platelet Volume, MPV) in mice with a genetic loss of TGR5 compared to controls (). No differences in glycoprotein expression at the platelet membrane such as integrin β3, integrin α5, GPVI and GPIbα and unaltered white and red blood cell counts and hemoglobin levels were detected in TGR5 deficient and control platelets (, Supplemental Figure S3A-C)). In contrast, we measured elevated P-selectin exposure at the membrane of TGR5 deficient platelets following platelet activation with ADP and U46619, the snake venom convulxin and PAR4 peptide at intermediate and high concentrations (). Enhanced P-selectin exposure was paralleled by elevated integrin αIIbβ3 activation under resting conditions, as well as after platelet stimulation with ADP, ADP and U46619, and an intermediate concentration with PAR4 peptide (). To investigate if altered activation of TGR5 deficient platelets leads to any differences in thrombus formation, we perfused whole blood from TGR5 deficient and control mice over a collagen-coated surface using different arterial shear rates (450 sec−1 and 1000 sec−1). We detected an enhanced surface coverage of the three-dimensional thrombi on collagen under a shear rate of 450 sec−1 but no differences at a shear rate of 1000 sec−1. However, the analysis of thrombus height by mean fluorescence intensity using fluorescently labeled platelets revealed a slightly but significantly enhanced thrombus volume using whole blood from TGR5 deficient mice compared to controls (). Thus, elevated platelet activation translates into elevated thrombus formation under flow conditions.
Figure 5. Genetic deficiency of TGR5 leads to increased platelet activation and thrombus formation. (A) platelet count and (B) mean platelet volume was measured by a hematology analyzer (n = 17). (C) Expression of indicated glycoproteins on the surface of platelets was measured via MFI in flow cytometric analysis in the platelet specific FSC/SSC gate (n = 7). For platelet activation analysis, (D) externalization of P-selectin and (E) activation of αIIbβ3 integrin on the platelet surface of WT and TGR5-/- platelets was recorded with indicated agonists (n = 10). Heparinized murine whole blood was observed regarding thrombus formation on a collagen matrix under arterial shear rates. Surface coverage (F) and thrombus size were calculated with (H) representative images (n = 3–5). Surface coverage and thrombus size was determined via detection of the fluorescent signal using Fiji software. (I-K) isolated murine platelets (20×106) were stimulated with indicated agonists and analyzed for the phosphorylation of AKT and ERK 1/2 in TGR5-/- platelets after stimulation with ADP or thrombin (n = 3). Phosphorylation state of (J) ERK1/2 and (K) AKT was analyzed by normalizing the specific relative density signal to the loading control GAPDH. Statistical significance was determined by unpaired student’s t-test. *p < .05, **p < .01, *p < .001. Data shown as mean + SEM. CRP = collagen-related peptide, thr. = thrombin.
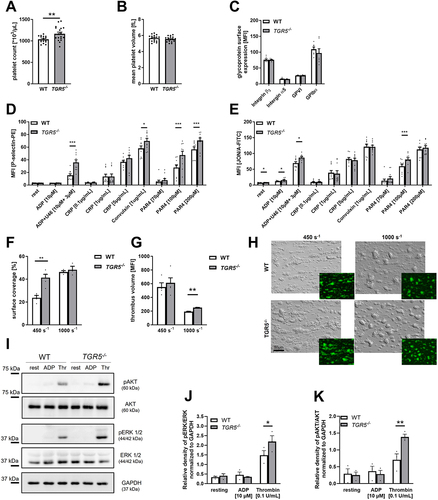
It is well known that TGR5 stimulates the MAPK signaling pathway in cholangiocytes.Citation21 In vascular endothelial cells, the phosphorylation of AKT is enhanced after TLCA induced activation of TGR5.Citation22 Therefore, we next explored the phosphorylation of ERK1/2 and AKT in platelets. In TGR5 deficient platelets, enhanced phosphorylation of ERK1/2 after thrombin stimulation was observed (, Supplemental Figure S3D). In line with enhanced ERK1/2 phosphorylation, we detected elevated AKT phosphorylation in response to thrombin (, Supplemental Figure S3D) suggesting that TGR5 deficiency enhances MAPK signaling in platelets.
