
Abstract
Genetic variations in the genes encoding G protein-coupled receptors (GPCRs) can disrupt receptor structure and function, which can result in human genetic diseases. Disease-causing mutations have been reported in at least 55 GPCRs for more than 66 monogenic diseases in humans. The spectrum of pathogenic and likely pathogenic variants includes loss of function variants that decrease receptor signaling on one extreme and gain of function that may result in biased signaling or constitutive activity, originally modeled on prototypical rhodopsin GPCR variants identified in retinitis pigmentosa, on the other. GPCR variants disrupt ligand binding, G protein coupling, accessory protein function, receptor desensitization and receptor recycling. Next generation sequencing has made it possible to identify variants of uncertain significance (VUS). We discuss variants in receptors known to result in disease and in silico strategies for disambiguation of VUS such as sorting intolerant from tolerant and polymorphism phenotyping. Modeling of variants has contributed to drug development and precision medicine, including drugs that target the melanocortin receptor in obesity and interventions that reverse loss of gonadotropin-releasing hormone receptor from the cell surface in idiopathic hypogonadotropic hypogonadism. Activating and inactivating variants of the calcium sensing receptor (CaSR) gene that are pathogenic in familial hypocalciuric hypercalcemia and autosomal dominant hypocalcemia have enabled the development of calcimimetics and calcilytics. Next generation sequencing has continued to identify variants in GPCR genes, including orphan receptors, that contribute to human phenotypes and may have therapeutic potential. Variants of the CaSR gene, some encoding an arginine-rich region that promotes receptor phosphorylation and intracellular retention, have been linked to an idiopathic epilepsy syndrome. Agnostic strategies have identified variants of the pyroglutamylated RF amide peptide receptor gene in intellectual disability and G protein-coupled receptor 39 identified in psoriatic arthropathy. Coding variants of the G protein-coupled receptor L1 (GPR37L1) orphan receptor gene have been identified in a rare familial progressive myoclonus epilepsy. The study of the role of GPCR variants in monogenic, Mendelian phenotypes has provided the basis of modeling the significance of more common variants of pharmacogenetic significance.
Table of contents
Introduction 2Pathogenic GPCR gene variants in disease 3Genetic variants contributing to diseases of the retina 5Rhodopsin biochemistry 5Pathogenesis of retinitis pigmentosa 5Night blindness 5Functional genomics 5Potential therapies 6Oguchi disease: GRK1 and rhodopsin 6Thyroid stimulating hormone receptor variants 6Activating TSHR variants in hyperfunctioning thyroid disorders 6Thyroid phenotype variability 6Grave’s disease 7Calcium-sensing receptor variants 7Neonatal severe hyperparathyroidism and familial hypocalciuric hypercalcemia 7Hypocalcemia, hypoparathyroidism, and hypocalcemic hypercalciuria 10Biased signaling and endosomal signaling 11Polymorphisms and non-calcitropic disease 12Parathyroid hormone receptor and bone metabolism 13GPCR variants and obesity 14Melanocortin 4 receptor variants 14Variants in the MC4R accessory protein MRAP2 15Pharmacological targeting of MC4R 15Familial glucocorticoid deficiency 16Follicle stimulating hormone 16Luteinizing hormone/choriogonadotropin receptor variants 17Constitutive LH receptor variants 17Gonadotropin releasing hormone receptor variants 17Idiopathic hypogonadotropic hypogonadism 18Drug development 18Variants identified by next generation sequencing 18Vasopressin V2 receptor variants 18Arginine vasopressin resistance 19Loss of function phenotype 19Population genetics 19Expression studies 20Autosomal recessive AVP-R 20Pharmacology 20AVPR2 and nephrogenic syndrome of inappropriate antidiuresis 21Endothelin and Hirschsprung’s disease 21Purinergic receptors and clotting 21GPCRs and monogenic asthma 22Orphan receptors 22Conclusions 22
Introduction
Genetic variation in G protein-coupled receptor (GPCR) genes has been associated with a spectrum of phenotypes, from altered drug responses and altered susceptibility to disease to Mendelian disease [Citation1,Citation2]. To date, pathogenic variants in at least 55 GPCR genes are known to be disease-causing (), and missense variants have been identified in 108 GPCR drug targets, with many known to be of pharmacological importance [Citation3]. Here we discuss recurrent pathogenic variants, mutations that are transmitted within families affected by disease, and compare them with de novo variants that may be very rare or lethal. Each naturally-occurring GPCR variant identified provides a window of opportunity for the study of receptor function that complements in vivo data on GPCR pharmacology and drug discovery. The signaling characteristics of mutant GPCRs can provide insight into the structure and function of variants associated with a phenotype that is primarily pharmacogenetic [Citation2–5].
Figure 1. Number of known disease mutations in GPCRs. Data obtained from DisGeNET v 7.0 on all missense variants with a level of evidence greater than or equal to 0.8.
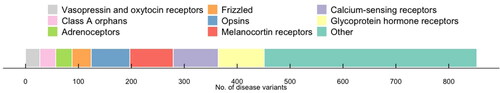
The study of variant GPCRs is of continuing relevance because there are more than 84 Class A orphan receptors for which the endogenous activator remains unknown [Citation4]. Based on data on GPCRs of known function, however, we know that recurrent variants may contribute to the polygenic risk for a complex phenotype and are less likely to result in monogenic disorders. Many variants are of pharmacogenetic relevance because they contribute to an altered drug response phenotype. For example, variants of the calcium-sensing receptor (CaSR) may be associated with monogenic disease, altered pharmacology or variants of uncertain significance (VUS) when they are not demonstrably benign [Citation6]. By contrast with variants associated with monogenic disease, pharmacogenetic variants may result in hypermorphic (partial gain), hypomorphic (partial loss), or biased signaling of a receptor where activation results in a bias toward other pathways. Alterations of efficacy or potency of the variant receptor compared to the wild-type receptor in pharmaco-genetics, therefore, suggests a continuum with those mutated in disease.
Pathogenic GPCR gene variants in disease
Expression of variant GPCRs in vitro has been shown to result in a spectrum of pathogenic and likely pathogenic variants. On one extreme, these include loss of function (LOF) variants that decrease receptor signaling. In some cases, LOF variants result in a dominant negative phenotype in which their expression in heterozygous individuals serves to impair the function of the wild-type receptor [Citation7]. A proposed and viable classification system for LOF variants, based on the life cycle of GPCRs, includes five subtypes: (1) defective GPCR synthesis; (2) defective trafficking to the cell surface, often resulting in retention within the endoplasmic reticulum (ER); (3) defective ligand binding; (4) defective receptor activation; and (5) mutants with no known effects or VUS [Citation7]. We discuss mutant GPCRs associated with disease in the context of these variant classifications as well as pharmacological interventions that may target each. These include pharmacological chaperones, which are small molecules that stabilize insufficient or misfolded receptors that would otherwise not reach the cell membrane [Citation8].
On the other extreme, gain of function (GOF) variants can result in constitutively activating mutations (CAMs) with autonomous signaling. The pharmacogenetics of GPCRs will be discussed in the context of altered response to ligands, and signaling will be discussed in the context of clinical efficacy. Variants identified in class A, B, C, and F GPCR families have been modeled on earlier in vitro studies [Citation1,Citation3–5]. The first naturally occurring CAM that causes disease in humans was discovered in rhodopsin, and it is discussed as the prototypical pathogenic variant of this class [Citation9].
The extreme receptor states of LOF and GOF, depicted schematically in , result in changes in ligand binding, coupling to G protein, GPCR desensitization, and GPCR trafficking [Citation2,Citation7–9]. Understanding the role of variant GPCRs in human genetic disease has led to drug discovery strategies that target variant GPCRs [Citation2–4]. Here, we discuss the wide variety of GPCR gene variants and signaling disruptions that have been shown to be associated with monogenic disease phenotypes [Citation1–9]. Knowledge of GPCR genes helps to define: (1) the genomic, proteomic, and signaling properties of the many GPCRs available for drug discovery using “reverse pharmacology”; (2) the signaling changes that result from pathogenic and likely pathogenic variants in identified GPCRs; and (3) the treatment implications for patients with pathogenic variants in GPCR genes. Selected examples of clinically actionable pathogenic and likely pathogenic variants (mutations) that are associated with human genetic disease are presented in .
Figure 2. Gain of function (GOF) and loss of function (LOF) mutations in GPCRs. General mechanisms of GOF mutations in GPCRs that include altered receptor selectivity for a specific probe, a gain or change in the preferred signaling pathway triggering constitutive activity, increased binding and/or signaling kinetics, a change in the ligand-modality, as well as increased transportation or recycling may all lead to an amplified receptor activity capacity. On the other hand, LOF mutations in GPCRs may result in an overall disruption or decrease of binding, potency, efficacy, or expression parameters including alterations in receptor stability, folding, trafficking, or the ability to interact with other scaffolding or signaling proteins.
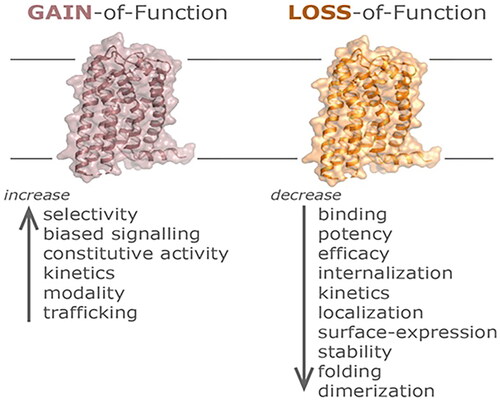
Table 1. Selected G protein-coupled receptors (GPCRs) associated with human genetic disease.
Genetic variants contributing to diseases of the retina
Studies of GPCR variants began with examining the properties of the prototypical GPCR, rhodopsin, in diseases of the retina. The study of rhodopsin sequence variants in diseases of the retina is nearing its fifth decade. The most common causes of retinitis pigmentosa (RP) are the more than 150 rhodopsin variants that have been identified in autosomal dominant RP, and less frequently, in autosomal recessive RP, because the rhodopsin p.Pro23His substitution was found to be pathogenic in disease [Citation10–12]. Subsequently, pathogenic rhodopsin variants have been identified in dominant congenital stationary night blindness [Citation11].
While recessive RP tends to be associated with LOF, the dominant form of RP results from constitutive activity and/or dominant negative pathology. Molecular studies have identified distinct consequences to rhodopsin structure and function that are associated with different variants [Citation10–19].
In addition to rhodopsin, variants in at least fifty genes contribute to non-syndromic RP. Variants in a further twelve genes contribute to syndromic RP such as Usher syndrome, and seventeen more harbor pathogenic variants in Bardet-Biedl syndrome [Citation10].
Rhodopsin biochemistry
The study of a specific amino acid sequence variation, the transmembrane spanning helix 3 (TM3) p.Leu125Arg variant, contributed to understanding the extent to which a rhodopsin can desensitize. An experiment in which the amino acid at position 125 of rhodopsin was serially substituted found that smaller residues were better able to activate transducin. For example, substitution of p.Leu125Trp and p.Leu125Tyr variants, which are bulky, was found to elicit little transducin signal. These results suggest that the steric effect of amino acid side chains can inhibit G protein activation [Citation19]. The data on p.Leu125 in rhodopsin’s TM3, located near the 11-cis-retinal binding pocket, helped to determine the chromophore-binding pocket structure [Citation19] of this prototypical GPCR, rhodopsin.
Pathogenesis of retinitis pigmentosa
The discovery of pathogenic rhodopsin CAMs provided the data that was used to form the basis of our understanding of the conformational changes that result in GPCR activation. These studies also identified inverse agonists, a class of ligands that restore basal levels of signaling due to conformational changes that uncouple CAMs from the G protein. For example, 11-cis-retinal, a retinoic acid derivative, is an inverse agonist that acts at rhodopsin [Citation4,Citation5,Citation10].
In addition to the pathology of rhodopsin phosphorylation and desensitization that is characteristic of rhodopsin variants [Citation10,Citation11], variants of G protein-coupled receptor kinase (GRK) genes are implicated in the pathology of RP. In the retina, GRK1 phosphorylation of rhodopsin contributes to rapid desensitizing after exposure to light. While pathogenic rhodopsin variants can constitutively activate the G-protein complex, transducin, they can also result in constitutive GRK1 phosphorylation of rhodopsin. This inappropriate rhodopsin phosphorylation can result in interaction with beta-arrestin, thereby quenching coupling to transducin and inhibiting signaling. These observations were foundational in understanding the reciprocal relationship between GPCR activation and the GRK phosphorylation that facilitates uncoupling from the G protein and resultant quenching of the signal [Citation10].
Night blindness
RP consists of a group of ocular anomalies that include night blindness and retinal dystrophies. There is considerable variability in the phenotype and pathology associated with variants, including the p.Thr4Lys [Citation14], p.Asn15Ser [Citation15], p.Thr17Met, p.Pro23His [Citation16,Citation17], p.Pro23Leu, p.Gln28His, p.Gly90Asp, p.Glu113Gln, p.Ala292Glu, and p.Lys296Glu [Citation10,Citation13]. For example, complete night blindness results from p.Gly90Asp and p.Ala292Glu variants, while RP can result from other rhodopsin variants [Citation13]. Different substitutions at p.Gly90 may result in distinct phenotypes [Citation18].
Functional genomics
Next generation sequencing (NGS) has enabled the identification of VUS in all GPCRs, including rhodopsin. Strategies for distinguishing benign from pathogenic or likely pathogenic variants are important in diagnostics as well as in selecting treatment modalities, including gene therapy. Strategies including deep mutational scanning have been developed to screen libraries of receptor variants expressed in human cell lines with G protein barcoded transcriptional reporters [Citation20]. These methods allow the screening of the human genome for GPCR variants that are relevant to pharmacogenetics.
One strategy that has been reported for characterizing GPCR variant function involves characterizing the surface expression of a rhodopsin variant library [Citation21]. The authors used NGS to inform a flow cytometry-based assay that potentially could be performed on all variants identified in the genome without barcoding. This approach identified variants with decreased expression, including p.Gly18Asp, p.Gly101Val, and p.Pro180Thr, that are transmitted in affected pedigrees [Citation21].
Potential therapies
Since 1998, many gene therapy approaches have been explored in preclinical animal studies. Models of autosomal dominant RP, including transgenic mice and pig models, have been used to evaluate gene knockdown, augmentation, editing, or a combination of these approaches. For example, in the rhodopsin T4R dog model, a replacement strategy with an adeno-associated virus vector has been shown to prevent photoreceptor degeneration.
Experimentally, CRISPR-Cas9 gene editing may be able to eliminate rhodopsin variants; however, ethical constraints make this impractical in human subjects. More practically, while gene augmentation with a wild-type copy of rhodopsin may overcome dominant negative variants, gene knockdown may be more useful for GOF variants. Heterogeneity in autosomal dominant RP and related disorders suggests that a mutation-independent strategy may be more successful if knockdown is combined with gene replacement [Citation13].
Oguchi disease: GRK1 and rhodopsin
In addition to GRK1 variants, autosomal recessive Oguchi disease is a rhodopsin-related disorder associated with night blindness that can result from S-antigen visual arrestin gene (SAG) variants. In the wild-type, the S-antigen quenches the signal of rhodopsin that has been phosphorylated upon photoactivation; however, its variants result in a disruption of phototransduction recovery. For example, nonsense variants in the SAG gene (e.g. c.916G>T; p.Glu306*) have been reported to impair GRK1-mediated desensitization of rhodopsin [Citation22]. SAG variants [Citation22], similar to variants of other genes [Citation23], disrupt the quenching associated with light induced phosphorylation [Citation23]. This reflects the fact that disruption of GPCR accessory proteins can result in disease phenotypes that resemble receptor dysregulation.
Thyroid stimulating hormone receptor variants
The thyroid stimulating hormone receptor (TSHR) is integral to the regulation of T3 and T4 production and secretion through a thyroid stimulating hormone (TSH) feedback loop. Activating and inactivating variants of the genes, TSH and TSHR, are associated with many cases of thyroid disease. The TSHR variants are expressed mainly on thyroid follicular cells. Variants of TSHR alter TSH signaling because they decrease Gαs adenylyl cyclase stimulation. The dysregulation of the TSH function resulting from disrupted TSHR is associated with abnormal growth of the cells responsible for secreting thyroid hormone.
For example, variants of the TSHR located in domains are reported to activate the receptor protein. By contrast, variants in other portions of the TSHR have been reported in somatic variants of multinodular goiter [Citation51–58]. The p.Gly431Ser position of TSHR, for example, has been reported to be constitutively active [Citation54].
Activating TSHR variants in hyperfunctioning thyroid disorders
Goiter refers to athyroid gland enlargement that can be due to a number of causes, including dietary iodine deficiency. Diffuse, multi-nodal goiter refers to a thyroid gland that is diffusely hyperplastic and overproduces thyroid hormone. Activating “germline” variants of TSHR cause non-autoimmune autosomal dominant hyperthyroidism. Somatic TSHR variants can be found in functioning thyroid adenoma.
Toxic thyroid hyperplasia, or non-autoimmune autosomal dominant hyperthyroidism, has been associated with similar variants [Citation51,Citation52,Citation54,Citation58,Citation59]. For example, TSHR variants have been identified in TM3 (p.Val509Ala) and TM7 (p.Cys672Thr), as well as the carboxyl tail (p.Asp727Glu) [Citation60]. The effects of these germ-line variants are similar with respect to somatic variants found in hyperfunctioning thyroid adenoma [Citation51,Citation59].
