
Abstract
Introduction
Herein, we developed an engineered extracellular vehicle (EV)-based method for ameliorating inflammatory responses in psoriasis.
Methods
EVs, derived from annexin A1 (ANXA1) overexpressing T cells, were co-extruded with M2 macrophage membrane to obtain engineered EVs. In vitro, the effect of engineered EVs on macrophage polarization was evaluated by real-time PCR. In imiquimod (IMQ)-induced psoriasis-like mouse model, the efficacy of engineered EVs in ameliorating psoriatic inflammation was evaluated by Psoriasis Area and Severity Index (PASI) score and immunohistochemistry staining after subcutaneous injection of EVs.
Results
The engineered EVs not only preserved the high stability of M2 macrophage membrane but also retained the macrophage reprogramming potential of ANXA1 overexpressed in T cells. In the psoriasis-like mouse model, subcutaneous injection of engineered EVs successfully reduced the PASI score and the levels of pro-inflammatory cytokines, including IL-1β, IL-6, and TNF-α. Along with high biosafety, the administration of EVs also rescued the histomorphological changes of spleen, liver, and kidney.
Conclusions
The engineered EVs exhibited the potential to alleviate inflammation of psoriasis, providing new insights and potential strategies for the immunotherapies of psoriasis.
1. Introduction
Psoriasis is a chronic inflammatory disease. Clinical treatments of psoriasis mainly include topical medications, phototherapy, immunosuppressive agents, and biological drug therapies (Citation1). However, these treatments have significant drawbacks, such as intolerable side effects or recurrence after drug withdrawal. Therefore, there is an urgent need to develop new treatment strategies that can address these drawbacks.
The cellular composition of the skin greatly affects the dermal inflammatory process. In psoriasis, the persistent high ratio of M1/M2 macrophages contributes to chronic inflammation (Citation2). Mechanistically, antigen-presenting cells (APCs), such as macrophages and dendritic cells (DCs), activate Th1, Th17, and Th23 cells by presenting skin self-antigens and producing inflammatory cytokines, thereby forming a positive feedback loop of inflammation (Citation3). Intriguingly, macrophages exhibit reprogramming potential. A recent study indicates that a circRNA (hsa_circ_0004287) can reduce the stability of MALAT1, thereby inhibiting M1 macrophage polarization and alleviating psoriatic inflammation (Citation4). Hence, exploring methods to intervene in macrophage polarization in the pathogenesis of psoriasis would provide new clinical applications for psoriasis.
Extracellular vesicles (EVs) include exosomes (EXOs), microvesicles (MVs), and apoptotic bodies (APOs) (Citation5). EVs contain different components (e.g., nucleic acids, proteins, and lipids), which are crucial for intercellular communication (Citation6–8). Thus far, many clinical trials have been conducted to assess the applicability of EV-based therapies for various pathological conditions including psoriasis (Citation9,Citation10). Zhang et al. demonstrate that subcutaneous injection of EXOs, derived from human umbilical cord blood mesenchymal stem cells (MSCs), inhibits the STAT3 signaling pathway in keratinocytes and suppresses the activation of DC cells, thereby alleviating inflammation severity in psoriasis (Citation10). These results suggested that the inflammatory responses in psoriasis can be regulated by exogenous EVs.
Although EV-based clinical approaches are being carried out in full swing for their low toxicity, high biocompatibility, and plasticity (Citation11), several caveats must be considered. For instance, the composition and targeting capacities of EVs vary depending on their cell sources (Citation12,Citation13). Besides, EVs cannot be stored at −80 °C for a long term. Desired characteristics of EVs, such as increased targeting ability, increased drug loading, and reduced immunogenicity, can be achieved by EV engineering based on their original properties (Citation14,Citation15). The membrane fusion-based hybrid technology is derived from the classical EXO separation technology. The biofilm used for EV hybridization and fusion can be either artificially modified liposome membrane or extracted complete cell membrane (Citation16–18). In this study, we have fused the EVs, derived from annexin A1 (ANXA1) overexpressing T cells with M2 macrophage membrane. Membrane fusion preserves the nature of EVs and M2 macrophage membrane, thereby retaining the anti-inflammatory properties of M2 macrophage as well as ANXA1 protein.
Our results showed that the engineered EVs could be effectively engulfed by macrophages, which promoted their M2 polarization in vitro. In addition, in vivo results showed that after subcutaneous injection of engineered EVs, the M2 macrophage biomarker Arg-1 was upregulated in lesional skin and the psoriasis-like skin inflammation was significantly ameliorated. We thus proposed a promising EV-based therapeutic approach for psoriasis.
2. Materials and methods
2.1. Human samples
In this study, the psoriatic skin biopsies were obtained from the lesional skin of psoriasis patients, while the normal skin biopsies were collected from discarded surgical specimens of healthy donors. Both patients and healthy donors underwent strict screening, and only subjects who had not received any medical treatment within 3 months prior to the recruitment were selected.
