
Abstract
In bacteria, K+ is used to maintain cell volume and osmotic potential. Homeostasis normally involves a network of constitutively expressed transport systems, but in K+ deficient environments, the KdpFABC complex uses ATP to pump K+ into the cell. This complex appears to be a hybrid of two types of transporters, with KdpA descending from the superfamily of K+ transporters and KdpB belonging to the superfamily of P-type ATPases. Studies of enzymatic activity documented a catalytic cycle with hallmarks of classical P-type ATPases and studies of ion transport indicated that K+ import into the cytosol occurred in the second half of this cycle in conjunction with hydrolysis of an aspartyl phosphate intermediate. Atomic structures of the KdpFABC complex from X-ray crystallography and cryo-EM have recently revealed conformations before and after formation of this aspartyl phosphate that appear to contradict the functional studies. Specifically, structural comparisons with the archetypal P-type ATPase, SERCA, suggest that K+ transport occurs in the first half of the cycle, accompanying formation of the aspartyl phosphate. Further controversy has arisen regarding the path by which K+ crosses the membrane. The X-ray structure supports the conventional view that KdpA provides the conduit, whereas cryo-EM structures suggest that K+ moves from KdpA through a long, intramembrane tunnel to reach canonical ion binding sites in KdpB from which they are released to the cytosol. This review discusses evidence supporting these contradictory models and identifies key experiments needed to resolve discrepancies and produce a unified model for this fascinating mechanistic hybrid.
K+ transport and the need for an active pump
The role of K+ in osmoregulation
All cells need to control the contents of their cytoplasm and to maintain their cellular volume. This is especially challenging for bacteria, as they must rapidly adapt to environments with widely varying compositions and concentrations of essential solutes. For bacteria, membrane transport represents the primary control mechanism. In the case of volume, the cell controls osmotic forces across the membrane to avoid rupture, on the one hand, or dehydration and collapse, on the other. Osmosis refers to the flow of water across the cell membrane and can be driven by gradients of impermeable solutes which generate osmotic pressure. Cells with a stabilizing cell wall, like Escherichia coli, maintain a strong positive osmotic pressure inside the cell, which is referred to as turgor. K+ is by far the most abundant cation in the cytosol and is thus the dominant contributor to turgor pressure (Epstein, Citation2003). The cell membrane is largely impermeable to K+ and, by establishing a K+ gradient, the cell generates osmotic pressure and an electrochemical membrane potential which are used to adapt to changing environmental osmolalities. As a cell grows, it must take up K+ to drive an expansion of cell surface and volume; this process requires energy. In non-growing cells there is a balance between uptake and release of K+ and a tight regulation of the relevant transport systems maintains homeostasis. Depending on extracellular osmolality and pH, the intracellular K+ concentrations in bacteria range between 0.2 and 1 M (Richey et al., Citation1987) and inside negative membrane potentials between −100 and −150 mV (Damper & Epstein, Citation1981; Krulwich et al., Citation2011).
Regulation of K+ concentration
Bacteria have a variety of uptake systems designed to maintain stable cytoplasmic K+ concentrations at different external K+ concentrations. In the case of decreased osmolality of the surrounding medium (osmotic downshock), the bacteria releases K+ in addition to other osmolytes like glutamate, trehalose, putrecine, and proline (Altendorf et al., Citation2009). The ions are released through K channels such as KefB and KefC (Epstein, Citation2003) and through the nonselective mechanosensitive channels MscS and MscL (Booth & Blount, Citation2012). In the case of increased osmolality (osmotic upshock), K+ accumulation rapidly occurs through the Trk potassium uptake system (Rhoads & Epstein, Citation1978). These systems have K+ affinities in the order of 0.5 mM (TrkG) and 2.5 mM (TrkH) and some might be energized by the proton-motive force and/or regulated by binding of ATP (Diskowski et al., Citation2015; Stewart et al., Citation1985). A further K+ uptake system, Kup (formerly TrkD) has a K+ affinity in the order of 0.4 mM (Bossemeyer et al., Citation1989), but is not involved in osmoregulation (Schleyer et al., Citation1993). These K+-uptake systems are constitutively expressed and are sufficient for most environments the bacteria encounter. In K+ deficient environments (<100 µM), however, expression of the KdpFABC complex is induced to actively pump K+ across the plasma membrane with an apparent K+ affinity of about 2 µM (Rhoads et al., Citation1976).
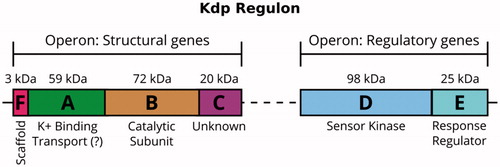
The Kdp system comprises two operons (), one of which contains the genes of proteins that sense and respond to environmental conditions (kdpDE) and the other provides the genes of the protein complex which carries out the transport process (kdpFABC) (Polarek et al., Citation1988). KdpD and KdpE form a two-component system able to sense K+-limiting conditions and then to trigger expression of the pump (Altendorf et al., Citation1994). Although the function of KdpD as a membrane-bound sensor kinase and KdpE as a soluble response regulator is generally agreed (Epstein, Citation2016; Heermann & Jung, Citation2010), the precise signal recognized by KdpD is still under discussion. The most recent proposal is that KdpD does not sense turgor pressure directly, but instead senses the concentration of two cations in the cytoplasm, Na+ and NH4+ (Epstein, Citation2016). Upon activation, KdpD autophosphorylates a histidine residue in its cytoplasmic tail, transfers this phosphate to KdpE, which binds to the promotor that activates kdpFABC transcription and expression of the KdpFABC complex (Heermann & Jung, Citation2010).
Figure 1. The Kdp K+-uptake system is closely regulated at the transcriptional level. The control is mediated by the sensor kinase KdpD and the response regulator KdpE, which are expressed constitutively. In an environment with K+ deficiency, KdpD phosphorylates KdpE, which then promotes transcription of the kdpFABC operon and expression of the KdpFABC complex.
Types of ion transport
In principle, there are two different modes of transport that account for the uptake and release of cations across the membranes of all living cells: passive and active (Gadsby et al., Citation2009). In passive transport, ions or other hydrophilic substances move “downhill”, following their electrochemical potential gradient across the membrane. The diffusion rate of cations through the hydrophobic core of the lipid bilayer is quite small, but is greatly facilitated by ion channels which the cell is able to regulate by a variety of different mechanisms (Hille, Citation2001). On their own, these passive transport systems would eventually equilibrate ion concentrations across the membrane, and therefore, are not able to maintain the ion gradients that produce turgor pressure and more generally sustain the cell. Therefore, active transport systems exist to transfer substrates “uphill” against their electrochemical gradients. The energy for such uphill transport is provided in two different ways. For the first, called primary active transport, energy is provided directly by light, redox energy or hydrolysis of energy-rich compounds such as ATP (Läuger, Citation1991). For the second, called secondary active transport, the uphill transport of one substrate is coupled to the downhill movement of a second substrate, whose electrochemical potential gradient drives the process (Stein, Citation1986).
The KdpFABC complex functions as a primary active transporter that uses ATP as an energy source. This complex is an unusual hybrid in which a subunit related to a family of K+ channels is associated with a subunit related to ATP-dependent ion pumps. The way in which these subunits work together to accomplish K+ transport has been a topic of great interest for at least 40 years.
K+ channels and the superfamily of K+ transporters
Ion channels are ubiquitous across the phylogenetic tree. Although initially discovered in nerve axons where they promote propagation of electrical signals, ion channels also play key roles in plants, fungi and prokaryotes. As illustrated by the bacterial archetype, KcsA, these molecules are capable of a high flux of a specific ionic species across the membrane. KcsA is composed of two transmembrane helices separated by a P-loop (M1PM2). This motif assembles into a homotetramer in which the P-loop crosses the extracellular leaflet of the membrane as “pore” helix and then folds back with a Gly-rich loop that provides oxygen ligands within a pore that also serves as a selectivity filter for K+. This basic architecture has diversified greatly to accommodate different ionic species and to incorporate a variety of different gating mechanisms to control transport. TrkH and KtrB are two closely related channels from the Trk protein family that helps to maintain K+ concentrations and turgor in bacteria. These channels are members of the Superfamily of K Transporters (SKT), which also includes HKT1,2 from plants and Trk1,2 from yeast (Diskowski et al., Citation2015; Waters et al., Citation2013). Members of the SKT superfamily are the product of gene duplications which have produced a four-fold genetic repeat of the original subunit represented by KscA. TrkH and KtrB retain the central selectivity filter, but have lost the strict four-fold symmetry of the homotetrameric KcsA and have added a distinctive kinked helix to the third repeat (D3), which has been proposed to function in gating (Cao et al., Citation2013). Both TrkH and KtrB are associated with soluble subunits (TrkA and KtrA, respectively) that control ion flux in response to ATP binding by interaction with the gate (Diskowski et al., Citation2015). Although the structural mechanisms are still under investigation (Diskowski et al., Citation2017), one hypothesis is that the oligomeric assembly of soluble domains undergoes a conformational change that pulls on the D3M2 helix to open the gate and allow K+ to flow across the membrane.