Although pathogenically heterogeneous, toxic multinodular goiter can result in hyperthyroidism. Further complicating the molecular diagnostics is the fact that activating variants of Gαs and the TSHR are reported in goiter. These variants are associated with autonomously hyperfunctioning thyroid adenomas [Citation52] and many non-adenomatous hyperfunctioning nodules identified in the thyroid gland of patients with toxic or functionally autonomous multinodular goiter [Citation61].
Thyroid phenotype variability
Even though TSHR variants are generally located within TM domains, the extent of their conservation is associated with phenotypic variability. The broad clinical phenotypes of single amino acid changes such as p.Ser505Arg are at odds with their putative constitutive activity with respect to second messenger production. However, their enhanced agonist sensitivity can be inferred to reflect their role in congenital non-autoimmune hyperthyroidism [Citation62]. In this context, the role of genomic background in modulating the clinical significance of some variants cannot be ignored [Citation53]. The highly variable thyroid phenotypes associated with TSHR variants may reflect the presence of co-repressor defects reported in many patients [Citation63].
Grave’s disease
The hallmark of Graves’ disease (GD) pathogenesis is the presence of activating TSHR antibodies. The association of TSHR variants with GD has been identified from genome-wide association studies. Thyroid autoimmunity associated with GD was narrowed to a 40 kb disease-susceptibility region in intron 1 [Citation64], which was reported to be in linkage disequilibrium with at least five single nucleotide variants (SNVs) [Citation65]. SNVs identified in TSHR intron 1 may result in a reduction of TSHR expression in the thymus, diminished central tolerance and TSHR autoimmunity [Citation65].
With respect to TSHR intron 1 GD-associated SNVs, Brand et al. reported that their putative regulation of mRNA spicing may result in a TSHR A-subunit that is autoantigenic [Citation65]. By contrast [Citation64], in genetically susceptible individuals, intron 1 SNVs may modulate negative selection of TSHR-autoreactive T cells in the thymus. Over expression of the protective allele in the thymus may help overcome central tolerance escape of TSHR-reactive T cells and impact phenotype severity. The expression and pathogenesis of TSHR variants in GD may be impacted by the environmental impact on epigenetic marks [Citation64].
Calcium-sensing receptor variants
The calcium-sensing receptor (CaSR), expressed on parathyroid gland cells secreting parathyroid hormone (PTH) and cells lining the renal tubule, detects extracellular calcium concentration (Ca2+e) [Citation107]. The CaSR is essential in maintaining Ca2+e by sensing changes in blood calcium levels. The conformational changes resulting from Ca2+ binding to the CaSR result in signaling that modifies PTH secretion and renal handling of cations [Citation107]. The calcium set-point (defined as the Ca2+ concentration that half-maximally inhibits PTH secretion) is dependent on CaSR expression and activity [Citation107]
Like the metabotropic glutamate receptors (mGluRs), the gamma aminobutyric acid receptor type B and the taste and odorant receptors, the CaSR is a member of the class C subfamily. Class C GPCRs share two common structural features: they form constitutive homodimers and have extensive extracellular domains (ECDs) comprising a bilobed Venus Flytrap domain (VFTD) that include the orthosteric ligand binding sites, and a cysteine-rich domain (). Structural studies have shown that the CaSR ECD harbors multiple Ca2+ binding sites as well as sites for phosphate ions and L-amino acids including tryptophan [Citation108]. Cryo-electron microscopy (EM) studies suggest that upon activation, the homodimer adopts a conformational asymmetry that prepares one protomer to interact with the G-protein. While calcimimetic drugs stabilize this process, calcilytic drugs lock the CaSR in an inactive symmetric configuration [Citation109].
Figure 3. Above, a schematic showing the major structural regions of the CaSR extracellular domain (ECD). the CaSR forms a functional homodimer (protomer 1 in brown and protomer 2 in gray) comprising a bilobed Venus Flytrap domain (VFTD) and a cysteine-domain (CRD). the VFTD contains the ligand-rich binding sites for Ca2+ and amino acids including LR-Trp. Structural studies indicate there are at least two Ca2+-binding sites in each protomer with the cartoon depicting those identified in the recent cryo-EM structures [Citation109]. Loop 1 and 2 reach across the homodimer interface to contact residues on the opposite protomer. Activating mutations have been identified in several residues within these loops. Both inactivating and activating mutations are concentrated in the region between the two lobes of the VFTD, and the CRD homodimer interface. Left, a recent structural representation of the active site (https://www.rcsb.org/structure/7M3F).
![Figure 3. Above, a schematic showing the major structural regions of the CaSR extracellular domain (ECD). the CaSR forms a functional homodimer (protomer 1 in brown and protomer 2 in gray) comprising a bilobed Venus Flytrap domain (VFTD) and a cysteine-domain (CRD). the VFTD contains the ligand-rich binding sites for Ca2+ and amino acids including LR-Trp. Structural studies indicate there are at least two Ca2+-binding sites in each protomer with the cartoon depicting those identified in the recent cryo-EM structures [Citation109]. Loop 1 and 2 reach across the homodimer interface to contact residues on the opposite protomer. Activating mutations have been identified in several residues within these loops. Both inactivating and activating mutations are concentrated in the region between the two lobes of the VFTD, and the CRD homodimer interface. Left, a recent structural representation of the active site (https://www.rcsb.org/structure/7M3F).](/cms/asset/895970c6-5d1e-41df-9c52-e224c632399c/ilab_a_2286606_f0003_c.jpg)
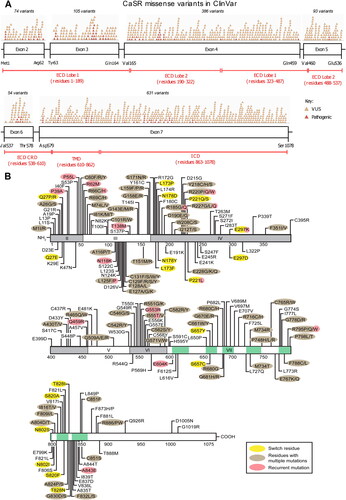
Inherited CaSR gene variants can result in a variety of mineral ion homeostasis disorders. Among individuals with calcium homeostasis disorders, over 460 germline variants have been identified [Citation110]. While inactivating CaSR variants are associated with familial hypocalciuric hypercalcemia type 1 and neonatal severe hyperparathyroidism (NSHPT), activating variants are associated with Bartter’s syndrome type V and autosomal dominant hypocalcemia type 1. outlines the location of selected CaSR variants [Citation111].
Figure 4. Missense mutations in the CaSR. (A) Schema of the CaSR (which has 1,078 amino acids) showing the relationship between exons 2-7 and the portions of the protein they encode. Exons 2-6 and the beginning of exon 7 encode the ECD comprising ∼610 amino acids, exon 7 encodes the TMD of ∼250 amino acids including membrane-spanning helices TM1-TM7, ECL1-ECL3, ICL1-ICL3, and the ICD of ∼200 amino acids. The locations of missense variants reported in ClinVar and annotated in GnomAD (version 4.0.0, October 2023) as either pathogenic (pink) or variants of unknown significance (orange) are shown above the bars representing exons. It should be noted that many ClinVar variants have not been functionally analysed and may represent benign non-disease causing variants. (B) Schema of the CaSR showing missense mutations that have been functionally characterized and therefore are known pathogenic variants. Multiple mutations have been reported at some of these residues. Please see online version for color figure.
ECD: extracellular domain; ECL: extracellular loop; ICD: intracellular domain; ICL: intracellular loop; TM: transmembrane spanning helix; TMD: transmembrane domain.
Neonatal severe hyperparathyroidism and familial hypocalciuric hypercalcemia
Neonatal severe hyperparathyroidism (NSHPT) presents with significant hypercalcemia (Ca2+ exceeding 4.5 mmol/L), skeletal demineralization, failure to thrive, hypotonia, lethargy, respiratory distress, and dehydration [Citation110,Citation112]. Delays in effective treatment, which often requires parathyroidectomy, can lead to a devastating if not fatal, neurodevelopmental disorder [Citation111] known as autosomal dominant familial hypocalciuric hypercalcemia (FHH). FHH is asymptomatic in more than 70% of cases. It consists of long-term elevation of serum Ca2+, high PTH levels and low excretion of Ca2+ from the kidneys [Citation110,Citation112]. Although the extent of hypercalcemia may resemble that found in mild primary hyperparathyroidism, hypercalcemia persists in FHH despite subtotal parathyroidectomy. As a result, in order to avoid unnecessary surgery, it is imperative to distinguish primary hyperparathyroidism and FHH. This can be achieved by genetic testing and assay for calcium/creatinine clearance ratio (less than 0.01 for FHH) and the identification of kidney stones, cardiovascular events, and fractures that more often present in primary hyperparathyroidism. Although FHH is generally considered benign, recurrent pancreatitis can be a rare presentation [Citation113,Citation114].
More than 85% of cases of NSHPT result from homozygous or compound heterozygous variants [Citation110,Citation112]. Heterozygous variants have been reported in fewer cases of NSHPT, within which they cluster in the ECD regions necessary for receptor activation. These include the region between VFTD lobes and the cysteine-rich domain homodimer interface [Citation108–110]. However, variants in these regions are not always associated with severe disease. Moreover, the same genetic variants have been identified in the heterozygous state in some individuals that present with FHH, while other patients develop severe hypercalcemia (e.g. p.Arg185Gln). The interaction of CaSR variants and exposure to maternal vitamin D and Ca2+ in utero, or other genetic variants, may influence disease severity [Citation115] in these patients.
More than 300 different CaSR variants have been associated with the familial hypocalciuric hypercalcemia type 1 (FHH1) [Citation116]. The majority (80%) of these inactivating variants are missense; however, truncation (nonsense or frameshift), indel, and splice-site variants have been reported [Citation110]. FHH1 variants have been identified in the ECD, transmembrane domain (TMD) and intracellular domain (ICD) (see ). Missense CaSR variants cluster in ECD regions involved in dimer formation, ligand-binding and ICL1 (ICL, intracellular loop) regions that are integral to the homodimer interface [Citation108,Citation110]. The location of variants throughout the CaSR is consistent with structural studies showing that the receptor requires conformational rearrangements within multiple regions of the ECD and TMD for ligand-induced activation [Citation108,Citation109].
Homozygous variants have been reported in a few individuals with FHH1 who present earlier with higher serum Ca2+ levels than FHH patients with heterozygous variants [Citation117,Citation118]. By contrast with dominant negative LOFs, homozygous FHH1 variants such as p.Gln159Arg are associated with a milder LOF [Citation117].
FHH is generally a heterogeneous disorder. FHH type 1 results from inactivating heterozygous variants in the Gα11 protein (encoded by GNA11), which impair G protein signaling. FHH type 2 results from variants of the adaptor protein-2 sigma subunit AP2σ (encoded by AP2S1), which enhance CaSR internalization [Citation117,Citation118]. Individuals with AP2σ variants have higher serum Ca2+ levels and may exhibit symptomatic hypercalcemia [Citation118].
Cinacalcet, a CaSR positive allosteric modulator that increases CaSR sensitivity to extracellular Ca2+, has been used to lower serum Ca2+ and PTH in some NSHPT patients, but not in those with more damaging variants [Citation119]. Moreover, cinacalcet may act to normalize serum Ca2+ and PTH, and resolve hypercalcemic symptoms such as vomiting, headaches, fatigue, abdominal pain, and musculoskeletal pain in patients with AP2S1 and GNA11 variants [Citation119]. However, many cases of asymptomatic FHH do not benefit from the use of cinacalcet, and it should be noted that it is not approved by the US Food and Drug Administration for use in pediatric patients.
Hypocalcemia, hypoparathyroidism, and hypocalcemic hypercalciuria
Pathogenic activating CaSR variants are responsible for hypocalcemic disorders including autosomal dominant hypocalcemia and Bartter’s syndrome type V. ADH1-associated CaSR mutations increase the sensitivity of the receptor to extracellular calcium and consequently, patients present with low or inappropriately normal levels of PTH [Citation110,Citation118]. Hyperphosphatemia, hypercalciuria and hypomagnesemia are also reported [Citation120]. More than 70% of ADH patients have hypocalcemic symptoms such as seizures, tetany, carpopedal spasms, and paresthesia. A third of patients exhibit signs of nephrocalcinosis and basal ganglia calcification [Citation110].
Over one hundred CaSR variants have been identified in ADH, most of them missense. Even proportions of these variants are located in the ECD and the TMD [Citation110]. ECD variants are located in three portions of the receptor: the “hinge” between lobes 1 and 2, the homodimer interface, and the loop 2 region involved with inter-protomer dimerization [Citation108,Citation110]. More than twenty variants are located in TM6 and TM7. Cryo-EM studies show that this region undergoes the largest structural rearrangement during the switch from the inactive-to-active state, predicting that ADH variants promote GOF through conformational change [Citation109].
Many CaSR variants associated with ADH are de novo or are inherited in the heterozygous state [Citation110]. Of the two homozygous variants identified in ADH1, one is a large deletion of 80 residues from the intracellular C-terminus that serves to truncate the cytoplasmic tail. Because degradation signals are lost during truncation, the truncated receptors are often found on the cell surface when expressed in cell lines [Citation121]. Another homozygous ADH1 mutation is a missense variant that has been identified as a heterozygous variant in the normal population. In vitro functional analyses showed that expression of the variant receptor results in reduced Ca2+ EC50 (drug concentration resulting in half-maximal response) values, but that simulation of the heterogeneous state by expression of equal amounts of wild-type and mutant receptor have no effect on receptor activity [Citation122]. This highlights the importance of functional investigation of heterozygous variants in patients with calcemic disorders.
Occasionally, ADH1 presents with features (renal salt wasting, metabolic alkalosis and hypokalemia) of Bartter’s syndrome. This heterogenous disorder results from variants that disrupt NaCl transport across the thick ascending limb of the loop of Henle. The consequent loss of magnesium and calcium to the urine may inhibit NaCl reabsorption in the loop of Henle of some individuals with CaSR GOF variants [Citation110].
ADH1 should be distinguished from other forms of hypoparathyroidism because treatment with vitamin D or its metabolites, while benefitting hypocalcemia, may worsen nephrocalcinosis and hypercalciuria [Citation120]. Loss of function of CaSR in the renal tissue may explain the relatively higher degree of calciuria and nephrocalcinosis in ADH1 compared to other forms of hypoparathyroidism. Recombinant PTH and/or thiazide diuretics that lower urinary Ca2+ may provide other treatments. In mouse models, negative allosteric modulators, often referred to as calcilytics, have been shown to increase serum Ca2+ and PTH while reducing urinary Ca2+. In human ADH1, there are data from clinical trials on the efficacy of calcilytics such as NPSP795 and encaleret. For example, PTH levels increased in a dose-dependent fashion and the urinary fractional excretion of calcium was reduced in response to NPSP795. No correlation between in vitro responses to NPSP795 and clinical parameters were identified, indicating that in vitro data cannot predict in vivo responses to calcilytics, and that genetic background or other factors may influence drug responses [Citation117]. A phase 2b open-label trial with encaleret in individuals with ADH1 is currently underway; preliminary results indicate that encaleret increases blood PTH and decreases serum phosphates [Citation117]. ADH is now recognized as a heterogenous disorder because activating variants in Gα11 have been reported in ADH type 2. This is associated with less pronounced hypercalciuria, raising the question of whether the CaSR in the renal tubules couples to G proteins other than G11 [Citation118].
Biased signaling and endosomal signaling
Following stimulation, the CaSR can couple with multiple pathways. While CaSR generally couples to Gαq/11, liberating intracellular calcium (Ca2+i) and activating mitogen-activated protein kinase (MAPK) pathways, coupling to Gαi/o has been described and results in a reduction of cAMP [Citation110,Citation118]. Functional analyses have shown that variants of CaSR can promote signaling bias. While FHH1 variants tend to couple preferentially to MAPK pathways or reduce both Ca2+i and MAPK pathways equally, ADH1 variants tend to favor activation of Ca2+i dependent pathways [Citation123]. Substitutions of different amino acids at specific positions of the CaSR have demonstrated the clustering of FHH and ADH variants. These amino acid “switch residues” are located in critical structures such as the VFTD and TM3-TM6 that are required for receptor activation and G protein coupling. Amino acid substitution can bias signaling toward pathways that act through Ca2+i or MAPK [Citation124].
Autoantibodies to the CaSR have been reported in some cases of acquired hypocalciuric hypercalcemia. Autoantibodies that interact with the VFTD as allosteric modulators have been described to elicit signaling bias by enhancing Ca2+i while reducing pERK (phospho-extracellular signal-regulated kinase) [Citation125]. It has been hypothesized that this unique effect on signaling may arise from preferential coupling to Gi-MAPK pathways, although how this signaling pattern results in AHH remains to be explained. Further studies of these autoantibodies could identify specific receptor conformations that are required for the activation of different G proteins or for activation of Ca2+i versus MAPK signaling. The p.Arg680Gly variant of ADH1, which disrupts a salt-bridge hypothesized to regulate G protein coupling, may result in bias in the signaling of CaSR toward a β-arrestin-mediated pathway [Citation110]. Studies of AP2σ variants show that CaSR signaling through long-acting MAPK signals do so by sustained signaling from internalized CaSR-G protein complexes at endosomes [Citation124]. An improved understanding of the physiological effects of these biased signaling pathways and of the activation mechanisms of CaSR will likely inform the design of the next generation of allosteric modulators targeting the CaSR.