2.2. Animal experiments
8-12 weeks old male C57BL/6 mice purchased from Shanghai Laboratory Animal Center (Shanghai, China) were maintained under pathogen‐free conditions with ad libitum access to food and water. The mice in the treatment group were treated daily with topical application of 62.5 mg imiquimod (IMQ) cream (5%) (Sichuan Mingxin Pharmaceutical Co., Ltd, Chengdu, China) on the backs, whereas the mice in the control group were treated with Vaseline Lanette cream. All mice were observed for the following seven consecutive days as previously described (Citation19). As for EV treatment, mice were subcutaneously injected with 50 μg EVs on day 0, 2, 4, and 6. On day 8, mice were sacrificed, and tissues were collected.
2.3. Cell culture
Human monocytic cell line THP-1, human peripheral leukemia T cell line Jurkat and mouse lymphoma cell line EL4 were cultured and maintained in complete RPMI 1640 (Gibco, Carlsbad, CA), supplemented with 10% (v/v) heat-inactivated fetal bovine serum (FBS) and 1% penicillin/streptomycin. Mouse macrophage cell line RAW264.7 and human embryonic kidney epithelial cell line 293T were cultured in DMEM (Gibco, Carlsbad, CA) containing 10% (v/v) heat-inactivated FBS and 1% penicillin/streptomycin.
THP-1 cells were differentiated into M0 macrophages by treating with 100 nM phorbol myristate acetate (PMA; Sigma-Aldrich, St. Louis, MO) for 24 h. The M0 macrophages were then polarized toward M2 macrophages by treating with 20 ng/mL of human IL-4 (R&D Systems, Minneapolis, MN) for 48 h. RAW264.7 macrophages were treated or untreated with 20 ng/mL of mouse IL-4 (R&D Systems, Minneapolis, MN) for 48 h to obtain M2 and M0 macrophages, respectively. For all experiments, cells were incubated at 37 °C in a humidified atmosphere containing 5% (v/v) CO2.
2.4. Cell infection
To achieve overexpression, the ANXA1 cDNA sequence was cloned into pHR lentiviral vector (pHR-ANXA1) along with the GFP gene. The empty pHR vector (pHR-NC) was used as a negative control. pHR-ANXA1 or pHR-NC was transfected into 293T cells along with delta 8.9 and pMD2.G using PEI reagent (PolySciences, Warrington, PA). The viruses were harvested at 24, 48, and 72 h post-transfection, respectively. T cells were expanded in culture media for 4 days and diluted to a density of 1 million cells/well before being infected with lentiviruses in the presence of 8 μg/mL polybrene (Sigma-Aldrich, St. Louis, MO) for 24 h. Then, the GFP-positive cells were sorted.
2.5. Isolation of EVs
FBS was ultracentrifuged at 100,000 × g at 4 °C for at least 4 h to obtain EV-depleted FBS. Cells were cultured in media containing EV-depleted FBS. Then, the cell culture supernatant was pre-cleared by centrifuging at 500 × g for 15 min followed by 10,000 × g for 20 min. To isolate EVs, the supernatant was further ultracentrifuged at 100,000 × g for more than 3 h, and centrifuged again under the same condition after being resuspended with PBS.
2.6. Fabrication of engineered EVs
Macrophages were collected with cell membrane extraction buffer (Beyotime, Beijing, China). Then, the cell membrane was separated by repeated freeze–thaw cycles, followed by centrifuging at 8000 × g for 15 min and sonicating for 2 min at a frequency of 42 kHz with a power input of 100 W (Citation20). The concentration of EVs and M2 macrophage membrane vesicles was measured by BCA Protein Assay Kit (Thermo Fisher Scientific, Waltham, MA), then they were extruded through a 100 nm polycarbonate porous membrane for 10 times using an Avanti mini extruder (Avanti Polar Lipids, Alabaster, AL).
2.7. Characterization of EVs
Nano tracking analysis (NTA) was conducted using NanoSight (Malvern Instruments Ltd, Malvern, UK) to measure the size and zeta potential of EVs. EVs were visualized using transmission electron microscopy (TEM) with a model JMPEG- PTMC-1230 (JEOL, Tokyo, Japan).
2.8. Cellular uptake assay
EVs were incubated in PBS with 1 μM 1,1′-dioctadecyl-3,3,3′,3′-tetramethylindocarbocyanine perchlorate (Dil; Sigma-Aldrich, St. Louis, MO) for 20 min. The excess dye was removed by centrifugation, and the pelleted EVs were resuspended in ∼100 μL PBS. Macrophages were seeded at a density of 2 × 104 cells/well in six-well plates and incubated overnight. After pretreatment with Hank’s balanced salt solution (HBSS) for 15 min, macrophages were co-incubated with the Dil-labeled EVs.
For the cellular uptake assay, macrophages were seeded in six-well plates. The inhibitors including 300 mM sucrose (Sangon, Shanghai, China), 100 μM 5-(N, N-dimethyl) amiloride hydrochloride (DMA; MCE, Monmouth Junction, NJ), and 200 μM genistein (MCE, Monmouth Junction, NJ) were added into each well and incubated for 30 min (Citation21). Then, the inhibitors were withdrawn, followed by the addition of Dil-labeled EVs. After incubation, the cells were visualized under the fluorescent microscope (Leica, Wetzlar, Germany).
