
Abstract
Many nongenotoxic chemicals have been shown to produce liver tumors in mice and/or rats by a mode of action (MOA) involving activation of the constitutive androstane receptor (CAR). Studies with phenobarbital (PB) and other compounds have identified the key events for this MOA: CAR activation; increased hepatocellular proliferation; altered foci formation; and ultimately the development of adenomas/carcinomas. In terms of human relevance, the pivotal species difference is that CAR activators are mitogenic agents in mouse and rat hepatocytes, but they do not stimulate increased hepatocellular proliferation in humans. This conclusion is supported by substantial in vitro studies with cultured rodent and human hepatocytes and also by in vivo studies with chimeric mice with human hepatocytes. Examination of the literature reveals many similarities in the hepatic effects and species differences between activators of rodent CAR and the peroxisome proliferator-activated receptor alpha (PPARα), with PPARα activators also not being mitogenic agents in human hepatocytes. Overall, a critical analysis of the available data demonstrates that the established MOA for rodent liver tumor formation by PB and other CAR activators is qualitatively not plausible for humans. This conclusion is supported by data from several human epidemiology studies.
Introduction
The constitutive androstane receptor (CAR; also known as NR113) is a member of the nuclear receptor superfamily (subfamily 1, group I), which is highly expressed in liver and at much lower levels in small intestine and kidney (The Human Protein Atlas – https://v18.proteinatlas.org/ENSG00000143257-NR1I3/tissue), and is involved in the regulation of xenobiotic metabolism, cell proliferation, apoptosis, energy metabolism, and lipid homeostasis (Omiecinski et al. Citation2011a; Yang and Wang Citation2014; Yoshinari Citation2019; Daujat-Chavanieu and Gerbal-Chaloin Citation2020). CAR can be activated by a large number of chemicals, either by direct ligand binding or by a ligand-independent mechanism resulting in nuclear translocation and heterodimerization with the retinoid X receptor (RXR), which is followed by binding to response elements in DNA (Omiecinski et al. Citation2011a; Yang and Wang Citation2014; Lynch et al. Citation2019; Negishi et al. Citation2020). Two other members of the nuclear receptor superfamily are the pregnane X receptor (PXR; also known as NR112) and the peroxisome proliferator-activated receptor alpha (PPARα; also known as NR1C1), which are also highly expressed in liver and are involved in xenobiotic and intermediary metabolism (Omiecinski et al. Citation2011a; Corton et al. Citation2014, Citation2018; Yoshinari Citation2019; Daujat-Chavanieu and Gerbal-Chaloin Citation2020). Activation of CAR, PXR, and PPARα is known to result in the induction of cytochrome P450 (CYP) enzymes in the CYP2B, CYP3A, and CYP4A subfamilies, respectively, but other CYP subfamily enzymes are also induced by activation of these and other hepatic receptors (Martignoni et al. Citation2006; Yoshinari et al. Citation2008; Omiecinski et al. Citation2011b). Several studies have shown that there can be considerable crosstalk between hepatic CAR and PXR receptors with some compounds being activators of both of these nuclear receptors (Maglich et al. Citation2002; Omiecinski et al. Citation2011a).
Phenobarbital (PB) and its sodium salt (sodium phenobarbital, NaPB) are known to activate CAR by a ligand-independent mechanism and are prototypical inducers of CYP2B and other CYP subfamily enzymes in both rodent and human liver. In this review studies with either PB or NaPB will just be referred to as PB only. Many studies have demonstrated that prolonged treatment with PB, a nongenotoxic chemical, can result in liver tumor formation in the mouse (Whysner et al. Citation1996; IARC Citation2001; Elcombe et al. Citation2014). Investigations employing mice lacking hepatic CAR (i.e. CAR knockout (KO) mice) have demonstrated the crucial role of hepatic CAR in mouse liver tumor formation. Unlike normal (i.e. wild-type) mice, the treatment of CAR KO mice with PB did not result in an increase in liver weight or evidence of liver centrilobular hepatocellular hypertrophy by morphological examination, did not result in an induction of Cyp2b subfamily enzymes, did not increase hepatocyte replicative DNA synthesis (RDS) and following initiation with the genotoxic agent diethylnitrosamine (DEN) did not promote liver tumor formation (Wei et al. Citation2000; Yamamoto et al. Citation2004; Huang et al. Citation2005; Scheer et al. Citation2008). In addition, unlike wild-type mice, no liver tumors were observed in CAR KO mice after treatment with 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene (TCPOBOP), which is a potent mouse hepatic CAR activator (Omiecinski et al. Citation2011a), either with or without prior DEN administration (Huang et al. Citation2005).