Dysregulated phosphorylation of PKA and vasodilator-stimulated phosphoprotein (VASP) in TGR5 deficient platelets is responsible for altered platelet activation
TGR5 couples to Gαs proteins and activates the adenylate cyclase (AC) leading to enhanced generation of cyclic adenosine monophosphate (cAMP) that alters PKA activation in the hepatic system.Citation21 In platelets, PKA activation is modulated by PGI2 leading to platelet inhibition.Citation23 Therefore, we next investigated, if TGR5 signaling affects PKA substrate serine phosphorylation after stimulation of platelets with thrombin and CRP, respectively. Immunoblot analysis of cell lysates revealed reduced serine phosphorylation of PKA substrates with a size of 110, 50 and 40 kDa following thrombin stimulation of platelets, while no alterations were detected with CRP (, Supplemental Figure S4A). Interestingly, under basal conditions, elevated serine phosphorylation of PKA substrates with a size of 110 and 50 kDa were detected, suggesting dysregulated phosphorylation of PKA already in resting platelets (, Supplemental Figure S4A).
Figure 6. Dysregulated phosphorylation of PKA and VASP in TGR5 deficient platelets and after bile acids stimulation is responsible for altered platelet activation. (A-E) isolated murine platelets (20 × 106) were stimulated with indicated agonists and analyzed for the phosphorylation of PKA substrates and VASP. (A) Analysis of PKA substrate phosphorylation after stimulation with collagen related peptide (CRP) or thrombin (thr) (n = 4). (B) Analysis of different phosphorylation substrates of PKA that were differentiated by their molecular weight. (C-D) analysis of VASP phosphorylation at Ser157 after stimulation with sodium nitroprusside (SNP; 5 µM, 5 min) or PGI (prostaglandin; 1 µM, 5 min). (E) Analysis of cAMP levels in platelet lysates and in the supernatant of thrombin stimulated platelets isolated from WT and TGR5-/- mice (n = 4–5). (F) Isolated human platelets (20 × 106) were incubated with indicated bile acid (50 µM) or DMSO (0.05%, control for TLC) for 1 h, and phospo-PKA substrate phosphorylation was analyzed. (G) Analysis of different phosphorylation substrates of PKA that were differentiated by their molecular weight (n = 5). Depicted are mean values + SEM; *p < .05, **p < .01 using two-way ANOVA followed by Sidak’s multiple comparison test (A-E) and RM one-way ANOVA with holm-Sidak multiple comparison (G).
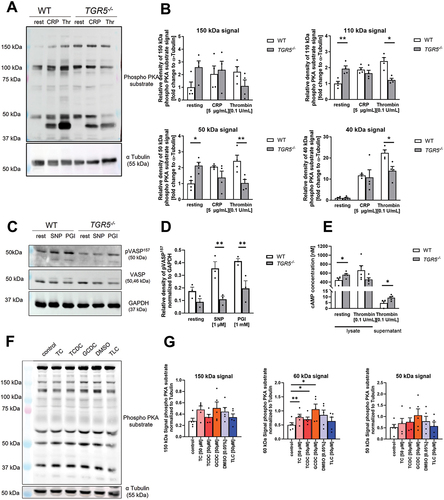
Next, we analyzed if reduced PKA activation in response to thrombin affects the phosphorylation of the vasodilator-stimulated phosphoprotein (VASP) in platelets from TGR5 deficient mice. To this end, we investigated the phosphorylation of VASP at serine 157 because PKA is known to target this phosphorylation site.Citation24 As shown in , VASP phosphorylation was reduced after treatment of TGR5 deficient platelets with SNP or PGI2 compared to controls (, Supplemental Figure S4B). To investigate if reduced PKA activation and VASP phosphorylation in the absence of TGR5 is mediated by cAMP, we analyzed the concentration of cAMP in lysates and the supernatant of platelets under resting and thrombin stimulated conditions (). In TGR5 deficient platelet lysates, elevated cAMP levels under resting but decreased (by trend) levels under thrombin stimulating conditions were detected. Furthermore, we found elevated cAMP levels in the supernatant of TGR5 deficient platelets compared to controls ().
