
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Small intestinal (SI) tumors are relatively uncommon outcomes in rodent cancer bioassays, and limited information regarding chemical-induced SI tumorigenesis has been reported in the published literature. Herein, we propose a cytotoxicity-mediated adverse outcome pathway (AOP) for SI tumors by leveraging extensive target species- and site-specific molecular, cellular, and histological mode of action (MOA) research for three reference chemicals, the fungicides captan and folpet and the transition metal hexavalent chromium (Cr(VI)). The gut barrier functions through highly efficient homeostatic regulation of SI epithelial cell sloughing, regenerative proliferation, and repair, which involves the replacement of up to 1011 cells per day. This dynamic turnover in the SI provides a unique local environment for a cytotoxicity mediated AOP/MOA. Upon entering the duodenum, cytotoxicity to the villous epithelium is the molecular initiating event, as indicated by crypt elongation, villous atrophy/blunting, and other morphologic changes. Over time, the regenerative capacity of the gut epithelium to compensate declines as epithelial loss accelerates, especially at higher exposures. The first key event (KE), sustained regenerative crypt proliferation/hyperplasia, requires sufficient durations, likely exceeding 6 or 12 months, due to extensive repair capacity, to create more opportunities for the second KE, spontaneous mutation/transformation, ultimately leading to proximal SI tumors. Per OECD guidance, biological plausibility, essentiality, and empirical support were assessed using modified Bradford Hill considerations. The weight-of-evidence also included a lack of induced mutations in the duodenum after up to 90 days of Cr(VI) or captan exposure. The extensive evidence for this AOP, along with the knowledge that human exposures are orders of magnitude below those associated with KEs in this AOP, supports its use for regulatory applications, including hazard identification and risk assessment.
1. Introduction
Human health risk assessment of potential carcinogens has evolved from simply counting tumors present in animals in long-term bioassays to understanding the mechanistic basis for chemical-induced cancers. With such information, a plausible mode of action (MOA) can be hypothesized and used to inform quantitative approaches for deriving safety criteria for environmental agents (Sonich-Mullin et al. Citation2001; U.S. EPA Citation2005; Boobis et al. Citation2008). Understandably, tumors that arise frequently in cancer bioassays tend to receive the most targeted research. For example, kidney tumors that are frequently observed in male rats are often, but not always, the result of α2 u-globulin-induced nephropathy leading to renal tubular hyperplasia and subsequently neoplasia (Hard et al. Citation1993). This process is an idiosyncratic response specific to male rats, and therefore these tumors are well recognized to have little relevance to human health. Intestinal cancers, by comparison, are rarely observed in rodent bioassays (Gold et al. Citation2001; Chandra et al. Citation2010) and, historically, have lacked a clear unified etiology. Thus, the relevance of rodent intestinal tumors to human health risk assessment has not received as much attention as other rodent tumors. Moreover, small intestine (SI) physiology and toxicokinetics (e.g. absorption and metabolic capability) have not been well studied in relation to human health risk assessment. At a regulatory level, this uncertainty has resulted in default assumptions about the MOA (e.g. mutagenicity) and shape of the dose-cancer relationship (e.g. low-dose linear) for certain intestinal carcinogens. Issues related to default assumptions about target-site mutagenicity and the low-dose linearity of the dose-response are highlighted by recent regulatory efforts to develop safe exposure levels to hexavalent chromium [Cr(VI)] in drinking water based on tumor responses in the proximal SI of mice.
In 2008, the National Toxicology Program (NTP) released the results of a 2-year drinking water study in mice and rats that reported two target sites for carcinogenicity – oral cavity tumors in F344 rats exposed to 180 ppm Cr(VI) and duodenal tumors in B6C3F1 mice exposed to ≥30 ppm Cr(VI) (NTP Citation2008; Stout et al. Citation2009). Despite clear indications of chronic cytotoxicity and regenerative hyperplasia in the duodenum, the MOA for these tumors was concluded by some regulators shortly thereafter to involve direct-acting mutagenicity or an unknown MOA, and a linear dose response analysis was proposed (McCarroll et al. Citation2010; Stern Citation2010; U.S. EPA Citation2010). These assumptions result in an extra risk of one person in one hundred thousand people developing intestinal cancer at drinking water exposures equating to 0.035 ppb Cr(VI),Footnote1 even though reported average concentration of Cr(VI) in US water sources ranges from 0.2 to 5 ppb (AWWA Citation2004; CDPH Citation2011; Moffat et al. Citation2018), with the 95th %-ile for Cr(VI) exposure in US water sources being 3 ppb (U.S. EPA Citation2017). The current maximum contaminant level for total chromium in water is 100 ppb (McNeill et al. Citation2012; Moffat et al. Citation2018). The Cr(VI) concentrations that induced tumors in mice (≥30 ppm) are four orders of magnitude higher than the 95th %-ile drinking water exposures.
These early assumptions about the MOA and shape of the dose-cancer relationship were, perhaps, prudent in the absence of targeted MOA research in the SI, and the fact that Cr(VI) is a genotoxicant, particularly in in vitro assays. A rather large number of studies targeted at informing risk assessment of Cr(VI) were conducted from ∼2010 to 2019, including pharmacokinetics (Kirman et al. Citation2012, Citation2013, Citation2016, Citation2017; Proctor et al. Citation2012; De Flora et al. Citation2016), in vivo genotoxicity assays (De Flora et al. Citation2008; O'Brien et al. 2013; Thompson, Seiter, et al. Citation2015; Thompson, Wolf, et al. Citation2015; Thompson, Young, et al. Citation2015; Citation2017; Aoki et al. Citation2019), transcriptomic analyses (Kopec, Kim, et al. Citation2012; Kopec, Thompson, et al. Citation2012; Thompson, Gregory, et al. Citation2012; Rager et al. Citation2017), and histopathological analyses (Thompson, Proctor, et al. Citation2011, Citation2012; Thompson, Seiter, et al. Citation2015; Thompson, Wolf, et al. Citation2015; Cullen et al. Citation2016). Based on this large body of new information, both the scientists directly involved in the research as well as others reviewing the work have concluded that Cr(VI) causes SI cancer through a non-mutagenic MOA and that non-linear risk assessment approaches are most appropriate (Thompson et al. Citation2014; Haney Citation2015; Health Canada Citation2016; TCEQ Citation2016; Thompson et al. Citation2018; FSCJ Citation2019; WHO Citation2019).
A cytotoxicity-mediated MOA for SI tumors in mice has clear precedent. Captan and the structurally similar fungicide folpet induce SI tumors in mice and have been hypothesized to cause these tumors through sustained damage to SI villi resulting in chronic epithelial hyperplasia within intestinal crypts (Gordon Citation2007; Cohen et al. Citation2010). The U.S. EPA Office of Pesticide Programs, for example, determined that captan is “not likely to be a human carcinogen at dose levels that do not cause cytotoxicity and regenerative cell hyperplasia” (U.S. EPA Citation2004) and that folpet is “not likely to be carcinogenic to humans at doses that do not cause an irritation response in the mucosal epithelium” (U.S. EPA Citation2012). Further, a series of studies have compared the SI effects of Cr(VI) and captan/folpet and concluded that they share many phenotypic responses despite having different chemical structures (Thompson, Suh, et al. Citation2017; Thompson, Wolf, et al. Citation2017; Chappell et al. Citation2019). This work suggests that these compounds have similar MOAs and provides a well-developed data set supporting a cytotoxicity-mediated adverse outcome pathway (AOP), as described herein.
The extensive molecular, cellular, tissue, and organ-level evidence that now exists for Cr(VI), captan, and folpet, over a broad range of exposure conditions and durations, is sufficient to construct, to our knowledge, the first AOP that supports SI tumor development in mice occurring via cytotoxicity-mediated initiating events. Across seven carcinogenicity bioassays identified in mice, the range of lifetime exposure concentrations associated with dose-related development of SI tumors was 30 to 180 ppm in drinking water for Cr(VI), 6000 ppm to 16 000 ppm in feed for captan, and 1000 ppm to 12 000 ppm in feed for folpet. As such, the objective of this manuscript was to document the weight of evidence (WOE) supporting (or refuting) the key events (KEs) and KE relationships (KERs) leading to SI tumors. This AOP can be used for regulatory applications including hazard identification and risk assessment.
2. AOP development
The proposed AOP generally conforms to Organization for Economic Co-Operation and Development (OECD) guidance (OECD Citation2018), albeit with a few practical modifications to address (1) a relatively rare tumor location in rodent bioassays, (2) carcinogens that likely have nonspecific initiating events (e.g. not receptor-mediated), and (3) avoidance of necessary redundancy inherent to many frameworks. The OECD Revised Guidance Document on Developing and Assessing AOP (OECD Citation2018) states that the AOP framework is a transparent and scientific-based means to organize and present current knowledge of predictable relationships between molecular initiating events (MIEs), subsequent KEs, and the adverse outcome (AO). The ultimate objective underlying AOP development is to support inference or extrapolation from one KE to another (OECD Citation2018).
Although AOPs are often characterized as “chemical agnostic,” it is recognized that our understanding must flow from basic research and chemical-specific information. As such, the proposed AOP is based on the MOA research for Cr(VI), captan, and folpet. As described in Chandra et al. (Citation2010), the only other NTP studies reporting SI tumors in mice are indium phosphide and methylene blue trihydride. Because the evidence for these two chemicals was characterized as uncertain or “some evidence” and only in male mice, these were not considered in the development of this AOP. Hydrogen peroxide-induced intestinal tumors were also considered; however, many of the available studies do not include images of the non-neoplastic duodenal lesions, thus making it difficult to compare the lesions to Cr(VI) and captan/folpet.
The AOP shown in proposes that, upon entering the duodenum, cytotoxicity to the villous epithelium is the MIE. Over time, the regenerative capacity of the gut epithelium to compensate declines as epithelial loss accelerates, especially at higher-dose exposures. The first KE, sustained regenerative crypt proliferation/hyperplasia, requires exposure durations, likely exceeding 6 or 12 months, due to extensive repair capacity, to create more opportunities for the second KE, spontaneous mutation/transformation, ultimately leading to proximal SI tumors.
Figure 1. Proposed AOP for cytotoxicity-mediated SI cancer in mice. (A) AOP diagram. See text for discussion regarding (M)IE. Future evolution of this AOP may better define the (M)IE associated with villous cytotoxicity. (B) Structure of intestinal mucosa. Villi are finger- or leaf-like projections into the lumen that are predominantly covered with mature, absorptive enterocytes, along with occasional mucus-secreting goblet cells. These cells live only for a few days, then die and slough into the lumen. The crypts, or glands of Lieberkühn, are tubular invaginations of the epithelium, lined largely with younger epithelial cells, which serve as a source of enterocytes to multiple villi. At the base of the crypts are stem cells, which divide continually and function as the source of all the epithelial cells in the crypts and on the villi. Adapted from O'Brien et al. (Citation2013).
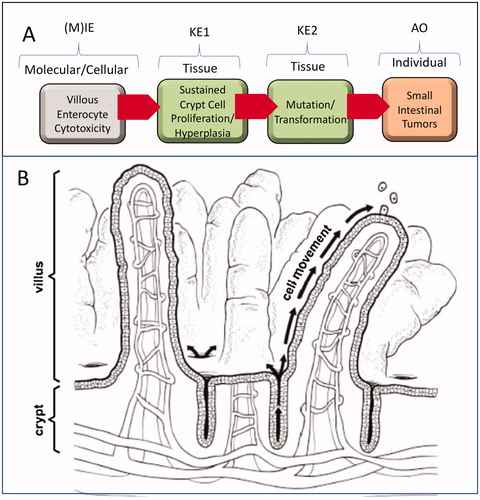
In developing an AOP it is important to understand the unique characteristics of the tissues of interest. The SI is divided into three contiguous segments comprised of the duodenum (proximal SI), the jejunum, and ileum (distal SI). The duodenum is the primary site of nutrient absorption, which is facilitated by the large surface area afforded by fingerlike projections from the mucosal surface called villi (DeSesso et al. Citation2000; Treuting et al. Citation2018). These villi are lined by mature (differentiated) enterocytes and supplied by stem cells that reside below the intestinal surface in invaginations called crypts or glands of Lieberkühn (). The intestinal mucosa is one of the most proliferative tissues in the body, with cells transiting from the crypt to villous tips along the crypt-villus axis within a span of approximately 2–3 days in rodents and 3–6 days in humans (van der Flier and Clevers Citation2009; Greaves Citation2012). The intestinal stem cells reside near the base of the crypt and divide asymmetrically to give rise to daughter cells that move through the transit-amplifying region of the crypt and up toward to the villous tips where the enterocytes will soon slough into the intestinal lumen (). As enterocytes move toward the villus, they become committed non-dividing enterocytes. The prevailing theory on intestinal cancer is that tumors arise from mutations that occur within the crypt stem cells that remain in the base of the crypt and thus have time to acquire multiple mutations leading to cell transformation (Potten and Loeffler Citation1990; Barker et al. Citation2009). As shown in , crypts are invaginations below the intestinal surface that contain mucus secreted from goblet cells. Overall, the SI and structure of the intestinal mucosa are similar in mice, rats, and humans (DeSesso et al. Citation2000; Nolte et al. Citation2016). A notable difference is plicae in humans, which are circular folds lined by villi that further increase the surface area of the intestine; rodents instead have taller villi to increase surface area (DeSesso et al. Citation2000; Treuting et al. Citation2018). In all three species, the duodenal villi are longer/taller than those in the jejunum and ileum. All three species possess large intestine and colon that contain crypts but lack villi (Nolte et al. Citation2016).
