
Abstract
Periodontitis is an immuno-inflammatory disease of the soft tissues surrounding the teeth. Periodontitis is linked to many communicable and non-communicable diseases such as diabetes, cardiovascular disease, rheumatoid arthritis, and cancers. The oral-systemic link between periodontal disease and systemic diseases is attributed to the spread of inflammation, microbial products and microbes to distant organ systems. Oral bacteria reach the gut via swallowed saliva, whereby they induce gut dysbiosis and gastrointestinal dysfunctions. Some periodontal pathogens like Porphyromonas. gingivalis, Klebsiella, Helicobacter. Pylori, Streptococcus, Veillonella, Parvimonas micra, Fusobacterium nucleatum, Peptostreptococcus, Haemophilus, Aggregatibacter actinomycetomcommitans and Streptococcus mutans can withstand the unfavorable acidic, survive in the gut and result in gut dysbiosis. Gut dysbiosis increases gut inflammation, and induce dysplastic changes that lead to gut dysfunction. Various studies have linked oral bacteria, and oral-gut axis to various GIT disorders like inflammatory bowel disease, liver diseases, hepatocellular and pancreatic ductal carcinoma, ulcerative colitis, and Crohn’s disease. Although the correlation between periodontitis and GIT disorders is well established, the intricate molecular mechanisms by which oral microflora induce these changes have not been discussed extensively. This review comprehensively discusses the intricate and unique molecular and immunological mechanisms by which periodontal pathogens can induce gut dysbiosis and dysfunction.
Introduction
Periodontitis is a chronic immuno-inflammatory disease of the soft and hard tissue caused by the interaction of the microbial biofilm formed around the teeth with the host. The most common microorganisms in the oral biofilm associated with periodontitis include Porphyromonas gingivalis, Tannerella forsythia, Aggregatibacter actinomycetemcomitans, Prevotella intermedia, Fusobacterium nucleatum, Capnocytophaga gingivalis, Treponema denticola, Treponema socranskii, Eubacterium nodatum and Campylobacter rectus (Teles et al. Citation2013). The interaction of oral microbes with the host triggers the release of many proinflammatory mediators (cytokines), reactive oxygen species (ROS), and acute phase proteins locally in the gingival and periodontal tissues (Kaur et al. Citation2018; Slots Citation2019). These inflammatory mediators along with bacteria and bacterial by-products enter the systemic circulation either via the blood vessels (bacteremia) or via the swallowed saliva. This increases the microbial and inflammatory burden in the systemic circulation and increases the oxidative stress (Casarin et al. Citation2010). It has been estimated that approximately 1010 oral microbes reach the gut per day through saliva (Li et al. Citation2000; Lockhart et al. Citation2009; Peng et al. Citation2022). The chances of bacteremia also increase in individuals with poor oral hygiene and the presence of inflamed gingival and periodontal tissues (Swidsinski et al. Citation2005; Parahitiyawa et al. Citation2009).
The entry of oral bacteria into the gut disturbs the homeostasis and functioning of the entire GIT system. Oral bacteria can survive in the stomach, penetrate the gut mucosa, and transverse via the duct of Wirsung and the biliary duct to other parts of GIT (Huh and Roh, Citation2020). Oral bacteria have been identified in the pancreas, liver, colon, and intestine (Qin et al. Citation2014). Oral bacteria survival in the gut shifts the pancreatic, biliary, and liver microbial profile, increases the influx of inflammation mediators into the gut mucosa, and activates the process of apoptosis and tumorigenic reaction (Chen et al. Citation2019). Based on these changes many studies have associated periodontal diseases with various GIT disorders such as gastritis (Maisonneuve et al. Citation2017; She et al. Citation2020), colon and pancreatic cancers (Chung et al. Citation2021; Read et al. Citation2021), inflammatory bowel disease (IBD), ulcerative colitis (UC), Crohn’s disease (CD) (Dowman et al. Citation2010), chronic liver disease (Hernandez et al. Citation2022), hepatocellular carcinoma (Hooper and Gordon Citation2001). However, limited papers comprehensively discuss the intricate molecular pathogenic mechanisms by which periodontitis induces GIT dysfunctions. This review paper provides an update on the molecular and immunological mechanisms by which periodontal pathogens induce gut dysfunction and dysbiosis.
Methodology
Search strategy, data extraction, eligibility criteria
The electronic databases MEDLINE (PubMed), SCOPUS, Web of Science, and EMBASE were searched on August 20, 2022, from their earliest records. The search was updated till October 2023. The following search terms were utilized for data collection: were as follows: (Gastrointestinal OR Crohn’s Disease[Title/Abstract] OR Inflammatory Bowel Disease[Title/Abstract] OR Lactose Intolerance[Title/Abstract] OR Diarrhoea[Title/Abstract] OR Constipation[Title/Abstract] OR Gall Stone[Title/Abstract] OR GERD[Title/Abstract] OR Peptic Ulcer[Title/Abstract] OR Celiac Disease[Title/Abstract] OR Ulcerative Colitis[Title/Abstract] OR Pancreatitis[Title/Abstract] OR Esophagitis[Title/Abstract] OR Gastritis[Title/Abstract] OR Achalasia[Title/Abstract] OR Enterocolitis[Title/Abstract] OR Mesenteric[Title/Abstract] OR Duodenitis[Title/Abstract] OR Duodenum[Title/Abstract] OR Duodenal[Title/Abstract] OR Jejunitis[Title/Abstract] OR Ileitis[Title/Abstract] OR Gastric Fluid[Title/Abstract] OR Stomach[Title/Abstract] OR Caecum[Title/Abstract] OR Sprue[Title/Abstract] OR Bowel[Title/Abstract] OR Appendicitis[Title/Abstract] OR Proctitis[Title/Abstract] OR Colon Intestine[Title/Abstract] OR Small sIntestine[Title/Abstract] OR Large Intestine[Title/Abstract] OR Anal[Title/Abstract] OR Anus[Title/Abstract] OR megacolon[Title/Abstract] OR Cholecystitis[Title/Abstract] OR Biliary Tract[Title/Abstract] OR Cholangitis[Title/Abstract] OR Intussusception[Title/Abstract] OR Steatorrhea[Title/Abstract] OR Malabsorption Syndrome[Title/Abstract] OR Belching[Title/Abstract] OR Flatulence[Title/Abstract]) AND (Periodontitis[Title/Abstract] OR Periodontal Inflammation[Title/Abstract] OR Adult Periodontitis[Title/Abstract] OR Gingivitis[Title/Abstract] OR Gingival Hyperplasia[Title/Abstract] OR Aggressive Periodontitis[Title/Abstract] OR Gingival Bleeding[Title/Abstract] OR Refractive Periodontitis[Title/Abstract] OR Periodontal disease[Title/Abstract] OR Gingival disease[Title/Abstract] OR Oral disease[Title/Abstract] OR Oral Microorganism[Title/Abstract] OR Oral bacteria[Title/Abstract] OR oral Microbiome[Title/Abstract]. Articles that are written only in the English language were selected. The details of articles retrieved from all databases are provided in Table S1. The search was complemented by hand-searching the reference lists of the included articles and performing a citation search for any additional reviews.
Inclusion and exclusion criteria
To answer the focused questions only articles analyzing the role of periodontitis, including the role of periodontal pathogens on the gut tissue, gut microflora, or explaining any pathogenic mechanism were included. All systematic and narrative review articles, human in vivo (observational studies, longitudinal study design, randomized clinical trial comprising of interventional trials, cohort study, case-control study, and cross-sectional study), in vitro, and animal studies evaluating the role of periodontitis or periodontal pathogens in the development of GIT dysfunction were included. All case reports, case series, letters to editors, and editorials were excluded.
Data collection
All articles were screened using Microsoft Excel. A total of 2828 articles were collected. After removing the duplicates, 1561 articles were taken up for title and abstract screening. After title abstract screening 354 articles were included for full-text screening. Finally, 91 were included in the review. The key features of identified relevant research studies were evaluated and are summarized in a narrative review as follows.
Results
Oral bacteria identified in the gut
The gut microbiota mostly comprises of anaerobes microorganisms, belonging to five major phyla: Bacteroidetes, Firmicutes, Actinobacteria, Proteobacteria, and Verrucomicrobia (Schmidt et al., Citation2019). Among these, many belong to the typical oral-resident species. Some of the common oral taxa detected in the gut are Parvimonas, Porphyromonas, Klebsiella Peptostreptococcus, Proteobacteria, Streptococcus, Veillonella, Haemophilus, Peptostreptococcus and Fusobacterium. (Huh and Roh, Citation2020). Some of the key oral species identified in the gut include Helicobacter Pylori, Parvimonas micra, P. gingivalis, T. forsythia, T. denticola, P. intermedia, F. necrophorum, A. actinomycetomcomitans, F. nucleatum, Haemophilus influenza. Escherichia coli, Klebsiella species, Filifactor alocius, Campylobacter rectus, and Streptococcus faecalis (Al Asqah et al. Citation2009; Iwauchi et al. Citation2019). Many oral bacteria, most commonly H. Pylori have been identified in the gut of patients with chronic gastritis, peptic ulcer, IBD, and gastric cancer (Li et al., Citation2019). Periodontal pathogens have also been identified in the fecal samples of patients with liver cirrhosis (Nakahara et al. Citation2018), non-alcoholic fatty liver disorder (NAFLD) (Li et al. Citation2021), hepatocellular carcinoma (Liu et al., Citation2020), Ulcerative colitis (UC), and Crohn’s disease (CD) (Ren et al. Citation2019). H. pylori, C. rectus, and Streptococcus faecalis have been identified in pancreatic tissues and ducts, liver, intestines, and colon of patients with pancreatic and colon cancer, liver disease, and IBD (Grąt et al. Citation2016; Ni et al. Citation2019). Patients with hepatocellular carcinoma with cirrhosis have higher levels of E. coli and Fusobacteria species compared to those with hepatocellular carcinoma (Ahn et al. Citation2012). P. gingivalis, a keystone pathogen for periodontal disease, has also been identified as a marker positively linked with higher risk and mortality in pancreatic ductal adenocarcinoma (PDAC) (Michaud et al. Citation2013; Rubinstein et al. Citation2013). F. nucleatum is also considered a potential marker for chronic liver diseases, liver cirrhosis, IBD, pancreatic, hepatocellular cancers, and colorectal cancers (CRC) (Castellarin et al. Citation2012; Dejea and Sears Citation2016; Chen et al. Citation2019). Dejea et al. also noted an enhanced presence of F. nucleatum in biofilms deriving CRC specimens (Drewes et al. Citation2017). F. nucleatum was absent in healthy colonic tissue and all the isolated biofilms were polymicrobial, with the matched biofilm-positive tumor and healthy tissue samples harboring invasive bacteria. Thus, biofilms associated with CRC present similar structure and pathogenic potential to those in oral diseases, possibly linked with CRC tumorigenesis. Other studies also reveal that intestinal biofilms contain oral commensal (Parvimonas, Peptostreptococcus, Prevotella) in addition to pathogenic (F. nucleatum, P. gingivalis) periodontal bacteria, which could result in CRC development (Warren et al. Citation2013). Warren et al. observed the coexistence of oral anaerobic bacteria including F. nucleatum, Campylobacter, and Leptotrichia in healthy and CRC tissues (Bajaj et al. Citation2015). These species were found to form a cooperative polymicrobial network in tumor tissue with other species such as P. gingivalis. Moreover, Campylobacter strains also showed the ability to co-aggregate with F. nucleatum, the latter serving as a bridging microorganism, colonizing the intestinal mucosa by attracting other compatible oral bacteria.
Patients with liver cirrhosis also have an increased level of fecal microbial species that are oral in origin (Lourenςo et al. Citation2018). describes the studies where various oral bacteria have been identified in the gut. Lourenço et al. assessed the gut microbiota in individuals with periodontal diseases (Gevers et al. Citation2014) and found an increase in the Firmicutes/Bacteroides ratio and enrichment of the following phylum: Euryarcheota, Verrucoccus, and Proteobacteria (Gevers et al. Citation2014). Besides, a variety of oral taxas that are associated with periodontal destruction and inflammation were found in the gut, irrespective of the periodontal status. Patients with IBD have a significant increase in the levels of Haemophilus and Veillonella in the gut mucosa (Grivennikov et al. Citation2012). Like IBD, colorectal tumors have increased intestinal colonization of oral microbiota (Seki and Brenner Citation2008).
Table 1. Studies associated with periodontal pathogens and GIT dysfunction.
Mechanisms by which oral bacteria increase the risk of GIT dysfunction
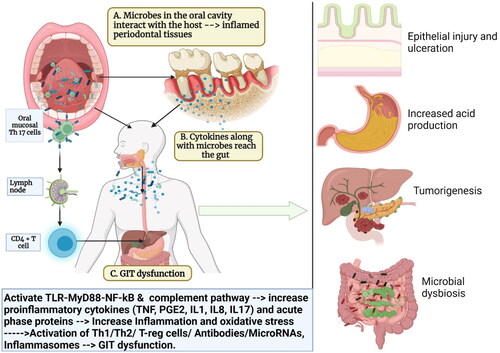
There are various mechanisms by which oral bacteria can induce changes in the gut. Some of the potential pathogenic mechanisms by which oral bacteria induce GUT dysfunction are discussed as follows ():
Figure 1. Schematic representation explaining the link between periodontal disease, oral bacteria, and gastrointestinal (GIT) dysfunction: (A) the oral cavity is a reservoir of millions of microbes that form a biofilm on the hard (teeth and restorations) and soft tissues (gums, tongues, palate, etc). The interaction of the microbes to the periodontal tissues initiates an inflammatory response that locally leads to periodontal inflammation; (B) the onset of periodontal inflammation (gingivitis and periodontitis) is marked by a massive release of proinflammatory mediators such as IL1, IL8, IL17, and TNF along with activation of various signaling pathways and host receptors such as toll-like receptors (TLRs), the complement system, nuclear factor kappa beta (NF-Kb); (C) these proinflammatory mediators enter the systemic circulation via blood vessels or via the saliva and reach the liver where they trigger the acute phase response and the release of acute phase proteins like C-reactive proteins (CRP). This increases the systemic inflammatory burden and oxidative stress. (D) The increased inflammatory burden in turn affects various parts of the gut and leads to GIT dysfunction (Created in Biorender).
Activation of Toll-like receptors (TLRs) and release of pro-inflammatory cytokines
Modulation of the complement system
Alteration of the oxidative stress and release of reactive oxygen species
Activation of inflammasome
Activation of acute phase response and release of acute phase proteins
Activation of microRNAs
Release of volatile sulfur compounds
Necrotoxicity of host cells
Activation of TLR receptors and release of proinflammatory cytokines
Oral bacteria after entering the gut attaches to various TLRs (TLR2, TLR3, TLR4, TLR5) that are located in the epithelium, lamina propria, and cells (macrophages, neutrophils, and Kuffer cells) of the gut (Alarcón et al. Citation2016). TLR2/4 are two of the most common TLRs activated during periodontal disease which are also associated with gastrointestinal dysfunction. TLRs act like sensors of the gut and play a key role in maintaining gut homeostasis, controlling the immune responses, and modulating its microbial profile. Genetic alterations of TLRs are associated with a change in the composition of gut microbiota. A change in the TLR function and TLR polymorphisms/mutations have been identified and directly linked to the immune response of the gut (gut immunity), the microbial profile of the gut, and gut inflammation (Bunte and Beikler Citation2019). The increased efflux of proinflammatory mediators and TLR activation alters the microbial profile and this is the primary cause of various gut disorders such as gastritis (Andrukhov et al. Citation2011; Miura and Ohnishi Citation2014), non-alcoholic fatty liver disease (Hausmann et al. Citation2002), IBD, CD and UC (Cario et al. Citation2004; Franchimont et al. Citation2004), peptic ulcers (Medzhitov et al. Citation1997; McClain et al. Citation1999; Seki et al. Citation2007; Sanchez-Munoz et al. Citation2008; Seiderer et al. Citation2008; Azer Citation2013; Hug et al. Citation2018), hepatic fibrosis and cancers of the gut including hepatic carcinomas, CRC, and pancreatic cancer (Liu et al. Citation2014) (Table S2:Role of different TLRs in the gut).
The interaction of oral bacteria with TLRs activates various inflammatory signaling pathways (NF-Kb, JNKs, PI3K, MAPK, activator protein 1 (AP-1), and p38) in the gut that activates the release of numerous pro-inflammatory (TNF-α, IL-1β, IL-6, IL-12, IL-18) and anti-inflammatory cytokines (IL-10) (Zhang and Ghosh Citation2001). Among all the signaling pathways, nuclear factor kappa beta (NF-kB-MyD88) is the most important inflammatory pathway triggered during gut inflammation and tumorigenesis (Seiderer et al. Citation2008; Jia et al. Citation2019). The activation of NF-kB involves the activation of TLR-MyD88 signaling by bacterial adhesions such as LPS. The LPS from oral bacteria attaches to the MD2/TLR4 (Myeloid differentiation protein 2/TLR 4 complex) and subsequently triggers the host’s innate immune response (Wang and Ohura Citation2002). LPS-binding protein (LPS-BP), a 60 kDa serum glycoprotein, is also present in the gut, liver, and gastrointestinal epithelial cells and is activated upon invasion of the gut by oral bacteria (Rogler et al. Citation1998).
Chronic NF-κB signaling is also the primary signaling pathway for the development of IBD (UC and CD), intestinal inflammation, and pancreatic, colon, and gastric cancer (Medzhitov et al. Citation1997; Bainbridge and Darveau Citation2001; Andresen et al. Citation2005; Visekruna et al. Citation2006; Berkovich et al. Citation2022). Mitogen-activated protein kinase (MAPK) pathway is a key inflammatory pathway increased with P. gingivalis invasion in the gut (van Deventer Citation1999). TNF is another potential mediator released during periodontitis that activates the mitogen-activated protein kinase (MAPK) pathway and increases the risk of CD and UC (Baert et al. Citation1999; Hietaranta et al. Citation2004). TNF can change the TLR4 expression in the pancreas and cause pancreatic inflammation (pancreatitis). TLR4 also causes a massive release of elastase and heparan sulfate in the pancreatic tissue and these enzymes in turn destroy the pancreatic parenchyma and subsequently increase pancreatic inflammation (Johnson et al. Citation2002; Zhang et al. Citation2018). Studies have noted that LPS-containing micro-vesicles from P. gingivalis can penetrate the gut mucosa like "micro bullets" and increase the inflammation of the gut (Liang et al. Citation2011). P. gingivalis can even crosstalk with the complement receptor (C5aR) and TLR2, which in turn upregulates the release of IL-1, IL-6, IL-17, and TNF (Groeger et al., Citation2017). It can even activate the complement receptors (CRs), and nucleotide oligomerization domain-like receptors (NOD receptors) that in turn increase the levels of cytokines in the gut (Zheng et al. Citation2018). The activation of NK-kB and TLR-induced cytokines such as IL-1β, IL-6, and IL-17 also induces CD4(+) T-cell proliferation by activating the STAT3 signaling pathway that increases the proliferation, translocation, and metastasis of cancer cells (Scholz et al. Citation2003). STAT3 activation has been linked to pancreatic cancer (Kanda et al. Citation2004), gastric cancer (Tsareva et al. Citation2007), and CRC (Nakajima et al. Citation2015).
Fukata et al. demonstrated that P. gingivalis activation of TLRs significantly alters the phylogenetic structure and diversity of the gut’s microbial communities. This change in the gut microbiota is accompanied by the spread of Enterobacteria to the liver, a decrease in the tight junction proteins (Tjp1 and Occluden) mRNA expression, and an increase in serum endotoxins (Fukata et al. Citation2006). The entry of P. gingivalis and increase in proinflammatory cytokines (TNF, IL-6) increase the permeability of the intestine and reduction in intestinal barrier function in both the small and large intestines (Fukata et al. Citation2006). Oral bacteria also activate TLR4 leading to Cox-2 receptor activation in the intestinal epithelial. This leads to proliferation in the injured intestine and the release of proinflammatory mediators such as tumor necrosis factor (TNF), PGG2, and PGE2 in the intestine (Liu et al. Citation2020). These pro-inflammatory cytokines along with microbial affect the tight junction proteins “zonula occludens-1” and “occludin” in the gut and lead to gut inflammation and permeability (Kato et al. Citation2018) (). Increased permeability of the gut epithelium favors the penetration of bacteria and cyclically increases inflammation. P. gingivalis-induced gut inflammation and alteration of the microbiota profiles also affect the metabolite profiles of the gut and increase the risk of calcification (Li et al. Citation2022). LPS and CpG (5′—C—phosphate—G—3′) DNA from oral bacteria can also affect the TLR4 and TLR9 receptors in patients and is a potential link leading to sclerosis changes seen in patients’ primary biliary cirrhosis and primary sclerosing cholangitis (Xiao et al. Citation2016; Rodrigues et al. Citation2018; Lv et al. Citation2021). Moreover, P. gingivalis-LPS can also stimulate M1 and M2 macrophages primarily through TLR2 activation, which in turn releases nitric oxide synthase and nitric oxide (NO) in gut tissues. The release of NO increases the permeability of the gut and allows more bacterial species to enter the gut tissues which increase gut inflammation (Uhrig et al. Citation2005). TLRs not only control innate immunity but also critically regulate adaptive immunity, such as T-cell activation in the gut.
