Abstract
The amyloidogenesis occurring in Alzheimer's disease represents a fundamental membrane-related pathology involving a membrane-bound substrate metabolized by integral membrane proteases (secretases). Thus, the amyloid-β peptide (Aβ), which accumulates extracellularly as plaques in the brains of Alzheimer's disease patients, is derived by sequential proteolytic cleavage of the integral transmembrane amyloid precursor protein (APP). Beta-Secretase or BACE-1 (β-site APP cleaving enzyme) is a transmembrane aspartic protease responsible for the first of these cleavage events, generating the soluble APP ectodomain sAPPβ, and a C-terminal fragment CTFβ. CTFβ is subsequently cleaved by the γ-secretase complex, of which presenilin is the catalytic core, to produce Aβ. A variety of studies indicate that cholesterol is an important factor in the regulation of Aβ production, with high cholesterol levels being linked to increased Aβ generation and deposition. However, the mechanism(s) underlying this effect are unclear at present. Recent evidence suggests that amyloidogenic APP processing may preferentially occur in the cholesterol-rich regions of membranes known as lipid rafts, and that changes in cholesterol levels could exert their effects by altering the distribution of APP-cleaving enzymes within the membrane. Rafts may be involved in the aggregation of Aβ and also in its clearance by amyloid-degrading enzymes such as plasmin or possibly neprilysin (NEP).
| Acronyms | ||
| amyloid-β peptide | = | Aβ |
| angiotensin-converting enzyme | = | ACE |
| Alzheimer's disease | = | AD |
| A disintegrin and metalloprotease | = | ADAM |
| apolipoprotein E | = | ApoE |
| amyloid precursor protein | = | APP |
| β-site APP cleaving enzyme | = | BACE-1 |
| cluster differentiation antigen | = | CD |
| cerebrospinal fluid | = | CSF |
| C-terminal fragment | = | CTF |
| endoplasmic reticulum | = | ER |
| endothelin-converting enzyme | = | ECE |
| fluorescence resonance energy transfer | = | FRET |
| glycosyl-phosphatidylinositol | = | GPI |
| insulysin (insulin-degrading enzyme) | = | IDE |
| low density lipoprotein | = | LDL |
| lipoprotein receptor-related protein | = | LRP |
| neprilysin | = | NEP |
| presenilin | = | PS |
| presenilin | = | PS |
| α-secretase derived soluble APP ectodomain | = | sAPPα |
| β-secretase derived soluble APP ectodomain | = | sAPPβ |
| TNF-α converting enzyme | = | TACE |
| trans-Golgi network | = | TGN |
| tumour necrosis factor | = | TNF |
Introduction
Alzheimer's disease (AD) is a devastating neurodegenerative disease, and the most common cause of dementia today. It is estimated that more than 10% of people over the age of 65 are affected and, with this sector of the population increasing more rapidly than any other, it is easy to see why determining the causes of the disease with a view to developing therapies is the focus of much current research.
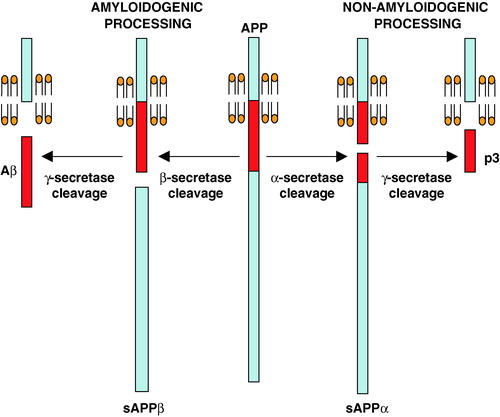
One of the pathological characteristics of AD is the development of extracellular senile plaques in the brain (Selkoe [Citation1994]; Yamaguchi et al. [Citation1988]). These are composed mainly of a small peptide known as amyloid-β (Aβ) (Masters et al. [Citation1985]), and, according to the ‘amyloid cascade hypothesis’, it is this peptide and/or the plaques, that are a major causative agent in the development of the disease (Hardy & Higgins [Citation1992]; Selkoe [Citation1999]). The Aβ peptide is generated by sequential proteolytic cleavage of the transmembrane amyloid precursor protein (APP) by two membrane-bound enzymes, β- and γ-secretase (). β-Secretase, a transmembrane aspartic protease known as BACE-1 or Asp-2 (Hussain et al. [Citation1999]; Sinha et al. [Citation1999]; Vassar et al. [Citation1999]; Yan et al. [Citation1999]), cleaves at the N-terminal end of the Aβ region, releasing the soluble sAPPβ, and this is followed by γ-secretase cleavage of the membrane-bound C-terminal fragment to release Aβ. γ-Secretase activity is known to be closely associated with the presenilin (PS) proteins (De Strooper et al. [Citation1998]; Wolfe et al. [Citation1999]), and it has been demonstrated that at least three other proteins, nicastrin, aph-1 and pen-2, are required to form an active γ-secretase complex (Francis et al. [Citation2002]; Goutte et al. [Citation2002]; Yu et al. [Citation2000]). A third enzyme, α-secretase, probably a member of the ADAMs (a disintegrin and metalloprotease) family of enzymes (Buxbaum et al. [Citation1998]; Lammich et al. [Citation1999]; reviewed in Allinson et al. [Citation2003]), is also capable of cleaving APP, but this occurs within the Aβ region and is therefore termed non-amyloidogenic ().
Figure 1. Amyloidogenic and non-amyloidogenic processing of APP. The alternative, competitive routes of processing of APP through the consecutive actions of β- and γ-secretases (amyloidogenic pathway) or α- and γ-secretases (non-amyloidogenic pathway) are indicated. This Figure is reproduced in colour in Molecular Membrane Biology online.
Over the past decade, an increasing amount of evidence derived from genetic, epidemiological and biochemical studies has pointed to a role for cholesterol in the development of AD, and particularly in the regulation of APP processing. The major risk factor for late-onset AD, after age, is the presence of a particular allele of the apolipoprotein E (ApoE) gene, which codes for a protein involved in cholesterol transport (Corder et al. [Citation1993]). Individuals carrying the ε4 allele of apoE have an increased risk of developing AD (Seshadri et al. [Citation1995]) and show a lower average age of onset of the disease (Corder et al. [Citation1993]; Strittmatter & Roses [Citation1995]). Other genes related to cholesterol metabolism have also been linked to AD, although none as unequivocally as apoE. These include the gene encoding the low-density lipoprotein receptor-related protein (LRP), which is a major receptor for apoE in the brain (Baum et al. [Citation1998]; Kang et al. [Citation1997]; Van Leuven et al. [Citation1998]), and cyp46, which encodes cholesterol 24-hydroxylase, the enzyme responsible for the catabolism of cholesterol to 24S-hydroxycholesterol (Kölsch et al. [Citation2002]; Papassotiropoulos et al. [Citation2003]).
Epidemiological evidence linking cholesterol and lipoprotein levels in plasma with AD development also exists. Several studies have found that high cholesterol levels in mid-life correlate strongly with amyloid deposition and the risk of developing AD in later life (Kivipelto et al. [Citation2001], [Citation2002]; Notkola et al. [Citation1998]; Pappolla et al. [Citation2003]). Levels of plasma low-density lipoprotein (LDL) at post-mortem have also been found to be higher in AD patients than in control subjects (Kuo et al. [Citation1998]). Conversely, patients taking cholesterol-lowering drugs, known as statins, have been found to have a lower incidence of AD than the general population (Jick et al. [Citation2000]; Wolozin et al. [Citation2000]), although more recent, prospective studies have produced mixed conclusions (Shepherd et al. [Citation2002]; Simons et al. [Citation2002]).
Studies examining the effects of cholesterol loading or depletion in both cultured cells and in vivo also demonstrate a strong link between high cholesterol levels and increased Aβ production. Animals fed a high cholesterol diet showed increased Aβ accumulation (Li et al. [Citation2003]; Refolo et al. [Citation2000]; Shie et al. [Citation2002]; Sparks et al. [Citation1994]), whereas treatment with cholesterol-lowering drugs resulted in lower Aβ levels (Fassbender et al. [Citation2001]; Refolo et al. [Citation2001]). These results have been replicated in cultured cells by removal of existing cholesterol or addition of exogenous cholesterol (Fassbender et al. [Citation2001]; Frears et al. [Citation1999]; Simons et al. [Citation1998]). The effects seen in these studies suggest that cholesterol is regulating APP processing, as changes in the levels of APP C-terminal fragments (CTFs) and soluble fragments were also observed (Refolo et al. [Citation2000], [Citation2001]; Simons et al. [Citation1998]).
