Abstract
The existence of thresholds for toxicants is a matter of debate in chemical risk assessment and regulation. Current risk assessment methods are based on the assumption that, in the absence of sufficient data, carcinogenesis does not have a threshold, while noncarcinogenic endpoints are assumed to be thresholded. Advances in our fundamental understanding of the events that underlie toxicity are providing opportunities to address these assumptions about thresholds. A key events dose-response analytic framework was used to evaluate three aspects of toxicity. The first section illustrates how a fundamental understanding of the mode of action for the hepatic toxicity and the hepatocarcinogenicity of chloroform in rodents can replace the assumption of low-dose linearity. The second section describes how advances in our understanding of the molecular aspects of carcinogenesis allow us to consider the critical steps in genotoxic carcinogenesis in a key events framework. The third section deals with the case of endocrine disrupters, where the most significant question regarding thresholds is the possible additivity to an endogenous background of hormonal activity. Each of the examples suggests that current assumptions about thresholds can be refined. Understanding inter-individual variability in the events involved in toxicological effects may enable a true population threshold(s) to be identified.
INTRODUCTION
The risk assessment of low molecular weight chemicals (which include many human and veterinary drugs, environmental contaminants, pesticides, industrial chemicals, food additives, and flavorings, etc.) depends considerably upon conclusions about the nature of the dose-response relationship for the critical toxicological effect. Specifically, the approach to chemicals for which there is no evidence of a threshold in their dose-response curve is different from that of chemicals where a threshold is observed or assumed. In the former situations, the approach also varies with jurisdiction. For example, in the United States, depending on the intended use of the chemical, linear extrapolation from a point of departure on the dose-response curve to a virtually safe dose may be undertaken (). In Europe, for substances whose exposure is avoidable, the ALARA (as-low-as-reasonably-achievable) principle may be invoked. Recently, for those substances where exposure may be difficult or impossible to avoid, a margin of exposure (MOE) between a point of departure and estimated human exposure is calculated to help provide advice on the relative magnitude of the risk and the priority for risk management action (CitationBarlow et al., 2006).
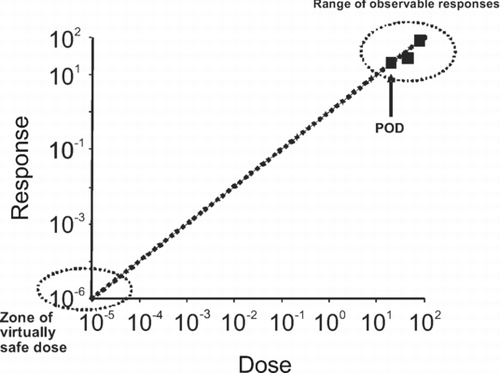
Figure 1 Extrapolation to human exposure when the dose-response curve cannot be assumed to exhibit a threshold. The data in the observable range (typically a response of 10–100% in a cancer bioassay) are used to determine a point of departure (POD) (e.g. NOAEL, BMDL10). The POD is used to extrapolate linearly to a virtually safe dose, that is usually associated with a risk of 1 in 105 or 1 in 106, the choice of value being a risk management decision. Note that the axes are plotted on logarithmic scales, because of the range over which extrapolation is necessary.
Where compounds are thought to exhibit a threshold in their dose-response curve, a deterministic approach is often adopted in assessing acceptability of exposure (CitationBoobis, 2007). A point of departure on the dose-response curve is divided by an uncertainty (or safety or adjustment) factor, to allow for inter-species and inter-individual differences, thereby identifying the equivalent point of departure on the dose-response curve for a sensitive individual (). In the absence of specific information, a default value is used for the uncertainty factor, while if specific information is available on either inter-species or inter-individual differences in toxicokinetics or toxicodynamics, this can be used to derive a chemical specific adjustment factor, as described by IPCS (CitationMeek et al., 2002).
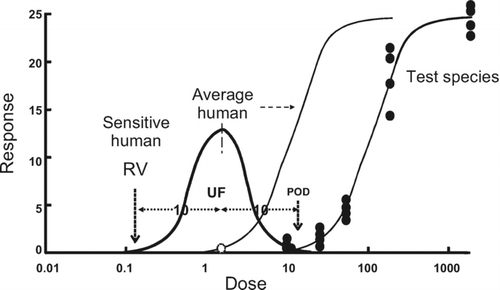
Figure 2 Extrapolation to human exposure when the dose response-curve can be assumed to exhibit a threshold. The data in the observable range are used to determine a point of departure (POD) (e.g. NOAEL, BMDL10). The POD is used to determine the equivalent dose in a sensitive human by dividing it by an uncertainty factor, to allow for possible inter-species and inter-individual differences. The default values are normally 10-fold. The inter-species factor is used to determine the POD in an average human (leftward shifted sigmoidal curve). Within the population there will be a range of sensitivities, reflected by a distribution of PODs (Gaussian curve). The factor for inter-individual sensitivity is used to determine the POD in a sensitive subject, which will lie towards the far left of the distribution curve. This POD has been termed a reference value (RV), for example the tolerable daily intake (TDI).
There is therefore considerable interest in improving methods for establishing whether a toxicological response exhibits a threshold and in developing procedures for identifying such thresholds. These thresholds in the dose-response curve for a toxicological response in a genetically (relatively) homogeneous species have been termed biological thresholds.Footnote 1 In contrast, within the human population each individual will exhibit a different sensitivity, giving rise to the concept of a population threshold, a dose below which no one is sensitive (see ).
The sensitivity of all experimental studies in laboratory species or humans and observational (epidemiological) studies in humans is limited by factors such as the number of observational units (e.g. volunteers), the frequency of the response, and the sensitivity (or reliability) of the detection methods. Hence, it is not possible to prove the existence of a threshold by empirical observation (although it may be possible to prove nonlinearity of the dose-response curve). Currently, conclusions on whether a specific toxicological response exhibits a threshold are based on a combination of observation, biological plausibility, and accumulated experience. However, the view has been expressed recently that the risk assessment of effects previously based on a threshold approach should move to linear low-dose extrapolation (e.g. CitationWhite et al., 2009). The development of a systematic approach to explore whether a given toxicological response does exhibit a biological threshold using a key events dose-response framework based on the mode of action responds to this concern.
The International Life Sciences Institute Research Foundation (ILSI RF) convened an international group of experts with tripartite composition, drawn from industry, academia, and government agencies, to address this question (see CitationJulien et al., 2009, in this issue). The group addressed a number of classes of substance—nutrients, allergens, microbial agents, and chemicals. As part of the exercise, case studies were developed to examine the utility of the approach proposed and to identify data gaps and research needs. This manuscript describes case studies applying the framework described below to chemicals, including chloroform as a member of the group of nonDNA-reactive, nonmutagenic carcinogens, DNA-reactive carcinogens, and endocrine-disrupting chemicals as a representative class of noncarcinogens.
THE KEY EVENTS DOSE-RESPONSE FRAMEWORK
As discussed above, due to limitations that all studies have, it is not possible to prove the existence of a biological threshold by observation alone. Hence, a mechanistic approach is required. The approach adopted was based on the concept of the mode of action (MOA), refined for chemical risk assessment over the past decade or so, particularly with the development by the WHO International Programme on Chemical Safety (IPCS) and ILSI (CitationSonich-Mullin et al., 2001; CitationMeek et al., 2003; CitationBoobis et al., 2006, Citation2008) of an analytic framework based on systematic evaluation of key events. A mode of action for a toxicological effect is a biologically plausible sequence of key events, starting with the interaction of an agent with a cell, through functional and anatomical changes, leading to an observed effect supported by robust experimental observations and mechanistic data. A key event is defined as an empirically observable, precursor step that is a necessary element of the mode of action, or is a marker for such an element (). The relevance of a key event in a particular mode of action, and of the mode of action overall, are evaluated using a weight of evidence approach based on the Bradford Hill criteria (CitationSonich-Mullin et al., 2001). The hypothesis is that by identifying the key events for a specific toxicological effect, these would be more amenable to the experimental demonstration of a threshold than the effect itself. It would be possible to utilize information on specific interventions, biological and chemical analogy, and the genetic differences in exploring the evidence for thresholds in a key event. Key events are, in principle, quantifiable by definition. An analysis of dose-response concordance should enable the identification of a rate limiting key event for the MOA and whether any one key event is the critical determinant of the response.
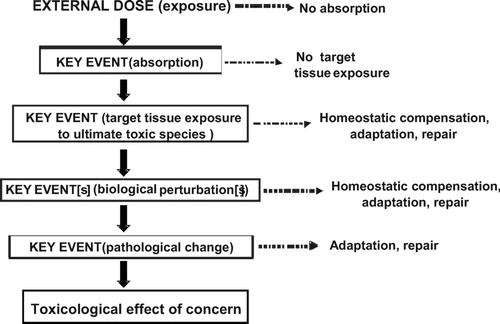
Figure 3 A generic representation of a mode of action for a toxicological effect as a series of key events. Note that often the early (upper) key events will be primarily toxicokinetic while the later (lower) ones will be largely toxicodynamic. Each key event is necessary but not sufficient to produce the toxicological effect. Each key event will exhibit its own dose-response curve, and possibly a threshold. The presence of a threshold, determined by the factors illustrated to the right hand side of the figure, will dictate whether there is progression to the next key event at a given dose. The toxicological effect will be manifest only if all key events operate.
The importance of this approach is reflected in the views of a number of regulatory agencies. For example, in the current US EPA Guidelines for Cancer Risk Assessment (CitationUS EPA, 2005), the following points are emphasized:
“When adequate data on mode of action provide sufficient evidence to support a nonlinear mode of action for the general population and/or any subpopulations of concern …—a reference dose/reference concentration that assumes that nonlinearity—is used.” “A sufficient basis to support this nonlinear procedure is likely to include data on responses that are key events integral to the carcinogenic process.” “This means that the POD may be based on these precursor response data, for example, hormone levels or mitogenic effects rather than tumor incidence data.”
This provides a real incentive to develop a robust approach for establishing nonlinearity of the dose-response curve using a mode of action, key events dose-response framework. Risk assessment based on demonstrable thresholds would increase the confidence of both risk managers and consumers. It would ensure the protection of public health, while not jeopardizing socio-economic development.
CASE STUDY: CHLOROFORM
Among the case studies considered by the ILSI RF working group, examples of target organ toxicity and cancer by a nongenotoxic mechanism were sought. Chloroform was considered a useful example, as it is both toxic to target organs and carcinogenic by a nongenotoxic mode of action. The evidence that chloroform causes cancer by a nongenotoxic mode of action has been reviewed on a number of occasions. In his review, CitationKomulainen (2004) stated that “The mainly negative and inconsistent genotoxicity data suggest that neither chloroform nor its metabolites react directly with DNA to cause DNA damage.” Similarly, in a comprehensive evaluation of the toxicity and carcinogenicity of chloroform published in 2004, IPCS concluded that “most studies did not identify genotoxic potential for chloroform. Results from a few, nonstandard studies indicate the possibility of a weak positive response in rats. Overall, however, the weight of evidence indicates that chloroform does not have significant genotoxic potential.”
Hence, chloroform can serve as a case study for both the toxicity to target organs and carcinogenicity by a nongenotoxic mode of action. In addition, it is a well-studied chemical, with a wealth of information available in the public domain. Chloroform is nephrotoxic, nephrocarcinogenic, hepatotoxic, and hepatocarcinogenic, depending on the species, the strain, and sex. There is evidence that all of these effects share key events and hence, will be considered together.