Figure 2. Schematic representation of how oral bacteria and their by-product injure the epithelium, increase gut permeability, and increase the risk of gut dysbiosis and inflammation: Oral bacteria interact with toll-like receptors (TLRs) in the gut and causes the release of proinflammatory cytokines (interleukins (IL1, IL8), prostaglandins (PGE2), tumor necrosis factors (TNF). Some of the oral bacteria also release volatile sulfur such as hydrogen sulfide, dimethyl sulfide, dimethyl disulfide, and methyl mercaptan which causes a reversible increase in epithelial permeability and loss of barrier function. Hydrogen sulfide has also been shown to increase crypt formation and ulceration by inducing DNA hypomethylation in the gut mucosa. These inflammatory mediators also affect the cell adhesion molecules such as zonula occludens and occludins and increase the permeability. This facilitates the easy invasion of the bacteria further into the gut tissues, increasing the risk of microbial dysbiosis in gut and gut inflammation (Created in Biorender).
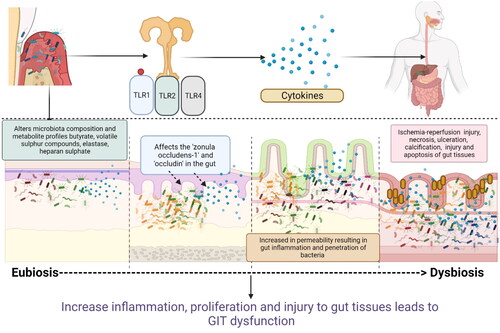
F. nucleatum, is another potential oral bacterium that can damage the gut epithelial integrity, activate TLRs, and increase the gut permeability (Ren et al. Citation2019). Studies have also noted that F. nucleatum-induced TLR4 activation can lead to liver sinusoidal endothelial cells producing more TNF-α and reactive oxygen species (ROS) via NF-κB activation resulting in liver dysfunction and proliferation of liver cells (Yang et al. Citation2017). When F. nucleatum activates the TLR4 signaling, Nf-kB and miRNA21 expression are also elevated. A study found that patients with CRC have higher levels of miRNA21 and F. nucleatum DNA in the tissues. These markers indicate a greater risk and poor prognosis for the CRC patients (Polak et al. Citation2012). F. nucleatum can co-aggregate and coinfect with oral pathogens like P. gingivalis and increase the inflammatory load in the gut tissues (Weber and Turner Citation2007).
The TLRs affect the CD4+ T cells and this in turn affects the growth of commensal bacteria in the gut. Alteration in the commensal microbial profile leads to the loss of integrity of the gut epithelium in IBD patients (Kitamoto et al. Citation2020). Commensal gut flora contributes to the expansion of the CD4+ Th17 cells leading to intestinal mucosal inflammation (Banchereau et al. Citation2000). TLRs activation are also leads to immature antigen-presenting cell maturation. The matured APC helps to present the pathogen-derived antigen to the TLRs and activate TLR-dependent co-stimulatory molecules, such as CD80/CD86. Activation of CD80/CD86 in turn increases the release of cytokines and increases gastric inflammation (Groeger et al., Citation2017). The activation of TLR4 by oral bacteria also impair epithelial proliferation and repair of ulcers in the gut (Kawasaki and Kawai Citation2014). Downstream of the TLR signaling pathways, activated NF-κB and IRF [interferon (IFN)] regulatory factor] also upregulate the expression of related genes responsible for phagocytosis and possess the ability to enhance phagocytic function and to kill microbesi (Lu et al. Citation2018). Apart from these functions, TLR signaling recruits activated natural killer cells (NK cells) and DCs. DCs are prompted by TLRs to present antigens to T-cells and initiate T-cell responses, thus providing a bridge between innate immunity and adaptive immunity (Hajishengallis et al. Citation2011). By restraining the TLR-induced over-immune responses, T-regulatory cells (T-regs) inhibit other T-cells from functioning effectively to maintain immune tolerance and have a key role in promoting tolerance at host–microbial interfaces. The balance between T-regs and effector T-cells is disturbed in patients with IBD.
Periodontal bacteria activation of the complement system and GIT dysfunction
Periodontal bacteria and periodontitis-induced inflammatory mediators are potential activators of complement receptors (CRs). Periodontal adhesion attaches to the complement receptors and activates the complement system with the massive release of C3a and C5a (potent chemoattractant) (Guo et al. Citation2004). C3a and C5a cause a massive influx of neutrophils, lymphocytes, and macrophages in the gut, thereby triggering the process of phagocytosis and activating the immune response in the gut (Schieferdecker et al. Citation1997). The products of the complement (C3a and C5a) interact with their cognate receptors, C3aR and C5aR in turn activate other signaling pathways of the innate and adaptive immune systems (Schieferdecker et al. Citation1997). C5aR is constitutively expressed in the Kupffer and stellate cells of the liver under normal conditions. However its expression is increased in response to inflammatory cytokines infiltration such as IL1, IL6, and TNF (Schieferdecker et al. Citation2001; Cao et al. Citation2012). The complement system can affect the microbes in the gut that indirectly affects the GIT function and gut homeostasis.
Any alteration in the complement system is linked with GIT disorders like CRC (Sina et al. Citation2018), UC, CD, liver fibrosis, viral hepatitis, alcoholic liver disease and hepatic ischemia/reperfusion injury (IRI) (Laufer et al., Citation2000). The components of the complement systems have been identified in the gut tissues of various GIT disorders. For example, a strong colonic C3 and IgG staining in the lamina propria and basolateral membrane of intestinal epithelial cells in patients with that UC (Ballard and Shiner Citation1974; Sugihara et al. Citation2010). The complement products (C4 and C3) have also been identified in the small intestinal crypts and within the villous and cryptic spaces of the intestine in patients with adenocarcinoma and CD (Feng et al. Citation2022). A study by Sugihara et al. observed that mucosal C3 messenger RNA is elevated, and it is correlated with IL-17 levels in IBD patients (Jain et al. Citation2014). IL-17 is a potential inflammatory mediator increased during periodontal inflammation, and it is positively correlated to C3 production in sub-epithelial myofibroblasts of the intestine (Ward and Eastwood Citation1975; Jain et al. Citation2014). The complement products such as C4 and C3 are also seen in the small intestinal crypts and within the villous and the crypt spaces of the intestine in patients with adenocarcinoma and CD (Richens et al. Citation1982; Feng et al. Citation2022). Studies found that C3 and C4 levels are elevated and correlated with CD and UC disease severity (Halstensen et al. Citation1989; Ahrenstedt et al. Citation1990). Furthermore, IL-17 upregulated C3 production in subepithelial myofibroblasts of the intestine leading to intestinal inflammation (Jain et al. Citation2014). A direct measure of jejunal secretions also showed increased levels of C3 and C4 in CD comapred to than in healthy patients (Halstensen et al. Citation1992). Cao et al. also observed an increased expression of C5aR1 and C5aR2 mRNA in CRC cell lines (Sina et al. Citation2018). The activation of this terminal complement cascade, specific for a neoepitope in the C9 part of the membrane attack complex (MAC), has been observed in the vascular MAC complex deposition correlated with IBD (Ueki et al. Citation1996). Unlike CD, patients with UC have only vascular MAC deposition in the colonic tissues (Pham et al. Citation1995). Studies have also found that IgG can trigger the classical complement activation in occur on the intestinal epithelial cells of UC patients. While no C1q or C4c deposition was noted, a strong apical C3b and MAC complex deposition on intstinal epithelial cells in the mucus was detected in ileal tissues of CD patients (Laufer et al., Citation2000). However, a study by Uekl et al. showed that only vascular but not epithelial MAC deposition could be detected in colonic tissues in UC patients (Pham et al. Citation1995). Based on these findings it can be hypothesized that activation of the classical pathway of the complement system is mainly activated in the epithelium of UC patients, while the activation of the alternative pathway of complement system is more dominant in CD patients. C4 mRNA expression has also been detected in all tissue samples from CD patients (Bao and Reinhardt Citation2015). Increased mucosal colonic C3 mRNA expression positively correlated with colonic interleukin 17 (IL-17) mRNA expression in IBD patients. Immunohistochemistry analyses performed in patients with fulminant and acute hepatitis have shown that the membrane attack complex (MAC) is deposited around necrotic areas, indicating activation of the complement system and its involvement in the pathogenesis of liver injury (Papadopoulos et al. Citation2021).
Periodontitis-induced IgG can even trigger the classical pathway of the complement system in the intestinal epithelial cells of UC patients and lead to C3b and MAC complex deposition on intestinal epithelial cells (Laufer et al. Citation2000). Periodontal pathogens like P. ginigvalis can downregulate the CR3a and CR5a receptors of the complement system for their survival and this in turn increases the inflammation and dysbiosis in the gut. P. gingivalis gingipains deconstruct C5 into C5a by directly promoting a C5 convertase-like function and activating thrombin to replace C5 convertase by promoting prothrombin (Ni et al. Citation2019). Through a C5a-mediated mechanism, gingipains cause M1 macrophages to polarize, which acts as a regulating factor and facilitates spread of P. gingivalis infection (Grąt et al. Citation2016). The likelihood of gut inflammation is increased when P. gingivalis-mediated C5a activates C5aR and promotes Gi-mediated intracellular Ca2+ signaling in the gut. This synergistically increases an otherwise weak cAMP response by P. gingivalis and leads to TLR2 activation and increased proinflammatory cytokine production (Ahn et al. Citation2012). Because of the ensuing crosstalk with the complement, P. gingivalis is less likely to be killed by nitric oxide when high levels of cAMP are sustained. This also triggers the activation of the cAMP-dependent protein kinase A, which in turn deactivates glycogen synthase kinase-3 (Michaud et al. Citation2013). In addition to inhibiting TLR2-induced IL-12p70, P. gingivalis-induced C5aR-TLR2 crosstalk also upregulates inflammatory cytokines such IL-1, IL-6, IL-17, and TNF (Castellarin et al. Citation2012; Rubinstein et al. Citation2013). Periodontitis induced activation of the complement system can lead to the creation of proinflammatory mediators and an altered immune response that result in inflammatory in the stomach and intestinal mucosa. Similar to gingipains released from P. gingivalis, the Interpains can activate the C1 complex in serum leading to the deposition of C1q on the surface of bacteria. The Interpain A (InpA), cysteine proteases released from Prevotella intermedia can also break down C3 of the complement system which increase the inflammation in the intestine (Dejea and Sears 2016). Interpains also cause the release of anaphylatoxin C3a, at sites with higher bacterial numbers can cause an increase in the systemic inflammatory load (Dejea and Sears Citation2016; Drewes et al. Citation2017).
Periodontitis-induced activation of T-cell and its subsets (Th1/ Th2/ Th17/T reg cells)
Periodontal pathogens, particularly P. gingivalis, increase the expression of co-stimulatory surface molecules (CD80 and CD86), which in turn activates various subsets of T-cells. Periodontitis-induced activation of the complement system induces Th1 responses, which in turn leads to the release of IL-12, IL-1, IFN, TNF, and IL-18) and Th2 response (IL-4, IL-5, IL-13, and IL-13) (Elvington et al. Citation2015). These ILs activate the many subsets of T-cells like T-helper cells (Th-cells), T-suppressor cells, T-regulatory cells (T-reg), and antigen-presenting cells (macrophages, dendritic cells, and Langerhans cells) in the host (Kubinak et al. Citation2015). The Th-cells lead to the formation of plasma cells that produce antibodies (IgG, IgA, and IgM antibodies) (Littman and Rudensky Citation2010). Th1 cells, which are a major source of IFN-γ and other important mediators that aid in the eradication of intracellular pathogens such as viruses and bacteria in the gut. Th cells help in differentiation of germinal center B-cells and production of high-affinity antibodies including intestinal IgA (Shale et al. Citation2013). The gut mucosa is enriched with unconventional T-cells, including the γδ-T cells, natural killer T (NKT) cells (Lynch et al. Citation2012; Toubal et al. Citation2020), and mucosal-associated invariant T (MAIT) cells. All these cells are critical regulators for maintaining gut barrier function and immune homeostasis (Ballard and Shiner Citation1974, Bykov et al., Citation2006, Adibrad et al. Citation2012; Moutsopoulos et al. Citation2012; Segata et al., Citation2012; CastañoRodríguez et al., Citation2017).
Th-17 cells are also known as major mediators of gut dysfunction. Inflamed periodontal tissue is a massive producer of Th-17 cells and it leads to an increase in Th-17 levels in the systemic circulation (Leal et al. Citation2015). Studies have also found that P. gingivalis through TLR 2/4 receptors and retinoid-related orphan nuclear receptor γt can boost the release of IL-17, IL-1β, IL-6, and IL-23, which in turn increases the differentiation of TH-17 cells (Crome et al. Citation2009). The activation of the Th17 pathway increase the release of IL-1β, IL-6, IL-17, IL-23, and IL-22 in the intestinal mucosal, and increased the risk of active UC and CD (Zhang et al., Citation2021). Enhanced IL-17 levels along with retinoic acid orphan receptor variant (RORC2) m-RNA expression increase the release of thrombospondin (TSP-1). Increased TSP-1 production increases the risk of IBD, CD, and UC (Yun et al. Citation2007). P. gingivalis-LPS can even increase the Th-17 cell response by interacting with delta-like ligand 4 expression in monocytes (Jia et al. Citation2020). This increases the release of Th17 cytokines that promote gut inflammation and tumorogenesis. P. gingivalis (gingipains) can directly induce the expression of CD69 and CD25 on the T-cells and cause TH17 mediated IL-17 production (Fujino et al. Citation2003). P. gingivalis is also known to activate IL-17 response through interaction with bradykinin B2 receptor or binding of protease-activated receptor-2 (Crome et al. Citation2009). Conversely, P. gingivalis has also been shown to downregulate the expression of T-reg transcription factors like Foxp3, TGF-β, and IL-10 via the TLR4 pathway (Ray Citation2020). IL-17 mRNA is highly expressed in inflamed mucosa of patients with UC and CD (Atarashi et al., Citation2017). P. gingvalis protease can even degrade Th1 and Th17 driving cytokines that help P. gingivalis to acquire nutrients and also facilitate the growth of other microbes in the gut. Apart from P. gingivalis, gut colonization by other oral bacteria like Enterobacter spp., and Klebsiella spp. has also been associated with an increase in the Th17 and Th1 cell accumulation (Rubin et al. Citation2012). Atarashi et al. found that Klebsiella pneumoniae ectopically colonizes the intestines via the oral cavity and significantly promotes Th1 cell responses (Hirsova et al. Citation2021).
T- cell mediated cytokines also increase mucosal inflammation and gastric permeability; change gastric motility; delay gastric emptying; and produce symptoms of colitis in patients with IBD, CD, and UC (Kempski et al. Citation2017). T-cell mediated immune response also increase the risk of gastric cancer and hepatocellular carcinoma (HCC) (Rismo et al. Citation2012). An immunohistochemistry study also observed an increased expression of CD68-positive cells, TH17, and IL-17 in inflamed intestinal mucosa in patients with IDB, UC, and CD (Hanna et al. Citation2022). Th17 cells were found to co-produce 5.5 times more IFNγ in the mucosa in IBD compared to healthy patients (Morton et al. Citation2014).
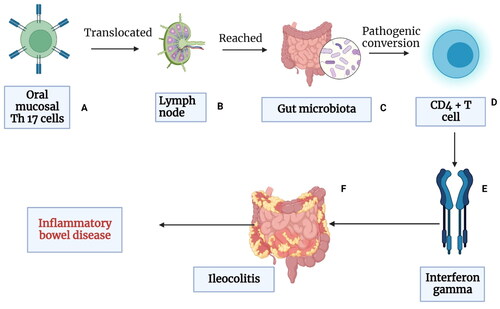
Since Th17 are activated immunologically by oral pathogens and locally produced Th17 cells can migrate from the mouth to the intestines via the lymph nodes, the role of periodontal inflammation induced Th17 production and intestinal inflammation should be noted (Hajishengallis and Chavakis Citation2021). The immune cells found in the oral draining lymph nodes can transmigrate to other lymphoid organs, including the gut. T-cells are “imprinted” for gut homing primarily in the mesenteric lymph nodes (MLNs), entering the intestine via recirculation from the blood, which is facilitated by mucosal addressing cell-adhesion molecule-1 and C-C motif chemokine ligand- 25 (Ivanov et al. Citation2009). Thus, it is possible that oral inflammation gives rise to the emergence of oral pathobiont-reactive T-cells. Pathogenic T-cells that transmigrate from the oral mucosa to the gut can activate gut bacteria and cause intestinal inflammation. Moreover, it is noteworthy that oral pathobionts do not colonize the GIT of healthy individuals. This evidence suggests that there are conditions that must be met for oral pathobionts to ectopically colonize the gut. Therefore, the colonization resistance of gut-resident microbiota must be disrupted, enabling the oral microbes to invade the gut. Gut inflammation perturbed colonization resistance mediated by the resident microbiota, makes it possible for the ingested oral pathobionts to outcompete and displace the resident bacteria (Banchereau et al. Citation2000). Gut colonization by Enterobacter spp.and Klebsiella spp. has also shown to increase the accumulation of TH17 and TH1 cells (Rubin et al. Citation2012). Experiments in mice have also revealed that oral TH17 cells induced during oral inflammation translocated to the gut, triggering gut inflammation (Banchereau et al. Citation2000). Another potential mechanism linking periodontal pathogens is the activation and migration of oral pathobionts into the gut that increases the conversion of oral Th17 TEM cells into the IFN-γ-producing Th17/Th1 cell (pathogenic Th17 cells) (Polepalle et al. Citation2015). Gut-migrated oral pathogenic Th17 cells increase the inflammation in the gut. and activate the gut-colonized oral microbiota, exacerbating colitis and other GIT diseases. Evidence has shown that oral pathobiont-reactive T cells that arise during periodontitis are colitogenic and are a potential cause of many GIT disorders (Banchereau et al. Citation2000)
Inflamed periodontal tissues activate the migration of oral pathobiont-reactive and pathogenic Th17 cells. The pathogenic Th17 cells that evolve during periodontitis reveal gut-homing markers e.g. - α4β7 integrin and CCR9, which causes the translocation of these cells to the gut mucosa. Gut-migrated oral pathogenic Th17 cells are then activated by gut-colonized oral microbiota, exacerbating colitis and other GIT diseases (Banchereau et al. Citation2000). The ectopically colonized oral pathobionts are required for expansion of gut-transmigrated oral TEM cells, possibly through antigen-specific proliferation. these two mechanisms may act synergistically. IL-1β induced by ectopically colonized oral pathobionts may also contribute to activation and/or expansion of oral TEM cells in the gut mucosa. Oral pathobiont-reactive T cells that arise duri coling periodontitis are togenic (Goto et al. Citation2014). Oral bacteria also result in the activation of inflammasome that can subsequently induce IL-1β by oral pathobionts. This plays a central role in the direct induction of colitis in the gut.
Recently, a special type of bacteria known as “segmented filamentous bacteria (SFB),” which lives in the intestine, has been identified as a powerful stimulant of the Th17 cell (Boirivant et al. Citation2008; Harbour et al. Citation2015). Few reports have shown that Th17 cells undergo pathogenic conversion in the gut, and this causes formation of IFN-γ-producing Th1-like CD4+ T cells. The ex-Th17 cells, which are produced by SFB also become pathogenic and cause severe intestinal inflammation (Strober Citation2010). Periodontal bacteria also increase the T-cell immune response in the gut through the activation of TLR-mediated release of CD4+ LAP + cells. LAP + CD4+ T cells are a newly identified subset of T-regulatory (T-reg) cells that express latent-associated peptide (LAP) and function within the latent transforming growth factor (TGF)-β complex to block the interaction between TGF-β and receptors on immune cells (Feng et al. Citation2011). The activation of CD4+ LAP + cells maintains homeostasis against intestinal bacterial invasion and maintain gut homeostasis (Sorensen et al. Citation2006). ( and ).