Active transport and the P-type ATPases
ATP hydrolyzing ion transporters (or ATPases) are divided in three subclasses, the F-type (called A-type in archaea), V-type and P-type ATPases. F-type, V-type and A-type ATPases have key sequence homologies and subunit architectures that underline their phylogenetic kinship (Coskun et al., Citation2004; Gogarten et al., Citation1989). P-type ATPases are the largest group of the ion-translocating ATPases that have diversified to perform many different and distinct roles in membranes (Kühlbrandt, Citation2004; Palmgren & Nissen, Citation2011). They are divided into ten families, members of which are found in almost all eukaryotic cells and in bacteria (Axelsen & Palmgren, Citation1998; Chan et al., Citation2010; Pedersen et al., Citation2014). The name is derived from the reaction intermediate formed when a highly conserved aspartic acid is phosphorylated (Pedersen & Carafoli, Citation1987). A common cycle of ATP hydrolysis incurs several intermediates with specific chemistry at the active site. As a result, P-type ATPases are often inhibited by characteristic transition state analogs such as vanadate, and fluoride derivatives of Al, Be and Mg.
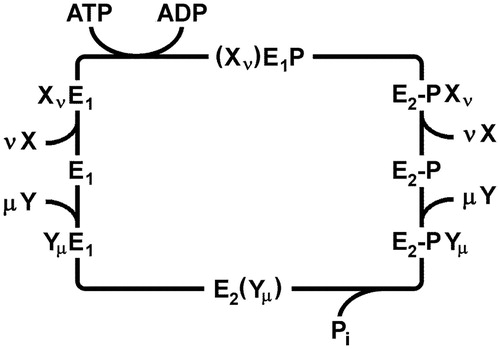
The first P-type ATPase studied in detail was the Na,K-ATPase. The pump cycle was named after Robert L. Post and R. Wayne Albers, who showed how enzyme and transport activities were related to each other (Albers, Citation1967; Post et al., Citation1972). The main features of this so-called Post-Albers cycle are common to all P-type ATPases investigated so far (. It defines two principal conformations: E1, in which the ion binding sites are accessible to the cytoplasm, and E2, in which the ion sites are accessible to the extracellular or luminal milieu. In the first half of the cycle, ATP hydrolysis and enzyme phosphorylation drive the protein conformation from E1 to E2–P. In the second half cycle, enzyme dephosphorylation accompanies return to the E1 conformation. These catalytic steps take place in the cytoplasmic domains and are allosterically coupled to ion transport in the transmembrane domain. Transport of two distinct ion species occurs according to a consecutive (or “ping-pong”) mechanism. In the first half cycle the first ion species is typically exported out of the cytoplasm, whereas in the second half cycle, the so-called counter ions are imported into the cytoplasm. During the transient, high-energy phosphorylated state known as E1–P, ion sites are occluded within the membrane domain as they are switched from one “access channel” to the other. This occluded state represents a critical distinction between channels and active transporters and precludes any unconstrained flow of ions through the protein.
Figure 2. Generalized Post-Albers cycle for P-type ATPases. E1 and E2 represent conformations with ion-binding sites facing the cytoplasm and extracellular medium, respectively. X and Y are ionic species that are transported consecutively out of and into the cytoplasm with stoichiometries of ν and µ, respectively. (Xν)E1P, and E2(Yµ) represent occluded states in which the ions are bound within the membrane domain and are unable to exchange with either aqueous phase. E1P and E2P represent states in which a conserved catalytic aspartate residue is phosphorylated. In general, formation of this phospho-enzyme accompanies transport of the X ions out of the cytoplasm in the first half cycle, whereas dephosphorylation and transport of the Y ions into the cytoplasm occurs in the second half cycle.
The KdpFABC complex
The KdpFABC transport system was discovered in the early 1970s (Epstein & Davies, Citation1970; Epstein & Kim, Citation1971) and has been generally described as P-type ATPase. This point of view arose because the KdpB subunit has all the hallmarks of a P-type ATPase; namely, KdpB has vanadate-sensitive ATPase activity, forms an acid-stable phosphoenzyme intermediate, and carries conserved signature sequences that are characteristic of this superfamily (Hesse et al., Citation1984). Nevertheless, a fundamental difference was established when transport-deficient mutations produced first by conventional UV irradiation (Buurman et al., Citation1995) and later by amber suppression scanning mutagenesis (Dorus et al., Citation2001) were all localized on the KdpA subunit. This result indicated that transport activity and ATP hydrolysis occurred on separate subunits, thus implying a completely novel mechanism for coupling these events. The KdpA subunit was shown to contain conserved signature sequences of SKT proteins (Durell et al., Citation2000), which includes TrkH and KtrB that are responsible for controlling cytoplasmic K+ concentrations under normal conditions. Although TrkH and KtrB function as channels, they are associated with accessory subunits (TrkA and KtrA, respectively) that control their gates. Thus, one could argue that KdpA recruited a P-type pump subunit in order to augment the physiological function of SKT proteins in K+ homeostasis.
Although it may be difficult to resolve the evolutionary progression, the KdpFABC complex presents an intriguing study in allostery, in which an ATPase subunit is used to convert a passive channel-like architecture into a coupled machine that is able to transport K+ ions against a concentration gradient spanning up to six orders of magnitude. A detailed understanding of this allostery is likely to enhance our appreciation of the individual components and thus shed light on mechanisms employed by other members of both SKT and P-type ATPase families.
Functional studies
Formation of the phosphorylated intermediate
Sequence analysis established KdpB as a P-type ATPase, and functional studies were used to show that it also employed a related mechanism. To start, Siebers and Altendorf demonstrated the formation of a covalently phosphorylated intermediate (Siebers & Altendorf, Citation1989). For this work, a mutant with lowered K+ affinity was used to explore the kinetics of reaction cycle and the dependence of the phosphorylation reaction on pump-specific substrates. Specifically, the KdpA mutation Q116R decreases the very high K+ affinity of wild type protein (Km of ∼2 μM increases to 6 mM), thus eliminating baseline activity due to unavoidable traces of K+ in working buffers. Experiments with E.coli plasma membrane fragments provided clear evidence for K+-dependent phosphorylation of KdpB and the ability of hydroxylamine to hydrolyze this phosphoenzyme suggested that phosphorylation occurred on a carboxyl group, which was subsequently shown to be D307 (Puppe et al., Citation1992). The highest steady-state level of phosphorylation was obtained in the absence of K+, which suggested that once formed, there was a very low rate of phosphoenzyme decomposition. In the presence of saturating 10 mM K+, the ATPase activity was maximal but the phosphorylation level was reduced to <40%. From this study a first reaction scheme was proposed (Siebers & Altendorf, Citation1989): (1) A K+-independent protein kinase activity led to the formation of the phosphorylated intermediate, E1 → E1–P. (2) A K+-stimulated phosphatase activity promoted the breakdown of the phosphointermediate, E2–P → E2. In addition, in the absence K+ a basal phosphatase activity was observed. High concentrations of K+ (>100 mM) reduced both the level of steady-state phosphorylation and the ATPase activity. Since open membrane fragments were used, information about ion transport across the membrane could not be obtained.
Inhibition by orthovanadate
A common inhibitor that was initially used to distinguish P-type from F-type and V-type ATPases is orthovanadate (Pedersen & Carafoli, Citation1987). The active species is VO4− which was first introduced in the study of Na,K-ATPase (Cantley et al., Citation1977). The vanadate adopts a stable trigonal bipyramidal structure and thereby is able to compete with phosphate binding in the transition state of phosphoenzyme hydrolysis. As a result, vanadate traps P-type ATPases in the E2–P state (Macara, Citation1980). Correspondingly, it was shown that the ATPase activity of the wild type Kdp complex can be inhibited by 1–10 µM orthovanadate (O’Neal et al., Citation1979; Siebers & Altendorf, Citation1989). In detail, it was shown that orthovanadate binds to the E2 conformation but did not displace 32 P from the phosphoprotein, similar to the SERCA pump (Siebers & Altendorf, Citation1989). Later it was found that it also inhibited ion transport (Fendler et al., Citation1996).