To date, the pathogenicity of CaSR variants has been investigated using largely second messenger readouts rather than direct measures of GPCR activation and G protein coupling. The most commonly-investigated pathway is generation of Ca2+i by transiently transfected plasmids encoding CaSR wild-type or mutant proteins. Ca2+i has been quantified using calcium sensors (e.g. fura-2) and fluorescent indicators (e.g. fluo-4), and occasionally using aequorin sensors [Citation123,Citation126,Citation127]. CaSR biased signaling has been assessed using a nuclear-factor of activated T cells response element (NFAT-RE) luciferase construct that can be activated by Ca2+i-calmodulin pathways [Citation124]. While investigation of CaSR-mediated Ca2+i signaling has successfully demonstrated the pathogenicity of many FHH- and ADH-associated variants, the assessment of a single signaling output can potentially miss important details about the effects of some CaSR variants, including those that bias receptor signaling and those that have no/moderate effects on Ca2+i. It has become more common in the last ten years for researchers to also investigate MAPK signaling. This is usually via generation of phosphorylated ERK1/2 by western blot, although quantitative AlphaScreen analysis (Amplified Luminescent Proximity Homogenous Assay Screen, Perkin Elmer, Waltham, MA, USA) and luciferase reporter assays measuring the serum response element, to which many ERK1/2 effectors bind, have been used [Citation126].
Assessment of phospholipase C activation by measuring inositol phosphate has also been increasingly used, as this measures Gq/11-mediated signaling in a quantitative manner and can easily be adapted for pharmacological screening platforms [Citation123]. The activation of other CaSR-mediated signaling pathways, including inhibition of cAMP by Gαi/o-coupling, has rarely been assessed. This is likely because of the difficulties associated with assessing a reduction in cAMP, which usually requires pretreatment of cells with forskolin to artificially elevate intracellular cAMP. Moreover, few reliable assays to quantify cAMP existed when CaSR variants were first described. However, researchers now have multiple assays to choose from, including end-point assays such as the LANCE (Perkin Elmer, Waltham, MA, USA) cAMP kit or live-cell kinetic assays such as those using Glosensor (Promega, Madison, Wisconsin, USA) cAMP. Therefore, it is likely that the effect of CaSR variants on cAMP and Gαi/o signaling will be explored in detail in future studies. Additionally, it is likely that the effects of CaSR mutants on G protein activation and coupling, rather than generation of downstream second messengers, will be assessed in the future as high-throughput screening assays using BRET (bioluminescence resonance energy transfer) and/or NanoBiT (NanoLuc® Binary Technology) are increasingly utilized.
One example of a BRET-based method is an open-source suite of 14 optimized BRET Gαβγ biosensors (named TRUPATH) that can be utilized for the interrogation of the transducerome down to single pathway resolution within cells. Biosensors such as Gα15 and Gα reveal coupling preferences for prototypic and understudied GPCRs [Citation124]. Other BRET tools can identify specific G proteins and β-arrestins that are integral to the signaling of a given GPCR. Translocation of effectors to the plasma membrane is the basis of monitoring G protein subtypes utilized by BRET. The advantage to this method is that it can detect not only biased signaling but also constitutive activity, and it can distinguish inverse agonism and polypharmacology [Citation127].
Polymorphisms and non-calcitropic disease
The increased availability of NGS datasets in the last decade has revealed the highly polymorphic nature of the CaSR gene. Missense variants in CaSR have been reported for gnomAD analysis of approximately 731,000 exomes and 76,000 genomes across diverse sub-populations. It is interesting to note that a null-model would have predicted more than 1420 missense variants; this suggests a strong evolutionary constraint on CaSR function (https://gnomad.broadinstitute.org/gene/ENSG00000036828?dataset=gnomad_r2_1).
As only approximately one-third of CaSR variants reported in association with FHH or ADH have been demonstrated to affect CaSR function, it is possible that some of these variants may not be pathogenic. This has been shown for some variants (e.g. p.Arg544Gln) that were reported in FHH cohorts, but were later discovered to be polymorphisms. Therefore, functional evaluation of CaSR variants by in vitro analysis identified in hyper/hypocalcemic individuals remains the gold standard to determine pathogenicity. However, the examination of CaSR variants in the over 51,000 individuals enrolled in the DiscovEHR cohort confirmed that variants predicted to be pathogenic had changes in serum calcium [Citation127]. While nonsense and frameshift CaSR variants were found to have aberrant Ca2+ concentrations, benign missense CaSR variants were predicted to be associated with a normocalcemic state. Thus, online pathogenicity tools such as PolyPhen (polymorphism phenotyping, http://genetics.bwh.harvard.edu/pph2/) and SIFT (sorting intolerant from tolerant, http://sift-dna.org/) may serve as initial predictors of CaSR variant effects on serum calcium concentrations and should be employed immediately following discovery of a CaSR variant in hyper/hypocalcemic individuals.
CaSR not only plays a key role in calcium homeostasis but also may contribute to a variety of non-calcitropic functions such as neurotransmission, glucose metabolism, hypertension, bronchoconstriction and wound healing. The extra-skeletal and extra-renal expression of CaSR in tissues is critical to maintaining basal Ca2+i. CaSR dysregulation in these tissues can contribute to neurological disease and smooth muscle pathology in bronchoconstriction [Citation110]. Targeting CaSR at these non-calcitropic tissues could provide novel treatments for disease. For example, increased expression of CaSR has been demonstrated in airway smooth muscle of allergen-sensitized mice and asthmatic patients. Treatment of mice with calcilytics reduced airway hyperresponsiveness and inflammation. Based on these observations, association studies have increasingly sought to establish if common or rare CaSR variants are themselves associated with a diverse range of pathologies.
The three most common carboxy terminal CaSR missense polymorphisms (encoding p.Ala986Ser, p.Arg990Gly, p.Glu1011Gln) have been found to contribute to a range of disorders including frequency of kidney stones, pancreatitis, primary hyperparathyroidism, and vascular calcification [Citation43]. However, these studies used small numbers of participants and different experimental parameters or statistical analyses and their findings were often inconsistent and difficult to interpret. Recent studies have attempted to combine multiple well-characterized population cohorts from diverse ethnic populations and have confirmed that CaSR polymorphisms influence serum calcium levels, but they did not find associations with other pathologies [Citation128]. Functional analyses of the three common polymorphisms showed that they may alter the CaSR Ca2+i EC50, although this appears to be in the opposite direction from that predicted to influence the implicated pathologies [Citation43]. Thus, while common polymorphisms influence serum calcium levels, it is unlikely that they have a major effect on other human disease pathologies in isolation.
The small changes in Ca2+i set point that result from CaSR variants in vitro may be too mild to be of clinical significance in some extra-skeletal tissues where receptor expression is much lower than that observed in parathyroid or kidney tissue. However, some associations between CaSR variants and non-calcitropic disease have been identified, including a rare missense CaSR variant identified in individuals with idiopathic generalized epilepsy. Functional studies have shown that the variant disrupts the proximal portion of the carboxyl tail of the CaSR; this includes an arginine-rich motif that promotes greater retention of the receptor at the cell-surface [Citation44]. Rare missense CaSR variants have been reported to be associated with cardiovascular disease, depression, dementia and alcohol abuse in the DiscovEHR cohort. Further studies in larger datasets are required to verify causation between CaSR variants and diverse pathologies [Citation127].
Parathyroid hormone receptor and bone metabolism
In humans, the parathyroid hormone 1 receptor gene (PTH1R), which has a wider tissue distribution than parathyroid hormone 2 receptor (PTH2R), encodes a receptor that binds both PTH and PTH-related protein (PTHrP). Because PTH2R has a narrower expression and different ligand rank order of potency than PTH1R, our discussion is focused on PTH1R. PTH regulates homeostasis of calcium and phosphate by enhancing bone resorption, modulating the synthesis of active vitamin D and modifying urinary excretion of calcium and phosphate, while PTHrP acts as a paracrine/autocrine regulator involved in vascular smooth muscle relaxation, transplacental calcium transport and enhanced bone remodeling [Citation42]. Like glucagon, glucagon-like peptide-1, growth hormone-releasing hormone, calcitonin, secretin and vasoactive intestinal peptide, PTH1R is a member of the class B GPCR family. PTH1R is highly expressed in metaphyseal growth plate, bone and kidney of fetal and adult tissues. PTH1R variants are associated with adult height and bone mineral density, consistent with its role in bone metabolism and endochondral bone formation [Citation42].
While the structurally similar PTHrP and PTH act at the PTH1R with identical pharmacological properties, PTH stimulates prolonged physiological responses (increases in active vitamin D and bone resorption markers) compared to PTHrP. Structural and kinetic signaling studies have revealed two ways in which the PTH1R ligands exert these distinct physiological profiles. PTH stabilizes the PTH1R in a pre-active state that interacts preferentially with a G-protein; this structural conformation is designated R0. Additionally, PTH mediates sustained PTH1R signaling through Gs-cAMP after internalization in endosomes, whereas PTHrP can signal only from the plasma membrane [Citation45].
The current generation of PTH1R-targeting compounds are designed with a focus on manipulation of these distinct receptor states. In animal models, long-acting-PTH, a PTH/PTHrP chimera with high R0 affinity, is associated with significantly longer endosomal cAMP response and prolonged hypercalcemia in animal models compared to PTH [Citation45]. Cryo-EM studies of the long-acting-PTH have demonstrated insertion of the N-terminus into the TMD core, leaving the C-terminal to bind rapidly and repeatedly to the PTH1R, thus enabling persistent activation [Citation45]. The synthetic N-terminal teriparatide (PTH 1-34), named for the first 34 amino acids of PTH, has anabolic effects on bone and osteoblast proliferation. Its effect on 1,25-dihydroxy-vitamin-D, calcium, and bone resorption markers makes it useful in treating osteoporosis [Citation45].
By contrast, abaloparatide, a human parathyroid hormone-related protein analog also designed to treat osteoporosis, binds PTH1R in a low affinity RG (receptor-G protein) conformation that has greater similarity to PTHrP binding but produces more transient cAMP responses [Citation42].
Germline variants in PTH1R cause rare skeletal dysplasias such as autosomal dominant Jansen-type metaphyseal chondrodysplasia, a rare short-limbed dwarfism resulting from growth plate abnormalities. Low or undetectable serum PTH in JMC is associated with chronic hypophosphatemia and hypercalcemia. Heterozygous PTH1R missense GOF variants, resulting in ligand-independent constitutively active receptors, are associated with the JMC phenotype [Citation46]. To date, there have been less than thirty cases reported. Although some cases of familial JMC have been reported, most variants arise de novo [Citation46]. Three amino acid residues located in the TMD and cytoplasmic face of PTH1R, p.His223, p.Thr410 and p.Ile458, allow the formation of a cavity necessary for G protein coupling by controlling the outward movement of the TMD regions of the PTH1R [Citation47]. Currently, JMC has no effective treatments; however, an inverse agonist of PTH1R has recently been described to significantly improve parameters such as bone marrow fibrosis, excess trabecular bone mass, and bone turnover in JMC-associated PTH1R-p.His223Arg mice [Citation47]. Future JMC treatments may include PTH1R inverse agonists.
Blomstrand lethal chondrodysplasia and related skeletal dysplasias and dental abnormalities are associated with homozygous or compound heterozygous LOF variants in PTH1R. Autosomal recessive BLC includes features of advanced endochondral bone maturation, short-limbed dwarfism, abnormal tooth morphogenesis and breast development associated with the increased calcification of advanced skeletal maturation. The fetal death associated with BLC resembles the phenotype of PTH1R knockout mice [Citation48]. Blomstrand disease has been subdivided into two groups, which, despite the nomenclature, can both be fatal. Severe type I BLC is the classical form and type II BLC is relatively milder. Complete or partial inactivation of the PTH1R, respectively, underlies the phenotypic difference attributable to type I versus type II [Citation48].
Homozygous PTH1R variants have also been found in Eiken syndrome, a milder form of inherited skeletal dysplasia associated with delayed bone ossification and bone remodeling abnormalities that is characterized by normal serum calcium, phosphate and vitamin D [Citation49]. Clinical phenotypes are variable, with some features, including primary failure of tooth eruption, being consistent with inactivating mutations, and other features, including delayed ossification, suggesting a GOF in chondrocytes. A recent study identified the PTH1R pathogenic variants in Eiken syndrome for the first time and provided some insight into why Eiken syndrome patients present with different phenotypes. The p.Arg485Stop variant increases basal cAMP signaling but reduces β-arrestin2 recruitment upon ligand stimulation. The missense variants, p.Glu35Lys and p.Tyr134Ser, impair PTHrP binding to PTH1R, and also impair β-arrestin2 recruitment and desensitization of cAMP signaling specifically in response to PTHrP but not to PTH. It has been hypothesized that β-arrestins may act to suppress basal PTH1R cAMP signaling under normal conditions and that reduced β-arrestin coupling could explain the delayed ossification observed in Eiken syndrome, whereas specific changes in response to PTHrP may result in the primary failure of tooth eruption [Citation50] Finally, a homozygous PTH1R variant has been reported in one individual with pseudohypoparathyroidism 1b. The residue affected, p.Arg186, contributes to ligand-receptor binding, and it is predicted that this mutation disrupts ligand binding affinity [Citation129].
Inactivating heterozygous PTH1R variants are present in enchondromas of approximately 10% of Ollier’s disease patients, a condition with multiple benign cartilage tumors, and a predisposition to malignant enchondromatosis [Citation130] that is associated with normal serum calcium and phosphate levels. Missense PTH1R variants reduce receptor activity by approximately 70%; while these have been suggested to contribute to Ollier’s disease, they are probably not entirely causative, the mode of inheritance being more likely a result of mosaicism [Citation130]. Possible germline transmission of a PTH1R variant has been reported in one case. Thus, the development of enchondromas in Ollier’s disease may arise from a combination of germline and somatic events [Citation130].
Aberrations of enchondromatosis result from abnormal PTH1R signaling, including autosomal dominant symmetrical enchondromatosis reported in patients with 12p11.23 to 12p11.22 duplications that contains the PTHLH gene encoding PTHrP. In addition, autosomal dominant inheritance of LOF variants in PTH1R, including those in BCL, can result in primary failure of tooth eruption. Haploinsufficiency of the receptor, resulting in loss of protein, may underlie the phenotype [Citation131].
GPCR variants and obesity
While feeding regulation, integration of satiety and body fat inputs are regulated by portions of the hypothalamus that signal through the orexin/hypocretin and leptin systems [Citation132], bodyweight regulation includes components of the leptin-melanocortin pathway. The first melanocortin receptors (MCR) reported were for the adrenocorticotropic hormone receptor (ACTHR or MC2R) and melanocyte-stimulating hormone receptor (MSHR or MC1R). While five MCRs are known, MC1R–MC5R, only MC3R and MC4R are expressed in the brain. The ligands for the MCRs are a group of peptide hormones that are produced as post-translational products from proopiomelanocortin (POMC), itself produced in the pituitary gland. The ligand-bound MCRs couple primarily to Gαs [Citation133–135].
Melanocortin 4 receptor variants
Melanocortin 4 receptor (MC4R) is expressed in the hypothalamic arcuate nucleus and has a central role in food intake. In response to signals such as leptin, POMC is released from neurons of the hypothalamic arcuate nucleus, and the biologically active products, αMSH and βMSH (MSH, melanocyte-stimulating hormone), act on MC4R at the paraventricular nucleus. Agouti-related protein (AgRP)-expressing arcuate neurons promote food intake by inhibiting MC4R [Citation135–146]. The balance between the activity of AgRP-containing neurons and αMSH-containing neurons determines melanocortin pathway activity [Citation140].
The study of MC4R knockout mice with an hyperphagic, overweight phenotype was foundational in the identification of the role of the MCH4-αMSH pathway in obesity [Citation142]. Subsequently, some forms of early-onset obesity have been reported to result from inheritance of variants in the gene encoding the MC4R. More than 200 variants have been identified to date although homozygous variants have also been reported and these individuals have an earlier onset of obesity. Most of the MC4R mutations are missense, although nonsense, frameshift and small insertions and deletions have been described [Citation144–147]. MC4R mutations impair cAMP generation, and cell surface expression is often defective due to retention in the ER.
Other mechanisms and loci have been proposed from the results of genome-wide association studies that identified the MC4R promoter SNP rs17066842 G > A variant as being associated with a lower likelihood of developing obesity and type 2 diabetes [Citation145]. Although the mechanism by which this SNP protects individuals from developing obesity and diabetes is incompletely understood, in vitro studies showed a complex pattern of receptor cell surface expression regulated by glucose concentration that varied across cell types. Further studies are required to understand how this SNP regulates MC4R surface expression in different cell types. The fine mapping of 39 loci that identified MC4R variants in tens of thousands of cases and controls also identified non-coding variants of other genes, including cyclin dependent kinase inhibitors, diacylglycerol kinase β and glucose-dependent insulinotropic polypeptide receptor in obesity and/or type 2 diabetes [Citation145]. These data suggest the MC4R signal is likely to be robust in this sample.
The contribution of LOF variants in MC4R to genetic adiposity may be greater than previously reported. A large study of the MC4R coding sequence in 5,724 participants enrolled in a UK longitudinal birth cohort study associated nonsynonymous MC4R variants with anthropometric phenotypes in childhood and early adulthood. There was a 0.30% frequency of LOF variants in MC4R such that carriers of these variants had a substantial risk of excess adiposity [Citation139].