Figure 3. Schematic representation of how periodontal pathogens activate various immune cells and increase gut inflammation: Periodontal pathogens activate the various signaling pathways of inflammations such as nuclear factor kappa beta (NF-kB), which in turn activates the expression of co-stimulatory surface molecules (CD80 and CD86) which in turn activates various subsets of T cells (T-helper cells, T-suppressor cells, T-regulatory cells) and antigen-presenting cells (macrophages, dendritic cells, and langerhans cells) in the host. Periodontitis activation of the complement system with the release of C3a and C5a (chemoattractant). Increased release of chemoattractant (IL8, TNF, C3a, C5a) induces the influx of neutrophils and macrophages into the gut tissues. These mediators also trigger Th1 responses (induce the release of IL-12, IL1, IFN, TNF alpha, and IL-18) and Th2 response (IL-4, IL-5, IL-13 and IL-13). These cytokines along with antibodies produce mucosal inflammation, increase gastric permeability, change gastric motility, delay gastric emptying, and produce symptoms of colitis in patients with IBD, CD, and UC. T-helper cells lead to the formation of plasma cells that produce antibodies (IgG, IgA, and IgM antibodies). Increased levels of IgA and IgG antibodies have been linked with gut inflammation and the development of GIT disorders such as IBD (Created in Biorender).
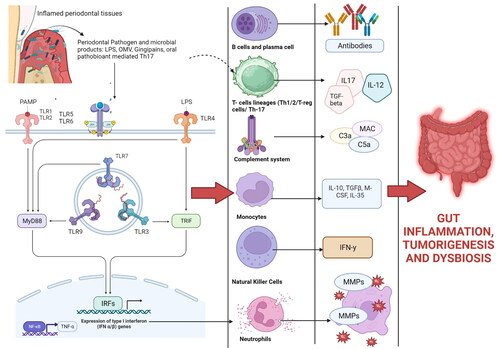
Figure 4. Periodontal tissues are massive producers of Th-17 cells which undergo pathogenic conversion in the gut, and this causes IFN-γ-producing Th1-like CD4+ T cells. Some oral bacteria that enter the gut increases the conversion of oral Th17 TEM cells into the IFN-γ-producing Th17/Th1 cell (pathogenic Th17 cells). Gut-migrated oral pathogenic Th17 cells in turn increase the inflammation in the gut and activate the gut-colonized oral microbiota, exacerbating colitis and other GIT diseases. Evidence has shown that oral pathobiont-reactive T cells that arise during periodontitis are colitogenic and are a potential cause of many GIT disorders such as inflammatory bowel disease (IBD) (Created in Biorender).
Th17 cells and T-reg cells are some of the most abundant CD4+ T-cells in the mucosal tissue of the gut. Th17 cells, induced by TGF-β and IL-6, through the master transcription factor RAR-related orphan receptor gamma (RORγt), play a critical role in protecting the gut against fungi and bacterial infection. These mediators also help in maintaining intestinal homeostasis through producing IL-17 and/or IL-22 (Lynch et al. Citation2012). An aberrant activation of Th17 cells is also associated with the pathogenesis of various autoimmune diseases of the gut (Lynch et al. Citation2012). CD25+Foxp3+ T-reg cells play a non-redundant role in the maintenance of intestinal homeostasis via IL-10 and TGF-β-dependent mechanisms (Toubal et al. Citation2020).
Periodontitis-induced acute phase response, release of acute phase protein and GIT dysfunction
Periodontitis-induced proinflammatory cytokines (IL-1, IL-6, IL-8, TNF) enter the liver and trigger the production and release of various acute phase proteins (APPs) (Slade et al. Citation2000). C-reactive proteins (CRP), fibrinogen, pentraxin, alpha-1 acid glycoprotein, alpha-1 protease inhibitor, alpha-1 anti-chymotrypsin, and haptoglobin are some of the common APPs (Slade et al. Citation2000; Megson et al. Citation2010). The APPs enter the systemic circulation and along with proinflammatory cytokines increase the overall oxidative stress and systemic inflammation. Periodontitis is considered as a risk factor for increased AAP production. Individuals with loss of teeth and severe periodontitis have a one-third more CRP levels compared to those without periodontitis. Those with periodontitis have two-times more levels of CRP than healthy subjects (Anderson and McCarty Citation1951; Fagan et al. Citation1982; Shojaee et al. Citation2013). The levels of CRP also increased in patients with CD, IBD, gastric inflammation, and pancreatic cancer. Fagan et al. observed that in patients with CD, serum levels of CRP, and higher CRP levels are noted in patients with severe CD compared with a moderate tomild CD. This indicates that periodontitis induced increased CRP production increases the risk of CD and UC (Volanakis and Kaplan Citation1971). CRP stimulates immune cells by binding to Fcγ receptors (FcγR) on leukocytes (monocytes, neutrophils and cells of a myeloid lineage) and increases production of IgG, linking the innate and adaptive immune system (Pohl et al. Citation2001). CRP binding to bacterial polysaccharides in the presence of Calcium activates compliment pathways and ultimately results in phagocytosis (Persson et al. Citation2005)
Increased APPs, particularly CRP, also stimulate neutrophils, lymphocytes, and macrophages in the epithelium and parenchymal tissue of the gut. The CRP binds to the bacterial polysaccharides in the presence of complement mediators C3a and C5a increases influx of neutrophils and exaggerates the process of phagocytosis. This increases the risk of injury in the gut and in turn worsen gut inflammation. The CRP can also recognize foreign pathogens and components of damaged cell walls of certain gram-positive oral bacteria, which increase the process of inflammation and oxidative stress. Apart from CRP, fibrinogen is another APP that is deposited in the gut during periodontitis. Periodontitis increases the expression of pro-inflammatory mediators in gut tissues and in turn increases the formation of fibrin-rich microthrombi. AAPs and proinflammatory mediators also increase the proliferation of liver cells and trigger the process of fibrosis (Sahingur et al. Citation2003). A study by Persson et al. found that periodontal pathogens increase the release of pro-inflammatory mediators (IL-1, IL6, TNF-alpha), which in turn activates hepatocytes to release CRP and fibrinogen (Ugarova and Yakubenko Citation2001). In addition, a relationship between the “455G/A gene polymorphism” and the β-fibrinogen gene promoter that increases the fibrinogen levels has been noted (Flick et al. Citation2007). Fibrinogen acts as a ligand for cell surface receptors (VE-cadherin, ICAM-1, αIIbβ3, α5β1, αVβ3, αMβ2, and αXβ2) on leukocytes, endothelial cells, platelets, fibroblasts, and smooth muscle cells (Adams et al. Citation2007). The activation of these receptors is a potent driver of local inflammation in the gut and tissue injury (Baruah et al. Citation2006; Gümüş et al. Citation2014).
Periodontitis-induced cytokines along with endotoxins (such as LPS and outer membrane proteins) activate the release of Pentraxins, another important APP. Gümüş et al. evaluated the PTX3 levels in saliva and serum levels of patients with periodontitis and found that periodontitis increases the pentraxin levels from 0.025 ng/mL to 49.2 ng/mL in saliva and 1.0 ng/mL in the serum (Deban et al. Citation2010). Pentraxins activate the macrophages, endothelial cells, myeloid cells, and dendritic cells by various cytokines. PTX3 is a TNF-stimulated gene that induces cellular proliferation, gastric inflammation, and cell injury. PTX3 also interacts with the complement C1q on the surface of dendritic cells and macrophages, and in turn, activates the classical pathway of the complement system (Kato et al. Citation2008; Savchenko et al. Citation2011). An increase in the pentraxin level increases the risk of UC and CD (Yilmaz et al. Citation2010; Kim et al. Citation2016). Kato et al. using plasma ELISA showed a higher PTX3 level in patients with active UC than in healthy subjects and patients with inactive UC, and this suggested that PTX3 was present in neutrophils in a crypt abscess in the gut (Yilmaz et al. Citation2010). Savchenko et al. in their study showed that the expression of PTX3 was observed mainly in neutrophils recruited to colonic mucosa obtained from UC patients. The numbers of PTX3-expressing cells and inflamed neutrophils were increased in the high histological grades of the inflammatory reaction suggesting that PTX3 expression depends on the colon tissue response to inflammation (Kim et al. Citation2016). They found that PTX3 protein may contribute to cell-mediated immune reaction in inflamed colon tissue, and in particular in crypt abscess lesions, of patients with UC (Kim et al. Citation2016).
Periodontitis induces inflammasome activation
The inflammasomes are innate immune system receptors/ multimeric protein structures that regulate apoptosis and induce inflammation in response to infectious microbes and molecules derived from host proteins. Inflammasome sensor molecules cross multiple pattern recognition receptors (PRR) families, including a nucleotide-binding domain, leucine-rich repeat-containing proteins (NLR, also known as NOD-like receptors). There are multiple inflammasomes that can be formed based are named after the sensor (PRR) they activate them. The different types of inflammasome are NLRP1, NLRP3, NLRP4, NLRP6, NLRP12, NLRC4, AIM2-Like Receptors and RIG-I-Like Receptors (435). Inflammasomes are implicated in various inflammatory disorders of the gut (Adibrad et al. Citation2012; Moutsopoulos et al. Citation2012).
Inflammasomes are triggered by many factors like chronic infections such as periodontitis, tissue damage, or metabolic imbalances. Periodontal bacteria (P. gingivalis (Zhao et al. Citation2014) A. actinomycetemcomitans (Ting et al. Citation2008; Hung et al. Citation2018), F. nucleatum are also known to be potent activators of inflammasomes. These bacteria interact with pattern recognition receptors (PRR) on host epithelial cells that lead to inflammasome formation via activations of NLRP signaling pathway (Zaki et al. Citation2011; de Zoete et al. Citation2014; Zhao et al. Citation2014). NLR family pyrin domain containing 3 (NLRP3) inflammasome is a complex inflammasome that is activated by various ligands including bacteria, bacterial toxins, and intracellular risk signals. Some of the different types of inflammasome like NLRP1, NLRP3, NLRP4, NLRP6, NLRP12, NLRC4, AIM2-Like Receptors and RIG-I-Like Receptors are activated during GIT dysfunction (Martinon and Tschopp Citation2007). Among all these NLRP3 is emerging as a crucial regulator of intestinal homeostasis (Franchi et al. Citation2009).
The activation of NLRP3 inflammasomes activates caspase 1, which proteolytically promotes the pro-inflammatory cytokines interleukin-1β (IL-1β) and IL-18 (Seo et al. Citation2015). Increased levels of IL-1β and IL-18 levels also increase the risk of IBD and gut tumorigenesis (Guo et al. Citation2014). Inflammasome-mediated secretion of IL-1β is also known to plays a central role in the pathogenesis of commensal pathobiont-driven IBD (Yamaguchi et al. Citation2015). A strong correlation between the NLRP3 inflammasome and periodontitis has also been confirmed by various studies, thereby confirming the link between periodontitis and GIT disorder (McCall et al. Citation2008). P. gingivalis’ gingipain and fimbriae play an important role in activating the NLRP3 inflammasome (Flood et al. Citation2015). Osteoblasts in the periodontal tissues also express NLRP3 inflammasome that increases caspase activation via activation of IL-1 and cell death by pyroptosis. This plays a central role in the pathogenesis of commensal pathobiont-driven IBD (Yamaguchi et al. Citation2015; Yuan et al. Citation2018). Inflammasome-mediated secretion of IL-1β and over-activation of pyroptosis play a pivotal role in the initiation and progression of IBD, UC, CD, and immune-medicated disease of GIT (Babbs Citation1992; Mangerich et al. Citation2013).
Periodontitis increase production of reactive oxygen species and GIT dysfunction
Reactive oxygen species (ROS) are molecules capable of independent existence, containing at least one oxygen atom and one or more unpaired electrons. Periodontitis is proven to be a potential source of many ROS. The influx of proinflammatory cytokines from the oral cavity to the gut along with various ROS causes influx of macrophages and neutrophils in the gut. Massive influx of these inflammatory cells in response to the chemotactic stimuli (IL-8, IL-1, IL-6, CRP, C3a, C5a) triggers the oxidative and non-oxidative pathways of phagocytosis that cause the production of more ROS. During the process of phagocytosis, many enzymatic contents from the granules of the neutrophils are released into the surrounding tissues (Swindle and Metcalfe Citation2007; Aviello and Knaus Citation2017). Many enzymes, such as peroxidases, NADPH oxidase (NOX), xanthine oxidase (XO), lipoxygenases (LOXs), glucose oxidase, myeloperoxidase (MPO), and cyclooxygenases (COXs), are also relesed from the nuetrophils along ROS in the gut (Sasaki and Joh Citation2007). Among these enzymes, mucosal NOXs (NOX1, NOX2, and dual oxidase 2 (DUOX2)), have been reported as novel IBD risk factors. These enzymes cause massive tissue destruction in the gut and increase the risk of gastric ulceration, gastritis, and IBD. Increased ROS formation also plays a central role in altering the microbiota of the gut (Thomas et al. Citation2014).
Xanthine oxidase can cause intestinal injury resulting from ischemia and reperfusion that in turn alters the intestinal functions leading to GIT injuries. Myeloperoxidase (MPO) is another active enzyme and biomarker for IBD. MPO along with hypochlorous acids (HOCl) leads to perpetual inflammation secondary tissue injury and cell death. Neutrophil Extracellular Trap (NET) is a specialized form of neutrophil death where MPO is entrapped in a DNA scaffold and continues to elicit HOCl activity that further contribute to host-tissue injury in the gut. MPO levels in serum and feces have been elevated in inflamed mucosa in UC and gastric malignancies (Pacher et al. Citation2007). Many studies have shown that periodontitis can increase the levels of superoxide dismutase, and MPO in the serum, and this could link between periodontal inflammation and GIT dysfunction (Lassègue et al. Citation2001). Studies have even noted that apart from ROS, reactive nitrogen oxides (RNS) are continuously formed as a result of a complex interplay between the host, commensal bacteria, and dietary factors. These compounds include nitric oxide (NO), nitrite, nitrate, peroxynitrite, S-nitrosothiols, nitrated fatty acids and N-nitrosamines, all of which are bioactive with the potential to affect physiological and pathological processes locally in the gut as well as systemically after absorption. The release of NO by NOX, NOS, and the production of the oxidant peroxynitrite are involved in the pathogenesis of IBD and CD (Chu et al. Citation2002; Swindle and Metcalfe Citation2007). Studies have also identified nitrosamines as a potential carcinogen for gastric and colon cancer. L-arginine-nitric oxide pathway and nitrate-nitrite-nitric oxide pathway are known to affect the local host defense and gut mucosal integrity, which in turn affects the systemic regulation of cardiovascular and metabolic function. NOX-4 activation leads to increased production of NADPH oxidases that in turn triggers the formation of superoxide ions. NOX4 also activates various signaling pathways like protein kinase C, protein kinase B, PDK-1, and c-Jun N-terminus kinase that increase the production of cytokines, exaggerate inflammation, and systemic oxidative stress (Desta and Graves Citation2007).
Some oral bacteria like Treponema denticola and P. gingivalis can endogenously increase the production of ROS/ RNS in the host. The entry of theseboral bacteria into the gut is by itself a potential source of oxidative stress. Treponema denticola can degrade glutathione, a natural antioxidant, and increase oxidative stress in the systemic circulation (Vona et al. Citation2021). P. gingivalis boosts oxidative stress by activating the NADPH-oxidase family member (NOX 4) receptors located in the host cells and tissues (Chiu et al. Citation2017). The increased production of ROS during periodontitis can also downregulate nuclear factor erythroid 2-related factor 2 (Nrf2), which is a ubiquitous master-transcription factor associated with the upregulation of antioxidant enzymes and cytoprotective proteins. Activation of Nrf2 provides a protective response against injury and its downregulation during periodontitis increases oxidative stress (Rao et al. Citation1997; McBee et al. Citation2017). The commensal bacteria in the gut particularly in the intestinal epithelial also produce ROS through activation of NOX1 and iNOS. The activation of iNOS causes an increase in the production of nitric oxide synthase (NOS) and facilitates the release of nitric oxide. ROS and NO damage the cytoskeleton proteins in the gut, dilate the blood vessels, alter the tight junctions that increase the epithelial permeability, and cause DNA damage (Anton and Shanahan Citation1998; Van Spaendonk et al. Citation2017). This promotes the entry of bacteria and bacterial toxins into the gut that can activate PRR signaling at the gut endothelium; disrupt the barrier function and induce GIT dysfunction. The protease enzyme secreted by bacteria also affects the junctional proteins in the gut epithelium by indirectly activating the proteinase-activated receptors (PARs) (Surrenti et al. Citation1993). The activation of PARs contributes to gastrointestinal disorders by affecting the tight junction and hemi desmosomes in the gut. Increased permeability also increases inflammation as it activates the TLRs and NOD expression in the endothelial cells and activates the release of cytokines.
Link between periodontitis, nervous system, and GIT dysfunction
The central nervous system (CNS) is linked with the gut via the hypothalamic–pituitary axis (HPA) (Goldin et al. Citation1989). Various neuropeptides such as Substance P, Vasoactive intestinal peptide (VIP), Neurokinin A (NKA), kininogens, and tachykinin serve as a major link between the enteric nervous system (ENS), CNS, and gut () (Goldin et al. Citation1989). Neuropeptides such as substance P, calcitonin gene-related peptide and neuropeptide Y (NPY), VIP, somatostatin, and corticotropin-releasing factor are important mediators for the bidirectional gut-brain communication. These neuropeptides also play a key role in modulating the gastrointestinal microbiota and its interaction with the gut-brain axis. Some of these neuropeptides act as intracellular signaling molecules and directly influence the production of Immunoglobulins (IgA, IgG, and IgM) in the gut. These neuropeptides bind to their corresponding receptors (G-protein coupled receptor superfamily, neurokinin (NK 1, 2, and 3) receptors) and increase cytokine production such as IL-1β, IL-6, IL-8, and TNF-α. The activation of these neuropeptides along with inflammatory mediators increases the process of lymphocyte mitogenesis, chemotaxis, phagocytosis, neutrophil lysosomal release and migration, and homing patterns of immune effector cells in the gut (Goode et al. Citation2000). When the neuropeptides are released at the nerve endings in the intestine, they diffuse into the surrounding tissues and affect the nearby muscles, epithelium, endothelium, and immune cells. The neuronal-mediated proinflammatory cytokines lead to the inflammation of intestinal/colonic mucosa (Fais et al. Citation1991; Holzer Citation1998). In patients with intestinal inflammation and IBD, colonic nerve damage and changes in mucosal innervation and neuropeptide expression have also been noted (Liu et al. Citation2017). Increased Substance P expression has been observed in both tissue and nerve fibers in the colon of patients with UC and CD (Fais et al. Citation1991). The perturbations of GIT motility in IBD are also influenced by Substance P and neurokinin 1 receptor (NK1) receptors. Various evidence has linked Substance P, NKA, calcitonin gene-related peptide, and VIP to the alteration of the motor activity of the GIT, intestinal anaphylaxis, infection, trauma, and stress (Lazaridis et al. Citation2017). Some microbes secrete neuropeptides in the gut. For example, Enterococcus, Candida, Escherichia, and Streptococcus synthesize 5-hydroxytryptamine; members of the genera Saccharomyces, Bacillus, and Escherichia generate dopamine and/or noradrenaline, members of the genus Lactobacillus produce acetylcholine; and members of the genera Lactobacillus and Bifidobacterium manufacture gamma-aminobutyric acid (GABA) (Györfi et al. Citation1992; Cryan and Dinan Citation2012; Nicholson et al. Citation2012). The microbiota-derived dopamine enters the lumen of the intestine and affects the adsorption capcity of the colon (Györfi et al. Citation1992). Lactobacillus, Bacteroides, Helicobacter pylori, Escherichia coli, and Candida species in the intestine also secrete proteins identical to the appetite-regulating peptides (Aliberti et al. Citation2003). Additionally, IgG and IgA autoantibodies against NPY, leptin, alpha-melanocyte-stimulating hormone, ghrelin, agouti-related protein (AgRP), and some other neuropeptides/peptides are involved in appetite control (Aliberti et al. Citation2003).
Figure 5. Schematic representation explaining how periodontal tissues are linked to the Central nervous system and gut: the Central nervous system (CNS) is linked with the gut via the hypothalamic–pituitary axis (HPA). Various neuropeptides such as substance P, vasoactive intestinal peptide (VIP), calcitonin gene-related peptides, neurokinin A (NKA), kininogens, and tachykinin serve as a major link between the enteric nervous system (ENS), CNS, and gut. Periodontal bacteria also increase the release of kininogens and substance P. These neuropeptides affect the enteric nervous systems (vagal and mesenteric nerves and affect gut mobility, gastric acid secretion, and release of inflammatory mediators. These neuropeptides act as intracellular signaling molecules and directly influence the production of immunoglobulin, lymphocyte mitogenesis, chemotaxis, phagocytosis, neutrophil lysosomal release and migration, and homing patterns of immune effector cells in the gut (created in Biorender).