Electrogenic K+ transport
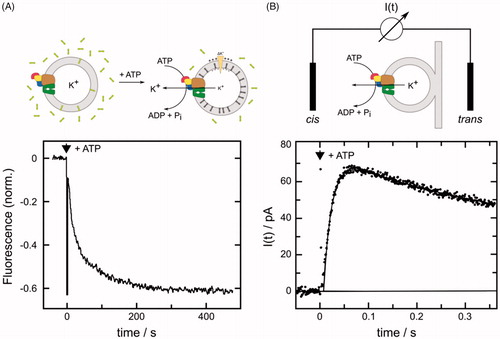
In early work, K+ transport was studied in E. coli cells (Rhoads et al., Citation1976). This technique was used to define apparent affinities for the different K+ transport systems and to screen Kdp mutants that were generated in strains lacking all other K+ transport systems (Buurman et al., Citation1995; Dorus et al., Citation2001).) . In this way, a wide array of mutations in KdpA was identified that reduce the K+ affinity of the complex. However, in order to study correlations between transport and ATPase activity, purified KdpFABC complexes were isolated and reconstituted into liposomes. After addition of ATP, inside-out oriented ion pumps, transported K+, thus generating an inside negative electric potential (Fendler et al., Citation1996). Two different approaches were used to monitor the electric potential and thus quantify the rate of transport: (1) A potential-sensitive fluorescence dye, DiSC3(5), was added to monitor the buildup of negative potential due to its increased repartitioning into the lipid phase (Apell & Damnjanovic, Citation2016) (). (2) Proteoliposomes were adsorbed to planar black lipid membrane (BLM) and external electrodes were used to record the electric potential across the vesicle membrane due to capacitive coupling between the two membranes (Fendler et al., Citation1996) (). In analogy to the Na,K-ATPase counter transport of a proton was considered, but the authors concluded that their experimental results did not support counter transport of any cation, thus contrasting KdpFABC with most other P-type ATPases. In the context of a Post-Albers cycle, these measurements of charge translocation suggested that K+ transport was associated with release to the cytoplasmic side of the membrane during the dephosphorylation half cycle. Further details were obtained with the low-affinity KdpFABC complex carrying the Q116R mutation in KdpA (Fendler et al., Citation1999). In the absence of K+, studies using BLMs revealed a minor charge translocation. This was assigned to the reorientation of a structural element of the protein: either a negative charge moving from the cytoplasmic to the periplasmic side of the dielectric field (as favored by these authors) or a positive charge moving in the opposite direction. Based on the experimental results the stoichiometry was finally refined to 2 K+/1 ATP. It was also shown that the small charge transient observed in the absence of K+ was not related to electrogenic conformational transitions in the KdpB subunit. All electrogenicity was assigned to the KdpA subunit.
Figure 3. Monitoring K+ transport by the KdpFABC. (A) The florescent dye DiSC3(5) redistributes between the aqueous phase and the inner leaflet of the vesicle membrane as K+ transport out of the vesicle generates a negative electric potential. The resulting increase in dye concentration in the inner leaflet causes fluorescence quenching. Accordingly, when ATP was added to a vesicle suspension, K+ transport is reflected by decreasing fluorescence as shown in the lower panel. The initial rate of decrease can be used to quantify transport and the exponential shape reflects inhibition of transport as the potential increases. Experimental data were adopted from Damnjanovic et al. (Citation2013). Lipid vesicles are depicted as gray annuli, Kdp as a multicolored complex and DiSC3(5) molecules as 3 rings. (B) Electric currents are generated by the Kdp complex upon release of a small amount of ATP from caged ATP and can be measured with a black lipid membrane setup with proteoliposomes tightly (and thus capacitively) coupled to the black lipid membrane (BLM). The lower panel shows the time course of the electric current detected by electrodes placed on both sides (cis and trans) of the black lipid membrane. The sign of the transient current (which ceases within a few seconds) indicates transport of positive charge out of the liposomes. Experimental data were adopted from Fendler et al. (Citation1996), Copyright 1996 American Chemical Society. I(t) represents a current meter connected to cis and trans electrodes.
The DiSC3(5) dye was used for in vitro characterization of ion selectivity, coupling and pH dependence of transport. In particular, the G232D mutation in KdpA was shown to alter the ion selectivity of the KdpFABC complex (Buurman et al., Citation1995; Schrader et al., Citation2000). This mutant pump transported various monovalent cations with decreasing efficiency, K+ > Rb+ > Na+ > Li+ > H+, and no Cs+, but the principal electrogenicity was preserved in this mutant. In a screen for K+ transport mutants, D583 and K586 in TM5 of KdpB were identified to play an important role in coupling ATPase activity to ion transport. Specifically, D583 and K586 were proposed to form a dipole and somehow mediate communication between KdpA and KdpB subunits (Bramkamp & Altendorf, Citation2005). When various mutations of these amino acids were investigated, it was found that any modification, including the reversal of the proposed dipole by the double mutant D583K:K586D, had deleterious impacts on K+ transport (Becker et al., Citation2007). It was concluded that this charge pair in KdpB is a requirement for energy transmission to facilitate active K+ transport and to trigger the ATP hydrolysis cycle in response to K+ binding.
With respect to pH, steady-state K+ currents were found to be maximal at pH 7.3–7.4 (Damnjanovic et al., Citation2013). At lower pH, binding of protons reduced the pump current and at higher pH the release of protons inhibited the electrogenic pump activity. In the absence of K+ on either side of the vesicle membrane, addition of ATP caused a minor H+ transport (∼8% of the K+ steady-state current under comparable conditions), which moved charge in the opposite direction from K+ transport (i.e., inside positive). The authors considered the possibility that this small current was an undesired leakage of bound protons to the extracellular side of the membrane during the ATP-triggered phosphorylation half cycle. However, they ultimately concluded that most of the bound protons are not actually transported, but rather are bound as allosteric factors in functional sites that facilitate the transport cycle.
Electrogenic partial reactions
More recently, an electrochromic fluorescence dye, RH 421, was introduced to study electrogenic effect in the KdpFABC complex (Damnjanovic & Apell, Citation2014b). This dye has been applied for more than two decades to resolve electrogenic partial reactions of the Na,K-ATPase, first in membrane fragments and later in solubilized preparations (Habeck et al., Citation2009). The dye molecules reside in the lipid phase of the membranes and respond to changes of the local electric field caused by uptake or release of ions from sites within the membrane domain of the protein. Unlike DiSC3(5), RH 421 allows the analysis of single reaction steps under non-turnover conditions.
When applied to the solubilized KdpFABC complex, RH 421 responded to both K+ and H+ binding to either the E1 or E2–P conformation, as stabilized by the absence or presence of ATP, respectively. The results indicate that these ions bind in an electrogenic manner to a site presumed to be in the membrane domain of the KdpFABC complex. The binding stoichiometry of each ion was relatively independent of the other ion, suggesting that there are independent sites (Damnjanovic et al., Citation2013). Studies of K+-binding indicated that the relevant sites inside KdpA do not move relative to the dielectric field during phosphorylation and dephosphorylation reactions. In the absence of K+, no significant electrogenic signals were induced by ATP binding, by enzyme phosphorylation (E1 → E1P), by the E1P → E2–P transition (Damnjanovic et al., Citation2013), or by backdoor phosphorylation (E2 → E2–P), consistent with the fact that these reactions take place in the cytoplasmic domain and do not induce charge movement in the membrane domain.
Conformational transitions were studied further with time-resolved ATP-concentration jump experiments on the millisecond time scale (Damnjanovic & Apell, Citation2014b) (. First, the results from these studies indicate that K+ transport occurred during the dephosphorylation part of the cycle (E2–P → E2). The amplitude of the fluorescence change was consistent with transport of 2 K+ per cycle. Second, the rate-limiting step of the cycle was determined to be the non-electrogenic conformational transition E1P → E2–P. Third, three protons were estimated to bind inside the membrane domain to so-far unidentified allosteric sites. These protons act as co-factors for K+ transport and may, at high concentrations, also function as weak competitors at the K+ transport sites. At low pH in the absence of K+, protons are translocated with very low efficiency, accounting for the observed inhibition of K+ transport at low pH. These findings led to the proposal of a modified Post-Albers scheme as discussed below ().
Figure 4. Electrogenic partial reactions of the KdpFABC complex monitored by RH421 fluorescence. (A) Fluorescence decrease induced by the release of ATP from caged ATP in the presence of a saturating concentration of K+. The red line represents the fit of an exponential function to the data. (B) The rate constant of the ATP-induced fluorescence decay was independent of the K+ concentration, thus indicating that the rate-limiting step occurred before ion binding. (C) The amplitude of the fluorescence decrease was dependent on K+ concentration with K1/2 of 6.8 ± 2.0 μM, which correlates with KM for the KdpFABC complex. The data were adapted from Damnjanovic and Apell (Citation2014b).
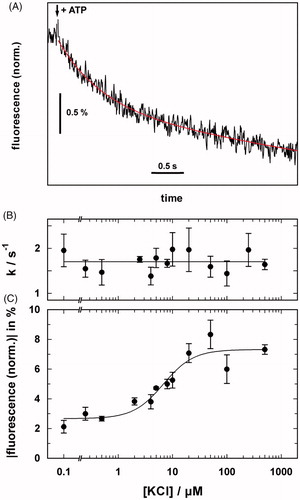
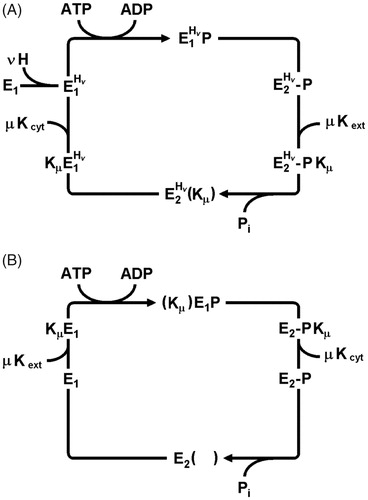
Figure 5. Conflicting Post-Albers reaction cycles based on functional and structural analyses of the KdpFABC complex. (A) Mechanism based on functional studies (Siebers & Altendorf, Citation1989; Damnjanovic & Apell, Citation2014b) that is in agreement with the “classical” cycle for P-type ATPases. In this case, K+ is imported in the dephosphorylating half cycle. These studies indicated that protons played an allosteric role during the transport cycle, but did not undergo transport across the membrane. (B) Mechanism based on structural studies (Haupt et al., Citation2004; Huang et al., Citation2017; Stock et al., Citation2018) in which K+ is transported in the phosphorylating half cycle.