2.1. (Molecular) initiating event [(M)IE]: villous enterocyte cytotoxicity
2.1.1. Overview
The OECD (Citation2018) defines a MIE as, “A specialized type of key event that represents the initial point of chemical/stressor interaction at the molecular level within the organism that results in a perturbation that starts the AOP.” Others have commented on the potential confusion of this terminology and have proposed additional or alternative terms such as initial molecular event (IME) or pre-MIE to clarify that detection of the MIE itself does not necessarily imply that an AO is imminent (Becker, Patlewicz, et al. Citation2015; Patlewicz et al. Citation2015). For example, in an AOP for rat liver tumors mediated by the aryl hydrocarbon receptor (AhR), binding to the AhR was a pre-MIE, whereas sustained AhR activation was the MIE (Becker, Ankley, et al. Citation2015). To date, there is no clear evidence for a specific initiating event for SI tumors at the molecular level; however, there is evidence for the general anatomical and cellular location of the initial point of interaction. In contrast to receptor-mediated signaling events, it is recognized that cytotoxicity is mediated by multiple cellular perturbations rather than a single molecular interaction resulting from a specific initiating event. While the exact molecular initiating mechanisms underlying cytotoxicity as the (M)IE have not yet been demonstrated, key evidence supports reactivity of bioactivated compounds with cellular components leading to insult and death. The question of whether identification of a single specific MIE is necessary for AOP development is beyond the scope of this article. However, there is broad precedent and regulatory acceptance for cytotoxicity as an initial or early KE in non-mutagenic MOAs by U.S. EPA and other health agencies internationally. The U.S. EPA Cancer Risk Assessment Guidelines consider cytotoxicity and regenerative proliferation as a reference MOA relevant to a wide variety of chemical agents, target tissues, and exposure routes (Wood et al. Citation2015). Captan and folpet are regarded as carcinogens that act via cytotoxic MOAs (U.S. EPA Citation2004, Citation2012; Eastmond Citation2012), as is Cr(VI) (Haney Citation2015; Health Canada Citation2016; TCEQ Citation2016; FSCJ Citation2019; WHO 2019). Cancer risk assessments for thiamethoxam-induced mouse liver tumors, chloroform-induced rat kidney tumors, acetochlor-induced nasal cavity tumors, and saccharin-induced rat bladder tumors are some other examples (referenced in Wood et al. Citation2015). Accordingly, we have characterized villous enterocyte cytotoxicity as a (M)IE, with parentheticals on “molecular” to denote that cytotoxicity as an initial or early KE in a non-mutagenic AOP/MOA may not involve or require a specific molecular event ().
Cr(VI), captan, and folpet share a common requirement to be transported to the SI lumen in a form capable of cellular uptake and thus inducing cytotoxicity to duodenal epithelial cells. High concentrations of ingested Cr(VI) that escape reduction to Cr(III) in the acidic environment of the stomach enter the higher pH of the intestinal lumen, which limits reduction of the Cr(VI). Luminal Cr(VI) then enters the duodenal villous enterocytes by ion mimicry, where it then reacts with cellular thiols (e.g. glutathione (GSH)) and other molecules including DNA (Salnikow and Zhitkovich Citation2008; Buttner and Beyersmann Citation1985). Ingested captan and folpet are relatively stable in the stomach but become unstable in the higher pH of the duodenum. This hydrolytic instability allows for rapid hydrolysis of captan/folpet via reaction with cellular thiols to form thiophosgene, the cytotoxic moiety (Wilkinson et al. Citation2004; Arce et al. Citation2010; Cohen et al. Citation2010). As such, the duodenum is susceptible to all three carcinogens. Recent transcriptomic comparisons between Cr(VI) and captan/folpet after 28 days of exposure indicate activation of hypoxia inducible factor 1 (HIF-1) and activator protein 1 (AP1) signaling pathways in the SI (Chappell et al. Citation2019). These biomarkers likely represent downstream responses secondary to cytotoxicity rather than specific MIEs.
Pharmacological depletion of GSH has been shown to shorten intestinal villi (an indication of cytotoxicity) (Martensson et al. Citation1990), and GSH supplementation and uptake from the basolateral membrane (i.e. via the blood) has been shown to mitigate intestinal injury (Lash et al. Citation1986). As such, chemicals that deplete GSH in the villi might accelerate villus enterocyte sloughing and stimulate regenerative hyperplasia. Cr(VI) and captan/folpet bind thiols and therefore high concentrations of these chemicals in the duodenum could lead to accelerated cell loss from perturbation of cellular thiol status in addition to more generalized cytotoxicity. However, the data on GSH effects of these chemicals are limited. For example, bolus dosing of folpet significantly decreased duodenal GSH when measured 0.5–2 h after exposure, but significantly increased GSH by 6 h (Arce et al. Citation2010). The effect of Cr(VI) on duodenal GSH immediately after exposure has not been investigated; however, longer-term (i.e. 13-week) exposures to Cr(VI) resulted in higher GSH levels and expression of genes involved in GSH synthesis (Thompson, Proctor, et al. Citation2011; Kopec, Kim, et al. Citation2012). These data suggest that acute exposures to Cr(VI) and captan/folpet may deplete GSH and subsequently induce a compensatory mechanism to increase GSH levels. More research is needed to establish whether changes in GSH could be an associative factor or specific MIE for these carcinogens.
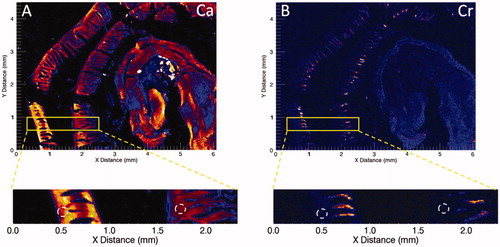
There is strong evidence that the duodenal villi are the initial target for SI tumorigenesis in mice. This is best demonstrated for Cr(VI), where synchrotron-based x-ray fluorescence (XRF) microscopy has shown that after 7 or 90 days of exposure to 180 ppm Cr(VI) in drinking water, Cr fluorescence is highest in the villous tips, present in the villi, and absent in the crypt compartment () (Thompson, Seiter, et al. Citation2015; Thompson, Wolf, et al. Citation2015). Wet tissue analyses indicate that the mg/kg Cr levels are higher in the duodenum than either the stomach or the jejunum/ileum following exposure up to 90 days (Thompson, Proctor, et al. Citation2011). This distribution is likely due to several factors, including slower reduction of Cr(VI) to Cr(III) in the higher pH of the SI relative to stomach, increased surface area of the duodenum, and transporter-mediated uptake of Cr(VI). Whether the lower levels of Cr in the jejunum and ileum are due to luminal reduction of Cr(VI) to Cr(III) or changes in transporter-mediated uptake is unknown.
Figure 2. Synchrotron-based XRF microscopy of Cr in Swiss roll preparations of a duodenum from a mouse exposed to 180 ppm Cr(VI) for 7 days. (A) XRF for calcium (Ca) indicates presence throughout the intestinal mucosa. (B) XRF for chromium (Cr) indicates presence in villi but not crypts (white dotted circles). Adapted from Thompson, Wolf, et al. (Citation2015).
The intestinal epithelium is a critical component of the gut barrier that prevents the transfer of harmful agents from the gut lumen into the circulation via the subjacent tissue, lymphatics, and blood vessels (Groschwitz and Hogan Citation2009; Williams et al. Citation2015). The gut barrier functions through highly efficient homeostatic regulation of SI epithelial cell sloughing, regenerative proliferation, and repair. The daily loss and replacement of SI epithelial cells has been estimated at 1011 cells per day in humans and up to 2 × 108 cells per day in mice (Williams et al. Citation2015). The distinct compartmentalization of villous and crypt epithelium is a key feature of this barrier. As highlighted by the XRF imaging for Cr(VI), the crypts are invaginations below the intestinal surface, and under normal circumstances, they are not directly exposed to the intestinal luminal contents (Cohen et al. Citation2010). This concept is supported by studies with the carcinogen benzo[a]pyrene (BaP), where immunohistochemical analyses have shown that BaP induces CYP1A1, a subtype of the cytochrome P450 family of metabolizing enzymes, in villi (but not crypts) following oral gavage, whereas CYP1A1 induction is observed in crypts (but not villous enterocytes) following intraperitoneal injection (Brooks et al. Citation1999). The study authors concluded from these results that intestinal crypts are part of the systemic compartment and not readily accessible from the lumen.
As mentioned, captan/folpet are relatively stable in the acidic stomach but rapidly hydrolyzed in the alkaline duodenum to tetrahydrophthalimide and thiophosgene. While tetrahydrophthalimide is of little toxicological concern, thiophosgene is an irritant to duodenal epithelial cells (Wilkinson et al. Citation2004). In contrast to Cr(VI), there are limited data demonstrating villus localization after captan/folpet exposure in mice. Following exposure to radiolabeled captan in diet, parent and metabolite(s) were detected in luminal contents (Wilkinson et al. Citation2004). Higher duodenal concentrations of captan were detected in mice compared to rats exposed to 250 mg/kg radiolabeled captan (Wong and Chang Citation1985). While no data examining the localization of folpet or its metabolites in the gastrointestinal tract of mice were identified, U.S. EPA (Citation2012) has posited that greater target tissue exposure in mice might explain why mice but not rats developed duodenal tumors. In addition to pharmacokinetic measurements or visualization (e.g. XRF microscopy), localization of agents within the mucosa can also be ascertained by presence or absence of histopathological lesions in the crypts and villi.
Cytotoxicity within the intestinal villi can be difficult to detect due to the efficient homeostatic regulation of the mucosa. While acute toxicity (e.g. cell death) might be detected as necrosis shortly after high-dose exposures, most bioassays examine tissues after several days, weeks, or months of exposure. When intestinal damage is mild, increased activity of the crypt replication zone compensates for the increased villous cell loss, and there may be little detectable change beyond an increase in mitotic activity and the length of the crypt zone (Brown Citation2013). Restated, crypt elongation can be an indicator of mild villus toxicity. When cell loss is more rapid or severe, crypt hyperplasia increases further and in some cases the villi become shorter and broader (villous blunting) due, in part, to the inability to fully compensate for increased cell death (Brown Citation2013). Without understanding of this fundamental intestinal biology, some researchers have misinterpreted villous atrophy/blunting observed after repeat-dose exposure as the first indicator of toxicity when, in fact, atrophy/blunting can indicate an inability of the crypt to compensate villous enterocyte loss. Instead, mild increases in crypt cell proliferation or crypt depth can indicate cell loss in the villus that is being replenished without atrophy/blunting. In response to cell death there is lateral migration of residual enterocytes to cover the basement membrane of the villus, as naked basement membrane is not tolerated. These residual enterocytes also flatten from a columnar outline to a cuboidal or squamous shape to increase their surface area. With increased loss of mature enterocytes, contraction of resident myofibroblasts within the villi is triggered along with apoptosis of stromal elements. This reduces villus height and surface area, enabling the flattened enterocytes to cover the surface of the villi (Uzal et al. Citation2016). It should be acknowledged that loss of crypt epithelial cells can also lead to severe villous changes due to reduced production of enterocytes; however, XRF imaging of Cr and clear evidence of healthy proliferating crypts indicate that direct crypt damage is not the cause of blunting for Cr(VI) and captan/folpet. As discussed in Section 2.6, certain viruses preferentially affect villous and crypt enterocytes, resulting in different histopathological characteristics. Features related to viral mediated villus toxicity are more similar to those induced by Cr(VI) and captan/folpet than viral mediated crypt toxicity.
As discussed, duodenal villi are the primary site of action for Cr(VI) and captan/folpet. Once inside cells, Cr(VI) does not undergo enzymatic metabolism; rather, it reacts with thiols, GSH, and ascorbate. Similarly, captan and folpet are not enzymatically metabolized, but instead undergo hydrolysis to thiophosgene – a process that also occurs more readily at higher pH than lower pH. Thiophosgene readily reacts with cellular thiols, which likely results in cytotoxicity. Although these agents react with DNA and other macromolecules, it is expected that their toxicity is mediated by nonspecific mechanisms such as protein binding and/or alteration of cellular redox status. Data regarding the interaction of Cr(VI) and captan/folpet with duodenal DNA are discussed as part of KE#2 (below). Importantly, villous enterocytes are committed, non-proliferating cells that slough into the intestinal lumen within 24–48 h of reaching the villi. Although dedifferentiation of these cells to more primitive states has been demonstrated in experimental models (Section 2.3), the available data for Cr(VI) and captan/folpet do not support tumors arising from genotoxicity and transformation of villous enterocytes.
2.1.2. Temporality
For the purpose of this AOP description, we have separated the cytotoxicity MIE from sustained crypt cell proliferation/hyperplasia (KE#1, see below) by distinguishing exposures of ≤1 week from longer exposure studies. This is consistent with both the importance of temporality in any MOA discussion, as well as the aforementioned attempts to distinguish transient molecular interactions from sustained interactions via the introduction of terms like “pre-MIE” and “IME” to the AOP paradigm. Villous epithelial cytotoxicity can be indicated by necrosis or other degenerative changes, villous blunting, and reactive secondary processes such as inflammation or immune cell infiltrates. For Cr(VI), three studies have examined the duodenum following 1 week of exposure (Thompson, Proctor, et al. Citation2011, Citation2012; Thompson, Wolf, et al. Citation2015). Thompson, Proctor, et al. (Citation2011) exposed mice to 0.1 to 180 ppm Cr(VI) and found evidence of villous cytoplasmic vacuolization in 3/5 mice at 60 ppm and 5/5 mice at 180 ppm, as well as crypt hyperplasia and villous atrophy in 3/5 mice at 180 ppm in H&E-stained slides (). In the jejunum, cytoplasmic vacuolization of villous epithelium was observed in 1/5 mice at both 60 and 180 ppm. Subsequent analyses using Feulgen’s stain to visualize nuclear material found no evidence of micronuclei (MN) or karyorrhectic nuclei in duodenal crypts at these high Cr(VI) concentrations; however, these lesions were occasionally observed at the villous tips (O'Brien et al. Citation2003).