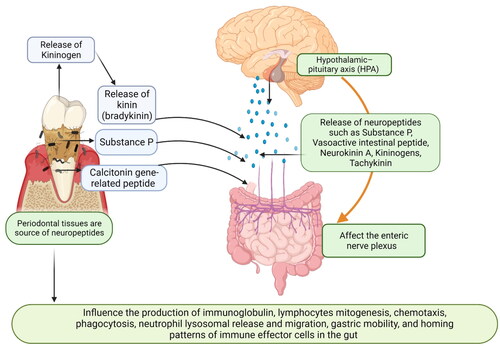
The entry of periodontal pathogens into the gut also increases the secretion of various neuropeptides, particularly Substance P, Kinin, and VIP (Azuma et al. Citation2004). Inflamed periodontal tissues act as an increased source of kinins (kininogen) (Pradeep et al. Citation2009). The kininogens released during periodontal inflammation are degraded to form bradykinin, which exaggerates the local and systemic inflammatory process. The release of bradykinin leads to vasodilatation, which in turn leads to plasma extravasation and the release of other mediators, notably the Substance P and calcitonin gene-related peptide, in the gut (Linden et al. Citation1997; Ku et al. Citation2020; Yan et al. Citation2020). Substance P released during periodontitis enters the systematic circulation and this can upregulate the hypoxia-inducible factor 1 alpha (HIF-1α) (Yin et al. Citation2022). The HIF-1α regulates the cellular adaptation to low oxygen levels, creates a hypoxic microenvironment, and alters the function of the gut barrier (Lembeck et al. Citation1992; Jeong et al. Citation2003). In chronic hypoxia, HIF-1α and some of its target gene products, such as iNOS, COX-2, IL-6, and IL-8 get activated, leading to increased inflammation, altered immunity, and apoptosis in the gut (Yiangou et al. Citation2001).
Studies have also found an increase in the levels of other neuropeptides such as calcitonin gene-related peptide, VIP, Neurokinin A, kininogens, and tachykinin in individuals with periodontitis. Tachykinins have a pro-inflammatory cytokine-like property, and increased tachykinin secretion is linked with increased microvascular permeability, plasma protein extravasation in the GIT, edema formation, altered motor functions and stimulation of contractions of the gut (Oztürk and Yildiz Citation2011). Vanilloid receptor-1 is another neuropeptide receptor, that is up-regulated during periodontal and can be a key link between periodontitis and IBD (Krongbaramee et al. Citation2021). Vallinoid receptors present in gingival tissues in both healthy and chronic periodontitis patients was found to increase the release of neuropeptides (Bazzoni et al. Citation2009). Vallinoid receptor activation in the sensory nerves of the stomach plays an important role in acute ethanol-mediated gastric mucosal injury.
Periodontitis-induced MicroRNAs (miRNAs) and their role in GIT dysfunction
Periodontitis can alter the levels of systemic miRNAs which in turn affects various gut functions (Yoneda et al. Citation2019). miRNA are a group of small non-coding RNAs, which negatively regulate protein expression and are implicated in several physiological and pathological mechanisms in the GIT. Periodontitis is considered a potential factor for increasing these miRNAs and inducing GIT dysfunction. miRNA-9 which is induced by TLR4-activated nuclear factor kappa B (NF-κB) in polymorphonuclear cells and monocytes during periodontitis can increase the gut mucosal permeability, and lead to edema formation (Bazzoni et al. Citation2009; Stoecklin-Wasmer et al., Citation2012). Studies have found variations in some of these miRNAs in patients with GIT disorders. For example, a study by Stoecklin et al. found that miRNA155 overlaps in patients with periodontal and liver diseases (O’Connell et al. Citation2007; Gonçalves Fernandes et al. Citation2020). miRNA-155, which is increased in periodontitis is involved in antigen presentation and activation of the TNF pathway in macrophages (Pirola et al. Citation2015).
Aberrant expression of miRNAs triggers dysregulation of multiple cellular processes involved in innate and adaptive immune responses. miRNAs act not only within the cell in which they were synthesized but, recently, they have been found outside cells bound to lipids and proteins. They can circulate throughout the body, transferring information between cells and altering gene expression in the recipient cells, as they can fuze with and be internalized by the recipient cells. The miRNA produced in inflamed periodontal tissues can enter the systemic circulation and lead to many systemic diseases. Some of the common miRNAs increased during periodontal inflammation include miRNA-9, miRNA-155, miRNA-203a, miRNA-147, miRNA-182, miRNA-183, miRNA-664a, miRNA501, miRNA-21 (O’Connell et al. Citation2007; Wong et al. Citation2018) (Table S3). Many of these mRNAs have been linked with the pathophysiology of IBD, chronic hepatitis, progression of nonalcoholic fatty liver to nonalcoholic steato-hepatogenesis and hepato-carcinogenesis (McKenna et al. Citation2010; Albuquerque-Souza and Sahingur Citation2022), tissue inflammation, and cancers of GIT (Albuquerque-Souza and Sahingur Citation2022). miRNAs are critical regulators of the immune response based on their ability to interfere with the post-transcriptional expression of specific target genes in the gut (Wu et al. Citation2010). Loss of the intestinal miRNAs impairs tissue integrity, alters barrier function, and increases gut inflammation (Paraskevi et al. Citation2012). Alteration in the ileac and colonic miRNA (miRNA-21, miRNA-23b, miRNA-29a, miRNA-106a, miRNA-107, miRNA-126, miRNA-191, and miRNA-200c) is noted in patients CD (Dalal and Kwon Citation2010). A study by Paraskevi et al. found an increase in levels of miRNA-16, miRNA-23a, miRNA-29a, miRNA-106a, miRNA-107, miRNA-126, miRNA-191, miRNA-199a-5p, miRNA-200c, miRNA-362-3p and miRNA-532-3p in the blood of patients with CD compared with the healthy controls; In the UC, three miRNAs (miRNA-16, miRNA-21, miRNA-28-5p, miRNA-151-5p, miRNA-155 and miRNA-199a-5p) were found to be significantly increased (Lyte Citation2011). miRNA-192 and miRNA-21 are also highly expressed of the active UC (Asano et al. Citation2012). Four miRNAs were found to be elevated in CD and chronic ileitis patients (miRNA-16, miRNA-21, miRNA-223, and miRNA-594) (Dalal and Kwon Citation2010). miR-375 regulates the proliferation of intestinal epithelial stem cells and miR-375 is notably under expressed in active UC and suppressed in intestinal epithelial stem cells by the presence of microbiota (Fetissov et al. Citation2008; Yang et al. Citation2013). In contrast, the overexpression of miR-21 in UC patients is related to intestinal epithelial barrier impairment through targeting Rho GTPase and ADP ribosylation factor 4 [ARF4] GTPase (Peck et al. Citation2017; Zhang et al. Citation2018). MiR-21 induces the degradation of RhoB mRNA and upregulation of ARF4. In this regard, miR-21 can decrease the transepithelial electrical resistance and increase the paracellular permeability (Zhang et al. Citation2018), which may be related to occludin, claudin 1, and claudin 4 decrease (Wang et al. Citation2016; Rodríguez-Nogales et al. Citation2017). Another miRNA related to the breach of the integrity of tight junctions is miR-223 (Li et al. Citation2020). Shown to be overexpressed in the serum and feces of CD and UC patients (Li et al. Citation2020) and DSS-induced colitis mice (James et al. Citation2020), miR-223 promotes IBD progression by downregulating the expression of the tight junction protein claudin 8 (Altaf-Ul-Amin et al. Citation2020).
Several studies have found significantly different profiles of hundreds of miRNAs (Malham et al. Citation2021; Zaki et al., Citation2022) between IBD and healthy individuals (Ji et al., Citation2018), pediatric and adult patients (Iborra et al. Citation2013), inflamed and non-inflamed mucosa, and in relation to disease activity (Thorlacius-Ussing et al. Citation2017; Schönauen et al. Citation2018; Zaki et al., Citation2022). One of the most studied miRNAs in IBD is miR-21. This miRNA is overexpressed systemically in the plasma as well as locally in colonic tissues (Shi et al. Citation2017; Yan et al. Citation2020). miR-21 is overexpressed in the lamina propria and in subsets of macrophages and T-cells of IBD patients compared with healthy controls (Shi et al. Citation2017). MiR-21 is also associated with UC, but not CD, as well as with disease activity and severity (Tian et al. Citation2019). Moreover, its levels are significantly increased in intestinal lesions of adults compared with pediatric UC patients (Tian et al. Citation2019). Thus, miR-21 is strongly recommended as a biomarker to distinguish between health and IBD, UC and CD, active and remission phases (Shi et al. Citation2017). Similarly, miR-31 levels also have been found to be increased in inflamed mucosa of IBD patients and mice with colitis compared with controls (Lu et al. Citation2005; Casado-Bed and Viennois Citation2022). Shi et al. demonstrated how miR-31 directly targets the expression of IL-25, a crucial counter-regulator cytokine of Th1/Th17 inflammatory responses. They also showed an inverse correlation between the anti-inflammatory cytokine IL-25 and miR-31 in the colon of both CD patients and mouse models (Casado-Bed and Viennois Citation2022). Moreover, the treatment with anti-miR-31 promoted IL-25 expression and improved significantly the induced colitis (Casado-Bed and Viennois Citation2022). Tian et al. recently showed that miR-31-knockout mice develop a more severe inflammatory response when inducing colitis. They demonstrated that miR-31, expressed predominantly in colonic epithelial cells, inhibits the expression of IL-17R and IL-17RA and GP130 inflammatory cytokine receptors and downregulates the pro-inflammatory nuclear factor-kB [NF-kB] and STAT3 signaling pathways (Lu et al. Citation2005). Despite the conflicting results, evidence indicates that miR-31 is a key regulatory factor of the Th17 response implicated in IBD (Schepeler et al. Citation2008). Differential expression of miRNAs in several malignancies, including colon cancer (Liu et al. Citation2016; Ji et al., Citation2018). A bidirectional interaction between host cells and gut microbiota via miRNA which participate in shaping the gut microbiota after being secreted from intestinal epithelial cells, and which accumulate in feces (Hu et al. Citation2015; Park et al. Citation2017). On the other hand, host miRNA expression can be influenced by the microbiota through microbe-derived metabolites that might potentially influence the host physiology (Temmerman et al. Citation2006). Moreover, the dysregulation of miRNA functions has been recently associated with IBD pathogenesis (Nandy et al. Citation2007).
Necrovirulence and increased cytotoxicity (necrotoxicity) of the gut by oral bacteria
Recently certain bacterial species were observed to induce death of the other microbial and host cells for their nutrition and survival. This process is called “necrovirulence” or “bacterial necrotrophy (Rodriguez Herrero et al. Citation2017). The nutrients released by this behavior from the killed siblings are being used to feed itself and other surviving bacteria under times of nutritional stress (Ma et al. Citation2023). It can be hypothesized that the increase in the amounts of dead bacteria provides multiple nutritional sources, such as C, N, P, and Fe, for the surviving bacteria in the oral ecosystem and increases biofilm formation (Ozdemir et al. Citation2001). The presence of dead bacteria stimulates the growth of other oral pathogens (necrotrophy) along with the induction of new phenotypes. This up-regulates genes for bacterial virulence (necrovirulence) and causes cytotoxicity toward host tissues (necrotoxicity).
Recently, studies have found that the oral cavity contains many dead bacteria, that act as a source of nutrients for live microbes. The daily use of various antimicrobial agents in the form of oral rinse, mouthwash, and kinds of toothpaste has resulted in an oral microbial massacre with high amounts of dead bacteria near few surviving bacteria. This creates a condition where the surviving pathogenic bacteria have access to a large amount of dead microbial biomass as a nutritional source for growth (necrotrophy). Herrero et al. (Rodriguez Herrero et al. Citation2017) demonstrated the necrotrophic growth of periodontal pathogens in the presence of different kinds of dead oral species. The presence of dead bacteria increased the growth of several periodontal pathogens in complex multi-species biofilms. The virulence genes of two periodontal pathogens “P. intermedia and P. gingivalis” also increased in the presence of dead bacteria (necrovirulence). The dead bacteria from the oral cavity also reach the gut via the swallowed saliva (Ozdemir et al. Citation2001). The swallowed dead bacteria from the oral cavity can stimulate various pathogens in the gut via necrotrophy and this leads to the development of new phenotypes and upregulation of bacterial virulence in the gut (Banchereau et al. Citation2000). A study by Kitamoto et al. also showed that P. gingivalis and P. intermedia exhibit a marked increase in growth when at least 10 dead bacterial cells were at their disposal for one living cell (Banchereau et al. Citation2000). A study also noted that the gingipain genes rgpA, rgpB, and kgp of P. gingivalis are upregulated in the presence of dead P. intermedia. Their increased expression can therefore be directly related to the observed necrotrophic behavior of P. gingivalis (Ozdemir et al. Citation2001). This process allows entry, promotion, and survival of many bacteria in the gut leading to exaggerated inflammation and dysbiosis.
Release of volatile sulfur compounds and GIT dysfunction
Microbial degradation of food particles produces volatile sulfur compounds (VSCs). The most important VSCs involved in halitosis are methyl mercaptan (CH3SH), hydrogen sulfide (H2S), and dimethyl sulfide (CH3)2S, diamines (indole and skatole) or polyamines (cadverin and putrescin). VSCs can enter the systemic circulation to various organ systems whereby they cause microbial dysbiosis, the metabolic activity of tissues and organs, the development of systemic diseases is a concern (Ng and Tonzetich Citation1984). Volatile sulfur compounds act as carcinogens and play a key role in tumorigenesis and cell function. Hydrogen sulfide, a genotoxic and inflammatory substance, is a VSC that enters the gut and causes a reversible increase in epithelial permeability and loss of barrier function (Pitcher and Cummings Citation1996; Medani et al. Citation2011). Hydrogen sulfide has also been shown to increase crypt formation by inducing DNA hypomethylation in gut mucosa (Deplancke and Gaskins Citation2003). Excessive amounts of hydrogen sulfide production lead to reduced clearance of hydrogen sulfide from the gut, which leads to the colonization of hydrogen sulfide leading to the etiology of UC (Ramakrishna et al. Citation1991). Hydrogen sulfide disrupts the delicate balance between apoptosis, proliferation, and differentiation in the intestinal epithelium (Yoo et al. Citation2008). An increased presence of hydrogen sulfide affects the functioning of Rhodanese (RHOD) and Thiol methyltransferase (TMT), enzymes present in the colon that help in the removal of hydrogen sulfide and these are some of the key factors for the onset of UC (Stummer et al. Citation2023). Hydrogen sulfide has also been shown to have a stimulatory effect on IL-10 production while decreasing pro-inflammatory cytokines such as IL-1β, IL-6, IL-8, IL-18, tumor necrosis factor-alpha (TNF-α) and interferon-gamma (IFN-γ). A link between IL-10 and IBD has already been shown in many studies, which confirm that impaired IL-10 secretion intensifies the inflammation in IBD. Additionally, Hydrogen sulfide protects the mucosa by increasing blood flow through vasodilation. This is important for decreasing mucosal damage through all erosive substances, like bile, acids and digestive enzymes, it faces and for fastening tissue repair after damage occurs (Koren et al. Citation2011; Lee et al. Citation2011; Xie et al. Citation2011; Loh and Blaut Citation2012; Stoecklin-Wasmer et al., Citation2012; Perri et al. Citation2012; Gao and Zheng Citation2013; Ogata et al. Citation2014; Kalea et al. Citation2015; Atarashi et al., Citation2017; Kang et al. Citation2017; Li et al., Citation2019; Schmidt et al., Citation2019; Rengara et al. Citation2020; Chu et al. Citation2021). Yoo et al. (Citation2008) observed that the levels of VSCs in exhaled breaths or aspirated gastric juices differed significantly between the non-erosive and erosive epithelial groups (p < 0.00001), suggesting that VSCs might reflect eroded epithelial damage induced by H. pylori infection. The expressions of cystathionine β-synthase (CBS) and cystathionine γ-lyase (CSE) were consistent with the degree of mucosal injury. Erosive changes in esophageal and gastroduodenal mucosa were strongly correlated with increased VSC levels, suggesting that halitosis might result from H. pylori-associated erosive lesions (Yoo et al. Citation2008).
Conclusion
Periodontitis-induced systemic inflammation and periodontal bacteria are potential risk factors for GIT dysfunction. Oral bacteria invasion into the GIT changes the microbial profile of the gut leading to gut dysbiosis and subsequent increase in gut inflammation (Banchereau et al. Citation2000). Oral bacteria adopt unique and intricate molecular mechanisms to alter gut function. The oral-gut axis plays a crucial role in the systemic dissemination of oral bacteria and devlopment of gut inflammation. Therefore, for effective management of GIT dysfunction, management of periodontal disease and establishing good oral health should be an integral part of the treatment plan.
Geolocation
India.
Supplemental Material
Download MS Word (42.9 KB)Disclosure statement
No potential conflict of interest was reported by the author(s).
Data availability statement
Data is available upon request via email to the corresponding author.
Correction Statement
This article has been corrected with minor changes. These changes do not impact the academic content of the article.
Additional information
Funding
References
- Adams RA, Bauer J, Flick MJ, Sikorski SL, Nuriel T, Lassmann H, Degen JL, Akassoglou K. 2007. The fibrin-derived gamma377-395 peptide inhibits microglia activation and suppresses relapsing paralysis in central nervous system autoimmune disease. J Exp Med. 204(3):571–582. doi: 10.1084/jem.20061931.
- Adibrad M, Deyhimi P, Ganjalikhani Hakemi M, Behfarnia P, Shahabuei M, Rafiee L. 2012. Signs of the presence of Th17 cells in chronic periodontal disease. J Periodontal Res. 47(4):525–531. doi: 10.1111/j.1600-0765.2011.01464.x.
- Ahn J, Segers S, Hayes RB. 2012. Periodontal disease, Porphyromonas gingivalis serum antibody levels and orodigestive cancer mortality. Carcinogenesis. 33(5):1055–1058. doi: 10.1093/carcin/bgs112.
- Ahrenstedt O, Knutson L, Nilsson B, Nilsson-Ekdahl K, Odlind B, Hällgren R. 1990. Enhanced local production of complement components in the small intestines of patients with Crohn’s disease. N Engl J Med. 322(19):1345–1349. doi: 10.1056/NEJM199005103221903.
- Al Asqah M, Al Hamoudi N, Anil S, Al Jebreen A, Al-Hamoudi WK. 2009. Is the presence of Helicobacter pylori in dental plaque of patients with chronic periodontitis a risk factor for gastric infection? Can J Gastroenterol. 23(3):177–179. doi: 10.1155/2009/950527.
- Alarcón P, González M, Castro É. 2016. Rol de la microbiota gastrointestinal en la regulación de la respuesta inmune [The role of gut microbiota in the regulation of the immune response]. Rev Med Chil. 144(7):910–916. Spanish. doi: 10.4067/S0034-98872016000700013.
- Albuquerque-Souza E, Sahingur SE. 2022. Periodontitis, chronic liver diseases, and the emerging oral-gut-liver axis. Periodontol 2000. 89(1):125–141. doi: 10.1111/prd.12427.
- Aliberti J, Viola JP, Vieira-de-Abreu A, Bozza PT, Sher A, Scharfstein J. 2003. Cutting edge: bradykinin induces IL-12 production by dendritic cells: a danger signal that drives Th1 polarization. J Immunol. 170(11):5349–5353. doi: 10.4049/jimmunol.170.11.5349.
- Altaf-Ul-Amin M, Karim MB, Hu P, Ono N, Kanaya S. 2020. Discovery of inflammatory bowel disease-associated miRNAs using a novel bipartite clustering approach. BMC Med Genomics. 13(Suppl 3):10. doi: 10.1186/s12920-020-0660-y.
- Anderson HC, McCARTY M. 1951. The occurrence in the rabbit of an acute phase protein analogous to human C reactive protein. J Exp Med. 93(1):25–36. doi: 10.1084/jem.93.1.25.
- Andresen L, Jørgensen VL, Perner A, Hansen A, Eugen-Olsen J, Rask-Madsen J. 2005. Activation of nuclear factor kappaB in colonic mucosa from patients with collagenous and ulcerative colitis. Gut. 54(4):503–509. doi: 10.1136/gut.2003.034165.
- Andrukhov O, Ulm C, Reischl H, Nguyen PQ, Matejka M, Rausch-Fan X. 2011. Serum cytokine levels in periodontitis patients in relation to the bacterial load. J Periodontol. 82(6):885–892. doi: 10.1902/jop.2010.100425.
- Ansari SA, Iqbal MUN, Khan TA, Kazmi SU. 2018. Association of oral Helicobacter pylori with gastric complications. Life Sci. 205:125–130. doi: 10.1016/j.lfs.2018.05.026.
- Anton PA, Shanahan F. 1998. Neuroimmunomodulation in inflammatory bowel disease. How far from "bench" to "bedside"? Ann N Y Acad Sci. 840(1):723–734. doi: 10.1111/j.1749-6632.1998.tb09611.x.
- Asano Y, Hiramoto T, Nishino R, Aiba Y, Kimura T, Yoshihara K, Koga Y, Sudo N. 2012. Critical role of gut microbiota in the production of biologically active, free catecholamines in the gut lumen of mice. Am J Physiol Gastrointest Liver Physiol. 303(11):G1288–95. doi: 10.1152/ajpgi.00341.2012.