Hypothesized Mode of Action
Chloroform is metabolized by CYP2E1 to a cytotoxic product (phosgene), which can bind covalently to tissue macromolecules, including proteins, and can generate reactive oxygen species. In those tissues with high CYP2E1 levels, the local formation of cytotoxic products leads to sustained cell death, with resultant organ toxicity. Persistent necrosis leads to regenerative hyperplasia which eventually results in the formation of tumors (). There are marked species, strain, and sex differences in susceptibility, both to organ toxicity and to carcinogenicity. The weight of evidence supporting this mode of action for chloroform has been reviewed in detail elsewhere (CitationMeek et al., 2003).
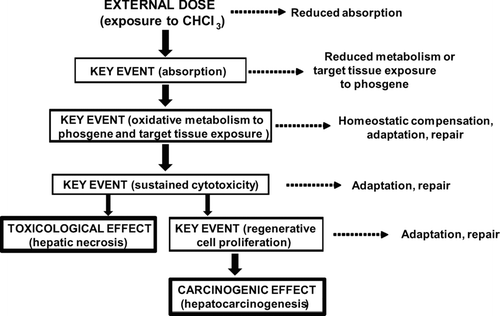
Figure 4 Postulated mode of action and key events for the hepatic toxicity and hepatocarcinogenicity of chloroform. Some of the factors that can influence the dose-response relationships of the key events, and whether they are likely to exhibit a threshold, are illustrated. The early (upper) key events are largely toxicokinetic while the later (lower) key events are largely toxicodynamic.
Key Events for Organ Toxicity and Tumor Development
-
Absorption and distribution of chloroform to target tissue
-
Generation of cytotoxic metabolite (phosgene) by P450 (CYP2E1)
-
Sustained cytotoxicity
-
Regeneration and proliferation
Each key event is considered with respect to what is known about the dose-response characteristics of the event, including whether the event may be thresholded. The roles of protective response mechanisms (e.g., detoxification, repair, adaptive mechanisms) are discussed, together with factors that may contribute to variability in the effectiveness of these mechanisms (e.g., environmental, genetic, etc.).
Key Event 1—Absorption and Distribution to Target Tissue
Chloroform is a low molecular weight (M R 119.4), lipid soluble (log P 1.97) compound that is a liquid at normal temperature and pressure. As such, it is readily absorbed from most sites of exposure, particularly the gastrointestinal tract and the lungs. However, the absorption rate is affected by the method of delivery, for example whether by oral gavage in corn oil or in drinking water. Distribution is rapid and extensive, with exposure of all tissues. Hence, target organ specificity is determined more by local metabolism than by systemic pharmacokinetics.
As chloroform is lipid soluble, its distribution into fat can influence systemic levels. Similarly, the rate of systemic removal can influence systemic levels. There is evidence that the elimination of chloroform at higher doses is saturable, so that systemic, and hence target tissue, exposure increases disproportionately at high doses (CitationAnand et al., 2003; Citation2006). This is particularly notable after a single exposure. It is very unlikely that accumulation in fat per se is saturable, even at doses used experimentally. Repeated exposure results in increased elimination of chloroform, at least in mice, by mechanisms that have yet to be determined (CitationAnand et al., 2006).
As a consequence of these phenomena, the AUC for chloroform in blood following a single oral aqueous gavage dose to mice increased 6-fold for doses of 150 mg/kg bw and 300 mg/kg bw, respectively. Target organ AUC increased to a similar extent, 8-fold in both liver and kidney. Cmax for chloroform also showed nonlinear increases with dose, 7-fold in blood, 8-fold in liver, and 9-fold in kidney. Following repeated exposure for 30 days, the target organ AUC increased only 3–4-fold over this dose range, while Cmax increased less than 2-fold (CitationAnand et al., 2006). As a consequence of these changes, while the target organ Cmax showed very little variation with repeated exposure at a dose of 150 mg/kg bw, at high doses of 300 mg/kg the target organ Cmax varied 8-fold in liver and 18-fold in kidney between one and 30 days exposure. AUC showed similar, though less marked differences, 4–6 fold at the low dose, and 9–13 fold at the high dose.
As noted above, exposure at high doses is influenced by saturation of elimination. However, repeated exposure at low doses appears to engage physiological response mechanisms that lead to increased elimination, reduction of AUC and Cmax, and decreased target tissue dose—i.e., changes that would be expected to affect the magnitude of the biological response, and there is evidence that this is so with tolerance to the target organ toxicity of chloroform developing on repeated exposures (CitationAnand et al., 2006). The effects may be of such a magnitude as to reduce target organ exposure to below that level necessary to provoke any biological response at all.
In summary, target tissue dose levels of chloroform are affected not only by administered dose, but by host response mechanisms that have not yet been elucidated. These host response mechanisms may include adaptive or protective response mechanisms that serve to increase elimination and thereby reduce target tissue dose with repeated exposure. An important unknown is the relationship between the dose level and frequency of dosing at which such adaptive response mechanisms are engaged, and indeed whether this is consistent across species. At high dose, elimination is saturable; however, at repeated low dose, elimination may be up-regulated. It is not known at what combination of dose level and frequency saturation of elimination begins to occur. Nor is information yet available on what conditions of dose level and frequency are required for up-regulation to occur.
Key Event 2—Generation of a Cytotoxic Metabolite (Phosgene) by P450 (CYP2E1)
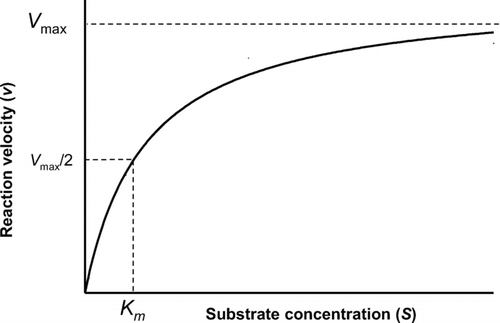
Chloroform itself is relatively unreactive but on P450-mediated oxidative metabolism, it forms one or more chemically unstable products that are cytotoxic (CitationPohl et al., 1977; CitationPohl and Krishna, 1978; CitationSmith and Hook, 1984). Most, if not all, of the metabolism of chloroform is catalyzed by the ethanol-inducible form of P450, CYP2E1. This enzyme exhibits classic Michaelis-Menten kinetics (CitationLipscomb et al., 2004), i.e.
Figure 5 Substrate concentration-reaction velocity curve for an enzyme reaction exhibiting Michaelis-Menten kinetics. The enzyme saturates at high concentrations, reaching a maximum velocity (Vmax). The concentration at which the enzyme velocity is 50% of its maximum is a measure of the enzyme affinity, K m . At low substrate concentrations (up to 50% saturation), the reaction is essentially linear with substrate concentration.
Chloroform is cytotoxic only in tissues in which CYP2E1 is expressed; moreover, the extent of tissue necrosis and the specific regions of tissue necrosis correlate with the extent of metabolic activation (as measured by covalent binding to protein). When CYP2E1 is inhibited chemically and in CYP2E1 knockout mice, null only for this form of P450, there is no toxicity to liver or kidney of mice in which chloroform is otherwise toxic (CitationConstan et al., 1999). Thus, the level of CYP2E1 expression is clearly a critical determining factor in whether, and the extent to which, chloroform is toxic to target tissues.
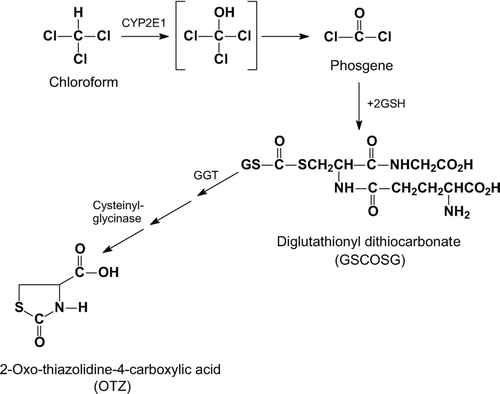
Quantitatively, the most important reactive metabolite of chloroform is phosgene (CitationPohl et al., 1984; CitationAmmann et al., 1998; CitationGemma et al., 2004). Phosgene is highly reactive and does not diffuse far from its site of formation before reacting with thiols, including the cellular nucleophile glutathione, which detoxifies phosgene, forming diglutathionyl dithiocarbonate () (CitationPohl et al., 1981). Glutathione may have a dual role in preventing the cytotoxicity of chloroform, through detoxification of phosgene and through the reduction of oxidized groups in critical macromolecular targets (CitationBurke et al., 2007). As a consequence of the reactivity of phosgene, the sites of toxicity for chloroform are determined by local metabolism to reactive products.
Figure 6 Metabolic activation of chloroform to phosgene and subsequent detoxication by glutathione. Chloroform is oxidized by CYP2E1 to an unstable hydroxylated intermediate, which spontaneously rearranges to the chemically reactive phosgene, the putative toxic species. Phosgene can react spontaneously with glutathione (GSH), the product of which is non-toxic. The glutathione conjugate is further metabolised via γ-glutamyltranspeptidase and cysteinylglycinase to yield 2-oxo-thiazolidine-4-carboxylic acid (OTZ), an unreactive product.
At low rates of chloroform metabolism, endogenous levels of glutathione will attenuate, if not prevent, the toxicological response. There is evidence, however, that exposure to chloroform at a sufficiently high dose results in a depletion of target organ glutathione levels (CitationWang et al., 1997, CitationBurke et al., 2007), but a threshold level of chloroform metabolism is necessary for such depletion to occur (CitationAmmann et al., 1998). Depletion of glutathione would be expected to increase available levels of reactive metabolite. (CitationStevens and Anders, 1981, CitationBeddowes et al., 2003), and in fact, when glutathione is depleted prior to the administration of chloroform, dose levels producing no observable toxicity in liver or kidney were toxic to both tissues in mice (Kluwe and Hook, 1981).
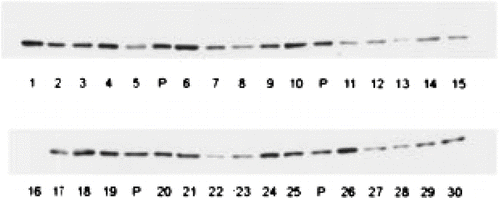
Tissue levels of CYP2E1 show appreciable interindividual variability (); for example, the specific hepatic content of CYP2E1 in adults varies by 10–12 fold (CitationEdwards et al., 1998; CitationLipscomb et al., 2004).Footnote 2 A variety of environmental and pathophysiological factors influence hepatic expression. CYP2E1 is inducible by low molecular weight solvents such as acetone and ethanol, by isoniazid therapy for tuberculosis, by certain physiological states such as fasting, and in some disease conditions such as diabetes (reviewed in CitationCederbaum, 2006). There is also indirect evidence for induction of liver CYP2E1 content in humans after drinking ethanol-containing beverages (CitationTsutsumi et al., 1989), or after treatment with isonizaid for some time. Paradoxically, CYP2E1 is inhibited by many of the compounds that can act as inducers, when exposure is acute, including ethanol, other solvents, and a variety of halogenated hydrocarbons (CitationRonis et al., 1996; CitationOhashi et al., 2005). A number of dietary constituents, such as diallyl sulphide, may also inhibit CYP2E1 to a greater or lesser extent (CitationLoizou and Cocker, 2001).
Figure 7 Interindividual variability in human hepatic CYP2E1 content. The illustration is of Western blots of equal protein loading from 30 microsomal samples of human liver, each from a different renal transplant donor, with a highly specific anti-CYP2E1 antibody. Donors had no evidence of liver pathology either histologically or by clinical chemistry. “P” represents a pooled sample used for quality control. (CitationEdwards et al.,1998).