2.9. Real-time PCR (RT-PCR)
Total RNA was extracted from cells using TRIzol (Ambion, Austin, TX). The isolated RNA was reverse transcribed using the Hiscript™ III 1st Strand cDNA Synthesis kit (Vazyme, Nanjing, China). RT-PCR was performed using the SYBR qPCR Master Mix Kit (Vazyme, Nanjing, China). The relative expression levels of selected genes were normalized to GAPDH by the 2–ΔΔCt method.
2.10. CCK8 assay
The cells were seeded in 96-well plates at a concentration of 2500 cells/well and incubated in 5% CO2 at 37 °C, followed by EV co-incubation. After incubation, 10 μL of CCK8 solution (Dojindo, Kumamoto, Japan) was added to each well and incubated for 2 h. The number of live cells was assessed by measuring the absorbance at 450 nm using a microplate reader (Thermo Fisher Scientific, Waltham, MA).
2.11. Western blotting (WB)
Cultured cells were lysed in radio-immunoprecipitation assay buffer and heated at 95 °C for 5 min. The protein samples were separated by SDS-polyacrylamide gels and transferred to PVDF membranes. The membranes were incubated in the blocking buffer containing 5% nonfat milk for 1 h followed by overnight incubation with primary antibodies against ANXA1 (1:1000, CST, Beverly, MA, #D5V2T) or alpha-tubulin (1:5000, Proteintech, Rosemont, IL, #11224-1-AP) at 4 °C. The membranes were then incubated with HRP-linked anti-rabbit IgG secondary antibody (CST, Beverly, MA) for 1 h. Protein expression levels were determined by densitometry analysis.
2.12. PASI scoring
We evaluated the severity of mouse model psoriasis according to the Psoriasis Area and Severity Index (PASI) score. The erythema score, infiltration score, desquamation score, and area score were independently evaluated. Each indicator has a five-point scale which goes from 0 to 4: 0 = none; 1 = slight; 2 = moderate; 3 = severe; 4 = very severe. The scores for each indicator were then summed to obtain the cumulative score (0–16 points).
2.13. Histological analysis
Patient biopsies and mouse tissue samples were fixed in paraformaldehyde at room temperature for 24 h and then embedded in paraffin. Six sections from the middle portion of the tissues were selected for hematoxylin and eosin (H&E) and standard immunohistochemistry (IHC) staining. Cell numbers were quantified as the mean number of positive cells in five fields (original magnification, ×400). Anti-CD68 (GB113109), anti-Arg-1 (GB11285), anti-TNF-α (GB13452), anti-IL-6 (GB11117), anti-IL-1β (GB11113), anti-IL-17A, and Anti-CD3 (GB111337) were obtained from Servicebio (Wuhan, China).
2.14. Statistical analysis
The results of in vitro reported here are representative of three independent experiments and the results of in vivo expressed as three mice per group. All data are presented as the mean ± standard error of the mean (SEM). Student’s t-test was used for comparisons between two groups. A p-value <0.05 was considered statistically significant.
3. Results
3.1. Macrophages accumulated in psoriatic skin
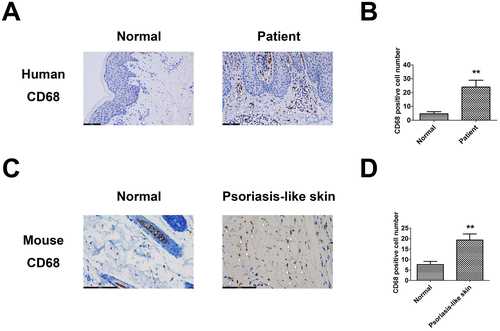
To identify whether macrophages had infiltrated into psoriasis skins, CD68+ macrophages from tissue samples were stained by IHC staining. The number of CD68+ macrophages was significantly higher in the patient group compared to the control group (). Similar results were observed in the IMQ-induced psoriasis-like mouse group and the control group (). These results demonstrated that proinflammatory macrophages significantly accumulated in psoriatic skin, suggesting the potential application of engineered EVs in ameliorating psoriatic skin inflammation by regulating the polarization of inflammatory macrophages.
Figure 1. Macrophages accumulated in psoriatic skin. (A, B) CD68+ cells in the skin of normal subjects and psoriasis patients. (C, D) CD68+ cells in normal and IMQ-induced psoriasis-like mice skin. **p < 0.01 versus the normal group.
3.2. Fabrication and characterization of engineered EVs
shows the preparation and optimization procedures of engineered EVs. T cells were first infected with ANXA1 overexpressing lentiviral vector, and the EVs were isolated by ultracentrifugation. Meanwhile, membrane vesicles of IL-4-induced M2 macrophage were separated by repeated freeze–thaw cycles. The engineered EVs were obtained by co-extruding the T cell-derived EVs with the M2 macrophage membrane. In the following part, the group name JA-EV and JC-EV would be used for the EVs isolated from Jurkat cells infected with either ANXA1 lentiviral vector or negative control lentiviral vector, respectively. The EVs isolated from EL4 cells infected with ANXA1 lentiviral vector or negative control lentiviral vector were named as EA-EV and EC-EV, respectively. At last, the engineered EVs generated by co-extruding JA-EV or EA-EV with M2 macrophage membrane were named as JAM and EAM, respectively.