Similar to PB, a number of other nongenotoxic chemicals have been shown to produce liver tumors in the mouse and/or rat by a mode of action (MOA) involving CAR activation. For PB and the chemicals listed in , the MOA for tumor formation has been established using the IPCS framework and the modified Bradford Hill considerations as described below (Sonich-Mullin et al. Citation2001; Meek et al. Citation2003; Seed et al. Citation2005; Boobis et al. Citation2006, Citation2008). In addition, based on more limited data (e.g. lack of hepatocyte RDS data at known carcinogenic dose levels), a number of other substances also appear to produce liver tumors in the mouse and/or rat by a CAR activation MOA. Examples include chlordane (Khasawinah and Grutsch Citation1989; Ross et al. Citation2010), dalcetrapib (Hoflack et al. Citation2012), diazepam (de la Iglesia et al. Citation1981; IARC Citation1996; Skoda et al. Citation2020), and ginkgo biloba extract (Maeda et al. Citation2015).
Table 1. Examples of compounds where a CAR activation MOA for mouse and/or rat liver tumor formation has been established.
Many chemicals have been identified as potential activators of human CAR (hCAR) from high-throughput screening assays employing either HepG2 or HepaRG cells (Bogen Citation2018; Lynch et al. Citation2019; Franzosa et al. Citation2021). It is thus very important to evaluate the potential risk of such chemicals for humans. The purpose of this review is to critically assess current experimental data concerning the human relevance of the MOA for rodent liver tumor formation by CAR activators. Due to the known crosstalk between hepatic CAR and PXR receptors, a number of studies with CAR KO animals have been performed with animals lacking both hepatic CAR and PXR receptors (CAR KO/PXR KO animals). The available data include studies with cultured rodent and human hepatocytes, studies with transgenic mice where either the mouse hepatic CAR or both the CAR and PXR receptors have been replaced with their human counterparts (i.e. hCAR or hCAR/hPXR mice), and investigations with chimeric mice with human or rat hepatocytes.
Liver tumor formation by PB and other CAR activators in experimental animals
Many studies have investigated liver tumor formation by PB in a number of mouse and rat strains. PB has been shown to produce hepatocellular adenomas and carcinomas in both high (e.g. C3H/He, B6C3F1) and low (e.g. C57BL/6, C57BL/10) spontaneous liver tumor incidence mouse strains (Whysner et al. Citation1996; IARC Citation2001; Elcombe et al. Citation2014). In contrast, while PB has been shown to increase the incidence of altered hepatic foci in the rat, only one study has reported an increase in the incidence of hepatocellular adenomas (IARC Citation2001; Elcombe et al. Citation2014). PB is also known to promote liver tumors in the mouse and rat after initiation with DEN or some other genotoxic carcinogens including 2-acetylaminofluorene and benzo[a]pyrene (Whysner et al. Citation1996; IARC Citation2001).
Although mice are more susceptible than rats to PB-induced liver tumor formation, this is not always the case for other rodent CAR activators. As shown in , some nongenotoxic rodent CAR activators produce liver tumors in both mice and rats, whereas other chemicals produce liver tumors in only one of these species. Differences between the mouse and rat in susceptibility to liver tumor formation may be due to a number of factors including species differences in receptor activation, the dose levels employed in the bioassays, and compound pharmacokinetic and/or metabolism differences. Thus, both the mouse and rat can be considered to be susceptible species to liver tumor formation induced by nongenotoxic CAR activators.