Certain genetic factors may also contribute to interindividual variability. CYP2E1 is genetically polymorphic (CitationHayashi et al., 1991; CitationHu et al., 1997) and some of the variants have been reported to affect CYP2E1 activity. However, the evidence regarding the impact of such polymorphism is somewhat inconsistent and any effect appears relatively modest (CitationIngelman-Sundberg, 2004).
Given the importance of CYP2E1 activity in the etiology of chloroform toxicity, variability in CYP2E1 levels may be a major contributing factor to inter- and intra-individual variability in response to chloroform. In general, CYP2E1 expression is not detectable during the first two trimesters of pregnancy (CitationJones et al., 1992, CitationJohnsrud et al., 2003), but expression increases during gestation and neonatal life, and reaches adult levels within 3 months of birth (CitationJohnsrud et al., 2003).
Enzyme (CYP2E1)-catalyzed formation of phosgene from chloroform obeys Michaelis-Menten kinetics, and hence there will be no dose below which phosgene is not produced. However, levels of phosgene can be detoxified by glutathione, although the efficiency of this process is not clear. Nevertheless, as CYP2E1 expression increases and/or GSH levels decrease, the location of the threshold for the effects of chloroform will decrease and the dose-response curve will become steeper. It is likely that loss of protection by GSH will not become apparent until there is marked depletion of this nucleophile. Given the large size of the GSH pool and its rapid rate of resynthesis, frequent high doses of chloroform or some other similar agent would be necessary to compromise GSH-mediated protection. In some individuals, susceptibility may be increased by rare inborn errors of GSH synthesis or by pathophysiological states which markedly affect GSH levels, e.g. hepatic failure, although any contrary effects on CYP2E1 activity would need to be taken into account.
The sources of variability in the expression level and activity of CYP2E1 will affect the rate of formation of phosgene from chloroform and hence, interindividual susceptibility to the effects of chloroform. The practical consequence of this would be that for a given individual, the threshold for generation of a critical concentration of phosgene would vary with their CYP2E1 phenotype, as determined by both genetic and environmental factors. It is anticipated that variations in CYP2E1 will be a greater determinant of interindividual differences in susceptibility to chloroform than variations in GSH concentration, for the reasons outlined above.
Key Event 3—Sustained Cytotoxicity
The toxic product(s) of chloroform metabolism, predominantly, if not exclusively, phosgene (CitationPohl et al., 1977; CitationPohl and Krishna, 1978; CitationPohl et al., 1984), is a highly reactive electrophile and readily reacts with nucelophiles, such as glutathione and macromolecules, including proteins, producing covalent adducts (CitationReynolds and Yee, 1967; CitationBranchlower et al., 1984). Modification and/or oxidation of critical macromolecules lead to cytotoxicity and necrosis (CitationConstan et al., 1999). The extent to which oxidative stress is involved in the toxicity of chloroform is unclear, the evidence being inconsistent (CitationAmmann et al., 1998; CitationBurke et al., 2007).
The mechanism of cell death has yet to be clearly determined, but there are data suggesting that it involves mitochondrial dysregulation leading to impairment of the mitochondrial membrane potential via effects on mitochondrial permeability transition (CitationHartig et al., 2005; CitationBurke et al., 2007).
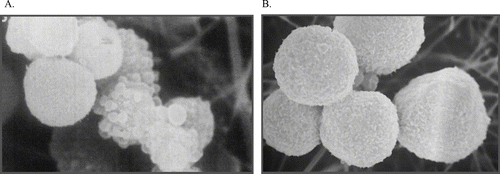
There is evidence that cell death (cytotoxicity) occurs only after a critical degree of damage to the cell and there is an observable threshold for cytotoxicity, both in vitro (CitationRuch et al., 1986; CitationAmmann et al., 1998; CitationSmith et al., 1985) and in vivo (CitationSmith et al., 1983; CitationLarson et al., 1994; CitationTemplin et al., 1998). Below certain chloroform levels, no toxicity is apparent in either liver or kidney. Further support for a threshold comes from the marked species, strain, sex, and tissue specificity of the toxicity of chloroform to the liver and kidney. For example, gavage dosing of Osborne-Mendel rats with chloroform to high dose levels, causes renal toxicity only in males (CitationTemplin et al., 1996a). No evidence for renal toxicity is found in females. Low levels of damage are not sufficient to cause cytotoxicity (CitationHartig et al., 2005), as repair and/or resynthesis of damaged proteins enables cellular recovery (). At present, there is empirical evidence for cellular repair of damage following exposure to chloroform (CitationLiao et al., 2007), but specific mechanistic information is not yet available. Such evidence is emerging for other toxicants causing damage to liver or kidney (CitationBang et al., 2008), but the extent to which the same processes extend to chloroform remains to be determined. In support of this, it has been shown that mitochondria can sustain a certain degree of insult before there is any change in membrane permeability, which is the determining factor in cytotoxicity (CitationZorov et al., 2006). These processes are ubiquitous, and human cells show similar threshold behavior to those from experimental animals (CitationTee et al., 1987). Cytotoxicity of chloroform to the liver and the kidney requires continuous exposure. When exposure stops, there is recovery from the tissue damage sustained (CitationTemplin et al., 1996b).
Figure 8 Reversibility of initial damage to hepatocytes. Freshly isolated hamster hepatocytes were exposed to acetaminophen for 90 min (A) and then incubated with the thiol reductant dithiothreitol (DTT) for a further 90 min (B). Despite morphological damage after 90 min, as evidenced by substantial plasma membrane blebbing, hepatocytes could be restored to normal morphology (and function) over the subsequent incubation with DTT, demonstrating that the effects observed at 90 min were fully reversible. If cells were incubated in buffer alone for the second 90 min period there was a marked loss of viability (CitationTee et al., 1986).
Key Event 4—Regeneration and Proliferation
Unless necrosis is so extensive that no viable cells remain, in many tissues the surviving cells will undergo compensatory proliferation to regenerate the tissue mass that is lost. This process is under tight control, so that apoptosis limits the extent of tissue growth to the extent necessary to restore the functional mass. This is a compensatory response, which occurs when the functional reserve of the tissue is compromised by the degree of damage. However, organs such as the liver and kidney have sufficient functional capacity to cope with a small amount of damage without mounting a restorative response. This results in a de facto threshold for proliferation. Proliferation, as measured by the bromodeoxyuridine (BrdU) labelling index, is dependent on both the duration and magnitude of exposure to chloroform. For example, in an inhalation study in F344 rats, it was necessary to expose the animals for more than 4 days to a dose of at least 30 ppm before any increase in proliferation could be detected in the kidney (CitationTemplin et al., 1996b). At 4 days, doses of up to 300 ppm (highest tested) had no effect on proliferation and similarly, with a dose of 10 ppm or less even after 13 weeks of exposure, there was no change in proliferation. The increase in proliferation was reversible, so that 6 weeks after exposure to 300 ppm for 13 weeks, the proliferation rate had returned to baseline values (CitationTemplin et al., 1996b). In the liver, no change in proliferation was observed at doses less than 300 ppm. Qualitatively similar results have been reported in B6C3F1 mice, exposed by the oral route (CitationLarson et al., 1994). In both liver and kidney, no proliferation was observed below a certain dose of chloroform, and the duration of exposure increased the response to a given dose.
As it is proliferation that leads to the selection or production of pre-cancerous cells, a necessary step in the progression to cancer, the absence of a proliferative response will result in a clear threshold for the carcinogenic response.
In addition to the extent of damage, the persistence of damage is an important determinant of the carcinogenic response. This needs to be sustained; otherwise there is insufficient time for selection/generation of altered cells.
The processes involved in tissue repair in humans are very similar to those in experimental animals. Hence, while there may be some differences in absolute sensitivity, there will still be a threshold in the response.
DISCUSSION
Absorption and Distribution of Chloroform to Target Tissue
Target tissue dose levels of chloroform can be affected by a variety of host factors, not all of which have been elucidated. These mechanisms may include adaptive or protective responses, which increase elimination. At high dose, the elimination of chloroform becomes saturated. While these factors will contribute to inter-species and interindividual differences in response, they do not represent an absolute barrier to the toxicity or carcinogenicity of chloroform. On the other hand, the systemic dose of chloroform, which depends upon the magnitude of the external dose and its rate of delivery to the systemic circulation, provides a clear biological threshold for the responses of concern. Hence, when administered by certain routes and/or below a certain dose, chloroform is not toxic.
Generation of Cytotoxic Metabolite (Phosgene) by P450 (CYP2E1)
The key event of generation of the cytotoxic metabolite (phosgene) is rate limiting, in that the location of the threshold for the response of concern, the shape of the overall dose-response curve, and the magnitude of the ultimate response will depend upon the levels of the causative agent that are produced, which in turn will depend upon the levels of expression of CYP2E1. If no phosgene is produced, there will be no organ toxicity and no tumorigenic response. Evidence for this is available from a number of studies, including those in CYP2E1 homozygous knockout mice. Many factors, both genetic and environmental, can influence the level of expression and activity of CYP2E1. These may affect the response qualitatively, as above, or quantitatively, in that susceptibility is altered but not eliminated, depending on the magnitude of the change in CYP2E1-dependent activity. While the response may also be modified by GSH concentration in target tissues, only when levels are substantially impaired is this likely to affect susceptibility to chloroform.
Sustained Cytotoxicity
Above a certain level of phosgene, damage to cells will result in cytotoxicity, i.e. cell death. However, repair and adaptive responses enable cells to sustain and recover from a certain degree of damage while maintaining their viability. These processes are not well defined for chloroform, but by analogy to similar agents, clearly play an important role in determining the biological threshold and interindividual variability in susceptibility. While host factors can undoubtedly influence these processes, specific information is not available. This is an aspect of chloroform toxicity that would be amenable to further research to fill the data gap.
Regeneration and Proliferation
Cytotoxicity must be of a certain magnitude and be sustained for a certain period of time before proliferation is stimulated. At lower doses or with shorter periods of exposure, adaptive and/or repair processes enable recovery of the organ without any increase in cell proliferation. This represents a biological threshold. The degree to which host factors influence the proliferative response following chloroform-induced cytotoxicity is not clear, and is an area that would benefit from further research.
Mode of Action
The earlier key events in the mode of action for the toxicity and carcinogenicity of chloroform, those relating to metabolism and disposition, have been studied in more detail than the later key events, particularly for the influence of host factors. In addition, while no mode of action is strictly sequential, it is clear that a progression from one key event to another is necessary to provoke a carcinogenic response from chloroform. The consequence is that it is not possible to determine the importance of the more distal key events from interference with proximal key events. For example, in CYP2E1 knockout mice, while the absence of a toxicological response is due to the inability to convert chloroform to phosgene, it is not clear how much of this is due to the fact that there is no cytotoxic effect in these animals. Such an analysis requires that each step can be studied independently of the earlier steps. For example, to avoid consideration of changes in CYP2E1 expression, could the effects of phosgene be studied directly (in this instance the experiment may be impractical)? Similarly, to determine the importance of proliferation, it would be necessary to induce cytotoxicity by some means that was independent of the precursor effects of chloroform. Hence, while it is apparent that the expression of CYP2E1 is a key determinant of the response, it is not possible to determine the relative importance of host factors on the other key events in this instance.