- Atarashi K, Suda W, Luo C, Kawaguchi T, Motoo I, Narushima S, Kiguchi Y, Yasuma K, Watanabe E, Tanoue T, et al. 2017. Ectopic colonization of oral bacteria in the intestine drives TH1 cell induction and inflammation. Science. 358(6361):359–365. doi: 10.1126/science.aan4526.
- Aviello G, Knaus UG. 2017. ROS in gastrointestinal inflammation: rescue Or Sabotage? Br J Pharmacol. 174(12):1704–1718. doi: 10.1111/bph.13428.
- Azer SA. 2013. Overview of molecular pathways in inflammatory bowel disease associated with colorectal cancer development. Eur J Gastroenterol Hepatol. 25(3):271–281. doi: 10.1097/MEG.0b013e32835b5803.
- Azuma H, Kido J, Ikedo D, Kataoka M, Nagata T. 2004. Substance P enhances the inhibition of osteoblastic cell differentiation induced by lipopolysaccharide from Porphyromonas gingivalis. J Periodontol. 75(7):974–981. doi: 10.1902/jop.2004.75.7.974.
- Babbs CF. 1992. Oxygen radicals in ulcerative colitis. Free Radic Biol Med. 13(2):169–181. doi: 10.1016/0891-5849(92)90079-v.
- Baert FJ, D’Haens GR, Peeters M, Hiele MI, Schaible TF, Shealy D, Geboes K, Rutgeerts PJ. 1999. Tumor necrosis factor alpha antibody (infliximab) therapy profoundly down-regulates the inflammation in Crohn’s ileocolitis. Gastroenterology. 116(1):22–28. doi: 10.1016/s0016-5085(99)70224-6.
- Bainbridge BW, Darveau RP. 2001. Porphyromonas gingivalis lipopolysaccharide: an unusual pattern recognition receptor ligand for the innate host defense system. Acta Odontol Scand. 59(3):131–138. doi: 10.1080/000163501750266710.
- Bajaj JS, Betrapally NS, Hylemon PB, Heuman DM, Daita K, White MB, Unser A, Thacker LR, Sanyal AJ, Kang DJ, et al. 2015. Salivary microbiota reflects changes in gut microbiota in cirrhosis with hepatic encephalopathy. Hepatology. 62(4):1260–1271. doi: 10.1002/hep.27819.
- Ballard J, Shiner M. 1974. Evidence of cytotoxicity in ulcerative colitis from immunofluorescent staining of the rectal mucosa. Lancet. 1(7865):1014–1017. doi: 10.1016/s0140-6736(74)90416-4.
- Banchereau J, Briere F, Caux C, Davoust J, Lebecque S, Liu YJ, Pulendran B, Palucka K. 2000. Immunobiology of dendritic cells. Annu Rev Immunol. 18(1):767–811. doi: 10.1146/annurev.immunol.18.1.767.
- Bao K, Reinhardt RL. 2015. The differential expression of IL-4 and IL-13 and its impact on type-2 immunity. Cytokine. 75(1):25–37. doi: 10.1016/j.cyto.2015.05.008.
- Baruah P, Dumitriu IE, Peri G, Russo V, Mantovani A, Manfredi AA, Rovere-Querini P. 2006. The tissue pentraxin PTX3 limits C1q-mediated complement activation and phagocytosis of apoptotic cells by dendritic cells. J Leukoc Biol. 80(1):87–95. doi: 10.1189/jlb.0805445.
- Bazzoni F, Rossato M, Fabbri M, Gaudiosi D, Mirolo M, Mori L, Tamassia N, Mantovani A, Cassatella MA, Locati M. 2009. Induction and regulatory function of miR-9 in human monocytes and neutrophils exposed to proinflammatory signals. Proc Natl Acad Sci U S A. 106(13):5282–5287. doi: 10.1073/pnas.0810909106.
- Berkovich L, Gerber M, Katzav A, Kidron D, Avital S. 2022. NF-kappa B expression in resected specimen of colonic cancer is higher compared to its expression in inflammatory bowel diseases and polyps. Sci Rep. 12(1):16645. doi: 10.1038/s41598-022-21078-7.
- Boirivant M, Amendola A, Butera A, Sanchez M, Xu L, Marinaro M, Kitani A, Di Giacinto C, Strober W, Fuss IJ. 2008. A transient breach in the epithelial barrier leads to regulatory T-cell generation and resistance to experimental colitis. Gastroenterology. 135(5):1612–1623.e5. doi: 10.1053/j.gastro.2008.07.028.
- Bunte K, Beikler T. 2019. Th17 Cells and the IL-23/IL-17 Axis in the Pathogenesis of Periodontitis and Immune-Mediated Inflammatory Diseases. Int J Mol Sci. 20(14):3394. doi: 10.3390/ijms20143394.
- Bykov I, Junnikkala S, Pekna M, Lindros KO, Meri S. 2006. Complement C3 contributes to ethanol-induced liver steatosis in mice. Ann Med. 38(4):280–286. doi: 10.1080/07853890600664608.
- Cao Q, McIsaac SM, Stadnyk AW. 2012. Human colonic epithelial cells detect and respond to C5a via apically expressed C5aR through the ERK pathway. Am J Physiol Cell Physiol. 302(12):C1731–40. doi: 10.1152/ajpcell.00213.2011.
- Cario E, Gerken G, Podolsky DK. 2004. Toll-like receptor 2 enhances ZO-1-associated intestinal epithelial barrier integrity via protein kinase C. Gastroenterology. 127(1):224–238. doi: 10.1053/j.gastro.2004.04.015.
- Casado-Bed M, Viennois E. 2022. MicroRNA and gut microbiota: tiny but mighty-novel insights into their cross-talk in inflammatory bowel disease pathogenesis and therapeutics. J Crohns Colitis. 16(6):992–1005. doi: 10.1093/ecco-jcc/jjab223.
- Casarin RCV, Ribeiro EDP, Mariano FS, Nociti FH, Casati MZ, Gonçalves RB. 2010. Levels of Aggregatibacter actinomycetemcomitans, Porphyromonas gingivalis, inflammatory cytokines and species-specific immunoglobulin G in generalized aggressive and chronic periodontitis. J Periodontal Res. 45(5):635–642. doi: 10.1111/j.1600-0765.2010.01278.x.
- Castaño-Rodríguez N, Goh KL, Fock KM, Mitchell HM, Kaakoush NO. 2017. Dysbiosis of the microbiome in gastric carcinogenesis. Sci Rep. 7(1):15957. doi: 10.1038/s41598-017-16289-2.
- Castellarin M, Warren RL, Freeman JD, Dreolini L, Krzywinski M, Strauss J, Barnes R, Watson P, Allen-Vercoe E, Moore RA, et al. 2012. Fusobacterium nucleatum infection is prevalent in human colorectal carcinoma. Genome Res. 22(2):299–306. doi: 10.1101/gr.126516.111.
- Chen Z, Cai J, Chen YM, Wei J, Li HB, Lu Y, Zhou Z, Chen XL. 2019. A meta-analysis of the association between the presence of Helicobacter pylori and periodontal diseases. Medicine (Baltimore). 98(22):e15922. doi: 10.1097/MD.0000000000015922.
- Chiu AV, Saigh MA, McCulloch CA, Glogauer M. 2017. The role of NrF2 in the regulation of periodontal health and disease. J Dent Res. 96(9):975–983. doi: 10.1177/0022034517715007.
- Chu L, Dong Z, Xu X, Cochran DL, Ebersole JL. 2002. Role of glutathione metabolism of Treponema denticola in bacterial growth and virulence expression. Infect Immun. 70(3):1113–1120. doi: 10.1128/IAI.70.3.1113-1120.2002.
- Chu XJ, Cao NW, Zhou HY, Meng X, Guo B, Zhang HY, Li BZ. 2021. The oral and gut microbiome in rheumatoid arthritis patients: a systematic review. Rheumatology (Oxford). 60(3):1054–1066. doi: 10.1093/rheumatology/keaa835.
- Chung M, Zhao N, Meier R, Koestler DC, Wu G, de Castillo E, Paster BJ, Charpentier K, Izard J, Kelsey KT, et al. 2021. Comparisons of oral, intestinal, and pancreatic bacterial microbiomes in patients with pancreatic cancer and other gastrointestinal diseases. J Oral Microbiol. 13(1):1887680. doi: 10.1080/20002297.2021.1887680.
- Crome SQ, Wang AY, Kang CY, Levings MK. 2009. The role of retinoic acid-related orphan receptor variant 2 and IL-17 in the development and function of human CD4+ T cells. Eur J Immunol. 39(6):1480–1493. doi: 10.1002/eji.200838908.
- Cryan JF, Dinan TG. 2012. Mind-altering microorganisms: the impact of the gut microbiota on brain and behaviour. Nat Rev Neurosci. 13(10):701–712. doi: 10.1038/nrn3346.
- Dalal SR, Kwon JH. 2010. The role of MicroRNA in inflammatory bowel disease. Gastroenterol Hepatol (N Y). 6(11):714–722.
- de Zoete MR, Palm NW, Zhu S, Flavell RA. 2014. Inflammasomes. Cold Spring Harb Perspect Biol. 6(12):a016287–a016287. doi: 10.1101/cshperspect.a016287.
- Deban L, Russo RC, Sironi M, Moalli F, Scanziani M, Zambelli V, Cuccovillo I, Bastone A, Gobbi M, Valentino S, et al. 2010. Regulation of leukocyte recruitment by the long pentraxin PTX3. Nat Immunol. 11(4):328–334. doi: 10.1038/ni.1854.
- Dejea CM, Sears CL. 2016. Do biofilms confer a pro-carcinogenic state? Gut Microbes. 7(1):54–57. Erratum in: doi: 10.1073/pnas.1406199111. Erratum in: doi: 10.1016/j.cmet.2015.04.011. doi: 10.1080/19490976.2015.1121363.
- Deplancke B, Gaskins HR. 2003. Hydrogen sulfide induces serum-independent cell cycle entry in nontransformed rat intestinal epithelial cells. Faseb J. 17(10):1310–1312. doi: 10.1096/fj.02-0883fje.
- Desta T, Graves DT. 2007. Fibroblast apoptosis induced by Porphyromonas gingivalis is stimulated by a gingipain and caspase-independent pathway that involves apoptosis-inducing factor. Cell Microbiol. 9(11):2667–2675. doi: 10.1111/j.1462-5822.2007.00987.x.
- Dowman JK, Tomlinson JW, Newsome PN. 2010. Pathogenesis of non-alcoholic fatty liver disease. QJM. 103(2):71–83. doi: 10.1093/qjmed/hcp158.
- Drewes JL, White JR, Dejea CM, Fathi P, Iyadorai T, Vadivelu J, Roslani AC, Wick EC, Mongodin EF, Loke MF, et al. 2017. High-resolution bacterial 16S rRNA gene profile meta-analysis and biofilm status reveal common colorectal cancer consortia. NPJ Biofilms Microbiomes. 3(1):34. Erratum in: NPJ Biofilms Microbiomes. 2019 9;5(1):2. doi: 10.1038/s41522-017-0040-3.
- Elvington M, Schepp-Berglind J, Tomlinson S. 2015. Regulation of the alternative pathway of complement modulates injury and immunity in a chronic model of dextran sulphate sodium-induced colitis. Clin Exp Immunol. 179(3):500–508. doi: 10.1111/cei.12464.
- Fagan EA, Dyck RF, Maton PN, Hodgson HJ, Chadwick VS, Petrie A, Pepys MB. 1982. Serum levels of C-reactive protein in Crohn’s disease and ulcerative colitis. Eur J Clin Invest. 12(4):351–359. doi: 10.1111/j.1365-2362.1982.tb02244.x.
- Fais S, Capobianchi MR, Pallone F, Di Marco P, Boirivant M, Dianzani F, Torsoli A. 1991. Spontaneous release of interferon gamma by intestinal lamina propria lymphocytes in Crohn’s disease. Kinetics of in vitro response to interferon gamma inducers. Gut. 32(4):403–407. doi: 10.1136/gut.32.4.403.
- Fan X, Alekseyenko AV, Wu J, Peters BA, Jacobs EJ, Gapstur SM, Purdue MP, Abnet CC, Stolzenberg-Solomon R, Miller G, et al. 2018. Human oral microbiome and prospective risk for pancreatic cancer: a population-based nested case-control study. Gut. 67(1):120–127. Jan doi: 10.1136/gutjnl-2016-312580.
- Feng T, Qin H, Wang L, Benveniste EN, Elson CO, Cong Y. 2011. Th17 cells induce colitis and promote Th1 cell responses through IL-17 induction of innate IL-12 and IL-23 production. J Immunol. 186(11):6313–6318. doi: 10.4049/jimmunol.1001454.
- Feng Y, Chen Z, Tu SQ, Wei JM, Hou YL, Kuang ZL, Kang XN, Ai H. 2022. Role of Interleukin-17A in the Pathomechanisms of Periodontitis and Related Systemic Chronic Inflammatory Diseases. Front Immunol. 13:862415. doi: 10.3389/fimmu.2022.862415.
- Fetissov SO, Hamze Sinno M, Coquerel Q, Do Rego JC, Coëffier M, Gilbert D, Hökfelt T, Déchelotte P. 2008. Emerging role of autoantibodies against appetite-regulating neuropeptides in eating disorders. Nutrition. 24(9):854–859. doi: 10.1016/j.nut.2008.06.021.
- Flick MJ, LaJeunesse CM, Talmage KE, Witte DP, Palumbo JS, Pinkerton MD, Thornton S, Degen JL. 2007. Fibrin(ogen) exacerbates inflammatory joint disease through a mechanism linked to the integrin alphaMbeta2 binding motif. J Clin Invest. 117(11):3224–3235. doi: 10.1172/JCI30134.
- Flood B, Oficjalska K, Laukens D, Fay J, O’Grady A, Caiazza F, Heetun Z, Mills KH, Sheahan K, Ryan EJ, et al. 2015. Altered expression of caspases-4 and -5 during inflammatory bowel disease and colorectal cancer: diagnostic and therapeutic potential. Clin Exp Immunol. 181(1):39–50. doi: 10.1111/cei.12617.
- Franchi L, Eigenbrod T, Muñoz-Planillo R, Nuñez G. 2009. The inflammasome: a caspase-1-activation platform that regulates immune responses and disease pathogenesis. Nat Immunol. 10(3):241–247. doi: 10.1038/ni.1703.
- Franchimont D, Vermeire S, El Housni H, Pierik M, Van Steen K, Gustot T, Quertinmont E, Abramowicz M, Van Gossum A, Devière J, et al. 2004. Deficient host-bacteria interactions in inflammatory bowel disease? The toll-like receptor (TLR)-4 Asp299gly polymorphism is associated with Crohn’s disease and ulcerative colitis. Gut. 53(7):987–992. doi: 10.1136/gut.2003.030205.
- Fujino S, Andoh A, Bamba S, Ogawa A, Hata K, Araki Y, Bamba T, Fujiyama Y. 2003. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 52(1):65–70. doi: 10.1136/gut.52.1.65.
- Fukata M, Chen A, Klepper A, Krishnareddy S, Vamadevan AS, Thomas LS, Xu R, Inoue H, Arditi M, Dannenberg AJ, et al. 2006. Cox-2 is regulated by Toll-like receptor-4 (TLR4) signaling: Role in proliferation and apoptosis in the intestine. Gastroenterology. 131(3):862–877. doi: 10.1053/j.gastro.2006.06.017.
- Gao B, Zheng L. 2013. microRNA expression in rat apical periodontitis bone lesion. Bone Res. 1(2):170–185. doi: 10.4248/BR201302006.
- Gao SG, Yang JQ, Ma ZK, Yuan X, Zhao C, Wang GC, Wei H, Feng XS, Qi YJ. Preoperative serum immunoglobulin G and A antibodies to Porphyromonas gingivalis are potential serum biomarkers for the diagnosis and prognosis of esophageal squamous cell carcinoma. BMC Cancer. 2018 3;18(1):17. doi: 10.1186/s12885-017-3905-1.
- Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, et al. 2014. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe. 15(3):382–392. doi: 10.1016/j.chom.2014.02.005.
- Goldin E, Karmeli F, Selinger Z, Rachmilewitz D. 1989. Colonic substance P levels are increased in ulcerative colitis and decreased in chronic severe constipation. Dig Dis Sci. 34(5):754–757. doi: 10.1007/BF01540348.
- Gonçalves Fernandes J, Morford LA, Harrison PL, Kompotiati T, Huang H, Aukhil I, Wallet SM, Macchion Shaddox L. 2020. Dysregulation of genes and microRNAs in localized aggressive periodontitis. J Clin Periodontol. 47(11):1317–1325. doi: 10.1111/jcpe.13361.
- Goode T, O’Connell J, Anton P, Wong H, Reeve J, O’Sullivan GC, Collins JK, Shanahan F. 2000. Neurokinin-1 receptor expression in inflammatory bowel disease: molecular quantitation and localisation. Gut. 47(3):387–396. doi: 10.1136/gut.47.3.387.
- Goto Y, Panea C, Nakato G, Cebula A, Lee C, Diez MG, Laufer TM, Ignatowicz L, Ivanov II. 2014. Segmented filamentous bacteria antigens presented by intestinal dendritic cells drive mucosal Th17 cell differentiation. Immunity. 40(4):594–607. doi: 10.1016/j.immuni.2014.03.005.
- Grąt M, Wronka KM, Krasnodębski M, Masior Ł, Lewandowski Z, Kosińska I, Grąt K, Stypułkowski J, Rejowski S, Wasilewicz M, et al. 2016. Profile of gut microbiota associated with the presence of hepatocellular cancer in patients with liver cirrhosis. Transplant Proc. 48(5):1687–1691. doi: 10.1016/j.transproceed.2016.01.077.
- Grivennikov SI, Wang K, Mucida D, Stewart CA, Schnabl B, Jauch D, Taniguchi K, Yu GY, Osterreicher CH, Hung KE, et al. 2012. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature. 491(7423):254–258. doi: 10.1038/nature11465.
- Groeger S, Jarzina F, Domann E, Meyle J. 2017. Porphyromonas gingivalis activates NFκB and MAPK pathways in human oral epithelial cells. BMC Immunol. 18(1):1. doi: 10.1186/s12865-016-0185-5.
- Gümüş P, Nizam N, Nalbantsoy A, Özçaka Ö, Buduneli N. 2014. Saliva and serum levels of pentraxin-3 and interleukin-1β in generalized aggressive or chronic periodontitis. J Periodontol. 85(3):e40-6–e46. doi: 10.1902/jop.2013.130281.
- Guo RF, Riedemann NC, Ward PA. 2004. Role of C5a–C5aR interaction in sepsis. Shock. 21(1):1–7. doi: 10.1097/01.shk.0000105502.75189.5e.
- Guo W, Ye P, Yu H, Liu Z, Yang P, Hunter N. 2014. CD24 activates the NLRP3 inflammasome through c-Src kinase activity in a model of the lining epithelium of inflamed periodontal tissues. Immun Inflamm Dis. 2(4):239–253. doi: 10.1002/iid3.40.
- Györfi A, Fazekas A, Rosivall L. 1992. Neurogenic inflammation and the oral mucosa. J Clin Periodontol. 19(10):731–736. doi: 10.1111/j.1600-051x.1992.tb02162.x.
- Hajishengallis G, Chavakis T. 2021. Local and systemic mechanisms linking periodontal disease and inflammatory comorbidities. Nat Rev Immunol. 21(7):426–440. doi: 10.1038/s41577-020-00488-6.
- Hajishengallis G, Liang S, Payne MA, Hashim A, Jotwani R, Eskan MA, McIntosh ML, Alsam A, Kirkwood KL, Lambris JD, et al. 2011. Low-abundance biofilm species orchestrates inflammatory periodontal disease through the commensal microbiota and complement. Cell Host Microbe. 10(5):497–506. doi: 10.1016/j.chom.2011.10.006.
- Halstensen TS, Mollnes TE, Fausa O, Brandtzaeg P. 1989. Deposits of terminal complement complex (TCC) in muscularis mucosae and submucosal vessels in ulcerative colitis and Crohn’s disease of the colon. Gut. 30(3):361–366. doi: 10.1136/gut.30.3.361.
- Halstensen TS, Mollnes TE, Garred P, Fausa O, Brandtzaeg P. 1992. Surface epithelium related activation of complement differs in Crohn’s disease and ulcerative colitis. Gut. 33(7):902–908. Erratum in: Gut 1992 Oct;33(10):1440. doi: 10.1136/gut.33.7.902.
- Hanna DN, Smith PM, Novitskiy SV, Washington MK, Zi J, Weaver CJ, Hamaamen JA, Lewis KB, Zhu J, Yang J, et al. 2022. SMAD4 suppresses colitis-associated carcinoma through inhibition of CCL20/CCR6-mediated inflammation. Gastroenterology. 163(5):1334–1350.e14. e14. doi: 10.1053/j.gastro.2022.07.016.