In a combined epidemiological and pharmacological study employing genotype information from 500,000 participants in the UK Biobank, 61 missense variants of MC4R were characterized in cAMP accumulation and β-arrestin signaling. Variants with a GOF in the arrestin pathway were associated with disease protective properties, while LOF variants in the Gs pathway were associated with increased body mass index and obesity-related cardiometabolic traits [Citation148]. In a follow-up study, variants that did not affect canonical Gs signaling and that were previously considered to be nonpathogenic were shown to disrupt other MC4 functions including trafficking to the plasma membrane, dimerization, and internalization, rendering these receptor functions to be physiologically relevant [Citation149]. This highlights the power of burden-testing of functionally related variants to increase power for phenotypic association testing. However, it should be stated that variants from genotype arrays, especially rare and imputed variants, can be unreliable, which can potentially lead to misinterpretation of causative relationships [Citation150,Citation151].
Several regions of ICL2 and adjacent regions of TM3 and TM4 may be involved with receptor dimerization formation. The signaling aberrations associated with MC4R variants may result from disruption of dimerization. For example, an ICL p.His158Arg activating MC4R mutation may disrupt dimer formation. Analysis of the consequences of naturally-occurring and engineered variants considerably facilitates MC4R drug design [Citation147,Citation152].
Variants in the MC4R accessory protein MRAP2
LOF variants of the accessory protein melanocortin-2 receptor accessory protein (MRAP) have been associated with monogenic hyperphagic obesity with hyperglycemia, hypertension and possibly female infertility, and global and Mc4r-neuron-specific inactivation of MRAP2 in mice increased food intake and weight gain [Citation153]. MRAP2 is a single-pass transmembrane protein that enhances the MC4 response to the agonist, αMSH [Citation154]. MRAP2 may potentially be targeted clinically, although the effect on other GPCRs, including the ghrelin and prokineticin receptors that have been reported to interact with MRAP2, would need to be considered.
Pharmacological targeting of MC4R
Small clinical trials of MC4R agonists such as setmelanotide have been reported in individuals with severe obesity due to either POMC deficiency obesity or leptin receptor (LEPR) deficiency obesity. The authors reported the results of a single-arm, open-label, multicenter, placebo-controlled phase 3 trial across North America and Western Europe. Ten participants were enrolled in the 12-week POMC trial and 11 participants were enrolled in the 12-week LEPR trial. For a four-week period, setmelanotide and placebo administration were blinded before switching to open-label for 32 weeks. The primary endpoint was 10% weight loss compared with baseline at approximately 1 year. The authors report that the majority of POMC trial participants and almost half of LEPR participants did achieve 10% weight loss in one year. Aside from injection site reactions, hyperpigmentation and transient nausea, the drug was well tolerated [Citation141]. Another MC4R agonist, bremelanotide, has also recently been approved in the United States for use in premenopausal women with low sexual desire. In small, randomized control studies of obese premenopausal women, bremelanotide reduced caloric intake [Citation155].
Familial glucocorticoid deficiency
Autosomal recessive familial glucocorticoid deficiency (FGD) disorder is distinguished by progressive primary adrenal insufficiency and normal mineralocorticoid metabolism. Type 1 FGD is associated with the inheritance of predominantly compound heterozygous variants of MC2R, the gene encoding ACTHR [Citation156,Citation157]. Missense and nonsense variants have been associated with the disease. For example, a germ-line nt.201C > T substitution, introducing a premature stop codon, TGA, results in the truncation of the entire carboxyl portion of the receptor. SNVs disrupting the sequence of TM2 and IC3 of ACTHR have been reported [Citation156]. Pathogenic variants of MC2R often result in defective trafficking of the receptor to cell surfaces due to retention at the ER. Other MC2R mutations affect the binding affinity of adrenocorticotropic hormone to the receptor.
Some variants of ACTHR, such as the p.Gly217 truncation, are relatively common polymorphisms that are pathogenic only when expressed in compound heterozygous form with a rare variant. Other factors that influence down-stream GPCR signaling include tissue expression and alternative splicing [Citation156,Citation158].
By contrast, type 2 FGD is generally an early onset disease (median 0.1 years), associated with variants in MRAP, the accessory protein required for surface expression of MC2R. The MRAP phenotype can resemble later-onset type 1 disease; however, it is consistent with in vitro work that demonstrates a right shift in the dose-response curve and decreased cAMP production. Type 3 FGD results from variants of the steroidogenic acute regulatory gene [Citation153,Citation154].
Follicle stimulating hormone
Follicle-stimulating hormone (FSH) is essential for ovarian development and follicle maturation in females and for Sertoli cell number and normal spermatogenesis in males. Inactivating or hyper-activating genetic variants have been identified in coding, regulatory, and splice sites of the follicle-stimulating hormone receptor gene (FSHR), leading to abnormalities of cell surface expression level and ligand binding that impacts female fertility. While inactivating variants can result in disorders such as premature ovarian insufficiency, polycystic ovary syndrome, ovarian hyper-stimulation syndrome (OHSS), as well as altered drug responses [Citation159], activating FSHR variants are associated with OHSS. Inactivating FSHR variants identified in female ovarian dysgenesis, however, appear to be benign in males, who instead occasionally harbor an asymptomatic, constitutively active FSHR mutation.
In the gnomAD (https://gnomad.broadinstitute.org/) database, there are at least 1,152 FSHR SNVs, of which 401 are missense. Exons 7 and to a greater extent, exon 10, harbor LOF variants, resulting in primary amenorrhea, elevated FSH levels, and infertility. For example, the p.Asn680Ser polymorphism of the FSHR has been associated with OHSS [Citation159].
While the phenotype associated with activating FSHR variants can manifest in heterozygotes, the phenotype associated with inactivating variants is present in homozygous or compound heterozygous patients. For example, the prototypical inactivating p.Ala189Val variant underlies a dominantly inherited pattern of ovarian dysgenesis. This variant disrupts the large ECD implicated in ligand binding while leaving the remaining TM-spanning domains and the carboxyl tail intact [Citation159].
Two FSHR variants located in exon 10, the p.Asn680Ser (rs6166G) variant and the p.Ala307Thr (rs6165) variant, may be of special interest because, in some populations, they are in linkage disequilibrium and are associated with ovarian response to stimulation by FSH. With respect to rs6166G carriers, resistance to clomiphene citrate and higher endogenous levels of base-line FSH levels were reported. Conversely, rs6166A allele carriers showed higher FSH sensitivity and an increased risk for developing OHSS syndrome. The phenotype may reflect the fact that, compared with rs6166A, rs6166G has greater resistance to FSH, slower kinetics of cAMP production, lower ERK1/2 activity, and less CREB phosphorylation [Citation159].
In addition, FSHR variants in the 5′-untranslated region of the gene are associated with ovarian response. The FSHR − 29 G > A (RS1394205) variant A allele may be associated with reduced expression and poor ovarian response. The evidence regarding the effect of the FSHR − 29 G > A (rs1394205) variant on the response to FSH administration was evaluated in a meta-analysis that suggested that higher doses were required in homozygotes for the A allele than carriers of the G allele [Citation159].
By contrast, male patients hypophysectomized because of a pituitary tumor can harbor constitutively active forms of the FSHR gene, a phenotype that is benign until unmasked by treatment [Citation155]. For example, the p.Asp567Gly variant located in IC3 results in a CAM characterized by elevated in vitro basal cAMP production. Hypophysecotomized males who are heterozygous for this CAM maintain normal spermatogenesis [Citation160,Citation161].
FSHR variants are relevant with respect to the treatment of infertility. A common variant, p.Asn680Ser, results in higher hormonal sensitivity and increased FSH production in normogonadotropic anovulatory women [Citation159]. While rare activating FSHR variants are benign in males, women with the p.Asn680Ser variant are at higher risk of OHSS, a complication of ovarian stimulation treatments that can be life-threatening [Citation157,Citation162]. The potential of pharmacogenomics includes the opportunity to predict side-effects of common interventions, and it could identify those at higher risk of developing OHSS during routine in vitro fertilization treatments.
Luteinizing hormone/choriogonadotropin receptor variants
The luteinizing hormone/choriogonadotropin receptor (LHCGR) couples to Gs to stimulate adenylyl cyclase. It is necessary for the function of the Leydig cells of the testicles in producing testosterone needed to maintain male secondary sex characteristics and male fertility [Citation163]. Constitutively active LHCGR variants result in autosomal, male-limited gonadotropin dependent disorders including familial male precocious puberty and testotoxicosis [Citation24,Citation25]. By contrast, in females, LOF variants of FSHR disrupt hormone binding and signaling and interfere with development and fertility. Disrupted ovarian function can cause amenorrhea, hypergonadotropic hypogonadism, ovarian failure, and/or dysgenesis [Citation26].
Constitutive LH receptor variants
While rare activating LHCGR variants have been found in men with normal spermatogenesis [Citation27], familial male precocious puberty associated with Leydig cell hyperplasia may result in low sperm cell counts. Affected males have been reported to harbor activating LHCGR variants that encode p.Asp567Gly in TM6 [Citation162,Citation163]. Other SNVs in the cytoplasmic portion of TM6 have also been reported. In each case, these CAMs were found to result in increased basal cAMP production. These variants have been reported in kindreds from diverse ethnic origins [Citation27,Citation28].
Constitutively active variants highlight structural components of GPCRs that are critical for G protein coupling. In vitro studies have highlighted regions of TM6 of the α1-adrenergic receptor that are homologous with 568Ala of FSHR. These engineered receptors share high basal adenylyl cyclase activation, suggesting that a TM6 alanine is integral to signal transduction [Citation27,Citation28].
Gonadotropin releasing hormone receptor variants
Activity of the hypothalamo-pituitary-gonadal axis is variable throughout life. An increase in the activity of this axis brings about puberty, resulting in sex-specific, secondary sexual features and a maturing reproductive system. Anatomical or functional defects may result in reduced gonadotropin releasing hormone (GNRH) and/or gonadotropin release, a condition known as hypogonadotropic hypogonadism (HH).
By contrast, idiopathic hypogonadotropic hypogonadism (IHH) is associated with adrenal insufficiency without anosmia (deficient sense of smell). IHH is divided into two categories: normosmic IHH (nIHH) and Kallmann syndrome (KS). It has been associated with inactivating variants in more than 50 genes. Many hereditary cases result from variants of the neurokinin receptor [Citation29–37].
Variants of the gonadotropin releasing hormone receptor gene (GNRHR) were first associated with reproductive failure in a subset of IHH [Citation38,Citation39]. and in isolated growth hormone deficiency 1B, associated with delayed puberty [Citation40]. The prokineticin receptor 1 and prokineticin receptor 2 have been implicated in a broader IHH phenotypic spectrum [Citation30]. In consanguineous families, variants of the kisspeptin (GPR54, metastin) receptor (KISS1R) gene have been identified [Citation31]. An analogous phenotype results from disruption of kisspeptin synthesis, due to its role in stimulating GNRH and gonadotropin, resulting in delayed sexual development and apulsatile gonadotropin levels. By contrast with other disorders that affect the hypothalamic-pituitary axis, impaired sexual development is not accompanied by anatomical abnormalities [Citation27–40].
In addition, KS is associated with variants in the PROKR1 (PROKR, prokineticin receptor) and PROKR2 genes. Together with their ligands, these GPCRs encode receptor-ligand pairs that regulate intestinal contraction, circadian rhythms and vascular function [Citation36]. Many missense variants in PROKR2 have been identified in patients with IHH or KS. While the phenotype is associated with compound heterozygous and homozygous GNRHR and KISS1R variants, PROKR2 variants are reported to be heterozygous and, in some cases, associated with digenic inheritance of WD repeat domain 11 (WDR11) variants. Similarly, the phenotype associated with semaphorin 3 A (SEMA3A) variants has been reported to be inherited in a digenic manner with variants in other KS-associated genes [Citation35].
The genes disrupted in KS and adrenal insufficiency subtypes of X-linked IHH are distinct from GNRHR variants. The abnormal development of the olfactory bulb is associated with intragenic microdeletions in the KAL gene [Citation32,Citation33]. By contrast, adrenal hypoplasia is associated with variants of the DAX1 gene that are inherited in an X-linked manner. The phenotype results from disruption of the nuclear hormone receptor function of the DAX1 protein that controls portions of reproductive development [Citation34,Citation35]. Subsequently, variants of many other genes have been associated with specific clinical features of IHH.
In addition, disruption of the anosmin 1 (ANOS1) gene that encodes an extracellular glycoprotein called anosmin-1 has been reported in more severe cases, and approximately 10% of patients with KS were found to have inactivating variants in FGFR1 or genes in the pathway [Citation35].
Idiopathic hypogonadotropic hypogonadism
Our understanding of how GNRHR variants result in idiopathic hypogonadotropic hypogonadism (IHH) has been steadily increasing [Citation29–41,Citation66–69]. At least 15 GNRHR variants have been described [Citation29]. Some of these encode p.Glu90Lys and p.Ser217Arg and are LOF variants. Other GNRHR variants, encoding p.Asn10Lys, p.Thr32Ile, and p.Gln10Arg, are partially inactivating with respect to eliciting an inositol phosphate response in vitro [Citation31].
The significance of GNRHR variants has been investigated in vitro. For example, the p.Glu90Ala and p.Arg139His [Citation41,Citation66] substitutions were found to abolish receptor activation. By contrast, the p.217Ser variant of TM5 is completely inactive, but substitutions of p.Ser217Gln and p.Ser217Tyr retain partial function. These studies have identified the role of steric hindrance that is disruptive to receptor structure [Citation41] and isolated residues critical to receptor function [Citation66].
High-throughput sequencing and subsequent experimental verification have allowed the identification of very rare pathogenic GNRNR variants. Whole exome sequencing (WES) has been used to identify a novel homozygous GNRHR mutation (NM_000406; c.364C > T, p.Leu122Phe) in two prepubertal boys with cryptorchidism and micro-penis. It was reported that mutant receptor expression on the cell membrane was lower than that of wild-type and that the mutation was LOF [Citation67]. Similarly, a novel homozygous missense mutation, c.521A > G (p.Q174R), in GNRHR was reported in two affected sisters. Disruption of the highly conserved p.Gln174Arg may impede the conformation that facilitates mobility of TMD4 and TMD5 [Citation68]. By contrast, partial LOF GNRHR variants have been identified that expanded the spectrum reported in IHH patients. For example, a heterozygous variant encoding an p.Arg240Gln variant of GNRHR that has no effect on mRNA and protein but demonstrates impaired ERK1/2 activation and inositol triphosphate/calcium signaling was identified in sporadic IHH patients [Citation69].
Drug development
The effect of GNRHR variants may be combatted by permeant antagonists such as the nonpeptide, IN3. A permeant drug is small enough to penetrate the cell membrane and bind to the GPCR intracellularly. These compounds, some peptide and some nonpeptide, act as pharmacological chaperones for GPCRs such as GNRHR that stabilize the receptor charge to the extent that its membrane expression and function is at least partly restored. For example, in HeLa cells, IN3 can rescue cell surface expression of hemagglutinin-tagged GNRHR expressed by adenovirus [Citation41]. These data suggest that the development of antagonists that target variant GNRHR and stabilize their expression in the cell membrane may serve to reverse the course of complex developmental disorders and may illustrate an important concept in drug design.
Variants identified by next generation sequencing
Access to WES or whole genome sequencing (WGS) has revealed variants such as tachykinin precursor 3 (TAC3) and tachykinin receptor 3 (TACR3) genes [Citation37] in family members of patients with IHH [Citation68] that may be associated with constitutional delay in growth and puberty. Although milder than IHH, variants associated with constitutional delay in growth and puberty may be part of the same spectrum [Citation37]. These methods have also identified homozygous p.Glu72* variants of GNRHR in very rare disorders such as isolated growth hormone deficiency type 4, a disorder manifesting growth hormone deficiency as delayed puberty and delayed bone maturation [Citation40].
Vasopressin V2 receptor variants
Arginine vasopressin resistance (AVP-R), known formerly as nephrogenic diabetes insipidus (NDI), is characterized by renal tubular resistance to the antidiuretic effect of arginine vasopressin (AVP), and can be inherited in X-linked, autosomal dominant or autosomal recessive manners. The X-linked patients, comprising at least 90% of cases, have been found to harbor variants of the vasopressin V2 receptor (AVPR2) gene [Citation70]. By contrast, autosomal recessive and dominant disease results from variants of the aquaporin 2 gene, a renal collecting duct water channel [Citation71]. While most AQP2 mutations are autosomal recessive, rare carboxy-terminal AQP2 mutations are dominant.
Arginine vasopressin resistance
X-linked AVP-R (OMIM 304800) results from variants of AVPR2, located in the Xq28 chromosome, a nonapeptide binding GPCR that belongs to the same subfamily as V1a and V1b vasopressin as well as the oxytocin receptors. AVPR2 primarily responds to AVP in the distal convoluted tubule and collecting ducts of the nephron by regulating the concentration of urine and maintaining water homeostasis [Citation72].
The signaling complex of the AVPR2 has been characterized using cryo-EM (). Because AVPR2 signals through β-arrestin promotes cAMP production, non-canonical signaling comprising retromers as well as arrestin has been posited [Citation73]. Noncanonical signaling of AVPR2 may explain why variants associated with AVP-R retained in the ER could still generate cAMP in response to a membrane permeant agonist [Citation74]. This suggests that AVP2R complexes can signal via cAMP/PKA (protein kinase A) pathways despite being internalized.