Despite some gaps in knowledge, particularly relating to the influence of host factors on later key events, the general conclusion is that the mode of action for chloroform is such that the dose-response would be nonlinear and a deterministic approach to the risk assessment of this compound has been accepted by a number of regulatory bodies, such as the US EPA (2006). In establishing a Maximum Contaminant Level Goal for chloroform in drinking water, the EPA concluded that “based on an analysis of the available scientific data on chloroform, EPA believes that the chloroform dose response is nonlinear and that chloroform is likely to be carcinogenic only under high exposure conditions” and as a consequence “chloroform is likely to be carcinogenic to humans only under high exposure conditions that lead to cytotoxicity and regenerative hyperplasia.”
It is clear that a key events analytic framework for a mode of action, such as the one outlined here, is of appreciable value in developing a science-based argument for moving from the default of no-threshold, low-dose extrapolation to an approach based on the concept of a biological threshold with a nonlinear dose-response relationship. Despite this, however, there is concern that because of inter-individual variability in sensitivity, it is not possible to identify a population threshold, and hence a precautionary approach is necessary. The key events analytic framework provides a rational basis for addressing this concern. By determining the mechanistic basis for the key events and characterizing the degree of human variability, and the rate limiting steps, in the underlying processes (for example see CitationHeinloth et al., 2004), it should be possible to establish the existence of population thresholds with the same confidence that it is now possible to establish the existence of biological thresholds.
CASE STUDY: DNA-REACTIVE CARCINOGENS
As discussed above, historically, health outcomes can differ substantially with regard to assumptions about dose-response at low doses. For the present discussion on approaches for establishing criteria for threshold responses for adverse outcomes of chemical exposures, it is convenient to consider chemicals in two separate health outcome related categories—carcinogens and noncarcinogens. In general terms, carcinogens produce linear or linear-quadratic tumor responses and noncarcinogens have threshold dose-responses for disease outcomes. However, this pragmatic description is being challenged, certainly at the population level (CitationNAS, 2008). Further, as discussed above, carcinogens are considered in two clearly distinguished groups based upon their MOA—DNA-reactive (mutagenic) and non-DNA-reactive (e.g. receptor-mediated, mitogenic, cytotoxic, oxidative stress). As described above, this results in a clear distinction being made in the U.S. EPA cancer risk assessment framework (CitationU.S. EPA, 2005) between these two broad groups—a mutagenic MOA results in a default linear extrapolation from the POD for human or rodent tumors, or other toxicological endpoints; a non-mutagenic MOA can lead to a nonlinear or threshold dose-response based upon the available supporting information. Thus, for the consideration of potential mechanisms for the development of threshold responses for chemical exposures, the ILSI RF working group developed a case study for DNA-reactive chemicals as a class rather than using a single chemical for a case study.
DNA-Reactive Carcinogens and Risk Assessment
As noted above, for cancer risk assessments, it has been common practice to presume that in the absence of data to the contrary, the default tumor dose-response curve for DNA-reactive (mutagenic) chemicals is linear at low doses (CitationU.S. EPA, 2005). This position is predicated on the assumption that even “one molecule” of a chemical has a finite probability of inducing cellular transformation and ultimately a tumor. The encouragement through the recently published U.S. EPA Guidelines for Carcinogen Risk Assessment (CitationU.S. EPA, 2005) to incorporate mechanistic data into the cancer risk assessment process has allowed for reconsideration of the default linear approach. Such reconsideration can be placed in the context of the new types of data that can be generated using the most recent molecular biology methods. These can inform the mode of action and help characterize the mechanisms of tumor formation (CitationEdwards and Preston, 2008).
Relationship of Initial DNA Damage to Mutagenic Events
DNA-reactive chemicals (or their metabolites) produce DNA damage (e.g., adducts, crosslinks) through the covalent binding of the reactive form of the chemical to DNA. In a general sense, DNA damage can be converted into a mutation by errors of replication on a damaged template or by errors of DNA repair. However, for the great majority of chemical carcinogens, these mutations are produced almost exclusively through errors of DNA replication (CitationPreston, 1991). It should be noted that while the dose-response curve for DNA adducts is generally linear, it does not follow that dose-response curves for gene mutations or chromosomal mutations are linear over a broad dose range (CitationSwenberg et al., 2008). Thus, the dose-response curve for mutations can be considered to be a probability curve denoting the probability as a function of dose (or DNA adduct level) that DNA damage can be converted into a mutation. The probability of converting an adduct into a mutation is not equally likely over a range of doses including low (environmental) ones (CitationOttender and Lutz, 1999).
The various steps from the initial formation of a critical mutation to a tumor are based on the generally accepted models of multistage carcinogenesis [for example, CitationFearon and Vogelstein (1990); CitationHanahan and Weinberg (2000); CitationMantovani, (2009)]. From this multistage characterization, the overall shape of the tumor dose-response can be considered to be an integration of the individual probability curves for the key events essential for tumor development following exposure to a DNA-reactive carcinogen.
Key Events Dose-Response Analysis
The particular pathway whereby a chemical can produce an adverse outcome is described by its mode of action (MOA). These pathways include DNA-reactivity, receptor-mediated responses, cytotoxicity with regenerative cell proliferation, and mitogenicity. In turn, a MOA is defined as a sequence of key events and processes, starting with interaction of an agent with a cell, proceeding through functional and anatomical changes, and resulting in cancer formation (or formation of a non-cancer health outcome). A key event is an empirically observable precursor step that is itself a necessary element of the MOA or is a biologically based marker for such an element. In the context of the dose-response characteristics for the adverse outcome, it is necessary to consider the plausible dose-response characteristics for each of the key events for a particular adverse outcome and further to establish if one or more of these is a determinant of the dose-response for the adverse outcome, either quantitatively or qualitatively. In this way it should be possible to establish if a real (or practical) threshold could be predicted for an adverse outcome. For the present discussion, the adverse outcome is cancer. The example that will be discussed here is that of DNA-reactive chemicals. The key events that lead along the path from uptake and distribution of the chemical though initial exposure of the target cells for cancer formation to final outcome of a metastatic tumor have been presented by CitationPreston and Williams (2005) and are:
-
Uptake and distribution of chemical within the body.
-
Exposure of target cells (e.g., stem cells) to ultimate DNA-reactive and mutagenic species, with or without metabolism.
-
Reaction with DNA in target cells to produce DNA damage.
-
Misreplication on damaged DNA template or misrepair of DNA damage.
-
Mutations in critical genes in replicating target cells.
-
Critical gene mutations result in initiation of new DNA/cell replication.
-
New cell replication leads to clonal expansion of mutant cells.
-
DNA replication can lead to further mutations in critical genes.
-
Imbalanced and uncontrolled clonal growth of mutant cells may lead to preneoplastic lesions.
-
Progression of preneoplastic cells results in emergence of overt neoplasms, solid tumors (which require neoangiogenesis), or leukemia.
-
Additional mutations in critical genes as a result of uncontrolled cell division results in malignancy.
There is relatively little information on the shape of the dose-response for any of these key events (including mutations and chromosome alterations) because so many studies are conducted for hazard identification using a limited number of doses and dose range or are for mechanistic studies where most frequently rather high exposures are used. In a few cases a broad range of concentrations has been used for in vitro mutation assays and there is some evidence for nonlinear responses, perhaps including threshold responses when viewed within the context of the background mutation frequency and the associated errors (CitationDoak et al., 2007; CitationSwenberg et al., 2008). There is a need to distinguish between gene mutations (involving 1 or 2 bases) and chromosomal mutations involving large segments of DNA (e.g. deletions, translocations, inversions). The reason is that on the basis of the “hit” theory, as usually described for ionizing radiation effects, a gene mutation would be predicted to require one hit whereas chromosome alterations would require two hits. The consequence is that the dose-response curve for gene mutations (above any threshold that might exist) would be linear with the dose, whereas that for chromosome mutations would be proportional to the (dose).2 For the majority of chemicals the overall tumorigenic response would be a combination of the two endpoints resulting in a “linear-quadratic” dose-response curve. The ratio of the linear and quadratic components would be chemical-specific, i.e. more linear or more quadratic. Whether a threshold would be predicted at low exposures is open to discussion; at least, an effective threshold, in the context of increase over background, can be predicted.
In the virtual absence of experimental data on the shape of the dose-response curve for these key events at low (environmental) exposure levels, it is appropriate to consider each key event in the context of what would be necessary for a threshold response to be predicted initially for an experimental animal MOA and then, by using a human relevance framework (CitationMeek et al., 2003), for exposed humans.
Key Event 1—Exposure of Target Cells to Ultimate DNA Reactive and Mutagenic Species—With or Without Metabolism
This Key Event is likely to be rate-limiting because without exposure to the target, other key events cannot occur. Such a failure of a chemical to reach the target cells could be exposure dependent. For a threshold response (over any particular concentration range) the inability of the parent chemical or the active metabolite to reach the target cells will result in the absence of an effective dose. It is important to use as much data for humans to establish whether or not a rodent response, as regards the target tissue dose, is plausible in humans. Similarly, the lack of the necessary metabolizing enzymes or enzyme activity for activating a specific chemical in a specific target cell will lead to the absence of an effective dose. Again, assessing plausibility in humans from available rodent data is necessary.
Key Event 2—Reaction with DNA in Target Cells to Produce DNA Damage
This Key Event is, of course, in part dependent on Key Event 1 for plausibility in humans. Of particular importance (for chronic exposures) is the steady-state level of DNA damage. There is a continual production and repair of DNA damage and at very low levels of exposure (and DNA damage) it is quite feasible that all DNA damage is repaired essentially immediately. The concern, in terms of eventual cancer outcome, is not the induction of DNA damage, but rather the accuracy and kinetics of its repair or the levels of damage that are present in any segment of DNA at the time of its replication. For a threshold response, DNA damage would need to be at a “zero” or “very, very low” steady state, or it would need to be repaired without error or not be present at the time of replication (CitationChan et al., 2006).
Key Event 3—Misreplication on a Damaged DNA Template or Misrepair of DNA Damage
This Key Event follows from the induction of DNA damage, and represents how the damage itself can be converted into a biological event, namely a mutation (point or chromosomal). DNA replication and repair processes have a background error rate that is low (CitationBudzowska and Kanaar, 2009). The addition of an additional low level of induced damage from a chemical exposure is unlikely to increase this rate. However, for a threshold response for this key event it is necessary that errors in replication be effectively “zero” above background. There is insufficient information available to reach such a conclusion, although there are mutation data that suggest this is the case (CitationDoak et al., 2007). Another consideration is that for DNA damage to be converted into a mutation, cell replication is required. DNA repair errors are considered to be even lower than replication errors (CitationPreston, 1991). Thus, if there is no replication in the target cells there will be no mutations produced. If there is a very low replication rate, then the race between repair and replication will be in favor of repair, thereby reducing the probability for producing a mutation and perhaps creating a practical threshold.
Key Event 4—Mutations in Critical Genes in Replicating Target Cells
For Key Event 3 described above, the mechanism by which mutations can be produced is discussed. The need for Key Event 4 is that such mutations be in critical genes. A prediction of this likelihood is, in part, dependent on the number of “cancer” genes present in the total genome. The most recent estimate is that there are about 120 “driver” genes for oncogenesis out of a total human genome of some 23,000 genes (CitationGreenman et al., 2007). Thus, the probability of inducing a critical mutation is very low at extremely low levels of replicating DNA damage. A calculation would almost certainly demonstrate a practical threshold for critical mutation induction.
