
Abstract
Hereditary progressive mucinous histiocytosis (HPMH) is an extremely rare progressive non-Langerhans cell histiocytic disorder presenting with only cutaneous manifestations. Patients typically present with multiple asymptomatic dome-shaped erythematous papules, usually involving the face and upper extremities. Twenty-six cases have been reported worldwide, with no spontaneous regression. Treatment with thalidomide stopped the progression of the disease in two cases. We report a case of progressive mucinous histiocytosis in a 31-year-old female patient with a history of tuberculosis who presented papular lesions on the face that later extended to the hands. She was treated with isoniazid for tuberculosis and isotretinoin for the skin lesions; the improvement was minimal during the next two months, with new lesions appearing on both hands. Thalidomide stopped the progression of the disease. The cause and pathogenesis of HPMH are undetermined. The pathogenesis of HPMH may be similar to that of lysosomal storage disease, considering the intra-cytoplasmic phospholipid deposition in both diseases, in addition to the likelihood of a role of macrophages in triggering the disease. In our patient, tuberculosis may have contributed.
Introduction
Hereditary progressive mucinous histiocytosis (HPMH) was first described in 1988 by Konrad Bork and Nikolaus Hoede in three females who suffered from generalized histiocytic tumors from early adolescence (Citation1). HPMH usually manifests with dome-shaped fleshy papules on the face, forearms, hands, and rarely on the legs, with no mucosal manifestations or systematic involvement (Citation2). HPMH is rare; 26 cases have been reported. Nodular accumulation of histiocytes and mucin is present in the dermis (Citation2). All cases presented with progressive disease without a tendency to resolve spontaneously. We report a 31-year-old patient with a history of tuberculosis (TB) who presented with a nine-month history of hereditary progressive erythematous papules that failed to respond to corticosteroids and isotretinoin. The patient was treated with thalidomide, and the lesions became smaller.
Case presentation
A 31-year-old female patient presented with multiple erythematous papular lesions on the face, appearing nine months beforehand (). There were no associated symptoms except for very mild pruritus. The patient had no systemic manifestations or mucosal involvement. During the previous few months, she was unsuccessfully treated with topical and intra-lesional corticosteroids. Two biopsy samples were taken from her face and showed similar results, with differential diagnoses for histiocytosis, papular mucinosis, pseudolymphoma, and lichen myxedematosus. Histological examination revealed an infiltrate involving the full thickness of the dermis composed of epithelioid and spindle-shaped histiocytes arranged in a collagenous stroma with abundant intervening mucin deposition with a vague storiform pattern. There was no grenz zone between the infiltrate and follicular epithelium. Perivascular infiltration of lymphocytes, plasma cells, and mast cells was also noted (). The histiocytes were negative for S100 and CD1a. CD68 was positive in at least some of the histiocytes. CD31 stained the histiocytes as well as vascular endothelial cells, and CD34 was positive in the spindle-shaped histiocytes (). The findings were consistent with progressive mucinous histiocytosis, a non-Langerhans cell histiocytosis ().
Figure 1. Erythematous papular lesions on the face.
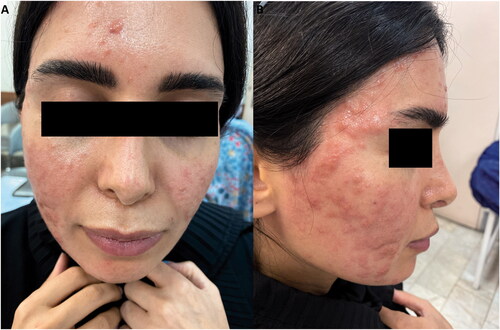
Figure 2. (A) H&E staining shows a dense infiltrate involving the full thickness of the dermis. There is no grenz zone between the infiltrate and surface/follicular epithelium. -The infiltrate has a vaguely storiform growth pattern (x100) and is composed of epithelioid and spindle-shaped histiocytes arranged in a collagenous stroma with mucin deposition. There is also a perivascular lymphoplasmacytic infiltration in the dermis (x400). (B) CD68; Immunohistochemistry staining for CD68 markers shows positive staining in most histiocytes. (C) CD1a; CD1a is negative in tumoral cells but positive in normal tissue Langerhans cells. (D) S100; S100 is negative in tumoral cells and highlights normal tissue dendritic cells and Langerhans cells (x100).