Despite a connection between cholesterol and AD having first been suggested more than a decade ago (Sparks et al. [Citation1990], [Citation1993]), molecular mechanisms that could explain this link have not been determined. One hypothesis, which will be discussed in this review, is that Aβ production, metabolism and aggregation depend upon cholesterol-rich regions of cell membranes known as lipid rafts.
Lipid rafts
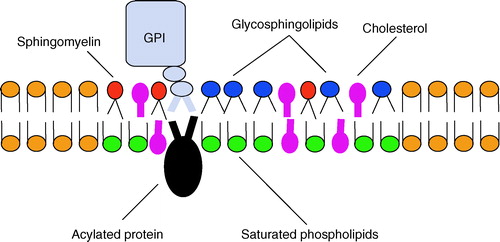
Lipid rafts are membrane microdomains which are enriched in cholesterol and sphingolipids. Phospholipids with saturated hydrocarbon chains are generally found in these regions, and these can pack together tightly with cholesterol to form highly ordered platforms which are distinct from the surrounding bilayer of largely unsaturated phospholipids (; for reviews see Brown & London [Citation1998]; Hooper [Citation1999]; Laude & Prior [Citation2004]; Simons & Ikonen [Citation1997]; Simons & Toomre [Citation2000]). Within these raft domains, certain types of protein tend to cluster, for example those with glycosyl-phosphatidylinositol (GPI) anchors and doubly acylated proteins. The lipid modifications on these proteins allow them to intercalate into the lipid raft structure, and therefore proteins with these types of anchor tend to be constitutively associated with raft domains (Simons & Ehehalt [Citation2002]). Other proteins are less closely associated, and their movement in and out of rafts can be controlled by factors such as ligand-binding or oligomerization (Harder et al. [Citation1998]; Zacharias et al. [Citation2002]).
Figure 2. Schematic diagram of a lipid raft domain. Lipid rafts represent sphingolipid and cholesterol rich regions of the membrane that selectively accumulate specific proteins, especially lipid-modified proteins. This Figure is reproduced in colour in Molecular Membrane Biology online.
Native rafts can only be detected in living cells (Simons & Toomre [Citation2000]) but most studies have taken advantage of cellular fractionation procedures to isolate and study rafts in vitro. One method of isolating raft domains in order to examine better their composition is based on work by Brown and Rose ([Citation1992]) demonstrating that sphingolipids and cholesterol from cell membranes are detergent-resistant at 4°C. Resident raft proteins such as GPI-anchored proteins are also insoluble in certain detergents, as originally shown for GPI-anchored hydrolases (alkaline phosphatase, aminopeptidase P, membrane dipeptidase, 5′-nucleotidase and trehalase) in renal microvillar membranes (Hooper & Turner [Citation1988a], Citation[b]). The most common detergent used to identify raft-resident proteins is Triton X-100, but many other detergents are also used, and studies of raft-localized proteins frequently yield diverse results, depending on the detergent used (Riddell et al. [Citation2001]; Röper et al. [Citation2000]; Taylor et al. [Citation2002]). The existence of different species of rafts with varied protein and/or lipid composition, or subcellular location, could account for the divergence seen in detergent insolubility (for review see Pike [Citation2004]). However, caution needs to be exercised when equating detergent-resistant membrane fractions with rafts in cell membranes (Lichtenberg et al. [Citation2005]).
Lipid rafts are believed to play a central role in several cellular processes, most notably in membrane sorting and trafficking, and in signal transduction (Laude & Prior [Citation2004]; Parton & Richards [Citation2003]; Simons & Toomre [Citation2000]). In these cases, rafts can act as platforms allowing particular proteins to cluster together to facilitate these processes. It has been hypothesized that raft domains could also provide an environment in which proteins involved in APP processing could cluster, thus increasing Aβ production. This will be discussed in the following sections.
APP processing and lipid rafts
There is growing evidence that amyloidogenic processing of APP may be facilitated in lipid raft domains, and may therefore be dependent on cholesterol levels. Conversely, non-amyloidogenic APP processing by α-secretase may occur in the non-raft regions of cell membranes. However, not all studies are consistent with these concepts.
Non-amyloidogenic processing
Few studies have investigated the association of α-secretase activity with lipid raft domains and/or cholesterol. The production of sAPPα has been shown to be down-regulated in APP transgenic mice fed a high-cholesterol diet (Howland et al. [Citation1998]; Refolo et al. [Citation2000]) and in cultured cells incubated with cholesterol (Bodovitz & Klein [Citation1996]). Depletion of cellular cholesterol, on the other hand, resulted in increased levels of sAPPα (Kojro et al. [Citation2001]), and the same effect was seen in mice treated with cholesterol-lowering drugs such as statins (Refolo et al. [Citation2001]).
As mentioned previously, α-secretase is believed to be a member of the ADAMs family of proteases, most likely ADAM10 and/or ADAM17/TACE (TNF-α converting enzyme; Buxbaum et al. [Citation1998]; Lammich et al. [Citation1999]; Postina et al. [Citation2004]; reviewed in Allinson et al. [Citation2003]). Kojro et al. ([Citation2001]) demonstrated that depleting cells of cholesterol resulted in higher expression of ADAM10, and therefore increased α-secretase activity. In the same study, a very small fraction of ADAM10 was shown to be raft-localized under normal conditions, and treatment of the cells with the raft disrupting agent filipin decreased this fraction while up-regulating sAPPα production. These results suggest that lowering cellular cholesterol could lead to a relocation of some ADAM10 from raft domains to non-raft regions of the membrane. This relocation appears to increase α-secretase cleavage of APP, indicating that it occurs more efficiently in non-raft domains.
Less is known about the relationship between ADAM17 and cholesterol, although one recent study has demonstrated that this enzyme is found in non-raft fractions of the membrane (von Tresckow et al. [Citation2004]). It has also been found that the shedding of two proteins, CD30 and the interleukin-6 receptor, by ADAM17 is increased following cholesterol depletion, suggesting that the activity of this enzyme can be regulated by cholesterol levels (Matthews et al. [Citation2003]; von Tresckow et al. [Citation2004]).
Amyloidogenic processing
Indirect evidence for the amyloidogenic processing of APP occurring in lipid rafts comes from a large number of studies looking at the localization of the β- and γ-secretase enzymes. A small but significant amount of BACE is found in raft domains (Cordy et al. [Citation2003]; Riddell et al. [Citation2001]), along with the presenilin proteins (Lee et al. [Citation1998]; Parkin et al. [Citation1999a]; Wahrle et al. [Citation2002]) and γ-secretase activity (Wahrle et al. [Citation2002]). Two recent reports have shown that the other components of the γ-secretase complex, nicastrin, aph-1 and pen-2, are also found in lipid rafts, and that the presence of all four components is required for the complex to become raft-associated and functionally active (Vetrivel et al. [Citation2004]; Urano et al. [Citation2005]). A subsequent study (Vetrivel et al. [Citation2005]) further suggests that there may be developmental differences in the location of the γ-secretase components. Vetrivel et al. ([Citation2005]) have proposed that γ-secretase residence in non-raft membranes may facilitate proteolysis of diverse substrates during embryonic development but translocation of γ-secretase to lipid rafts in adults ensures processing of certain substrates, including APP CTFs, while limiting processing of other potential substrates. In addition, Aβ itself is found in raft domains (Kawarabayashi et al. [Citation2004]; Lee et al. [Citation1998]). APP is believed to exist in two separate pools within the cell membrane, one associated with rafts and the other in the phospholipid domain (Bouillot et al. [Citation1996]; Ehehalt et al. [Citation2003]; Lee et al. [Citation1998]; Parkin et al. [Citation1999b]), and this would therefore allow it to be accessed by both α-secretase and β- and γ-secretases. According to this hypothesis, Aβ production would be favoured by high cholesterol levels, whereas a reduction in cholesterol levels would lead to a decrease in Aβ production. Several studies examining the effects of high and low cholesterol levels on APP processing in vivo and in cultured cells tend to support this hypothesis, and have been discussed above.
Studies examining the raft-association of AD-related proteins, and cholesterol loading and depletion studies, provide indirect evidence that lipid raft domains are important for the amyloidogenic processing of APP. A more direct connection between raft domains and Aβ production has been demonstrated by two recent studies. Ehehalt et al. ([Citation2003]) used antibodies to cross-link APP and BACE at the cell surface and showed that these two proteins co-patched with known raft markers but segregated away from non-raft markers. This cross-linking dramatically increased Aβ production, while the increase was not observed under conditions where rafts were disrupted, suggesting that the integrity of raft domains is required for Aβ production to take place. In another study, a GPI anchor was added to BACE in order to target it virtually exclusively to lipid raft domains (Cordy et al. [Citation2003]). This movement of BACE from non-raft to raft domains caused a substantial up-regulation in sAPPβ and Aβ production, indicating that cleavage of APP by BACE occurs more efficiently within raft domains.