- Harbour SN, Maynard CL, Zindl CL, Schoeb TR, Weaver CT. 2015. Th17 cells give rise to Th1 cells that are required for the pathogenesis of colitis. Proc Natl Acad Sci U S A. 112(22):7061–7066. doi: 10.1073/pnas.1415675112.
- Hausmann M, Kiessling S, Mestermann S, Webb G, Spöttl T, Andus T, Schölmerich J, Herfarth H, Ray K, Falk W, et al. 2002. Toll-like receptors 2 and 4 are up-regulated during intestinal inflammation. Gastroenterology. 122(7):1987–2000. doi: 10.1053/gast.2002.33662.
- Hernandez BY, Zhu X, Risch HA, Lu L, Ma X, Irwin ML, Lim JK, Taddei TH, Pawlish KS, Stroup AM, et al. 2022. Oral Cyanobacteria and Hepatocellular Carcinoma. Cancer Epidemiol Biomarkers Prev. 31(1):221–229. doi: 10.1158/1055-9965.EPI-21-0804.
- Hietaranta A, Mustonen H, Puolakkainen P, Haapiainen R, Kemppainen E. 2004. Proinflammatory effects of pancreatic elastase are mediated through TLR4 and NF-kappaB. Biochem Biophys Res Commun. 323(1):192–196. doi: 10.1016/j.bbrc.2004.08.077.
- Hirsova P, Bamidele AO, Wang H, Povero D, Revelo XS. 2021. Emerging roles of T cells in the pathogenesis of nonalcoholic steatohepatitis and hepatocellular carcinoma. Front Endocrinol (Lausanne). 12:760860. doi: 10.3389/fendo.2021.760860.
- Holzer P. 1998. Jul-Aug Implications of tachykinins and calcitonin gene-related peptide in inflammatory bowel disease. Digestion. 59(4):269–283. doi: 10.1159/000007504.
- Hooper LV, Gordon JI. 2001. Commensal host-bacterial relationships in the gut. Science. 292(5519):1115–1118. doi: 10.1126/science.1058709.
- Hu S, Liu L, Chang EB, Wang JY, Raufman JP. 2015. Butyrate inhibits pro-proliferative miR-92a by diminishing c-Myc-induced miR-17-92a cluster transcription in human colon cancer cells. Mol Cancer. 14(1):180. doi: 10.1186/s12943-015-0450-x.
- Hug H, Mohajeri MH, La Fata G. 2018. Toll-Like Receptors: Regulators of the Immune Response in the Human Gut. Nutrients. 10(2):203. doi: 10.3390/nu10020203.
- Huh JW, Roh TY. 2020. Opportunistic detection of Fusobacterium nucleatum as a marker for the early gut microbial dysbiosis. BMC Microbiol. 20(1):208. doi: 10.1186/s12866-020-01887-4.
- Hung SC, Huang PR, Almeida-da-Silva CLC, Atanasova KR, Yilmaz O, Ojcius DM. 2018. Oct-Nov NLRX1 modulates differentially NLRP3 inflammasome activation and NF-κB signaling during Fusobacterium nucleatum infection. Microbes Infect. 20(9-10):615–625. doi: 10.1016/j.micinf.2017.09.014.
- Iborra M, Bernuzzi F, Correale C, Vetrano S, Fiorino G, Beltrán B, Marabita F, Locati M, Spinelli A, Nos P, et al. 2013. Identification of serum and tissue micro-RNA expression profiles in different stages of inflammatory bowel disease. Clin Exp Immunol. 173(2):250–258. doi: 10.1111/cei.12104.
- Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, et al. 2009. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell. 139(3):485–498. doi: 10.1016/j.cell.2009.09.033.
- Iwauchi M, Horigome A, Ishikawa K, Mikuni A, Nakano M, Xiao JZ, Odamaki T, Hironaka S. 2019. Relationship between oral and gut microbiota in elderly people. Immun Inflamm Dis. 7(3):229–236. doi: 10.1002/iid3.266.
- Jain U, Otley AR, Van Limbergen J, Stadnyk AW. 2014. The complement system in inflammatory bowel disease. Inflamm Bowel Dis. 20(9):1628–1637. doi: 10.1097/MIB.0000000000000056.
- James JP, Riis LB, Malham M, Høgdall E, Langholz E, Nielsen BS. 2020. MicroRNA biomarkers in IBD-differential diagnosis and prediction of colitis-associated cancer. Int J Mol Sci. 21(21):7893. doi: 10.3390/ijms21217893.
- Jeong HJ, Chung HS, Lee BR, Kim SJ, Yoo SJ, Hong SH, Kim HM. 2003. Expression of proinflammatory cytokines via HIF-1alpha and NF-kappaB activation on desferrioxamine-stimulated HMC-1 cells. Biochem Biophys Res Commun. 306(4):805–811. doi: 10.1016/s0006-291x(03)01073-8.
- Ji Y, Li X, Zhu Y, Li N, Zhang N, Niu M. 2018. Faecal microRNA as a biomarker of the activity and prognosis of inflammatory bowel diseases. Biochem Biophys Res Commun. 503(4):2443–2450. doi: 10.1016/j.bbrc.2018.06.174.
- Jia L, Han N, Du J, Guo L, Luo Z, Liu Y. 2019. Pathogenesis of important virulence factors of Porphyromonas gingivalis via toll-like receptors. Front Cell Infect Microbiol. 9:262. doi: 10.3389/fcimb.2019.00262.
- Jia L, Wu R, Han N, Fu J, Luo Z, Guo L, Su Y, Du J, Liu Y. 2020. Porphyromonas gingivalis and Lactobacillus rhamnosus GG regulate the Th17/Treg balance in colitis via TLR4 and TLR2. Clin Transl Immunology. 9(11):e1213. doi: 10.1002/cti2.1213.
- Johnson GB, Brunn GJ, Kodaira Y, Platt JL. 2002. Receptor-mediated monitoring of tissue well-being via detection of soluble heparan sulfate by Toll-like receptor 4. J Immunol. 168(10):5233–5239. doi: 10.4049/jimmunol.168.10.5233.
- Kalea AZ, Hoteit R, Suvan J, Lovering RC, Palmen J, Cooper JA, Khodiyar VK, Harrington Z, Humphries SE, D’Aiuto F. 2015. Upregulation of gingival tissue miR-200b in obese periodontitis subjects. J Dent Res. 94(3 Suppl):59S–69S. doi: 10.1177/0022034514568197.
- Kanda N, Seno H, Konda Y, Marusawa H, Kanai M, Nakajima T, Kawashima T, Nanakin A, Sawabu T, Uenoyama Y, et al. 2004. STAT3 is constitutively activated and supports cell survival in association with survivin expression in gastric cancer cells. Oncogene. 23(28):4921–4929. doi: 10.1038/sj.onc.1207606.
- Kang DW, Adams JB, Gregory AC, Borody T, Chittick L, Fasano A, Khoruts A, Geis E, Maldonado J, McDonough-Means S, et al. 2017. Microbiota transfer therapy alters gut ecosystem and improves gastrointestinal and autism symptoms: an open-label study. Microbiome. 5(1):10. doi: 10.1186/s40168-016-0225-7.
- Kato S, Ochiai M, Sakurada T, Ohno S, Miyamoto K, Sagara M, Ito M, Takeuchi K, Imaki J, Itoh K, et al. 2008. Increased expression of long pentraxin PTX3 in inflammatory bowel diseases. Dig Dis Sci. 53(7):1910–1916. doi: 10.1007/s10620-007-0075-z.
- Kato T, Yamazaki K, Nakajima M, Date Y, Kikuchi J, Hase K, Ohno H, Yamazaki K. 2018. Oral Administration of Porphyromonas gingivalis Alters the Gut Microbiome and Serum Metabolome. mSphere. 3(5):e00460-18. doi: 10.1128/mSphere.00460-18.
- Kaur G, Grover V, Bhaskar N, Kaur RK, Jain A. 2018. Periodontal Infectogenomics. Inflamm Regen. 38(1):8. doi: 10.1186/s41232-018-0065-x.
- Kawasaki T, Kawai T. 2014. Toll-like receptor signaling pathways. Front Immunol. 5:461. doi: 10.3389/fimmu.2014.00461.
- Kempski J, Brockmann L, Gagliani N, Huber S. 2017. TH17 Cell and epithelial cell crosstalk during inflammatory bowel disease and carcinogenesis. Front Immunol. 8:1373. doi: 10.3389/fimmu.2017.01373.
- Kharaghani AA, Harzandi N, Khorsand B, Rajabnia M, Kharaghani AA, Houri H. 2023. High prevalence of Mucosa-Associated extended-spectrum β-Lactamase-producing Escherichia coli and Klebsiella pneumoniae among Iranain patients with inflammatory bowel disease (IBD). Ann Clin Microbiol Antimicrob. 22(1):86. ; 5. doi: 10.1186/s12941-023-00630-x.
- Khorsand B, Asadzadeh Aghdaei H, Nazemalhosseini-Mojarad E, Nadalian B, Nadalian B, Houri H. 2022. Overrepresentation of Enterobacteriaceae and Escherichia coli is the major gut microbiome signature in Crohn’s disease and ulcerative colitis; a comprehensive metagenomic analysis of IBDMDB datasets. Front Cell Infect Microbiol. 12:1015890. doi: 10.3389/fcimb.2022.1015890.
- Kim S, Park MH, Song YR, Na HS, Chung J. 2016. Aggregatibacter actinomycetemcomitans-Induced AIM2 Inflammasome Activation Is Suppressed by Xylitol in Differentiated THP-1 Macrophages. J Periodontol. 87(6):e116-26–e126. doi: 10.1902/jop.2016.150477.
- Kirk KF, Nielsen HL, Thorlacius-Ussing O, Nielsen H. 2016. Optimized cultivation of Campylobacter concisus from gut mucosal biopsies in inflammatory bowel disease. Gut Pathog. 8(1):27. doi: 10.1186/s13099-016-0111-7.
- Kitamoto S, Nagao-Kitamoto H, Jiao Y, Gillilland MG, 3rd, Hayashi A, Imai J, Sugihara K, Miyoshi M, Brazil JC, Kuffa P, et al. 2020. The intermucosal connection between the mouth and gut in commensal pathobiont-driven colitis. Cell. 182(2):447–462.e14. doi: 10.1016/j.cell.2020.05.048.
- Kong J, Yuan X, Wang J, Liu Y, Sun W, Gu B, Lan Z, Gao S. 2021. Frequencies of Porphyromonas gingivalis detection in oral-digestive tract tumors. Pathol Oncol Res. 27:628942. 1 Retraction in: Pathol Oncol Res. 2023 Dec 19;29:1611616. doi: 10.3389/pore.2021.628942.
- Koren O, Spor A, Felin J, Fåk F, Stombaugh J, Tremaroli V, Behre CJ, Knight R, Fagerberg B, Ley RE, et al. 2011. Human oral, gut, and plaque microbiota in patients with atherosclerosis. Proc Natl Acad Sci U S A. 108 Suppl 1(Suppl 1):4592–4598. doi: 10.1073/pnas.1011383107.
- Krongbaramee T, Zhu M, Qian Q, Zhang Z, Eliason S, Shu Y, Qian F, Akkouch A, Su D, Amendt BA, et al. 2021. Plasmid encoding microRNA-200c ameliorates periodontitis and systemic inflammation in obese mice. Mol Ther Nucleic Acids. 23:1204–1216. doi: 10.1016/j.omtn.2021.01.030.
- Kubinak JL, Petersen C, Stephens WZ, Soto R, Bake E, O’Connell RM, Round JL. 2015. MyD88 signaling in T cells directs IgA-mediated control of the microbiota to promote health. Cell Host Microbe. 17(2):153–163. doi: 10.1016/j.chom.2014.12.009.
- Ku T, Pandey R, Chauhan NS. 2020. Hypoxia inducible factor-1α: the curator of gut homeostasis. Front Cell Infect Microbiol. 10:227. doi: 10.3389/fcimb.2020.00227.
- Lassègue B, Sorescu D, Szöcs K, Yin Q, Akers M, Zhang Y, Grant SL, Lambeth JD, Griendling KK. 2001. Novel gp91(phox) homologues in vascular smooth muscle cells : nox1 mediates angiotensin II-induced superoxide formation and redox-sensitive signaling pathways. Circ Res. 88(9):888–894. doi: 10.1161/hh0901.090299.
- Laufer J, Oren R, Goldberg I, Horwitz A, Kopolovic J, Chowers Y, Passwell JH. 2000. Cellular localization of complement C3 and C4 transcripts in intestinal specimens from patients with Crohn’s disease. Clin Exp Immunol. 120(1):30–37. doi: 10.1046/j.1365-2249.2000.01168.x.
- Lazaridis LD, Pistiki A, Giamarellos-Bourboulis EJ, Georgitsi M, Damoraki G, Polymeros D, Dimitriadis GD, Triantafyllou K. 2017. Activation of NLRP3 inflammasome in inflammatory bowel disease: differences between crohn’s disease and ulcerative colitis. Dig Dis Sci. 62(9):2348–2356. doi: 10.1007/s10620-017-4609-8.
- Leal RF, Planell N, Kajekar R, Lozano JJ, Ordás I, Dotti I, Esteller M, Masamunt MC, Par H, Ricart E, et al. 2015. Identification of inflammatory mediators in patients with Crohn’s disease unresponsive to anti-TNFα therapy. Gut. 64(2):233–242. doi: 10.1136/gutjnl-2013-306518.
- Lee YH, Na HS, Jeong SY, Jeong SH, Park HR, Chung J. 2011. Comparison of inflammatory microRNA expression in healthy and periodontitis tissues. Biocell. 35(2):43–49. 22128589.
- Lembeck F, Donnerer J, Tsuchiya M, Nagahisa A. 1992. The non-peptide tachykinin antagonist, CP-96,345, is a potent inhibitor of neurogenic inflammation. Br J Pharmacol. 105(3):527–530. doi: 10.1111/j.1476-5381.1992.tb09013.x.
- Li B, Ge Y, Cheng L, Zeng B, Yu J, Peng X, Zhao J, Li W, Ren B, Li M, et al. 2019. Oral bacteria colonize and compete with gut microbiota in gnotobiotic mice. Int J Oral Sci. 11(1):10. doi: 10.1038/s41368-018-0043-9.
- Li M, Zhao J, Cao M, Liu R, Chen G, Li S, Xie Y, Xie J, Cheng Y, Huang L, et al. 2020. Mast cells-derived MiR-223 destroys intestinal barrier function by inhibition of CLDN8 expression in intestinal epithelial cells. Biol Res. 53(1):12. doi: 10.1186/s40659-020-00279-2.
- Li W, Xu J, Zhang R, Li Y, Wang J, Zhang X, Lin L. 2021. Is periodontal disease a risk indicator for colorectal cancer? A systematic review and meta-analysis. J Clin Periodontol. 48(3):336–347. doi: 10.1111/jcpe.13402.
- Li X, Kolltveit KM, Tronstad L, Olsen I. 2000. Systemic diseases caused by oral infection. Clin Microbiol Rev. 13(4):547–558. doi: 10.1128/CMR.13.4.547.
- Li ZJ, Gou HZ, Zhang YL, Song XJ, Zhang L. 2022. Role of intestinal flora in primary sclerosing cholangitis and its potential therapeutic value. World J Gastroenterol. 28(44):6213–6229. doi: 10.3748/wjg.v28.i44.6213.
- Liang Q, Chiu J, Chen Y, Huang Y, Higashimori A, Fang J, Brim H, Ashktorab H, Ng SC, Ng SSM, et al. 2017. Fecal bacteria act as novel biomarkers for noninvasive diagnosis of colorectal cancer. Clin Cancer Res. 23(8):2061–2070. doi: 10.1158/1078-0432.CCR-16-1599.
- Liang S, Krauss JL, Domon H, McIntosh ML, Hosur KB, Qu H, Li F, Tzekou A, Lambris JD, Hajishengallis G. 2011. The C5a receptor impairs IL-12-dependent clearance of Porphyromonas gingivalis and is required for induction of periodontal bone loss. J Immunol. 186(2):869–877. doi: 10.4049/jimmunol.1003252.
- Linden GJ, McKinnell J, Shaw C, Lundy FT. 1997. Substance P and neurokinin A in gingival crevicular fluid in periodontal health and disease. J Clin Periodontol. 24(11):799–803. doi: 10.1111/j.1600-051x.1997.tb01192.x.
- Littman DR, Rudensky AY. 2010. Th17 and regulatory T cells in mediating and restraining inflammation. Cell. 140(6):845–858. doi: 10.1016/j.cell.2010.02.021.
- Liu H, Hong XL, Sun TT, Huang XW, Wang JL, Xiong H. 2020. Fusobacterium nucleatum exacerbates colitis by damaging epithelial barriers and inducing aberrant inflammation. J Dig Dis. 21(7):385–398. doi: 10.1111/1751-2980.12909.
- Liu L, Dong Y, Ye M, Jin S, Yang J, Joosse ME, Sun Y, Zhang J, Lazarev M, Brant SR, et al. 2017. The pathogenic role of NLRP3 inflammasome activation in inflammatory bowel diseases of both mice and humans. J Crohns Colitis. 11(6):737–750. doi: 10.1093/ecco-jcc/jjw219.
- Liu S, da Cunha AP, Rezende RM, Cialic R, Wei Z, Bry L, Comstock LE, Gandhi R, Weiner HL. 2016. The host shapes the gut microbiota via fecal MicroRNA. Cell Host Microbe. 19(1):32–43. doi: 10.1016/j.chom.2015.12.005.
- Liu Y, Yin H, Zhao M, Lu Q. 2014. TLR2 and TLR4 in autoimmune diseases: a comprehensive review. Clin Rev Allergy Immunol. 47(2):136–147. doi: 10.1007/s12016-013-8402-y.
- Lockhart PB, Brennan MT, Thornhill M, Michalowicz BS, Noll J, Bahrani-Mougeot FK, Sasser HC. 2009. Poor oral hygiene as a risk factor for infective endocarditis-related bacteremia. J Am Dent Assoc. 140(10):1238–1244. doi: 10.14219/jada.archive.2009.0046.
- Loh G, Blaut M. 2012. Role of commensal gut bacteria in inflammatory bowel diseases. Gut Microbes. 3(6):544–555. doi: 10.4161/gmic.22156.
- Lourenςo TGB, Spencer SJ, Alm EJ, Colombo APV. 2018. Defining the gut microbiota in individuals with periodontal diseases: an exploratory study. J Oral Microbiol. 10(1):1487741. doi: 10.1080/20002297.2018.1487741.
- Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, Sweet-Cordero A, Ebert BL, Mak RH, Ferrando AA, et al. 2005. MicroRNA expression profiles classify human cancers. Nature. 435(7043):834–838. doi: 10.1038/nature03702.
- Lu Y, Li X, Liu S, Zhang Y, Zhang D. 2018. Toll-like receptors and inflammatory bowel disease. Front Immunol. 9:72. doi: 10.3389/fimmu.2018.00072.
- Lv L, Jiang H, Chen X, Wang Q, Wang K, Ye J, Li Y, Fang D, Lu Y, Yang L, et al. 2021. The salivary microbiota of patients with primary biliary cholangitis is distinctive and pathogenic. Front Immunol. 12:713647. doi: 10.3389/fimmu.2021.713647.
- Lynch L, Nowak M, Varghese B, Clark J, Hogan AE, Toxavidis V, Balk SP, O’Shea D, O’Farrelly C, Exley MA. 2012. Adipose tissue invariant NKT cells protect against diet-induced obesity and metabolic disorder through regulatory cytokine production. Immunity. 37(3):574–587. doi: 10.1016/j.immuni.2012.06.016.
- Lyte M. 2011. Probiotics function mechanistically as delivery vehicles for neuroactive compounds: microbial endocrinology in the design and use of probiotics. Bioessays. 33(8):574–581. doi: 10.1002/bies.201100024.
- Ma L, Pang C, Yan C, Chen J, Wang X, Hui J, Zhou L, Zhang X. 2023. Effect of lemon essential oil on halitosis. Oral Dis. 29(4):1845–1854. doi: 10.1111/odi.14140.
- Maisonneuve P, A S, Lowenfels AB. 2017. Periodontal disease, edentulism, and pancreatic cancer: a meta-analysis. Ann Oncol. 28(5):985–995. doi: 10.1093/annonc/mdx019.
- Malham M, James J, Jakobsen C, Høgdall E, Holmstroem K, Wewer V, Nielsen BS, Riis L. 2021. The mucosal microRNA profile relates to age and severity of disease in patients with ulcerative colitis. Gastroenterology. 160(3):S15. doi: 10.1053/j.gastro.2021.01.065.
- Mangerich A, Dedon PC, Fox JG, Tannenbaum SR, Wogan GN. 2013. Chemistry meets biology in colitis-associated carcinogenesis. Free Radic Res. 47(11):958–986. doi: 10.3109/10715762.2013.832239.