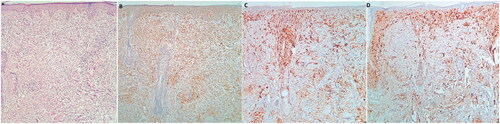
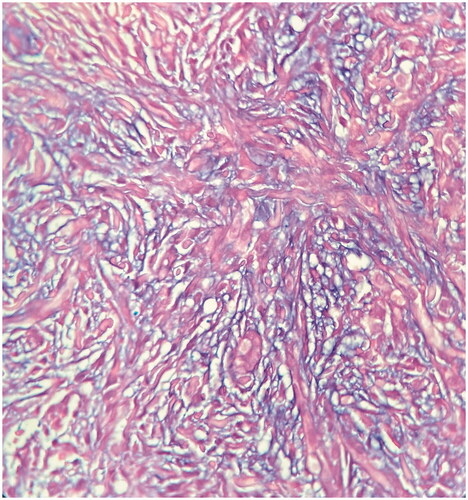
Figure 3. There is abundant mucin deposition in the dermis (alcian blue stain, x400).
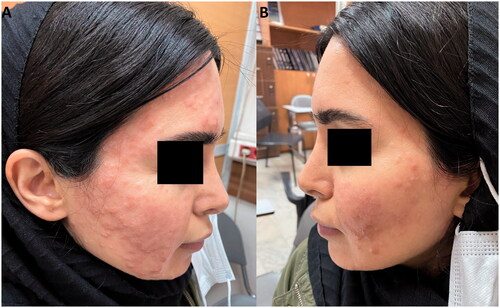
Laboratory findings included a white blood cell count of 4.48 × 103/uL, hemoglobin 13.2 g/dL, and a platelet count of 248 × 103/uL, with a normal liver function test and lipid profile. The patient had a positive tuberculin test (27 mm). Following consultation with an infectious disease specialist, isoniazid was initiated. Isotretinoin 20 mg daily was also prescribed for her skin lesions. The improvement was minimal during the next two months, with new skin lesions appearing on both hands. Later, thalidomide 50 mg daily was initiated, and during the follow-up in the next month, thalidomide led to a decrease in the size of the lesions, and no new papules developed during the treatment duration ().
Figure 4. (A/B) Decrease in number and size of papules following one month of treatment with thalidomide.
Discussion
Histiocytosis is divided into two sub-groups: Langerhans and non-Langerhans cell histiocytosis, distinguished by the immunophenotype of the affected cells (Citation3). Langerhans cell histiocytosis (LCH) includes Letterer–Siwe disease, Hand-Schüller-Christian disease, eosinophilic granuloma, congenital self-healing, and adult LCH, staining positively for CD1a, S100 protein, and CD207 (langerin), and negatively for CD68, CD163, and factor XIIIa (Citation3). Non-Langerhans cell histiocytosis (non-LCH) is divided into three subgroups: primarily cutaneous, cutaneous with systematic involvement, and primarily extra-cutaneous in addition to skin involvement (Citation4). Non-LCH includes juvenile xanthogranuloma, xanthoma disseminatum, benign cephalic histiocytosis, progressive nodular histiocytosis, spindle cell xanthogranuloma, and generalized eruptive histiocytosis, which are of dendritic origin; HPMH was later added to this group (Citation5).
Hereditary progressive mucinous histiocytosis is a very rare disease that occurs most often during childhood or adolescence and has no accompanying systematic manifestations or mucosal involvement (Citation2). It is characterized by multiple asymptomatic red or skin-colored papules, most often appearing on the face and extremities (Citation2). The histopathology for the lesions includes the collection of histiocytes in the dermis with the accumulation of mucin (Citation6). Scattered mast cells are also seen, in addition to increased vascularization in the dermis (Citation7). Under the electron microscope, the histiocytes are large and round or spindle in shape and contain many myelin and zebra bodies in addition to an extensive endoplasmic reticulum (Citation7). It is suggested that HPMH is similar to lysosomal storage disease because of the gradual progression in both diseases and histopathological similarities like intracytoplasmic phospholipid deposition (Citation6).
In our case, the patient had a past medical history of asymptomatic tuberculosis, diagnosed with a purified protein derivative skin test of 27 mm. In tuberculosis, the innate immune response is triggered by macrophages (Citation8). Pattern Recognition Receptors (PRRs) cause phagocytosis of the bacteria, which are damaged later by acidification and phagosome-lysosome fusion (Citation8). In our case, the proliferation of macrophages was attributed to tuberculosis, and the presence of mast cells in the dermis may have been related to the pathogenesis in this sporadic case of progressive mucinous histiocytosis. Hence, infectious causes should also be considered.