β-Secretase activity
There is some evidence to suggest that disrupting cholesterol transport can alter presenilin distribution within cells and affect APP processing (Burns et al. [Citation2003]; Runz et al. [Citation2002]), but the majority of available evidence indicates that alterations in β-secretase activity, rather than the other two secretase enzymes, are largely responsible for the dependence of Aβ production on cholesterol levels. Studies examining a variety of APP cleavage products have found that cholesterol depletion leads to a decrease in CTFβ production (Refolo et al. [Citation2001]; Simons et al. [Citation1998]), rather than an increase as would be expected if γ-secretase were being inhibited. Raised cholesterol levels result in an increase in CTFβ (Refolo et al. [Citation2000]), which again would not be seen if γ-secretase was being upregulated. In addition, cross-linking APP and BACE in raft domains (Ehehalt et al. [Citation2003]), or targeting BACE exclusively to rafts (Cordy et al. [Citation2003]), have a dramatic, and cholesterol-dependent, effect on Aβ production, indicating that the activity of this enzyme closely depends on cholesterol levels and on its localization within cholesterol-rich domains.
Despite the fact that β-secretase processing of APP has been the focus of a large amount of research, both before and since the identification of BACE, there is still some controversy regarding the subcellular compartment(s) in which BACE acts. The acidic pH optimum of BACE (Lin et al. [Citation2000]; Vassar et al. [Citation1999]) perhaps makes the endosomal/lysosomal system the most obvious location for its activity, and the involvement of this pathway was proposed long before the identification of BACE, when it was demonstrated that the internalization of APP from the cell surface is required for Aβ release (Koo & Squazzo [Citation1994]). Several groups have shown that labelled APP is internalized and cleaved to produce Aβ (Haass et al. [Citation1992]; Jung & Cashman [Citation1999]; Koo et al. [Citation1996]), and in addition, it has been demonstrated using a fluorescence resonance energy transfer (FRET) approach, that BACE and APP show a very close interaction at the cell surface and in early endosomal compartments (Kinoshita et al. [Citation2003]). Ehehalt et al. ([Citation2003]) have recently shown that inhibition of endocytosis by two different methods blocks β- but not α-secretase cleavage of APP, suggesting that most BACE activity occurs after internalization, rather than at the cell surface. Conversely, α-secretase activity was demonstrated to occur mainly at the cell-surface by using a cell-impenetrant biotinylated α-secretase inhibitor (Parvathy et al. [Citation1999]).
Other evidence however, indicates that the cell surface may be a significant site of Aβ generation. In the same study, Ehehalt et al. ([Citation2003]) demonstrated that while endocytosis was blocked, the co-patching of APP and BACE in rafts using antibodies still increased Aβ secretion, suggesting that cell surface rafts must be a suitable environment for Aβ production. In another study, using the same dynamin mutant to inhibit endocytosis, similar results were obtained (Chyung & Selkoe [Citation2003]).
Of course it is quite possible, even likely, that BACE can act at more than one site within the cell, and indeed Aβ has been reported to be produced in sites as varied as the ER (Cook et al. [Citation1997]; Hartmann et al. [Citation1997]) and the TGN (Hartmann et al. [Citation1997]). Aβ from the ER appears to be retained intracellularly, rather than being secreted (Lee et al. [Citation2003]), therefore the main sites of production of secreted Aβ are likely to be the TGN, plasma membrane and endosomes/lysosomes. Lipid rafts are present in all these regions (Mukherjee & Maxfield [Citation2000]; Simons & Ikonen [Citation1997]), suggesting they could be involved in the production of all secreted Aβ.
Following their work examining the dependence of BACE activity on raft localization and internalization from the plasma membrane, Ehehalt and colleagues (2003) hypothesized that endocytosis of lipid raft domains from the cell surface is required in order for BACE and APP to interact. They suggested that APP and BACE would be unlikely to be present in the same rafts at the cell surface, as each individual raft is thought to contain fewer than thirty protein molecules (Pralle et al. [Citation2000]), but following endocytosis individual rafts could cluster together, allowing BACE and APP to interact.
This model may provide a picture of APP processing under normal conditions, but it is possible that during ageing or AD other factors cause BACE and APP to co-localize to a greater extent, therefore accelerating the rate of Aβ production and progression of the disease. Changes in the distribution of lipids and cholesterol within cell membranes are known to occur during ageing (Wood et al. [Citation2002]), and in particular, ageing has been shown to alter the transbilayer distribution of cholesterol in mice (Igbavboa et al. [Citation1996]), with aged mice having significantly more cholesterol in the outer leaflet of the bilayer than younger animals. This could be important in regulating changes in the proportion of cell membranes containing rafts in young and aged brains. Currently, no data exist regarding changes in number, size or composition of lipid rafts that may occur during ageing, or during the progression of AD, but an increase in the number and size of rafts during ageing would increase the likelihood of BACE and APP interacting to produce Aβ both at the cell surface and in endosomes/lysosomes. Additionally, an increase in membrane cholesterol levels at any age may cause the same effect, encouraging the partitioning of BACE and APP into rafts and an associated increase in Aβ production and risk of AD. These scenarios are depicted schematically in .
Figure 3. Schematic diagram showing how APP and BACE could interact through raft clustering or through an increase in the number or size of rafts present. Under normal conditions some APP (light blue: online) and BACE (yellow: online) molecules are present in cholesterol-rich lipid rafts (depicted in blue: online) while some are in the phospholipid domains (orange: online). Due to the small size of the rafts, individual APP and BACE molecules are rarely present in the same raft. (A) Following endocytosis, rafts cluster together bringing APP and BACE molecules into contact, as proposed by Ehehalt et al. ([Citation2003]). (B) During aging, or if cholesterol levels are high, the proportion of the membrane forming raft domains may be increased. This would increase the likelihood of APP and BACE being present in the same raft, and allow them to interact to a greater extent. This Figure is reproduced in colour in Molecular Membrane Biology online.
![Figure 3. Schematic diagram showing how APP and BACE could interact through raft clustering or through an increase in the number or size of rafts present. Under normal conditions some APP (light blue: online) and BACE (yellow: online) molecules are present in cholesterol-rich lipid rafts (depicted in blue: online) while some are in the phospholipid domains (orange: online). Due to the small size of the rafts, individual APP and BACE molecules are rarely present in the same raft. (A) Following endocytosis, rafts cluster together bringing APP and BACE molecules into contact, as proposed by Ehehalt et al. ([Citation2003]). (B) During aging, or if cholesterol levels are high, the proportion of the membrane forming raft domains may be increased. This would increase the likelihood of APP and BACE being present in the same raft, and allow them to interact to a greater extent. This Figure is reproduced in colour in Molecular Membrane Biology online.](/cms/asset/5a725a45-049d-4322-9add-4a48c7a2d77c/imbc_a_149624_f0003_b.jpg)
An alternative hypothesis
Despite the large body of evidence that has been discussed above, indicating that low cholesterol levels may be beneficial for the prevention and/or treatment of AD, there are some data to suggest that low brain cholesterol can actually lead to neurodegeneration. Park et al. ([Citation2003]) found that female rodents treated with lovastatin showed reduced cholesterol levels together with an increase in amyloid production and plaque load. In addition, several genetic disorders of brain cholesterol metabolism result in neurodegeneration. Niemann Pick disease type C is characterized by an accumulation of cholesterol in endosomes and lysosomes but a reduction of cholesterol synthesis and transport to the axonal membrane (Karten et al. [Citation2002]). Aggregation of Aβ has also been reported in this disease (Burns & Duff [Citation2002]). In another disorder, RSH/Smith-Lemli-Opitz syndrome, lack of cholesterol and a build-up of 7-dehydrocholesterol lead to neurodegeneration and death (Porter [Citation2000]). In a recent study, both Aβ and APP were shown to oxidize cholesterol to 7-β-hydroxycholesterol (Nelson & Alkon [Citation2005]).
The data indicating that low cholesterol levels decrease amyloid production must also be interpreted with some caution. While cyclodextrins are commonly used to lower cholesterol in cell membranes, these compounds have other biological effects, which may lead to misinterpretation of the results. Also, many of the studies have been carried out in non-neuronal cell lines, possible affecting the conclusions.