Finally, a role for Mg2+ in facilitating K+ release at high K+ concentrations was investigated. Because the cytoplasmic K+ concentration in E. coli is generally >200 mM, the ion-binding sites are expected to be occupied by “K+ back binding” to the E1 state, thus inhibiting pump turnover (Damnjanovic et al., Citation2013). Although previous studies showed that K+ concentrations above 100 mM caused a reduction of the pump current (Fendler et al., Citation1996), the inhibition was not as strong as expected. Data from the DiSC3(5) experiments indicated that Mg2+ ions bind close to the entrance to the ion-binding sites and lead to a significant reduction of the local K+ concentration due to the Gouy-Chapman effect. As a result, release of K+ into the cytoplasm is facilitated, and in consequence, the turnover rate of the pump is promoted at physiological concentrations of Mg2+ (Damnjanovic & Apell, Citation2014a).
Summary
These biochemical and spectroscopic studies of the KdpFABC complex provided strong evidence for a mechanism conforming to the Post-Albers cycle of P-type ATPases used by Na,K-ATPase or H,K-ATPase (cf. ), in which K+ is transported in the second, dephosphorylation half of the cycle (E2–P → E1). The ion pump probably translocates 2 K+ ions per turnover and there is no counter ion transported in the first, phosphorylation half cycle (E1 → E2–P). Protons have allosteric effects and appear to be required for transport under physiological conditions. The electrogenic reaction steps are restricted to the diffusion of K+ ions into and out of their binding sites on either side of the membrane. There is no significant movement of the ions within the membrane domain of KdpFABC complex during the phosphorylation, dephosphorylation or the conformation transition steps.
Structure studies
Subunit composition and topology
The functional association between the subunits of the Kdp complex was initially tested by combinatorial expression and purification of subcomplexes (Gassel et al., Citation1998). For a functional complex, A, B and C subunits must be present. However, co-expression of KdpA and KdpC produces a subcomplex stable enough to survive detergent solubilization and purification, suggesting that they have a strong physical interaction. Given the respective roles of KdpA and KdpB in K+ transport and ATP-hydrolysis, respectively, physical interaction between these subunits seems to be a logical requirement, but the inability to co-purify these two subunits (unlike KdpA and KdpC) indicates a less stable interaction that might depend on the specific state of the transport cycle. Initially, the kdp operon was thought to comprise only three open reading frames, corresponding to A, B, and C subunits (Laimins et al., Citation1978). Later, an additional, very small hydrophobic subunit (KdpF) was discovered, which is expressed from the 5' end of the operon (Altendorf et al., Citation1998; Gassel et al., Citation1999). Although KdpF is absent in a significant number of bacterial genomes and is not essential for activity of the E. coli complex, it does contribute to the stability and activity of the KdpFABC complex in the detergent-solubilized state (Gassel & Altendorf, Citation2001; Gassel et al., Citation1999).
All four subunits contain one or more transmembrane helices that firmly anchor the complex in the membrane. Hydropathy plots for KdpB initially indicated 6 TM helices, with cytoplasmic domains characteristic of P-type ATPases occurring between TM2-3 and between TM4-5 (Axelsen & Palmgren, Citation1998; Serrano, Citation1988). A seventh TM helix was later proposed from homology modeling based on the X-ray structure of SERCA (Bramkamp, Citation2003; Haupt et al., Citation2004). Initial predictions for KdpA were based on fusions with PhoA and LacZ, which suggested the presence of 10 TM helices (Buurman et al., Citation1995), and on sequence comparison with K channels, which suggested the presence of at least two repeats of the MPM motif characterizing their selectivity filters (Jan & Jan, Citation1994). Homology with the SKT superfamily was later extended to produce a model with four MPM repeats (Durell et al., Citation2000). This model nicely explained the distribution of K+ affinity mutants that had previously been characterized (Buurman et al., Citation1995; Dorus et al., Citation2001). KdpC has a single TM helix at its N-terminus and no known homologs outside of the Kdp system. The 150 residue C-terminal domain of KdpC was shown to bind and be labeled by azido-ATP with relatively low affinity (Ahnert et al., Citation2006; Altendorf et al., Citation1992). This observation led to a model in which the large C-terminal domain resided within the cytoplasm and was proposed to help the nucleotide-binding domain of KdpB bind ATP (Bramkamp et al., Citation2007). Although NMR data was indeed consistent with this interpretation (Irzik et al., Citation2011), the recent X-ray structure discussed below revealed the opposite topology, in which the C-terminal domain of KdpC is associated with the periplasmic loops of KdpA. Finally, the 29-residue KdpF subunit comprises a single TM helix, reminiscent of the small regulatory subunits associated with SERCA (phospholamban and sarcolipin) (Primeau et al., Citation2018) and Na,K-ATPase (FXYD proteins) (Garty & Karlish, Citation2006). However, KdpF does not show homology with these other proteins and appears to be associated with a different region of KdpB.
Negative stain electron microscopy studies
As with many P-type ATPases, studies of the 3 D structure began with electron microscopy of two-dimensional crystals. An initial study overexpressed the Q116R low-K affinity mutant and used orthovanadate to induce two-dimensional crystallization of KdpFABC within inside-out vesicles (Iwane et al., Citation1996). A low-resolution projection structure from negatively stained crystals showed a monomer with three main domains composing the asymmetric unit. Subsequently, WT KdpFABC purified in detergent was shown to form a mixture of monomers and dimers using size-exclusion chromatography, cross-linking and imaging of negatively stained samples (Heitkamp et al., Citation2008). 3 D reconstruction of monomeric complexes using single-particle analysis resulted in a structure at 19 Å resolution that was used to produce an atomic model based on homology modeling and rigid body fitting (Heitkamp et al., Citation2009). Around the same time, another group reconstituted WT KdpFABC into liposomes, used orthovanadate to induce two-dimensional crystals, and used electron tomography and subtomogram averaging to generate a structure from negatively stained samples at 24 Å resolution (Hu et al., Citation2008). This structure revealed dimeric units composing the crystal lattice, though the intramembrane portions of the complex were not visible due to exclusion of stain from the bilayer.
X-ray crystallographic structure
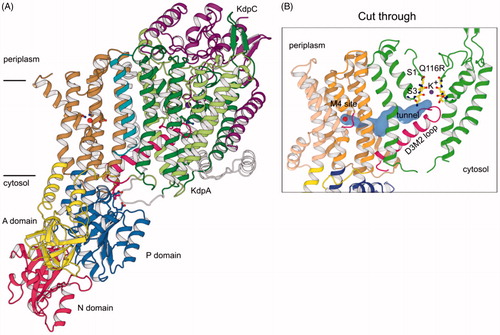
Structural understanding of the KdpFABC complex was advanced in 2017 by a X-ray crystallographic structure that revealed atomic details of the complex with a resolution of 2.9 Å (Huang et al., Citation2017). This structure confirmed the expected folds for KdpA, as a channel-like subunit from the SKT superfamily, and KdpB, as a pump-like subunit from the P-type ATPase superfamily. KdpC and KdpF both had the expected single TM helix (). The C-terminal domain of KdpC was docked between two periplasmic loops from KdpA, thus ruling out its proposed role as a catalytic chaperone for ATP (Irzik et al., Citation2011). Intriguingly, the position of this C-terminal domain near the entrance to the ion binding pathway is reminiscent of the β subunit of the Na,K-ATPase (Morth et al., Citation2007). Although these extracellular domains share no structural or sequence homology, they are both indispensable for function and may play some role in regulating ion binding. KdpF was located at the interface between the KdpA and KdpB subunits, consistent with its role in stabilizing the complex in detergent. Gel filtration of the detergent-solubilized complex was consistent with a monomeric state in detergent and molecular contacts in the crystal were not consistent with the formation of a physiologically relevant oligomer.
Figure 6. X-ray crystal structure of the KdpFABC complex in an E1-like conformation (PDB accession code 5MRW). (A) Overview of the structure, which had a resolution of 2.9 Å. The KdpF subunit (cyan) consists in a single membrane spanning helix at the interface of KdpA (green) and KdpB (transmembrane helices brown, P domain blue, A domain yellow, N domain red). KdpC (purple) consists of a single transmembrane helix and a periplasmic domain near the entrance to the selectivity filter of KdpA. (B) A cavity (blue surface) runs between the K+ ion (purple sphere) in the selectivity filter of KdpA and the water molecule in the canonical ion binding site in KdpB (occupied by a water molecule, red sphere). The cavity has a diameter ≥1.4 Å, which is consistent with its occupancy by water.