In reviewing the literature, the treatment of HPMH seems to be difficult and unpromising (). In 2013, a case reported by Verónica Narváez-Rosales was successfully treated with thalidomide, and no recurrence or development of new lesions was reported during the following year (Citation13). In 2016, another patient was successfully treated with thalidomide: the lesions regressed and no new papules developed during the treatment duration (Citation12). In our case, thalidomide stopped the progression and decreased the severity of the disease within a month.
Table 1. Review of literature on progressive mucinous histiocytosis hereditary and sporadic cases in chronological order.
Patient consent for publication
Written informed consent was obtained from the patient for publication of this case report and any accompanying images
Author contributions
MSD was involved in the diagnosis and management of the patients. AR reported the result of the histopathological evaluation. FA, RD, and AK did the literature review and drafted the manuscript. FA was responsible for the final editing of the manuscript and coordinated the study. All authors read and approved the final manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
References
- Konrad Bork MD, Nikolaus Hoede MD. Hereditary progressive mucinous histiocytosis in women, report of three members in a family. Arch Dermatol. August. 1988;124:1225–1229.
- INSERM. Orphanet: an online rare disease and orphan drug data base. Hereditary progressive mucinous histiocytosis. ORPHA. 1999;2020:158025.
- Goodman WT, Barrett TL. Histiocytoses. Dermatol Bolognia Fourth Edition. 2018;91:1614–1633.
- Weitzman S, Jaffe R. Uncommon histiocytic disorders: the non-Langerhans cell histiocytoses. Pediatr Blood Cancer. 2005;2005:pbc.20246.
- Nguyen NV, Prok L, Burgos A, et al. Hereditary progressive mucinous histiocytosis: new insights into a rare disease. Pediatr Dermatol. 2015;32(6):e273–e276.
- Cascarinohas Y, Caronb C, Butnaruc F, et al. Sporadic progressive mucinous histiocytosis. Ann Dermatol Venereol. 2017;144(3):33.
- Hemmati I, McLeod WA, Crawford RI. Progressive mucinous histiocytosis: importance of electron microscopy to confirm diagnosis. J Cutan Med Surg. 2010;14(5):245–248.
- Heemskerk D, Caws M, Marais B, et al. Tuberculosis in adults and children. London: Springer. 2015.
- Hobbs MM, Hall M, Sluzevich JC. Progressive papular eruption of the distal upper extremities in a patient with asymptomatic papules. JAMA Dermatol. 2021;157(5):591.
- Onoufriadis A, Boulouadnine B, Dachy G, et al. A germline mutation in the platelet-derived growth factor receptor beta gene may be implicated in hereditary progressive mucinous histiocytosis. Br J Dermatol. 2021;184(5):967–970.
- Requena C, Requena L, Traves V, et al. Hereditary progressive mucinous histiocytosis: three different phenotypes in three family members. J Cutan Pathol. 2017;44(9):781–785.
- Abou Rahal J, Cogrel O, Cribier B. Facial erythematous papules in a young woman. JAMA Dermatol. 2016;152(12):1377.
- Narváez-Rosales V, Sáez-de-Ocariz M, Toussaint-Caire S, et al. Sporadic progressive mucinous histiocytosis in a Mexican patient. Skin Med. 2013;11(3):175–178.
- Schlegel C, Metzler G, Burgdorf W, et al. Hereditary progressive mucinous histiocytosis: first report in a male patient. Acta Derm Venereol. 2010;90:65–67.
- Young A, Olivere J, Yoo S, et al. Two sporadic cases of adult-onset progressive mucinous histiocytosis. J Cutan Pathol. 2006;33(2):166–170.
- Antoni-Bach N, Pfister R, Grosshans E, et al. Hereditary progressive mucinous histiocytosis. Ann Dermatol Venereol. 2000;127(4):400–404.
- Sass U, André J, Song M. A sporadic case of progressive mucinous histiocytosis. Br J Dermatol. 2000;142(1):133–137.
- Wong D, Killingsworth M, Crosland G, et al. Hereditary progressive mucinous histiocytosis. Br J Dermatol. 1999;141(6):1101–1105.
- Mizushima J, Nogita T, Higaki Y, et al. Hereditary progressive mucinous histiocytosis. Int J Dermatol. 1997;36(12):958–960.
- Schröder K, Hettmannsperger U, Schmuth M, et al. Hereditary progressive mucinous histiocytosis. J Am Acad Dermatol. 1996;35(2):298–303.
- Bork K. Hereditary progressive mucinous histiocytosis. Immunohistochemical and ultrastructural studies in an additional family. Arch Dermatol. 1994;130(10):1300–1304.