Figure 2. Fabrication and characterization of engineered EVs. (A) Schematic diagram illustration of the fabrication procedures of engineered EVs. (B, C) Expression levels of ANXA1 protein in infected Jurkat and EL4 T cells. (D, E) Optimization of manufacturing procedures for engineered EVs with macrophage membrane fusion. (F, G) Stability assessment of EVs. (H) Zeta potentials of original EVs, engineered EVs, and M2 macrophage membrane. (I) TEM photos of engineered EVs. **p < 0.01 versus the normal group.
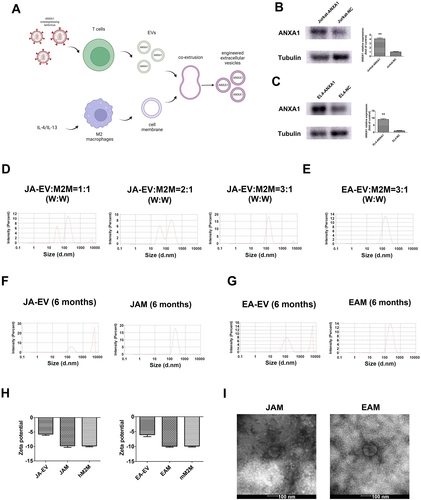
First, WB results revealed that ANXA1 proteins were successfully overexpressed in EVs from Jurkat and EL4 cells (). To optimize manufacturing procedures, the EVs and the M2 macrophage membrane were co-extruded at different ratios. The EVs were added with 1, 2, or 3 μg, and the cell membrane was set as 1 μg. NTA revealed two peaks in the size distribution when the vesicles were co-extruded at the 1:1 and 2:1 weight ratio. When the weight ratio reached 3:1, there was only one peak (). These results implied that the two kinds of membranes could not fuse well at a low weight ratio. Hence, the weight ratio 3:1 was adopted for further experiments. The results also showed that the particle size of JAM was about 144 nm (), and EAM was about 169 nm ().
For the stability evaluation of engineered EVs, JAM and EAM were stored at −80 °C for 6 months. NTA showed the size distribution of JA-EV and EA-EV increased significantly, indicating the fusion between EVs. However, the size of JAM and EAM slightly increased over time (). Zeta potential is an electrochemical property that can be used to measure the stability of EVs. A high absolute zeta potential value represents more electrostatic repulsion between particles, leading to higher stability. In our study, the zeta potentials of the EVs, engineered EVs, and cell membrane vesicles were approximately −5, −11, and −10 mV, respectively (), which indicated that co-extrusion improved the stability of EVs.
TEM photos showed that the sizes of JAM and EAM were slightly smaller than their estimated sizes in the NTA (). It was possibly because the samples were dried out and stained before TEM scanning. All these data indicated that we had successfully constructed the engineered EVs.
3.3. Cellular uptake and endocytic mechanism of EVs by macrophages
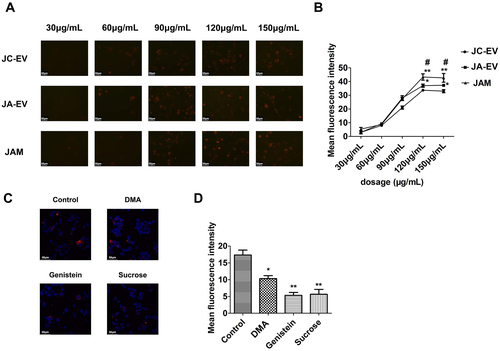
To identify whether the engineered EVs could be internalized by macrophages, JC-EV, JA-EV, and JAM were labeled with Dil dye. Macrophages were then incubated with Dil-labeled engineered EVs at different concentrations. Among the groups, JAM-treated cells exhibited the highest mean fluorescence intensity (MFI) when the EV concentration exceeded 120 μg/mL (). The internalization was also observed to be time-dependent, although, there was no difference in MFI among the groups (Supple Figure 1(A,B)). To further investigate the endocytic mechanism of EVs in macrophages, the cells were pretreated with inhibitory reagents DMA, genistein, or sucrose for 30 min to evaluate the effects of micropinocytosis, caveolae, and clathrin-mediated endocytosis on the internalization of EVs, respectively. Our results showed that both genistein and sucrose significantly reduced the intensity of red fluorescence in the macrophages, while DMA could also partly reduce macrophages’ uptake of EVs ().
Figure 3. Cellular uptake and endocytic mechanism of EVs by macrophages. (A, B) Macrophages were incubated with Dil-labeled EVs at indicated concentrations. *p < 0.05, **p < 0.01 versus JC-EV group. #p < 0.05 versus JA-EV group. (C, D) Macrophages were treated with DMA, genistein, or sucrose before incubating with Dil-labeled EVs. *p < 0.05, **p < 0.01 versus the control group.