A recent study has investigated the effect of low membrane cholesterol on Aβ generation in hippocampal neurons (Abad-Rodriguez et al. [Citation2004]), and found that a moderate decrease in cholesterol levels (<25% reduction) resulted in increased Aβ production, whereas a more severe reduction in cholesterol (>35%) led to a significant down-regulation of Aβ generation, as observed in previous studies (Cordy et al. [Citation2003]; Fassbender et al. [Citation2001]; Simons et al. [Citation1998]). The authors suggested that, in contrast to previous hypotheses, cleavage of APP by BACE may actually take place outside lipid raft domains, and a small reduction in cholesterol levels would allow BACE to exit the rafts and access the APP present in the detergent-soluble regions of the membrane. One possible explanation put forward to explain the discrepancy between this and the previously discussed view of β-secretase activity occurring in raft domains, was that previous studies have been carried out using cells overexpressing APP, and this APP may be misdirected to rafts where it can be cleaved by BACE. This is not true of all studies, however, and we have previously shown that in human neuroblastoma cells expressing endogenous levels of APP, BACE cleaves its substrate much more effectively when it is targeted to raft domains by the addition of a GPI anchor (Cordy et al. [Citation2003]). Very little GPI-anchored BACE was found in non-raft domains in this study, therefore it was concluded that the cleavage of APP was occurring in the rafts.
The dual effects of statins on cells also appear to have two opposing actions on Aβ metabolism (Cole et al. [Citation2005]). Reduced cholesterol levels as a consequence of statin treatment of cells favoured the α-secretase pathway and decreased Aβ secretion whereas the cholesterol-independent effects of statins, decreasing isoprenoid levels, resulted in the accumulation of APP, amyloidogenic fragments, and intracellular Aβ. Thus, low cholesterol and low isoprenoid levels appeared to have completely independent effects on APP metabolism and Aβ formation.
Although these recent results suggest that lowering brain cholesterol levels may be at best ineffective against AD and at worst, damaging to neuronal function, there is much evidence from animal studies that this is not the case (Fassbender et al. [Citation2001], Hutter-Paier et al. [Citation2004]; Refolo et al. [Citation2001]). In addition, no side effects relating to dementia or amyloid generation have ever been reported from the numerous patients around the world taking cholesterol-lowering drugs.
Other roles for lipid rafts in AD
The majority of work that has been carried out to investigate the connection between cholesterol/rafts and AD has focused on Aβ production. A few studies, however, have found that cholesterol and cholesterol-rich domains are also implicated in Aβ aggregation and toxicity. Aβ has been found to bind to GM1 ganglioside to form a species of peptide which appears to act as a ‘seed’ to accelerate amyloid fibril formation (Choo-Smith et al. [Citation1997]; Kakio et al. [Citation2002]; Yanagisawa et al. [Citation1995]). The initial binding is increased by clustering of GM1 molecules, which occurs in cholesterol-rich environments, such as lipid rafts (Kakio et al. [Citation2001]), and depleting cellular cholesterol levels has been reported to be able to protect cells from Aβ toxicity (Wang et al. [Citation2001]). More recent results have confirmed this by demonstrating that both membrane-binding and toxicity of Aβ were reduced by the removal of cholesterol (Subasinghe et al. [Citation2003]).
Although the evidence discussed so far suggests that lipid rafts play an important role in generation and toxicity of Aβ, there is some indication that rafts may also be involved in degradation of the peptide. Several proteases have been shown to be capable of degrading Aβ (Carson & Turner [Citation2002]), including neprilysin (NEP; Howell et al. [Citation1995]; Iwata et al. [Citation2000], [Citation2001]), its homologues, the endothelin converting enzymes (ECE-1 and ECE-2; Eckman et al. [Citation2003]), insulin degrading enzyme (IDE; Farris et al. [Citation2003]; Kurochkin & Goto [Citation1994]), angiotensin-converting enzyme (ACE; Oba et al. [Citation2005]; Hemming & Selkoe [Citation2005]) and the serine protease, plasmin (Ledesma et al. [Citation2000]; Tucker et al. [Citation2000]). Plasmin and, to a lesser extent, neprilysin, have both been shown to be raft-associated (Angelisova et al. [Citation1999], Ledesma et al. [Citation2000]), although recent work targeting neprilysin to rafts found that it was no more efficient at degrading Aβ in this fraction (Hama et al. [Citation2004]). The activity of plasmin, on the other hand, was found to be reduced under conditions in which rafts were disrupted (Ledesma et al. [Citation2003]), although the effect of this on Aβ degradation has not been directly investigated. Interestingly, levels of plasmin in AD brains are low when compared with control subjects (Ledesma et al. [Citation2000]), and a more recent study has suggested that low cholesterol levels and raft disorganization in AD patients may cause this effect and lead to a down-regulation of Aβ clearance (Ledesma et al. [Citation2003]).
Implications for Alzheimer's treatment
Although the exact nature of the complex link between cholesterol, lipid rafts and Alzheimer's disease is only beginning to be appreciated, the vast amount of knowledge gained in recent years provides hope that this relationship can be exploited in the development of AD therapies. If, as the evidence discussed so far suggests, Aβ production depends on the interaction of APP and BACE within lipid raft domains, then strategies such as cholesterol reduction, aimed at segregating BACE away from APP, may be effective. However, as mentioned previously, studies examining the effects of cholesterol-lowering drugs on Aβ production and the development of AD have produced mixed results. Indeed, the degree to which the level of cellular cholesterol is reduced may be the critical factor in modulating amyloidogenesis. In addition, it is generally believed that cholesterol is unable to cross the blood-brain barrier (BBB), meaning that neuronal cholesterol levels would be unaffected by modulating blood cholesterol levels via diet. This, however, may not be completely true, as mice given a high-lipid diet have been shown to have increased cholesterol levels in the CNS as well as in plasma (Refolo et al. [Citation2000]), and brain cholesterol levels are increased in hypercholesterolaemic transgenic mice (Accad et al. [Citation2000]).
The two initial retrospective studies that examined AD amongst patients taking statins (Jick et al. [Citation2000]; Wolozin et al. [Citation2000]) found that the prevalence of AD was reduced by almost 70% in this group, compared to control subjects. These findings have been corroborated by more recent, similar studies (Rockwood et al. [Citation2002]; Yaffe et al. [Citation2002]). Since these observations were made, several prospective trials of statin therapy in humans have been carried out, with variable results. In a study by Simons and colleagues (2002), the level of Aβ40 in the CSF of patients with mild AD was found to be decreased following simvastatin treatment for 26 weeks, while patients with more severe AD showed no change in Aβ levels. Cognitive decline was slower in both groups than in the placebo group. In another study, treatment with the same drug, but at a lower dose, resulted in a decrease in CSF sAPPα and sAPPβ levels, but no change in Aβ42 (Sjogren et al. [Citation2003]). A different drug, lovastatin, was used by Buxbaum et al. ([Citation2002]) to treat non-AD subjects, and found to decrease serum Aβ levels in a dose-dependent manner. Two larger studies, designed to look primarily at the cardiovascular benefits of long-term statin treatment, found that cognitive decline was not prevented by either simvastatin (Heart Protection Study Collaborative Group [Citation2002]) or pravastatin (Shepherd et al. [Citation2002]). The reasons for the discrepancies seen in these results are not known. Some statins, such as lovastatin and simvastatin, are able to cross the BBB, while others, such as atorvastatin and pravastatin, cannot (Caballero [Citation2004]), but as the studies described above have produced mixed results even using the same class of drug, it is unlikely that this factor is responsible.
From these data it is not possible to draw a definitive conclusion regarding the ability of statins to modulate AD progression. To add to the uncertainty, it has been proposed that any neuroprotective effects of the drugs could be due to their anti-oxidant and anti-inflammatory properties (Cucchiara & Kasner [Citation2001]; Zamvil & Steinman [Citation2002]), rather than to their ability to inhibit cholesterol synthesis. Another issue that has recently been brought to light is a gender-related difference in the response of mice to statin treatment. In a study by Park and colleagues (2003), female mice treated with lovastatin showed an increase in Aβ production and plaque load despite having reduced cholesterol levels. Male mice showed the same reduction in cholesterol but no change in Aβ production. If these results can be corroborated then it may be necessary to re-examine the data previously collected from statin trials to take gender into account.
Conclusions and future directions
The majority of the evidence discussed in this review suggests that Aβ generation is likely to take place, at least in part, within cholesterol-rich membrane domains, and that strategies such as drug-treatment or dietary modulation aimed at lowering cholesterol levels may be beneficial as an AD therapy. However, cholesterol metabolism and its regulation is complex and multi-factorial and, at present, some caution must be exhibited in recommending such therapeutic strategies. Alternative pathways, such as isoprenoid metabolism, may counterbalance cholesterol effects and cell-based studies do not always reflect changes seen in animal models. Nevertheless, the interactions between cholesterol levels, lipid rafts and amyloid production in the brain remain fertile territory for inter-disciplinary studies into the mechanisms of neurodegeneration and its treatment.