Despite more than 211 variants reported in 326 different families, few phenotypic differences have been noted [Citation70]. AVPR2 variants, some of which are shown in , are encoded by missense and nonsense variants as well as small indel variants [Citation70]. For example, substitutions of p.Ser167Thr and p.Leu44Pro, residues conserved in many GPCRs, result in familial AVP-R.
Figure 5. Schematic representation of the V2 receptor and identification of 193 putative disease-causing arginine vasopressin R2 (AVPR2) mutations. Predicted amino acids are shown as the one-letter amino acid code. A solid symbol indicates a codon with a missense or nonsense mutation; a number indicates more than one mutation in the same codon; other types of mutations are not indicated on the figure. There are 95 missense, 18 nonsense, 46 frameshift deletion or insertion, 7 in-frame deletion or insertion, 4 splice-site, 22 large deletion mutations, and 1 complex mutation. The gain of function mutations affecting codons R137 and F229 are indicated. Extracellular, intracellular and transmembrane domains are annotated as reported previously [Citation166].
![Figure 5. Schematic representation of the V2 receptor and identification of 193 putative disease-causing arginine vasopressin R2 (AVPR2) mutations. Predicted amino acids are shown as the one-letter amino acid code. A solid symbol indicates a codon with a missense or nonsense mutation; a number indicates more than one mutation in the same codon; other types of mutations are not indicated on the figure. There are 95 missense, 18 nonsense, 46 frameshift deletion or insertion, 7 in-frame deletion or insertion, 4 splice-site, 22 large deletion mutations, and 1 complex mutation. The gain of function mutations affecting codons R137 and F229 are indicated. Extracellular, intracellular and transmembrane domains are annotated as reported previously [Citation166].](/cms/asset/de6a1fd3-a2ea-4702-87c2-e496534e9e30/ilab_a_2286606_f0005_c.jpg)
Loss of function phenotype
The phenotype of X-linked AVP-R [Citation75] includes dehydration episodes, hypernatremia, and hyperthermia with onset as early as the first week of life. The dehydration associated with AVPR2 variants can lower arterial blood pressure below that sufficient to oxygenate the brain, kidneys, and other organs. The classic consequences of late diagnosis and inadequate treatment include mental and physical retardation that accompanies renal failure [Citation76]. Variable degrees of polyuria and polydipsia can be features exhibited by heterozygous females due to skewed X chromosome inactivation [Citation77,Citation78].
Population genetics
AVPR2 variants may be present due to a founder effect [Citation79]. The Hopewell truncation mutation (p.Trp71X), seen mostly in descendants of Ulster Scot immigrants to Colchester County, Nova Scotia, Canada, is associated with AVP-R in the largest (known) North American pedigree. A link with the Cannon pedigree, a large Utah kindred, which has a high prevalence of X-linked AVP-R, has been suggested [Citation79]. These variants have been found in many populations in North America. In Quebec, Canada, the incidence of AVP-R in live male births is estimated to be 8.8 in 1,000,000 [Citation77].
Currently there are approximately 98 living males from the Hopewell kindred and 18 from the Cannon pedigree. Forty-two affected males in Nova Scotia and New Brunswick, Canada, and eight affected males from the central United States have been genotyped for the p.Trp71X variant.
From the point of view of genetic and genomic counseling, families with a history of diabetes insipidus may be offered molecular testing if they grant informed consent. The clear benefit to patients and families of identifying pathogenic AVPR2 variants in those at risk of X-linked AVP-R is demonstrable by the prevention of physical developmental sequelae and intellectual disability associated with repeated episodes of dehydration.
Expression studies
Data from transiently transfecting variant AVPR2 cDNA in heterologous expression systems has formed the basis of classifying variants on the spectrum of pathogenic to benign based on evaluation of receptor function and subcellular localization [Citation80]. Those AVPR2 variant proteins that reach the cell surface but demonstrate ligand binding impairment and do not generate adequate cAMP are classified as type 1 mutants. In vitro studies determining AVPR2 cell surface expression and binding affinity established criteria for pathogenicity. As a result of this body of in vitro work, naturally-occurring AVPR2 variants can increasingly be classified in silico using an approach similar to that used for the LDL receptor. Pathogenic variants that have characteristically defective intracellular transport can be classified as type 2 mutants. Immunodetection strategies can be used to visualize epitope-tagged AVPR2 in whole cells in order to assess whether receptor numbers are reduced at the cell surface. Parallel immunofluorescence experiments can be conducted on intact cells (to quantitate receptors on the cell surfaces) or permeabilized (to isolate intracellular pools). There is evidence of pre-Golgi accumulation of type 2 pathogenic AVPR2 receptors because their initial glycosylation fails to undergo glycosyl-trimming in the Golgi. Enzyme-linked immunosorbent assay or assays of protein expression by western blot have been used to identify AVPR2 in membrane preparations.
In contrast to types 1 and 2 AVPR2 mutants, type 3 pathogenic variants are rare. Type 3 variants are not transcribed effectively due to mRNA instability and degradation. In general, northern blot analysis of AVPR2 expression suggested that for the majority of pathogenic variants (i.e. types 1 and 2), mRNA was not normal in quantity or size. Most pathogenic AVPR2 variants are classified as type 2 mutant receptors that are trapped in the cell’s interior and are not expressed on the cell surface [Citation81–84]. Like other pathogenic GPCRs [Citation85], the gene products associated with disease are notably beset by protein misfolding. In general, LOF phenotypes result from misfolding of variant proteins that prevents receptors from carrying out their function on the cell surface although the receptor is otherwise fully functional, a phenomenon that is also observed for secretory integral membranes or enzyme proteins processed in the ER, Golgi complex or lysosomes [Citation86]. ER folding is critical because misfolded proteins are retained in the ER before degradation.
Autosomal recessive AVP-R
The mechanisms associated with protein misfolding belie the disruption of urine countercurrent mechanisms present in autosomal recessive AVP-R. Pathogenic aquaporin 2 variants are expressed as misfolded proteins that are trapped in the ER [Citation87]. The misfolded membrane proteins that result in Gitelman syndrome [Citation88], Bartter syndrome [Citation89,Citation90], and cystinuria [Citation91] may share this ER-trapping mechanism.
Pharmacology
If misfolded AVPR2 type 2 can be overcome, the otherwise functional GPCR may be rescued. AVPR2 missense variants that impair folding may not accumulate as toxic aggregates so long as the functions of the collecting duct cells are normal. Some markers, such as the epithelial sodium channel, that are impaired in a sodium-losing state [Citation92] are normal in patients with AVPR2 variants. However, other consequences of type 2 AVPR2 pathogenic variants may include the insertion of one portion of a protein into the destabilized β sheet of a second molecule. This effect may be similar to that described for the common Z mutation in α1-antitrypsin deficiency [Citation93], which putatively “clogs” the ER [Citation94]. If the misfolded GPCR can be overcome by transporting it out of the ER to the target membrane, these receptors retain sufficient functionality to mitigate the pathology associated with the NDI phenotype [Citation95]. One therapeutic strategy would be to make use of pharmacological chaperones to promote AVPR2 escape from the ER [Citation84,Citation95,Citation96]. For example, permeant nonpeptide V2 and V1 receptor antagonists have been used to stabilize misfolded AVPR2 receptors, thereby rescuing them to the cell surface [Citation83]. The constitutive endocytosis associated with the R137H mutant has been rescued by non-peptide V2 antagonists [Citation98]. The constitutive p.Arg137His β-arrestin-promoted endocytosis itself was not prevented in vitro by the antagonist [Citation97], however, suggesting that, like other AVPR2 variants, effective treatment relies on pharmacological chaperone action [Citation74].
These strategies may be incorporated into new therapeutics for hereditary disease such as NDI that often results from protein misfolding and kinesis [Citation95,Citation96]. For example, five NDI patients with del62_64, p.Arg137His and p.Trp164Ser variants were administered SR49059, a non-peptide vasopressin antagonist. The non-peptide antagonist decreased urine volume and water intake and increasing urine osmolality without impacting sodium, potassium, and creatinine excretion or plasma sodium levels [Citation83]. This clinical study suggests a therapeutic approach that could be applied to the treatment of errors in protein folding [Citation95,Citation96].
AVPR2 and nephrogenic syndrome of inappropriate antidiuresis
The nephrogenic syndrome of inappropriate antidiuresis (NSIDA) phenotype, consisting of hyponatremia, concentrated urine, and suppressed AVP plasma concentrations, is a rare disorder of water balance that contrasts with AVP-R. Pathogenic variants of the AVPR2 gene that encode variants such as p.Arg137Cys, p.Arg137Leu [Citation99,Citation100,Citation164,Citation165] and p.Phe229Val [Citation166] have been identified in NSIDA. Structural studies of the antidiuretic hormone arginine-vasopressin V2 receptor signaling complex have been reported [Citation167]. In vitro, expression of both p.Arg137Cys and p.Arg137Leu variants were reported to have elevated basal cAMP signaling and agonist independent interaction with β-arrestins [Citation167].
Using the Ballesteros-Weinstein numbering system for GPCRs [Citation168], we can express the location of the structure disrupted by amino acid substitutions in terms of the most conserved residue in each of the transmembrane alpha-helices. In this numbering system, the most conserved residue is numbered 50, and amino acid numbers count up or down, depending on their relative location. The functional breakage resulting from the substitution of p.Arg137 can therefore be expressed with respect to the disruption of the Arg1373.50-Asp1363.49 ionic lock (see https://docs.gpcrdb.org/generic_numbering.html for information about this system). The p.Arg137Cys and p.Arg137Leu mutant structures have been suggested by cryo-EM results (see legend) [Citation166]. AVPR2 variants with GOF are extremely rare. While many patients with hyponatremia have been sequenced, classical AVP-R is associated with new and recurrent pathogenic LOF variants of the AVPR2 gene.
Endothelin and Hirschsprung’s disease
The enlarged colon in Hirschsprung’s disease is notable for the absence of gastrointestinal ganglion cells in the myenteric and submucosal plexuses. Variants of at least ten genes have been found to cause Hirschsprung’s disease and several loci have been implicated by linkage and other means. Study of the disorder is complicated by incomplete penetrance and variable expressivity [Citation169]. The largest number of familial and sporadic cases of Hirschsprung’s disease result from RET (ret proto-oncogene) gene variants [Citation170]. However, variants in the endothelin-β (ETB) receptor gene (EDNRB) and endothelin 3 (EDN3) ligand genes are potentially of wider relevance because they disrupt development [Citation171]. The three peptide ligands of the endothelin system act at two GPCRs, endothelin-α (ETA) and ETB receptors [Citation171]. Potential therapeutic applications of EDNRB may reflect its role in development of the enteric nervous system [Citation171].
While ETB receptor variants such as the p.Trp276Cys are most commonly associated with Hirschsprung’s disease, sporadic cases of Hirschsprung have been attributed to very rare activating ETB receptor variants, including p.Gly57Ser, p.Arg319Trp, and p.Pro383Leu [Citation172]. Rare variants in the EDN3 ligand gene [Citation103,Citation104,Citation171,Citation173] and endothelin-converting enzyme 1 [Citation105] are also implicated [Citation169].
The molecular pathology of Hirschsprung has been advanced from studying the ETB p.Trp276Cys receptor. Evolutionary conservation [Citation174] facilitated the study of the homologous p.257Trp ETA and p.258Trp ETB receptors in vitro with respect to coupling Gi, Go, and Gq. This showed reduced G coupling of engineered variants compared with the naturally-occurring p.Trp276Cys ETB receptor variant that is pathogenic in Hirschsprung’s disease.
Purinergic receptors and clotting
Purinergic receptors, themselves comprising a large subfamily, have overlapping selectivity for purinergic nucleotides. Because of its involvement in platelet aggregation, the purinergic receptor (P2RY12) is a target for treatment of thromboembolism and other clotting disorders. The anticlotting agents, triclopine and clopidogrel, target this receptor.
Originally identified through linkage analysis of a pedigree with a severe bleeding disorder that proved refractors to many pharmaceuticals, its discovery clarified the pharmacology of anticlotting agents such as triclopine and clopidogrel. A two-nucleotide P2RY12 deletion affecting the region encoding TM6 of P2RY12 was found to have reduced efficacy and potency for these anticlotting agents. Pharmacological agents with efficacy refractory bleeding disorders may be screened, in vitro, against expressed P2RY12 variants [Citation175].
Among patients not affected by classic bleeding disorders, the identification of purinergic receptors succeeded in refining the pharmacology of anticlotting agents. For example, patients with acute coronary syndromes subjected to percutaneous coronary intervention responded better to dual antiplatelet therapy with clopidogrel, an antagonist of P2RY12, and aspirin [Citation102]. This increased efficacy with dual therapy may reflect the three-fold increase in aspirin resistance in male Caucasians with a history of myocardial infarct that is attributable to the p.Cys893Thr P2Y purinoceptor 1 (P2RY1) variants [Citation102].
In addition to P2RY12, other genes have been implicated in receptor function and drug efficacy. Platelet aggregation pathways dependent on thromboxane have been shown to involve P2RY12 receptors [Citation102,Citation175]. In coronary heart disease, the rs6809699 variant of P2RY12 has been associated with resistance to the anticoagulant, clopidogrel. While its metabolism may be disrupted by cytochrome P2C19 LOF polymorphisms, the rs6809699 AA P2RY12 genotype can increase efficacy independent of cytochrome polymorphisms [Citation175].
GPCRs and monogenic asthma
The G protein-coupled receptor associated with asthma (GPRA) gene was positionally cloned to chromosome 7p13 in a large linkage study of monogenic asthma in Finland and in other European populations [Citation176–179]. De-orphanization studies subsequently identified that GPRA was the neuropeptide S receptor 1 (NPSR1) that may be implicated in disease mediated by immunoglobulin E. Although neuropeptide S acts at the NSPR1, it may bind other endogenous ligands and provide a potential drug target.
NSPR1 is part of a distinct signaling pathway that depends on the variability in expression of common isoforms of the receptor. This pathway can be dysregulated in asthma [Citation179]. NPSR1-A has a shorter, 29-residue C-terminus, whereas NSPR1-B has an alternate 3′ exon (E9b) encoding a longer 35-residue C-terminus [Citation179].
While the A isoform exhibits stronger signaling, the B isoform is expressed on bronchial epithelia and smooth muscle in asthmatics [Citation179]. The alternate carboxy-termini, containing unique phosphorylation sites, are likely to underpin this functional difference. While the A isoform of the NPPR1 receptor, therefore, may be promising due to its potential for use on drug screens for asthma, the difference in its tissue expression compared with the B isoform may be informative to drug design [Citation179].
Orphan receptors
De-orphanized GPCRs may be targeted clinically by small molecules even when an endogenous ligand has not been identified. NGS has identified orphan GPCR variants that are implicated in a variety of rare phenotypes [Citation180]. A study by Dershen examined 85 orphan GPCRs in 51,289 individuals; among this unselected cohort, strong, phenome-wide disease associations were identified with at least two variant categories for 39% of GPCRs. SNP-set (Sequence) Kernel Association Test (SKAT) was used to identify rare missense variants in GPR39 as well as synonymous variants that are associated with phenotypes, including psoriatic arthropathy, that alter receptor expression and function. Because rare synonymous variants may influence the expression and function of many proteins with a frequency similar to that of nonsynonymous variants, they may be increasingly recognized as being of significance in disease [Citation181]. For example, sequencing of 640,000 exomes resulted in the identification of GPR75 variants associated with protection from obesity [Citation182].
Conclusions
A wide spectrum of disease phenotypes, predispositions and traits are associated with variants in GPCRs. This includes disease-causing variants reported in at least 55 GPCRs for more than 66 monogenic diseases in human [Citation181]. Rare variants include LOF variants such as truncation and frameshift variants, missense variants with pathogenicity that can reflect constitutive activity, and synonymous variants that result in codon bias [Citation182]. Data on the pathogenic mechanisms of GPCR variant signaling in monogenic disease complements that from studies of engineered variant receptors. Collectively, these studies may suggest novel pharmaceutical interventions [Citation180] because variant receptors provide data for drug development specific to distinct phenotypes associated with GPCR disruption.
By contrast, with variants that are pathogenic in monogenic disease, many other variants confer risk for a disease or a phenotype consisting of altered pharmaceutical response [Citation3–5]. In this context, WES and WGS data will facilitate the analysis of rare variants associated with human phenotypes which have not been amenable to study so far [Citation183]. For example, two variants in the orphan QRFPR receptor for 26Rfa, c.202 T > C (p.Tyr68His) and c.1111C > T (p.Arg371Trp), were identified in two unrelated individuals with intellectual disability. Evaluation of these genetic variants by expressing them in vitro suggests that QRFPR variants, p.Tyr68His and p.Arg371Trp, reduce the mobilization of intracellular Ca2+ and down-stream signaling [Citation106]. The pathogenicity of these variants may result from the altered chain structure of adjacent amino acids that causes reduced binding of QRFPR to 26Rfa.