3.4. JAM reprogramed macrophages into M2 phenotype in vitro
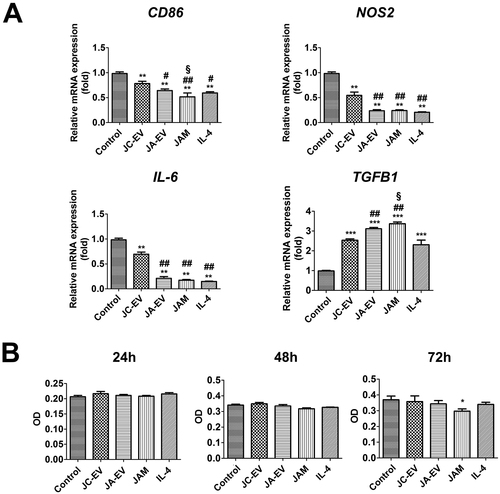
To evaluate the effect of JAM on the polarization of macrophages, macrophages were treated with PBS, JC-EV, JA-EV, or JAM, and the IL-4-stimulated macrophages were served as positive control. The phenotype of macrophages was determined by RT-PCR. The results indicated that JC-EV, JA-EV, and JAM significantly downregulated the transcription of M1 markers (CD86, NOS2, and IL-6), while upregulating the transcription of M2 marker TGFB1 (). These results revealed that JAM could effectively reprogram macrophages toward a reparative phenotype in vitro. In addition, the CCK8 assay revealed that only JAM significantly reduced the viable cell number of THP-1-derived macrophages after long-term treatment (72 h) ().
Figure 4. JAM reprogramed macrophages into M2 phenotype in vitro. (A) The relative mRNA levels of CD86, NOS2, IL-6, and TGFB1 in each group were measured by RT-PCR. (B) The number of viable macrophages was determined by CCK8 assay after incubating with EVs. *p < 0.05, **p < 0.01, ***p < 0.001 versus the control group. #p < 0.05, ##p < 0.01 versus JC-EV group. §p < 0.05 versus JA-EV group.
3.5. EAM ameliorated psoriasis-like skin inflammation in mice
To validate the in vitro data, we investigated the therapeutic effects of EAM in the IMQ-induced psoriasis-like mouse model. After the daily application of IMQ cream, mice were subcutaneously injected with PBS, EC-EV, EA-EV, or EAM every other day (). On day 8, the lesion scores and spleen size were evaluated.
Figure 5. EAM ameliorated psoriasis-like skin inflammation in mice. (A) Experimental design for EV treatment in psoriasis-like mice. (B) Representative photos of mice and spleen after treatment. (C) PASI score. (D) H&E staining of the lesion in each group. (E) IHC staining for CD68. (F) IHC staining for Arg-1. (G) IHC staining for IL-1β. (H) IHC staining for IL-6. (I) IHC staining for TNF-α. (J) IHC staining for IL-17A. *p < 0.05, **p < 0.01 versus the control group. #p < 0.05, ##p < 0.01 versus PBS group. §p < 0.05, §§p < 0.01 versus EC-EV group. &p < 0.05 versus EA-EV group.
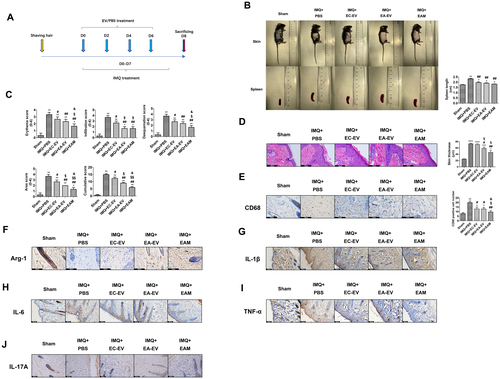
We observed that the PBS-treated group exhibited the largest spleen size (>2 cm). EV treatment dramatically relieved inflammation in the spleen, as reflected by the spleen size (<2 cm). However, there were no significant differences in the spleen size among the EC-EV, EA-EV, and EAM groups (). Besides, the PBS-treated mice also had the highest PASI score. EV-based treatment especially EAM could alleviate inflammatory responses in the skin as evidenced by a decreased PASI score (). The effect of engineered EVs on epidermal hyperplasia was further evaluated by H&E stained slides of the skin lesion. The results showed that IMQ treatment resulted in increased epidermal thickness, while treatment with EA-EV or EAM significantly relieved the abnormality ().
IHC staining of skin sections with the anti-CD68 antibody demonstrated that EAM treatment dramatically reduced the macrophage infiltration in skin lesions (). We observed that IMQ induced a significant decrease in the level of the M2 macrophage marker Arg-1 in the skin, and it was restored by EAM treatment (). Inflammatory factors, including IL-1β, IL-6, TNF-α, and IL-17A, are strongly associated with psoriasis prognosis. In our study, the inflammatory factors, especially IL-1β, IL-6, and TNF-α, were reduced in EAM group, compared with the PBS-treated group ().
3.6. Effects of EV treatment on major organs in the mouse model
According to previous studies, IMQ may cause systemic response (Citation22). To evaluate the histopathological changes in the major organs after IMQ administration, the histomorphological changes of the major organs on day 8 were evaluated by H&E and IHC staining. After IMQ treatment, the tissue morphologies of the spleen, liver, and kidney were significantly different from that in the control group, indicating the presence of acute systemic inflammation. In detail, the number of CD3+ cells in the spleen decreased after IMQ treatment, suggesting their recruitment into the circulation. However, the administration of EVs partially rescued the CD3+ T cell population ().
Figure 6. Effects of EV treatment on major organs in the psoriasis-like mouse model. (A) H&E and CD3 IHC staining of the spleen. (B) H&E staining of the liver. (C) H&E staining of the kidney.