To investigate, if bile acids have any impact on PKA substrate phosphorylation in human platelets, we incubated platelets from healthy volunteers with indicated bile acids or DMSO (control for TLC) for 1 h. The analysis of different phosphorylation substrates of PKA as differentiated by their molecular weight revealed that bile acids induce elevated phosphorylation of PKA substrates, either by trend (150 kDa and 50 kDa signal) or with statistical significance (60 kDa signal) (, Supplemental Figure S4C).
Discussion
In patients with cirrhosis, impaired inositol and arachidonic acid metabolism and altered platelet-vessel wall interaction have been described.Citation25,Citation26 Dysregulated signal transduction in platelets, an acquired storage pool deficiency and reduced production of arachidonic acid and thromboxane might account for reduced platelet activation and adhesion in cirrhotic patients.Citation27,Citation28 In addition, increased plasma levels of cyclic nucleotides and nitric oxide (NO) are thought to be responsible for impaired platelet activation in patients with liver disease.Citation29,Citation30 Interestingly, the correction of hemostatic abnormalities in patients with liver transplantation failed because bleeding tendencies were even enhanced but not reduced or avoided.Citation4 Many studies provide evidence that thrombocytopenia and platelet activation defects are responsible for defective hemostasis in patients with liver disease, but it is still under debate how cholestatic liver disease affects platelet activation and hemostasis. Hyper- and hypoactivity of platelets and hypo- and hypercoagulation have been described in patients with cholestatic liver disease.Citation31–33
In in vitro and in vivo rat and mouse models, the role of bile acids for altered platelet activation and aggregation has been studied.Citation10,Citation12,Citation34 In BDL mice, defective platelet activation originates from enhanced NO and prostacyclin plasma levels that are causative for elevated phosphorylation of VASP in platelets from BDL mice. Elevated bile acid plasma levels account for prolonged platelet activation defects in these mice, but how they affect platelet activation was unclear. In the present study, we provide evidence that the bile acid receptor TGR5 is responsible for reduced platelet activation upon high bile acid plasma levels because in the presence of TCDC or TLC, platelet adhesion to fibrinogen was unaltered in TGR5 deficient platelets but reduced with control platelets (). Furthermore, we detected elevated platelet activation and increased thrombus formation in the absence of TGR5 (). Mechanistically, the phosphorylation of PKA substrates was reduced in TGR5 deficient platelets (). To date, it is well accepted that activation of the adenylate cyclase (AC) by PGI2 increases cAMP levels leading to PKA activation in platelets and thus platelet inhibition.Citation35 However, TGR5 is a G-protein coupled receptor that activates the AC leading to PKA activation, e.g., in cholangiocytes.Citation21 Thus, our data strongly suggest that TGR5 at the platelet membrane activates PKA and VASP via AC as described for the PGI2-dependent signaling pathway in platelets. In addition, our data provides evidence for TGR5 to regulate MAPK signaling in platelets as already shown in cholangiocytes and vascular endothelial cells.Citation21,Citation22 Furthermore, the presented results indicate that enhanced platelet activation is not only due to reduced NO plasma levels. NO is able to trigger the activation of the soluble guanylate cyclase (sGC) leading to elevated cyclic guanosine monophosphate (cGMP) plasma levels and increased PKG activation but not to induce PKA activation in platelets.Citation35 However, we cannot exclude that reduced NO plasma levels might account for elevated platelet activation in TGR5 deficient mice since TGR5 agonism induces the production of NO in vascular endothelial cells.Citation22