Table 1. Incidence of Cr(VI)-induced crypt hyperplasia in mice.
As further evidence of acute villous toxicity, a subsequent Good Laboratory Practice (GLP)-compliant duodenal MN assay found that crypt enterocyte counts were increased from 39.9 ± 3.5 per crypt in control mice to 51.9 ± 2.8 per crypt at 21 ppm, and 67.1 ± 55 per crypt at 180 ppm (Thompson, Wolf, et al. Citation2015). Transcriptomic analyses have also indicated ≥2-fold increases in gene expression for the cell proliferation marker Ki67 in the duodenum at ≥20 ppm after one week of exposure (Kopec, Kim, et al. Citation2012; Rager et al. Citation2017; Thompson, Suh, et al. Citation2017).
Although previous studies with Cr(VI) did not report intestinal hyperplasia or villous atrophy in F344 rats exposed to up to 180 ppm Cr(VI) for 13 weeks or 2 years (NTP Citation2007 Citation2008), Thompson, Proctor, et al. (Citation2012) observed villous atrophy, enterocyte apoptosis, and crypt hyperplasia at 60 and 180 ppm in F344 rats exposed to Cr(VI) for one week. (These effects diminished somewhat by 13 weeks of exposure.) The effects in rats will be discussed further for KE#1 (below), but one interpretation of these findings is that F344 rats might have developed intestinal tumors in the NTP bioassay if there had been sustained severe tissue damage, consistent with that observed in mice.
Studies with captan show crypt hyperplasia and villous epithelial hypertrophy in male and female mice after only 1 week of exposure (Gordon et al. Citation2012). As discussed below, these effects persisted with longer exposures and dissipated after cessation of exposure. Unpublished studies with captan also report increased crypt hyperplasia in mice exposed to 3000 ppm in diet after only 3 days of exposure, with 100% incidence by day 7 (). Notably, hyperplasia was not yet evident at 1 day after exposure. Duodenal epithelial erosion and degeneration, but not crypt hyperplasia, were also seen 24 h after a single high (1430 mg/kg) bolus dose of folpet in mice (JMPR Citation2004a). The extensive WOE supporting villous enterocyte cytotoxicity after Cr(VI), captan, or folpet exposure has been critically reviewed elsewhere (Wilkinson et al. Citation2004; JMPR Citation2004a, Citation2004b; Cohen et al. Citation2010; Thompson et al. Citation2013; Health Canada Citation2016).
Table 2. Incidence of folpet- and captan-induced crypt hyperplasia in mice*.
In summary, there is consistent and direct cellular and histopathological evidence of villous enterocyte cytotoxicity as early as 1 to 7 days after oral exposures to a chemical stressor, based on evidence from Cr(VI), captan, and folpet exposures at tumorigenic concentrations. Extended exposure durations do not appear to appreciably impact the magnitude of the cytotoxic response, potentially due to the efficient regenerative and repair capacity of the intestinal epithelium.
2.2. KE#1: sustained crypt cell proliferation/hyperplasia (organ level)
As mentioned above, we selectively separate the villous enterocyte cytotoxicity (MIE) from sustained crypt cell proliferation/hyperplasia (KE#1) by considering exposures longer than 1 week as evidence for sustained crypt hyperplasia. Herein, we define sustained crypt hyperplasia as clear evidence of an elongated crypt compartment and potentially altered villus structure. In the case of Cr(VI), crypt hyperplasia has been observed at 1 week, 4 weeks, 13 weeks, and 104 weeks of exposure. In addition to diagnosis of crypt hyperplasia on histopathological evaluation, the number of crypt enterocytes and crypt length (or depth) have been quantified and shown to increase after 1 and 13 weeks of exposure (O'Brien et al. Citation2003; Thompson, Seiter, et al. Citation2015). Thus, qualitative and quantitative analyses demonstrate a dose and temporal progression, with crypt hyperplasia occurring after 7–90 days of exposure to carcinogenic concentrations of Cr(VI) (), and then at 2 years by qualitative histopathological evaluation (). Similarly, a temporal progression of crypt hyperplasia occurs after 3–90 days of exposure to carcinogenic concentrations of captan and folpet (, ). Crypt hyperplasia is evident as early as 3 days (but not 1 day) of dietary captan exposure at 3000 ppm in mice (JMPR Citation2004b).
Figure 3. Qualitative and quantitative evidence for sustained crypt hyperplasia following exposure to Cr(VI). Full transverse sections indicate the extent of villous blunting in mice exposed to 180 ppm Cr(VI) for 90 days (B) relative to controls (A). Higher magnification demonstrates the extent of crypt elongation in exposed mice (D) relative to controls (C). The arrows demonstrate blunting of villi, and the brackets indicate crypt expansion. (E) Cells per crypt and the incidence of crypt hyperplasia in mice exposed to various concentrations of Cr(VI) for 7 days. (F) Cells per crypt and the incidence of crypt hyperplasia in mice exposed to various concentrations of Cr(VI) for 90 days. Adapted from Thompson, Seiter, et al. (Citation2015); Thompson, Wolf, et al. (Citation2015); Thompson, Suh, et al. (Citation2017).
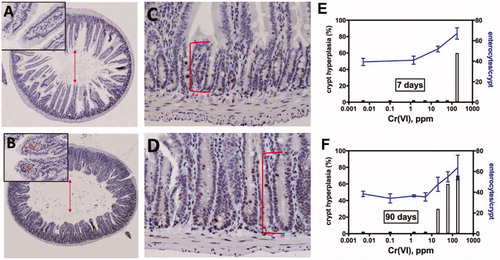
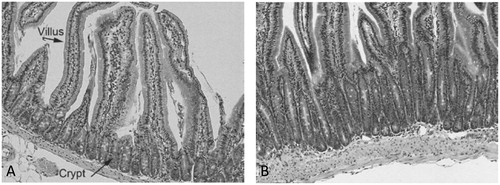
Figure 4. Qualitative evidence for sustained crypt hyperplasia following exposure to captan for 28 days. (A) Representative transverse section of control mouse SI. (B) Representative transverse section of SI in a mouse exposed to 6000 ppm folpet in diet. Folpet-induced short term cytotoxic and proliferative changes in the mouse duodenum, Gordon et al., Toxicology Mechanisms and Methods, reprinted by permission of Informa UK Limited, trading as Taylor & Francis Group, www.tandfonline.com
Increased cell proliferation in the mouse duodenal mucosa, measured as an increase in labeling indices for either bromodeoxyuridine (BrdU) or proliferating cell nuclear antigen (PCNA), is seen after folpet exposures of unspecified duration (as reviewed by Cohen et al. Citation2010). The PCNA labeling index was higher in the mouse duodenum after dietary captan exposure to 6000 ppm for 28, 56, or 91 days; this effect was most pronounced at 28 days and diminished thereafter (as reviewed by IPCS, 1996). Increased Ki67 mRNA expression was observed in the mouse duodenum after 7 and 90 days of exposure to ≥60 ppm Cr(VI) (Thompson et al. Citation2013).
OECD (Citation2018) states that reversibility of a KE is direct evidence of its essentiality and suggests that it is necessary but not sufficient to result in the AO. When sustained hyperplasia is a KE, it is difficult for reversibility to be proven essential unless animals are exposed to an agent for some length of time, exposure removed, and then animals monitored for aberrant cell proliferation or tumor formation several months after exposure. Such studies have been conducted for captan, where it was concluded that ≥6 months of dosing was necessary to induce tumors, but that diffuse hyperplasia would resolve after several months without exposure (Wilkinson et al. Citation2004). In these studies, 6-, 12-, or 18-month studies with dietary captan at 6000 ppm demonstrated that hyperplasia and tumors decreased to almost control levels after a 6-month recovery period. The epithelium in some of the recovery animals, however, differed from the concurrent controls; while incidence of epithelial hyperplasia decreased, the SI exhibited muscular hypertrophy and submucosal fibrosis and ectasia of submucosal vessels. These lesions appeared to be resolving foci of focal hyperplasia in which the extent of epithelial repair exceeded the repair of mesenchymal tissues.
These types of stop-dose studies are relatively uncommon. More typical short-term studies with a 2- to 4-week recovery period can show reversibility but do not fully address essentiality in tumor formation. Rather, these assays can be useful to demonstrate that permanent changes, possibly indicative of cell transformation, have not been induced in the tissue. In this regard, one published MOA for Cr(VI)-induced SI tumors lists mutational events prior to cell proliferation and hyperplasia (McCarroll et al. Citation2010), which seems to suggest that the observed hyperplasia is a result of genetic damage leading to dysregulated cell growth. If true, then stop-dose studies should not exhibit some level of recovery after dosing ceases. When Gordon et al. (Citation2012) exposed ICR mice to 6000 ppm folpet in diet for 4 weeks with and without a 2-week recovery, the incidence of crypt hyperplasia was reduced by ∼50% in the recovery group (). The authors speculated that a longer recovery period would have resulted in complete reversal of the hyperplasia. Gordon et al. (Citation2012) also concluded that these “early histologic changes support a non-DNA reactive mode of action for folpet carcinogenicity.” Thompson, Wolf, et al. (Citation2017) conducted a 4-week recovery study with the highest carcinogenic concentrations of Cr(VI), captan, and folpet used in their respective bioassays. Similar reductions in the incidence (∼50%) and severity of intestinal lesions were observed as in Gordon et al. (Citation2012) for all three agents.
Notably, Cr(VI), captan, and folpet do not induce SI tumors in rats. In the 2-year bioassay for Cr(VI), rats did not develop SI tumors, and the only reported non-neoplastic lesion in the rat SI was histiocytic infiltration. Crypt hyperplasia and villous atrophy/blunting were not noted in rats (NTP Citation2008; Stout et al. Citation2009; Witt et al. Citation2013), but it should be recognized that tissues not exhibiting tumors in bioassays may receive less scrutiny in evaluation of non-neoplastic lesions. As part of a comparison of mouse and rat SI lesions between the NTP bioassays and those in the studies conducted by Thompson et al., it was recognized that rats in the NTP bioassays exhibited some signs of crypt hyperplasia and villous damage (Cullen et al. Citation2016). This reevaluation corroborated earlier reports of such lesions in F344 rats exposed to Cr(VI) (Thompson, Proctor, et al. Citation2012). Overall, Cullen et al. concluded that the severity of SI lesions was much greater in mice than rats. In a more recent study, albino rats were exposed to “sodium dichromate” at 3 mg/ml (or 3000 ppm) for 30–60 days (Zainab et al. Citation2016). This dose is ∼1200 ppm Cr(VI), which is ∼6 times higher than the highest concentration in the 2-year Cr(VI) cancer bioassay (i.e. 180 ppm). The animals were administered doses of 3 or 9 mg/100 g body weight, or ∼2.4 and 7 mg Cr(VI)/kg body weight.Footnote2 While these mg/kg doses were comparable to those estimated from drinking water studies, the administered gavage concentration of Cr(VI) in the test article was extremely high. Zainab et al. (Citation2016) focused solely on villous effects, and reported that atrophy/blunting was clearly evident in the groups exposed for 60 days.
Taken together, these data indicate that the AOP might also be relevant to rats receiving sufficiently high concentrations of a chemical stressor to induce severe chronic cytoxicity and regenerative hyperplasia. The top concentration in the 2-year rat bioassay (180 ppm Cr(VI) or 17 mg Cr(VI)/kg-day in males and 20 mg Cr(VI)/kg-day in females) was selected based on reductions in body weight, water consumption, and increased glandular stomach ulceration at higher concentrations in shorter-term assays, whereas the top concentration in the mouse 2-year bioassay was selected based on reductions in body weight and water consumption in shorter-term assays (NTP Citation2008). Notably, the transit rate from the stomach to the duodenum is ∼4 times slower in rats than mice (Kirman et al. Citation2012), which might not only increase time for reduction of Cr(VI) to Cr(III) in the stomach but also increase the residence time of Cr(VI) to irritate the stomach mucosa of rats as compared to mice. If not for stomach irritation (or palatability issues), rats might have tolerated higher Cr(VI) concentrations in the NTP bioassay, potentially resulting in greater intestinal damage and intestinal tumor formation. Transcriptomic analyses indicate that mice and rats show similar molecular responses in the duodenum at comparable Cr tissue concentrations, but that mice ultimately achieved higher tissue concentrations of Cr (Kopec, Thompson, et al. Citation2012; Thompson et al. Citation2016).
In summary, there is consistent and direct cellular and histopathological evidence of crypt cell proliferation/regenerative hyperplasia occurring in mice as early as 1 to 7 days after oral exposure to a chemical stressor, based on empirical evidence from Cr(VI), captan, and folpet exposures at tumorigenic concentrations. Crypt hyperplasia must be sufficiently sustained across dose and time to result in tumorigenesis. Reversibility studies suggest that sufficient daily exposure durations may exceed 6 months, based on limited evidence of near complete recovery of crypt hyperplasia after exposures to tumorigenic doses for at least 6 months. Cr(VI) appears to induce milder KE phenotypes in rats, that do not progress to SI tumors. Thus, this AOP is specific to SI tumors in mice, as the AO has not been fully observed in rats exposed to Cr(VI), captan, or folpet.