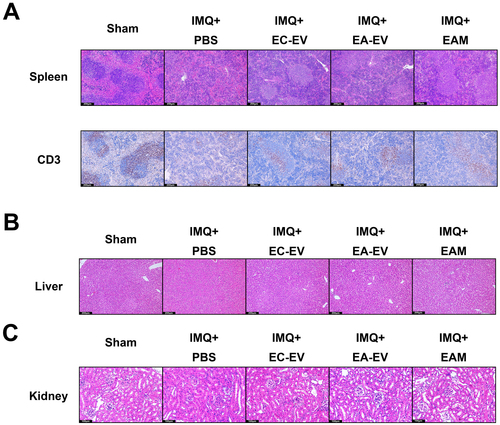
In addition, the liver morphology of hepatocytes indicated a severe organ injury in the PBS-treated group, manifested by disorganized cristae and damaged hepatic lobules. In contrast, EV treatment dramatically restored the integrity of hepatic lobules (). Similarly, significant renal interstitial injury was also found in the PBS-treated group, while EV treatment efficiently rescued these structural anomalies ().
Regarding toxicity to other organs, no histopathological changes were observed in the lung, brain, and heart of mice treated with or without EVs compared to the control group (Supple Figure 2(A-C)). Thus, EAM is generally biologically safe, which allows the potential clinical application.
4. Discussion
In this study, we demonstrated that engineered T cell-derived EVs could efficiently ameliorate psoriasis-like skin inflammation by regulating the polarization of macrophages and reducing the secretion of inflammatory cytokines in the skin. Many recent studies have focused on fusogenic nanoparticle-based anti-inflammatory therapies. To the best of our knowledge, the present study is the first to apply engineered T cell-derived EVs in psoriasis treatment.
We choose T cells overexpressing ANXA1 as the source of EVs because T cells naturally interact extensively with macrophages during process like antigen presentation. A previous study has confirmed that T cell-derived EVs possess tropism for macrophages (Citation23). ANXA1, a crucial anti-inflammatory regulator belonging to the calcium-dependent phospholipid-binding protein family, can decrease inflammation by reducing leukocyte adhesion and limiting the activity of cPLA2 (Citation24–27). ANXA1 also promotes the expression of IL-10 by activating the formyl peptide receptor type 2 (FRP2) on macrophages, which in turn induces the polarization of macrophages toward M2 phenotype in an autocrine manner (Citation28–30). In addition, ANXA1 can promote the generation of EXOs by facilitating the cholesterol transfer from endoplasmic reticulum to multivesicular body (MVB) (Citation31). Additionally, a proteomics study reveals the abundance of ANXA1 in the EXOs derived from T cells (Citation32). These results provide a biological basis for loading ANXA1 into EVs.
We choose M2 macrophage membrane as the fusion partner for EVs because it is enriched with proteins such as scavenger receptors CD163 and CD206. Activation of these receptors can promote the release of IL-10 or induce the polarization of M2 macrophages (Citation33,Citation34). IL-4, IL-13, and other ‘M2’ anti-inflammatory stimuli also induce the expression of IL-1 receptor type 2 (IL-1R2) in macrophages (Citation35). The binding of IL-1 to the IL-1R2, which lacks the Toll/IL-1 receptor (TIR) domain, attenuates subsequent signaling transduction by neutralizing IL-1 (Citation36). As EVs can fuse with target cells, we hypothesize that those recipient cells will inherit these molecules at the membrane surfaces (Citation37). To preserve the properties of the EVs and M2 macrophage membrane, we fused them together.
Intravenous injection would lead to the deposition of EVs in several organs (Citation38). Our local administration endows EVs with high targeting capacity as the internalization of EVs is much more efficient in phagocytes compared to non-phagocytic cells (Citation38). Our results revealed that the uptake of engineered EVs in macrophages was regulated by multiple pathways. Interestingly, the fusion with M2 macrophage membrane resulted in increased uptake of EVs by macrophages. As is well-known, macrophages infected with Mycobacterium tuberculosis or osteoclasts under physiological conditions tend to fuse with themselves, forming multinucleated cells. Previous results indicate that the fusion can be induced by the stimulation of IL-4 and IL-13, which promotes the expression of the fusion proteins called fusogens on the surface of macrophages (Citation39). This may explain why engineered EVs are internalized more efficiently.
In the in vivo experiments, we noticed that EV treatment effectively ameliorated skin inflammation and reduced macrophage infiltration. In vitro experiments showed that EAM treatment led to slight decrease of macrophage population after prolonged treatment. Thus, the reduced macrophage infiltration was likely due to the decreased recruitment of macrophages to the lesional skin. As expected, after M2 polarization, we found that EV treatment significantly downregulated the IMQ primed IL-1β, IL-6, and TNF-α accumulation in skin. All these results suggested that the engineered EVs effectively interrupted the positive feedback loop of inflammation in psoriasis. We also found that EV treatment, especially EAM, restored liver sinusoid structure and alleviated renal injury induced by IMQ. Psoriasis is not restricted to the skin. The cytokine levels in serum significantly elevate in psoriasis patients (Citation40), and psoriatic arthritis, a common complication of psoriasis, can cause great damage to distal joints (Citation41). Recently, Van Raemdonck et al. have found that macrophages in the skin lesion are capable to trigger skin-to-joint crosstalk in psoriatic arthritis (Citation42). All these results indicated the potential therapeutic application of our engineered EVs in the more extensive immune system.