While PB has been shown to produce liver tumors in a number of mouse strains, two studies have demonstrated that PB did not produce liver tumors in the Syrian hamster (Diwan et al. Citation1986; Stenbäck et al. Citation1986). In addition, unlike the mouse and rat, PB has been shown not to promote liver tumors in the Syrian hamster after initiation with DEN or other genotoxic carcinogens (Tanaka et al. Citation1987). While PB is known to stimulate RDS in cultured mouse and rat hepatocytes, PB has been shown not to stimulate RDS in cultured Syrian hamster and guinea pig hepatocytes (James and Roberts Citation1996). Like PB, chronic studies with four other CAR activators, namely 1,1′-(2,2,2-trichloroethylidene)-bis[4-chlorobenzene] (DDT) (Graillot et al. Citation1975; Cabral et al. Citation1982; Rossi et al. Citation1983), dieldrin (Stevenson et al. Citation1999; Wang et al. Citation2020), toxaphene (Goodman et al. Citation2000), and diazepam (IARC Citation1996), performed in the Syrian hamster did not result in liver tumor formation. However, one metabolite of DDT, namely 1,1′-(2,2-dichloroethenylidene)-bis[4-chlorobenzene] (DDE), was found to be weakly carcinogenic to the liver in Syrian golden hamsters whereas DDT was negative in the same study (Rossi et al. Citation1983). A chronic study with DDT was also performed in primates (24 animals comprising 13 Cynomolgus and 11 Rhesus monkeys) where only one Cynomolgus monkey developed hepatocellular carcinoma (HCC) after an observation period of almost 20 years and total dose of 292 g DDT, with no tumors being detected in the control group of 17 monkeys, comprising nine Cynomolgus and eight Rhesus monkeys (Takayama et al. Citation1999). However, as noted by the authors, it is unclear whether the single case of HCC with such long latent period was actually associated with DDT treatment.
Human epidemiology studies with rodent CAR activators
Due to the therapeutic uses of PB in humans as a sedative, hypnotic and antiepileptic agent for many years, data from a number of epidemiological studies are available. PB does not appear to increase the risk of liver tumors (or other tumors) in humans even at therapeutic doses (e.g. 3–6 mg/kg) producing blood levels similar to those that are carcinogenic in mice (Monro Citation1993; Whysner et al. Citation1996; IARC Citation2001; Elcombe et al. Citation2014; La Vecchia and Negri Citation2014). While IARC (IARC Citation2001) evaluated available human epidemiological studies up to 1995, a more recent analysis of the available literature concluded that “epidemiological data on PB and liver cancer are limited, but indicate an absence of any specific association and, in any case, allow the exclusion of an appreciable excess risk” (La Vecchia and Negri Citation2014). Thus, while the power of the studies conducted is limited by the low observed incidence of human liver tumors in the populations evaluated, the number of studies that have been performed would have sufficient power to detect an increase in liver tumors in human subjects given PB.
In a recent epidemiological study (Stritzelberger et al. Citation2021), a number of antiepileptic drugs were shown not to increase cancer incidence in human subjects. The antiepileptic agents examined included PB and carbamazepine; the latter being a known CAR activator which induces liver CYP2B enzymes and tumors in the rat (IPCS Citation1999; Tateishi et al. Citation1999; Cui et al. Citation2005; Faucette et al. Citation2007).