Key Events 5–11
The other Key Events (#s 5–11) naturally follow from the first 4. Since each Key Event is dependent on the qualitative and quantitative features of the dose-response for Key Events 1-4, any prediction of a threshold (real or practical) from Key Events 1-4 will influence discussion of subsequent Key Events in the context of a threshold. To obtain multiple mutations in a single cell which is necessary for the multistage tumor model that is currently the accepted one [including that developed in Hanahan and Weinberg's Hallmarks of Cancer (2000)] will further reduce the probability of producing Key Event 7. The probability is very low for producing critical mutations in target cells from low level exposures in repair proficient, replication efficient cells; this probability is reduced dramatically by requiring that these multiple mutations be in a single cell. A linear low dose-response for a single mutation cannot be used to predict the response for multiple mutations in a single cell over the same dose range.
Exposure-To-Outcome/Effect Continuum
It is of particular importance to note that the set of key events developed for tumors (discussed above) are a dependent set and not an independent one. Each step along the pathway from a normal cell to a transformed cell phenotype and ultimately to a tumor is dependent on the previous step. As described, the quantitative nature of each step (or key event) can be considered as the probability that a particular key event is converted into the subsequent key event. Certainly this continuum and the probabilistic nature of key event production would allow for the presence of threshold responses even for DNA-reactive chemicals.
Characteristics of Stressor/Host/Exposure Matrix
For DNA-reactive chemicals, the assessment of risk is broadly conducted for the “average” individual (in a population) in the absence of other confounding exposures. Clearly, risk will be influenced by other exposures, either increasing or decreasing ultimate tumor response to the chemical whose risk is being assessed. While potential sensitivity (susceptibility) factors (especially early life stage exposures) are considered, they are generally covered by default application. It could be argued that susceptible individuals will always fall outside any threshold response because threshold responses are always considered in terms of background levels and the background level for susceptible individuals is (by definition) above the normal background frequency of an adverse outcome, such as cancer.
In a general sense, susceptibility factors per se can have an influence at any or all key events, depending on the nature of the susceptibility, for example, for DNA repair effectiveness, the extent of metabolism, the specific metabolic pathways, and cytotoxicity.
Our understanding of how to incorporate chemical mixtures that include DNA-reactive carcinogens and susceptibility into the cancer risk assessment process is still very much an imperfect science and is an area that is ripe for research efforts.
Principles and Implications for Thresholds
For DNA-reactive chemicals, any assessment of low-dose response has to be put in the context of the background frequency of DNA damage and of genetic alterations. The low-dose response also has to include considerations of the efficiency of DNA repair—is its error rate dependent on dose; i.e., the level of DNA damage? The underlying principle is, given the relatively high levels of background DNA damage [e.g. the steady-state level of 500 base alterations per cell (CitationBarnes and Lindahl, 2004)], what is the likelihood that a small amount of additional (induced) DNA damage will elicit an adverse response, namely genetic alterations and tumors? A second principle is how dependent is an increase in the level of genetic alterations at low doses on an increased (induced) level of cell proliferation? A third principle is, given that tumor formation depends on the production of genomic instability, how likely is this to be induced by chemical exposures at low doses?
All of these principles impact the likelihood of there being threshold responses (or nonlinear responses) for genetic alterations and/or tumors at low levels of exposure to DNA-reactive chemicals.
Identification of the Types of Data Required to Characterize Low Dose Dose-Response Relationships for DNA-Reactive Chemicals
The types of study that can have an impact on our understanding of the possibility of threshold responses for tumors induced by DNA-reactive chemicals will be those that address the issue of the low-dose response of the key events and factors that either increase or decrease these. The following is a list of possible data needs:
-
Background levels of DNA damage.
-
Background levels of genetic alterations.
-
Dose-response curves for DNA adducts and genetic alterations over a low dose range.
-
DNA repair characteristics at low doses.
-
Rate of induced cell proliferation at low doses.
-
Identification of reliable biomarkers of response based on key events for characterizing low dose effects.
Application of New Technologies for Understanding Low Dose Dose-Responses and Key Current Gaps in Knowledge
The likely progress leading to a better understanding of the dose-response for adverse health outcomes following exposure to chemicals will be based on the knowledge of key events along the pathway from a normal to a pathologic state. The identification of informative markers of key events can be greatly enhanced by the use of whole genome techniques such as gene expression assessments [using microarrays or preferably mRNA counting techniques such as SAGE or ultra-high throughput sequencing (CitationMardis, 2008)], or proteomic assessment [using mass spectrometry based techniques (CitationLatterich et al., 2008)]. These approaches have the great advantage of being able to use data from the whole genome that will include gene-gene, protein-protein, and cell-cell interactions, for example. The ability to conduct these types of studies on very small tissue samples will enhance the ability to focus on altered cells without the confounding from normal cells. It is essential that computational approaches be used in parallel with data gathering exercises in order to be able to interpret the molecular data in a whole cell (or tissue) context (for example, CitationWu and Dewey, 2006). In this regard, molecular modeling techniques are an essential component of assessing low-dose responses where it is very difficult to obtain all the necessary informative experimental data. The majority of mutations associated with diseases are at nonselectable loci that makes their quantitation difficult in a technical sense.
There are a number of molecular techniques, especially those based on PCR technology, that are reaching sensitivity levels in the range required (1 in 107 cells) for the study of induced mutations (CitationMorlan et al., 2009). These will certainly be perfected in the near future.
CASE STUDY: ENDOCRINE-ACTIVE AGENTS
Endocrine disruption is a term that was coined to describe the adverse effects produced by toxicants that interfere with normal endocrine function. Endocrine disrupters can act by interfering with the synthesis or elimination of hormones, but probably the most important mechanisms are those involving interaction (either agonism or antagonism) with steroid hormone receptors. Steroid hormone receptors and other receptors within the nuclear hormone receptor family have small endogenous ligands that are stable and capable of diffusing across cell membranes to interact with their receptor, which is located in the cytoplasm or nucleus. Unlike membrane-bound receptors, which tend to have a rapid signal transduction process (e.g., the opening of a membrane channel, the phosphorylation of a protein), the signal transduction process for ligands of nuclear hormone receptors is slower and involves a specific pattern of gene expression. The receptor-ligand complex binds to specific response elements on the genome, which brings about selective up- and down-regulation of gene expression. The end result of this gene expression, and subsequent translation into proteins is to change the function and/or state of differentiation of the cell over a time course ranging from hours to days. Exogenous compounds that mimic the endogenous hormone can elicit inappropriate responses either by acting as agonists or antagonists. Although nuclear hormone receptors have high affinity and specificity for their endogenous ligand, some, such as the estrogen receptor, are capable of accommodating, albeit weakly, many other chemical structures in their binding sites.
There are a large number of steroid hormone receptors, with effects on virtually every organ system, although most operational definitions of endocrine disrupters concentrate on sex steroids and thyroid hormones. Endocrine disrupters have the potential to cause reproductive toxicity, developmental toxicity, or certain types of cancer in hormonally responsive organs, such as the uterus, the mammary gland, or the testis. Developmental effects of some of these toxicants, particularly those that interact with the estrogen receptor, may be latent, with few obvious structural effects in the fetus or neonate, but persistent effects on reproductive function when the organism reaches sexual maturity.
For the purposes of this review, we will use estrogens and interaction with the estrogen receptor as an example system by which to assess the notion of thresholds for endocrine disrupters. Estrogens influence a number of systems, but are particularly important in the development of the reproductive system and in the regulation of normal female reproductive function. The normal function of hormones is to bring about changes in the state of differentiation or function of cells for a period of hours to days. An excellent example is the rodent estrus cycle (or its corollary, the human menstrual cycle) in which the synthesis and release of estrogens, followed by progestagens, results in a predictable cycle of changes in the ovary, the uterus, the vagina, the pituitary, and CNS, returning to the initial state at the end of each cycle. The rodent uterotrophic response driven by estrogens is a consequence of a characteristic profile of gene expression changes that occurs in a temporal pattern progressing through transcription factors that initiate changes in the state of cellular differentiation, induction of cell cycle machinery that drives proliferation, and eventually the induction of inflammatory and pro-apoptotic factors that drive the return of the organ to its uninduced state (CitationMoggs et al., 2004, CitationFertuck et al., 2003, CitationNaciff et al., 2007). Although these responses are mediated at the level of the receptor and gene, they require the coordinated action of a large number of genes within each cell, and the change in function of large numbers of cells in different tissues in order to produce a physiological response. It seems unlikely that the estrus cycle of rodents could be perturbed in a nonthresholded way. High concentrations of endogenous estrogen are needed to elicit the ovarian, uterine, and vaginal changes that are integral to the estrus cycle. The response is generally maximal; i.e., adding more estrogen does not increase the mass of the uterus or make the vaginal epithelium more cornified. Antagonists of the estrogen receptor have the potential to decrease the magnitude of the response, but because of the high level of endogenous estrogen that is present to drive the onset of estrus, it is not possible that the displacement of a single agonist molecule from a single receptor (which is the definition of a non-threshold response) would have any effect, even at the level of a single cell, on the response to estrogens.
While the changes elicited by estrogens and progestagens may be dramatic (e.g., a several-fold increase in uterine weight, marked changes in the cytology of the vaginal epithelium) they represent normal physiological states. This range of normality needs to be taken into account when assessing whether hormonal effects are adverse, either by elicitation of a sub- (or supra-) normal response, or persistence in a physiological state that is intended to be transitory. The relevance of this distinction for thresholds is that the threshold must lie above the physiological range of function for the hormone. A similar concept was developed for toxic effects for a neurological function which can perform normally within a range of values for neurochemical concentrations, behaviors, etc. (CitationGaylor and Slikker, 1994).
There are a couple of points to consider in determining whether thresholds exist for hormones. The first is to determine whether there are circumstances in human physiology where the action of a single molecule of a hormone, or the inhibition of the action of a single hormone molecule, could change a biological response. The second, arguably more important, question is whether addition to the existing background levels of a hormone would be sufficient to drive a biological response. We can consider examples of both using the key events dose-response framework.
For the first case, let us consider the role of estrogens and luteinizing hormone in stimulating ovulation. During the late diestrus and proestrus phases of the estrus cycle, the ovary releases a large quantity of estradiol. Although estrogens generally inhibit GnRH secretion, the high levels of estrogens present during proestrus have the opposite effect, stimulating GnRH secretion by the hypothalamus. This in turn stimulates a spike of LH and FSH secretion by the pituitary. The LH surge induces a number of biochemical and structural changes in mature ovarian follicles that result in their rupture and release of ova, as well as the differentiation of granulosa cells into luteal cells. (In reality, there are more steps and more hormones involved in ovulation, but for the sake of illustration we can keep the example simple.) We can consider there to be several key events in this example—the synthesis of a sufficiently high level of estradiol by the ovary to produce a stimulatory effect on the hypothalamus; the release of GnRH from the hypothalamus and its action on the anterior pituitary; and the release of LH from the pituitary in sufficient quantity, at the right time, to stimulate ovulation. Each of these steps can be analyzed to determine whether it is likely to be thresholded.
The first step, the synthesis of a sufficient quantity of estradiol by the ovary during late diestrus/ early proestrus, could ostensibly be affected by treatment with a steroid synthesis inhibitor. Steroid synthesis inhibitors can affect fertility; however, it is unlikely that this effect would be nonthresholded. The synthesis of estradiol in the ovary takes place in a large number of cells, such that a reduction in the capability of one would have no measurable effect. There is also likely to be plasticity in the quantity of steroidogenic enzymes in a cell, in that inhibition of enzyme activity would also lead to rapid induction of enzyme activity. There is some empirical evidence that the effect of steroid synthesis inhibitors on ovarian estrogen synthesis is nonlinear (i.e., thresholded). CitationPowlin et al. (1998) reported no effect of up to 1 μM aminoglutethamide, an aromatase inhibitor, (with significant decreases starting at 10 μM) on estradiol synthesis by the explanted rat ovary. CitationShin et al. (2006) reported prolongation of the estrus cycle in a 28-day study, with associated increases in estradiol, LH and FSH (which were likely compensatory to steroid inhibition), but with clear nonlinearity in the dose-response relationship.