Ligands were bound at functional sites in the membrane domains of both KdpA and KdpB. Specifically, densities were seen within the selectivity filter of KdpA and at the canonical cation binding site of P-type ATPases, next to the M4 helix of KdpB; the former was modeled as K+ and the latter as a water molecule based on the presence and absence of anomalous scattering, respectively (). In the cytoplasmic domains of KdpB, the structural elements responsible for ATP hydrolysis were all present. Specifically, the P domain harbors the aspartate residue that is transiently phosphorylated, the N domain has the ATP binding pocket, and the A domain has a conserved TGES motif that hydrolyzes the aspartyl phosphate. Given the dramatic changes seen in the cytoplasmic domains of SERCA between different conformational states (Møller et al., Citation2010), their configuration can be used to deduce the enzymatic state; thus, this X-ray structure appears most similar to the E1 state, in which the catalytic aspartate in the P domain is facing the N domain and the A domain is disengaged from the catalytic site. Furthermore, the M4 helix, which harbors the canonical cation site, is in an extended conformation consistent with allosteric signals that characterize the E1 state and that initiate the phosphorylation of the catalytic aspartate. Based on these observations, the authors speculated that the water molecule bound at the canonical site in some way senses the binding of K+ by KdpA and initiates the ATPase cycle of KdpB.
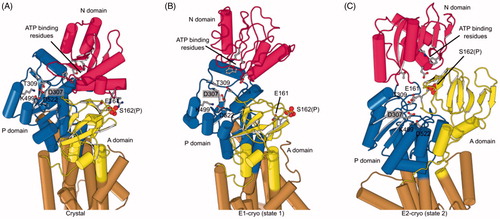
Despite the overall similarities to other P-type ATPases, KdpB has some unique features. The A domain is known for its flexibility, but in KdpB, it is rotated ∼180° relative to the E1 conformation in SERCA and the conserved TGES sequence interacts with the N domain via an unprecedented phosphoserine (Ser162) (. Also, the N domain is so close to the P domain that ATP is sterically excluded from its binding site. These unusual features suggested that constraints imposed by the phosphoserine might be inhibitory, an idea that was supported by a stimulatory effect of lambda phosphatase on ATPase activity.
Figure 7. Phosphorylation and structural environment of Ser162. This residue is part of the conserved TGES motif in the A domain (yellow), which occupies different configurations in the three structures. (A) Crystal structure which represents the E1 enzymatic state. (B) State 1 structure from cryo-EM which also represents the E1 state. (C) State 2 from cryo-EM which represents the E2 state. The phosphor-serine, S162(P), mediates a salt bridge with two non-conserved residues in the N domain (red) in the X-ray structure. The A domain is known to be highly mobile and although its resolution in cryo-EM maps was not sufficient to see the phosphate, the juxtaposition of A and N domains in these two structures indicated that the salt bridge could not be formed. In state 2, E161 is seen approaching the catalytic aspartate (D307) in the P-domain (blue), which reflects its role in hydrolysis of the phosphoenzyme in the E2 state.
KdpA has an architecture related to K channels, which is represented by KcsA. This architecture centers on a selectivity filter built from four symmetric, glycine-rich loops that use carbonyl oxygens to coordinate K+ within the conductance pore. There are four primary sites denoted S1–S4 that form a linear array of well-ordered densities in KcsA and other homotetrameric channels. This linear array is essential to their selectivity, which is governed by the kinetics of ion binding and release rather than by equilibrium binding (Liu & Lockless, Citation2013). In contrast, KdpA and its relatives TrkH and KtrB are a single polypeptide chain and the filter has only approximate four fold symmetry. The oxygen ligands that coordinate ions are more poorly ordered and only a single K+ is seen at the S3 site () (Cao et al., Citation2013; Vieira-Pires et al., Citation2013). In the case of TrkH and KtrB, which function as channels with a high flux of ions, loss of the linear array on ions leads to poor selectivity for K+ over Na+ (Diskowski et al., Citation2015). KdpFABC, however, operates as a pump with a much slower turnover rate (∼100 s−1). As a result, equilibrium binding governs selectivity and the innate preference of the filter architecture explains the observed ∼100-fold selectivity for K+ over Na+ (Liu & Lockless, Citation2013).
The X-ray structure also showed that the K+ site in KdpA is connected to the canonical cation site in KdpB by a tunnel that is completely buried within the hydrophobic core of the protein. Although similar tunnels have never been seen in P-type ATPases, the extraordinary length of over 40 Å spanning two subunits and connecting the two functional sites suggested that this tunnel serves a functional purpose in the mechanism of KdpFABC. Based on the 1.4 Å diameter and the fact that the tunnel has access to the aqueous phase via the selectivity filter suggests that it is filled with water, similar to cavities described in other proteins, including channels. The authors suggested that this tunnel would accommodate a single line of water molecules, which could serve as a “wire” to transmit protons between the two sites, as is discussed in the next section.
Although the pseudo four-fold repeat of KdpA appears to be a gene duplication of the M1PM2 fold from KcsA, the third repeat features a distinctive kink in the M2 helix that has also been seen in TrkH and KtrB. The intramembrane loop connecting the two halves of this M2 helix lies directly above the selectivity filter and blocks escape of ions toward cytoplasmic side of the membrane, suggesting that it represents a gate (. For TrkH and KtrB, associated cytoplasmic subunits (TrkA and KtrA) have been proposed to open this gate by pulling on the distal part of the M2 helix (Diskowski et al., Citation2015). In the Kdp complex, similar control was ascribed to KdpB, which is connected to the kinked M2 helix via a network of interactions with the P domain. Based on these features, the paper concluded that the KdpA subunit provided a pathway for K+ that was consistent with that proposed for SKT channels. The function of the KdpB subunit was to sense K+ binding through water mediated interactions and then to drive opening and closure of the channel to produce active transport (Huang et al., Citation2017).
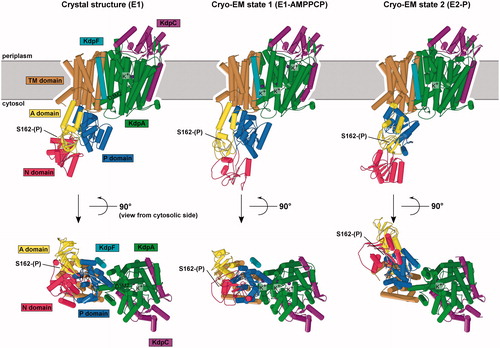
Figure 8. Comparison of KdpFABC structures. (A) X-ray crystal structure. (B) State 1 from cryo-EM. (C) State 2 from cryo-EM. Only minor changes were observed in KdpA, KdpC and KdpF. However, the arrangements of N, P and A domains of KdpB have significant differences in E1-like states and the E2-P state. The apparent loss of secondary structure in the N-domain from the cryo-EM state 1 is likely to be an artifact of the real-space fitting to the relatively low resolution densities in this region of that map.
Cryo-Electron microscopy studies
In 2018, two cryo-EM structures emerged to show several new features with intriguing mechanistic implications. The new structures were derived from a single detergent-solubilized sample containing K+, AMPPCP and AlF4− and represent two distinct conformational states (termed state 1 and state 2) (Stock et al., Citation2018) that were resolved to 3.7 Å and 4.0 Å respectively (. Overall these structures confirmed the general topology and organization of the KdpFABC complex and offered no evidence for oligomerization. Neither state showed significant differences in the KdpA, KdpC and KdpF subunits.
Major structural changes were seen, however, in the cytoplasmic domains of KdpB. Comparison with structures of SERCA showed that state 1 resembled an E1–AMPPCP state, whereas state 2 was most similar to an E2–AlF4, or E2–P state. Because the resolution near the catalytic site was in the 5–6 A range, AMPPCP and AlF4− could not be directly visualized; nevertheless, these ligands could be docked into their expected binding sites without clashes. For both states extra density at Ser162 indicated that, like in the crystal structure, this residue was phosphorylated, and this conclusion was supported by data from mass spectrometry. It is still unclear how the Ser162-phosphorylated state affects transport activity, but the EM work clearly shows that the interaction observed in the crystal structure between the phosphorylated Ser162 and the N domain is not stable in solution ().
This new E1 state was overall very similar to the crystal structure, despite differences in the positions of A and N domains. In particular, the N domain is poised differently above the P domain in a position that presumably facilitates transfer of Pi to the catalytic aspartate. Although the A domain has translated by ∼14 Å, its unusual angle relative to the P domain is similar to the crystal structure. In the membrane domain, a tunnel was described running from the M4 site of KdpB to the selectivity filter of KdpA, though it narrows significantly below 1.4 Å at the subunit interface, and the gating loop was found to be blocking the classical channel exit pathway through KdpA, just as in the crystallographic structure (.
The new putative E2–P state showed some surprises. Although the positions of the P domain and A domain were consistent with crystallographic work on SERCA, the coupling helix and gating loop from KdpA were unmoved (. These elements of D3M2 had been proposed to be the cytoplasmic gate to the selectivity filter and interactions with the P-domain suggested a mechanism to operate this gate. Instead, this structure suggested that K+ followed a completely different path through the protein. Although the tunnel is observed in KdpA, it ends at the interface between KdpA and KdpB subunits and a new channel leads from the cytosol towards the canonical binding site in the KdpB, thus providing a potential exit path running between the now disconnected coupling helix and the P domain (. This exit channel is completely different from the cytosolic entry/exit channel observed in SERCA, which runs between M1, M2 and M3.