Agnostic strategies have also resulted in the identification of a c.1047G > T, p.Lys349Asn variant in GPR37L1, an orphan GPCR, in a rare familial progressive myoclonus epilepsy. These patients experience progressive neurological deterioration associated with refractory myoclonic and generalized seizures. Examination of a mouse model of this variant suggests that it results in loss of GPR37L1 function associated with elevated susceptibility to seizures [Citation184]. The agnostic strategies for the identification of variants contributing to human phenotypes made possible by NGS have facilitated not only the disambiguation of orphan receptors but also the identification of variants in those receptors that suggest potential interventions.
| Abbreviations | ||
| ACTHR | = | adrenocorticotrophic hormone receptor, MC2R |
| ADH | = | autosomal dominant hypocalcemia |
| AgRP | = | agouti-related protein |
| BRET | = | bioluminescence resonance energy transfer |
| CAM | = | constitutively active mutant |
| Ca2+e | = | extracellular calcium concentration |
| Ca2+i | = | ionized calcium concentration |
| CaSR | = | calcium-sensing receptor |
| EC50 | = | drug concentration resulting in half-maximal response |
| ECD | = | extracellular domain |
| EDN3 | = | endothelin 3 |
| EDNRB | = | endothelin-β receptor gene |
| EM | = | electron microscopy |
| ER | = | endoplasmic reticulum |
| ETA | = | endothelin-α |
| ETB | = | endothelin-β |
| FGD | = | familial glucocorticoid deficiency |
| FHH | = | familial hypocalciuric hypercalcemia |
| FSH | = | follicle stimulating hormone |
| FSHR | = | follicle stimulating hormone receptor |
| GNRH | = | gonadotropin releasing hormone |
| GNRHR | = | gonadotropin releasing hormone receptor |
| GOF | = | gain of function |
| GPCR | = | G protein-coupled receptor |
| GPR37L1 | = | G protein-coupled receptor L1 |
| GRK | = | G protein-coupled receptor kinase |
| ICD | = | intracellular domain |
| ICL | = | intracellular loop |
| IHH | = | idiopathic hypogonadotropic hypogonadism |
| JMC | = | Jansen-type metaphyseal chondrodysplasia |
| KS | = | Kallmann syndrome |
| LHCGR | = | luteinizing hormone/choriogonadotropin receptor |
| LOF | = | loss of function |
| MAPK | = | mitogen-activated protein kinase |
| MCR | = | melanocortin receptor |
| MC1R | = | melanocortin 1 receptor, melanocyte-stimulating hormone receptor |
| MC4R | = | melanocortin 4 receptor |
| MSH | = | melanocyte-stimulating hormone |
| NanoBiT | = | NanoLuc® Binary Technology |
| NDI | = | nephrogenic diabetes insipidus |
| NGS | = | next generation sequencing |
| NPSR1 | = | neuropeptide S receptor 1 |
| NSHPT | = | neonatal severe hyperparathyroidism |
| OHSS | = | ovarian hyper-stimulation syndrome |
| P2RY12 | = | purinergic receptor 12 |
| POMC | = | proopiomelanocortin |
| PROKR | = | prokineticin receptor |
| PTH | = | parathyroid hormone |
| PTH1R | = | parathyroid hormone 1 receptor |
| PTH2R | = | parathyroid hormone 1 receptor |
| PTHrP | = | PTH-related protein |
| RP | = | retinitis pigmentosa |
| SNV | = | single nucleotide variant |
| TM | = | transmembrane spanning helix, e.g. TM3 |
| TMD | = | transmembrane domain |
| TSH | = | thyroid stimulating hormone |
| TSHR | = | thyroid stimulating hormone receptor |
| VFTD | = | bilobed Venus Flytrap domain |
| VUS | = | variant of unknown significance |
| WES | = | whole exome sequencing |
| WGS | = | whole genome sequencing |
Acknowledgments
We dedicate this work to the memory of a “fine chap”, Dr. Geoff Hendy, Department of Medicine, McGill University, and Calcium Research Laboratory, Royal Victoria Hospital, Montreal, Quebec, Canada.
Disclosure statement
The authors have no conflicts of interest to declare.
Additional information
Funding
References
- Spiegel AM. Introduction to G-protein coupled signal transduction and human disease. In: Spiegel AM, editor. G proteins, receptors, and disease. Totowa (NJ): Humana Press; 1998:1–21.
- Hauser AS, Attwood MM, Rask-Andersen M, et al. Trends in GPCR drug discovery: new agents, targets and indications. Nat Rev Drug Discov. 2017;16(12):829–842. doi: 10.1038/nrd.2017.178.
- Hauser AS, Chavali S, Masuho I, et al. Pharmacogenomics of GPCR drug targets. Cell. 2018;172(1–2):41–54.e19. doi: 10.1016/j.cell.2017.11.033.
- Hauser AS. Personalized medicine through GPCR pharmacogenomics. In: Kenakin T, editor. Comprehensive pharmacology. Amsterdam (NL): Elsevier; 2021. p. 2022191–2022219.
- Hauser AS, Gloriam DE, Bräuner-Osborne H, et al. Novel approaches leading towards peptide GPCR de-orphanisation. Br J Pharmacol. 2019;177(5):961–968. doi: 10.1111/bph.14950.
- Shrivastava A, Mathur K, Verma RK, et al. Molecular dynamics study of tropical calcific pancreatitis (TCP) associated calcium-sensing receptor single nucleotide variation. Front Mol Biosci. 2022;9:982831. doi: 10.3389/fmolb.2022.982831.
- Tao Y-T. Inactivating mutations of G protein-coupled receptors and diseases: structure-function insights and therapeutic implications. Pharmacol Ther. 2006;111(3):949–973. doi: 10.1016/j.pharmthera.2006.02.008.
- Tao Y-T, Conn PM. Chaperoning G protein-coupled receptors: from cell biology to therapeutics. Endocr Rev. 2014;35(4):602–647. doi: 10.1210/er.2013-1121.
- Tao Y-X. Constitutive activation of G protein-coupled receptors and diseases: insights into mechanisms of activation and therapeutics. Pharmacol Ther. 2008;120(2):129–148. doi: 10.1016/j.pharmthera.2008.07.005.
- Daiger SP, Sullivan LS, Bowne SJ. Genes and mutations causing retinitis pigmentosa. Clin Genet. 2013;84(2):132–141. doi: 10.1111/cge.12203.
- Roushar FJ, McKee AG, Kuntz CP, et al. Molecular basis for variations in the sensitivity of pathogenic rhodopsin variants to 9-cis-retinal. J Biol Chem. 2022;298(8):102266. doi: 10.1016/j.jbc.2022.102266.
- Dryja TP, McGee TL, Reichel E, et al. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990;343(6256):364–366. doi: 10.1038/343364a0.
- Sudharsan R, Beltran WA. Progress in gene therapy for rhodopsin autosomal dominant retinitis pigmentosa. Adv Exp Med Biol. 2019;1185:113–118. doi: 10.1007/978-3-030-27378-1_19.
- Rim J, Oprian DD. Constitutive activation of opsin—interaction of mutants with rhodopsin kinase and arrestin. Biochemistry. 1995;34(37):11938–11945. doi: 10.1021/bi00037a035.
- Bunge S, Wedemann H, David D, et al. Molecular analysis and genetic-mapping of the rhodopsin gene in families with autosomal dominant retinitis pigmentosa. Genomics. 1993;17(1):230–233. doi: 10.1006/geno.1993.1309.
- Farrar GJ, Mcwilliam P, Bradley DG, et al. Autosomal dominant retinitis pigmentosa—linkage to rhodopsin and evidence for genetic-heterogeneity. Genomics. 1990;8(1):35–40. doi: 10.1016/0888-7543(90)90223-h.
- Inglehearn CF, Lester DH, Bashir R, et al. Recombination between rhodopsin and locus D3S47 (C17) in rhodopsin retinitis pigmentosa families. Am J Hum Genet. 1992;50(3):590–597.
- Neidhardt J, Barthelmes D, Farahmand F, et al. Different amino acid substitutions at the same position in rhodopsin lead to distinct phenotypes. Invest Ophthalmol Vis Sci. 2006;47(4):1630–1635. doi: 10.1167/iovs.05-1317.
- Andres A, Kosoy A, Garriga P, et al. Mutations at position 125 in transmembrane helix III of rhodopsin affect the structure and signalling of the receptor. Eur J Biochem. 2001;268(22):5696–5704. doi: 10.1046/j.0014-2956.2001.02509.x.
- Jones EM, Lubock NB, Venkatakrishnan AJ, et al. Structural and functional characterization of G protein-coupled receptors with deep mutational scanning. Elife. 2020;9:e54895. doi: 10.7554/eLife.54895.
- Wan A, Place E, Pierce EA, et al. Characterizing variants of unknown significance in rhodopsin: a functional genomics approach. Hum Mutat. 2019;40(8):1127–1144. doi: 10.1002/humu.23762.
- Waheed NK, Qavi AH, Malik SN, et al. A nonsense mutation in S-antigen (p.Glu306*) causes Oguchi disease. Mol Vis. 2012;18:1253–1259.
- Fuchs S, Nakazawa M, Maw M, et al. A homozygous 1-base pair deletion in the Arrestin gene is a frequent cause of Oguchi disease in Japanese. Nat Genet. 1995;10(3):360–362. doi: 10.1038/ng0795-360.
- Iiri T, Herzmark P, Nakamoto JM, et al. Rapid gdp release from G(S-alpha) in patients with gain and loss of endocrine function. Nature. 1994;371(6493):164–168. doi: 10.1038/371164a0.
- Gromoll J, Schulz A, Borta H, et al. Homozygous mutation within the conserved Ala-Phe-Asn-Glu-Thr motif of exon 7 of the LH receptor causes male pseudohermaphroditism. Eur J Endocrinol. 2002;147(5):597–608. doi: 10.1530/eje.0.1470597.
- Althumairy A, Zhang X, Baez N, et al. Glycoprotein G-protein coupled receptors in disease: luteinizing hormone receptors and follicle stimulating hormone receptors. Diseases. 2020;8(3):35. doi: 10.3390/diseases8030035.
- Casas-González P, Scaglia HE, Pérez-Solís MA, et al. Normal testicular function without detectable follicle-stimulating hormone. A novel mutation in the follicle stimulating hormone receptor gene leading to apparent constitutive activity and impaired agonist-induced desensitization and internalization. Mol Cell Endocrinol. 2012;364(1-2):71–82. doi: 10.1016/j.mce.2012.08.011.
- Kosugi K, Van Dop C, Geffner ME, et al. Characterization of heterogeneous mutations causing constitutive activation of the luteinizing hormone receptor in familial male precocious puberty. Hum Mol Genet. 2017;4(2):183–188. doi: 10.1093/hmg/4.2.183.
- Topaloğlu AK. Update on the genetics of idiopathic hypogonadotropic hypogonadism. J Clin Res Pediatr Endocrinol. 2017;9(Suppl 2):113–122. doi: 10.4274/jcrpe.2017.S010.
- Zhao Y, Wu J, Wang X, et al. Prokineticins and their G protein-coupled receptors in health and disease. Prog Mol Biol Transl Sci. 2019;161:149–179.
- de Roux N, Genin E, Carel JC, et al. Hypogonadotropic hypogonadism due to loss of function of the KiSS1-derived peptide receptor GPR54. Proc Natl Acad Sci U S A. 2003;100(19):10972–10976. doi: 10.1073/pnas.1834399100.
- Franco B, Guioli S, Pragliola A, et al. A gene deleted in kallmanns syndrome shares homology with neural cell-adhesion and axonal path-fi nding molecules. Nature. 1991;353(6344):529–536. doi: 10.1038/353529a0.
- Legouis R, Hardelin JP, Levilliers J, et al. The candidate gene for the X-linked kallmann syndrome encodes a protein related to adhesion molecules. Cell. 1991;67(2):423–435. doi: 10.1016/0092-8674(91)90193-3.
- Habiby RL, Boepple P, Nachtigall L, et al. Adrenal hypoplasia congenita with hypogonadotropic hypogonadism—evidence that DAX-1 mutations lead to combined hypothalamic and pituitary defects in gonadotropin production. J Clin Invest. 1996;98(4):1055–1062. doi: 10.1172/JCI118866.
- Cham G, O'Brien B, Kimble RM. Idiopathic hypogonadotropic hypogonadism: a rare cause of primary amenorrhoea in adolescence-a review and update on diagnosis, management and advances in genetic understanding. BMJ Case Rep. 2021;14(4):e239495. doi: 10.1136/bcr-2020-239495.
- Monnier C, Dodé C, Fabre L, et al. PROKR2 missense mutations associated with kallmann syndrome impair receptor signalling activity. Hum Mol Genet. 2009;18(1):75–81. doi: 10.1093/hmg/ddn318.
- Topaloglu AK, Reimann F, Guclu M, et al. TAC3 and TACR3 mutations in familial hypogonadotropic hypogonadism reveal a key role for neurokinin B in the Central control of reproduction. Nat Genet. 2009;41(3):354–358. doi: 10.1038/ng.306.
- de Roux N, Young J, Misrahi M, et al. A family with hypogonadotropic hypogonadism and mutations in the gonadotropin-releasing hormone receptor. N Engl J Med. 1997;337(22):1597–1602. doi: 10.1056/NEJM199711273372205.
- Layman LC, Cohen DP, Jin M, et al. Mutations in gonadotropin-releasing hormone receptor gene cause hypogonadotropic hypogonadism. Nat Genet. 1998;18(1):14–15. doi: 10.1038/ng0198-14.
- Ahmad S, Ali MZ, Abbasi SW, et al. A GHRHR founder mutation causes isolated growth hormone deficiency type IV in a consanguineous pakistani family. Front Endocrinol (Lausanne). 2023;14:1066182. doi: 10.3389/fendo.2023.1066182.
- Finch AR, Caunt CJ, Armstrong SP, et al. Plasma membrane expression of gonadotropin-releasing hormone receptors: regulation by peptide and nonpeptide antagonists. Mol Endocrinol. 2010;24(2):423–435. doi: 10.1210/me.2009-0343.
- Martin TJ, Sims NA, Seeman E. Physiological and pharmacological roles of PTH and PTHrP in bone using their shared receptor, PTH1R. Endocr Rev. 2021;42(4):383–406. doi: 10.1210/endrev/bnab005.
- Ewers M, Canaff L, Weh AE, et al. The three common polymorphisms p.A986S, p.R990G and p.Q1011E in the calcium sensing receptor (CASR) are not associated with chronic pancreatitis. Pancreatology. 2021;21(7):1299–1304. doi: 10.1016/j.pan.2021.08.008.
- Stepanchick A, McKenna J, McGovern O, et al. Calcium sensing receptor mutations implicated in pancreatitis and idiopathic epilepsy syndrome disrupt an arginine-rich retention motif. Cell Physiol Biochem. 2010;26(3):363–374. doi: 10.1159/000320560.
- Sutkeviciute I, Clark LJ, White AD, et al. PTH/PTHrP receptor signaling, allostery, and structures. Trends Endocrinol Metab. 2019;30(11):860–874. doi: 10.1016/j.tem.2019.07.011.
- Saito H, Noda H, Gatault P, et al. Progression of mineral ion abnormalities in patients with jansen metaphyseal chondrodysplasia. J Clin Endocrinol Metab. 2018;103(7):2660–2669. doi: 10.1210/jc.2018-00332.
- Noda H, Guo J, Khatri A, et al. An inverse agonist ligand of the PTH receptor partially rescues skeletal defects in a mouse model of jansen’s metaphyseal chondrodysplasia. J Bone Miner Res. 2020;35(3):540–549. doi: 10.1002/jbmr.3913.
- Hoogendam J, Farih-Sips H, Wÿnaendts LC, et al. Novel mutations in the parathyroid hormone (PTH)/PTH-related peptide receptor type 1 causing blomstrand osteochondrodysplasia types I and II. J Clin Endocrinol Metab. 2007;92(3):1088–1095. doi: 10.1210/jc.2006-0300.
- Duchatelet S, Ostergaard E, Cortes D, et al. Recessive mutations in PTHR1 cause contrasting skeletal dysplasias in eiken and blomstrand syndromes. Hum Mol Genet. 2005;14(1):1–5. doi: 10.1093/hmg/ddi001.
- Portales-Castillo I, Dean T, Cheloha RW, et al. Altered signaling and desensitization responses in PTH1R mutants associated with eiken syndrome. Commun Biol. 2023;6(1):599. doi: 10.1038/s42003-023-04966-0.
- Duprez L, Parma J, Van Sande J, et al. Germline mutations in the thyrotropin receptor gene cause non-autoimmune autosomal dominant hyperthyroidism. Nat Genet. 1994;7(3):396–401. doi: 10.1038/ng0794-396.
- Arturi F, Capula C, Chiefari E, et al. Thyroid hyperfunctioning adenomas with and without gsp/TSH receptor mutations show similar clinical features. Exp Clin Endocrinol Diabetes. 1998;106(3):234–236. doi: 10.1055/s-0029-1211982.
- Larrivée-Vanier S, Magne F, Hamdoun E, et al. Severe congenital hypothyroidism due to a novel deep intronic mutation in the TSH receptor gene causing intron retention. J Endocr Soc. 2020;5(3):bvaa183. doi: 10.1210/jendso/bvaa183.
- Biebermann H, Schoneberg T, Hess C, et al. The first activating TSH receptor mutation in transmembrane domain 1 identified in a family with nonautoimmune hyperthyroidism. J Clin Endocrinol Metab. 2001;86(9):4429–4433. doi: 10.1210/jcem.86.9.7888.
- Jordan N, Williams N, Gregory JW, et al. The W546X mutation of the thyrotropin receptor gene: potential major contributor to thyroid dysfunction in a caucasian population. J Clin Endocrinol Metab. 2003;88(3):1002–1005. doi: 10.1210/jc.2002-021301.
- Biebermann H, Winkler F, Kleinau G. Genetic defects, thyroid growth and malfunctions of the TSHR in pediatric patients. Front Biosci (Landmark Ed). 2010;15(3):913–933. doi: 10.2741/3654.