Lastly, even though the overall components of the engineered EVs have not been evaluated in our study, there have been convincing results supporting the anti-inflammatory responses are deeply dependent on the pre-loaded ANXA1 protein and M2 macrophage membrane in the EVs, which potently regulate macrophage polarization. All the compelling evidence has suggested that we successfully built a bridge between constructing a fusogenic nanoparticle-delivery system and psoriasis therapy.
Ethical approval
The study was approved by the Ethics Committee of Shanghai Tenth People’s Hospital affiliated to Tongji University School of Medicine (Shanghai, China) and was performed in accordance with the Declaration of Helsinki.
Consent form
Informed consents were signed by all patients and donors. All animal experiments were approved by the Animal Care and Use Committee of Shanghai Jiao Tong University School of Medicine (no. A-2017-009) and were performed according to the committee-approved guidelines.
Author contributions
Zeng Wang designed the experiment and analyzed the data. Zeng Wang, Zhizhen Qin, Jiadie Wang, Xinqi Xu, Mengxin Zhang, Yuyue Liang performed the experiments. Yukun Huang conducted NTA analysis. Zengyang Yu and Yu Gong provided the human sample. Luxian Zhou provided the conception. Yiran Qiu and Minglu Ma performed the WB. The manuscript was written by Zeng Wang and Zhizhen Qin. Zhizhen Qin, Dan Li and Bin Li critically reviewed the article. All the authors read and approved the manuscript.
Supplemental Material
Download Zip (3.5 MB)Acknowledgements
The authors thank the Shanghai Collaborative Innovation Center of Cellular Homeostasis Regulation and Human Diseases for providing constructive comments on our work and Jieqiong Chen, Le Kuai, Shouyu Ke, Xiaoxia Wang, Hao Cheng, and Qianru Huang for their valuable suggestions.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
Data will be made available on reasonable request.
Additional information
Funding
References
- Griffiths CEM, Armstrong AW, Gudjonsson JE, et al. Psoriasis. Lancet. 2021;397(10281):1–10.
- Lin SH, Chuang HY, Ho JC, et al. Treatment with TNF-alpha inhibitor rectifies M1 macrophage polarization from blood CD14+ monocytes in patients with psoriasis independent of STAT1 and IRF-1 activation. J Dermatol Sci. 2018;91(3):276–284.
- Hu P, Wang M, Gao H, et al. The role of helper T cells in psoriasis. Front Immunol. 2021;12:788940.
- Yang L, Fu J, Han X, et al. Hsa_circ_0004287 inhibits macrophage-mediated inflammation in an N(6)-methyladenosine-dependent manner in atopic dermatitis and psoriasis. J Allergy Clin Immunol. 2022;149(6):2021–2033.
- Willms E, Cabanas C, Mager I, et al. Extracellular vesicle heterogeneity: subpopulations, isolation techniques, and diverse functions in cancer progression. Front Immunol. 2018;9:738.
- Kalluri R, LeBleu VS. The biology, function, and biomedical applications of exosomes. Science. 2020;367(6478):eaau6977.
- Pegtel DM, Gould SJ. Exosomes. Annu Rev Biochem. 2019;88:487–514.
- O’Brien K, Breyne K, Ughetto S, et al. RNA delivery by extracellular vesicles in mammalian cells and its applications. Nat Rev Mol Cell Biol. 2020;21(10):585–606.
- Zhang B, Lai RC, Sim WK, et al. Topical application of mesenchymal stem cell exosomes alleviates the imiquimod induced psoriasis-like inflammation. Int J Mol Sci. 2021;22(2):720.
- Zhang Y, Yan J, Li Z, et al. Exosomes derived from human umbilical cord mesenchymal stem cells alleviate psoriasis-like skin inflammation. J Interferon Cytokine Res. 2022;42(1):8–18.
- Cheng L, Hill AF. Therapeutically harnessing extracellular vesicles. Nat Rev Drug Discov. 2022;21(5):379–399.
- Garcia-Martin R, Wang G, Brandao BB, et al. MicroRNA sequence codes for small extracellular vesicle release and cellular retention. Nature. 2022;601(7893):446–451.
- Sun T, Kwong CHT, Gao C, et al. Amelioration of ulcerative colitis via inflammatory regulation by macrophage-biomimetic nanomedicine. Theranostics. 2020;10(22):10106–10119.
- Sadeghi S, Tehrani FR, Tahmasebi S, et al. Exosome engineering in cell therapy and drug delivery. Inflammopharmacology. 2023;31(1):145–169.
- Herrmann IK, Wood MJA, Fuhrmann G. Extracellular vesicles as a next-generation drug delivery platform. Nat Nanotechnol. 2021;16(7):748–759.
- Sato YT, Umezaki K, Sawada S, et al. Engineering hybrid exosomes by membrane fusion with liposomes. Sci Rep. 2016;6:21933.
- Hu S, Wang X, Li Z, et al. Platelet membrane and stem cell exosome hybrid enhances cellular uptake and targeting to heart injury. Nano Today. 2021;39:101210.