This paper was first published online on prEview on 27 January 2006.
We thank the Medical Research Council for support of this work.
References
- Abad-Rodriguez J, Ledesma MD, Craessaerts K, Perga S, Medina M, Delacourte A, Dingwall C, De Strooper B, Dotti CG. Neuronal membrane cholesterol loss enhances amyloid peptide generation. J Cell Biol 2004; 167: 953–960
- Accad M, Smith SJ, Newland DL, Sanan DA, King LE, Jr, Linton MF, Fazio S, Farese RV, Jr. Massive xanthomatosis and altered composition of atherosclerotic lesions in hyperlipidemic mice lacking acyl CoA:cholesterol acyltransferase 1. J Clin Invest 2000; 105: 711–719
- Allinson TM, Parkin ET, Turner AJ, Hooper NM. ADAMs family members as amyloid precursor protein alpha-secretases. J Neurosci Res 2003; 74: 342–352
- Angelisova P, Drbal K, Horejsi V, Cerny J. Association of CD10/neutral endopeptidase-24.11 with membrane micro domains rich in glycosylphosphatidylinositol-anchored proteins and Lyn kinase. Blood 1999; 93: 1437–1439
- Baum L, Chen L, Ng HK, Chan YS, Mak YT, Woo J, Chiu HF, Pang CP. Low density lipoprotein receptor related protein gene exon 3 polymorphism association with Alzheimer's disease in Chinese. Neurosci Lett 1998; 247: 33–36
- Bodovitz S, Klein WL. Cholesterol modulates alpha-secretase cleavage of amyloid precursor protein. J Biol Chem 1996; 271: 4436–4440
- Bouillot C, Prochiantz A, Rougon G, Allinquant B. Axonal amyloid precursor protein expressed by neurons in vitro is present in a membrane fraction with caveolae-like properties. J Biol Chem 1996; 271: 7640–7644
- Brown DA, London E. Functions of lipid rafts in biological membranes. Ann Rev Cell Dev Biol 1998; 14: 111–136
- Brown DA, Rose JK. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 1992; 68: 533–544
- Burns M, Duff K. Cholesterol in Alzheimer's disease and tauopathies. Ann NY Acad Sci 2002; 977: 367–375
- Burns M, Gaynor K, Olm V, Mercken M, LaFrancois J, Wang L, Mathews PM, Noble W, Matsuoka Y, Duff K. Presenilin redistribution associated with aberrant cholesterol transport enhances beta-amyloid production in vivo. J Neurosci 2003; 23: 5645–5649
- Buxbaum JD, Cullen EI, Friedhoff LT. Pharmacological concentrations of the HMG-CoA reductase inhibitor lovastatin decrease the formation of the Alzheimer beta-amyloid peptide in vitro and in patients. Front Biosci 2002; 7: a50–a59
- Buxbaum JD, Liu KN, Luo Y, Slack JL, Stocking KL, Peschon JJ, Johnson RS, Castner BJ, Cerretti DP, Black RA. Evidence that tumour necrosis factor alpha-converting enzyme is involved in regulated alpha-secretase cleavage of the Alzheimer amyloid protein precursor. J Biol Chem 1998; 273: 27765–27767
- Caballero J. Do statins slow down Alzheimer's disease? A review. J Clin Pharm Ther 2004; 29: 209–213
- Carson JA, Turner AJ. Beta-amyloid catabolism: roles for neprilysin (NEP) and other metallopeptidases?. J Neurochem 2002; 81: 1–8
- Choo-Smith LP, Garzon-Rodriguez W, Glabe CG, Surewicz WK. Acceleration of amyloid fibril formation by specific binding of Abeta-(1-40) peptide to ganglioside-containing membrane vesicles. J Biol Chem 1997; 272: 22987–22990
- Chyung JH, Selkoe DJ. Inhibition of receptor-mediated endocytosis demonstrates generation of amyloid beta-protein at the cell surface. J Biol Chem 2003; 278: 51035–51043
- Cole SL, Grudzien A, Manhart IO, Kelly BL, Oakley H, Vassar R. Statins cause intracellular accumulation of amyloid precursor protein, beta-secretase-cleaved fragments, and amyloid beta-peptide via an isoprenoid-dependent mechanism. J Biol Chem 2005; 280: 18755–18770
- Cook DG, Forman MS, Sung JC, Leight S, Kolson DL, Iwatsubo T, Lee VM, Doms RW. Alzheimer's A beta(1-42) is generated in the endoplasmic reticulum/intermediate compartment of NT2N cells. Nature Med 1997; 3: 1021–1023
- Corder EH, Saunders AM, Strittmatter WJ, Schmechel DE, Gaskell PC, Small GW, Roses AD, Haines JL, Pericak-Vance MA. Gene dose of apolipoprotein E type 4 allele and the risk of Alzheimer's disease in late onset families. Science 1993; 261: 921–923
- Cordy JM, Hussain I, Dingwall C, Hooper NM, Turner AJ. Exclusively targeting β-secretase to lipid rafts by GPI-anchor addition upregulates β-site processing of the amyloid precursor protein. Proc Natl Acad Sci USA 2003; 100: 11735–11740
- Cucchiara B, Kasner SE. Use of statins in CNS disorders. J Neurol Sci 2001; 187: 81–89
- De Strooper B, Saftig P, Craessaerts K, Vanderstichele H, Guhde G, Annaert W, Von Figura K, Van Leuven F. Deficiency of presenilin-1 inhibits the normal cleavage of amyloid precursor protein. Nature 1998; 391: 387–390
- Eckman EA, Watson M, Marlow L, Sambamurti K, Eckman CB. Alzheimer's disease beta-amyloid peptide is increased in mice deficient in endothelin-converting enzyme. J Biol Chem 2003; 278: 2081–2084
- Ehehalt R, Keller P, Haass C, Thiele C, Simons K. Amyloidogenic processing of the Alzheimer β-amyloid precursor protein depends on lipid rafts. J Cell Biol 2003; 160: 113–123
- Farris W, Mansourian S, Chang Y, Lindsley L, Eckman EA, Frosch M, Eckman CB, Tanzi RE, Selkoe DJ, Guénette S. Insulin-degrading enzyme regulates the levels of insulin, amyloid beta-protein, and the beta-amyloid precursor protein intracellular domain in vivo. Proc Natl Acad Sci USA 2003; 100: 4162–4167
- Fassbender K, Simons M, Bergmann C, Stroick M, Lütjohann D, Keller P, Runz H, Kühl S, Bertsch T, von Bergmann K, Hennerici M, Beyreuther K, Hartmann T. Simvastatin strongly reduces levels of Alzheimer's disease β-amyloid peptides Aβ42 and Aβ40 in vitro and in vivo. Proc Natl Acad Sci USA 2001; 98: 5856–5861
- Francis R, McGrath G, Zhang J, Ruddy DA, Sym M, Apfeld J, Nicoll M, Maxwell M, Hai B, Ellis MC, Parks AL, Xu W, Li J, Gurney M, Myers RL, Himes CS, Hiebsch R, Ruble C, Nye JS, Curtis D. Aph-1 and pen-2 are required for Notch pathway signaling, gamma-secretase cleavage of betaAPP, and presenilin protein accumulation. Dev Cell 2002; 3: 85–97
- Frears ER, Stephens DJ, Walters CE, Davies H, Austen BM. The role of cholesterol in the biosynthesis of β-amyloid. Mol Neurosci 1999; 10: 1699–1705
- Goutte C, Tsunozaki M, Hale VA, Priess JR. APH-1 is a multipass membrane protein essential for the Notch signaling pathway in Caenorhabditis elegans embryos. Proc Natl Acad Sci USA 2002; 99: 775–779
- Haass C, Koo EH, Mellon A, Hung AY, Selkoe DJ. Targeting of cell-surface beta-amyloid precursor protein to lysosomes: alternative processing into amyloid-bearing fragments. Nature 1992; 357: 500–503
- Hama E, Shirotani K, Iwata N, Saido TC. Effects of neprilysin chimeric proteins targeted to subcellular compartments on amyloid-β-peptide clearance in primary neurons. J Biol Chem 2004; 279: 30259–30264
- Harder T, Scheiffele P, Verkade P, Simons K. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J Cell Biol 1998; 141: 929–942
- Hardy JA, Higgins GA. Alzheimer's disease: the amyloid cascade hypothesis. Science 1992; 256: 184–185
- Hartmann T, Bieger SC, Bruhl B, Tienari PJ, Ida N, Allsop D, Roberts GW, Masters CL, Dotti CG, Unsicker K, Beyreuther K. Distinct sites of intracellular production for Alzheimer's disease A beta40/42 amyloid peptides. Nature Med 1997; 3: 1016–1020
- Heart Protection Study Collaborative Group. MRC/BHF Heart Protection Study of cholesterol lowering with simvastatin in 20,536 high-risk individuals: a randomised placebo-controlled trial. Lancet 2002; 360: 7–22
- Hemming, ML, Selkoe, DJ. 2005. Amyloid beta-protein is degraded by cellular angiotensin-converting enzyme (ACE) and elevated by an ACE inhibitor. J Biol Chem, 280:37644–37650.