- Hébrant A, Van Staveren WCG, Maenhaut C, et al. Genetic hyperthyroidism: hyperthyroidism due to activating TSHR mutations. Eur J Endocrinol. 2011;164(1):1–9. doi: 10.1530/EJE-10-0775.
- Gabriel EM, Bergert ER, Grant CS, et al. Germline polymorphism of codon 727 of human thyroid-stimulating hormone receptor is associated with toxic multinodular goiter. J Clin Endocrinol Metab. 1999;84(9):3328–3335. doi: 10.1210/jc.84.9.3328.
- Biebermann H, Schoneberg T, Krude H, et al. Constitutively activating TSH receptor mutations as a molecular cause of non-autoimmune hyperthyroidism in childhood. Langenbecks Arch Surg. 2000;385(6):390–392. doi: 10.1007/s004230000145.
- Karges B, Krause G, Homoki J, et al. TSH receptor mutation V509A causes familial hyperthyroidism by release of interhelical constraints between transmembrane helices TMH3 and TMH5. J Endocrinol. 2005;186(2):377–385. doi: 10.1677/joe.1.06208.
- Tonacchera M, Agretti P, Chiovato L, et al. Activating thyrotropin receptor mutations are present in nonadenomatous hyperfunctioning nodules of toxic or autonomous multinodular goiter. J Clin Endocrinol Metab. 2000;85(6):2270–2274. doi: 10.1210/jcem.85.6.6634.
- Lueblinghoff J, Eszlinger M, Jaeschke H, et al. Shared sporadic and somatic thyrotropin receptor mutations display more active in vitro activities than familial thyrotropin receptor mutations. Thyroid. 2011;21(3):221–229. doi: 10.1089/thy.2010.0312.
- Boelen A, Kwakkel J, Fliers E. Thyroid hormone receptors in health and disease. Minerva Endocrinol. 2012;37(4):291–304.
- Marín-Sánchez A, Álvarez-Sierra D, González O, et al. Regulation of TSHR expression in the thyroid and thymus may contribute to TSHR tolerance failure in graves’ disease patients via two distinct mechanisms. Front Immunol. 2019;10:1695. doi: 10.3389/fimmu.2019.01695.
- Brand OJ, Barrett JC, Simmonds MJ, et al. Association of the thyroid stimulating hormone receptor gene (TSHR) with Graves’ disease. Hum Mol Genet. 2009;18(9):1704–1713. doi: 10.1093/hmg/ddp087.
- Noel SD, Kaiser UB. G protein-coupled receptors involved in GnRH regulation: molecular insights from human disease. Mol Cell Endocrinol. 2011;346(1-2):91–101. doi: 10.1016/j.mce.2011.06.022.
- Zhang R, Linpeng S, Li Z, et al. Deficiency in GnRH receptor trafficking due to a novel homozygous mutation causes idiopathic hypogonadotropic hypogonadism in three prepubertal siblings. Gene. 2018;669:42–46. doi: 10.1016/j.gene.2018.05.050.
- Wang L, Lin W, Li X, et al. A case report of congenital idiopathic hypogonadotropic hypogonadism caused by novel mutation of GNRHR gene. Medicine (Baltimore). 2021;2021;100(5):e24007. doi: 10.1097/MD.0000000000024007.
- Chen Y, Sun T, Niu Y, et al. A partial loss-of-function variant in GNRNR gene in a chinese cohort with idiopathic hypogonadotropic hypogonadism. Transl Androl Urol. 2021;10(4):1676–1687. doi: 10.21037/tau-20-1390.
- Spanakis E, Milord E, Gragnoli C. AVPR2 variants and mutations in nephrogenic diabetes insipidus: review and missense mutation significance. J Cell Physiol. 2008;217(3):605–617. doi: 10.1002/jcp.21552.
- Bichet DG, El Tarazi A, Matar J, et al. Aquaporin-2: new mutations responsible for autosomal recessive nephrogenic diabetes insipidus—update and epidemiology. Clin Kidney J. 2012;5(3):195–202. doi: 10.1093/ckj/sfs029.
- Rosenthal W, Seibold A, Antaramian A, et al. Molecular identification of the gene responsible for congenital nephrogenic diabetes insipidus. Nature. 1992;359(6392):233–235. doi: 10.1038/359233a0.
- Feinstein TN, Yui N, Webber MJ, et al. Noncanonical control of vasopressin receptor type 2 signaling by retromer and arrestin. J Biol Chem. 2013;288(39):27849–27860. doi: 10.1074/jbc.M112.445098.
- Los EL, Deen PM, Robben JH. Potential of nonpeptide (ant)agonists to rescue vasopressin V2 receptor mutants for the treatment of X-linked nephrogenic diabetes insipidus. J Neuroendocrinol. 2010;22(5):393–399. doi: 10.1111/j.1365-2826.2010.01983.x.
- McKusick VA. Mendelian inheritance in man and its online version, OMIM. Am J Hum Genet. 2007;80(4):588–604. doi: 10.1086/514346.
- Fujiwara TM, Bichet DG. Molecular biology of hereditary diabetes insipidus. J Am Soc Nephrol. 2005;16(10):2836–2846. doi: 10.1681/ASN.2005040371.
- Arthus M-F, Lonergan M, Crumley MJ, et al. Report of 33 novel AVPR2 mutations and analysis of 117 families with X-linked nephrogenic diabetes insipidus. J Am Soc Nephrol. 2000;11(6):1044–1054. doi: 10.1681/ASN.V1161044.
- Nomura Y, Onigata K, Nagashima T, et al. Detection of skewed X-inactivation in two female carriers of vasopressin type 2 receptor gene mutation. J Clin Endocrinol Metab. 1997;82(10):3434–3437. doi: 10.1210/jcem.82.10.4312.
- Bichet DG, Arthus M-F, Lonergan M, et al. X-linked nephrogenic diabetes insipidus mutations in North america and the hopewell hypothesis. J Clin Invest. 1993;92(3):1262–1268. doi: 10.1172/JCI116698.
- Hobbs HH, Russell DW, Brown MS, et al. The LDL receptor locus in familial hypercholesterolemia: mutational analysis of a membrane protein. Annu Rev Genet. 1990;24(1):133–170. doi: 10.1146/annurev.ge.24.120190.001025.
- Wuller S, Wiesner B, Loffl Er A, et al. Pharmacochaperones post-translationally enhance cell surface expression by increasing conformational stability of wild-type and mutant vasopressin V2 receptors. J Biol Chem. 2004;279(45):47254–47263. doi: 10.1074/jbc.M408154200.
- Hermosilla R, Oueslati M, Donalies U, et al. Disease-causing V(2) vasopressin receptors are retained in different compartments of the early secretory pathway. Traffic. 2004;5(12):993–1005. doi: 10.1111/j.1600-0854.2004.00239.x.
- Bernier V, Morello JP, Zarruk A, et al. Pharmacologic chaperones as a potential treatment for X-linked nephrogenic diabetes insipidus. J Am Soc Nephrol. 2006;17(1):232–243. doi: 10.1681/ASN.2005080854.
- Morello JP, Salahpour A, Laperrière A, et al. Pharmacological chaperones rescue cell-surface expression and function of misfolded V2 vasopressin receptor mutants. J Clin Invest. 2000;105(7):887–895. doi: 10.1172/JCI8688.
- Schoneberg T, Schulz A, Biebermann H, et al. Mutant G-protein-coupled receptors as a cause of human diseases. Pharmacol Ther. 2004;104(3):173–206. doi: 10.1016/j.pharmthera.2004.08.008.
- Romisch K. A cure for traffic jams: small molecule chaperones in the endoplasmic reticulum. Traffic. 2004;5(11):815–820. doi: 10.1111/j.1600-0854.2004.00231.x.
- Tamarappoo BK, Verkman AS. Defective aquaporin-2 trafficking in nephrogenic diabetes insipidus and correction by chemical chaperones. J Clin Invest. 1998;101(10):2257–2267. doi: 10.1172/JCI2303.
- Kunchaparty S, Palcso M, Berkman J, et al. Defective processing and expression of thiazide-sensitive Na-Cl cotransporter as a cause of gitelman’s syndrome. Am J Physiol. 1999;277(4):F643–F649. doi: 10.1152/ajprenal.1999.277.4.F643.
- Hayama A, Rai T, Sasaki S, et al. Molecular mechanisms of bartter syndrome caused by mutations in the BSND gene. Histochem Cell Biol. 2003;119(6):485–493. doi: 10.1007/s00418-003-0535-2.
- Peters M, Ermert S, Jeck N, et al. Classification and rescue of ROMK mutations underlying hyperprostaglandin E syndrome/antenatal bartter syndrome. Kidney Int. 2003;64(3):923–932. doi: 10.1046/j.1523-1755.2003.00153.x.
- Chillaron J, Estevez R, Samarzija I, et al. An intracellular trafficking defect in type I cystinuria rBAT mutants M467T and M467K. J Biol Chem. 1997;272(14):9543–9549. doi: 10.1074/jbc.272.14.9543.
- Bonnardeaux A, Bichet DG. Inherited disorders of the renal tubule. In: Taal MW, Chertow GM, Marsden PA, Sko K, Yu ASL, Brenner BM, editors. Brenner & rector’s the kidney. Vol. 2. 9th ed. Philadelphia (PA): Elsevier Saunders; 2012:1584–1625.
- Lomas DA, Evans DL, Finch JT, et al. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature. 1992;357(6379):605–607. doi: 10.1038/357605a0.
- Lawless MW, Greene CM, Mulgrew A, et al. Activation of endoplasmic reticulum specific stress responses associated with the conformational disease Z alpha 1-antitrypsin deficiency. J Immunol. 2004;172(9):5722–5726. doi: 10.4049/jimmunol.172.9.5722.
- Cohen FE, Kelly JW. Therapeutic approaches to protein-misfolding diseases. Nature. 2003;426(6968):905–909. doi: 10.1038/nature02265.
- Ulloa-Aguirre A, Janovick JA, Brothers SP, et al. Pharmacologic rescue of conformationally- defective proteins: implications for the treatment of human disease. Traffic. 2004;5(11):821–837. doi: 10.1111/j.1600-0854.2004.00232.x.
- Bernier V, Lagace M, Lonergan M, et al. Functional rescue of the constitutively internalized V2 vasopressin receptor mutant R137H by the pharmacological chaperone action of SR49059. Mol Endocrinol. 2004;18(8):2074–2084. doi: 10.1210/me.2004-0080.
- Barak LS, Oakley RH, Laporte SA, et al. Constitutive arrestin-mediated desensitization of a human vasopressin receptor mutant associated with nephrogenic diabetes insipidus. Proc Natl Acad Sci U S A. 2001;98(1):93–98. doi: 10.1073/pnas.98.1.93.
- Soule S, Florkowski C, Potter H, et al. Intermittent severe, symptomatic hyponatraemia due to the nephrogenic syndrome of inappropriate antidiuresis. Ann Clin Biochem. 2008;45(Pt 5):520–523. doi: 10.1258/acb.2007.007211.
- Feldman BJ, Rosenthal SM, Vargas GA, et al. Nephrogenic syndrome of inappropriate antidiuresis. N Engl J Med. 2005;352(18):1884–1890. doi: 10.1056/NEJMoa042743.
- Marcialis MA, Faa V, Fanos V, et al. Neonatal onset of nephrogenic syndrome of inappropriate antidiuresis. Pediatr Nephrol. 2008;23(12):2267–2271. doi: 10.1007/s00467-008-0913-z.
- Zhang Y-J, Li D-J, Li Z-Y, et al. Influence of genetic polymorphisms in P2Y12 receptor signaling pathway on antiplatelet response to clopidogrel in coronary heart disease. BMC Cardiovasc Disord. 2022;22(1):575. doi: 10.1186/s12872-022-02988-w.
- Hofstra RM, Osinga J, Tan-Sindhunata G, et al. A homozygous mutation in the endothelin-3 gene associated with a combined waardenburg type 2 and hirschsprung phenotype (Shah-Waardenburg syndrome). Nat Genet. 1996;12(4):445–447. doi: 10.1038/ng0496-445.
- Hofstra RMW, Valdenaire O, Arch E, et al. A loss-of-function mutation in the endothelin-converting enzyme 1 (ECE-1) associated with hirschsprung disease, cardiac defects, and autonomic dysfunction. Am J Hum Genet. 1999;64(1):304–308. doi: 10.1086/302184.
- Fuchs S, Amiel J, Claudel S, et al. Functional characterization of three mutations of the endothelin B receptor gene in patients with hirschsprung’s disease: evidence for selective loss of G(i) coupling. Mol Med. 2001;7(2):115–124. doi: 10.1007/BF03401945.
- Li H, Lou R, Xu X, et al. The variations in human orphan G protein-coupled receptor QRFPR affect PI3K-AKT-mTOR signaling. J Clin Lab Anal. 2021;35(7):e23822. doi: 10.1002/jcla.23822.
- Brown EM. Role of the calcium-sensing receptor in extracellular calcium homeostasis. Best Pract Res Clin Endocrinol Metab. 2013;27(3):333–343. doi: 10.1016/j.beem.2013.02.006.
- Deng X, Xin Y, Miller CL, et al. Structural mechanism of cooperative regulation of calcium-sensing receptor-mediated cellular signaling. Curr Opin Physiol. 2020;17:269–277. doi: 10.1016/j.cophys.2020.08.020.
- Gao Y, Robertson MJ, Rahman SN, et al. Asymmetric activation of the calcium-sensing receptor homodimer. Nature. 2021;595(7867):455–459. doi: 10.1038/s41586-021-03691-0.
- Gorvin CM. Molecular and clinical insights from studies of calcium-sensing receptor mutations. J Mol Endocrinol. 2019;63(2):R1–R16. doi: 10.1530/JME-19-0104.
- Pidasheva S, D'Souza-Li L, Canaff L, et al. CASRdb: calcium-sensing receptor locus-specific database for mutations causing familial (benign) hypocalciuric hypercalcemia, neonatal severe hyperparathyroidism, and autosomal dominant hypocalcemia. Hum Mutat. 2004;24(2):107–111. doi: 10.1002/humu.20067.
- Marx SJ, Sinaii N. Neonatal severe hyperparathyroidism: novel insights from calcium, PTH, and the CASR gene. J Clin Endocrinol Metab. 2020;105(4):1061–1078. doi: 10.1210/clinem/dgz233.
- Christensen SE, Nissen PH, Vestergaard P, et al. Discriminative power of three indices of renal calcium excretion for the distinction between familial hypocalciuric hypercalcaemia and primary hyperparathyroidism: a follow-up study on methods. Clin Endocrinol (Oxf). 2008;69(5):713–720. doi: 10.1111/j.1365-2265.2008.03259.x.
- Roszko KL, Stapleton Smith LM, Sridhar AV, et al. Autosomal dominant hypocalcemia type 1: a systematic review. J Bone Miner Res. 2022;37(10):1926–1935. doi: 10.1002/jbmr.4659.
- Marx SJ. Calcimimetic use in familial hypocalciuric hypercalcemia-A perspective in endocrinology. J Clin Endocrinol Metab. 2017;102(11):3933–3936. doi: 10.1210/jc.2017-01606.
- Vahe C, Benomar K, Espiard S, et al. Diseases associated with calcium-sensing receptor. Orphanet J Rare Dis. 2017;12(1):19. doi: 10.1186/s13023-017-0570-z.
- Roberts MS, Gafni RI, Brillante B, et al. Treatment of autosomal dominant hypocalcemia type 1 with the calcilytic NPSP795 (SHP635). J Bone Miner Res. 2019;34(9):1609–1618. doi: 10.1002/jbmr.3747.
- Hannan FM, Babinsky VN, Thakker RV. Disorders of the calcium-sensing receptor and partner proteins: insights into the molecular basis of calcium homeostasis. J Mol Endocrinol. 2016;57(3):R127–R142. doi: 10.1530/JME-16-0124.
- Mayr B, Schnabel D, Dörr H-G, et al. Genetics in endocrinology: gain and loss of function mutations of the calcium-sensing receptor and associated proteins: current treatment concepts. Eur J Endocrinol. 2016;174(5):R189–R208. doi: 10.1530/EJE-15-1028.
- Roszko KL, Bi RD, Mannstadt M. Autosomal dominant hypocalcemia (hypoparathyroidism) types 1 and 2. Front Physiol. 2016;7:458. doi: 10.3389/fphys.2016.00458.
- Obermannova B, Sumnik Z, Dusatkova P, et al. Novel calcium-sensing receptor cytoplasmic tail deletion mutation causing autosomal dominant hypocalcemia: molecular and clinical study. Eur J Endocrinol. 2016;174(4):K1–K11. doi: 10.1530/EJE-15-1216.
- Cavaco BM, Canaff L, Nolin-Lapalme A, et al. Homozygous calcium-sensing receptor polymorphism R544Q presents as hypocalcemic hypoparathyroidism. J Clin Endocrinol Metab. 2018;103(8):2879–2888. doi: 10.1210/jc.2017-02407.
- Leach K, Wen A, Davey AE, et al. Identification of molecular phenotypes and biased signaling induced by naturally occurring mutations of the human calcium-sensing receptor. Endocrinology. 2012;153(9):4304–4316. doi: 10.1210/en.2012-1449.
- Gorvin CM, Frost M, Malinauskas T, et al. Calcium-sensing receptor residues with loss- and gain-of-function mutations are located in regions of conformational change and cause signalling bias. Hum Mol Genet. 2018;27(21):3720–3733. doi: 10.1093/hmg/ddy263.