In contrast, the bile acid receptor FXR has been shown to modulate cGMP levels without affecting cAMP levels in mice.Citation15 Here, we show that TGR5 signaling in platelets affects cAMP levels. However, engagement of both receptors, FXR and TGR5, results in the activation of VASP, an important inhibitor of platelet aggregation. Thus, these bile acid receptors directly affect platelet activation, but differ in the signaling mechanism because TGR5 induces phosphorylation of VASP at Ser157 via activation of the AC and PKA while the FXR affects the phosphorylation of VASP at Ser239 via activation of the sGC. The FXR is a ligand-activated transcription factor that has been identified in platelets. Importantly, engagement of the FXR inhibits platelet activation. Thus, the atheroprotective effect of the FXR ligand GW4064 might be due –at least in part- to antiplatelet effects. In line with the FXR, we propose that also TGR5 is a potential target for the prevention of atherothrombotic diseases. Activation of the TGR5 by TGR5 agonists might reduce platelet activation and inflammation in patients with myocardial infarction or stroke and thus is a promising new approach for the treatment of thrombo-inflammatory diseases. Thus, it will be of utmost interest to analyze ligand binding to TGR5 and how it affects arterial thrombosis, myocardial infarction, stroke and other thrombo-inflammatory diseases such as abdominal aortic aneurysm (AAA) in the near future.
Authorship contributions
FR and ME designed the study. FR, MR and DH performed experiments. FR, VK-A and ME analyzed and interpreted data. FR and ME wrote the manuscript with all authors providing feedback.
Supplemental Material
Download PDF (596.8 KB)Acknowledgments
We thank Martina Spelleken for excellent technical assistance and Niloofar Salehzadeh for the evaluation of Western blots.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Supplementary material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/09537104.2024.2322733.
Additional information
Funding
References
- Ruggeri ZM, Mendolicchio GL. Adhesion mechanisms in platelet function. Circ Res. 2007;100(12):1673–13. doi:10.1161/01.RES.0000267878.97021.ab.
- Savage B, Almus-Jacobs F, Ruggeri ZM. Specific synergy of multiple substrate–receptor interactions in platelet thrombus formation under flow. Cell. 1998;94(5):657–666. doi:10.1016/S0092-8674(00)81607-4.
- Lisman T, Leebeek FWG, de Groot PG. Haemostatic abnormalities in patients with liver disease. J Hepatol. 2002;37(2):280–287. doi:10.1016/S0168-8278(02)00199-X.
- Lisman T, Porte RJ. Activation and regulation of hemostasis in acute liver failure and acute pancreatitis. Semin Thromb Hemost. 2010;36(4):437–443. doi:10.1055/s-0030-1254052.
- Ben-Ari Z, Osman E, Hutton RA, Burroughs AK. Disseminated intravascular coagulation in liver cirrhosis: fact or fiction? Am J Gastroenterol. 1999;94(10):2977–82. doi:10.1111/j.1572-0241.1999.01446.x.
- Carr JM. Disseminated intravascular coagulation in cirrhosis. Hepatology. 1989;10(1):103–10. doi:10.1002/hep.1840100120.
- Stein SF, Harker LA. Kinetic and functional studies of platelets, fibrinogen, and plasminogen in patients with hepatic cirrhosis. J Lab Clin Med. 1982;99:217–30.
- Baele G, Beke R, Barbier F. In vitro inhibition of platelet aggregation by bile salts. Thromb Haemost. 1980;44(2):62–4. doi:10.1055/s-0038-1650084.
- Takano S, Suzuki T. Effect of bile on aggregation and morphology of rabbit platelets. Tohoku J Exp Med. 1980;131(1):71–8. doi:10.1620/tjem.131.71.
- Pereira J, Accatino L, Pizarro M, Mezzano V, Ibañez A, Mezzano D. In vivo effect of bile salts on platelet aggregation in rats. Thromb Res. 1995;80(4):357–62. doi:10.1016/0049-3848(95)00187-V.