2.3. KE#2: mutation/transformation (organ level)
It is well accepted that each round of cell division contains a small probability of spontaneous mutation (Moolgavkar and Knudson Citation1981; Greenfield et al. Citation1984; Cohen and Ellwein Citation1990; Tomasetti and Vogelstein Citation2015). In the normal SI, epithelial cell proliferation is limited to the crypt compartment. Intestinal stem cells reside near the base of the crypt, and divide asymmetrically to give rise to daughter cells that move through the transit-amplifying region of the crypt and ultimately onto the villi, all within a span of 2–3 days in rodents and 3–6 days in humans (van der Flier and Clevers Citation2009; Greaves Citation2012). It is conceivable that under high proliferative pressure, some villous enterocytes may not be fully differentiated upon reaching the villus and thus might occasionally undergo cell division. However, the high turnover rate of villous epithelium, exacerbated by the toxic assault from the lumen and the migratory pressure from the crypt, would most likely ensure rapid death and/or sloughing of such cells. Although dedifferentiation of villous enterocytes has been demonstrated experimentally in mice, these were genetically engineered mice harboring dual activating mutations in both Wnt/β-catenin and NF-κB signaling pathways (Schwitalla et al. Citation2013). As discussed in detail by Thompson, Seiter, et al. (Citation2015), reprogrammed/de-differentiated villous enterocytes can form crypt-like foci on villi (Schwitalla et al. 2013) and no such foci were observed in the Thompson, Seiter, et al. (Citation2015) study, nor in other Cr(VI) drinking water studies (NTP Citation2007, Citation2008; Stout et al. Citation2009). Thus, while dedifferentiation of villous enterocytes is possible, the prevailing theory on intestinal cancer is that mutations occur/accumulate in crypt stem cells (Potten and Loeffler Citation1990; Barker et al. Citation2009). For example, Barker et al. (Citation2009) have shown that controlled deletion of the adenomatous polyposis coli (Apc) gene in crypt stem cells results in rapid transformation and adenoma formation with the stem cell remaining in the base of the crypt, whereas targeted Apc deletion initiated within the transit amplifying daughter cells leads to stalled microadenomas. Beyond the SI, it is generally recognized that DNA damage or spontaneous mutations leading to transformation must occur in progenitor/stem cells (Wood et al. Citation2015).
Based on prevailing data, we believe that the most likely path to SI tumors is through mutation of crypt stem cells, whether induced by direct DNA damage or secondary to errors during normal DNA replication. Intestinal stem cells (i.e. Lgr5+ cells) are reported to divide once per day, thereby undergoing several hundred divisions in a mouse lifespan (Barker et al. Citation2008). In mice, each crypt has approximately six stem cells (Barker et al. Citation2009; van der Flier and Clevers Citation2009), indicating that each crypt normally undergoes thousands of stem cell divisions in a lifetime. Under sustained proliferative pressure, it is conceivable that stem cells undergo more asymmetric replications over time, or possibly undergo more symmetric divisions to create additional stem cells within the crypt. Either way, sustained crypt hyperplasia is likely accompanied by a significant increase in stem cell divisions and thus potential for mutation and transformation. This effect is evident histologically as elongation of the crypt, which is potentially associated with an increase in the number of stem cells, all dividing at the usual rapid rate of the SI. The results with Apc deletion indicate that a very few specific mutations are needed for SI neoplasia, and it has been reported that perhaps only two or three driver mutations are required to induce colorectal and other cancers in humans (Tomasetti et al. Citation2015).
Current genotoxicity testing paradigms do not contain methods/assays for measuring spontaneous mutations that arise from prolonged increases in cell proliferation. Notably, the standard duration for an OECD guideline transgenic rodent (TGR) mutation assay is 28 days in a rapidly proliferating tissue, and long-term exposures are not recommended (OECD Citation2013). An apparent requirement for KEs in the AOP framework is that KEs are measurable. This generally implies that positive results are necessary to support the role of each KE. We propose a logical extension of this concept of measurability to include acknowledgement that measured negative findings are, in fact, critical indirect evidence that can support a KE.
SI tumors provide a unique case for genotoxicity testing within an AOP or MOA framework. An international expert working group on genotoxicity testing has stated that in vivo genotoxicity assays should ideally be conducted in (1) tissues that receive high doses of the active parent compound or metabolite, (2) the site of carcinogenic action, and (3) a highly proliferative tissue (MacGregor et al. Citation2015). The duodenum fulfills all of these criteria for Cr(VI), captan, and folpet, and negative results in well-conducted genotoxicity assays should thus be considered as strong evidence against early (i.e. upstream) genotoxic initiating events and strong support for downstream KEs related to spontaneous mutation and transformation. As such, the AOP shown in requires both positive evidence for KEs related to cytotoxicity and regenerative hyperplasia as well as negative genotoxicity results in the duodenal crypt cells. On the other hand, positive genotoxicity results in the duodenal crypt cells would make it difficult to rule out genotoxicity as an early upstream KE, and likely indicate involvement of an alternative AOP – perhaps involving direct DNA damage.
Assays directly measuring genotoxic responses in the duodenum following exposure to Cr(VI), captan, and folpet have resulted in negative findings. As shown in , all three carcinogens have negative MN results (Chidiac and Goldberg Citation1987; Gudi and Krsmanovic Citation2001; O'Brien et al. Citation2003; Thompson, Wolf, et al. Citation2015). Multiple TGR assays for Cr(VI) have reported no changes in mutation frequency (MF) in the duodenum or oral mucosa (Thompson, Young, et al. Citation2015; Citation2017; Aoki et al. Citation2019), and an unpublished TGR assay for captan was negative for mutation in the target tissue, thereby prompting regulators in Japan to develop threshold toxicity values for captan (FSCJ Citation2017).
Table 3. Summary of in vivo duodenal genotoxicity assays for Cr(VI), captan, and folpet.
While in vivo evidence of genotoxicity is negative for Cr(VI), captan, and folpet, all three are positive for genotoxicity in vitro (De Flora et al. Citation1984; Arce et al. Citation2010; Zhitkovich Citation2011). Captan and folpet have also been reported to bind to DNA associated proteins. Cr-DNA adducts in homogenized duodenal tissue following Cr(VI) exposure have been reported, albeit with potential ex vivo contamination during processing (O'Brien et al. Citation2003). However, XRF mapping of Cr fluorescence in intact fixed duodenal sections indicates that the Cr-DNA binding would be almost entirely limited to the intestinal villi, i.e. committed non-proliferative and non-pluripotent enterocytes (). As for other Cr(VI)-related DNA lesions, Cr(VI) does not induce DNA-protein crosslinks (DPC) or oxidative DNA damage in the duodenum (De Flora et al. Citation2008). Overall, direct DNA binding capability does not preclude this AOP from being operational for some agents, provided that there is sufficient evidence against in vivo genotoxicity.
With regard to the importance of assessing in vivo genotoxicity, it is well documented that in vivo genotoxicity tests in rodent species are prescribed as follow up assays for chemicals that have positive in vitro genotoxicity results, and that negative in vivo results are generally interpreted as indicating that the chemicals of interest are safe for humans (e.g. EFSA and OECD). In vivo blood MN assays and either a TGR or comet assay are widely recommended as follow up in vivo studies. In addition to the aforementioned duodenal MN assays, blood MN assays are generally negative for Cr(VI), captan, and folpet (Arce et al. Citation2010, and Thompson, Suh, et al. Citation2017). Recent recommendations by the International Working Group on Genotoxicity Testing concluded that the TGR and comet assays were essentially equivalent for detecting genotoxicity when one of these studies were coupled with an in vivo MN assay (Kirkland et al. Citation2019). In addition, they noted that the TGR assay was preferable for MOA investigation, especially when conducted in target tissues. It is curious that negative in vivo follow up genotoxicity assay results (i.e. evidence of absence) are useful for determining safety in some areas of risk assessment, but are often viewed with skepticism (e.g. absence of evidence is not evidence of absence) when the same assays are negative in the target tissue and species where carcinogenicity is observed at extremely high and/or cytotoxic concentrations. Notwithstanding the above considerations, it should also be recognized that many risk assessors have interest in moving away from binary yes/no genotoxicity determinations to understanding that even some DNA-reactive compounds have thresholds for inducing genotoxicity (Johnson et al. Citation2014; Clewell et al. Citation2019).
In summary, there is strong evidence that early genotoxic events are not involved in the carcinogenic processes in the duodenum following exposure to Cr(VI), captan, or folpet. While positive results would indicate the potential for direct acting genotoxicity, consistent negative results should be viewed as supporting spontaneous mutation resulting from chronic increases in cell proliferation.
2.4. AO: SI tumors in mice (individual/population level)
Collectively, there are seven carcinogenicity bioassays in mice: three each for folpet or captan and one for Cr(VI), all of which have been reviewed elsewhere (U.S. EPA Citation2004, Citation2012; Wilkinson et al. Citation2004; Cohen et al. Citation2010; Health Canada Citation2016; FSCJ Citation2017, Citation2019). Duodenal tumors are seen after lifetime exposure of B6C3F1 mice to Cr(VI), captan, or folpet. Captan and folpet exposure were also associated with duodenal tumors in CD-1 mice. Folpet resulted in a higher incidence of jejunal tumors in mice at very high doses (12 000 ppm) compared to the LOAEL for duodenal tumors (5000 ppm; FSCJ Citation2017), while Cr(VI) was also associated with a low incidence of jejunal tumors at 180 ppm Cr(VI) (NTP Citation2008). The proximal jejunum is the continuation of the SI from the duodenum, and tumors arising in it would be expected to have the same MOA as those in the duodenum. Of the three identified bioassays for folpet, only one did not report SI tumors; however, the exposure levels tested were lower than in the positive studies. In this study, the highest tested concentration was 1350 ppm, which is comparable to the lowest concentration in the other two studies, in which the duodenal tumor LOAEL was 1000 or 5000 ppm. Thus, the lack of tumors in this study of folpet is attributed to study design (i.e. dose selection) rather than an inconsistency in the WOE.
For Cr(VI), the duodenal tumors had a long latency considering that the MIE and regenerative hyperplasia were evident as early as 1 week of exposure (O’Brien et al. Citation2013; Thompson, Proctor, et al. Citation2011). In mice that died prior to scheduled sacrifice, the first incidence of adenoma or carcinoma occurred at 451 days (∼15 months) or terminal sacrifice at 729 days (∼18 months), respectively, in males, or 693 days (∼23 months) or 625 days (20 months), respectively, in females (NTP Citation2008). No information to assess the latency of duodenal tumor formation were identified for captan or folpet. As discussed above, SI tumors were not seen in any tested rat strain to date, including F344, CD, SD, Wistar, and/or Osborne-Mendel rats, after lifetime exposure to Cr(VI), captan, or folpet. For Cr(VI), detailed investigations indicted that rats exhibited similar albeit milder intestinal effects compared to mice and without tumor formation.
In summary, there is consistent and direct histological evidence of SI tumors in mice, but not rats, orally exposed to comparable tumorigenic doses of chemical stressor for a lifetime. The AO was concurrent with or preceded by each of the AOP KEs in a dose and/or temporal fashion. There is no evidence of the AO occurring in mice at exposure conditions not associated with measured KEs in the AOP.
2.5. Stressors
According to OECD (Citation2018), stressors are chemical or non-chemical (e.g. genetic and environmental) factors that might trigger an AOP. The AOP described herein shares features with celiac disease, which itself has different origins (Dickson et al. Citation2006; Kamboj and Oxentenko Citation2017). Celiac disease, which typically manifests as gastrointestinal distress, is believed to result from dietary gluten-induced inflammatory responses in the proximal SI. Definitive diagnosis requires duodenal biopsies interpreted in context of other factors. Grading of the disease is based on histopathological characteristics progressing from increased intraepithelial lymphocytes, to increased intraepithelial lymphocytes with mild crypt hyperplasia, intraepithelial lymphocytes with crypt hyperplasia and villous blunting, and increased intraepithelial lymphocytes with normal crypt depth but flattened mucosa (Dickson et al. Citation2006). Many drugs induce similar pathology albeit without involvement of intraepithelial lymphocytes; for example, metabolites of non-steroidal anti-inflammatory drugs (NSAIDs) excreted in the bile are thought to have direct toxic effects in the SI (Kamboj and Oxentenko Citation2017). Captan and folpet, but not Cr(VI), are reported to increase intraepithelial lymphocytes in the mouse duodenum.
Viral stressors also provide important and well-established insight into the AOP described herein. Cytopathic viruses, such as coronavirus infection in piglets, have a tropism for mature enterocytes, where infection leads to blunting of villi due to death of mature enterocytes and compensatory expansion of the proliferative compartment (i.e. hyperplasia of the crypt epithelium) in response to the demand for additional mature enterocytes (Uzal et al. Citation2016). These changes are essentially the same features seen following exposure to Cr(VI) and captan/folpet in mice. Rotavirus similarly infects and damages differentiated enterocytes of villous tips leading to crypt proliferation (Zou et al. Citation2018). Parvoviruses, in contrast, have a tropism for crypt cells. Infection kills the crypt cells and villi become blunted due to the lack of replacement of mature enterocytes, but without evidence of crypt hyperplasia during the acute infection phase. Parvovirus-induced injury is termed “radiomimetic” because the infection reproduces the effect seen in cases of radiation exposure where the rapidly mitotic cells are killed more readily than others, resulting in villous blunting from death of the crypt epithelium (Uzal et al. Citation2016). These viruses provide a model for distinguishing different pathways to a similar histologic picture. In the first pathway, damage to mature enterocytes leads to villous blunting and crypt hyperplasia, while in the second pathway, damage to crypt cells also leads to villous blunting but occurs without evidence of crypt hyperplasia.
In addition to chemical and viral stressors, there is evidence for physical damage to villi from digesta. For example, digesta containing abrasive grasses and sedges can induce villous damage (e.g. blunting) in rodents (Wieczorek et al. Citation2015), and dietary fiber can alter proliferative characteristics of the rodent SI (Greaves Citation2012). In an unpublished pilot study, mice exposed to 180 ppm Cr(VI) for 28 days had higher severity scores when fed the same chow in pellet form than meal form, despite measured Cr levels being slightly higher in meal-fed mice than pellet-fed mice (). Considering further that water intake (and thus water content in digesta) is reduced by up to 30% in rodents exposed to high concentrations of Cr(VI) (NTP Citation2007, Citation2008; Thompson, Proctor, et al. Citation2011, Citation2012; Thompson, Wolf, et al. Citation2015, Citation2017; Thompson, Young, et al. Citation2015), there may be a synergy between chemical and physical damage to the intestinal villi.