- Makita N, Ando T, Sato J, et al. Cinacalcet corrects biased allosteric modulation of CaSR by AHH autoantibody. JCI Insight. 2019;4(8):e126449. doi: 10.1172/jci.insight.126449.
- Gorvin CM, Rogers A, Hastoy B, et al. AP2 sigma mutations impair calcium-sensing receptor trafficking and signaling, and show an endosomal pathway to spatially direct G-protein selectivity. Cell Rep. 2018;22(4):1054–1066. doi: 10.1016/j.celrep.2017.12.089.
- Dershem R, Gorvin CM, Metpally RPR, et al. Familial hypocalciuric hypercalcemia type 1 and autosomal-dominant hypocalcemia type 1: prevalence in a large healthcare population. Am J Hum Genet. 2020;106(6):734–747. doi: 10.1016/j.ajhg.2020.04.006.
- Cerani A, Zhou S, Forgetta V, et al. Genetic predisposition to increased serum calcium, bone mineral density, and fracture risk in individuals with normal calcium levels: mendelian randomisation study. BMJ. 2019;366:l4410. doi: 10.1136/bmj.l4410.
- Guerreiro R, Brás J, Batista S, et al. Pseudohypoparathyroidism type I-b with neurological involvement is associated with a homozygous PTH1R mutation. Genes Brain Behav. 2016;15(7):669–677. doi: 10.1111/gbb.12308.
- Couvineau A, Wouters V, Bertrand G, et al. PTHR1 mutations associated with ollier disease result in receptor loss of function. Hum Mol Genet. 2008;17(18):2766–2775. doi: 10.1093/hmg/ddn176.
- Roth H, Fritsche LG, Meier C, et al. Expanding the spectrum of PTH1R mutations in patients with primary failure of tooth eruption. Clin Oral Investig. 2014;18(2):377–384. doi: 10.1007/s00784-013-1014-3.
- Thompson MD, Sakurai T, Rainero I, et al. Orexin receptor multimerization versus functional interactions: neuropharmacological implications for opioid and cannabinoid signalling and pharmacogenetics. Pharmaceuticals (Basel). 2017;10(4):79. doi: 10.3390/ph10040079.
- Calbet-Llopart N, Combalia M, Kiroglu A, et al. Common genetic variants associated with melanoma risk or naevus count in patients with wildtype MC1R melanoma. Br J Dermatol. 2022;187(5):753–764. doi: 10.1111/bjd.21707.
- Tao Y-X. The melanocortin-4 receptor: physiology, pharmacology, and pathophysiology. Endocr Rev. 2010;31(4):506–543. doi: 10.1210/er.2009-0037.
- Hinney A, Volckmar A-L, Knoll N. Melanocortin-4 receptor in energy homeostasis and obesity pathogenesis. Prog Mol Biol Transl Sci. 2013;114:147–191. doi: 10.1016/B978-0-12-386933-3.00005-4.
- Qin L, Tiwari AK, Zai CC, et al. Regulation of melanocortin-4-receptor (MC4R) expression by SNP rs17066842 is dependent on glucose concentration. Eur Neuropsychopharmacol. 2020;37:39–48. doi: 10.1016/j.euroneuro.2020.05.008.
- Yeo GSH, Farooqi IS, Aminian S, et al. A frameshift mutation in MC4R associated with dominantly inherited human obesity. Nat Genet. 1998;20(2):111–112. doi: 10.1038/2404.
- Lubrano-Berthelier C, Le Stunff C, Bougneres P, et al. Clinical case seminar—a homozygous null mutation delineates the role of the melanocortin-4 receptor in humans. J Clin Endocrinol Metab. 2004;89(5):2028–2032. doi: 10.1210/jc.2003-031993.
- Wade KH, Lam BYH, Melvin A, et al. Loss-of-function mutations in the melanocortin 4 receptor in a UK birth cohort. Nat Med. 2021;27(6):1088–1096. doi: 10.1038/s41591-021-01349-y.
- Adan RAH, Kas MJH. Inverse agonism gains weight. Trends Pharmacol Sci. 2003;24(6):315–321. doi: 10.1016/S0165-6147(03)00130-5.
- Clément K, van den Akker E, Argente J; Setmelanotide POMC and LEPR Phase 3 Trial Investigators., et al. Efficacy and safety of setmelanotide, an MC4R agonist, in individuals with severe obesity due to LEPR or POMC deficiency: single-arm, open-label, multicentre, phase 3 trials. Lancet Diabetes Endocrinol. 2020;8(12):960–970. doi: 10.1016/S2213-8587(20)30364-8.
- Marsh DJ, Hollopeter G, Huszar D, et al. Response of melanocortin-4 receptor deficient mice to anorectic and orexigenic peptides. Nat Genet. 1999;21(1):119–122. doi: 10.1038/5070.
- Branson R, Potoczna N, Kral JG, et al. Binge eating as a major phenotype of melanocortin 4 receptor gene mutations. N Engl J Med. 2003;348(12):1096–1103. doi: 10.1056/NEJMoa021971.
- List JF, Habener JF. Defective melanocortin 4 receptors in hyperphagia and morbid obesity. N Engl J Med. 2003;348(12):1160–1163. doi: 10.1056/NEJMe030013.
- Gaulton KJ, Ferreira T, Lee Y, DIAbetes Genetics Replication And Meta-analysis (DIAGRAM) Consortium., et al. Genetic fine mapping and genomic annotation defines causal mechanisms at type 2 diabetes susceptibility loci. Nat Genet. 2015;47(12):1415–1425. doi: 10.1038/ng.3437.
- Vaisse C, Clement K, Durand E, et al. Melanocortin-4 receptor mutations are a frequent and heterogeneous cause of morbid obesity. J Clin Invest. 2000;106(2):253–262. doi: 10.1172/JCI9238.
- Clément K, Mosbah H, Poitou C. Rare genetic forms of obesity: from gene to therapy. Physiol Behav. 2020;227:113–134.
- Lotta LA, Mokrosiński J, Mendes de Oliveira E, et al. Human gain-of-function MC4R variants show signaling bias and protect against obesity. Cell. 2019;177(3):597–607.e9. doi: 10.1016/j.cell.2019.03.044.
- Brouwers B, Mendes de Oliveira E, Marti-Solano M, et al. Human MC4R variants affect endocytosis, trafficking and dimerization revealing multiple cellular mechanisms involved in weight regulation. Cell Rep. 2021;34(12):108862. doi: 10.1016/j.celrep.2021.108862.
- Weedon MN, Wright CF, Patel KA, et al. Unreliability of genotyping arrays for detecting very rare variants in human genetic studies: example from a recent study of MC4R. Cell. 2021;184(7):1651. doi: 10.1016/j.cell.2021.03.015.
- Lotta L, Langenberg C, Wareham NJ, et al. Reply to unreliability of genotyping arrays for detecting very rare variants in human genetic studies: example from a recent study of MC4R. Cell. 2021;184(7):1652–1653. doi: 10.1016/j.cell.2021.03.014.
- Piechowski CL, Rediger A, Lagemann C, et al. Inhibition of melanocortin-4 receptor dimerization by substitutions in intracellular loop 2. J Mol Endocrinol. 2013;51(1):109–118. doi: 10.1530/JME-13-0061.
- Baron M, Maillet J, Huyvaert M, et al. Loss-of-function mutations in MRAP2 are pathogenic in hyperphagic obesity with hyperglycemia and hypertension. Nat Med. 2019;25(11):1733–1738. doi: 10.1038/s41591-019-0622-0.
- Berruien NNA, Smith CL. Emerging roles of melanocortin receptor accessory proteins (MRAP and MRAP2) in physiology and pathophysiology. Gene. 2020;757:144949. doi: 10.1016/j.gene.2020.144949.
- Spana C, Jordan R, Fischkoff S. Of bremelanotide on body weight of obese women: data from two phase 1 randomized controlled trials. Diabetes Obes Metab. 2022Effect;24(6):1084–1093. doi: 10.1111/dom.14672.
- Tsigos C, Tsiotra P, Garibaldi LR, et al. Mutations of the ACTH receptor gene in a new family with isolated glucocorticoid deficiency. Mol Genet Metab. 2000;71(4):646–650. doi: 10.1006/mgme.2000.3090.
- Metherell LA, Naville D, Halaby G, et al. Nonclassic lipoid congenital adrenal hyperplasia masquerading as familial glucocorticoid deficiency. J Clin Endocrinol Metab. 2009;94(10):3865–3871. doi: 10.1210/jc.2009-0467.
- Marti-Solano M, Crilly SE, Malinverni D, et al. Combinatorial expression of GPCR isoforms affects signalling and drug responses. Nature. 2020;587(7835):650–656. doi: 10.1038/s41586-020-2888-2.
- Alviggi C, Conforti A, Santi D, et al. Clinical relevance of genetic variants of gonadotrophins and their receptors in controlled ovarian stimulation: a systematic review and meta-analysis. Hum Reprod Update. 2018;24(5):599–614. doi: 10.1093/humupd/dmy019.
- Gromoll J, Simoni M, Nieschlag E. An activating mutation of the follicle-stimulating hormone receptor autonomously sustains spermatogenesis in a hypophysectomized man. J Clin Endocrinol Metab. 1996;81(4):1367–1370. doi: 10.1210/jcem.81.4.8636335.
- Ulloa-Aguirre A, Zariñán T, Dias JA, et al. Mutations in G protein-coupled receptors that impact receptor trafficking and reproductive function. Mol Cell Endocrinol. 2014;382(1):411–423. doi: 10.1016/j.mce.2013.06.024.
- De LA, Montanelli L, Van DJ, et al. Presence and absence of follicle-stimulating hormone receptor mutations provide some insights into spontaneous ovarian hyperstimulation syndrome physiopathology. J Clin Endocrinol Metab. 2006;91(2):555–562. doi: 10.1210/jc.2005-1580.
- Meehan TP, Narayan P. Constitutively active luteinizing hormone receptors: consequences of in vivo expression. Mol Cell Endocrinol. 2007;260-262:294–300. doi: 10.1016/j.mce.2006.03.045.
- Decaux G, Vandergheynst F, Bouko Y, et al. Nephrogenic syndrome of inappropriate antidiuresis in adults: high phenotypic variability in men and women from a large pedigree. J Am Soc Nephrol. 2007;18(2):606–612. doi: 10.1681/ASN.2006090987.
- Carpentier E, Greenbaum LA, Rochdi D, et al. Identification and characterization of an activating F229V substitution in the V2 vasopressin receptor in an infant with NSIAD. J Am Soc Nephrol. 2012;23(10):1635–1640. doi: 10.1681/ASN.2012010077.
- Bous J, Orcel H, Floquet N, et al. Cryo-electron microscopy structure of the antidiuretic hormone arginine-vasopressin V2 receptor signaling complex. Sci Adv. 2021;7(21):eabg5628. doi: 10.1126/sciadv.abg5628.
- Kocan M, See HB, Sampaio NG, et al. Agonist-independent interactions between beta-arrestins and mutant vasopressin type II receptors associated with nephrogenic syndrome of inappropriate antidiuresis. Mol Endocrinol. 2009;23(4):559–571. doi: 10.1210/me.2008-0321.
- Ballesteros JA, Weinstein H. Integrated methods for the construction of three-dimensional models and computational probing of structure-function relations in G protein-coupled receptors. Methods Neurosci. 1995;25:366–428.
- Brooks A, Oostra BA, Hofstra RM. Studying the genetics of hirschsprung’s disease: unraveling an oligogenic disorder. Clin Genet. 2005;67(1):6–14. doi: 10.1111/j.1399-0004.2004.00319.x.
- Sánchez-Mejías A, Fernández RM, López Alonso M, et al. New roles of EDNRB and EDN3 in the pathogenesis of hirschsprung disease. Genet Med. 2010;12(1):39–43. doi: 10.1097/GIM.0b013e3181c371b0.
- Puffenberger EG, Hosoda K, Washington SS, et al. A missense mutation of the endothelin- B receptor gene in multigenic hirschsprung’s disease. Cell. 1994;79(7):1257–1266. doi: 10.1016/0092-8674(94)90016-7.
- Imamura F, Arimoto I, Fujiyoshi Y, et al. W276 mutation in the endothelin receptor subtype B impairs G(q) but not G(i) or G(o) coupling. Biochemistry. 2000;39(4):686–692. doi: 10.1021/bi991981z.
- Attie T, Till M, Pelet A, et al. Mutation of the endothelin-receptor-B gene in Waardenburg-Hirschsprung-disease. Hum Mol Genet. 1995;4(12):2407–2409. doi: 10.1093/hmg/4.12.2407.
- Hollopeter G, Jantzen HM, Vincent D, et al. Identification of the platelet ADP receptor targeted by antithrombotic drugs. Nature. 2001;409(6817):202–207. doi: 10.1038/35051599.
- Jefferson BK, Foster JH, McCarthy JJ, et al. Aspirin resistance and a single gene. Am J Cardiol. 2005;95(6):805–808. doi: 10.1016/j.amjcard.2004.11.045.
- Laitinen T, Polvi A, Rydman P, et al. Characterization of a common susceptibility locus for asthma-related traits. Science. 2004;304(5668):300–304. doi: 10.1126/science.1090010.
- Kormann MSD, Carr D, Klopp N, et al. G-protein-coupled receptor polymorphisms are associated with asthma in a large german population. Am J Respir Crit Care Med. 2005;171(12):1358–1362. doi: 10.1164/rccm.200410-1312OC.
- Melen E, Bruce S, Doekes G, et al. Haplotypes of G protein-coupled receptor 154 are associated with childhood allergy and asthma. Am J Respir Crit Care Med. 2005;171(10):1089–1095. doi: 10.1164/rccm.200410-1317OC.
- Pietras CO, Vendelin J, Anedda F, et al. The asthma candidate gene NPSR1 mediates isoform specific downstream signalling. BMC Pulm Med. 2011;11(1):39. doi: 10.1186/1471-2466-11-39.
- Schöneberg T, Liebscher I. Mutations in G protein–coupled receptors: mechanisms, pathophysiology and potential therapeutic approaches. Pharmacol Rev. 2021;73(1):89–119. doi: 10.1124/pharmrev.120.000011.
- Dershem R, Metpally RPR, Jeffreys K, et al. Rare-variant pathogenicity triage and inclusion of synonymous variants improves analysis of disease associations of orphan G protein-coupled receptors. J Biol Chem. 2019;294(48):18109–18121. doi: 10.1074/jbc.RA119.009253.
- Akbari P, Gilani A, Sosina O, et al. Sequencing of 640,000 exomes identifies GPR75 variants associated with protection from obesity. Science. 2021;373(6550):eabf8683. doi: 10.1126/science.abf8683.
- Thompson MD, Hendy GN, Percy ME, et al. G protein-coupled receptor mutations and human genetic disease. Methods Mol Biol. 2014;1175:153–187.
- Giddens MM, Wong JC, Schroeder JP, et al. GPR37L1 modulates seizure susceptibility: evidence from mouse studies and analyses of a human GPR37L1 variant. Neurobiol Dis. 2017;106:181–190. doi: 10.1016/j.nbd.2017.07.006.
- Socin H, Rubio Almanza M, Tomé Fernández-Ladreda M. Reproduction, smell, and neurodevelopmental disorders: genetic defects in different hypogonadotropic hypogonadal syndromes. Front Endocrinol (Lausanne). 2014;5:109. doi: 10.3389/fendo.2014.00109.
- Weston MD, Luijendijk MW, Humphrey KD, et al. Mutations in the VLGR1 gene implicate G-protein signaling in the pathogenesis of Usher syndrome type II. Am J Hum Genet. 2004;74:357–366. doi: 10.1086/381685.
- Zhou P, Meng H, Liang X, et al. ADGRV1 variants in febrile seizures/epilepsy with antecedent febrile seizures and their associations with audio-visual abnormalities. Front Mol Neurosci. 2022,15:864074. doi: 10.3389/fnmol.2022.864074.
- Wang Y, Huso D, Cahill H, et al. Progressive cerebellar, auditory, and esophageal dysfunction caused by targeted disruption of the frizzled-4 gene. J Neurosci. 2001;21:4761–4771.
- Xu Q, Wang Y, Dabdoub A, et al. Vascular development in the retina and inner ear: control by norrin and frizzled-4, a high-affinity ligand-receptor pair. Cell. 2004;116:883–895.
- Drenser KA, Dailey W, Vinekar A, et al. Clinical presentation and genetic correlation of patients with mutations affecting the FZD4 gene. Arch Ophthalmol. 2009;127(12):1649–1654. doi: 10.1001/archophthalmol.2009.322.
- van der Ende SR, Meyers BS, Capasso JE, et al. Severe familial exudative vitreoretinopathy, congenital hearing loss, and developmental delay in a child with biallelic variants in FZD4. JAMA Ophthalmol. 2022;140(9):889–893. doi: 10.1001/jamaophthalmol.2022.2914.
- Huang Y, Paxton WA, Wolinsky SM, et al. The role of a mutant CCR5 allele in HIV-1 transmission and disease progression. Nat Med. 1996;2(11):1240–1243. doi: 10.1038/nm1196-1240.
- Mohamed H, Gurrola T, Berman R, et al. Targeting CCR5 as a component of an HIV-1 therapeutic strategy. Wigdahl B Front Immunol. 2022;12:816515. doi: 10.3389/fimmu.2021.816515.