- Hooper NM. Detergent-insoluble glycosphingolipid/cholesterol-rich membrane domains, lipid rafts and caveolae. Mol Memb Biol 1999; 16: 145–156
- Hooper NM, Turner AJ. Ectoenzymes of the kidney microvillar membrane: differential solubilisation by detergents can predict a glycosyl-phosphatidylinositol anchor. Biochem J 1988a; 250: 865–869
- Hooper NM, Turner AJ. Ectoenzymes of the kidney microvillar membrane. Aminopeptidase P is anchored by a glycosyl-phosphatidylinositol moiety. FEBS Lett 1988b; 229: 340–344
- Howell S, Nalbantoglu J, Crine P. Neutral endopeptidase can hydrolyze beta-amyloid (1-40) but shows no effect on beta-amyloid precursor protein metabolism. Peptides 1995; 16: 647–652
- Howland DS, Trusko SP, Savage MJ, Reaume AG, Lang DM, Hirsch JD, Maeda N, Siman R, Greenberg BD, Scott RW, Flood DG. Modulation of secreted beta-amyloid precursor protein and amyloid beta-peptide in brain by cholesterol. J Biol Chem 1998; 273: 16576–16582
- Hussain I, Powell DJ, Howlett DR, Tew DG, Meek TD, Chapman C, Gloger IS, Murphy KE, Southan C, Ryan DM, Smith TS, Simmons DL, Walsh FS, Dingwall C, Christie G. Identification of a novel aspartic protease (Asp 2) as β-secretase. Mol Cell Neurosci 1999; 14: 419–427
- Hutter-Paier B, Huttunen HJ, Puglielli L, Eckman CB, Kim DY, Hofmeister A, Moir RD, Domnitz SB, Frosch MP, Windisch M, Kovacs DM. The ACAT inhibitor CP-113,818 markedly reduces amyloid pathology in a mouse model of Alzheimer's disease. Neuron 2004; 44: 227–238
- Igbavboa U, Avdulov NA, Schroeder F, Wood WG. Increasing age alters transbilayer fluidity and cholesterol asymmetry in synaptic plasma membranes of mice. J Neurochem 1996; 66: 1717–1725
- Iwata N, Tsubuki S, Takaki Y, Shirotani K, Lu B, Gerard NP, Gerard C, Hama E, Lee HJ, Saido TC. Metabolic regulation of brain Abeta by neprilysin. Science 2001; 292: 1550–1552
- Iwata N, Tsubuki S, Takaki Y, Watanabe K, Sekiguchi M, Hosoki E, Kawashima-Morishima M, Lee HJ, Hama E, Sekine-Aizawa Y, Saido TC. Identification of the major Abeta1-42-degrading catabolic pathway in brain parenchyma: suppression leads to biochemical and pathological deposition. Nature Med 2000; 6: 143–150
- Jick H, Zornberg GL, Jick SS, Seshadri S, Drachman DA. Statins and the risk of dementia. Lancet 2000; 356: 1627–1631
- Jung SS, Cashman NR. Processing of the beta-amyloid precursor protein in ex vivo human brain cells. Neuroreport 1999; 10: 3875–3879
- Kakio A, Nishimoto S, Yanagisawa K, Kozutsumi Y, Matsuzaki K. Cholesterol-dependent formation of GM1 ganglioside-bound amyloid beta-protein, an endogenous seed for Alzheimer amyloid. J Biol Chem 2001; 276: 24985–24990
- Kakio A, Nishimoto S, Yanagisawa K, Kozutsumi Y, Matsuzaki K. Interactions of amyloid beta-protein with various gangliosides in raft-like membranes: importance of GM1 ganglioside-bound form as an endogenous seed for Alzheimer amyloid. Biochemistry 2002; 41: 7385–7390
- Kang DE, Saitoh T, Chen X, Xia Y, Masliah E, Hansen LA, Thomas RG, Thal LJ, Katzman R. Genetic association of the low-density lipoprotein receptor-related protein gene (LRP), an apolipoprotein E receptor, with late-onset Alzheimer's disease. Neurology 1997; 49: 56–61
- Karten B, Vance DE, Campenot RB, Vance JE. Cholesterol accumulates in cell bodies, but is decreased in distal axons, of Niemann-Pick C1-deficient neurons. J Neurochem 2002; 83: 1154–1163
- Kawarabayashi T, Shoji M, Younkin LH, Wen-Lang L, Dickson DW, Murakami T, Matsubara E, Abe K, Ashe KH, Younkin SG. Dimeric amyloid beta protein rapidly accumulates in lipid rafts followed by apolipoprotein E and phosphorylated tau accumulation in the Tg2576 mouse model of Alzheimer's disease. J Neurosci 2004; 24: 3801–3809
- Kinoshita A, Fukumoto H, Shah T, Whelan CM, Irizarry MC, Hyman BT. Demonstration by FRET of BACE interaction with the amyloid precursor protein at the cell surface and in early endosomes. J Cell Sci 2003; 116: 3339–3346
- Kivipelto M, Helkala EL, Laakso MP, Hanninen T, Hallikainen M, Alhainen K, Soininen H, Tuomilehto J, Nissinen A. Midlife vascular risk factors and Alzheimer's disease in later life: longitudinal, population based study. Brit Med J 2001; 322: 1447–1451
- Kivipelto M, Helkala EL, Laakso MP, Hanninen T, Hallikainen M, Alhainen K, Iivonen S, Mannermaa A, Tuomilehto J, Nissinen A, Soininen H. Apolipoprotein E epsilon4 allele, elevated midlife total cholesterol level, and high midlife systolic blood pressure are independent risk factors for late-life Alzheimer disease. Ann Intern Med 2002; 137: 149–155
- Kojro E, Gimpl G, Lammich S, Marz W, Fahrenholz F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha -secretase ADAM 10. Proc Natl Acad Sci USA 2001; 98: 5815–5820
- Kölsch H, Lütjohann D, Ludwig M, Schulte A, Ptok U, Jessen F, von Bergmann K, Rao ML, Maier W, Heun R. Polymorphism in the cholesterol 24S-hydroxylase gene is associated with Alzheimer's disease. Mol Psychiatry 2002; 7: 899–902
- Koo EH, Squazzo SL. Evidence that production and release of amyloid β-protein involves the endocytic pathway. J Biol Chem 1994; 269: 17386–17389
- Koo EH, Squazzo SL, Selkoe DJ, Koo CH. Trafficking of cell-surface amyloid beta-protein precursor. I. Secretion, endocytosis and recycling as detected by labeled monoclonal antibody. J Cell Sci 1996; 109: 991–998
- Kuo YM, Emmerling MR, Bisgaier CL, Essenburg AD, Lampert HC, Drumm D, Roher AE. Elevated low-density lipoprotein in Alzheimer's disease correlates with brain Aβ 1-42 levels. Biochem Biophys Res Commun 1998; 252: 711–715
- Kurochkin IV, Goto S. Alzheimer's beta-amyloid peptide specifically interacts with and is degraded by insulin degrading enzyme. FEBS Letts 1994; 345: 33–37
- Lammich S, Kojro E, Postina R, Gilbert S, Pfeiffer R, Jasionowski M, Haass C, Fahrenholz F. Constitutive and regulated alpha-secretase cleavage of Alzheimer's amyloid precursor protein by a disintegrin and metalloprotease. Proc Natl Acad Sci USA 1999; 96: 3922–3927
- Laude AJ, Prior IA. Plasma membrane microdomains: Organization, function and trafficking. Mol Membr Biol 2004; 21: 193–205
- Ledesma MN, Abad-Rodriguez J, Galvan C, Biondi E, Navarro P, Delacourte A, Dingwall C, Dotti CG. Raft disorganization leads to reduced plasmin activity in Alzheimer's disease brains. EMBO Rep 2003; 4: 1190–1196
- Ledesma MD, Da Silva JS, Crassaerts K, Delacourte A, De Strooper B, Dotti CG. Brain plasmin enhances APP alpha-cleavage and Abeta degradation and is reduced in Alzheimer's disease brains. EMBO Rep 2000; 1: 530–535
- Lee EB, Skovronsky DM, Abtahian F, Doms RW, Lee VM. Secretion and intracellular generation of truncated Abeta in beta-site amyloid-beta precursor protein-cleaving enzyme expressing human neurons. J Biol Chem 2003; 278: 4458–4466