- Martinon F, Tschopp J. 2007. Inflammatory caspases and inflammasomes: master switches of inflammation. Cell Death Differ. 14(1):10–22. doi: 10.1038/sj.cdd.4402038.
- McBee ME, Chionh YH, Sharaf ML, Ho P, Cai MW, Dedon PC. 2017. Production of superoxide in bacteria is stress- and cell state-dependent: a gating-optimized flow cytometry method that minimizes ros measurement artifacts with fluorescent dyes. Front Microbiol. 8:459. doi: 10.3389/fmicb.2017.00459.
- McCall SH, Sahraei M, Young AB, Worley CS, Duncan JA, Ting JP, Marriott I. 2008. Osteoblasts express NLRP3, a nucleotide-binding domain and leucine-rich repeat region containing receptor implicated in bacterially induced cell death. J Bone Miner Res. 23(1):30–40. doi: 10.1359/jbmr.071002.
- McClain CJ, Barve S, Deaciuc I, Kugelmas M, Hill D. 1999. Cytokines in alcoholic liver disease. Semin Liver Dis. 19(2):205–219. doi: 10.1055/s-2007-1007110.
- McKenna LB, Schug J, Vourekas A, McKenna JB, Bramswig NC, Friedman JR, Kaestner KH. 2010. MicroRNAs control intestinal epithelial differentiation, architecture, and barrier function. Gastroenterology. 139(5):1654–1664, 1664.e1. doi: 10.1053/j.gastro.2010.07.040.
- Medani M, Collins D, Docherty NG, Baird AW, O’Connell PR, Winter DC. 2011. Emerging role of hydrogen sulfide in colonic physiology and pathophysiology. Inflamm Bowel Dis. 17(7):1620–1625. doi: 10.1002/ibd.21528.
- Medzhitov R, Preston-Hurlburt P, Janeway CA.Jr. 1997. A human homologue of the Drosophila Toll protein signals activation of adaptive immunity. Nature. 388(6640):394–397. doi: 10.1038/41131.
- Megson E, Fitzsimmons T, Dharmapatni K, Bartold PM. 2010. C-reactive protein in gingival crevicular fluid may be indicative of systemic inflammation. J Clin Periodontol. 37(9):797–804. doi: 10.1111/j.1600-051X.2010.01603.x.
- Michaud DS, Izard J, Wilhelm-Benartzi CS, You DH, Grote VA, Tjønneland A, Dahm CC, Overvad K, Jenab M, Fedirko V, et al. 2013. Plasma antibodies to oral bacteria and risk of pancreatic cancer in a large European prospective cohort study. Gut. 62(12):1764–1770. doi: 10.1136/gutjnl-2012-303006.
- Mima K, Nishihara R, Qian ZR, Cao Y, Sukawa Y, Nowak JA, Yang J, Dou R, Masugi Y, Song M, et al. 2016. Fusobacterium nucleatum in colorectal carcinoma tissue and patient prognosis. Gut. 65(12):1973–1980. doi: 10.1136/gutjnl-2015-310101.
- Mitsuhashi K, Nosho K, Sukawa Y, Matsunaga Y, Ito M, Kurihara H, Kanno S, Igarashi H, Naito T, Adachi Y, et al. 2015. Association of Fusobacterium species in pancreatic cancer tissues with molecular features and prognosis. Oncotarget. 6(9):7209–7220. doi: 10.18632/oncotarget.3109.
- Miura K, Ohnishi H. 2014. Role of gut microbiota and Toll-like receptors in nonalcoholic fatty liver disease. World J Gastroenterol. 20(23):7381–7391. doi: 10.3748/wjg.v20.i23.7381.
- Morton AM, Sefik E, Upadhyay R, Weissleder R, Benoist C, Mathis D. 2014. Endoscopic photoconversion reveals unexpectedly broad leukocyte trafficking to and from the gut. Proc Natl Acad Sci U S A. 111(18):6696–6701. doi: 10.1073/pnas.1405634111.
- Moutsopoulos NM, Kling HM, Angelov N, Jin W, Palmer RJ, Nares S, Osorio M, Wahl SM. 2012. Porphyromonas gingivalis promotes Th17 inducing pathways in chronic periodontitis. J Autoimmun. 39(4):294–303. doi: 10.1016/j.jaut.2012.03.003.
- Nakahara T, Hyogo H, Ono A, Nagaoki Y, Kawaoka T, Miki D, Tsuge M, Hiraga N, Hayes CN, Hiramatsu A, et al. 2018. Involvement of Porphyromonas gingivalis in the progression of non-alcoholic fatty liver disease. J Gastroenterol. 53(2):269–280. doi: 10.1007/s00535-017-1368-4.
- Nakajima M, Arimatsu K, Kato T, Matsuda Y, Minagawa T, Takahashi N, Ohno H, Yamazaki K. 2015. Oral administration of P. gingivalis induces dysbiosis of gut microbiota and impaired barrier function leading to dissemination of enterobacteria to the liver. PLoS One. 10(7):e0134234. doi: 10.1371/journal.pone.0134234.
- Nandy SK, Bapat PM, Venkatesh KV. 2007. Sporulating bacteria prefers predation to cannibalism in mixed cultures. FEBS Lett. 581(1):151–156. doi: 10.1016/j.febslet.2006.12.011.
- Ng W, Tonzetich J. 1984. Effect of hydrogen sulfide and methyl mercaptan on the permeability of oral mucosa. J Dent Res. 63(7):994–997. doi: 10.1177/00220345840630071701.
- Ni J, Huang R, Zhou H, Xu X, Li Y, Cao P, Zhong K, Ge M, Chen X, Hou B, et al. 2019. Analysis of the relationship between the degree of dysbiosis in gut microbiota and prognosis at different stages of primary hepatocellular carcinoma. Front Microbiol. 10:1458. doi: 10.3389/fmicb.2019.01458.
- Nicholson JK, Holmes E, Kinross J, Burcelin R, Gibson G, Jia W, Pettersson S. 2012. Host-gut microbiota metabolic interactions. Science. 336(6086):1262–1267. doi: 10.1126/science.1223813.
- O’Connell RM, Taganov KD, Boldin MP, Cheng G, Baltimore D. 2007. MicroRNA-155 is induced during the macrophage inflammatory response. Proc Natl Acad Sci U S A. 104(5):1604–1609. doi: 10.1073/pnas.0610731104.
- Ogata Y, Matsui S, Kato A, Zhou L, Nakayama Y, Takai H. 2014. MicroRNA expression in inflamed and noninflamed gingival tissues from Japanese patients. J Oral Sci. 56(4):253–260. doi: 10.2334/josnusd.56.253.
- Ozdemir A, Mas MR, Sahin S, Sağlamkaya U, Ateşkan U. 2001. Detection of Helicobacter pylori colonization in dental plaques and tongue scrapings of patients with chronic gastritis. Quintessence Int. 32(2):131–134.
- Oztürk A, Yildiz L. 2011. Expression of transient receptor potential vanilloid receptor 1 and toll-like receptor 4 in aggressive periodontitis and in chronic periodontitis. J Periodontal Res. 46(4):475–482. doi: 10.1111/j.1600-0765.2011.01363.x.
- Pacher P, Beckman JS, Liaudet L. 2007. Nitric oxide and peroxynitrite in health and disease. Physiol Rev. 87(1):315–424. doi: 10.1152/physrev.00029.2006.
- Papadopoulos G, Berland R, Sunkavalli A, Coats SR, Darveau RP, Genco CA. 2021. Microbial Lipid A Remodeling Controls Cross-Presentation Efficiency and CD8 T Cell Priming by Modulating Dendritic Cell Function. Infect Immun. 89(2):e00335-20. doi: 10.1128/IAI.00335-20.
- Parahitiyawa NB, Jin LJ, Leung WK, Yam WC, Samaranayake LP. 2009. Microbiology of odontogenic bacteremia: beyond endocarditis. Clin Microbiol Rev. 22(1):46–64. Erratum in: Clin Microbiol Rev. 2009 Apr;22(2):386. doi: 10.1128/CMR.00028-08.
- Paraskevi A, Theodoropoulos G, Papaconstantinou I, Mantzaris G, Nikiteas N, Gazouli M. 2012. Circulating MicroRNA in inflammatory bowel disease. J Crohns Colitis. 6(9):900–904. doi: 10.1016/j.crohns.2012.02.006.
- Park JH, Peyrin-Biroulet L, Eisenhut M, Shin JI. 2017. IBD immunopathogenesis: A comprehensive review of inflammatory molecules. Autoimmun Rev. 16(4):416–426. doi: 10.1016/j.autrev.2017.02.013.
- Peck BC, Mah AT, Pitman WA, Ding S, Lund PK, Sethupathy P. 2017. Functional transcriptomics in diverse intestinal epithelial cell types reveals robust MicroRNA sensitivity in intestinal stem cells to microbial status. J Biol Chem. 292(7):2586–2600. doi: 10.1074/jbc.M116.770099.
- Peng X, Cheng L, You Y, Tang C, Ren B, Li Y, Xu X, Zhou X. 2022. Oral microbiota in human systematic diseases. Int J Oral Sci. 14(1):14. doi: 10.1038/s41368-022-00163-7.
- Perri R, Nares S, Zhang S, Barros SP, Offenbacher S. 2012. MicroRNA modulation in obesity and periodontitis. J Dent Res. 91(1):33–38. doi: 10.1177/0022034511425045.
- Persson GR, Pettersson T, Ohlsson O, Renvert S. 2005. High‐sensitivity serum C‐reactive protein levels in subjects with or without myocardial infarction or periodontitis. J Clin Periodontol. 32(3):219–224. doi: 10.1111/j.1600-051X.2005.00648.x.
- Peters BA, Wu J, Pei Z, Yang L, Purdue MP, Freedman ND, Jacobs EJ, Gapstur SM, Hayes RB, Ahn J. 2017. Oral microbiome composition reflects prospective risk for esophageal cancers. Cancer Res. 77(23):6777–6787. doi: 10.1158/0008-5472.CAN-17-1296.
- Pham BN, Mosnier JF, Durand F, Scoazec JY, Chazouilleres O, Degos F, Belghiti J, Degott C, Benhamou JP, Erlinger S. 1995. Immunostaining for membrane attack complex of complement is related to cell necrosis in fulminant and acute hepatitis. Gastroenterology. 108(2):495–504. doi: 10.1016/0016-5085(95)90079-9.
- Pirola CJ, Fernández Gianotti T, Castaño GO, Mallardi P, San Martino J, Mora Gonzalez Lopez Ledesma M, Flichman D, Mirshahi F, Sanyal AJ, Sookoian S. 2015. Circulating microRNA signature in non-alcoholic fatty liver disease: from serum non-coding RNAs to liver histology and disease pathogenesis. Gut. 64(5):800–812. doi: 10.1136/gutjnl-2014-306996.
- Pitcher MC, Cummings JH. 1996. Hydrogen sulphide: a bacterial toxin in ulcerative colitis? Gut. 39(1):1–4. doi: 10.1136/gut.39.1.1.
- Pohl JF, Melin-Aldana H, Sabla G, Degen JL, Bezerra JA. 2001. Plasminogen deficiency leads to impaired lobular reorganization and matrix accumulation after chronic liver injury. Am J Pathol. 159(6):2179–2186. doi: 10.1016/S0002-9440(10)63069-6.
- Polak D, Shapira L, Weiss EI, Houri-Haddad Y. 2012. The role of coaggregation between Porphyromonas gingivalis and Fusobacterium nucleatum on the host response to mixed infection. J Clin Periodontol. 39(7):617–625. doi: 10.1111/j.1600-051X.2012.01889.x.
- Polepalle T, Moogala S, Boggarapu S, Pesala DS, Palagi FB. 2015. Acute phase proteins and their role in periodontitis: a review. J Clin Diagn Res. 9(11):ZE01–5. doi: 10.7860/JCDR/2015/15692.6728.
- Pradeep AR, Raj S, Aruna G, Chowdhry S. 2009. Gingival crevicular fluid and plasma levels of neuropeptide Substance-P in periodontal health, disease and after nonsurgical therapy. J Periodontal Res. 44(2):232–237. doi: 10.1111/j.1600-0765.2008.01138.x.
- Qi Y, Zang SQ, Wei J, Yu HC, Yang Z, Wu HM, Kang Y, Tao H, Yang MF, Jin L, et al. 2021. High-throughput sequencing provides insights into oral microbiota dysbiosis in association with inflammatory bowel disease. Genomics. 113(1):664–676. doi: 10.1016/j.ygeno.2020.09.063.
- Qin N, Yang F, Li A, Prifti E, Chen Y, Shao L, Guo J, Le Chatelier E, Yao J, Wu L, et al. 2014. Alterations of the human gut microbiome in liver cirrhosis. Nature. 513(7516):59–64. doi: 10.1038/nature13568.
- Ramakrishna BS, Roberts-Thomson IC, Pannall PR, Roediger WE. 1991. Impaired sulphation of phenol by the colonic mucosa in quiescent and active ulcerative colitis. Gut. 32(1):46–49. doi: 10.1136/gut.32.1.46.
- Rao RK, Baker RD, Baker SS, Gupta A, Holycross M. 1997. Oxidant-induced disruption of intestinal epithelial barrier function: role of protein tyrosine phosphorylation. Am J Physiol. 273(4):G812–23. doi: 10.1152/ajpgi.1997.273.4.G812.
- Ray K. 2020. The oral-gut axis in IBD. Nat Rev Gastroenterol Hepatol. 17(9):532–532. doi: 10.1038/s41575-020-0346-0.
- Read E, Curtis MA, Neves JF. 2021. The role of oral bacteria in inflammatory bowel disease. Nat Rev Gastroenterol Hepatol. 18(10):731–742. doi: 10.1038/s41575-021-00488-4.
- Ren Z, Li A, Jiang J, Zhou L, Yu Z, Lu H, Xie H, Chen X, Shao L, Zhang R, et al. 2019. Gut microbiome analysis as a tool towards targeted non-invasive biomarkers for early hepatocellular carcinoma. Gut. 68(6):1014–1023. doi: 10.1136/gutjnl-2017-315084.
- Rengara S, Vivio EE, Parkes M, Peterson DA, Roberson EDO, Newberry RD, Ciorba MA, Hsieh CS. 2020. Dynamic immunoglobulin responses to gut bacteria during inflammatory bowel disease. Gut Microbes. 11(3):405–420. doi: 10.1080/19490976.2019.1626683.
- Richens ER, Thorp CM, Bland PW, Hall ND. 1982. Circulating immune complexes in Crohn’s disease. Their characterization and interrelationship with components of the complement system. Dig Dis Sci. 27(2):129–138. doi: 10.1007/BF01311706.
- Rismo R, Olsen T, Cui G, Christiansen I, Florholmen J, Goll R. 2012. Mucosal cytokine gene expression profiles as biomarkers of response to infliximab in ulcerative colitis. Scand J Gastroenterol. 47(5):538–547. doi: 10.3109/00365521.2012.667146.
- Rodrigues PM, Perugorria MJ, Santos-Laso A, Bujanda L, Beuers U, Banales JM. 2018. Primary biliary cholangitis: A tale of epigenetically-induced secretory failure? J Hepatol. 69(6):1371–1383. doi: 10.1016/j.jhep.2018.08.020.
- Rodriguez Herrero E, Boon N, Pauwels M, Bernaerts K, Slomka V, Quirynen M, Teughels W. 2017. Necrotrophic growth of periodontopathogens is a novel virulence factor in oral biofilms. Sci Rep. 7(1):1107. doi: 10.1038/s41598-017-01239-9.
- Rodríguez-Nogales A, Algieri F, Garrido-Mesa J, Vezza T, Utrilla MP, Chueca N, Garcia F, Olivares M, Rodríguez-Cabezas ME, Gálvez J. 2017. Differential intestinal anti-inflammatory effects of Lactobacillus fermentum and Lactobacillus salivarius in DSS mouse colitis: impact on microRNAs expression and microbiota composition. Mol Nutr Food Res. 61(11) doi: 10.1002/mnfr.201700144.
- Rogler G, Brand K, Vogl D, Page S, Hofmeister R, Andus T, Knuechel R, Baeuerle PA, Schölmerich J, Gross V. 1998. Nuclear factor kappaB is activated in macrophages and epithelial cells of inflamed intestinal mucosa. Gastroenterology. 115(2):357–369. doi: 10.1016/s0016-5085(98)70202-1.
- Rubin DT, Uluscu O, Sederman R. 2012. Response to biologic therapy in Crohn’s disease is improved with early treatment: an analysis of health claims data. Inflamm Bowel Dis. 18(12):2225–2231. doi: 10.1002/ibd.22925.
- Rubinstein MR, Wang X, Liu W, Hao Y, Cai G, Han YW. 2013. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe. 14(2):195–206. doi: 10.1016/j.chom.2013.07.012.
- Sahingur SE, Sharma A, Genco RJ, De Nardin E. 2003. Association of increased levels of fibrinogen and the -455G/A fibrinogen gene polymorphism with chronic periodontitis. J Periodontol. 74(3):329–337. doi: 10.1902/jop.2003.74.3.329.
- Sanchez-Munoz F, Dominguez-Lopez A, Yamamoto-Furusho JK. 2008. Role of cytokines in inflammatory bowel disease. World J Gastroenterol. 14(27):4280–4288. doi: 10.3748/wjg.14.4280.
- Sasaki M, Joh T. 2007. Oxidative stress and ischemia-reperfusion injury in gastrointestinal tract and antioxidant, protective agents. J Clin Biochem Nutr. 40(1):1–12. doi: 10.3164/jcbn.40.1.
- Savchenko AS, Inoue A, Ohashi R, Jiang S, Hasegawa G, Tanaka T, Hamakubo T, Kodama T, Aoyagi Y, Ushiki T, et al. 2011. Long pentraxin 3 (PTX3) expression and release by neutrophils in vitro and in ulcerative colitis. Pathol Int. 61(5):290–297. doi: 10.1111/j.1440-1827.2011.02651.x.
- Schepeler T, Reinert JT, Ostenfeld MS, Christensen LL, Silahtaroglu AN, Dyrskjøt L, Wiuf C, Sørensen FJ, Kruhøffer M, Laurberg S, et al. 2008. Diagnostic and prognostic microRNAs in stage II colon cancer. Cancer Res. 68(15):6416–6424. doi: 10.1158/0008-5472.CAN-07-6110.
- Schieferdecker HL, Rothermel E, Timmermann A, Götze O, Jungermann K. 1997. Anaphylatoxin C5a receptor mRNA is strongly expressed in Kupffer and stellate cells and weakly in sinusoidal endothelial cells but not in hepatocytes of normal rat liver. FEBS Lett. 406(3):305–309. doi: 10.1016/s0014-5793(97)00292-5.
- Schieferdecker HL, Schlaf G, Jungermann K, Götze O. 2001. Functions of anaphylatoxin C5a in rat liver: direct and indirect actions on nonparenchymal and parenchymal cells. Int Immunopharmacol. 1(3):469–481. doi: 10.1016/s1567-5769(00)00038-2.
- Schmidt TS, Hayward MR, Coelho LP, Li SS, Costea PI, Voigt AY, Wirbel J, Maistrenko OM, Alves RJ, Bergsten E, et al. 2019. Extensive transmission of microbes along the gastrointestinal tract. Elife. 8:e42693. doi: 10.7554/eLife.42693.
- Scholz A, Heinze S, Detjen KM, Peters M, Welzel M, Hauff P, Schirner M, Wiedenmann B, Rosewicz S. 2003. Activated signal transducer and activator of transcription 3 (STAT3) supports the malignant phenotype of human pancreatic cancer. Gastroenterology. 125(3):891–905. doi: 10.1016/s0016-5085(03)01064-3.
- Schönauen K, Le N, von Arnim U, Schulz C, Malfertheiner P, Link A. 2018. Circulating and fecal microRNAs as biomarkers for inflammatory bowel diseases. Inflamm Bowel Dis. 24(7):1547–1557. doi: 10.1093/ibd/izy046.
- Segata N, Haake SK, Mannon P, Lemon KP, Waldron L, Gevers D, Huttenhower C, Izard J. 2012. Composition of the adult digestive tract bacterial microbiome based on seven mouth surfaces, tonsils, throat and stool samples. Genome Biol. 13(6):R42. doi: 10.1186/gb-2012-13-6-r42.
- Seiderer J, Elben I, Diegelmann J, Glas J, Stallhofer J, Tillack C, Pfennig S, Jürgens M, Schmechel S, Konrad A, et al. 2008. Role of the novel Th17 cytokine IL-17F in inflammatory bowel disease (IBD): upregulated colonic IL-17F expression in active Crohn’s disease and analysis of the IL17F p.His161Arg polymorphism in IBD. Inflamm Bowel Dis. 14(4):437–445. doi: 10.1002/ibd.20339.
- Seki E, Brenner DA. 2008. Toll-like receptors and adaptor molecules in liver disease: update. Hepatology. 48(1):322–335. doi: 10.1002/hep.22306.