Similarly, it has been proposed that duodenal tumors induced by hydrogen peroxide (HP) in drinking water are due, in part, to physical stress resulting from ∼50% reductions in water intake due to poor palatability (DeSesso et al. Citation2000). Ito et al. (Citation1981, Citation1982) reported duodenal tumors in mice but not rats after exposure to 0.4% HP, along with duodenal hyperplasia. Glandular stomach erosion was also reported, indicating that these concentrations were damaging to the gastric mucosa. Notably, Ito et al. (Citation1982) discuss the observed intestinal lesions as similar to those that were previously hypothesized to have “arisen through continuing multiplication of crypt epithelial cells, combined with a failure of their differentiation into mature villus cells.” Subsequent work demonstrated a correlation between the tumorigenic potency of HP in different strains of mice and the amount of duodenal catalase activity (Ito et al. Citation1984). These findings indicate an important role for HP detoxification in the MOA for HP-induced duodenal tumors, and potential involvement of oxidative stress in cytotoxicity and perhaps genotoxicity. An OECD GLP-compliant 90-day drinking water study with HP later showed complete reversibility of duodenal hyperplasia following a six week recovery time (Weiner et al. Citation2000). Based on their results as well as unpublished genotoxicity findings, Weiner et al. posited that “any long term effects of HP are related to an irritation response, rather than genotoxic response.” Unfortunately, many of these cited HP studies did not include images of the non-neoplastic duodenal lesions, thus making it difficult to compare the lesions to Cr(VI), captan, and folpet. Overall, the potential interactions between chemical and physical stress on duodenal histopathology deserve more investigation.
Chappell et al. (Citation2019) demonstrated similarities in the transcriptomic responses in the duodenum among mice exposed to tumorigenic concentrations of Cr(VI), captan, or folpet for 28 days which could serve as biomarkers for identifying other stressors that might operate via this AOP. Gene-level comparisons identifying 126/546 (23%) differentially expressed genes altered in the same direction and 25 upregulated pathways. These changes related to cellular metabolism, stress, inflammatory/immune cell response, and cell proliferation, including upregulation in HIF-1 and AP1 signaling pathways, which have been linked to intestinal injury and angiogenesis/carcinogenesis.
2.6. Alternative modes of action
To date, two published articles have proposed a mutagenic MOA for SI tumors from Cr(VI) exposure (McCarroll et al. Citation2010; Zhitkovich Citation2011). No alternative MOAs were found for captan or folpet, although the issue of DNA reactivity was discussed at length in Cohen et al. (Citation2010) and Arce et al. (Citation2010). McCarroll et al. (Citation2010) describe a mutagenic MOA for Cr(VI) in the mouse SI based on available evidence prior to the initiation of much of the research described herein. Much of the evidence was comprised of non-target tissue data and in vitro studies. Evidence for a mutagenic MOA was based on results from a comet assay showing dose-dependent increases in comet tail length in leukocytes after 1 day of Cr(VI) exposure at gavage doses as low as 0.6 mg/kg-day (Dana Devi et al. Citation2001). Notably, achieving this dose via gavage implies the administration of ∼60 ppm Cr(VI). As discussed above, Cr(VI) does not appear to reach or cause DNA damage within the duodenal crypt compartment after prolonged drinking water exposures up to 180 ppm. McCarroll et al. (Citation2010) did not differentiate potential effects in the crypt and villus, implying that the location of DNA damage within the SI tissue structure was not considered. Finally, McCarroll et al. (Citation2010) considered the primary data gap supporting a cytotoxicity-mediated MOA to be the lack of histological evidence of cytotoxicity in either the 90-day or 2-year rat or mouse NTP (Citation2007, Citation2008) bioassays. Similarly, Zhitkovich (Citation2011) states that “no overt toxicity (necrosis) was found in the SI of any Cr(VI)-exposed groups of mice and rats.” Both McCarroll et al. (Citation2010) and Zhitkovich (Citation2011) did not integrate the intestinal histopathology and biology described herein when making their MOA conclusions.
Due to reactivity of Cr(VI) with GSH and ascorbate, it was initially hypothesized that oxidative stress might play a prominent role in the MOA (Thompson, Haws, et al. Citation2011). Although there is some transcriptomic evidence for this idea, such as induction of genes involved in GSH synthesis (Kopec, Kim, et al. Citation2012), oxidative DNA damage has not been observed in the mouse duodenum following Cr(VI) exposure in drinking water at up to 180 ppm for up to 9 months (De Flora et al. Citation2008; Thompson, Proctor, et al. Citation2011). Interestingly, recent in vivo mutation studies with potassium bromate (KBrO3) have provided evidence that oxidative stress can result in increased MF in gpt delta mice (Aoki et al. Citation2020). KBrO3 induces tumors in the proximal SI (and other organs), and Aoki et al. (Citation2020) have shown that KBrO3 increased oxidative DNA damage and G to T transversions in a threshold manner consistent with an observed threshold in MF in the duodenum. These findings not only provide insight into the MOA for bromate-induced SI cancer in mice but also demonstrate that the gpt delta TGR model might have detected increases in MF for Cr(VI) if increased oxidative stress were a driver in the MOAs. Even with an increase in MF, Aoki et al. (Citation2020) proposed a threshold no-observed-genotoxic-effect-level level for KBrO3 protective of SI cancer.
It should be pointed out that no agents have definitively been identified as causing mouse (or human) SI tumors via a mutagenic MOA. Genotoxic carcinogens such as x-ray irradiation (XR), benzo(a)pyrene (BaP), n-methyl-N-nitrosourea (MNU), and dimethylhydrazine (DMH) have caused mouse SI tumors after oral exposure among multiple other local and systemic tumor sites which were affected at higher rates and/or lower doses (IARC Citation1978, Citation2012b, Citation1999a, Citation1999b). The SI tumors were observed after short exposure durations (e.g. days or weeks,) and latencies (e.g. <6 months). Key information for assessing a MOA in the SI for XR, BaP, and MNU include a dose-related increase in nuclear aberrations (NA) in the duodenal crypt after a single gavage dose (Goldberg and Chidiac Citation1986), providing evidence for a mutagenic MIE in the target tissue. DMH caused duodenal NA after intraperitoneal exposure and was not tested via gavage exposure. These studies were not designed to examine intermediate KEs in the SI, given the relative insensitivity of the SI, in terms of dose and/or incidence, compared to other tumor sites. These agents also affect a wider variety of tissues and at higher tumor rates and/or lower doses compared to SI tumors and with relatively short exposure durations and latency for SI tumors. Given these distinctions, data for Cr(VI) alone (or together with captan and folpet) would be characterized as moderate to strong “counter evidence” in a mutagenic MOA, in contrast to stressors like XR, BaP, NMNU, and DMH.
3. Overall assessment of the AOP
According to OECD (Citation2018) guidance, the Overall Assessment Summary addresses the WOE for the AOP as a basis to consider appropriate regulatory application (e.g. priority setting, testing strategies, or risk assessment). The assessment of the overall confidence in an AOP and its associated KEs is based primarily on the “essentiality” of the KEs, “biological plausibility” of the KERs, and the “empirical support” for the KERs (OECD Citation2018). A summary of the evidence supporting Essentiality (), Biological Plausibility (), and Empirical Support () has been tabulated, as recommended by OECD. Based on these criteria, the overall confidence in the AOP is considered high due to well-developed and broadly accepted evidence streams.
Table 4. Support for essentiality of key events (KEs).
Table 5. Support for biological plausibility of key event relationships (KERs).
Table 6. Empirical support for key event relationships (KERs).
Various regulatory authorities have accepted a cytotoxicity-mediated regenerative hyperplasia-based MOA for Cr(VI), captan, and folpet (U.S. EPA Citation2004, Citation2012; Health Canada Citation2016; FSCJ Citation2017, Citation2019). For Cr(VI), Health Canada (Citation2016) noted that no studies have identified the essentiality of early KEs (i.e. studies measuring the presence or absence of tumors after one of the early KEs, such as cytotoxicity, is prevented). However, there is evidence of reversibility in shorter-term bioassays, an absence of genotoxicity in the target tissue, and consistent and reproducible non-neoplastic lesions in the duodenum. While no major gaps in the MOA were identified, Health Canada (Citation2016) noted that additional measures might provide more detailed mechanistic understanding of (1) Cr(III) and Cr(VI) speciation in biological samples, (2) oxidative status (in addition to GSH/GSSG) in SI epithelial tissues, (3) differentiation of chromium–DNA adducts between crypt and villi enterocytes in vivo, and (4) DNA methylation. Health Canada (Citation2016) noted that although this type of information might provide useful context for chromium toxicity, it would not change the non-mutagenic MOA. Health Canada (Citation2016) considered the lack of mutagenicity in target tissues to be a major weakness in the WOE supporting a mutagenic MOA. No significant gaps of weaknesses in the MOA were noted by regulators for captan or folpet. Overall, while there are no perceived significant gaps or weaknesses in the KEs or KERs supporting this AOP, identification of the exact molecular mechanisms underlying cytotoxicity as the (M)IE would better conform to the AOP framework; however, it is not clear to these authors that cytotoxic MOAs/AOPs necessarily have a single MIE.
3.1. Biological domain of applicability
According to OECD (Citation2018), the biological domain of applicability for an AOP is defined based on the groups of organisms for which the measurements represented by the KEs and KERs are operative. For Cr(VI) captan, and folpet, there appear to be no obvious sex differences in susceptibility. There are inadequate data to examine potential differential susceptibility based on life-stage, as nearly all of the identified rodent studies have commenced exposure at 6–8 weeks of age. While there is no evidence of the AO in rats, we do note evidence for milder non-neoplastic KEs in rats, and data suggest that the entire AOP might be relevant to rats under higher exposure conditions.
With respect to humans, the relevance of this MOA/AOP in relation to Cr(VI) or folpet exposure has been previously reviewed (Cohen et al. Citation2010; Thompson et al. Citation2013) and is updated here, again using the World Health Organization/International Programme on Chemical Safety (WHO/IPCS) human relevance framework, which addresses the following fundamental criteria: (1) Is the WOE sufficient to establish the MOA in animals? (2) Can human relevance of the MOA be reasonably excluded on the basis of fundamental, qualitative differences in KEs between experimental animals and humans? (3) Can human relevance of the MOA be reasonably excluded on the basis of quantitative differences in either kinetic or dynamic factors between experimental animals and humans and (4) Conclusion: Statement of confidence, analysis, and implications (Boobis et al. Citation2008). These criteria are briefly discussed below.
(1) The WOE is sufficient to establish the AOP/MOA in mice and (2) the human relevance of the MOA cannot be reasonably excluded on the basis of fundamental, qualitative interspecies differences. However, based on Cr(VI), captan and folpet, cytotoxic exposure levels in mice (e.g. 30–180 ppm in drinking water for Cr(VI), 6000 to 16 000 ppm in feed for captan, and 1000 to 12 000 ppm for folpet) were required to be sustained for long periods of time (e.g. at least 6–12 months). For Cr(VI), the human equivalent dose (HED) in mg/kg for duodenal hyperplasia was estimated from mice to be 0.02 mg/kg-day (Thompson et al. Citation2018), which is equivalent to 0.54 ppm of Cr(VI) in drinking water daily for a lifetime, based on the 95th %-ile drinking water intake of 37 ml/kg-day (U.S. EPA Citation2019). This is 180-fold higher than the 95th %-ile Cr(VI) concentration in U.S. drinking water of 3 ppb. Importantly, the HED estimate only includes the induction of hyperplasia, which itself is not sufficient to induce SI cancer. In the NTP (Citation2008) cancer bioassay, mice exposed to 5–20 ppm Cr(VI) exhibited increased hyperplasia without SI tumor development. Again, there is a dose-dependent relationship between exposure and the level of hyperplasia (), as well as a KER between the amount of hyperplasia and tumor risk. Given the highly efficient homeostatic regulation of SI epithelial cell sloughing, regenerative proliferation, and repair, a mathematical or computational model is needed to quantitatively describe these KERs. This type quantitative AOP (qAOP) model requires detailed response–response relationship and incorporation of such compensatory responses to allow one to infer the magnitude or probability of an AO (Perkins et al. Citation2019).
(3) There are quantitative differences in kinetic factors between experimental animals and humans and also nonlinearity in the pharmacokinetic reduction of Cr(VI) to inert Cr(III) (Kirman et al. Citation2012, Citation2013, Citation2017). Although the gastrointestinal tract is generally similar between rodents and humans, species-specific differences in pH in various sections of the gastrointestinal tract influence the pH-dependent Cr(VI) reduction rate, as do feeding patterns and gastric motility (McConnell et al. Citation2008; Kirman et al. Citation2012, Citation2013). Physiologically based pharmacokinetic (PBPK) models integrating gastric transit and reduction collectively and consistently predict that a higher percentage of Cr(VI) may escape stomach reduction in mice relative to humans, which exhibit the highest extent of reduction at low doses compared to rats and mice (Kirman et al. Citation2012, Citation2013, Citation2017; Proctor et al. Citation2012; Sasso and Schlosser Citation2015). For example, nonlinear kinetics are predicted to begin in humans at doses >0.01 mg/kg-day or in mice and rats at doses >0.1 mg/kg-day, largely due to differences in reduction capacity (Kirman et al. Citation2017). Over the concentration range tested in the NTP bioassay (∼0.5–10 mg/kg-day), dose-dependent delivery of Cr(VI) from the stomach to the SI was predicted to rise from ∼30 to 70% in mice, ∼40 to 80% in rats, or ∼6 to 40% in humans (Kirman et al. Citation2017). Comparable PBPK models for captan or folpet were not identified; however, aggregate exposures to captan or folpet through food or drinking water are, similar to Cr(VI), at least three orders of magnitude lower than exposures causing SI tumors and thus not a concern for SI carcinogenicity risk assessment (U.S. EPA Citation2004, Citation1999). (4) To conclude, in the context of the WHO/IPCS human relevance framework, the WOE is sufficient to establish the MOA in animals, with qualitatively plausible KEs in humans. However, the KEs become quantitively implausible in humans after accounting for interspecies toxicokinetic differences, as well as background levels of human exposure. Confidence in this AOP/MOA is high (as addressed later in Section 3.3) and the implications suggest that the AOP/MOA is unlikely to be quantitatively relevant to humans. As mentioned previously, the AOP/MOA may be relevant to rats, if sufficient exposures across dose and time are achieved, since the KEs (but not the AO) have been observed in rats exposed to Cr(VI).