These observations led the authors to suggest that K+ might enter the selectivity filter in KdpA, move through the tunnel to the canonical binding site in KdpB, and then be released to the cytosol (. In support of this idea, 4 density peaks were observed along this pathway and were modeled as K ions. In state 2, a peak was found between the selectivity filter and the gating loop of KdpA right next to the mouth of the tunnel. In state 1, 3 peaks were observed within the proposed pathway. One was at the S1 position in the selectivity filter (not S3 as in the crystal structure, and the TrkH and KtrB structures). The second peak was in the same position as in state 2, just above the filter in the KdpA subunit below the coupling helix. The last peak in state 1 was inside the tunnel near the subunit interface. In this last position, the peak is 4.5 Å from the nearest polar moiety (carbonyl oxygen on Ala227 of KdpB), but within van der Waals binding distance (<4 Å) of carbon-atoms from Val538 (KdpA), Ile421 (KdpA) and Val231 (KdpB). There is no density at the canonical site in KdpB, where a distinct peak in the crystal structure was modeled as water. Although there may be some uncertainty in assignment of these peaks, collapse of the tunnel in KdpA and opening of a potential exit channel in KdpB in the E2–P state led to the novel idea that ions are shuttled from KdpA across to KdpB, which then exports them to the cytosol. The derived mechanism completely redefines the location of the two gates needed for transport and suggests that KdpA may simply select K+ ions from the media and hand them to KdpB for energy-coupled transport (.
Figure 9. Schematic representation of the two mechanisms proposed for the KdpFABC complex based on structural studies. (A) Mechanism in which K+ travels through KdpA and the tunnel serves as a signaling conduit. K+ is bound from the periplasm in the selectivity filter of KdpA in the E1 conformation. A charge migrates through the tunnel and stimulates the ATPase cycle in KdpB. Conformational changes result in movement of the coupling helix (pink) that allows release of K+ to the cytoplasm in E2P. (B) Mechanism in which K+ travels from the periplasm to the selectivity filter and through the tunnel from KdpA to KdpB in the E1 conformation and is then released to the cytosol by KdpB in the P-E2 conformation.
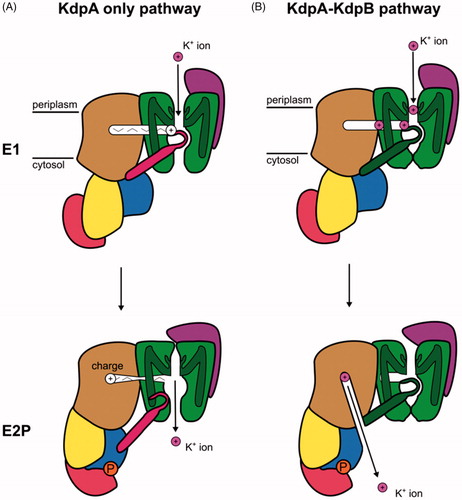
Approach to an unified mechanistic model
Despite the sustained effort over several decades and the recent breakthrough in structural analysis, the results of functional and structural studies only lead to contradictions about the order of events in the reaction mechanism that underlies transport by the KdpFABC complex. Rather than try to reconcile these contradictions we will use the following section to identify key issues with the hope to guide future experiments that will eventually clarify the inner workings of this fascinating molecular machine.
The pump cycle
As introduced above, the Post-Albers cycle represents an archetype that has been adapted to all P-type ATPases studied so far. The cycle is characterized by two fundamental observations (): (1) Transport is performed in a consecutive mode in which the primary ion species is bound from the cytoplasm, then, after release to the extracellular or luminal milieu, a second ion species is bound at these same sites and imported to the cytoplasm. (2) Export of the primary ions from the cell is performed in the first half cycle in which the enzyme is phosphorylated by ATP and import of secondary, counter-ions (either H+ or K+) occurs during the second, dephosphorylation half cycle. Exceptions include H-ATPases, Cu-ATPases, lipid flippases and KdpFABC, which do not appear to transport any counter-ion. In the case of H-ATPase, H+ is exported in the first half of the cycle, suggesting that H+ act as the primary ion according to point 2 above. Although the polar lipids carried by lipid flippases represent a fundamentally different substrate, these lipids are moved from the outer to the inner leaflet of the bilayer during the second half cycle, making them analogous to a counter ion in the Post-Albers cycle. In the case of KdpFABC, K+ is imported into the cell with functional studies indicating that this occurs during the first half cycle while structural studies indicate the opposite, thus bringing uncertainty to the order of events in the cycle
Evidence for K+ transport in the enzyme dephosphorylating half-cycle
Evidence for K+ transport in the second half of the cycle () comes from one of the first functional studies of phosphorylated intermediates, which found maximal steady-state levels of phosphorylation in the absence of K+ (Siebers & Altendorf, Citation1989). Upon addition of up to millimolar K+, ATPase activity increased to maximal levels, while the level of steady-state phosphoenzyme decreased to a basal level. The authors concluded that formation of the phosphoenzyme from the E1 conformation is not dependent on K+ binding, but that K+ stimulates dephosphorylation which would in turn lead to its transport in the second half of the cycle. The next study to investigate ion-moving partial reactions responsible for ion movement used fluorescence dyes that are sensitive to the local electric-field (Damnjanovic & Apell, Citation2014b). In time-resolved experiments the RH21 dye reported on the electrogenic response of the KdpFABC complex to an ATP-concentration jump. Whereas the rate of the response was independent of K+, the magnitude of the response depended on K+ concentration with a K1/2 of ∼5.3 µM, consistent with the Km for transport. These observations suggested that K+ binding sites were not occupied during the rate-limiting formation of E1∼P, but bound after the enzyme transitioned to the deoccluded E2–P state, thus also supporting a mechanism in which transport occurs during the second, dephosphorylating half of the cycle.
Evidence for K+ transport in the enzyme phosphorylating half-cycle
Proposals for K+ transport during the phosphorylation half cycle () first arose from NMR studies of the isolated nucleotide-binding domain (Haupt et al., Citation2004). The authors pointed out that as a general principle, ion binding to P-type ATPases typically gave rise to a high ATP affinity conformation and that ion-binding sites were always occupied when ATPase activity was observed. This point of view was reiterated a year later in interpreting results on single amino-acid substitutions in KdpB (Bramkamp & Altendorf, Citation2005). These authors argued that enzyme activity and ion transport require an interplay between the KdpA and KdpB subunits and that the energy provided by ATP hydrolysis in KdpB is needed to fuel K+ transport in KdpA. They proposed that ATP-driven reorientation of a dipole formed by D583 and K586 at the interface between KdpB and KdpA mediated this exchange of energy. This “phase shifted” Post-Albers cycle also appeared to be consistent with structural analyses. In the crystal structure of the KdpFABC complex the arrangement of the cytoplasmic domains of the KdpB subunit indicated an E1 conformation and the KdpA subunit revealed an ion-binding site that was occupied and open to the extracellular milieu (Huang et al., Citation2017). This conformation was seen again in the cryo-EM structures (Stock et al., Citation2018), resembling an E1 state, whereas in the other, E2-like state, KdpB had blocked access from the extracellular side and provided a channel to allow ions to escape toward the cytoplasm. At face value, these structural studies are in agreement with a Post-Albers cycle in which K+ inward transport occurs in the first half cycle that accompanies phosphorylation.
Future directions
At this point in time, additional information from both functional and structural studies are required to clarify the intriguing structural mechanism of KdpFABC. Some of the issues that need to be addressed are as follows. Does K+ move through the tunnel from KdpA to KdpB, as suggested by the cryo-EM structures, or does it move exclusively through KdpA, as was previously proposed. Although a density is seen in the cryo-EM structure of state 1, polar ligands necessary to coordinate a dehydrated ion are not present along this tunnel making it important to consider the energetic feasibility of this pathway. Furthermore, the tunnel diameter in the presented E1 structure from cryo-EM falls in KdpB well below that of K+ and the exit channel in the E2 structure does not appear to reach the canonical ion binding site in KdpB. Clarification of the densities both within the tunnel and at the canonical ion site in KdpB, which was modeled as a water molecule in the crystal structure, would help address the path taken by K+ during transport.
In addition, more information about the intracellular and extracellular gates is needed. Does the “gating loop” formed by the D3M2 element on KdpA play an active role in transport, or a static element that redirects K+ into the tunnel? The idea that this loop plays a role in gating originates with work on TrkH and KtrB, but the most recent cryo-EM structures of KtrB in the open state show a completely different conformational change without displacement of this loop, suggesting that it may be a static element (Diskowski et al., Citation2017). Where is the extracellular gate required to occlude K+? Does the soluble domain of KdpC play a role in blocking the extracellular entrance to the selectivity filter, or are ions trapped at the canonical site in KdpB by collapse of the tunnel? The location of KdpC near the mouth of the selectivity filter is intriguing, but the mechanistic model derived from the cryo-EM structures suggests that KdpA is relatively static and that conformational changes in KdpB controls access to the canonical ion binding site on M4.