- Rayamajhi S, Nguyen TDT, Marasini R, et al. Macrophage-derived exosome-mimetic hybrid vesicles for tumor targeted drug delivery. Acta Biomater. 2019;94:482–494.
- Peng S, Cao M, Sun X, et al. Recombinant programmed cell death 1 inhibits psoriatic inflammation in imiquimod‑treated mice. Int J Mol Med. 2020;46(2):869–879.
- Zhang N, Song Y, Huang Z, et al. Monocyte mimics improve mesenchymal stem cell-derived extracellular vesicle homing in a mouse MI/RI model. Biomaterials. 2020;255:120168.
- Wang Z, Zhu H, Shi H, et al. Exosomes derived from M1 macrophages aggravate neointimal hyperplasia following carotid artery injuries in mice through miR-222/CDKN1B/CDKN1C pathway. Cell Death Dis. 2019;10(6):422.
- Grine L, Steeland S, Van Ryckeghem S, et al. Topical imiquimod yields systemic effects due to unintended oral uptake. Sci Rep. 2016;6:20134.
- Feng D, Zhao WL, Ye YY, et al. Cellular internalization of exosomes occurs through phagocytosis. Traffic. 2010;11(5):675–687.
- Araujo TG, Mota STS, Ferreira HSV, et al. Annexin A1 as a regulator of immune response in cancer. Cells. 2021;10(9):2245.
- de Coupade C, Solito E, Levine JD. Dexamethasone enhances interaction of endogenous annexin 1 with l-selectin and triggers shedding of l-selectin in the monocytic cell line U-937. Br J Pharmacol. 2003;140(1):133–145.
- Kim KM, Kim DK, Park YM, et al. Annexin-I inhibits phospholipase A2 by specific interaction, not by substrate depletion. FEBS Lett. 1994;343(3):251–255.
- Solito E, Romero IA, Marullo S, et al. Annexin 1 binds to U937 monocytic cells and inhibits their adhesion to microvascular endothelium: involvement of the alpha 4 beta 1 integrin. J Immunol. 2000;165(3):1573–1581.
- Parente L, Solito E. Annexin 1: more than an anti-phospholipase protein. Inflamm Res. 2004;53(4):125–132.
- Ferlazzo V, D’Agostino P, Milano S, et al. Anti-inflammatory effects of annexin-1: stimulation of IL-10 release and inhibition of nitric oxide synthesis. Int Immunopharmacol. 2003;3(10–11):1363–1369.
- Li Y, Cai L, Wang H, et al. Pleiotropic regulation of macrophage polarization and tumorigenesis by formyl peptide receptor-2. Oncogene. 2011;30(36):3887–3899.
- Eden ER, Sanchez-Heras E, Tsapara A, et al. Annexin A1 tethers membrane contact sites that mediate ER to endosome cholesterol transport. Dev Cell. 2016;37(5):473–483.
- Bosque A, Dietz L, Gallego-Lleyda A, et al. Comparative proteomics of exosomes secreted by tumoral Jurkat T cells and normal human T cell blasts unravels a potential tumorigenic role for valosin-containing protein. Oncotarget. 2016;7(20):29287–29305.
- Hagert C, Sareila O, Kelkka T, et al. The macrophage mannose receptor regulate mannan-induced psoriasis, psoriatic arthritis, and rheumatoid arthritis-like disease models. Front Immunol. 2018;9:114.
- Landis RC, Philippidis P, Domin J, et al. Haptoglobin genotype-dependent anti-inflammatory signaling in CD163(+) macrophages. Int J Inflam. 2013;2013:980327.
- Supino D, Minute L, Mariancini A, et al. Negative regulation of the IL-1 system by IL-1R2 and IL-1R8: relevance in pathophysiology and disease. Front Immunol. 2022;13:804641.
- Zhou N, Chen LL, Chen J, et al. Molecular characterization and expression analysis of IL-1beta and two types of IL-1 receptor in barbel steed (Hemibarbus labeo). Comp Biochem Physiol B Biochem Mol Biol. 2020;241:110393.
- Jadli AS, Ballasy N, Edalat P, et al. Inside(sight) of tiny communicator: exosome biogenesis, secretion, and uptake. Mol Cell Biochem. 2020;467(1–2):77–94.
- Gurung S, Perocheau D, Touramanidou L, et al. The exosome journey: from biogenesis to uptake and intracellular signalling. Cell Commun Signal. 2021;19(1):47.
- Pereira M, Petretto E, Gordon S, et al. Common signalling pathways in macrophage and osteoclast multinucleation. J Cell Sci. 2018;131(11):jcs216267.
- Bai F, Zheng W, Dong Y, et al. Serum levels of adipokines and cytokines in psoriasis patients: a systematic review and meta-analysis. Oncotarget. 2018;9(1):1266–1278.
- Gladman DD, Antoni C, Mease P, et al. Psoriatic arthritis: epidemiology, clinical features, course, and outcome. Ann Rheum Dis. 2005;64(Suppl. 2):ii14–ii17.
- Van Raemdonck K, Umar S, Palasiewicz K, et al. TLR7 endogenous ligands remodel glycolytic macrophages and trigger skin-to-joint crosstalk in psoriatic arthritis. Eur J Immunol. 2021;51(3):714–720.