The International Agency for Research on Cancer (IARC) has labeled captan as “not classifiable as to its carcinogenicity in humans” (IARC Citation1987) but has not classified folpet. IARC classified Cr(VI) as “carcinogenic to humans” based on lung and nasal sinus cancers in humans, with the underlying evidence derived from occupational and animal inhalation studies (IARC Citation2012a). IARC concluded that there was insufficient evidence of effects on gastrointestinal sites, mainly stomach (IARC Citation2012a). A recent comprehensive systematic review on epidemiological and animal evidence for Cr(VI) found no evidence for an increased risk of stomach cancer in humans (Suh et al. Citation2019), and an earlier meta-analysis of cancer risk among Cr(VI) exposed workers found very few cases among 36 studies included, and no evidence of an increased risk in SI tumors (Gatto et al. Citation2010).
In summary, the biological domain of applicability for this AOP is mice, as mice are the only species in which indices that represent the KEs and KERs, whether molecular, cellular, or tissue-based, were measured and found to be operative in a manner sufficient to result in the AO. A key criterion for defining an AO is its relevance for regulatory decision-making (OECD Citation2018). While the AOP is qualitatively plausible in humans (or rats), the exposure levels sufficiently high and prolonged to result in the AO in a species other than mice have not been observed or defined for any known chemical stressor. As mentioned previously, interspecies toxicokinetic differences in gastric transit and reduction of Cr(VI) as well as environmental exposure levels of Cr(VI), captan or folpet, in relation to carcinogenic exposures, suggest that this AOP is unlikely to be quantitatively plausible in humans.
3.2. Essentiality of the KEs
According to OECD (Citation2018), the essentiality of KEs can “only be assessed relative to manipulation of a given KE (e.g. experimentally blocking or exacerbating the event) on the downstream sequence of KEs…”. Essentiality and counterfactual studies are often difficult to conduct. For example, it would be difficult to conduct a study where Cr(VI) villous uptake is inhibited because a mouse model that is null for the requisite transporter(s) would likely have confounding effects as a result of the loss of the transporter. Nevertheless, presents data that inform essentiality of KEs to the extent that is feasible. For example, while studies inhibiting Cr(VI) uptake have not been conducted, the NTP has conducted a 2-year bioassay with trivalent chromium (Cr(III)). Despite much higher dietary exposures in feed relative to Cr(VI) doses, bioavailability of Cr(III) is much lower than that of Cr(VI) and no tumors or SI lesions were observed. Notwithstanding pharmacokinetic differences between Cr(VI) and Cr(III), the studies do support a requirement for villous uptake of Cr(VI). One can also view the lack of tumors further down the GI tract as evidence of the essentiality of villous cytotoxicity and absence of direct crypt exposure. The colon contains crypts (but not villi), yet no tumors were observed. The ileum contains villi, but measured Cr levels were lower than in the duodenum (Thompson, Proctor, et al. Citation2011), potentially indicating decreased ability to absorb Cr(VI) and thus initiate KEs.
Long-term (i.e. ≥6-month) exposure and recovery studies with captan have demonstrated the essentiality of sustained crypt hyperplasia in tumor formation, and by extension, more opportunities for spontaneous mutation and transformation leading to such tumors. The inability to detect induced mutations in the duodenum of transgenic mice exposed for up to 90 days to carcinogenic exposures to Cr(VI) in drinking water could be interpreted as evidence that crypt hyperplasia must similarly be sustained for prolonged durations to induce mutations leading duodenal tumors.
Previous WOE evaluations by others have judged the overall empirical support for this AOP, based on Cr(VI) data, to be moderate to high (strong) (Becker, Ankley, et al. Citation2015). “Biological plausibility” was considered high, while the evidence for “essentiality” was considered moderate due to the lack of reversibility studies. “Empirical support” for crypt hyperplasia increasing the potential for spontaneous mutations was considered moderate (i.e. indirect) since methodologies to measure spontaneous mutations are lacking. Relevant new data that have become available since this previous WOE evaluation help inform some of the identified data gaps. For example, as mentioned, no significant increase in gpt mutant frequency occurred in the duodenum of transgenic C57BL/6J mice exposed to tumorigenic Cr(VI) concentrations in both 28- and 90-day studies (Aoki et al. Citation2019). Further, comparable transcriptomic responses in the duodenum of mice exposed for 28 days to Cr(VI), captan or folpet provided additional molecular support for a cytotoxicity-mediated MOA (Chappell et al. Citation2019), consistent with gene expression pathways relevant to p53/DNA damage being enriched in vitro, but not after in vivo exposures for 13 weeks at tumorigenic concentrations (Thompson, Proctor, et al. Citation2012; Rager et al. Citation2017). The presence of Cr and elevated γ-H2AX immunoreactivity in villus tips, without aberrant foci indicative of transformation, after 13 weeks of exposure to tumorigenic concentrations in mice (Thompson, Seiter, et al. Citation2015), also provides further support for cytotoxicity-mediated mechanisms. The reversibility of early KEs in mice exposed to Cr(VI) (or captan or folpet) via drinking water for 28 days followed by a 28-day recovery period also provides additional direct evidence of the essentiality of the KEs (Thompson, Wolf, et al. Citation2017).
3.3. Evidence assessment and quantitative considerations for KERs
The biological plausibility, empirical support, and quantitative understanding from each KER in an AOP are assessed together in this portion of the AOP (OECD Citation2018). Biological plausibility of each of the KERs in the AOP is the most influential consideration in assessing WOE or degree of confidence in an AOP for potential regulatory application. While less influential than biological plausibility of the KERs and essentiality of the KEs (), empirical support can increase confidence in the relationships included in an AOP (). As such, OECD (Citation2018) considers that a quantitative weighting scheme may be useful to assess confidence in the AOP, based on the rank ordering of the relevant Bradford Hill considerations, and cites some proof-of- concept examples, described elsewhere (Becker, Ankley, et al. Citation2015; Collier et al. Citation2016; Becker et al. Citation2017).
Using the proposed quantitative scoring and weighting approach, the AOP and its proposed KEs were considered to be highly supported by (i.e. 95% confidence in) the WOE, when assessed toward the “evolved” Bradford Hill considerationsFootnote3 of biological plausibility, essentiality, and empirical support per OECD (Citation2018) guidance (). The primary considerations that impacted (i.e. lowered) the confidence score were the relatively few evaluations directly measuring the IE and the structural analogy (or lack thereof) of chemical stressors that operate via this AOP, as Cr(VI) has no chemical, physical, or structural analogy to captan/folpet, which are structurally related and share a common major metabolite.
Table 7. Evolved Bradford Hill causal considerations, defining questions, and body of evidence per OECD (Citation2018).
4. Conclusions
The utility of AOPs for regulatory application is defined, to a large extent, by the confidence and precision with which they facilitate extrapolation of data measured at low levels of biological organization to predicted outcomes at higher levels of organization and the extent to which they can link biological effect measurements to their specific causes (OECD Citation2018). This AOP proposes that duodenal tumors in mice occur secondary to chronic villous enterocyte cytotoxicity and regenerative repair-driven proliferation. The supporting KEs in this AOP have been measured at the molecular, cellular, and tissue level within the dose range and time course of the observed AO for Cr(VI), captan, and folpet, the reference chemical stressors. As such, the biological plausibility, essentiality, and empirical support for the IE and KEs was judged to be strong and to provide high confidence according to OECD (Citation2018) guidelines. Evidence for a primary mutagenic MOA for the reference chemicals was considered weak, given that positive responses were obtained either in vitro or in non-target tissues after high and/or non-physiological routes of exposure. Genotoxicity evaluations using exposure conditions consistent with the measured KEs and AO, including in the target tissue, were negative. Based on the overall WOE for this AOP and its associated KEs, it can be used for regulatory applications including hazard identification and risk assessment.
Supplemental Material
Download MS Word (13.9 KB)Acknowledgements
The authors greatly appreciate the valuable comments from reviewers selected by the Editor and anonymous to the authors which helped improve the manuscript.
Declaration of interest
The authors’ employment affiliations are shown in the title block. ToxStrategies is a private consulting firm providing services to private and public organizations on toxicology and risk assessment. Boehringer Ingelheim Pharmaceuticals, Inc. is a private pharmaceutical company. This work was supported by the Hexavalent Chromium Science Panel of the American Chemistry Council (ACC, https://www.americanchemistry.com/Hexavalent-Chromium/) after it was conceived and presented by ToxStrategies to this Panel in 2018, as it represented a data gap in the science for intestinal carcinogens. The ACC is America’s oldest trade association of its kind, representing more than 170 companies engaged in the production, manufacture and use of chemicals. Members of this Panel, under the direction and coordination of Ms. Eileen Conneely, were given the opportunity to review the draft manuscript to provide feedback to the authors regarding the clarity of the science presented but not on the interpretation of research findings. No comments were received or provided in any form. The contents of this manuscript reflect solely the view of the authors.
The project was funded through contracts between ACC and ToxStrategies. ToxStrategies is currently contracted with multiple chemical panels at ACC to provide scientific consulting support. ToxStrategies is also currently contracted by ACC on other projects involving the evaluation of the available science and a risk assessment for hexavalent chromium [Cr(VI)]. All the scientists at ToxStrategies (VB, MH, DP, CT) involved in the development of the current manuscript were provided salary compensation as part of their normal employment as scientific consultants. JC and EG were subcontracted by ToxStrategies. SC and CW graciously donated their time. SC receives support from the Havlik-Wall Professorship from the University of Nebraska Medical Center. EG formerly studied captan on folpet while employed by Makhteshim-Agan of North America, Inc.
There are no conflicts of interest for any of the authors to disclose related to the submission of this manuscript. None of the authors are currently engaged to testify as experts on behalf of the sponsors in litigation related to Cr(VI). CT, MH, and DP have participated in meetings with the EPA and other agencies, on behalf of ACC, to discuss the current state of the science for Cr(VI) and the need to consider mode of action data in a risk assessment for Cr(VI). It is anticipated that regulatory authorities will consider the contents of this review in making regulatory decisions regarding potential health effects of Cr(VI), captan or folpet.
Supplemental material
Supplemental material for this article is available online here.
Notes
2 Note: Zainab et al. (Citation2016) has several reporting deficiencies.
3 Considerations modified from those proposed by Bradford Hill (Hill Citation1965) for assessment of causality in epidemiological studies
References
- Aoki Y, Matsumoto M, Matsumoto M, Masumura K, Nohmi T. 2019. Mutant frequency is not increased in mice orally exposed to sodium dichromate. Food Saf. 7:2–10.
- Aoki Y, Taniguchi Y, Matsumoto M, Matsumoto M, Ohno M, Masumura K, Sasaki S, Tsuzuki T, Yamamoto M, Nohmi T. 2020. Oxidative-stress-driven mutagenesis in the small intestine of the gpt delta mouse induced by oral administration of potassium bromate. Mutat Res. 850–851:503136.
- Arce GT, Gordon EB, Cohen SM, Singh P. 2010. Genetic toxicology of folpet and captan. Crit Rev Toxicol. 40:546–574.
- AWWA. 2004. Occurrence survey of boron and hexavalent chromium. Denver (CO): American Water Works Association Research Foundation.
- Barker N, van de Wetering M, Clevers H. 2008. The intestinal stem cell. Genes Dev. 22:1856–1864.
- Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. 2009. Crypt stem cells as the cells-of-origin of intestinal cancer. Nature. 457:608–611.
- Becker RA, Ankley GT, Edwards SW, Kennedy SW, Linkov I, Meek B, Sachana M, Segner H, Van Der Burg B, Villeneuve DL, et al. 2015. Increasing scientific confidence in adverse outcome pathways: application of tailored Bradford-Hill considerations for evaluating weight of evidence. Regul Toxicol Pharmacol. 72:514–537.
- Becker RA, Dellarco V, Seed J, Kronenberg JM, Meek B, Foreman J, Palermo C, Kirman C, Linkov I, Schoeny R, et al. 2017. Quantitative weight of evidence to assess confidence in potential modes of action. Regul Toxicol Pharmacol. 86:205–220.
- Becker RA, Patlewicz G, Simon TW, Rowlands JC, Budinsky RA. 2015. The adverse outcome pathway for rodent liver tumor promotion by sustained activation of the aryl hydrocarbon receptor. Regul Toxicol Pharmacol. 73:172–190.
- Boobis AR, Doe JE, Heinrich-Hirsch B, Meek ME, Munn S, Ruchirawat M, Schlatter J, Seed J, Vickers C. 2008. IPCS framework for analyzing the relevance of a cancer mode of action for humans. Crit Rev Toxicol. 38:87–96.
- Brooks RA, Gooderham NJ, Edwards RJ, Boobis AR, Winton DJ. 1999. The mutagenicity of benzo[a]pyrene in mouse small intestine. Carcinogenesis. 20:109–114.
- Brown I. 2013. The pathology of malnutrition and malabsorption. In: Shepherd NA, Warren BF, Williams GT, Greenson JK, Lauwers GY, Novelli MR, editors. Morson and Dawson's gastrointestinal pathology. 5th ed. Chichester (UK): Wiley-Blackwell.