Secretion of GnRH and the elicitation of LH release is also a multi-cellular process and therefore unlikely to be affected by the activity of a single molecule of toxicant in a single cell. However, the LH surge needs to reach a certain magnitude in order to effectively stimulate ovulation, possibly making it more vulnerable to small changes in GnRH. Curtailing the LH surge prevents ovulation; delay of the surge may also hinder ovulation, and results in ovulation being out of synchrony with the estrus cycle, either of which can impair fertility and fecundity. However, studies with a variety of different agents that can blunt the LH surge at sufficiently high dose levels, suggest a nonlinearity in the dose-response curve (e.g., CitationLaws et al., 2000). From a theoretical standpoint it makes sense that this response would be thresholded, as it is controlled by a group of cells, so that an effect of a single molecule or dysfunction of a single cell is unlikely to have a noticeable impact on the end result. However, the fact that the increase in LH does not have to be fully inhibited to have an adverse effect on ovulation does indicate that it is likely to be a sensitive step in the control of ovulation, although not necessarily a nonthresholded step.
Probably the more important theoretical consideration relevant to thresholds is that of additivity to an existing background. Generally, the endogenous ligand for a hormone receptor is present at some concentration. Is it possible for the endogenous hormone to be present at sufficiently high levels during certain physiological states that the addition of even a single molecule of an exogenous ligand is sufficient to drive the response from one that is physiological to one that is adverse?
The aspects of development that are controlled by estrogens may be the most sensitive to adverse effects of estrogen agonists. Is it possible that these effects are nonthresholded? There are some specialized cases in which minute changes in the estrogen level can lead to a change in the developmental outcome. An example of this is the temperature-sensitive sex determination in red-eared slider turtles, in which the temperature determines the aromatase activity, which in turn mediates estrogen synthesis, and the level of estrogen in the egg determines sex, a phenomenon that appears to be best explained as nonthresholded (CitationSheehan, 2006). Exogenous estrogens or aromatase inhibitors can substitute for temperature in modulating the sex ratio. In this example, the level of estrogen synthesized in the eggs is already sufficient to drive sexual differentiation at the temperatures typically encountered by this species; in other words, the level of endogenous estrogen under normal conditions for this species is already midway up the dose-response curve, and any change in estrogen levels would lead to changes in the developmental outcome.
Sex determination is not quite as plastic in mammals. However, although the chromosomal aspect of sex determination is hardwired, the androgens can significantly masculinize the female fetus and antiandrogens can drive the morphological development of the male reproductive system in a female direction. For example, vinclozolin, which has antiandrogenic activity, has been shown to decrease the anogenital distance in male rats in a linear fashion down to fairly low dosages (CitationGray et al., 1999). It is not possible to determine whether this effect is thresholded by morphology alone, as the variability in the measurement would overwhelm the ability to detect very small changes in a reasonable number of animals.
This problem of methodological limitations has made it difficult to reach conclusions about additivity to the background because it has not been possible to design experiments that have sufficient sensitivity to determine whether very small doses of a compound have any effect. New technology in the form of genome-wide gene expression analysis has the potential to remedy this problem. Gene expression analysis has the sensitivity to determine whether these purported effects are real. Available methods like microarray techniques, real-time RT-PCR, and bead-based oligonucleotide methods have sensitivities in the subattomole range (CitationNaciff et al., 2005a). When combined with the power to query the entire genome (by microarray) to ensure that unexpected changes in gene expression are not missed, this is a powerful tool to address the potential low-dose effects of agents like estrogens for which the signal transduction process is dependent on gene expression. Gene expression has been analyzed in fetal rat testis exposed transplacentally to three different compounds with estrogenic activity—ethinyl estradiol, genistein, and bisphenol A. Doses for each compound spanned five or six orders of magnitude, starting from a dosage known to have pharmacological activity, down to extremely low dose levels. All three compounds have effects on gene expression at the higher dose levels, and there are still some effects on gene expression at doses lower than those that had morphological effects; however, at the lowest dose levels of these compounds there were no changes from control in gene expression (CitationNaciff et al., 2005b). This result strongly suggests a threshold for activity of estrogens on gene expression during development. More work needs to be done to provide additional data sets that are comparably comprehensive, but gene expression analysis appears to be a promising experimental approach. While the methodology is still not sensitive enough to definitively demonstrate a threshold, it may be sufficiently sensitive to eliminate the difference between real thresholds and the pragmatic thresholds that represent ambient environmental exposure levels. That, plus the advantage that genome-wide microarrays cover the entire possible range of gene expression, makes microarray technology an attractive option for plumbing the depths of the dose-response curve.
CONCLUSIONS
This paper presents different approaches to evaluate the nature of the low end of the dose-response curve and the possible existence of thresholds for toxic agents, using a key events dose-response framework as a consistent way for presenting and analyzing the critical steps in a toxic process. The general conclusion is that there are rate-limiting key events that, if not met, can lead to a threshold for the dose-response, irrespective of the MOA involved.
To further address this issue, advantage can be taken of scientific advances that have made it possible to address the question of thresholds in a variety of ways. For example, it is possible to conduct research that provides a deep and thorough mechanistic understanding of the critical events in the toxicity of a particular agent thereby leading to qualitative and quantitative considerations of key events. As examples of this approach, in this paper we use the extensive literature on chloroform to analyze the biological basis for the nonlinearity in dose-response for chloroform, a hepatotoxicant and nongenotoxic carcinogen in rodents. It is also feasible to use a fundamental understanding of the biology underlying a disease process as the basis for constructing a key events framework for that process. In this paper, we take advantage of a more profound understanding of the molecular events that drive carcinogenesis to describe a key events framework for genotoxic carcinogens. A third approach is to use more sensitive technology to elucidate the low end of the dose-response curve. We illustrate this for endocrine-active agents, which are likely to be inherently thresholded (except in a few unusual cases) but for which the main concern is additivity to the endogenous background activity of hormones. Microarray technology for the global analysis of gene expression may be a sensitive enough tool to address the low end of the dose-response curve down to and below ambient exposure levels, thereby providing a basis for establishing pragmatic thresholds. These case studies contribute to a more robust basis for the assumption that many, if not all, toxicological effects exhibit biological thresholds and provide guidance on research needs to further address this question.
ACKNOWLEDGMENTS
This paper is one of the work products of an international group of experts convened by the International Life Sciences Institute (ILSI) Research Foundation which is a nonprofit, worldwide foundation established in 1978 to advance the understanding of scientific issues relating to nutrition, food safety, toxicology, risk assessment, and the environment. The Research Foundation was established in 1984 to create a vehicle for ILSI to support research. Its risk assessment program sponsors, organizes, and participates in a wide range of activities to develop and disseminate new scientific knowledge, encourage exchange of ideas, and build consensus among scientists from academia, industry, government, and public interest groups in its efforts to improve the scientific basis of risk assessment.
Financial support for this project from the following sources is gratefully acknowledged: ILSI Research Foundation, Health Canada, Ajinomoto, Coca-Cola Company, Groupe Danone, Kellogg Company, Kraft Foods, Inc., Mars, Inc., Mead Johnson Nutritionals, Monsanto Company, Nestlé, PepsiCo, Inc., The Procter & Gamble Company, Syngenta Ltd., Flavor Extract Manufacturers Association, and Grocery Manufacturers Association.
Declaration of Interests : Dr. Daston has received grant funding from the Cefic Long-range Research Initiative relating to signal transduction pathways in developmental toxicology. All other authors declare that they have no relevant interests to disclose.
This manuscript has been reviewed according to U.S. EPA guidelines although it does not necessarily reflect agency policy.
Notes
1 Where the term “threshold” is used on its own in this document, it refers to a biological threshold.
2 While low levels of CYP2E1 can be detected in rodent kidney (CitationWilke et al., 1994; CitationPoloyac et al., 2004), CYP2E1 was not detectable in samples of human kidney (CitationAmet et al., 1997; CitationBaker et al., 2005)
REFERENCES
- Amet , Y. , Berthou , F. , Fournier , G. , Dreano , Y. , Bardou , L. , Cledes , J. and Menez , J-F. 1997 . Cytochrome P450 4A and 2E1 expression in human kidney microsomes . Biochem Pharmacol. , 53 : 765 – 771 .
- Anand , S. S. , Soni , M. G. , Vaidya , V. S. , Murthy , S. N. , Mumtaz , M. M. and Mehendale , H. M. 2003 . Extent and timeliness of tissue repair determines the dose-related hepatotoxicity of chloroform . Int J Toxicol. , 22 ( 1 ) : 25 – 33 .
- Anand , S. S. , Philip , B. K. , Palkar , P. S. , Mumtaz , M. M. , Latendresse , J. R. and Mehendale , H. M. 2006 . Adaptive tolerance in mice upon subchronic exposure to chloroform: Increased exhalation and target tissue regeneration . Toxicol Appl Pharmacol. , 213 ( 3 ) : 267 – 281 .
- Ammann , P. , Laethem , C. L. and Kedderis , G. L. 1998 . Chloroform-induced cytolethality in freshly isolated male B6C3F1 mouse and F-344 rat hepatocytes . Toxicol Appl Pharmacol. , 149 : 217 – 225 .
- Baker , J. R. , Edwards , R. J. , Lasker , J. M. , Moore , M. R. and Satarug , S. 2005 . Renal and hepatic accumulation of cadmium and lead in the expression of CYP4F2 and CYP2E1 . Toxicol Lett. , 159 : 182 – 191 .
- Bang , J. W. , Crockford , D. J. , Holmes , E. , Pazos , F. , Sternberg , M. J. , Muggleton , S. H. and Nicholson , J. K. 2008 . Integrative top-down system metabolic modeling in experimental disease states via data-driven Bayesian methods . J Proteome Res. , 7 : 497 – 503 .
- Barlow , S. , Renwick , A. G. , Kleiner , J. , Bridges , J. W. , Busk , L. , Dybing , E. , Edler , L. , Eisenbrand , G. , Fink-Gremmels , J. , Knaap , A. , Kroes , R. , Liem , D. , Müller , D. J. , Page , S. , Rolland , V. , Schlatter , J. , Tritscher , A. , Tueting , W. and Würtzen , G. 2006 . Risk assessment of substances that are both genotoxic and carcinogenic report of an International Conference organized by EFSA and WHO with support of ILSI Europe . Food Chem Toxicol. , 44 : 1636 – 1650 .
- Barnes , D. E. and Lindahl , T. 2004 . Repair and genetic consequences of endogenous DNA base damage in mammalian cells . Annu Rev Genet. , 38 : 445 – 476 .
- Beddowes , E. J. , Faux , S. P. and Chipman , J. K. 2003 . Chloroform, carbon tetrachloride and glutathione depletion induce secondary genotoxicity in liver cells via oxidative stress . Toxicology , 187 ( 2–3 ) : 101 – 115 .
- Boobis , A. R. 2007 . Risk assessment of dietary supplements . Novartis Found Symp , 282 : 3 – 25 .
- Boobis , A. R. , Cohen , S. M. , Dellarco , V. , McGregor , D. , Meek , M. E. , Vickers , C. , Willcocks , D. and Farland , W. 2006 . IPCS framework for analyzing the relevance of a cancer mode of action for humans . Crit Rev Toxicol. , 36 : 781 – 792 .
- Boobis , A. R. , Doe , J. E. , Heinrich-Hirsch , B. , Meek , M. E. , Munn , S. , Ruchirawat , M. , Schlatter , J. , Seed , J. and Vickers , C. 2008 . IPCS framework for analyzing the relevance of a noncancer mode of action for humans . Crit Rev Toxicol. , 38 : 87 – 96 .
- Branchflower , R. V. , Nunn , D. S. , Highet , R. J. , Smith , J. H. , Hook , J. B. and Pohl , L. R. 1984 . Nephrotoxicity of chloroform: Metabolism to phosgene by the mouse kidney . Toxicol Appl Pharmacol. , 72 : 159 – 168 .
- Budzowska , M. and Kanaar , R. 2009 . Mechanisms of dealing with DNA damage-induced replication problems . Cell Biochem Biophys. , 53 : 17 – 31 .
- Burke , A. S. , Redeker , K. , Kurten , R. C. , James , L. P. and Hinson , J. A. 2007 . Mechanisms of chloroform-induced hepatotoxicity: Oxidative stress and mitochondrial permeability transition in freshly isolated mouse hepatocytes . J Toxicol Environ Health, Part A. , 70 : 1936 – 1945 .
- Cederbaum , A. I. 2006 . CYP2E1–biochemical and toxicological aspects and role in alcohol-induced liver injury . Mt Sinai J Med. , 73 : 657 – 672 .
- Chan , K. K. , Zhang , Q. M. and Dianov , G. L. 2006 . Base excision repair fidelity in normal and cancer cells . Mutagenesis , 21 : 173 – 178 .
- Constan , A. A. , Sprankle , C. S. , Peters , J. M. , Kedderis , G. L. , Everitt , J. I. , Wong , B. A. , Gonzalez , F. L. and Butterworth , B. E. 1999 . Metabolism of chloroform by cytochrome P450 2E1 is required for induction of toxicity in the liver, kidney, and nose of male mice . Toxicol Appl Pharmacol. , 160 : 120 – 126 .
- Doak , S. H. , Jenkins , G. J. , Johnson , G. E. , Quick , E. , Parry , E. M. and Parry , J. M. 2007 . Mechanistic influences for mutation induction curves after exposure to DNA-reactive carcinogens . Cancer Res. , 67 : 3904 – 3911 .
- Edwards , R. J. , Adams , D. A. , Watts , P. S. , Davies , D. S. and Boobis , A. R. 1998 . Development of a comprehensive panel of antibodies against the major xenobiotic metabolising forms of cytochrome P450 in humans . Biochem Pharmacol. , 56 : 377 – 387 .
- Edwards , S. W. and Preston , R. J. 2008 . Systems biology and mode of action based risk assessment . Toxicol Sci. , 106 : 312 – 318 .
- Fearon , E. R. and Vogelstein , B. 1990 . A genetic model for colorectal tumorigenesis . Cell , 61 : 759 – 767 .
- Fertuck , K. C. , Eckel , J. E. , Gennings , C. and Zacharewski , T. R. 2003 . Identification of temporal patterns of gene expression in the uteri of immature, ovariectomized mice following exposure to ethynylestradiol . Physiol Genom. , 15 : 127 – 141 .
- Gaylor , D. W. and Slikker , W. 1994 . Modeling for risk assessment of neurotoxic effects . Risk Anal. , 14 ( 3 ) : 333 – 338 .
- Gemma , S. , Testai , E. , Chieco , P. and Vittozzi , L. 2004 . Bioactivation, toxicokinetics and acute effects of chloroform in Fisher 344 and Osborne Mendel male rats . J Appl Toxicol. , 24 : 203 – 210 .
- Gray , L. E. Jr. , Ostby , J. , Monosson , E. and Kelce , W. R. 1999 . Environmental antiandrogens: low doses of the fungicide vinclozolin alter sexual differentiation of the male rat . Toxicol Ind Health. , 15 : 48 – 64 .
- Greenman , C. , Stephens , P. , Smith , R. , Dalgliesh , G. L. , Hunter , C. , Bignell , G. , Davies , H. , Teague , J. , Butler , A. , Stevens , C. , Edkins , S. , O'Meara , S. , Vastrik , I. , Schmidt , E. E. , Avis , T. , Barthorpe , S. , Bhamra , G. , Buck , G. , Choudhury , B. , Clements , J. , Cole , J. , Dicks , E. , Forbes , S. , Gray , K. , Halliday , K. , Harrison , R. , Hills , K. , Hinton , J. , Jenkinson , A. , Jones , D. , Menzies , A. , Mironenko , T. , Perry , J. , Raine , K. , Richardson , D. , Shepherd , R. , Small , A. , Tofts , C. , Varian , J. , Webb , T. , West , S. , Widaa , S. , Yates , A. , Cahill , D. P. , Louis , D. N. , Goldstraw , P. , Nicholson , A. G. , Brasseur , F. , Looijenga , L. , Weber , B. L. , Chiew , Y. E. , DeFazio , A. , Greaves , M. F. , Green , A. R. , Campbell , P. , Birney , E. , Easton , D. F. , Chenevix-Trench , G. , Tan , M. H. , Khoo , S. K. , The , B. T. , Yuen , S. T. , Leung , S. Y. , Wooster , R. , Futreal , P. A. and Stratton , M. R. 2007 . Patterns of somatic mutations in human cancer genomes . Nature , 446 : 153 – 158 .
- Hanahan , D. and Weinberg , R. A. 2000 . The hallmarks of cancer . Cell , 100 : 57 – 70 .
- Hartig , S. , Fries , S. and Balcarcel , R. R. 2005 . Reduced mitochondrial membrane potential and metabolism correspond to acute chloroform toxicity of in vitro hepatocytes . J Appl Toxicol. , 25 : 310 – 317 .
- Hayashi , S. , Watanabe , J. and Kawajiri , K. 1991 . Genetic polymorphisms in the 59-flanking region change transcriptional regulation of the human cytochrome P450IIE1 gene . J Biochem Tokyo , 110 : 559 – 565 .
- Heinloth , A. N. , Irwin , R. D. , Boorman , G. A. , Nettesheim , P. , Fannin , R. D. , Sieber , S. O. , Snell , M. L. , Tucker , C. J. , Li , L. , Travlos , G. S. , Vansant , G. , Blackshear , P. E. , Tennant , R. W. , Cunningham , M. L. and Paules , R. S. 2004 . Gene expression profiling of rat livers reveals indicators of potential adverse effects . Toxicol Sci. , 80 : 193 – 202 .
- Hu , Y. , Oscarson , M. , Johansson , I. , Yue , Q. Y. , Dahl , M. L. , Tabone , M. , Arinco , S. , Albano , E. and Ingelman Sundberg , M. 1997 . Genetic polymorphism of human CYP2E1: characterization of two variant alleles . Mol Pharmacol. , 51 : 370 – 376 .
- Ingelman-Sundberg , M. 2004 . Human drug metabolising cytochrome P450 enzymes: Properties and polymorphisms . Naunyn Schmiedebergs Arch Pharmacol , 369 : 89 – 104 .
- Ingelman-Sundberg , M. , Johansson , I. , Penttila , K. E. , Glaumann , H. and Lindros , K. O. 1988 . Centrilobular expression of ethanol-inducible cytochrome P-450 (IIE1) in rat liver . Biochem Biophys Res Commun. , 157 : 55 – 60 .
- IPCS (International Programme on Chemical Safety) . 2004 . Concise International Chemical Safety Assessment (CICAD) , Geneva : WHO . No. 58: Chloroform
- Johnsrud , E. K. , Koukouritaki , S. B. , Divakaran , K. , Brunengraber , L. L. , Hines , R. N. and McCarver , D. G. 2003 . Human hepatic CYP2E1 expression during development . J Pharmacol Exp Ther. , 307 : 402 – 407 .
- Jones , S. M. , Boobis , A. R. , Moore , G. E. and Stanier , P. M. 1992 . Expression of CYP2E1 during human fetal development: methylation of the CYP2E1 gene in human fetal and adult liver samples . Biochem Pharmacol. , 43 : 876 – 1879 .
- Julien , E. , Boobis , A. R. , Olin , S. S. and the ILSI Research Foundation Threshold Working Group . 2009 . The Key Events Dose-Response Framework: A cross-disciplinary mode-of-action based approach to examining dose-response and thresholds . Crit. Rev. Food Sci. Nutr. , 49 : 682 – 689 .
- Komulainen , H. 2004 . Experimental cancer studies of chlorinated by-products . Toxicology , 198 : 239 – 248 .
- Larson , J. L. , Wolf , D. C. and Butterworth , B. E. 1994 . Induced cytotoxicity and cell proliferation in the hepatocarcinogenicity of chloroform in female B6C3F1 mice: Comparison of administration by gavage in corn oil vs. ad libitum in drinking water . Fundam Appl Toxicol. , 22 : 90 – 102 .
- Latterich , M. , Abramovitz , M. and Leyland-Jones , B. 2008 . Proteomics: new technologies and clinical applications . Eur J Cancer , 44 : 2737 – 2741 .
- Laws , S. C. , Ferrell , J. M. , Stoker , T. E. , Schmid , J. and Cooper , R. L. 2000 . The effects of atrazine on female wistar rats: An evaluation of the protocol for assessing pubertal development and thyroid function . Toxicol. Sci. , 58 : 366 – 376 .
- Liao , K. H. , Tan , Y. M. , Conolly , R. B. , Borghoff , S. J. , Gargas , M. L. , Andersen , M. E. and Clewell , H. J. III . 2007 . Bayesian estimation of pharmacokinetic and pharmacodynamic parameters in a mode-of-action-based cancer risk assessment for chloroform . Risk Anal. , 27 : 1535 – 1551 .
- Lipscomb , J. C. , Barton , H. A. , Tornero-Velez , R. , Evans , M. V. , Alcasey , S. , Snawder , J. E. and Laskey , J. 2004 . The metabolic rate constants and specific activity of human and rat hepatic cytochrome P-450 2E1 toward toluene and chloroform . J Toxicol Environ Health, Part A , 67 : 537 – 553 .
- Loizou , G. D. and Cocker , J. 2001 . The effects of alcohol and diallyl sulphide on CYP2E1 activity in humans: A phenotyping study using chlorzoxazone . Hum Exp Toxicol. , 20 : 321 – 327 .
- Mantovani , A. 2009 . Cancer: Inflaming metastasis . Nature , 457 : 36 – 37 .
- Mardis , E. R. 2008 . Next-generation DNA sequencing methods . Ann Rev Genomics and Human Genet. , 9 : 387 – 402 .
- Meek , M. E. , Bucher , J. R. , Cohen , S. M. , Dellarco , V. , Hill , R. N. , Lehman-McKeeman , L. D. , Longfellow , D. G. , Pastoor , T. , Seed , J. and Patton , D. E. 2003 . A framework for human relevance analysis of information on carcinogenic modes of action . Crit Rev Toxicol. , 33 : 591 – 653 .
- Meek , M. E. , Renwick , A. , Ohanian , E. , Dourson , M. , Lake , B. , Naumann , B. D. and Vu , V. 2002 . Guidelines for application of chemical-specific adjustment factors in dose/concentration-response assessment . Toxicology , 181–182 : 115 – 120 .
- Moggs , J. G. , Tinwell , H. , Spurway , T. , Chang , H. S. , Pate , I. , Lim , F. L. , Moore , D. J. , Soames , A. , Stuckey , R. , Currie , R. , Zhu , T. , Kimber , I. , Ashby , J. and Orphanides , G. 2004 . Phenotypic anchoring of gene expression changes during estrogen-induced uterine growth . Environ. Health Perspect. , 112 : 1589 – 1606 .
- Morlan , J. , Baker , J. and Sinicropi , D. 2009 . Mutation detection by Real-time PCR: A simple, robust and highly selective method . PLoS ONE , 4:e4584 : 1 – 11 .
- Naciff , J. M. , Overmann , G. J. , Torontali , S. M. , Carr , G. J. , Khambatta , Z. S. , Tiesman , J. P. , Richardson , B. D. and Daston , G. P. 2007 . Uterine temporal responses to acute exposurto 17alpha-ethinyl estradiol in the immature rat . Toxicol. Sci. , 97 : 467 – 490 .
- Naciff , J. M. , Richardson , B. D. , Oliver , K. G. , Jump , L. M. , Torontali , S. M. , Juhlin , K. D. , Carr , G. J. , Paine , J. R. , Tiesman , J. P. and Daston , G. P. 2005a . Design of a microsphere-based high-throughput gene expression assay to determine estrogenic potential . Environ. Health Perspect. , 113 : 1164 – 1171 .
- Naciff , J. M. , Hess , K. A. , Overmann , G. J. , Torontali , S. M. , Carr , G. J. , Tiesman , J. P. , Foertsch , L. M. , Richardson , B. D. , Martinez , J. E. and Daston , G. P. 2005b . Gene expression changes induced in the testis by transplacental exposure to high and low doses of 17-alpha-ethynyl estradiol, genistein, or bisphenol A . Toxicol. Sci. , 86 : 396 – 416 .
- NAS (National Academies of Science) . 2008 . Science and Decisions: Advancing Risk Assessment , Washington, DC : National Academies of Science, National Academies Press .
- Ohashi , Y. , Yamada , K. , Takemoto , I. , Mizutani , T. and Saeki , K. 2005 . Inhibition of human cytochrome P450 2E1 by halogenated anilines, phenols, and thiophenols . Biol Pharm Bull. , 28 : 1221 – 1223 .
- Ottender , M. and Lutz , W. K. 1999 . Correlation of DNA adduct levels with tumor incidence: carcinogenic potency of DNA adducts . Mutat Res. , 424 : 237 – 247 .
- Pohl , L. R. and Krishna , G. 1978 . Deuterium isotope effect in bioactivation and hepatotoxicity of chloroform . Life Sci. , 23 : 1067 – 1072 .
- Pohl , L. R. , Bhooshan , B. , Whittaker , N. F. and Krishna , G. 1977 . Phosgene: A metabolite of chloroform . Biochem Biophys Res Commun. , 79 : 684 – 691 .
- Pohl , L. R. , Branchflower , R. V. , Highet , R. J. , Martin , J. L. , Nunn , D. S. , Monks , T. J. , George , J. W. and Hinson , J. A. 1981 . The formation of diglutathionyl dithiocarbonate as a metabolite of chloroform, bromotrichloromethane and carbon tetrachloride . Drug Metab Dispos. , 9 : 334 – 339 .
- Pohl , L. R. , George , J. W. and Satoh , H. 1984 . Strain differences in chloroform-induced nephrotoxicity: Different rates of metabolism of chloroform to phosgene by the mouse kidney . Drug Metab Dispos. , 12 : 304 – 308 .
- Poloyac , S. M. , Tortorici , M. A. , Przychodzin , D. I. , Reynolds , R. B. , Xie , W. , Frye , R. F. and Zemaitis , M. A. 2004 . The effect of isoniazid on CYP2E1- and CYP4A-mediated hydroxylation of arachidonic acid in the rat liver and kidney . Drug Metab Dispos. , 32 : 727 – 733 .
- Powlin , S. S. , Cook , J. C. , Novak , S. and O'Connor , J. C. 1998 . Ex vivo and in vitro testis and ovary explants: Utility for identifying steroid biosynthesis inhibitors and comparison to a Tier I screening battery . Toxicol. Sci. , 46 : 61 – 74 .
- Preston , R. J. 1991 . “ Mechanisms of induction of chromosomal alterations and sister chromatid exchanges: Presentation of a generalized hypothesis ” . In Genetic Toxicology: A Treatise , Edited by: Li , A. P. and Heflich , R. H. Boca Raton, FL : CRC Press .
- Preston , R. J. and Williams , G. M. 2005 . DNA-reactive carcinogens: mode of action and human cancer hazard . Crit Rev Toxicol. , 35 : 673 – 683 .
- Reynolds , E. S. and Yee , A. G. 1967 . Liver parenchymal injury. V. Relationship between patterns of chloromethane-C14 incorporation into constituents of liver in vivo and cellular injury . Lab Invest. , 16 : 591 – 603 .
- Ronis , M. J. J. , Lindros , K. O. and Ingelman-Sundberg , M. 1996 . “ The cytochrome P4502E subfamily ” . In Ioannides I (ed.) Cytochromes P450, pharmacological and toxicological aspects , Edited by: Ioannides , I. 211 – 239 . Boca Raton : CRC Press .
- Ruch , R. J. , Klaunig , J. E. , Schultz , N. E. , Askari , A. B. , Lacher , D. A. , Pereira , M. S. and Goldblatt , P. J. 1986 . Mechanisms of chloroform and carbon tetrachioride toxicity in primary cultured mouse hepatocytes . Environ Health Perspect. , 69 : 301 – 305 .
- Sheehan , D. M. 2006 . No-threshold dose-response curves for nongenotoxic chemicals: Findings and applications for risk assessment . Environ. Res. , 100 : 93 – 99 .
- Shin , J. H. , Moon , H. J. , Kang , I. H. , Kim , T. S. , Kim , I. Y. , Park , I. S. , Kim , H. S. , Jeung , E. B. and Han , S. Y. 2006 . Repeated 28-day oral toxicity study of ketoconazole in rats based on the draft protocol for the “Enhanced OECD Test Guideline No. 407” to detect endocrine effects . Arch. Toxicol. , 80 : 797 – 803 .
- Smith , J. H. and Hook , J. B. 1983 . Mechanism of chloroform nephrotoxicity. II. In vitro evidence for renal metabolism of chloroform in mice . Toxicol Appl Pharmacol. , 70 : 480 – 485 .
- Smith , J. H. and Hook , J. B. 1984 . Mechanism of chloroform nephrotoxicity: III. Renal and hepatic microsomal metabolism of chloroform in mice . Toxicol Appl Pharmacol. , 73 : 511 – 524 .
- Smith , J. H. , Hewitt , W. R. and Hook , J. B. 1985 . Role of intrarenal biotransformation in chloroform-induced nephrotoxicity in rats . Toxicol Appl Pharmacol. , 79 : 166 – 174 .
- Smith , J. H. , Maita , K. , Sleight , S. D. and Hook , J. B. 1983 . Mechanism of chloroform nephrotoxicity. I. Time course of chloroform toxicity in male and female mice . Toxicol Appl Pharmacol , 70 : 467 – 479 .
- Sonich-Mullin , C. , Fielder , R. , Wiltse , J. , Baetcke , K. , Dempsey , J. , Fenner-Crisp , P. , Grant , D. , Hartley , M. , Knaap , A. , Kroese , D. , Mangelsdorf , I. , Meek , M. E. , Rice , J. M. and Younes , M. 2001 . IPCS conceptual framework for evaluating a mode of action for chemical carcinogenesis . Regul Toxicol Pharmacol. , 34 : 146 – 152 .
- Stevens , J. L. and Anders , M. W. 1981 . Effect of cysteine, diethylmaleate and phenobarbital treatments on the hepatotoxicity of (1H)- and (2H)-chloroform . Chem-Biol Interact , 37 : 207 – 217 .
- Swenberg , J. A. , Fryar-Tita , E. , Jeong , Y. C. , Boysen , G. , Starr , T. , Walker , V. E. and Albertini , R. J. 2008 . Biomarkers in toxicology and risk assessment: Informing critical dose-response relationships . Chem Res Toxicol. , 21 : 253 – 265 .
- Tee , L. B. , Boobis , A. R. , Huggett , A. C. and Davies , D. S. 1986 . Reversal of acetaminophen toxicity in isolated hamster hepatocytes by dithiothreitol . Toxicol Appl Pharmacol. , 83 : 294 – 314 .
- Tee , L. B. , Davies , D. S. , Seddon , C. E. and Boobis , A. R. 1987 . Species differences in the hepatotoxicity of paracetamol are due to differences in the rate of conversion to its cytotoxic metabolite . Biochem Pharmacol. , 36 : 1041 – 1052 .
- Templin , M. V. , Constan , A. A. , Wolf , D. C. , Wong , B. A. and Butterworth , B. E. 1998 . Patterns of chloroform-induced regenerative cell proliferation in BDF1 mice correlate with organ specificity and dose-response of tumor formation . Carcinogenesis , 19 : 187 – 193 .
- Templin , M. V. , Jamison , K. C. , Wolf , D. C. , Morgan , K. T. and Butterworth , B. E. 1996a . Comparison of chloroform-induced toxicity in the kidneys, liver and nasal passages of male Osborne–Mendel and F-344 rats . Cancer Lett. , 104 : 71 – 78 .
- Templin , M. V. , Larson , J. L. , Butterworth , B. E. , Jamison , K. , Wong , B. A. and Wolf , D. C. 1996b . A 90-day chloroform inhalation study in F-344 rats: Profile of toxicity and relevance to cancer studies . Fund Appl Toxicol. , 32 : 109 – 125 .
- Tsutsumi , M. , Lasker , J. M. , Shimizu , M. , Rosman , A. S. and Lieber , C. S. 1989 . The intralobular distribution of ethanol-inducible P450IIE1 in rat and human liver . Hepatology , 10 : 437 – 446 .
- U.S. EPA (U.S. Environmental Protection Agency) . 2005 . Guidelines for Carcinogen Risk Assessment, Risk Assessment Forum , Washington, DC : U.S. Environmental Protection Agency . Available from: http://www.epa.gov/cancerguidelines
- U.S. EPA . 2006 . Chloroform MCLG . Fed Reg , 71 : 407
- Wang , P. Y. , Kaneko , T. , Tsukada , H. , Nakano , M. , Nakajima , T. and Sato , A. 1997 . Time courses of hepatic injuries induced by chloroform and by carbon tetrachloride: Comparison of biochemical and histopathological changes . Arch Toxicol. , 71 ( 10 ) : 638 – 645 .
- White , R. H. , Cote , I. , Zeise , L. , Fox , M. , Dominici , F. , Burke , T. A. , White , P. D. , Hattis , D. B. and Samet , J. M. 2009 . State-of-the-science workshop report: Issues and approaches in low-dose–response extrapolation for environmental health risk assessment . Environ Health Perspect. , 117 : 283 – 287 .
- Wilke , A. V. , Clay , A. S. and Borghoff , S. J. 1994 . Cytochrome P450 2E1 (P450 2E1) levels in male and female rat kidney: Implications for xenobiotic metabolism (abstract) . Toxicologist , 14 : 73 (Abstract #199)
- Wu , X. and Dewey , T. G. 2006 . From microarray to biological networks: Analysis of gene expression profiles . Methods Mol Biol. , 316 : 35 – 48 .
- Zorov , D. B. , Juhaszova , M. and Sollott , S. J. 2006 . Mitochondrial ROS-induced ROS release: An update and review . Biochim Biophys Acta. , 1757 : 509 – 517 .