These fundamental questions indicate that we still do not understand the design principles for this hybrid pumping system and its relationship to the SKT and P-type ATPase families. KdpB has been described as the evolutionary precursor for the P-type superfamily with the implication that other branches of the family gained the ability to select and directly bind their respective substrates (Axelsen & Palmgren, Citation1998). In the Kdp complex, the KdpA subunit appears to fill that role, making KdpB the only P-type pump where selection of substrate does not occur in the primary and occluded binding site. Why does KdpB require lateral transfer of K+ from a selectivity filter rather than binding K directly from the extracellular side like Na/K-ATPase? Are there other members of the P-type ATPase superfamily, such as those Type V ATPases with unknown function, that partner with other subunits to bind and select novel substrates? Similarly, the proposed channel toward the cytosol seen in the cryo-EM structure of KdpFABC is so far unique amongst known P-type ATPases and calls for future study.
An important unanswered question relates to the effect of serine phosphorylation on KdpFABC. The discovery of this posttranslational modification in the crystal structure was a surprise and evidence was presented that phosphorylation of KdpB-Ser162 is inhibitory (Huang et al., Citation2017). Considering evidence that E. coli can rapidly shut down transport by the Kdp complex when placed in K+ rich media (Roe et al., Citation2000), it seems reasonable to hypothesize that this inhibition may have physiological importance. In fact, maximal ATPase activities reported in the literature vary by almost two orders of magnitude, which might be explained by differing levels of serine phosphorylation. It is therefore important to determine the effects that this phosphorylation has on the observed structural conformations and on the functional data relating K+ transport to phosphoenzyme formation and hydrolysis. For a start, the orientation of the A-domain in the crystal structure and in the cryo-EM state 1 are uncharacteristic of P-type ATPases and the presence of this additional charge near the catalytic site in the E2 state seems likely to have some effect on the dephosphorylation step. Further structural and functional studies on unphosphorylated and fully active KdpFABC preparations will be important to try to resolve the current contradictions about the causality between K+ binding and phosphorylation of the catalytic aspartate in the P domain, thus producing a mechanistic model that everyone can agree on.
| Abbreviations | ||
| AMPPNP | = | Adenylyl-imidodiphosphate |
| BLM | = | black lipid membrane |
| DiSC3(5) | = | 3,3'-dipropylthiadicarbocyanine iodide |
| PMCA | = | plasma membrane Ca-ATPase |
| RH421 | = | N-(4-sulfobutyl)-4-(4-(p-(dipentylamino)phenyl)butadienyl)-pyridinium inner salt |
| SERCA | = | sarcoplasmic, endoplasmic reticulum Ca-ATPase |
| SKT | = | superfamily of K transporters |
| TM | = | transmembrane |
Disclosure statement
The authors report no conflicts of interest.
Additional information
Funding
References
- Ahnert F, Schmid R, Altendorf K, Greie JC. 2006. ATP binding properties of the soluble part of the KdpC subunit from the Escherichia coli K(+)-transporting KdpFABC P-type ATPase. Biochemistry 45:11038–11046.
- Albers RW. 1967. Biochemical aspects of active transport. Annu Rev Biochem 36:727–756.
- Altendorf K, Booth IR, Gralla J, Greie JC, Rosenthal AZ, Wood JM. 2009. Osmotic stress. EcoSal Plus [Epub ahead of print]. doi:10.1128/ecosalplus.5.4.5.
- Altendorf K, Gassel M, Puppe W, Möllenkamp T, Zeeck A, Boddien C. 1998. Structure and function of the Kdp-ATPase of Escherichia coli. Acta Physiol Scand Suppl 643:137–146.
- Altendorf K, Siebers A, Epstein W. 1992. The KDP ATPase of Escherichia coli. Ann N Y Acad Sci 671:228–243.
- Altendorf K, Voelkner P, Puppe W. 1994. The sensor kinase KdpD and the response regulator KdpE control expression of the kdpFABC operon in Escherichia coli. Res Microbiol 145:374–381.
- Apell HJ, Damnjanovic B. 2016. Assaying P-type ATPases reconstituted in liposomes. Methods Mol Biol 1377:127–156.
- Axelsen KB, Palmgren MG. 1998. Evolution of substrate specificities in the P-type ATPase superfamily. J Mol Evol 46:84–101.
- Becker D, Fendler K, Altendorf K, Greie JC. 2007. The conserved dipole in transmembrane helix 5 of KdpB in the Escherichia coli KdpFABC P-type ATPase is crucial for coupling and the electrogenic K+-translocation step. Biochemistry 46:13920–13928.
- Booth IR, Blount P. 2012. The MscS and MscL families of mechanosensitive channels act as microbial emergency release valves. J Bacteriol 194:4802–4809.
- Bossemeyer D, Schlosser A, Bakker EP. 1989. Specific cesium transport via the Escherichia coli Kup (TrkD) K+ uptake system. J Bacteriol 171:2219–2221.
- Bramkamp M. 2003. Characterization of the KdpFABC complex from Escherichia coli, of soluble subdomains from KdpB, and of a homologous protein of Methanococcus jannaschii [dissertation]. Osnabrück: University of Osnabrück. 1–126.
- Bramkamp M, Altendorf K. 2005. Single amino acid substitution in the putative transmembrane helix V in KdpB of the KdpFABC complex of Escherichia coli uncouples ATPase activity and ion transport. Biochemistry 44:8260–8266.
- Bramkamp M, Altendorf K, Greie JC. 2007. Common patterns and unique features of P-type ATPases: a comparative view on the KdpFABC complex from Escherichia coli (Review). Mol Membr Biol 24:375–386.
- Buurman ET, Kim KT, Epstein W. 1995. Genetic evidence for two sequentially occupied K+ binding sites in the Kdp transport ATPase. J Biol Chem 270:6678–6685.
- Cantley LC, Josephson L, Warner R, Yanagisawa M, Lechene C, Guidotti G. 1977. Vanadate is a potent (Na,K)-ATPase inhibitor found in ATP derived from muscle. J Biol Chem 252:7421–7423.
- Cao Y, Pan Y, Huang H, Jin X, Levin EJ, Kloss B. 2013. Gating of the TrkH ion channel by its associated RCK protein TrkA. Nature 496:317–322.
- Chan H, Babayan V, Blyumin E, Gandhi C, Hak K, Harake D. 2010. The p-type ATPase superfamily. J Mol Microbiol Biotechnol 19:5–104.
- Coskun Ü, Chaban YL, Lingl A, Müller V, Keegstra W, Boekema EJ, et al. 2004. Structure and subunit arrangement of the A-type ATP synthase complex from the archaeon Methanococcus jannaschii visualized by electron microscopy. J Biol Chem 279:38644–38648.
- Damnjanovic B, Apell H-J. 2014a. KdpFABC reconstituted in Escherichia coli lipid vesicles: substrate dependence of the transport rate. Biochemistry 53:5674–5682.
- Damnjanovic B, Apell H-J. 2014b. Role of protons in the pump cycle of KdpFABC investigated by time-resolved kinetic experiments. Biochemistry 53:3218–3228.
- Damnjanovic B, Weber A, Potschies M, Greie JC, Apell H-J. 2013. Mechanistic analysis of the pump cycle of the KdpFABC P-type ATPase. Biochemistry 52:5563–5576.
- Damper PD, Epstein W. 1981. Role of the membrane potential in bacterial resistance to aminoglycoside antibiotics. Antimicrob Agents Chemother 20:803–808.
- Diskowski M, Mehdipour AR, Wunnicke D, Mills DJ, Mikusevic V, Barland N, et al. 2017. Helical jackknives control the gates of the double-pore K + uptake system KtrAB. Elife 6:e24303.
- Diskowski M, Mikusevic V, Stock C, Hänelt I. 2015. Functional diversity of the superfamily of K+ transporters to meet various requirements. Biol Chem 396:1003–1014.
- Dorus S, Mimura H, Epstein W. 2001. Substrate-binding clusters of the K+-transporting Kdp ATPase of Escherichia coli Investigated by amber suppression scanning mutagenesis. J Biol Sci 276:9590–9598.
- Durell SR, Bakker EP, Guy HR. 2000. Does the KdpA subunit from the high affinity K(+)-translocating P-type KDP-ATPase have a structure similar to that of K(+) channels? Biophys J 78:188–199.
- Epstein W. 2003. The roles and regulation of potassium in bacteria. Prog Nucleic Acid Res Mol Biol 75:293–320.
- Epstein W. 2016. The KdpD sensor Kinase of Escherichia coli responds to several distinct signals to turn on expression of the Kdp transport system. J Bacteriol 198:212–220.
- Epstein W, Davies M. 1970. Potassium-dependant mutants of Escherichia coli K-12. J Bacteriol 101:836–843.
- Epstein W, Kim BS. 1971. Potassium transport loci in Escherichia coli K-12. J Bacteriol 108:639–644.
- Fendler K, Dröse S, Altendorf K, Bamberg E. 1996. Electrogenic K+ transport by the Kdp-ATPase of Escherichia coli. Biochemistry 35:8009–8017.
- Fendler K, Dröse S, Epstein W, Bamberg E, Altendorf K. 1999. The Kdp-ATPase of Escherichia coli mediates an ATP-dependent, K+-independent electrogenic partial reaction. Biochemistry 38:1850–1856.
- Gadsby DC, Takeuchi A, Artigas P, Reyes N. 2009. Review. Peering into an ATPase ion pump with single-channel recordings. Philos. Trans. R. Soc. Lond., B, Biol. Sci 364:229–238.