- Buttner B, Beyersmann D. 1985. Modification of the erythrocyte anion carrier by chromate. Xenobiotica. 15(8–9):735–741.
- CDPH. 2011. Chromium-6 in drinking water: an overview of sampling results. Sacramento (CA): California Department of Public Health.
- Chandra SA, Nolan MW, Malarkey DE. 2010. Chemical carcinogenesis of the gastrointestinal tract in rodents: an overview with emphasis on NTP carcinogenesis bioassays. Toxicol Pathol. 38:188–197.
- Chappell GA, Rager JE, Wolf J, Babic M, LeBlanc KJ, Ring CL, Harris MA, Thompson CM. 2019. Comparison of gene expression responses in the small intestine of mice following exposure to 3 carcinogens using the S1500+ gene set informs a potential common adverse outcome pathway. Toxicol Pathol. 47:851–864.
- Chidiac P, Goldberg MT. 1987. Lack of induction of nuclear aberrations by captan in mouse duodenum. Environ Mutagen. 9:297–306.
- Clay PP. 2004. Evaluation of the in vivo comet assay in the mouse duodenum. Alderley Park, Macclesfield, Cheshire, UK: Central Toxicology Laboratory. Sponsor: Beer Sheva, Israel: Makhteshim Chemical Works, Ltd. Report no. SM1245. September 22, 2004. MRID 47605801. As cited by Arce (2010).
- Clewell RA, Thompson CM, Clewell HJ III. 2019. Dose-dependence of chemical carcinogenicity: biological mechanisms for thresholds and implications for risk assessment. Chem Biol Interact. 301:112–127.
- Cohen SM, Ellwein LB. 1990. Cell proliferation in carcinogenesis. Science. 249:1007–1011.
- Cohen SM, Gordon EB, Singh P, Arce GT, Nyska A. 2010. Carcinogenic mode of action of folpet in mice and evaluation of its relevance to humans. Crit Rev Toxicol. 40:531–545.
- Collier ZA, Gust KA, Gonzalez-Morales B, Gong P, Wilbanks MS, Linkov I, Perkins EJ. 2016. A weight of evidence assessment approach for adverse outcome pathways. Regul Toxicol Pharmacol. 75:46–57.
- Cullen JM, Ward JM, Thompson CM. 2016. Reevaluation and classification of duodenal lesions in B6C3F1 mice and F344 rats from 4 studies of hexavalent chromium in drinking water. Toxicol Pathol. 44:279–289.
- Dana Devi K, Rozati R, Saleha Banu B, Jamil K, Grover P. 2001. In vivo genotoxic effect of potassium dichromate in mice leukocytes using comet assay. Food Chem Toxicol. 39:859–865.
- De Flora S, D'Agostini F, Balansky R, Micale R, Baluce B, Izzotti A. 2008. Lack of genotoxic effects in hematopoietic and gastrointestinal cells of mice receiving chromium(VI) with the drinking water. Mutat Res. 659:60–67.
- De Flora S, Camoirano A, Micale RT, La Maestra S, Savarino V, Zentilin P, Marabotto E, Suh M, Proctor DM. 2016. Reduction of hexavalent chromium by fasted and fed human gastric fluid. I. Chemical reduction and mitigation of mutagenicity. Toxicol Appl Pharmacol. 306:113–119.
- De Flora S, Zanacchi P, Camoirano A, Bennicelli C, Badolati GS. 1984. Genotoxic activity and potency of 135 compounds in the Ames reversion test and in a bacterial DNA-repair test. Mutat Res Rev Genet Toxicol. 133(3):161–198.
- DeSesso JM, Lavin AL, Hsia SM, Mavis RD. 2000. Assessment of the carcinogenicity associated with oral exposures to hydrogen peroxide. Food Chem Toxicol. 38:1021–1041.
- Dickson BC, Streutker CJ, Chetty R. 2006. Coeliac disease: an update for pathologists. J Clin Pathol. 59:1008–1016.
- Eastmond DA. 2012. Factors influencing mutagenic mode of action determinations of regulatory and advisory agencies. Mutat Res Rev Mutat Res. 751(1):49–63.
- FSCJ. 2017. Risk assessment report captan (pesticides). Food Saf Comm Jpn. 5:61–66.
- FSCJ. 2019. Risk assessment report hexavalent chromium (beverages). Food Saf Comm Jpn. 7:56–57.
- Gatto NM, Kelsh MA, Mai DH, Suh M, Proctor DM. 2010. Occupational exposure to hexavalent chromium and cancers of the gastrointestinal tract: a meta-analysis. Cancer Epidemiol. 34(4):388–399.
- Gold LS, Manley NB, Slone TH, Ward JM. 2001. Compendium of chemical carcinogens by target organ: results of chronic bioassays in rats, mice, hamsters, dogs, and monkeys. Toxicol Pathol. 29:639–652.
- Goldberg MT, Chidiac P. 1986. An in vivo assay for small intestine genotoxicity. Mutat Res. 164:209–215.
- Gordon E. 2007. Captan: transition from 'B2' to 'not likely'. How pesticide registrants affected the EPA cancer classification update. J Appl Toxicol. 27:519–526.
- Gordon E, Cohen SM, Singh P. 2012. Folpet-induced short term cytotoxic and proliferative changes in the mouse duodenum. Toxicol Mechan Methods. 22:54–59.
- Greaves P. 2012. Histopathology of preclinical toxicity studies. 4th ed. London (UK): Elsevier-Academic Press.
- Greenfield RE, Ellwein LB, Cohen SM. 1984. A general probabilistic model of carcinogenesis: analysis of experimental urinary bladder cancer. Carcinogenesis. 5:437–445.
- Groschwitz KR, Hogan SP. 2009. Intestinal barrier function: molecular regulation and disease pathogenesis. J Allergy Clin Immunol. 124(1):3–20.
- Gudi R, Krsmanovic L. 2001. Nuclear aberration test in the mouse duo- denum (folpet) and photomicrographs. Rockville (MD): BioReliance. Report no. AA31SK.123005.BTL. MRID 45745105.
- Haney J Jr. 2015. Consideration of non-linear, non-threshold and threshold approaches for assessing the carcinogenicity of oral exposure to hexavalent chromium. Regul Toxicol Pharmacol. 73:834–852.
- Hard GC, Rodgers IS, Baetcke KP, Richards WL, McGaughy RE, Valcovic LR. 1993. Hazard evaluation of chemicals that cause accumulation of alpha 2u-globulin, hyaline droplet nephropathy, and tubule neoplasia in the kidneys of male rats. Environ Health Perspect. 99:313–349.
- Health Canada. 2016. Guidelines for Canadian drinking water quality: guideline technical document – chromium. Ottawa (ON): Health Environment and Consumer Safety Branch, Water and Air Quality Bureau, Health Canada.
- Hill AB. 1965. The environment and disease: association or causation? Proc Royal Soc Med. 58(5):295–300.
- [IARC] International Agency for Research on Cancer. 1978. Overall evaluations of carcinogenicity: some N-nitroso compounds 17. International Agency for Research on Cancer.
- IARC 1987. Overall evaluations of carcinogenicity: an updating of IARC monographs Volumes 1–42. IARC Monographs Supplement 7. International Agency for Research on Cancer.
- IARC. 1999a. Some Chemicals that Cause Tumours of the Kidney or Urinary Bladder in Rodents and Some Other Substances 73. International Agency for Research on Cancer.
- IARC. 1999b. Re-evaluation of Some Organic Chemicals, Hydrazine and Hydrogen Peroxide (Part 1, Part 2, Part 3) 71. International Agency for Research on Cancer.
- IARC. 2012a. A review of human carcinogens: arsenic, metals, fibres, and dusts. IARC Monographs on the Evaluation of Carcinogenic Risks to Humans 100C. International Agency for Research on Cancer.
- IARC. 2012b. Radiation 100D. International Agency for Research on Cancer.
- Ito A, Naito M, Naito Y, Watanabe H. 1982. Induction and characterization of gastro-duodenal lesions in mice given continuous oral administration of hydrogen peroxide. Gan. 73:315–322.
- Ito A, Watanabe H, Naito M, Naito Y. 1981. Induction of duodenal tumors in mice by oral administration of hydrogen peroxide. Gan. 72:174–175.
- Ito A, Watanabe H, Naito M, Naito Y, Kawashima K. 1984. Correlation between induction of duodenal tumor by hydrogen peroxide and catalase activity in mice. Gan. 75:17–21.
- JMPR. 1996. Joint FAO/WHO meeting on pesticide residues. Tox Monograph: Folpet.
- JMPR. 2004a. Joint FAO/WHO meeting on pesticide residues. Tox Monograph: Folpet. JMPR.
- JMPR. 2004b. Joint FAO/WHO meeting on pesticide residues. Tox Monograph: Captan. JMPR.
- Johnson GE, Soeteman-Hernandez LG, Gollapudi BB, Bodger OG, Dearfield KL, Heflich RH, Hixon JG, Lovell DP, MacGregor JT, Pottenger LH, et al. 2014. Derivation of point of departure (PoD) estimates in genetic toxicology studies and their potential applications in risk assessment. Environ Mol Mutagen. 55:609–623.
- Kamboj AK, Oxentenko AS. 2017. Clinical and histologic mimickers of celiac disease. Clin Transl Gastroenterol. 8:e114.
- Kirkland D, Uno Y, Luijten M, Beevers C, van Benthem J, Burlinson B, Dertinger S, Douglas GR, Hamada S, Horibata K, et al. 2019. In vivo genotoxicity testing strategies: report from the 7th International workshop on genotoxicity testing (IWGT). Mutat Res. 847:403035
- Kirman CR, Aylward LL, Suh M, Harris MA, Thompson CM, Haws LC, Proctor DM, Lin SS, Parker W, Hays SM. 2013. Physiologically based pharmacokinetic model for humans orally exposed to chromium. Chem Biol Interact. 204:13–27.
- Kirman CR, Hays SM, Aylward LL, Suh M, Harris MA, Thompson CM, Haws LC, Proctor DM. 2012. Physiologically based pharmacokinetic model for rats and mice orally exposed to chromium. Chem Biol Interact. 200:45–64.
- Kirman CR, Suh M, Hays SM, Gurleyuk H, Gerads R, De Flora S, Parker W, Lin S, Haws LC, Harris MA, et al. 2016. Reduction of hexavalent chromium by fasted and fed human gastric fluid. II. Ex vivo gastric reduction modeling. Toxicol Appl Pharmacol. 306:120–133.
- Kirman CR, Suh M, Proctor DM, Hays SM. 2017. Improved physiologically based pharmacokinetic model for oral exposures to chromium in mice, rats, and humans to address temporal variation and sensitive populations. Toxicol Appl Pharmacol. 325:9–17.
- Kopec AK, Kim S, Forgacs AL, Zacharewski TR, Proctor DM, Harris MA, Haws LC, Thompson CM. 2012. Genome-wide gene expression effects in B6C3F1 mouse intestinal epithelia following 7 and 90days of exposure to hexavalent chromium in drinking water. Toxicol Appl Pharmacol. 259:13–26.
- Kopec AK, Thompson CM, Kim S, Forgacs AL, Zacharewski TR. 2012. Comparative toxicogenomic analysis of oral Cr(VI) exposure effects in rat and mouse small intestinal epithelia. Toxicol Appl Pharmacol. 262:124–138.
- Lash LH, Hagen TM, Jones DP. 1986. Exogenous glutathione protects intestinal epithelial cells from oxidative injury. Proc Natl Acad Sci USA. 83:4641–4645.
- MacGregor JT, Frotschl R, White PA, Crump KS, Eastmond DA, Fukushima S, Guerard M, Hayashi M, Soeteman-Hernandez LG, Johnson GE, et al. 2015. IWGT report on quantitative approaches to genotoxicity risk assessment II. Use of point-of-departure (PoD) metrics in defining acceptable exposure limits and assessing human risk. Mutat Res Genet Toxicol Environ Mutag. 783:66–78.
- Martensson J, Jain A, Meister A. 1990. Glutathione is required for intestinal function. Proc Natl Acad Sci USA. 87:1715–1719.
- McCarroll N, Keshava N, Chen J, Akerman G, Kligerman A, Rinde E. 2010. An evaluation of the mode of action framework for mutagenic carcinogens case study II: chromium (VI). Environ Mol Mutagen. 51:89–111.
- McConnell EL, Fadda HM, Basit AW. 2008. Gut instincts: explorations in intestinal physiology and drug delivery. Int J Pharm. 364(2):213–226.
- McNeill LS, Mclean JE, Parks JL, Edwards MA. 2012. Hexavalent chromium review, part 2: chemistry, occurrence, and treatment. J Am Waterworks Assoc. 104:E395–E405.
- Moffat I, Martinova N, Seidel C, Thompson CM. 2018. Hexavalent chromium in drinking water. J Am Waterworks Assoc. 110:E22–E35.
- Moolgavkar SH, Knudson AG Jr. 1981. Mutation and cancer: a model for human carcinogenesis. J Natl Cancer Inst. 66:1037–1052.
- Nolte T, Brander-Weber P, Dangler C, Deschl U, Elwell MR, Greaves P, Hailey R, Leach MW, Pandiri AR, Rogers A, et al. 2016. Nonproliferative and proliferative lesions ofthe gastrointestinal tract, pancreas andSalivary glands of the rat and mouse. J Toxicol Pathol. 29:1S–125S.
- NTP. 2007. National Toxicology Program technical report on the toxicity studies of sodium dichromate dihydrate (CAS No. 7789-12-0) administered in drinking water to male and female F344/N rats and B6C3F1 mice and male BALB/c and am3-C57BL/6 mice. NTP. NTP Toxicity Report Series Number 72, NIH Publication No. 07-5964.