- Li L, Cao D, Garber DW, Kim H, Fukuchi K. Association of aortic atherosclerosis with cerebral beta-amyloidosis and learning deficits in a mouse model of Alzheimer's disease. Am J Pathol 2003; 163: 2155–2164
- Lichtenberg D, Goni FM, Heerklotz H. Detergent-resistant membranes should not be identified with membrane rafts. Trends Biochem Sci 2005; 30: 430–436
- Lin X, Koelsch G, Wu S, Downs D, Dashti A, Tang J. Human aspartic protease memapsin 2 cleaves the β-secretase site of β-amyloid precursor protein. Proc Natl Acad Sci USA 2000; 97: 1456–1460
- Lee S, Liyanage U, Bickel PE, Xia W, Lansbury PT, Kosik KS. A detergent-insoluble membrane compartment contains Aβ in vivo. Nature Med 1998; 4: 730–734
- Masters CL, Simms G, Weinman NA, Multhaup G, McDonald BL, Beyreuther K. Amyloid plaque core protein in Alzheimer disease and Down syndrome. Proc Natl Acad Sci USA 1985; 82: 4245–4249
- Matthews V, Schuster B, Schutze S, Bussmeyer I, Ludwig A, Hundhausen C, Sadowski T, Saftig P, Hartmann D, Kallen KJ, Rose-John S. Cellular cholesterol depletion triggers shedding of the human interleukin-6 receptor by ADAM10 and ADAM17 (TACE). J Biol Chem 2003; 278: 38829–38839
- Mukherjee S, Maxfield FR. Role of membrane organisation and membrane domains in endocytic lipid trafficking. Traffic 2000; 1: 203–211
- Nelson TJ, Alkon DL. Oxidation of cholesterol by amyloid precursor protein and β-amyloid peptide. J Biol Chem 2005; 280: 7377–7387
- Notkola IL, Sulkava R, Pekkanen J, Erkinjuntti T, Ehnholm C, Kivinen P, Tuomilehto J, Nissinen A. Serum total cholesterol, apolipoprotein E epsilon 4 allele, and Alzheimer's disease. Neuroepidemiology 1998; 17: 14–20
- Oba R, Igarashi A, Kamata M, Nagata K, Takano S, Nakagawa H. The N-terminal active centre of human angiotensin-converting enzyme degrades Alzheimer amyloid beta-peptide. Eur J Neurosci 2005; 21: 733–740
- Papassotiropoulos A, Streffer JR, Tsolaki M, Schmid S, Thal D, Nicosia F, Iakovidou V, Maddalena A, Lütjohann D, Ghebremedhin E, Hegi T, Pasch T, Traxler M, Bruhl A, Benussi L, Binetti G, Braak H, Nitsch RM, Hock C. Increased brain beta-amyloid load, phosphorylated tau, and risk of Alzheimer disease associated with an intronic CYP46 polymorphism. Arch Neurol 2003; 60: 29–35
- Pappolla MA, Bryant-Thomas TK, Herbert D, Pacheco J, Fabra Garcia M, Manjon M, Girones X, Henry TL, Matsubara E, Zambon D, Wolozin B, Sano M, Cruz-Sanchez FF, Thal LJ, Petanceska SS, Refolo LM. Mild hypercholesterolemia is an early risk factor for the development of Alzheimer amyloid pathology. Neurology 2003; 61: 199–205
- Park I-H, Hwang EM, Hong HS, Boo JH, Oh SS, Lee J, Jung MW, Bang OY, Kim SU, Mook-Jung I. Lovastatin enhances Aβ production and senile plaque deposition in female Tg2576 mice. Neurobiol Aging 2003; 24: 637–643
- Parkin ET, Hussain I, Karran EH, Turner AJ, Hooper NM. Characterization of detergent-insoluble complexes containing the familial Alzheimer's disease-associated presenilins. J Neurochem 1999a; 72: 1534–1543
- Parkin ET, Turner AJ, Hooper NM. Amyloid precursor protein, although partially detergent-insoluble in mouse cerebral cortex, behaves as an atypical lipid raft protein. Biochem J 1999b; 344: 23–30
- Parton RG, Richards AA. Lipid rafts and caveolae as portals for endocytosis: New insights and common mechanisms. Traffic 2003; 4: 724–738
- Parvathy S, Hussain I, Karran EH, Turner AJ, Hooper NM. Cleavage of the Alzheimer's amyloid precursor protein by α-secretase occurs at the surface of neuronal cells. Biochemistry 1999; 38: 9728–9734
- Pike LJ. Lipid rafts: Heterogeneity on the high seas. Biochem J 2004; 378: 281–292
- Postina R, Schroeder A, Dewachter I, Bohl J, Schmitt U, Kojro E, Prinzen C, Endres K, Hiemke C, Blessing M, Flamez P, Dequenne A, Godaux E, van Leuven F, Fahrenholz F. A disintegrin-metalloproteinase prevents amyloid plaque formation and hippocampal defects in an Alzheimer disease mouse model. J Clin Invest 2004; 113: 1456–1464
- Pralle A, Keller P, Florin EL, Simons K, Horber JK. Sphingolipid-cholesterol rafts diffuse as small entities in the plasma membrane of mammalian cells. J Cell Biol 2000; 148: 997–1008
- Porter FD. RSH/Smith-Lemli-Opitz syndrome: A multiple congenital anomaly/mental retardation syndrome due to an inborn error of cholesterol biosynthesis. Mol Genet Metab 2000; 71: 163–174
- Refolo LM, Malester B, LaFrancois J, Bryant-Thomas T, Wang R, Tint GS, Sambamurti K, Duff K, Pappolla MA. Hypercholesterolemia accelerates the Alzheimer's amyloid pathology in a transgenic mouse model. Neurobiol Dis 2000; 4: 321–331
- Refolo LM, Pappolla MA, LaFrancois J, Malester B, Schmidt SD, Thomas-Bryant T, Tint GS, Wang R, Mercken M, Petanceska SS, Duff KE. A cholesterol-lowering drug reduces beta-amyloid pathology in a transgenic mouse model of Alzheimer's disease. Neurobiol Dis 2001; 8: 890–899
- Riddell DR, Christie G, Hussain I, Dingwall C. Compartmentalization of β-secretase (Asp2) into low-buoyant density, non-caveolar lipid rafts. Curr Biol 2001; 11: 1288–1293
- Rockwood K, Kirkland S, Hogan DB, MacKnight C, Merry H, Verreault R, Wolfson C, McDowell I. Use of lipid-lowering agents, indication bias, and the risk of dementia in community-dwelling elderly people. Arch Neurol 2002; 59: 223–227
- Röper K, Corbeil D, Huttner WB. Retention of prominin in microvilli reveals distinct cholesterol-based lipid microdomains in the apical plasma membrane. Nature Cell Biol 2000; 2: 582–592
- Runz H, Rietdorf J, Tomic I, de Bernard M, Beyreuther K, Pepperkok R, Hartmann T. Inhibition of intracellular cholesterol transport alters presenilin localization and amyloid precursor protein processing in neuronal cells. J Neurosci 2002; 22: 1679–1689
- Selkoe DJ. Normal and abnormal biology of the β-amyloid precursor protein. Ann Rev Neurosci 1994; 17: 489–517
- Selkoe DJ. Translating cell biology into therapeutic advances in Alzheimer's disease. Nature 1999; 399: A23–A31
- Seshadri S, Drachman DA, Lippa CF. Apolipoprotein E epsilon 4 allele and the lifetime risk of Alzheimer's disease. What physicians know, and what they should know. Arch Neurol 1995; 52: 1074–1079
- Shepherd J, Blauw GJ, Murphy MB, Bollen EL, Buckley BM, Cobbe SM, Ford I, Gaw A, Hyland M, Jukema JW, Kamper AM, Macfarlane PW, Meinders AE, Norrie J, Packard CJ, Perry IJ, Stott DJ, Sweeney BJ, Twomey C, Westendorp RG. Pravastatin in elderly individuals at risk of vascular disease (PROSPER): a randomised controlled trial. Lancet 2002; 360: 1623–1630
- Shie FS, Jin LW, Cook DG, Leverenz JB, LeBoeuf RC. Diet-induced hypercholesterolemia enhances brain Abeta accumulation in transgenic mice. Neuroreport 2002; 25: 455–459
- Simons K, Ehehalt R. Cholesterol, lipid rafts, and disease. J Clin Invest 2002; 110: 597–603
- Simons K, Ikonen E. Functional rafts in cell membranes. Nature 1997; 387: 569–572
- Simons K, Toomre D. Lipid rafts and signal transduction. Nature Rev Mol Cell Biol 2000; 1: 31–39
- Simons M, Keller P, De Strooper B, Beyreuther K, Dotti CG, Simons K. Cholesterol depletion inhibits the generation of β-amyloid in hippocampal neurons. Proc Natl Acad Sci USA 1998; 95: 6460–6464
- Simons M, Schwarzler F, Lütjohann D, von Bergmann K, Beyreuther K, Dichgans J, Wormstall H, Hartmann T, Schulz JB. Treatment with simvastatin in normocholesterolemic patients with Alzheimer's disease: a 26-week randomized, placebo-controlled, double-blind trial. Ann Neurol 2002; 52: 346–350
- Sinha S, Anderson JP, Barbour R, Basi GS, Caccavello R, Davis D, Doan M, Dovey HF, Frigon N, Hong J, Jacobson-Croak K, Jewett N, Keim P, Knops J, Lieberburg I, Power M, Tan H, Tatsuno G, Tung J, Schenk D, Seubert P, Suomensaari SM, Wang S, Walker D, Zhao J, McConlogue L, John V. Purification and cloning of amyloid precursor protein β-secretase from human brain. Nature 1999; 402: 537–540
- Sjogren M, Gustafsson K, Syversen S, Olsson A, Edman A, Davidsson P, Wallin A, Blennow K. Treatment with simvastatin in patients with Alzheimer's disease lowers both alpha- and beta-cleaved amyloid precursor protein. Dement Geriatr Cogn Disord 2003; 16: 25–30
- Sparks DL, Hunsaker JC, Scheff SW, Kryscio RJ, Henson JL, Markesbery WR. Cortical senile plaques in coronary artery disease, aging and Alzheimer's disease. Neurobiol Aging 1990; 11: 601–607
- Sparks DL, Liu H, Scheff SW, Coyne CM, Hunsaker JC. Temporal sequence of plaque formation in the cerebral cortex of non-demented individuals. J Neuropathol Exp Neurol 1993; 52: 135–142
- Sparks DL, Scheff SW, Hunsaker JC, Liu H, Landers T, Gross DR. Induction of Alzheimer-like beta-amyloid immunoreactivity in the brains of rabbits with dietary cholesterol. Exp Neurol 1994; 126: 88–94
- Strittmatter WJ, Roses AD. Apolipoprotein E and Alzheimer disease. Proc Natl Acad Sci USA 1995; 92: 4725–4727
- Subasinghe S, Unabia S, Barrow CJ, Mok SS, Aguilar MI, Small DH. Cholesterol is necessary both for the toxic effect of Abeta peptides on vascular smooth muscle cells and for Abeta binding to vascular smooth muscle cell membranes. J Neurochem 2003; 84: 471–479
- Taylor CM, Coetzee T, Pfeiffer SE. Detergent-insoluble glycosphingolipid/cholesterol microdomains of the myelin membrane. J Neurochem 2002; 81: 993–1004
- Tucker HM, Kihiko M, Caldwell JN, Wright S, Kawarabayashi T, Price D, Walker D, Scheff S, McGillis JP, Rydel RE, Estus S. The plasmin system is induced by and degrades amyloid-beta aggregates. J Neurosci 2000; 20: 3937–3946
- Urano Y, Hayashi I, Isoo N, Reid PC, Shibasaki Y, Noguchi N, Tomita T, Iwatsubo T, Hamakubo T, Kodama T. Association of active gamma-secretase complex with lipid rafts. J Lipid Res 2005; 46: 904–912
- Van Leuven F, Thiry E, Stas L, Nelissen B. Analysis of the human LRPAP1 gene coding for the lipoprotein receptor-associated protein: identification of 22 polymorphisms and one mutation. Genomics 1998; 52: 145–151
- Vassar R, Bennett BD, Babu-Khan S, Kahn S, Mendiaz EA, Denis P, Teplow DB, Ross S, Amarante P, Loeloff R, Luo Y, Fisher S, Fuller J, Edenson S, Lile J, Jarosinski MA, Biere AL, Curran E, Burgess T, Louis J, Collins F, Treanor J, Rogers G, Citron M. β-secretase cleavage of Alzheimer's amyloid precursor protein by the transmembrane aspartic protease BACE. Science 1999; 286: 735–741
- Vetrivel KS, Cheng H, Lin W, Sakurai T, Li T, Nukina N, Wong PC, Xu H, Thinakaran G. Association of γ-secretase with lipid rafts in post-golgi and endosome membranes. J Biol Chem 2004; 279: 44945–44954
- Vetrivel KS, Cheng H, Kim SH, Chen Y, Barnes NY, Parent AT, Sisodia SS, Thinakaran G. Spatial segregation of gamma-secretase and substrates in distinct membrane domains. J Biol Chem 2005; 280: 25892–25900
- von Tresckow B, Kallen KJ, von Strandmann EP, Borchmann P, Lange H, Engert A, Hansen HP. Depletion of cellular cholesterol and lipid rafts increases shedding of CD30. J Immunol 2004; 172: 4324–4331
- Wahrle S, Das P, Nyborg AC, McLendon C, Shoji M, Kawarabayashi T, Younkin LH, Younkin SG, Golde TE. Cholesterol-dependent γ-secretase activity in buoyant cholesterol-rich membrane microdomains. Neurobiol Dis 2002; 9: 11–23
- Wang SS, Rymer DL, Good TA. Reduction in cholesterol and sialic acid content protects cells from the toxic effects of beta-amyloid peptides. J Biol Chem 2001; 276: 42027–42034
- Wolfe MS, Xia W, Ostaszewski BL, Diehl TS, Kimberly WT, Selkoe DJ. Two transmembrane aspartates in presenilin-1 required for presenilin endoproteolysis and gamma-secretase activity. Nature 1999; 398: 513–517
- Wolozin B, Kellman W, Ruosseau P, Celesia GG, Siegel G. Decreased prevalence of Alzheimer disease associated with 3-hydroxy-3-methyglutaryl coenzyme A reductase inhibitors. Arch Neurol 2000; 57: 1439–1443
- Wood WG, Schroeder F, Igbavboa U, Avdulov NA, Chochina SV. Brain membrane cholesterol domains, aging and amyloid beta-peptides. Neurobiol Aging 2002; 23: 685–694
- Yaffe K, Barrett-Connor E, Lin F, Grady D. Serum lipoprotein levels, statin use, and cognitive function in older women. Arch Neurol 2002; 59: 378–384
- Yamaguchi H, Hirai S, Morimatsu M, Shoji M, Harigaya Y. Diffuse type of senile plaques in the brains of Alzheimer-type dementia. Acta Neuropathol 1988; 77: 113–119
- Yan R, Bienkowski MJ, Shuck ME, Miao H, Tory MC, Pauley AM, Brashler JR, Stratman NC, Matthews WR, Buhl AE, Carter DB, Tomasselli AG, Parodi LA, Heinrikson RL, Gurney ME. Membrane-anchored aspartyl protease with Alzheimer's disease β-secretase activity. Nature 1999; 402: 533–537
- Yanagisawa K, Odaka A, Suzuki N, Ihara Y. GM1 ganglioside-bound amyloid beta-protein (A beta): a possible form of preamyloid in Alzheimer's disease. Nature Med 1995; 1: 1062–1066
- Yu G, Nishimura M, Arawaka S, Levitan D, Zhang L, Tandon A, Song Y-Q, Rogaeva E, Chen F, Kawarai T, Supala A, Levesque L, Yu H, Yang D-S, Holmes E, Milman P, Liang Y, Zhang DM, Xu DH, Sato C, Rogaev E, Smith M, Janus C, Zhang Y, Aebersold R, Farrer L, Sorbi S, Bruni A, Fraser P, St George-Hislop P. Nicastrin modulates presenilin-mediated notch/glp-1 signal transduction and βAPP processing. Nature 2000; 407: 48–54
- Zacharias DA, Violin JD, Newton AC, Tsien RY. Partitioning of lipid-modified monomeric GFPs into membrane microdomains of live cells. Science 2002; 296: 913–916
- Zamvil SS, Steinman L. Cholesterol-lowering statins possess anti-inflammatory activity that might be useful for treatment of MS. Neurology 2002; 59: 970–971