- Garty H, Karlish SJ. 2006. Role of FXYD proteins in ion transport. Annu Rev Physiol 68:431–459.
- Gassel M, Altendorf K. 2001. Analysis of KdpC of the K+-transporting KdpFABC complex of Escherichia coli. Eur J Biochem 268:1772–1781.
- Gassel M, Möllenkamp T, Puppe W, Altendorf K. 1999. The KdpF subunit is part of the K(+)-translocating Kdp complex of Escherichia coli and is responsible for stabilization of the complex in vitro. J Biol Chem 274:37901–37907.
- Gassel M, Siebers A, Epstein W, Altendorf K. 1998. Assembly of the Kdp complex, the multi-subunit K+-transport ATPase of Escherichia coli. Biochim Biophys Acta 1415:77–84.
- Gogarten JP, Kibak H, Dittrich P, Taiz L, Bowman EJ, Bowman BJ, et al. 1989. Evolution of the vacuolar H+-ATPase: implications for the origin of eukaryotes. Proc Natl Acad Sci USA 86:6661–6665.
- Habeck M, Cirri E, Katz A, Karlish SJ, Apell H-J. 2009. Investigation of electrogenic partial reactions in detergent-solubilized Na,K-ATPase. Biochemistry 48:9147–9155.
- Haupt M, Bramkamp M, Coles M, Altendorf K, Kessler H. 2004. Inter-domain motions of the N-domain of the KdpFABC complex, a P-type ATPase, are not driven by ATP-induced conformational changes. J Mol Biol 342:1547–1558.
- Heermann R, Jung K. 2010. The complexity of the 'simple' two-component system KdpD/KdpE in Escherichia coli. FEMS Microbiol Lett 304:97–106.
- Heitkamp T, Böttcher B, Greie JC. 2009. Solution structure of the KdpFABC P-type ATPase from Escherichia coli by electron microscopic single particle analysis. J Struct Biol 166:295–302.
- Heitkamp T, Kalinowski R, Böttcher B, Borsch M, Altendorf K, Greie JC. 2008. K+-Translocating KdpFABC P-type ATPase from Escherichia coli acts as a functional and structural dimer. Biochemistry 47:3564–3575.
- Hesse JE, Wieczorek L, Altendorf K, Reicin AS, Dorus E, Epstein W. 1984. Sequence homology between two membrane transport ATPases, the Kdp-ATPase of Escherichia coli and the Ca2+-ATPase of sarcoplasmic reticulum. Proc Natl Acad Sci USA 81:4746–4750.
- Hille B. 2001. Ionic channels of excitable membranes. Sunderland (MA): Sinauer Associates, Inc., 1–814.
- Hu GB, Rice WJ, Dröse S, Altendorf K, Stokes DL. 2008. Three-dimensional structure of the KdpFABC complex of Escherichia coli by electron tomography of two-dimensional crystals. J Struct Biol 161:411–418.
- Huang CS, Pedersen BP, Stokes DL. 2017. Crystal structure of the potassium-importing KdpFABC membrane complex. Nature 546:681–685.
- Irzik K, Pfrotzschner J, Goss T, Ahnert F, Haupt M, Greie JC. 2011. The KdpC subunit of the Escherichia coli K+-transporting KdpB P-type ATPase acts as a catalytic chaperone. Febs J 278:3041–3053.
- Iwane AH, Ikeda I, Kimura Y, Fujiyoshi Y, Altendorf K, Epstein W. 1996. Two-dimensional crystals of the Kdp-ATPase of Escherichia coli. FEBS Lett 396:172–176.
- Jan LY, Jan YN. 1994. Potassium channels and their evolving gates. Nature 371:119–122.
- Krulwich TA, Sachs G, Padan E. 2011. Molecular aspects of bacterial pH sensing and homeostasis. Nat Rev Microbiol 9:330–343.
- Kühlbrandt W. 2004. Biology, structure and mechanism of P-type ATPases. Nat Rev Mol Cell Biol 5:282–295.
- Laimins LA, Rhoads DB, Altendorf K, Epstein W. 1978. Identification of the structural proteins of an ATP-driven potassium transport system in Escherichia coli. Proc Natl Acad Sci USA 75:3216–3219.
- Läuger P. 1991. Electrogenic ion pumps. Sunderland (MA): Sinauer Associates, Inc., 1–313.
- Liu S, Lockless SW. 2013. Equilibrium selectivity alone does not create K+-selective ion conduction in K+ channels. Nat Commun 4:2746.
- Macara IG. 1980. Vanadium – an element in search of a role. Trends Biochem Sci 5:92–94.
- Møller JV, Olesen C, Winther AM, Nissen P. 2010. The sarcoplasmic Ca2+-ATPase: design of a perfect chemi-osmotic pump. Q Rev Biophys 43:501–566.
- Morth JP, Pedersen BP, Toustrup-Jensen MS, Sørensen TL-M, Petersen J, Andersen JP, et al. 2007. Crystal structure of the sodium-potassium pump. Nature 450:1043–1049.
- O'Neal SG, Rhoads DB, Racker E. 1979. Vanadate inhibition of sarcoplasmic reticulum Ca2+-ATPase and other ATPases. Biochem Biophys Res Commun 89:845–850.
- Palmgren MG, Nissen P. 2011. P-type ATPases. Annu Rev Biophys 40:243–266.
- Pedersen PL, Carafoli E. 1987. Ion motive ATPases. I. Ubiquity, properties, and significance to cell function. TIBS 12:146–150.
- Pedersen BP, Ifrim G, Liboriussen P, Axelsen KB, Palmgren MG, Nissen P, et al. 2014. Large scale identification and categorization of protein sequences using structured logistic regression. PLoS One 9:e85139.
- Polarek JW, Walderhaug MO, Epstein W. 1988. Genetics of Kdp, the K+-transport ATPase of Escherichia coli. Meth. Enzymol 157:655–667.
- Post RL, Hegyvary C, Kume S. 1972. Activation by adenosine triphosphate in the phosphorylation kinetics of sodium and potassium ion transport adenosine triphosphatase. J Biol Chem 247:6530–6540.
- Primeau JO, Armanious GP, Fisher ME, Young HS. 2018. The SarcoEndoplasmic reticulum calcium ATPase. Subcell Biochem 87:229–258.
- Puppe W, Siebers A, Altendorf K. 1992. The phosphorylation site of the Kdp-ATPase of Escherichia coli: site-directed mutagenesis of the aspartic acid residues 300 and 307 of the KdpB subunit. Mol Microbiol 6:3511–3520.
- Rhoads DB, Epstein W. 1978. Cation transport in Escherichia coli. IX. Regulation of K transport. J Gen Physiol 72:283–295.
- Rhoads DB, Waters FB, Epstein W. 1976. Cation transport in Escherichia coli. VIII. Potassium transport mutants. J Gen Physiol 67:325–341.
- Richey B, Cayley DS, Mossing MC, Kolka C, Anderson CF, Farrar TC, et al. 1987. Variability of the intracellular ionic environment of Escherichia coli. Differences between in vitro and in vivo effects of ion concentrations on protein-DNA interactions and gene expression. J Biol Chem 262:7157–7164.
- Roe AJ, McLaggan D, O’Byrne CP, Booth IR. 2000. Rapid inactivation of the Escherichia coli Kdp K+ uptake system by high potassium concentrations. Mol Microbiol 35:1235–1243.
- Schleyer M, Schmid R, Bakker EP. 1993. Transient, specific and extremely rapid release of osmolytes from growing cells of Escherichia coli K-12 exposed to hypoosmotic shock. Arch Microbiol 160:424–431.
- Schrader M, Fendler K, Bamberg E, Gassel M, Epstein W, Altendorf K, et al. 2000. Replacement of glycine 232 by aspartic acid in the KdpA subunit broadens the ion specificity of the K(+)-translocating KdpFABC complex. Biophys J 79:802–813.
- Serrano R. 1988. Structure and function of proton translocating ATPase in plasma membranes of plants and fungi. Biochim Biophys Acta 947:1–28.
- Siebers A, Altendorf K. 1989. Characterization of the phosphorylated intermediate of the K+-translocating Kdp-ATPase from Escherichia coli. J Biol Chem 264:5831–5838.
- Stein WD. 1986. Transport and diffusion across cell membranes. London: Academic Press Inc., 1–685.
- Stewart LM, Bakker EP, Booth IR. 1985. Energy coupling to K+ uptake via the Trk system in Escherichia coli: the role of ATP. J Gen Microbiol 131:77–85.
- Stock C, Hielkema L, Tascón I, Wunnicke D, Oostergetel GT, Azkargorta M, et al. 2018. Cryo-EM structures of KdpFABC suggest a K+ transport mechanism via two inter-subunit half-channels. Nat Commun 9:4971.
- Vieira-Pires RS, Szollosi A, Morais-Cabral JH. 2013. The structure of the KtrAB potassium transporter. Nature 496:323–328.
- Waters S, Gilliham M, Hrmova M. 2013. Plant High-Affinity Potassium (HKT) Transporters involved in salinity tolerance: structural insights to probe differences in ion selectivity. Int J Mol Sci 14:7660–7680.