- NTP. 2008. National Toxicology Program technical report on the toxicology and carcinogenesis studies of sodium dichromate dihydrate (CAS No. 7789-12-0) in F344/N rats and B6C3F1 mice (drinking water studies). NTP. NTP TR 546. NIH Publication No. 08-5887.
- O'Brien TJ, Ceryak S, Patierno SR. 2003. Complexities of chromium carcinogenesis: role of cellular response, repair and recovery mechanisms. Mutat Res. 533:3–36.
- O’Brien TJ, Ding H, Suh M, Thompson CM, Parsons BL, Harris MA, Winkelman WA, Wolf JC, Hixon JG, Schwartz AM, et al. 2013. Assessment of K-Ras mutant frequency and micronucleus incidence in the mouse duodenum following 90-days of exposure to Cr(VI) in drinking water. Mutat Res. 754:15–21.
- [OECD] Organisation for Economic Co-operation and Development. 2013. Guideline for testing of chemicals. 2013. Test Guideline 488, - Transgenic rodent somatic and germ cell gene mutation assays, adopted July 28. Paris (France): OECD.
- OECD. 2018. User’s handbook supplement to the guidance document for developing and assessing AOPs. Paris (France): OECD.
- Patlewicz G, Simon TW, Rowlands JC, Budinsky RA, Becker RA. 2015. Proposing a scientific confidence framework to help support the application of adverse outcome pathways for regulatory purposes. Regul Toxicol Pharmacol. 71:463–477.
- Perkins EJ, Ashauer R, Burgoon L, Conolly R, Landesmann B, Mackay C, Murphy CA, Pollesch N, Wheeler JR, Zupanic A, et al. 2019. Building and applying quantitative adverse outcome pathway models for chemical hazard and risk assessment. Environ Toxicol Chem. 38:1850–1865.
- Potten CS, Loeffler M. 1990. Stem cells: attributes, cycles, spirals, pitfalls and uncertainties. Lessons for and from the crypt. Development. 110:1001–1020.
- Proctor DM, Suh M, Aylward LL, Kirman CR, Harris MA, Thompson CM, Gurleyuk H, Gerads R, Haws LC, Hays SM. 2012. Hexavalent chromium reduction kinetics in rodent stomach contents. Chemosphere. 89:487–493.
- Rager JE, Ring CL, Fry RC, Suh M, Proctor DM, Haws LC, Harris MA, Thompson CM. 2017. High-throughput screening data interpretation in the context of in vivo transcriptomic responses to oral Cr(VI) exposure. Toxicol Sci. 158:199–212.
- Salnikow K, Zhitkovich A. 2008. Genetic and epigenetic mechanisms in metal carcinogenesis and cocarcinogenesis: nickel, arsenic, and chromium. Chem Res Toxicol. 21(1):28–44.
- Sasso AF, Schlosser PM. 2015. An evaluation of in vivo models for toxicokinetics of hexavalent chromium in the stomach. Toxicol Appl Pharmacol. 287:293–298.
- Schwitalla S, Fingerle AA, Cammareri P, Nebelsiek T, Goktuna SI, Ziegler PK, Canli O, Heijmans J, Huels DJ, Moreaux G, et al. 2013. Intestinal tumorigenesis initiated by dedifferentiation and acquisition of stem-cell-like properties. Cell. 152:25–38.
- Sonich-Mullin C, Fielder R, Wiltse J, Baetcke K, Dempsey J, Fenner-Crisp P, Grant D, Hartley M, Knaap A, Kroese D, et al.; International Programme on Chemical Safety. 2001. IPCS conceptual framework for evaluating a mode of action for chemical carcinogenesis. Regul Toxicol Pharmacol. 34:146–152.
- Stern AH. 2010. A quantitative assessment of the carcinogenicity of hexavalent chromium by the oral route and its relevance to human exposure. Environ Res. 110:798–807.
- Stout MD, Herbert RA, Kissling GE, Collins BJ, Travlos GS, Witt KL, Melnick RL, Abdo KM, Malarkey DE, Hooth MJ. 2009. Hexavalent chromium is carcinogenic to F344/N rats and B6C3F1 mice after chronic oral exposure. Environ Health Perspect. 117:716–722.
- Suh M, Wikoff D, Lipworth L, Goodman M, Fitch S, Mittal L, Ring C, Proctor D. 2019. Hexavalent chromium and stomach cancer: a systematic review and meta-analysis. Crit Rev Toxicol. 49:140–159.
- TCEQ. 2016. Hexavalent chromium oral reference dose: development support document (Final). TCEH.
- Thompson CM, Gregory Hixon J, Proctor DM, Haws LC, Suh M, Urban JD, Harris MA. 2012. Assessment of genotoxic potential of Cr(VI) in the mouse duodenum: an in silico comparison with mutagenic and nonmutagenic carcinogens across tissues. Regul Toxicol Pharmacol. 64:68–76.
- Thompson CM, Haws LC, Harris MA, Gatto NM, Proctor DM. 2011. Application of the U.S. EPA mode of action Framework for purposes of guiding future research: a case study involving the oral carcinogenicity of hexavalent chromium. Toxicol Sci. 119:20–40.
- Thompson CM, Kirman CR, Hays SM, Suh M, Harvey SE, Proctor DM, Rager JE, Haws LC, Harris MA. 2018. Integration of mechanistic and pharmacokinetic information to derive oral reference dose and margin-of-exposure values for hexavalent chromium. J Appl Toxicol. 38:351–365.
- Thompson CM, Kirman CR, Proctor DM, Haws LC, Suh M, Hays SM, Hixon JG, Harris MA. 2014. A chronic oral reference dose for hexavalent chromium-induced intestinal cancer. J Appl Toxicol. 34:525–536.
- Thompson CM, Proctor DM, Haws LC, Hebert CD, Grimes SD, Shertzer HG, Kopec AK, Hixon JG, Zacharewski TR, Harris MA. 2011. Investigation of the mode of action underlying the tumorigenic response induced in B6C3F1 mice exposed orally to hexavalent chromium. Toxicol Sci. 123:58–70.
- Thompson CM, Proctor DM, Suh M, Haws LC, Hebert CD, Mann JF, Shertzer HG, Hixon JG, Harris MA. 2012. Comparison of the effects of hexavalent chromium in the alimentary canal of F344 rats and B6C3F1 mice following exposure in drinking water: implications for carcinogenic modes of action. Toxicol Sci. 125:79–90.
- Thompson CM, Proctor DM, Suh M, Haws LC, Kirman CR, Harris MA. 2013. Assessment of the mode of action underlying development of rodent small intestinal tumors following oral exposure to hexavalent chromium and relevance to humans. Crit Rev Toxicol. 43:244–274.
- Thompson CM, Rager JE, Suh M, Ring CL, Proctor DM, Haws LC, Fry RC, Harris MA. 2016. Transcriptomic responses in the oral cavity of F344 rats and B6C3F1 mice following exposure to Cr(VI): implications for risk assessment. Environ Mol Mutagen. 57:706–716.
- Thompson CM, Seiter J, Chappell MA, Tappero RV, Proctor DM, Suh M, Wolf JC, Haws LC, Vitale R, Mittal L, et al. 2015. Synchrotron-based imaging of chromium and gamma-H2AX immunostaining in the duodenum following repeated exposure to cr(VI) in drinking water. Toxicol Sci. 143:16–25.
- Thompson CM, Suh M, Proctor DM, Haws LC, Harris MA. 2017. Ten factors for considering the mode of action of Cr(VI)-induced gastrointestinal tumors in rodents. Mutat Res. 823:45–57.
- Thompson CM, Wolf JC, Elbekai RH, Paranjpe MG, Seiter JM, Chappell MA, Tappero RV, Suh M, Proctor DM, Bichteler A, et al. 2015. Duodenal crypt health following exposure to Cr(VI): micronucleus scoring, gamma-H2AX immunostaining, and synchrotron X-ray fluorescence microscopy. Mutat Res Genet Toxicol Environ Mutag. 789–790:61–66.
- Thompson CM, Wolf JC, McCoy A, Suh M, Proctor DM, Kirman CR, Haws LC, Harris MA. 2017. Comparison of toxicity and recovery in the duodenum of B6C3F1 mice following treatment with intestinal carcinogens captan, folpet, and hexavalent chromium. Toxicol Pathol. 45:1091–1101.
- Thompson CM, Young RR, Dinesdurage H, Suh M, Harris MA, Rohr AC, Proctor DM. 2017. Assessment of the mutagenic potential of hexavalent chromium in the duodenum of big blue(R) rats. Toxicol Appl Pharmacol. 330:48–52.
- Thompson CM, Young RR, Suh M, Dinesdurage HR, Elbekai RH, Harris MA, Rohr AC, Proctor DM. 2015. Assessment of the mutagenic potential of Cr(VI) in the oral mucosa of Big Blue((R)) transgenic F344 rats. Environ Mol Mutagen. 56:621–628.
- Tomasetti C, Marchionni L, Nowak MA, Parmigiani G, Vogelstein B. 2015. Only three driver gene mutations are required for the development of lung and colorectal cancers. Proc Natl Acad Sci USA. 112:118–123.
- Tomasetti C, Vogelstein B. 2015. Cancer etiology. Variation in cancer risk among tissues can be explained by the number of stem cell divisions. Science. 347:78–81.
- Treuting PM, Arends MJ, Dintzis SM. 2018. Upper gastrointestinal tract. In: Treuting PM, Dintzis SM, Montine KS, editors. Comparative anatomy and histology. London (UK): Academic Press; p. 191–211.
- U.S. EPA. 1995. Science assessment of captan. [Memorandum dated 1995 Feb 21]. Washington (DC): U.S. Environmental Protection Agency.
- U.S. EPA. 1999. Reregistration eligibility decision (RED). Folpet. Washington (DC): U.S. Environmental Protection Agency. EPA/738/R-99/011.
- U.S. EPA. 2004. Captan; Cancer reclassification; Amendment of reregistration eligibility decision; Notice of availability. Fed Regist. 69:68357–68360.
- U.S. EPA. 2005. Guidelines for carcinogen risk assessment. Risk Assessment Forum. Washington (DC): U.S. Environmental Protection Agency. EPA/630/P-03/001F.
- U.S. EPA. 2010. Toxicological review of hexavalent chromium in support of summary information on the integrated risk information system (IRIS). External Review DRAFT. Washington (DC): U.S. Environmental Protection Agency.
- U.S. EPA. 2012. Folpet: human health risk scoping document in support of registration review. [Memorandum dated 2012 Nov 28]. Washington (DC): U.S. Environmental Protection Agency.
- U.S. EPA. 2017. Data summary of the third unregulated contaminant monitoring rule (UCMR3). Washington (DC): U.S. Environmental Protection Agency. EPA 815‐S‐17‐001.
- U.S. EPA. 2019. Exposure Factors Handbook 2011 Edition, Chapter 3: ingestion of water and other select liquids (February 2019 Update). Washington (DC): U.S. Environmental Protection Agency. EPA/600/R-18/259F.
- Uzal FA, Plattner BL, Hostetter JM. 2016. Alimentary system. In: Maxie MG, editor. Pathology of domestic animals. 6th ed. St. Louis (MO): Elsevier.
- van der Flier LG, Clevers H. 2009. Stem cells, self-renewal, and differentiation in the intestinal epithelium. Annu Rev Physiol. 71:241–260.
- Weiner ML, Freeman C, Trochimowicz H, de Gerlache J, Jacobi S, Malinverno G, Mayr W, Regnier JF. 2000. 13-week drinking water toxicity study of hydrogen peroxide with 6-week recovery period in catalase-deficient mice. Food Chem Toxicol. 38:607–615.
- [WHO] World Health Organization. 2019. Chromium in drinking water: draft background document for development of WHO guidelines for drinking-water quality. WHO.
- Wieczorek M, Szafrańska PA, Labecka AM, Lázaro J, Konarzewski M. 2015. Effect of the abrasive properties of sedges on the intestinal absorptive surface and resting metabolic rate of root voles. J Exp Biol. 218:309–315.
- Wilkinson CF, Arce G, Gordon EB. 2004. Scientific analysis of the data relating to the reclassification of captan under EPA’s new guidelines for carcinogen risk assessment. Captan Task Force. CTF 0104.
- Williams JM, Duckworth CA, Burkitt MD, Watson AJM, Campbell BJ, Pritchard DM. 2015. Epithelial cell shedding and barrier function: a matter of life and death at the small intestinal villus tip. Vet Pathol. 52(3):445–455.
- Witt KL, Stout MD, Herbert RA, Travlos GS, Kissling GE, Collins BJ, Hooth MJ. 2013. Mechanistic insights from the NTP studies of chromium. Toxicol Pathol. 41:326–342.
- Wong ZA, Chang HM. 1985. The comparative metabolism of captan in the rat and mouse. Richmond (CA): Chevron Chemical Company. [Dated 1985 Nov 14]. Unpublished study No. SOCAL 1992, S-2163. Accession No. 262633.
- Wood CE, Hukkanen RR, Sura R, Jacobson-Kram D, Nolte T, Odin M, Cohen SM. 2015. Scientific and Regulatory Policy Committee (SRPC) review: interpretation and use of cell proliferation data in cancer risk assessment. Toxicol Pathol. 43:760–775.
- Zainab JM, Jihad AA, Assel KH. 2016. Toxicopathological effects of sodium dichromate (chromium CrVI) on small intestine of laboratory albino rats (Rattus norvegicus). Al-Qadisiyah J Vet Med Sci. 15:5.
- Zhitkovich A. 2011. Chromium in drinking water: sources, metabolism, and cancer risks. Chem Res Toxicol. 24:1617–1629.
- Zou WY, Blutt SE, Zeng XL, Chen MS, Lo YH, Castillo-Azofeifa D, Klein OD, Shroyer NF, Donowitz M, Estes MK. 2018. Epithelial WNT liagnds are essential drivers of intestinal stem cell activation. Cell Rep. 22:1003–1015.