- Tag CG, Sauer-Lehnen S, Weiskirchen S, Borkham-Kamphorst E, Tolba RH, Tacke F, Weiskirchen R. Bile duct ligation in mice: induction of inflammatory liver injury and fibrosis by obstructive cholestasis. J Vis Exp. 2015;(96). doi:10.3791/52438-v.
- Gowert NS, Klier M, Reich M, Reusswig F, Donner L, Keitel V, Häussinger D, Elvers M. Defective platelet activation and bleeding complications upon cholestasis in mice. Cell Physiol Biochem. 2017;41(6):2133–49. doi:10.1159/000475566.
- Walter U, Eigenthaler M, Geiger J, Reinhard M. Role of cyclic nucleotide-dependent protein kinases and their common substrate VASP in the regulation of human platelets. Adv Exp Med Biol. 1993;344:237–49.
- Perino A, Demagny H, Velazquez-Villegas L, Schoonjans K. Molecular physiology of bile acid signaling in health, disease, and aging. Physiol Rev. 2021;101(2):683–731. doi:10.1152/physrev.00049.2019.
- Moraes LA, Unsworth AJ, Vaiyapuri S, Ali MS, Sasikumar P, Sage T, Flora GD, Bye AP, Kriek N, Dorchies E, et al. Farnesoid X receptor and its ligands inhibit the function of platelets. Arterioscler Thromb Vasc Biol. 2016;36(12):2324–2333. doi:10.1161/ATVBAHA.116.308093.
- Keitel V, Häussinger D. Role of TGR5 (GPBAR1) in liver disease. Semin Liver Dis. 2018;38(4):333–339. doi:10.1055/s-0038-1669940.
- Keitel V, Häussinger D. Perspective: TGR5 (Gpbar-1) in liver physiology and disease. Clin Res Hepatol Gastroenterol. 2012;36(5):412–9. doi:10.1016/j.clinre.2012.03.008.
- Reich M, Deutschmann K, Sommerfeld A, Klindt C, Kluge S, Kubitz R, Ullmer C, Knoefel WT, Herebian D, Mayatepek E, et al. TGR5 is essential for bile acid-dependent cholangiocyte proliferation in vivo and in vitro. Gut. 2016;65(3):487–501. doi:10.1136/gutjnl-2015-309458.
- Keitel V, Cupisti K, Ullmer C, Knoefel WT, Kubitz R, Häussinger D. The membrane-bound bile acid receptor TGR5 is localized in the epithelium of human gallbladders. Hepatology. 2009;50(3):861–70. doi:10.1002/hep.23032.
- Reusswig F, Fazel Modares N, Brechtenkamp M, Wienands L, Krüger I, Behnke K, Lee‐Sundlov MM, Herebian D, Scheller J, Hoffmeister KM, et al. Efficiently restored thrombopoietin production by Ashwell-Morell receptor and IL-6R induced Janus Kinase 2/Signal transducer and activator of transcription signaling early after partial hepatectomy. Hepatology. 2021;74(1):411–427. doi:10.1002/hep.31698.
- Deutschmann K, Reich M, Klindt C, Dröge C, Spomer L, Häussinger D, Keitel V. Bile acid receptors in the biliary tree: TGR5 in physiology and disease. Biochim Biophys Acta Mol Basis Dis. 2018;1864(4):1319–25. doi:10.1016/j.bbadis.2017.08.021.
- Kida T, Tsubosaka Y, Hori M, Ozaki H, Murata T. Bile acid receptor TGR5 agonism induces NO production and reduces monocyte adhesion in vascular endothelial cells. Arterioscler Thromb Vasc Biol. 2013;33(7):1663–9. doi:10.1161/ATVBAHA.113.301565.
- Nagy Z, Smolenski A. Cyclic nucleotide-dependent inhibitory signaling interweaves with activating pathways to determine platelet responses. Res Pract Thromb Haemost. 2018;2(3):558–71. doi:10.1002/rth2.12122.