- Seki E, De Minicis S, Osterreicher CH, Kluwe J, Osawa Y, Brenner DA, Schwabe RF. 2007. TLR4 enhances TGF-beta signaling and hepatic fibrosis. Nat Med. 13(11):1324–1332. doi: 10.1038/nm1663.
- Seo SU, Kamada N, Muñoz-Planillo R, Kim YG, Kim D, Koizumi Y, Hasegawa M, Himpsl SD, Browne HP, Lawley TD, et al. 2015. Distinct commensals induce interleukin-1β via NLRP3 inflammasome in inflammatory monocytes to promote intestinal inflammation in response to injury. Immunity. 42(4):744–755. doi: 10.1016/j.immuni.2015.03.004.
- Shale M, Schiering C, Powrie F. 2013. CD4(+) T-cell subsets in intestinal inflammation. Immunol Rev. 252(1):164–182. doi: 10.1111/imr.12039.
- She YY, Kong XB, Ge YP, Liu ZY, Chen JY, Jiang JW, Jiang HB, Fang SL. 2020. Periodontitis and inflammatory bowel disease: a meta-analysis. BMC Oral Health. 20(1):67. doi: 10.1186/s12903-020-1053-5.
- Shi T, Xie Y, Fu Y, Zhou Q, Ma Z, Ma J, Huang Z, Zhang J, Chen J. 2017. The signaling axis of microRNA-31/interleukin-25 regulates Th1/Th17-mediated inflammation response in colitis. Mucosal Immunol. 10(4):983–995. doi: 10.1038/mi.2016.102.
- Shojaee M, Fereydooni Golpasha M, Maliji G, Bijani A, Aghajanpour Mir SM, Mousavi Kani SN. 2013. C - reactive protein levels in patients with periodontal disease and normal subjects. Int J Mol Cell Med. 2(3):151–155.
- Sina C, Kemper C, Derer S. 2018. The intestinal complement system in inflammatory bowel disease: Shaping intestinal barrier function. Semin Immunol. 37:66–73. doi: 10.1016/j.smim.2018.02.008.
- Slade GD, Offenbacher S, Beck JD, Heiss G, Pankow JS. 2000. Acute-phase inflammatory response to periodontal disease in the US population. J Dent Res. 79(1):49–57. doi: 10.1177/00220345000790010701.
- Slots J. 2019. Focal infection of periodontal origin. Periodontol 2000. 79(1):233–235. doi: 10.1111/prd.12258.
- Sorensen NS, Tegtmeier C, Andresen LO, Piñeiro M, Toussaint MJ, Campbell FM, Lampreave F, Heegaard PM. 2006. The porcine acute phase protein response to acute clinical and subclinical experimental infection with Streptococcus suis. Vet Immunol Immunopathol. 113(1-2):157–168. doi: 10.1016/j.vetimm.2006.04.008.
- Stoecklin-Wasmer C, Guarnieri P, Celenti R, Demmer RT, Kebschull M, Papapanou PN. 2012. MicroRNAs and their target genes in gingival tissues. J Dent Res. 91(10):934–940. doi: 10.1177/0022034512456551.
- Strober W. 2010. Inside the microbial and immune labyrinth: gut microbes: friends or fiends? Nat Med. 16(11):1195–1197. doi: 10.1038/nm1110-1195.
- Stummer N, Feichtinger RG, Weghuber D, Kofler B, Schneider AM. 2023. Role of hydrogen sulfide in inflammatory bowel disease. Antioxidants (Basel). 12(8):1570. doi: 10.3390/antiox12081570.
- Sugihara T, Kobori A, Imaeda H, Tsujikawa T, Amagase K, Takeuchi K, Fujiyama Y, Andoh A. 2010. The increased mucosal mRNA expressions of complement C3 and interleukin-17 in inflammatory bowel disease. Clin Exp Immunol. 160(3):386–393. doi: 10.1111/j.1365-2249.2010.04093.x.
- Surrenti C, Renzi D, Garcea MR, Surrenti E, Salvadori G. 1993. Colonic vasoactive intestinal polypeptide in ulcerative colitis. J Physiol Paris. 87(5):307–311. doi: 10.1016/0928-4257(93)90037-t.
- Swidsinski A, Schlien P, Pernthaler A, Gottschalk U, Bärlehner E, Decker G, Swidsinski S, Strassburg J, Loening-Baucke V, Hoffmann U, et al. 2005. Bacterial biofilm within diseased pancreatic and biliary tracts. Gut. 54(3):388–395. doi: 10.1136/gut.2004.043059.
- Swindle EJ, Metcalfe DD. 2007. The role of reactive oxygen species and nitric oxide in mast cell-dependent inflammatory processes. Immunol Rev. 217(1):186–205. doi: 10.1111/j.1600-065X.2007.00513.x.
- Takuma R, Morozumi T, Yamamoto Y, Kobayashi T, Matsui T, Yoneda M, Kessoku T, Nogami A, Tamura M, Kamata Y, et al. 2023. Association between non-alcoholic steatohepatitis-related hepatocellular carcinoma and periodontopathic bacteria: a cross-sectional pilot study. Applied Sciences. 13(6):3893. 19doi: 10.3390/app13063893.
- Teles R, Teles F, Frias-Lopez J, Paster B, Haffajee A. 2013. Lessons learned and unlearned in periodontal microbiology. Periodontol 2000. 62(1):95–162. doi: 10.1111/prd.12010.
- Temmerman R, Vervaeren H, Noseda B, Boon N, Verstraete W. 2006. Necrotrophic growth of Legionella pneumophila. Appl Environ Microbiol. 72(6):4323–4328. doi: 10.1128/AEM.00070-06.
- Thomas B, Rao A, Prasad BR, Kumari S. 2014. Serum levels of antioxidants and superoxide dismutase in periodontitis patients with diabetes type 2. J Indian Soc Periodontol. 18(4):451–455. doi: 10.4103/0972-124X.138686.
- Thorlacius-Ussing G, Schnack Nielsen B, Andersen V, Holmstrøm K, Pedersen AE. 2017. Expression and localization of miR-21 and miR-126 in mucosal tissue from patients with inflammatory bowel disease. Inflamm Bowel Dis. 23(5):739–752. doi: 10.1097/MIB.0000000000001086.
- Tian Y, Xu J, Li Y, Zhao R, Du S, Lv C, Wu W, Liu R, Sheng X, Song Y, et al. 2019. MicroRNA-31 reduces inflammatory signaling and promotes regeneration in colon epithelium, and delivery of mimics in microspheres reduces colitis in mice. Gastroenterology. 156(8):2281–2296.e6. doi: 10.1053/j.gastro.2019.02.023.
- Ting JP, Lovering RC, Alnemri ES, Bertin J, Boss JM, Davis BK, Flavell RA, Girardin SE, Godzik A, Harton JA, et al. 2008. The NLR gene family: a standard nomenclature. Immunity. 28(3):285–287. doi: 10.1016/j.immuni.2008.02.005.
- Toubal A, Kiaf B, Beaudoin L, Cagninacci L, Rhimi M, Fruchet B, da Silva J, Corbett AJ, Simoni Y, Lantz O, et al. 2020. Mucosal-associated invariant T cells promote inflammation and intestinal dysbiosis leading to metabolic dysfunction during obesity. Nat Commun. 11(1):3755. doi: 10.1038/s41467-020-17307-0.
- Tsareva SA, Moriggl R, Corvinus FM, Wiederanders B, Schütz A, Kovacic B, Friedrich K. 2007. Signal transducer and activator of transcription 3 activation promotes invasive growth of colon carcinomas through matrix metalloproteinase induction. Neoplasia. 9(4):279–291. doi: 10.1593/neo.06820.
- Ueki T, Mizuno M, Uesu T, Kiso T, Nasu J, Inaba T, Kihara Y, Matsuoka Y, Okada H, Fujita T, et al. 1996. Distribution of activated complement, C3b, and its degraded fragments, iC3b/C3dg, in the colonic mucosa of ulcerative colitis (UC). Clin Exp Immunol. 104(2):286–292. doi: 10.1046/j.1365-2249.1996.17721.x.
- Ugarova TP, Yakubenko VP. 2001. Recognition of fibrinogen by leukocyte integrins. Ann N Y Acad Sci. 936(1):368–385. doi: 10.1111/j.1749-6632.2001.tb03523.x.
- Uhrig A, Banafsche R, Kremer M, Hegenbarth S, Hamann A, Neurath M, Gerken G, Limmer A, Knolle PA. 2005. Development and functional consequences of LPS tolerance in sinusoidal endothelial cells of the liver. J Leukoc Biol. 77(5):626–633. Erratum in: J Leukoc Biol. 2005 Jul;78(1):5A. doi: 10.1189/jlb.0604332.
- van Deventer SJ. 1999. Review article: targeting TNF alpha as a key cytokine in the inflammatory processes of Crohn’s disease–the mechanisms of action of infliximab. Aliment Pharmacol Ther. 13 Suppl 4:3–8; discussion 38. doi: 10.1046/j.1365-2036.1999.00024.x.
- Van Spaendonk H, Ceuleers H, Witters L, Patteet E, Joossens J, Augustyns K, Lambeir AM, De Meester I, De Man JG, De Winter BY. 2017. Regulation of intestinal permeability: the role of proteases. World J Gastroenterol. 23(12):2106–2123. doi: 10.3748/wjg.v23.i12.2106.
- Visekruna A, Joeris T, Seidel D, Kroesen A, Loddenkemper C, Zeitz M, Kaufmann SH, Schmidt-Ullrich R, Steinhoff U. 2006. Proteasome-mediated degradation of IkappaBalpha and processing of p105 in Crohn disease and ulcerative colitis. J Clin Invest. 116(12):3195–3203. doi: 10.1172/JCI28804.
- Volanakis JE, Kaplan MH. 1971. Specificity of C-reactive protein for choline phosphate residues of pneumococcal C-polysaccharide. Proc Soc Exp Biol Med. 136(2):612–614. doi: 10.3181/00379727-136-35323.
- Vona R, Pallotta L, Cappelletti M, Severi C, Matarrese P. 2021. The impact of oxidative stress in human pathology: focus on gastrointestinal disorders. Antioxidants (Basel). 10(2):201. doi: 10.3390/antiox10020201.
- Wang H, Chao K, Ng SC, Bai AH, Yu Q, Yu J, Li M, Cui Y, Chen M, Hu JF, et al. 2016. Pro-inflammatory miR-223 mediates the cross-talk between the IL23 pathway and the intestinal barrier in inflammatory bowel disease. Genome Biol. 17(1):58. doi: 10.1186/s13059-016-0901-8.
- Wang PL, Ohura K. 2002. Porphyromonas gingivalis lipopolysaccharide signaling in gingival fibroblasts-CD14 and Toll-like receptors. Crit Rev Oral Biol Med. 13(2):132–142. doi: 10.1177/154411130201300204.
- Ward M, Eastwood MA. 1975. Serum C3 and C4 complement components in ulcerative colitis and Crohn’s disease. Digestion. 13(1-2):100–103. doi: 10.1159/000197698.
- Warren RL, Freeman DJ, Pleasance S, Watson P, Moore RA, Cochrane K, Allen-Vercoe E, Holt RA. 2013. Co-occurrence of anaerobic bacteria in colorectal carcinomas. Microbiome. 1(1):16. doi: 10.1186/2049-2618-1-16.
- Weber CR, Turner JR. 2007. Inflammatory bowel disease: is it really just another break in the wall? Gut. 56(1):6–8. doi: 10.1136/gut.2006.104182.
- Wong CM, Tsang FH, Ng IO. 2018. Non-coding RNAs in hepatocellular carcinoma: molecular functions and pathological implications. Nat Rev Gastroenterol Hepatol. 15(3):137–151. doi: 10.1038/nrgastro.2017.169.
- Wong SH, Kwong TNY, Chow TC, Luk AKC, Dai RZW, Nakatsu G, Lam TYT, Zhang L, Wu JCY, Chan FKL, et al. 2017. Quantitation of faecal Fusobacterium improves faecal immunochemical test in detecting advanced colorectal neoplasia. Gut. 66(8):1441–1448. doi: 10.1136/gutjnl-2016-312766.
- Wu F, Zhang S, Dassopoulos T, Harris ML, Bayless TM, Meltzer SJ, Brant SR, Kwon JH. 2010. Identification of microRNAs associated with ileal and colonic Crohn’s disease. Inflamm Bowel Dis. 16(10):1729–1738. doi: 10.1002/ibd.21267.
- Xiao YT, Yan WH, Cao Y, Yan JK, Cai W. 2016. Neutralization of IL-6 and TNF-α ameliorates intestinal permeability in DSS-induced colitis. Cytokine. 83:189–192. doi: 10.1016/j.cyto.2016.04.012.
- Xie YF, Shu R, Jiang SY, Liu DL, Zhang XL. 2011. Comparison of microRNA profiles of human periodontal diseased and healthy gingival tissues. Int J Oral Sci. 3(3):125–134. doi: 10.4248/IJOS11046.
- Yamaguchi Y, Kurita-Ochiai T, Kobayashi R, Suzuki T, Ando T. 2015. Activation of the NLRP3 inflammasome in Porphyromonas gingivalis-accelerated atherosclerosis. Pathog Dis. 73(4):ftv011. doi: 10.1093/femspd/ftv011.
- Yamamura K, Baba Y, Nakagawa S, Mima K, Miyake K, Nakamura K, Sawayama H, Kinoshita K, Ishimoto T, Iwatsuki M, et al. 2016. Human microbiome fusobacterium nucleatum in esophageal cancer tissue is associated with prognosis. Clin Cancer Res. 22(22):5574–5581. doi: 10.1158/1078-0432.CCR-16-1786.
- Yamaoka Y, Suehiro Y, Hashimoto S, Hoshida T, Fujimoto M, Watanabe M, Imanaga D, Sakai K, Matsumoto T, Nishioka M, et al. 2018. Fusobacterium nucleatum as a prognostic marker of colorectal cancer in a Japanese population. J Gastroenterol. 53(4):517–524. doi: 10.1007/s00535-017-1382-6.
- Yan H, Zhang X, Xu Y. 2020. Aberrant expression of miR-21 in patients with inflammatory bowel disease: A protocol for systematic review and meta analysis. Medicine (Baltimore). 99(17):e19693.). doi: 10.1097/MD.0000000000019693.
- Yan K, Lin Q, Tang K, Liu S, Du Y, Yu X, Li S. 2020. Substance P participates in periodontitis by upregulating HIF-1α and RANKL/OPG ratio. BMC Oral Health. 20(1):27. doi: 10.1186/s12903-020-1017-9.
- Yang Y, Ma Y, Shi C, Chen H, Zhang H, Chen N, Zhang P, Wang F, Yang J, Yang J, et al. 2013. Overexpression of miR-21 in patients with ulcerative colitis impairs intestinal epithelial barrier function through targeting the Rho GTPase RhoB. Biochem Biophys Res Commun. 434(4):746–752. doi: 10.1016/j.bbrc.2013.03.122.
- Yang Y, Weng W, Peng J, Hong L, Yang L, Toiyama Y, Gao R, Liu M, Yin M, Pan C, et al. 2017. Fusobacterium nucleatum Increases Proliferation of Colorectal Cancer Cells and Tumor Development in Mice by Activating Toll-Like Receptor 4 Signaling to Nuclear Factor-κB, and Up-regulating Expression of MicroRNA-21. Gastroenterology. 152(4):851–866.e24. doi: 10.1053/j.gastro.2016.11.018.
- Yde Aagaard ME, Frahm Kirk K, Linde Nielsen H, Harder Tarpgaard I, Bach Hansen J, Nielsen H. 2020. Campylobacter concisus is prevalent in the gastrointestinal tract of patients with microscopic colitis. Scand J Gastroenterol. 55(8):924–930. doi: 10.1080/00365521.2020.1792976.
- Yiangou Y, Facer P, Dyer NH, Chan CL, Knowles C, Williams NS, Anand P. 2001. Vanilloid receptor 1 immunoreactivity in inflamed human bowel. Lancet. 357(9265):1338–1339. doi: 10.1016/s0140-6736(00)04503-7.
- Yilmaz O, Sater AA, Yao L, Koutouzis T, Pettengill M, Ojcius DM. 2010. ATP-dependent activation of an inflammasome in primary gingival epithelial cells infected by Porphyromonas gingivalis. Cell Microbiol. 12(2):188–198. 98. PMID: 11501 doi: 10.1111/j.1462-5822.2009.01390.x.
- Yin J, Ren Y, Yang K, Wang W, Wang T, Xiao W, Yang H. 2022. The role of hypoxia-inducible factor 1-alpha in inflammatory bowel disease. Cell Biol Int. 46(1):46–51. doi: 10.1002/cbin.11712.
- Yoneda T, Tomofuji T, Ekuni D, Azuma T, Maruyama T, Fujimori K, Sugiura Y, Morita M. 2019. Serum microRNAs and chronic periodontitis: a case-control study. Arch Oral Biol. 101:57–63. doi: 10.1016/j.archoralbio.2019.03.009.
- Yoo SH, Jung HS, Sohn WS, Kim BH, Ku BH, Kim YS, Park SW, Hahm KB. 2008. Volatile sulfur compounds as a predictor for esophagogastroduodenal mucosal injury. Gut Liver. 2(2):113–118. doi: 10.5009/gnl.2008.2.2.113.
- Yuan J, Chen C, Cui J, Lu J, Yan C, Wei X, Zhao X, Li N, Li S, Xue G, et al. 2019. Fatty Liver Disease Caused by High-Alcohol-Producing Klebsiella pneumoniae. Cell Metab. 30(4):675–688.e7. Erratum in: cell Metab. 2019 Dec 3;30(6):1172. doi: 10.1016/j.cmet.2019.08.018.
- Yuan YY, Xie KX, Wang SL, Yuan LW. 2018. Inflammatory caspase-related pyroptosis: mechanism, regulation and therapeutic potential for inflammatory bowel disease. Gastroenterol Rep (Oxf). 6(3):167–176. doi: 10.1093/gastro/goy011.
- Yun LW, Decarlo AA, Hunter N. 2007. Blockade of protease-activated receptors on T cells correlates with altered proteolysis of CD27 by gingipains of Porphyromonas gingivalis. Clin Exp Immunol. 150(2):217–229. doi: 10.1111/j.1365-2249.2007.03488.x.
- Zaki A, Fawzy A, Akel SY, Gamal H, Elshimy RAA. 2022. Evaluation of microRNA 92a expression and its target protein bim in colorectal cancer. Asian Pac J Cancer Prev. 23(2):723–730. doi: 10.31557/APJCP.2022.23.2.723.
- Zaki MH, Lamkanfi M, Kanneganti TD. 2011. The Nlrp3 inflammasome: contributions to intestinal homeostasis. Trends Immunol. 32(4):171–179. doi: 10.1016/j.it.2011.02.002.
- Zhang C, Xu C, Gao L, Li X, Zhao C. 2021. Porphyromonas gingivalis lipopolysaccharide promotes T-hel per17 cell differentiation by upregulating Delta-like ligand 4 expression on CD14+ monocytes. PeerJ. 9:e11094. doi: 10.7717/peerj.11094.
- Zhang G, Ghosh S. 2001. Toll-like receptor-mediated NF-kappaB activation: a phylogenetically conserved paradigm in innate immunity. J Clin Invest. 107(1):13–19. doi: 10.1172/JCI11837.
- Zhang J, Yu C, Zhang X, Chen H, Dong J, Lu W, Song Z, Zhou W. 2018. Porphyromonas gingivalis lipopolysaccharide induces cognitive dysfunction, mediated by neuronal inflammation via activation of the TLR4 signaling pathway in C57BL/6 mice. J Neuroinflammation. 15(1):37. doi: 10.1186/s12974-017-1052-x.
- Zhang L, Gao X, Zhou J, Chen S, Zhang J, Zhang Y, Chen B, Yang J. 2020. Increased risks of dental caries and periodontal disease in Chinese patients with inflammatory bowel disease. Int Dent J. 70(3):227–236. doi: 10.1111/idj.12542.
- Zhang L, Zhang F, He DK, Fan XM, Shen J. 2018. MicroRNA-21 is upregulated during intestinal barrier dysfunction induced by ischemia reperfusion. Kaohsiung J Med Sci. 34(10):556–563. doi: 10.1016/j.kjms.2018.05.006.
- Zhao P, Liu J, Pan C, Pan Y. 2014. NLRP3 inflammasome is required for apoptosis of Aggregatibacter actinomycetemcomitans-infected human osteoblastic MG63 cells. Acta Histochem. 116(7):1119–1124. doi: 10.1016/j.acthis.2014.05.008.
- Zheng T, Ma G, Tang M, Li Z, Xu R. 2018. IL-8 Secreted from M2 Macrophages Promoted Prostate Tumorigenesis via STAT3/MALAT1 Pathway. Int J Mol Sci. 20(1):98. doi: 10.3390/ijms20010098.