The benzodiazepine drug oxazepam has been shown to produce liver tumors in the mouse (Bucher et al. Citation1994). Short-term studies have shown that oxazepam produces similar effects on CYP enzymes and hepatocyte RDS in mouse liver to those produced by PB (Cunningham et al. Citation1994; Griffin et al. Citation1995, Citation1996). While oxazepam has also been reported to induce mouse hepatic Cyp4a enzymes (Parkinson et al. Citation2006), the results of gene array studies suggest this compound is primarily a CAR activator in rodent liver (C. Corton, personal communication; Rooney et al. Citation2019). In human epidemiological studies, no association was observed between oxazepam treatment and liver tumor formation in one study (Friedman et al. Citation2009), and oxazepam was shown not to increase the incidence of all cancers in another study (Iqbal et al. Citation2015).
MOA analysis and human risk assessment
Currently, long-term bioassays in the mouse and rat are employed to screen for chemicals which may be carcinogenic to humans. Analysis of tumor data demonstrates that the liver is the most common site of tumor formation in both the mouse and rat (Huff et al. Citation1991; Gold et al. Citation2001; Thoolen et al. Citation2010). The potential usefulness and difficulties of such long-term bioassays for human risk assessment have been evaluated in many publications (Cohen Citation2004, Citation2010, Citation2017; Cohen and Arnold Citation2011; Osimitz et al. Citation2013; Goodman Citation2018; Cohen et al. Citation2019; Doe et al. Citation2019; Heusinkveld et al. Citation2020; Luijten et al. Citation2020). Such bioassays have two fundamental assumptions: first, rodent carcinogens are human carcinogens (i.e. interspecies extrapolation); and second, results obtained in rodents at high dose levels will be indicative of potential effects in humans at environmentally relevant exposure levels (i.e. dose extrapolation) (Cohen Citation2010, Citation2017; Goodman Citation2018). Certainly for most nongenotoxic rodent carcinogens, one or both of these assumptions are not correct. Moreover, such long-term bioassays are expensive, time consuming and require large numbers of animals. Short-term screens have been proposed based on evaluation of MOA to avoid the use of the long-term bioassays (Cohen Citation2010; Cohen et al. Citation2019).
MOA studies are now employed to help ascertain the human relevance of tumors produced in rodents by nongenotoxic carcinogens. A framework for MOA analysis of rodent tumor and non-tumor toxicity, together with assessment of human relevance, was established by the International Life Sciences Institute (ILSI) (supported by the United States Environmental Protection Agency (US.EPA) and Health Canada) and the International Programme on Chemical Safety (IPCS) of the World Health Organization (WHO) and is described in a number of publications (Sonich-Mullin et al. Citation2001; Meek et al. Citation2003, Citation2014a, Citation2014b; Cohen et al. Citation2004; Seed et al. Citation2005; Boobis et al. Citation2006, Citation2008; Holsapple et al. Citation2006). For carcinogenicity, the first stage is to evaluate whether it is possible to establish a MOA for tumor formation in experimental animals by identifying a series of key and associative events using a weight-of-evidence approach based on the modified Bradford Hill considerations (Sonich-Mullin et al. Citation2001; Meek et al. Citation2003, Citation2014a; Cohen et al. Citation2004; Boobis et al. Citation2006; Andersen et al. Citation2014). Once a robust MOA is established, the key and associative events are compared, first qualitatively and then quantitatively between effects in experimental animals and humans. MOAs have been established for tumor formation by nongenotoxic chemicals for various rodent tissues. For example, a recent analysis of 411 unique agrochemicals that have been evaluated for carcinogenicity by US.EPA and the European Chemicals Agency (ECHA) identified 170 chemicals as nongenotoxic carcinogens, which produced 340 cases of treatment-related tumor formation, of which MOAs or MOA networks could be identified in 224 instances (Heusinkveld et al. Citation2020). The further development of innovative test methods and enhanced understanding of carcinogenic processes will permit a better understanding of tumor formation in rodents and an evaluation of the relevance of such rodent tumors to humans (Rooney et al. Citation2018; Cohen et al. Citation2019; Heusinkveld et al. Citation2020; Luijten et al. Citation2020).
MOA for rodent liver tumor formation by PB and other CAR activators
As shown in , a number of MOAs have been established for liver tumor formation, both in humans and in rodent models (Cohen Citation2004; Cohen and Arnold Citation2016). Based on an evaluation of literature data, the key and associative events (Andersen et al. Citation2014) for the CAR-mediated MOA for PB-induced rodent liver tumor formation were established by Elcombe et al. (Citation2014) and are shown in . Activation of CAR, altered gene expression specific to CAR activation, increased cell proliferation, clonal expansion leading to altered hepatic foci, and ultimately liver tumor formation are considered to be key events, as they constitute necessary steps in the MOA (Elcombe et al. Citation2014). In addition, induction of hepatic CYP2B enzymes and liver hepatocellular centrilobular or panlobular hypertrophy (both morphological changes and increases in liver weight) are considered associative events and as such represent reliable markers of CAR activation (Elcombe et al. Citation2014). While other associative events and/or modulating factors (e.g. decreased apoptosis, inhibition of gap junctional intercellular communication) may also be involved in tumor formation, data on such endpoints are not specific to CAR activation and are not required to establish a CAR-dependent MOA for rodent liver tumor formation (Elcombe et al. Citation2014; Lake Citation2018; Peffer et al. Citation2018b). Similarly, while much MOA data can be obtained from short-term studies, data on clonal expansion to altered hepatic foci and formation of liver tumors (adenomas and carcinomas) can only be obtained in long-term studies, and, depending on the time points selected, such increases may not be observed. However, the absence of such data does not detract from establishing a CAR-dependent rodent liver tumor MOA, as altered liver foci are considered to be precursor lesions for subsequent liver tumor formation (Thoolen et al. Citation2012). However, it should be noted that an increased incidence of hepatocellular foci does not always result in subsequent liver tumor formation (Sistare et al. Citation2011). The minimum data set required to establish a CAR-activation MOA for rodent liver tumor formation is discussed below.
Table 2. Some MOAs for hepatocellular carcinogenesis.
Table 3. Key and associative events for rodent liver tumor formation by PB and other CAR activators.
As shown in , a number of nongenotoxic chemicals which are CAR activators have been shown to produce liver tumors in the mouse and/or rat. Robust MOAs have been established for these compounds, which have similar key and associative events to those identified for PB (Holsapple et al. Citation2006; Lake Citation2009, Citation2018; Cohen Citation2010; Elcombe et al. Citation2014; Yamada Citation2018).
CAR-dependent hepatocyte proliferation
An evaluation of the key and associative events in the established MOA for rodent liver tumor formation by PB and other CAR activators demonstrates the pivotal role of the stimulation of hepatocyte RDS. Increased cell proliferation represents an essential preneoplastic step in carcinogenesis by most nongenotoxic substances (Cohen and Arnold Citation2011; Wood et al. Citation2015; Wolf et al. Citation2019). Hepatocyte RDS in rodent liver can be determined by various techniques (Elcombe et al. Citation2014; Wood et al. Citation2015) including administering a DNA precursor (e.g. 5-bromo-2′-deoxyuridine (BRDU), [3H]thymidine) either as a single dose or continuously for several days via an osmotic pump and also by performing immunohistochemistry of liver sections for DNA replication markers such as Ki-67 (MKI67) and proliferating cell nuclear antigen (PCNA). Cell proliferation in rodent liver is normally quantified as the hepatocyte labeling index (i.e. the percentage of hepatocyte nuclei undergoing RDS).
In studies performed in the mouse and rat, CAR activators produce significant increases in hepatocyte RDS, expressed as the labeling index, at early time points (e.g. 3, 7, or 14 days), but generally not at longer time points (e.g. 28 or 90 days). However, this does not mean that effects of CAR activators on hepatocyte RDS in rodent liver are only transient, that is, an increase in the rate of hepatocyte RDS is only observed after short-term treatment with CAR activators. As demonstrated in many studies, the treatment of mice and rats with PB and other nongenotoxic CAR activators results in a sustained increase in liver weight (Whysner et al. Citation1996; Elcombe et al. Citation2014; Lake Citation2018). The hepatocyte labeling index is only a measure of the percentage of hepatocyte nuclei undergoing RDS and takes no account of the increase in liver weight and hence the total number of hepatocytes per animal. Indeed, the treatment of rats with PB has been shown to result in a significant increase in the total number of hepatocytes per animal (Carthew et al. Citation1998). Hence, because of the increase in liver size and the number of hepatocytes per animal, PB and other nongenotoxic CAR activators produce a sustained increase of cell proliferation in rodent liver taking into account the number of hepatocyte replications, not just the rate (Lake Citation2009, Citation2018; Cohen Citation2010, Citation2017; Cohen and Arnold Citation2011; Elcombe et al. Citation2014; Yamada Citation2018).
It is not the rate of cell replication that is important but the number of DNA replications (Moolgavkar and Knudson Citation1981; Greenfield et al. Citation1984; Cohen and Ellwein Citation1990), which will be increased if there is an increase in cell number, even if the rate is the same as controls. Furthermore, even a brief increase in cell replication early followed by control numbers of replications can be adequate to increase cancer risk over the span of a lifetime study (Ellwein and Cohen Citation1988). The liver is replicating all of the time, albeit at relatively low rates, but is adequate for generating an increased risk over time building on the increased number of replications occurring early. Thus, increased cell replication (even as reported as a tangent change) has been widely accepted as a key event in non-genotoxic carcinogenesis (Wood et al. Citation2015) including for CAR activators (Elcombe et al. Citation2014; Lake Citation2018) and PPARα activators (Corton et al. Citation2014, Citation2018).
In contrast to that the key event is the increase in the number of cell replications, increased liver weight can serve as an associative event as an indicator of increased RDS, especially after determining an increased labeling index rate at an early time point (Elcombe et al. Citation2014; Lake Citation2018). This is similar to what has been described for PPARα activators (Corton et al. Citation2014, Citation2018).
As with any key event, significant increases in hepatocyte RDS must be observed at dose levels which subsequently result in liver tumor formation in mice and/or rats. For example, PB has been shown to increase the incidence of hepatocellular adenomas and carcinomas in male mice of the low spontaneous tumor incidence C57BL/10J strain at a dietary level of 1000 ppm, but not at 200 ppm (Jones et al. Citation2009). MOA studies demonstrated that while treatment with 1000 ppm PB for 3, 8, or 15 days resulted in significant increases in hepatocyte labeling index values, no such effects were observed at a dietary level of 200 ppm. In another study, treatment with 3000 ppm, but not 100 ppm, of the natural pyrethrins in the diet increased the incidence of hepatocellular adenoma in female Sprague-Dawley rats (Osimitz and Lake Citation2009), with MOA studies demonstrating that treatment with 3000 ppm, but not 100 ppm, pyrethrins for 7 and 14 days resulted in significant increases in hepatocyte labeling index values (Price et al. Citation2007).
That hepatic CAR activation is pivotal to the stimulation of hepatocyte RDS in mice and rats treated with PB and other nongenotoxic CAR activators has been demonstrated in many studies with animals lacking either hepatic CAR or hepatic CAR and PXR. For example, while PB has been shown to induce hepatocyte RDS in wild-type (i.e. normal) mice and rats, no increases in hepatocyte RDS were observed following the treatment of CAR KO mice (Wei et al. Citation2000; Huang et al. Citation2005), CAR KO/PXR KO mice (Ross et al. Citation2010; Haines et al. Citation2019), CAR KO rats (Okuda et al. Citation2017b; Haines et al. Citation2018a; Yamada et al. Citation2020), and CAR KO/PXR KO rats (Haines et al. Citation2019) with PB. The lack of effect of PB on hepatocyte RDS in receptor KO mice and rats is not due to PB exposure as in studies where terminal plasma PB levels were analyzed, these were significantly higher in mice and rats lacking either hepatic CAR or CAR and PXR (Luisier et al. Citation2014; Haines et al. Citation2018a; Haines et al. Citation2019; Yamada et al. Citation2020). Presumably, the explanation for this effect is that the absence of the nuclear receptor(s) prevents the downstream effects on liver hypertrophy and induction of enzymes which metabolize PB. Apart from PB, as shown in , a number of other studies have demonstrated that nongenotoxic CAR activators produce a stimulation of hepatocyte RDS in wild-type mice and/or rats, but do not produce activation of RDS in animals lacking either hepatic CAR or CAR and PXR.
The results of these in vivo studies are supported by investigations with cultured rodent hepatocytes. For example, while PB, metazachlor and other CAR activators increased RDS in cultured wild-type rat hepatocytes, no such effects were observed in rat CAR KO hepatocytes (Wiemann et al. Citation2019; Goettel et al. Citation2020). Similarly, while PB and benfluralin induced RDS in cultured wild-type rat hepatocytes, no increase in RDS was observed in CAR KO/PXR KO rat hepatocytes (Strupp et al. Citation2020). In these in vitro studies, the functional viability of the rat hepatocyte preparations and their responsiveness to a known mitogen was confirmed by significant increases in RDS being produced in wild-type, CAR KO and CAR KO/PXR KO rat hepatocytes by treatment with the known hepatocyte mitogen, epidermal growth factor (EGF) (Wiemann et al. Citation2019; Goettel et al. Citation2020; Strupp et al. Citation2020).
Comparison of the effects of PB and other CAR activators on RDS and other endpoints in rodent and human hepatocytes
As described above, both the mouse and rat are susceptible species to the stimulation of hepatocyte RDS and also to liver tumor formation following prolonged treatment with nongenotoxic CAR activators. For the human risk assessment of rodent liver tumor formation produced by nongenotoxic CAR activators, it is clearly important to evaluate whether rodent liver CAR activators can stimulate RDS in human hepatocytes. As it is not possible to directly assess the effects of rodent CAR activators on cell proliferation in human liver, a number of experimental systems utilizing either human tissue or use of human hepatic nuclear receptors have been developed. These systems comprise in vitro studies with cultured human hepatocytes, together with in vivo studies with transgenic mice containing human hepatic PXR and/or CAR and in chimeric mice with human hepatocytes. Data from studies with these three experimental models are reviewed below.
Studies in cultured human hepatocytes
Primary cultures of animal and human hepatocytes have been extensively used for in vitro testing (e.g. cytotoxicity, CYP enzyme induction, and RDS studies) as they can maintain functional activities for at least 24–72 h. Such cultures permit the medium-throughput screening of compounds and are thus useful for examining inter-species and inter-individual differences. While PB has been shown to stimulate RDS in cultured mouse and rat hepatocytes, many studies from a number of different laboratories have demonstrated that PB does not increase RDS in cultured human hepatocytes (Parzefall et al. Citation1991; Hirose et al. Citation2009; Yamada et al. Citation2015; Okuda et al. Citation2017a; Haines et al. Citation2018b; Peffer et al. Citation2018a; Wiemann et al. Citation2019; Kondo et al. Citation2020; Lake et al. Citation2020; Strupp et al. Citation2020). For example, in both published (Hirose et al. Citation2009; Yamada et al. Citation2015; Okuda et al. Citation2017a; Kondo et al. Citation2020) studies (five male and nine female donors), and unpublished studies (six male and three female donors) from our laboratory, overall PB was shown not to increase hepatocyte RDS in cultured primary human hepatocytes from a total of 23 human donors (11 males and 12 females) with ages ranging from 10 months to 80 years. In addition to studies with PB, a number of other nongenotoxic rodent CAR activators, including benfluralin, metazachlor, metofluthrin, momfluorothrin, the natural pyrethrins, nitrapyrin, sedaxane, and TCPOBOP have also been shown not to increase RDS in cultured human hepatocytes (Hirose et al. Citation2009; Osimitz and Lake Citation2009; Yamada et al. