Abstract
Stress is one of the factors provoking cardiovascular complications. The purpose of the study was to explore the role of vasopressin (VP) in central control of arterial blood pressure and heart rate under resting conditions and during stimulation by an alarming stress (air jet stress) in myocardial infarct-induced cardiac failure. Six groups of male Sprague Dawley (SD) rats were subjected either to sham surgery (sham rats) or to ligation of a left coronary artery (infarcted rats). After 5 weeks both infarcted and sham rats were subjected either to intracerebroventricular infusion of artificial cerebrospinal fluid (aCSF) (sham aCSF and infarcted aCSF), [Arg8]-VP (sham VP and infarcted VP) or VP V1a receptor antagonist (d(CH2)5[Tyr(Me)2Ala-]VP, sham V1ANT and infarcted V1ANT). Air jet stress elicited significantly greater increases in mean arterial blood pressure (MABP) and heart rate in the infarcted aCSF than in the sham aCSF rats. Intracerebroventricular infusion of V1ANT significantly reduced resting MABP and MABP and heart rate increases in response to stress in the infarcted but not in the sham rats. Intracerebroventricular infusion of VP elicited a significant increase in resting MABP in the infarcted VP but not in the sham VP rats. The results provide evidence for enhanced engagement of the brain V1 VP receptors in regulation of resting MABP and in generation of exaggerated cardiovascular responses to air jet stress during the post-infarct state.
Introduction
Stress is considered to be one of the factors which may provoke cardiovascular complications, especially in patients with coronary artery disease (Kario et al. Citation2003; Fleet et al. Citation2005). This raises the possibility that regulation of the cardiovascular system by stressors may be altered in patients with cardiac failure. However, the mechanism serving as the link between a neurogenic stress and the exaggerated cardiovascular responses is not yet well characterized. Several lines of evidence support a significant role of vasopressin (VP) in neurogenic control of the cardiovascular system. Brain VP neurones innervate the most important cardiovascular control regions of the central nervous system (Bujis and Kalsbeck Citation1995), and acute disturbances in haemodynamics exert significant influence on the release of VP not only into the systemic circulation but also into the brain (Szczepanska-Sadowska et al. Citation1983; Demotes-Mainard et al. Citation1986). Moreover, experimental studies strongly suggest significant reorganisation of the brain VP system in chronic cardiovascular disorders, as manifested by changes in VP content and/or expression of V1 VP receptors (V1R) or V1R mRNA in the brain (Rascher et al. Citation1982; Sladek et al. Citation1988; Muders et al. Citation2002; Góźdź et al. Citation2003), as well as by significant alterations in the control of blood pressure by centrally acting VP (Budzikowski et al. Citation1996; Paczwa et al. Citation1997; Szczepanska-Sadowska et al. Citation1998; Paczwa et al. Citation1999). Positive effects of administration of VP antagonists in heart failure as well as availability of non peptide antagonists of VP receptors have renewed interest in the role of VP in cardiovascular pathology (Burrell et al. Citation2000). However, until now the main interest of investigators has been focused on cardiovascular effects of systemic administration of VP or VP antagonists, while relatively little attention has been paid to the central neuroregulatory effects of this peptide. The evidence that heart failure produced by constriction of the abdominal aorta significantly influences VP content in specific brain cardiovascular control regions (Muders et al. Citation2002), strongly suggests that the involvement of VP in central regulation of the cardiovascular system may be altered in heart failure. The specific purpose of the present study was to test the hypothesis that myocardial infarction is associated with an altered control of blood pressure and heart rate by VP acting in the brain under resting conditions and during application of an environmental alarming stressor (air jet stress). The rationale for investigating the role of VP in regulation of the cardiovascular parameters during stress was based on the following lines of evidence. First, it has been reported that blood pressure and heart rate responses to the air jet stress are exaggerated in rats with myocardial infarction (Zhang et al. Citation1999). Second, one of the mechanisms responsible for this phenomenon may be an enhanced anxiety evoked by a novel stimulus. It is well recognised that stress associated with anxiety is one important factor provoking severe disturbances in regulation of the cardiovascular system (Lampert et al. Citation2000; Grippo et al. Citation2003; Kario et al. Citation2003; Grippo et al. Citation2004; Fleet et al. Citation2005) while depression, which is frequently manifested by human subjects who have experienced myocardial infarction is regarded as a negative prognostic feature for the cardiovascular outcome (Clarke et al. Citation2000; Lesperance et al. Citation2002). Third, VP is one of the most extensively studied neuropeptides in pathophysiology of anxiety disorders and depression (Purba et al. Citation1996; Carrasco and van de Kar Citation2003; Griebel et al. Citation2003; Landgraf Citation2003; Bielsky et al. Citation2004; Wigger et al. Citation2004). Therefore, it appeared essential to elucidate whether VP, through its action in the brain may regulate cardiovascular responses to stress in the post-infarct state. In the present study, we provide evidence for greater involvement of brain V1a receptors in the regulation of MABP and heart rate in rats with myocardial infarction than in control rats, both under resting conditions and during an alarming stress.
Methods
Animals and surgical procedures
Animals
The study was carried out on male Sprague Dawley (SD) rats obtained from the animal house of the Medical University of Warsaw. The rats were maintained on a 12 h/12 h light–dark cycle (light on at 7.00 am) and fed ad libitum with a commercial rat diet containing 0.45% NaCl. The rats had free access to tap water, except during the experimental session. All surgical procedures and experimental protocols were approved by the Ethical Committee on Animal Research of the Warsaw Medical University and in accordance with international/EU guidelines and regulations.
Myocardial infarct and sham surgery
At the beginning of the study, 10–12 weeks old rats were subjected either to the myocardial infarct (MI) (infarcted rats) or to the sham surgery (sham rats). Myocardial infarction was produced by ligation of the left coronary artery according to our own modification of the procedure discribed by Selye et al. (Citation1960). The surgery was performed under chloral hydrate anaesthesia (36 mg/100 g b wt, ip). The surgical incision was made between the fourth and the fifth intercostal space and the heart was exteriorized, while ventilation of the lungs was maintained by frequent administration of air puffs by means of a small rubber balloon connected with the rats nose by means of a plastic tube. The left coronary artery was ligated with a 6-0 prolen suture thread (Ethicon). Subsequently, the heart was replaced in the thoracic cavity and the wound was closed with surgical sutures (Ethicon 4.0). The whole procedure lasted 14–16 min. In Group 2, the surgical procedure was similar, except that the coronary artery was not ligated and the pericardium was only touched with the suture needle. After surgery, the rats were given antibiotic (Taromentin, Polfa, 3 mg/100 g b wt, ip) and 30 mg of buprenorphine sulphate (Polfa) and placed in their home cages. In each rat the size of the infarct was evaluated during the post mortem examination, as described below. The rate of mortality of all rats subjected to open chest surgery amounted to 27% (48 and 5% in the infarcted and sham rats, respectively). The non-surviving rats died either during the surgery or during the following 24 h.
Implantation of the intracranial cannula
Four weeks after the coronary artery ligation or sham surgery the rats were anaesthetised with chloral hydrate (36 mg/100 g b wt, ip) and a guide tube was implanted in the left lateral cerebral ventricle. The rat's head was placed in the stereotaxic apparatus (Kopff) and levelled between lambda and bregma. The scalp was incised 2 mm lateral to the midsagittal suture and an opening was burred 1.3 mm posterior and 2.0 mm lateral to bregma. Subsequently, the stainless steel guide tube (o.d. − 0.81 mm, MIFAM) was introduced vertically into the brain to 4.5 mm below the surface of the skull and fixed to the skull with acrylic cement (Duracryl, SPOFA-DENTAL). A stainless steel stylet (o.d. − 0.46 mm) was inserted into the tube and the scalp wound was closed with 2–3 surgical sutures. After surgery the rat was given antibiotic (Taromentin, Polfa, 3 mg/100 g b wt, ip). The position of the guide tube in the lateral ventricle was verified post mortem. To this end Evans blue (5 μl) was injected through the guide tube via the intracerebroventricular (i.c.v.) infusion cannula. The brain was removed without fixation and sectioned sagittally to inspect the ventricular system for presence of the dye. Two out of 51 rats subjected to the experimental sessions, in which the dye has not stained the ventricular system was confined to the brain tissue around the cannula, were not included in the statistical analysis.
Implantation of the arterial catheter
Seven days after the cranial surgery the rats were anaesthetized with chloral hydrate (36 mg/100 g b wt, ip). An intraarterial catheter filled with 0.9% NaCl containing 500 U of heparin/ml was introduced into the aorta through the left femoral artery. The catheter consisted of two parts. The intraarterial part comprised a 3.5–4.0 cm-long tube (i.d. 0.5 mm, o.d. 0.8 mm; Dural Plastics and Engineering, Auburn, Australia) placed in the aorta 2 cm below the renal arteries, connected with the external part made from polyvinyl tubing. This tubing was tunnelled under the skin and exteriorised dorsally on the neck. The skin was closed with sutures and the rats were given Taromentin (Polfa, 3 mg/100 g b wt ip). The experiments were performed 24–48 h after this surgery when the rats were fully recovered from anaesthesia and could move without apparent discomfort.
Experimental groups and experimental design
Six groups of experiments were performed on 49 rats with successful implantation of the i.c.v. cannula. All experiments were performed at the same time of day between 16.00–19.00 h.
Intracerebroventricular infusion of artificial cerebrospinal fluid
Nine sham-operated and eight infarcted rats were given an i.c.v. infusion of artificial cerebrospinal fluid (aCSF) (sham aCSF and infarcted aCSF groups) to evaluate changes in resting mean arterial blood pressure (MABP) and heart rate, and to compare MABP and heart rate responses to air jet stress under control conditions. The composition of aCSF was: Na+ − 156.0 mmol/l, K+ − 3.0 mmol/l, Ca2+ − 1.5 mmol/l, Cl− − 147.0 mmol/l and HC − 15.0 mmol/l. At the beginning of each experiment the rat was placed in the experimental cage in which it could move freely but did not have access to food and water. The intraarterial catheter was connected to the arterial blood pressure and heart rate recording system (BIOPAC, MP100, Santa Barbara CA, USA). MABP was calculated by the computer program as the area under the arterial pressure curve divided by the cardiac cycle duration. After 10–15 min to allow stabilisation of MABP and heart rate an aCSF i.c.v. infusion was started at a rate of 5 μl/h (Harvard 22 Infusion Pump, Harvard, Smith Natick, MA). The i.c.v. infusion and measurements of MABP and heart rate were continued until the end of the experiment. After 40 min of aCSF infusion under resting conditions the air jet was applied. The air jet stress comprised compressed air blown on the top of the rat's head for 1 s via a laboratory made device. This consisted of a tank containing compressed air (10 atmospheres) connected by a plastic tube (i.d. 3 mm) to a funnel (i.d. 41.5 mm) held above the rat's head. The air jet stress was applied only once to each rat to avoid the possibility of adaptation. To evaluate the cardiovascular responses to air jet stress, MABP and heart rate were recorded before, during application of the stressor and over the next 10 min. The maximum increases in MABP and heart rate from the level immediately preceding application of the stressor were used to evaluate the magnitude of the response. The times between the onset of application of the air jet stress and the maximum pressor and tachycardic responses (time zero to peak latency) were measured on the recordings by two investigators.
Intracerebroventricular infusion of vasopressin
In the sham VP (n = 8) and infarcted VP (n = 8) groups we sought to determine whether the MI results in altered responsiveness to the central pressor effect of VP under resting conditions, and whether i.c.v. administration of VP may influence the cardiovascular responses to air jet stress in the sham and infarcted VP rats. The experimental design was similar to that in the sham aCSF and infarcted aCSF groups, except that VP ([Arg8]-VP, Sigma) was i.c.v. infused at 100 ng/5 μl aCSF/h (92.3 pmol/h–1.54 pmol/min). The rate of VP infusion was based on the results of our previous study (Stępniakowski et al. Citation1994), in which a single i.c.v. injection of 1 pmole of VP was found to produce a threshold pressor effect in conscious normotensive WKY rats. Preliminary experiments performed in the present study revealed that i.c.v. infusion of VP at the rate of 100 pg/h elevated MABP in all infarcted rats while in the sham rats, the pressor response was present only in some of the animals. Therefore, it was assumed that VP infused at this rate exerts a threshold pressor effect.
Intracerebroventricular infusion of V1 antagonist
The sham V1 antagonist (sham V1ANT) (n = 8) and infarcted V1ANT (n = 8) groups of experiments were performed to investigate the role of the brain V1 VP receptors in the regulation of the cardiovascular parameters under resting conditions and during stress. To this end the rats were subjected to a similar experimental procedure as the sham aCSF and the infarcted aCSF rats, except that the brain V1 VP receptors were blocked by i.c.v. administration of a V1a receptors antagonist d(CH2)5[Tyr(Me)2Ala-]VP; its V1b properties are not known (Manning et al. Citation1992 and Manning M.–personal communication). The antagonist was infused at a rate of 216 ng/5 μl aCSF/h (185 pmol/h–3.1 pmol/min) during the whole experiment. The rate of infusion of the V1ANT was established on the basis of our previous study (Budzikowski et al. Citation1996), in which i.c.v. infusion of another V1 receptors antagonist [d(Et2)Tyr(Me)]DVP at a rate of 300 ng/h (263 pmol/h) effectively prevented the pressor response to i.c.v. injection of a suprathreshold dose of 50 ng (46 pmol) of VP but did not abolish the pressor response to intravenous injection of a suprathreshold dose of 5 ng (4.6 pmol) of VP. Based on the effective pressor doses of both antagonists it was established that infusion of [d(CH2)5[Tyr(Me)2Ala-
]VP at a rate of 216 ng/h was expected to be equipotent with infusion of [d(Et2)Tyr(Me)]DVP at a rate of 300 ng/h, and to effectively block central V1 VP receptors without affecting the peripheral pressor effect of VP.
Evaluation of infarct size
After the experiments the rats were killed by an overdose of chloral hydrate, the heart was excised from the thorax, gently washed with saline and wiped with a tissue. The left ventricle, including the septum was separated from the right ventricle and both ventricles were weighed. To determine the size of the infarct the left ventricle was cut along the longitudinal axis and placed flat on transparent paper. The circumference of the infarct on the internal and external surface of the ventricle was outlined and these two measurements were averaged and expressed as a percentage of the size of the left ventricle wall. Presence and dimensions of the infarct scar were also confirmed by histological examination. To this end, fragments of the infarcted and control hearts were placed in 10% formaldehyde solution. Fixed tissue blocks from each heart were paraffin-embedded and sectioned at 4 μm. Sections were processed for hematoxylin and eosin staining for routine morphologic examination and evaluation of fibrosis. Sirius Red (0.1%) in saturated aqueous picric acid was used to detect collagen in the heart. The fractional volume of collagen was determined using a computer-based semi-automated image-analysis system AnalySIS (Zeiss) coupled to a CCD camera (PAL, 768 × 576 fps). The total surface of all structures positive for collagen staining was expressed as a fraction of the total area within the field.
Statistical analysis
Statistica software (release 5) was used. All values presented in the text, Figures and Tables are means ± standard errors. The cardiovascular parameters were recorded per minute during each experiment. In each group 5 min averages covering the first 35 min of the infusion under resting conditions were used in the statistical analysis. The overall analysis of significance of changes in MABP and heart rate from baseline was performed using three-way ANOVA with two levels of rats (sham vs infarct) × three levels of type of experiment (aCSF, VP, V1ANT) × seven levels of repeated measurements design. Horizontal and vertical multiple pair wise comparisons (Curran-Everett and Benos Citation2004; Ludbrook Citation1994) were made using the Tukey test. This was followed by two-way ANOVA with three levels of types of experiments and seven levels of repeated measurements as factors. The two-way ANOVA was performed separately for the infarcted and sham rats. Detailed comparisons to isolate significant differences in changes of MABP and heart rate were made using the Tukey test. To evaluate the stress-induced blood pressure and heart rate responses the maximum changes in MABP or heart rate found during the first 5 min after application of the air jet stress were compared with the values found immediately before application of the air jet stress in the same group. The significance of differences between the air jet stress-induced maximum changes of the cardiovascular parameters in different groups of experiments was evaluated with two-way ANOVA according to a design with two levels of rats (sham vs infarct) and three levels of experimental groups (aCSF, VP, V1ANT), followed by horizontal and vertical Tukey comparisons. The same approach was used to evaluate significance of differences between the time zero to peak latencies of the maximum MABP and heart rate responses in the different groups of experiments.
Results
As shown in the sham-operated and the infarcted rats had similar body weight, MABP and heart rate at the beginning of the experiments. Resting values of MABP and heart rate before the i.c.v. infusions in the individual groups of experiments are shown in . The post mortem examination revealed that the average surface of the infarct amounted to 35.6 ± 1.6% of the LV size (range: 25.0–46.5%). Dimensions of the post-infarct scar in the individual groups of infracted rats are presented in . In each case, histological examination confirmed presence of the fibrous scar in the region of the heart that was identified as being infarcted during prior visual inspection. Total collagen fraction in the infarct scar amounted to 0.29 ± 0.03. In the regions not affected by the infarct and in the hearts harvested from the sham-operated rats collagen fraction was too low to be measured.
Table I. Characteristics of sham-operated and infarcted SD rats.
Table II. infarct Size And Resting Values Of Mabp And Heart Rate In Sham-operated And Infarcted Rats Before Intracerebroventricular Infusion Of Acsf, Vp Or—{d(ch2)5[tyr(me)2ala-]vp} (V1ant).
Cardiovascular responses under resting conditions
Mean arterial blood pressure
The overall three-way ANOVA on changes in MABP from baseline revealed significant differences between the types of experiments [F(2,43) = 19.97; P < 0.0001], significant interaction of the types of experiment with changes in MABP in the infarcted- and the sham-operated rats [F(2,43) = 6.0; P < 0.005] as well as significant interaction of the type of experiments with the repeated measurements [F(12,258) = 4.59; P < 0.0001]. Detailed differences between and within the sham-operated and the infarcted groups during infusions of aCSF, VP and V1ANT were isolated by means of two-way ANOVA. This revealed significant differences in MABP between the infarcted and the sham rats during i.c.v. infusion of V1ANT [F(1,14) = 18.5; P < 0.0007] (). Moreover, changes in MABP in the infarcted and sham rats during administration of V1ANT significantly interacted [F (6,84) = 2.53; P < 0.03]. Specifically, significant differences in decrease of MABP between the sham and the infarcted rats were found between 20 and 35 min of i.c.v. V1ANT infusion by the Tukey test (). In contrast, changes in MABP during i.c.v. infusions of aCSF and VP in the infarcted- and the sham-operated rats did not differ significantly.
Figure 1 (A) Comparison of changes in MABP from baseline in the sham-operated and the infarcted (MI) rats during i.c.v infusions of VP V1 receptor antagonist (V1ANT), *—significant differences between the corresponding means in the rats with and without the infarct, #—significant difference from baseline. (B) Changes in resting MABP from baseline during i.c.v. infusion of aCSF, arginine VP and VP V1ANT in the sham-operated (upper part) and the infarcted (MI, lower part) rats. Number of rats in the groups: sham CSF—9, sham VP—8, sham V1ANT—8, infarct CSF—8, infarct VP—8, infarct V1ANT—8. Means ± standard errors are shown. The symbols indicate significant differences between the corresponding means in different groups of experiments:+ − significant difference between aCSF and VP groups, #—significant difference between aCSF and V1ANT groups, *—significant difference between VP and V1ANT groups. The results were considered significant if P < 0.05.
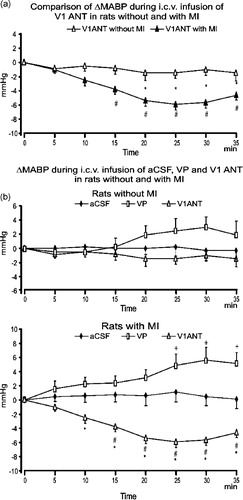
Two-way ANOVA performed on MABP and HR changes in the sham-operated rats did not reveal significant differences between the sham aCSF, sham VP and sham V1ANT groups. No significant differences were found for MABP and HR changes from baseline in individual groups of sham-operated rats by one-way ANOVA.
In the infarcted rats two-way ANOVA revealed significant differences for MABP changes between the groups of experiments [F(2,21) = 22.91; P < 0.0001] as well as significant interaction between the groups and repeated measurements [F(12,126) = 4.51; P < 0.0001]. Specifically, significant differences were found by Tukey tests in the infarcted rats between the aCSF and VP groups, the aCSF and V1ANT groups, and between the VP and V1ANT groups (, lower part). Blockade of brain V1 receptors and administration of VP in the infarcted rats elicited also significant changes in MABP from baseline. Significant decline of MABP was observed between 20 and 35 min of i.c.v. V1ANT infusion (P < 0.001, Tukey test, data not shown) while a significant increase in MABP was found during 25–30 min of VP infusion (P < 0.01, Tukey test, data not shown). In spite of continuation of V1ANT and VP infusions changes in MABP in the infarcted rats were transient and at 40 min of infusion they were below the level of significance. During infusion of aCSF in the infarcted rats MABP was not altered in comparison to baseline.
Heart rate
Resting heart rate was not significantly affected by i.c.v. infusions of aCSF, VP or V1ANT either in the sham-operated or in the infarcted rats (data not shown). Nor were significant differences found in fluctuations of heart rate between the different experimental groups.
Cardiovascular responses to stress
Mean arterial blood pressure
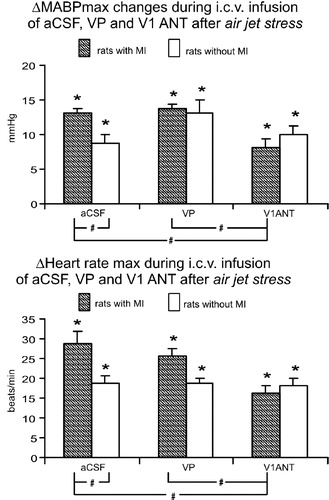
A significant, short-lasting increase in MABP was observed in each group of experiments after application of air jet stress. Representative tracings of MABP and changes evoked by air jet stress are shown in . The overall two-way ANOVA performed on the air jet stress-induced maximum changes in MABP during infusion of aCSF, VP and V1ANT in the infarcted and sham-operated rats disclosed significant interaction for infarcted and sham rats with the type of experiment [F(2,43) = 4.34; P < 0.02]. Detailed analysis revealed a significant difference between the sham aCSF and infarcted aCSF groups (P < 0.05; Tukey test, , upper part), while the differences between the sham VP and infarcted VP, as well as between the sham V1ANT and infarcted V1ANT groups were not significant. One-way ANOVA performed on data from sham rats did not show significant differences between the maximum changes in MABP after application of air jet stress during i.c.v. infusions of aCSF, VP or V1ANT (). In contrast, a significant difference between different types of experiments was found when one-way ANOVA was performed on data from the infarcted rats [F(2,21) = 20.41; P < 0.0001]. Detailed analysis disclosed significant differences in MABP responses between the infarcted aCSF and infarcted V1ANT groups (P < 0.001, Tukey test) and between the infarcted VP and infarcted V1ANT groups (P < 0.0001, Tukey test), while the difference in the maximum changes in MABP between the infarcted VP and infarcted aCSF groups was not significant.
Figure 2 Representative tracings of air jet stress-induced MABP and heart rate changes in the sham-operated (left) and the infarcted (MI, right) rats. The rectangle indicates the time of air jet stress application.
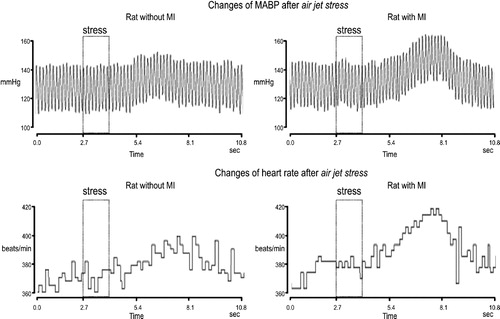
Figure 3 Maximum changes in mean arterial pressure (ΔMABPmax) and heart rate in response to the air jet stress in rats with and without the MI subjected to i.c.v. infusions of aCSF, arginine VP or VP V1ANT. Number of rats in the groups: sham CSF—9, sham VP—8, sham V1ANT—8, infarct CSF—8, infarct VP—8, infarct V1ANT—8. Means ± standard errors are shown. *—significant difference from baseline, #—significant difference between the corresponding groups. The results were considered significant if P < 0.05.
Heart rate
Two way ANOVA revealed a significant difference for the maximum air jet stress-induced changes of heart rate between the sham-operated and the infarcted rats [F(1,43) = 8, 28; P < 0.006], as well as significant interaction for the infarcted and sham rats with the type of experiment [F(2,43) = 3.44; P < 0.04]. Representative tracings of heart rate changes in the sham-operated and the infarcted rats subjected to stress are shown in . As shown in CitationFigure 3, the maximum tachycardic response elicited by application of air jet stress during i.c.v. infusion of aCSF was significantly greater in the infarcted than in the sham rats (P < 0.05, Tukey test). The maximum changes in heart rate in the infarcted VP and sham VP groups did not differ, nor were significant differences found between the maximum changes of heart rate in the infarcted V1ANT and the sham V1ANT groups. One-way ANOVA, performed on air jet stress-induced maximum changes in heart rate in the infarcted and sham-operated rats separately, revealed significant differences in the infarcted rats [F(2,21) = 8.18; P < 0.002], but not in the sham-operated rats. Specifically, significant differences were found between the infarcted aCSF and infarcted V1ANT groups (P < 0.003, Tukey test), and between the infarcted VP and infarcted V1ANT groups (P < 0.01, Tukey test).
Latency to stress responses
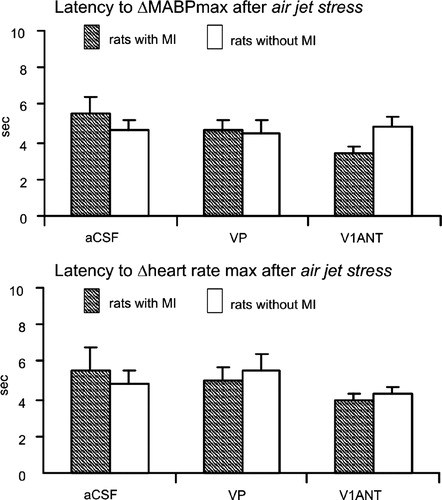
The time zero to peak latencies for the air jet stress-induced maximum increases in MABP and for the maximum increases in heart rate in the infarcted and sham-operated rats did not differ by two way ANOVA. Neither were significant differences found during i.c.v. infusions of aCSF, VP and V1ANT when MABP and heart rate responses to air jet stress were analyzed separately in the sham-operated and the infarcted rats by one way ANOVA (.)
Figure 4 The time zero-peak latency between application of air jet stress and MABP and heart rate during i.c.v. infusions of cerebrospinal fluid (aCSF), VP and VP V1ANT. Number of rats in the group: sham CSF—9, sham VP—8, sham V1ANT—8, infarct CSF—8, infarct VP—8, infarct V1ANT—8.
Discussion
A major new finding of the present study is a demonstration that the myocardial infarction results in enhanced involvement of the brain VP V1 receptors in the regulation of blood pressure. Specifically, we have found that in the infarcted rats blockade of V1 VP receptors in the brain caused significant reduction of the baseline MABP and abolished potentiation of the cardiovascular responses to an alarming air jet stress. These effects were not present in the sham-operated rats.
Recent investigations have provided evidence that myocardial infarction provokes significant changes in the central control of the cardiovascular system, as manifested by increased renal sympathetic nerve activity and an enhanced stimulation of the brain angiotensin 1 (AT1) and mineralocorticoid receptors during the post-infarct state (DiBona et al. Citation1995; Leenen et al. Citation1999; Zhang et al. Citation1999; Francis et al. Citation2001; Zhang et al. Citation2002; Felder et al. Citation2003; Zhu et al. Citation2004). The present study extends these observations by uncovering an increased role of brain VP in regulation of blood pressure and heart rate in the infarcted rats.
Post mortem examination of the heart in the infarcted rats revealed that the infarct size amounted to 35.6% of the total surface of the left ventricle wall. According to the data reported by the other authors (Leenen et al. Citation1999; Zhu et al. Citation2004) a MI of similar size significantly increases the left ventricle end-diastolic pressure. We have measured the left ventricle diastolic pressure at the end of experiments in some of our rats and found that it was significantly greater in the infarcted (15–20 mm Hg) than in the sham-operated rats (1–3 mm Hg). Thus, it can be assumed that the MI resulted in development of heart failure in the present study. The MI was also associated with hypertrophy of the right ventricle. This finding is in agreement with the data reported by other authors (Leenen et al. Citation1999; Zhang et al.Citation1999) for infarcts of similar size and suggests overloading of the right ventricle, most likely due to congestion of the pulmonary circulation caused by left ventricle failure. The left ventricle weight in the infarcted rats was not changed 5 weeks after infarction as compared to the sham rats. Similarly, it was not different in the study of Zhang et al. (Citation1999) at 4 week after infarction, although it was increased at 8 week after ligation of the coronary artery.
The baseline MABP found in our study at the beginning of the terminal experiments did not differ in the infarcted and in the sham-operated rats. It was lower than in the rats with MI in the study of DiBona et al. (Citation1995) but higher than in the infarcted rats in the study of Zhang et al. (Citation1999). Intracerebroventricular administration of a specific V1 receptor antagonist under resting conditions significantly decreased the baseline MABP in the infarcted rats, but had no influence on haemodynamic parameters in the sham operated animals. Absence of a significant hypotensive effect after blockade of brain V1 VP receptors in the sham-operated rats is in accordance with our previous studies (Budzikowski et al. Citation1996; Szczepanska-Sadowska et al. Citation1998; Paczwa et al. Citation1999). It is interesting that in spite of an apparently greater role of activation of V1 receptors in regulation of blood pressure in the infarcted rats as compared to the sham rats the baseline blood pressure was similar in both groups. Maintenance of the baseline blood pressure at a similar level in the infarcted and the sham rats was possibly due to activation of compensatory mechanisms. In this regard, the central pressor effect of VP is effectively buffered by the opposite action of nitric oxide and atrial natriuretic peptide in the brain (Stępniakowski et al. Citation1994; Paczwa et al. Citation1997). The question arises whether the hypotensive effect of i.c.v. applied V1ANT observed in the infarcted rats could be caused by leakage of the peptide into the systemic circulation. This does not appear to be the case because in our previous study (Budzikowski et al. Citation1996) we found that i.c.v. administration of a similar V1 antagonist [d(Et2)Tyr(Me)]DVP at an equivalent rate (based on the peripheral pressor effectiveness of both peptides), abolishes the increase of blood pressure following the suprathreshold dose of i.c.v. administered VP, but does not influence the magnitude of the pressor response to the suprathreshold dose of intravenously applied VP (see also “Methods” section). The present study discloses also an increased sensitivity of the infarcted rats to the pressor effect of i.c.v. administration of VP as indicated by significant differences in changes of MABP between the infarcted VP and infarcted aCSF groups and between infarcted VP and infarcted V1ANT groups. These differences were absent in the sham groups.
The most interesting finding of the present study is a demonstration of a significant involvement of brain VP in generation of the exaggerated responses of the cardiovascular system to stress. Increased pressor and tachycardic responses after application of air jet stress were also described in the study of Zhang et al. (Citation1999). However, in the latter study, the experimental design was different because the air jet was blown onto the rat's mouth and therefore could stimulate also nasal mechanoreceptors. Our results provide evidence that blockade of V1 receptors in the brain normalises the exaggerated cardiovascular responses to stress in the infarcted rats. For the heart an inevitable consequence of stress-induced increase in heart rate and MABP is a sudden increase in work load. For the infarcted heart this may cause a significant mismatch between the oxygen requirements and supply and may be therefore considered as a detrimental effect. Although, it is known that stress can accelerate development of fatal cardiovascular disorders in patients suffering from coronary artery disease (Kario et al. Citation2003; Fleet et al. Citation2005), the mechanisms underlying this negative effect are only poorly understood. In particular, little is known about whether the MI may result in chronic resetting of regulation of the cardiovascular system by the brain neurotransmitters involved in generation of behavioural and cardiovascular responses to stress. In the study of Zhang et al. (Citation2002), the exaggerated cardiovascular responses to air jet in the infarcted rats were abolished by chronic i.c.v. administration of the AT1 receptor antagonist losartan. However, it is known that angiotensin II stimulates release of VP by paraventricular nucleus (PVN) neurons (Veltmar et al. Citation1992) and that it mediates central cardiovascular effects of angiotensin II (Haack and Möhring Citation1978; Łoń et al. Citation1996). Therefore, it is likely that activation of AT1 receptors on VP neurons by angiotensin II is a necessary step for potentiation of the cardioacceleratory and pressor responses to stress. Interestingly, in the present study central administration of exogenous VP did not markedly modify the pressor and tachycardic responses to stress either in the sham or in the infarcted rats. This may indicate that both in the sham and in the infarcted rats endogenous VP exerts a maximum stimulatory effect on V1 receptors located on neurons involved in regulation of MABP and heart rate during stress. In this context, the results with administration of V1ANT under resting conditions and during stress may suggest that the number, occupancy or effective coupling of V1 receptors in the brain may be greater in the infarcted than in the sham rats. Involvement of V1 receptors in intensification of the cardiovascular responses to the alarming stress in the infarcted rats appears to be of particular interest in view of the data showing that VP is an important neuromodulator of behavioural responses to stress (Holmes et al. Citation2003; Landgraf Citation2003; Bielsky et al. Citation2004; Wigger et al. Citation2004). A growing number of experimental studies provide substantial evidence for a significant role of VP parvocellular neurons, and of V1a and V1b receptors in arousing an anxiogenic and depression-like behaviour in animals (Carrasco and van de Kar Citation2003; Landgraf Citation2003; Bielsky et al. Citation2004; Wigger et al. Citation2004). There is also substantial evidence for a profound reorganisation of the brain VP system in humans suffering from depressive disorders, as manifested by elevated levels of VP in CSF and the PVN and by an increased number of V1 receptors in the hypothalamus (Purba et al. Citation1996; Griebel et al. Citation2003; Holmes et al. Citation2003). Parvocellular VP neurons of the PVN innervate the stressor-processing regions of the amygdala, septum, bed nucleus of the stria terminalis and cortex (Holmes et al. Citation2003). Also, PVN VP neurones send projections to the cardiovascular control regions of the brain (Bujis and Kalsbeck Citation1995; Holmes et al. Citation2003).
The question arises of how selective was blockade of V1 receptors by the V1antagonist used in the present study. It is known that d(CH2)5[Tyr(Me)2,Ala-]AVP is an effective antagonist of V1a receptors, however, its V1b properties have not been evaluated (Manning et al. Citation1992 and Manning, personal communication). It has been also established that d(CH2)5[Tyr(Me)2,Ala-
]AVP has much greater selectivity for V1 receptors than for V2 receptors, its anti-vasopressor effective dose being about 590 times less than the anti-antidiuretic dose (Manning et al. Citation1992). In addition, our previous study questions a role for brain V2 receptors in the regulation of cardiovascular functions in the rat (Budzikowski et al. Citation1996). Antioxytocic properties of d(CH2)5[Tyr(Me)2,Ala-
]AVP are not known (Manning, personal communication). Thus, it cannot be excluded that d(CH2)5[Tyr(Me)2,Ala-
]AVP could interact to some extent with the oxytocin receptor. Previous studies on oxytocin knockout mice (Bernatova et al. Citation2004) and the effects of stimulation and blockade of oxytocin receptors in the brain stem of the rat suggest that oxytocin may reduce pressor responses to stress and increase baroreceptor gain (Higa et al. Citation2002). Therefore, it does not seem likely that reduction of the pressor and heart rate responses to stress after i.c.v. administration of d(CH2)5[Tyr(Me)2,Ala-
]AVP in the present study could result from blockade of oxytocin receptors. On the other hand, it cannot be excluded that simultaneous nonselective blockade of oxytocin receptors by d(CH2)5[Tyr(Me)2,Ala-
]AVP could attenuate hypotensive effects of blockade of V1 receptors. Thus, although our results provide evidence for an enhanced involvement of brain V1a receptors in regulation of cardiovascular parameters in the infarcted rats, an interaction of V1b or oxytocin receptors with this effect cannot be excluded at the present stage.
Several mechanisms may account for activation of the brain VP system in the infarcted rats. It may be caused by neurogenic or neurohormonal signals arising from the ischaemic heart, from the other parts of the cardiovascular system or from other organs that are inadequately perfused. The MI results also in an activation of the systemic and brain renin-angiotensin-aldosterone systems (Leenen et al. Citation1999; Zhang et al. Citation1999; Francis et al. Citation2001; Zhang et al. Citation2002; Felder et al. Citation2003; Zhu et al. Citation2004). Angiotensin II is one of the most potent stimulators of VP release (Haack and Möhring Citation1978; Veltmar et al. Citation1992), while mineralocorticoids increase expression of VP receptors in the brain (Swords et al. Citation1991). Myocardial infarction is also associated with an increased release of cytokines (Ono et al. Citation1998; Francis et al. Citation2004), which potentially may be involved in stimulation of VP synthesis and enhanced release in the brain (Pardy et al. Citation1993; Wilkinson et al. Citation1994). At present, it is difficult to speculate what is the primary cause of an increased involvement of the brain VP system in regulation of the cardiovascular responses to stress in the post-infarct state. It can only be hypothesised that signals generated in peripheral receptors by the post-infarct stimuli and/or action of some neurohumoral factors may finally affect the same vasopressinergic neurons that are involved in the central regulation of cardiovascular responses to stress.
In conclusion, the results of our investigation demonstrate an enhanced involvement of brain VP V1 receptors in regulation of the cardiovascular system after the myocardial infarction. This is manifested by a greater role of central VP V1a receptors and of centrally released VP in the maintenance of arterial blood pressure at a resting level and in the generation of exaggerated cardiovascular responses to an alarming, emotional stressor in the infarcted rats. Further studies are needed to identify the specific groups of neurones responsible for enhanced involvement of V1a receptors in cardiovascular responses in the infarcted rats as well as any role of the other types of VP receptors and oxytocin receptors in these responses.
Acknowledgements
The authors wish to express their gratitude to Professor Maurice Manning from the Department of Biochemistry and Molecular Biology, Medical College of Ohio, Toledo, for the generous supply of VP antagonist used in the present study, to Mrs Barbara Nowacka, Marzanna Tkaczyk and Mr Marcin Kumosa for their skilful technical assistance and to Dr Bogdan Sadowski for his expert help in the statistical analysis of the data. This study was supported by a grant from the Medical University of Warsaw (1MAW2/2002–2004), the State Committee for Scientific Research in Poland (KBN, grant 4P05A 074) and Centre of Excellence CICERO.
References
- Bernatova I, Rigatto KV, Key MP, Morris M. Stress-induced pressor and corticosterone responses in oxytocin-deficient mice. Exp Physiol 2004; 89: 549–557
- Bielsky IF, Hu S‐B, Szegda KL, Westphal H, Young LJ. Profound impairment in social recognition and reduction in anxiety-like behavior in vasopressin V1a receptor knockout mice. Neuropsychopharmacology 2004; 29: 483–493
- Budzikowski AS, Paczwa P, Szczepanska-Sadowska E. Central V1 AVP receptors are involved in cardiovascular adaptation to hypovolemia in WKY but not in SHR. Am J Physiol Heart Circ Physiol 1996; 271: H1057–H1064
- Bujis RM, Kalsbeck A. Anatomy and physiology of vasopressin pathways: From temperature regulation to corticotrophin-inhibiting neurotransmission. Neurohypophysis. Recent progress of vasopressin and oxytocin research. Elsevier, Amsterdam 1995; 57–65
- Burrell LM, Risvanis J, Johnston CI, Naitoh M, Balding LC. Vasopressin receptor antagonism—a therapeutic option in heart failure and hypertension. Exp Physiol 2000; 85: 259S–265S
- Carrasco GA, van de Kar LD. Neuroendocrine pharmacology of stress. Eur J Pharmacol 2003; 463: 235–272
- Clarke SP, Frasure-Smith N, Lesperance F, Bourassa MG. Psychosocial factors as predictors of functional status at 1 year patients with left ventricular dysfunction. Res Nurs Health 2000; 23: 290–300
- Curran-Everett D, Benos DJ. Guidelines for reporting statistics in journals published by the American Physiological Society. Physiol Genomics 2004; 18: 249–251
- Demotes-Mainard J, Chauveau J, Rodriguez F, Vincent JD, Paulain DA. Septal release of vasopressin in response to osmotic, hypovolemic and electrical stimulation in rats. Brain Res 1986; 381: 314–321
- DiBona GF, Jones SJ, Brooks VL. Ang II receptor blockade and arterial baroreflex regulation of renal nerve activity in cardiac failure. Am J Physiol Regul Integr Comp Physiol 1995; 269: R1189–R1196
- Felder RB, Francis J, Zhang Z-H, Wie S-G, Weiss RM, Johnson AK. Heart failure and the brain: New perspectives. Am J Physiol Regul Integr Comp Physiol 2003; 284: R259–R276
- Fleet R, Lesperance F, Arsenault A, Gregoire J, Lavoie K, Laurin C, Harel F, Burelle D, Lambert J, Beitman B, Frasure-Smith N. Myocardial perfusion study of panic attacks in patients with coronary artery disease. Am J Cardiol 2005; 96: 1064–1068
- Francis J, Weiss RM, Wei S-G, Johnson AK, Beltz TG, Zimmerman K, Felder RB. Central mineralocorticoid receptor blockade improves volume regulation and reduces sympathetic drive in heart failure. Am J Physiol Heart Circ Physiol 2001; 281: H2241–H2251
- Francis J, Zhang Z-H, Weiss RM, Felder R. Neural regulation of the proinflamatory cytokine response to acute myocardial infarction. Am J Physiol Heart Circ Physiol 2004; 287: H791–H797
- Góźdź A, Szczepanska-Sadowska E, Maśliński W, Kumosa M, Szczepanska K, Dobruch J. Differential expression of vasopressin V1a and V1b receptors mRNA in the brain of renin transgenic TGRmRen(2)27 and Sprague Dawley rats. Brain Res Bull 2003; 59: 399–403
- Griebel G, Simiand J, Stemmelin J, Serradeil L, Gal C, Steinberg R. The vasopressin V1b receptor as a therapeutic target in stress-related disorders. Curr Drug Targets CNS Neurol Disord 2003; 2: 191–200
- Grippo AJ, Beltz TG, Johnson AK. Behavioral and cardiovascular changes in the chronic mild stress model of depression. Physiol Behav 2003; 78: 703–710
- Grippo AJ, Santos CM, Johnson RF, Beltz TG, Martins JB, Felder RB, Johnson AK. Increased susceptibility to ventricular arrhythmias in a rodent model of experimental depression. Am J Physiol Heart Circ Physiol 2004; 286: H619–H626
- Haack D, Möhring J. Vasopressin mediated blood pressure response to intraventricular injection of angiotensin II in the rat. Pflügers Arch 1978; 373: 167–173
- Higa KT, Mori E, Viana FF, Morris M, Michelini LC. Baroreflex control of heart rate by oxytocin in the solitary-vagal complex. Am J Physiol Regul Integr Comp Physiol 2002; 282: R537–R545
- Holmes A, Heilig M, Rupniak NMJ, Steckler T, Griebel G. Neuropeptide systems as novel therapeutic targets for depression and anxiety disorders. Trends Pharmacol Sci 2003; 24: 580–588
- Kario K, McEwen BS, Pickering TG. Disasters and the heart: A review of the effects of earthquake induced stress on cardiovascular disease. Hypertens Res 2003; 26: 355–367
- Lampert R, Jain D, Burg MM, Batsford WP, McPherson CA. Destabilizing effects of mental stress on ventricular arrhythmia in patients with implantable cardioverter-defibrillators. Circulation 2000; 101: 158–164
- Landgraf R. HAB/LAB rats: An animal model of extremes in trait anxiety and depression. Clin Neurosci Res 2003; 3: 239–244
- Leenen FHH, Yuan B, Huang BS. Brain “ouabain” and angiotensin II contribute to cardiac dysfunction after myocardial infarction. Am J Physiol Heart Circ Physiol 1999; 277: H1786–H1792
- Lesperance F, Frasure-Smith N, Talajic M, Bourassa MG. Five-year risk of mortality in relation to initial severity and one-year changes in depression symptoms after myocardial infarction. Circulation 2002; 105: 1049–1053
- Łoń S, Szczepańska-Sadowska E, Szczypaczewska M. Evidence that centrally released arginine vasopressin is involved in central pressor action of angiotensin II. Am J Physiol Heart Circ Physiol 1996; 270: H167–H173
- Ludbrook J. Repeated measurements and multiple comparisons in cardiovascular research. Cardiovasc Res 1994; 28: 303–311
- Manning M, Stoev S, Bankowski K, Misicka A, Lammek B, Wo NC, Sawyer WH. Synthesis and some pharmacological properties of potent and selective antagonists of the vasopressor (V1-receptor) response to arginine vasopressin. J Med Chem 1992; 35: 382–388
- Muders F, Riegger GA, Bahner U, Palkovits M. The central vasopressinergic system in experimental left ventricular hypertrophy and dysfunction. Prog Brain Res 2002; 139: 275–279
- Ono K, Matsumori A, Shioi T, Furokawa Y, Sasayama S. Cytokine gene expression after myocardial infarction in rat hearts: Possible implication in left ventricular remodeling. Circulation 1998; 98: 149–156
- Paczwa P, Budzikowski AS, Szczepanska-Sadowska E. Enhancement of central pressor effect of AVP in SHR and WKY rats by intracranial NG-nitro-l-arginine. Brain Res 1997; 748: 51–61
- Paczwa P, Szczepańska-Sadowska E, łoń S, Ganten U, Ganten D. Role of central AT1 and V1 receptors in cardiovascular adaptation to hemorrhage in SD and renin TGR rats. Am J Physiol Heart Circ Physiol 1999; 276: H1918–H1926
- Pardy K, Murphy D, Carter D, Hui KM. The influence of interleukin-2 on vasopressin and oxytocin gene expression in the rodent hypothalamus. J Neuroimmunol 1993; 42: 131–138
- Purba JS, Hoogendijk WJ, Hofman MA, Swaab DF. Increased number of vasopressin- and oxytocin-expressing neurons in the paraventricular nucleus of the hypothalamus in depression. Arch Gen Psychiatry 1996; 53: 137–143
- Rascher W, Lang RE, Unger T, Ganten D, Gross F. Vasopressin in brain of spontaneously hypertensive rats. Am J Physiol Heart Circ Physiol 1982; 242: H496–H499
- Selye H, Bajusz E, Grasso S, Mendell P. Simple techniques for the surgical occlusion of coronary vessels in the rat. Angiology 1960; 11: 398–407
- Sladek CD, Devine MA, Felten SY, Aravich PF, Blair ML. Abnormalities in hypothalamic and neurohypophysial vasopressin content are not a consequence of hypertension in the spontaneously hypertensive rat. Brain Res 1988; 445: 39–46
- Stępniakowski K, Budzikowski A, Łoń S, Szczepańska-Sadowska E. Central cardiovascular effects of AVP and ANP in normotensive and spontaneously hypertensive rats. J Auton Nerv Syst 1994; 47: 33–43
- Swords BH, Wyss JM, Berecek KH. Central vasopressin receptors are upregulated by deoxycorticosterone acetate. Brain Res 1991; 559: 10–16
- Szczepanska-Sadowska E, Gray D, Simon-Opperman C. Vasopressin in blood and third ventricle aCSF during dehydration, thirst, and hemorrhage. Am J Physiol Regul Comp Physiol 1983; 245: R549–R555
- Szczepanska-Sadowska E, Paczwa P, Łoń S, Ganten D. Increased pressor function of central vasopressinergic system in hypertensive renin transgenic rats. J Hypertens 1998; 16: 1505–1514
- Veltmar A, Culman J, Quadri F, Rascher W, Unger T. Involvement of adrenergic and angiotensinergic receptors in the paraventricular nucleus in the angiotensin II-induced vasopressin release. J Pharmacol Exp Ther 1992; 263: 1253–1260
- Wigger A, Sanchez MM, Mathys KC, Ebner K, Frank E, Liu D, Kresse A, Neumann ID, Holsboer F, Plotsky PM, Landgraf R. Alterations in central neuropeptide expression, release and receptor binding in rats bred for high anxiety: Critical role of vasopressin. Neuropsychopharmacology 2004; 29: 1–14
- Wilkinson MF, Horn TF, Kasting NW, Pittman QJ. Central interleukin-1 beta stimulation of vasopressin release in the rat brain: Activation of an antipyretic pathway. J Physiol 1994; 481: 641–646
- Zhang W, Huang BS, Leenen FH. Brain renin-angiotensin system and sympathetic hyperactivity in rats after myocardial infarction. Am J Physiol Heart Circ Physiol 1999; 276: H1608–H1615
- Zhang Z-H, Francis J, Weiss RM, Felder RB. The renin-angiotensin-aldosterone system excites hypothalamic paraventricular nucleus neurons in heart failure. Am J Physiol Heart Circ Physiol 2002; 283: H423–H433
- Zhu G-Q, Gao L, Li V, Patel KP, Zucker IH, Wang W. AT1 receptor mRNA antisense normalizes enhanced cardiac sympathetic afferent reflex in rats with chronic heart failure. Am J Physiol Heart Circ Physiol 2004; 287: H1828–H1835