- Aktas B, Utz A, Hoenig-Liedl P, Walter U, Geiger J. Dipyridamole enhances NO/cGMP-mediated vasodilator-stimulated phosphoprotein phosphorylation and signaling in human platelets: in vitro and in vivo/ex vivo studies. Stroke. 2003;34(3):764–769. doi:10.1161/01.STR.0000056527.34434.59PMID:12624305.
- Laffi G, Cominelli F, Ruggiero M, Fedi S, Chiarugi VP, La Villa G, Pinzani M, Gentilini P. Altered platelet function in cirrhosis of the liver: impairment of inositol lipid and arachidonic acid metabolism in response to agonists. Hepatology. 1988;8(6):1620–6. doi:10.1002/hep.1840080625.
- Ordinas A, Escolar G, Cirera I, Vinas M, Cobo F, Bosch J, Teres J, Rodés J. Existence of a platelet-adhesion defect in patients with cirrhosis independent of hematocrit: studies under flow conditions. Hepatology. 1996;24(5):1137–42. doi:10.1002/hep.510240526.
- Laffi G, Marra F, Gresele P, Romagnoli P, Palermo A, Bartolini O, Simoni A, Orlandi L, Selli ML, Nenci GG, et al. Evidence for a storage pool defect in platelets from cirrhotic patients with defective aggregation. Gastroenterology. 1992;103(2):641–6. doi:10.1016/0016-5085(92)90859-W.
- Owen JS, Hutton RA, Day RC, Bruckdorfer KR, McIntyre N. Platelet lipid composition and platelet aggregation in human liver disease. J Lipid Res. 1981;22(3):423–30. doi:10.1016/S0022-2275(20)34955-5.
- Albornoz L, Bandi JC, Otaso JC, Laudanno O, Mastai R. Prolonged bleeding time in experimental cirrhosis: role of nitric oxide. J Hepatol. 1999;30(3):456–60. doi:10.1016/S0168-8278(99)80105-6.
- Laffi G, Marra F, Failli P, Ruggiero M, Cecchi E, Carloni V, Giotti A, Gentilini P. Defective signal transduction in platelets from cirrhotics is associated with increased cyclic nucleotides. Gastroenterology. 1993;105(1):148–56. doi:10.1016/0016-5085(93)90020-D.
- Biagini MR, Tozzi A, Marcucci R, Paniccia R, Fedi S, Milani S, Galli A, Ceni E, Capanni M, Manta R, et al. Hyperhomocysteinemia and hypercoagulability in primary biliary cirrhosis. World J Gastroenterol. 2006;12(10):1607–12. doi:10.3748/wjg.v12.i10.1607.
- Ingeberg S, Jacobsen P, Fischer E, Bentsen KD. Platelet aggregation and release of ATP in patients with hepatic cirrhosis. Scand J Gastroenterol. 1985;20(3):285–8. doi:10.3109/00365528509091651.
- Pihusch R, Salat C, Göhring P, Hentrich M, Wegner H, Pihusch M, Hiller E, Kolb H-J, Ostermann H. Factor XIII activity levels in patients with allogeneic haematopoietic stem cell transplantation and acute graft-versus-host disease of the gut. Br J Haematol. 2002;117(2):469–76. doi:10.1046/j.1365-2141.2002.03420.x.
- Atucha NM, Iyú D, Alcaraz A, Rosa V, Martínez-Prieto C, Ortiz M, Rosado J, García-Estañ J. Altered calcium signalling in platelets from bile-duct-ligated rats. Clin Sci (Lond). 2007;112(3):167–74. doi:10.1042/CS20060226.
- Chirkov YY, Nguyen TH, Horowitz JD. Impairment of anti-aggregatory responses to nitric oxide and prostacyclin: mechanisms and clinical implications in cardiovascular disease. Int J Mol Sci. 2022;23(3):1042. doi:10.3390/ijms23031042.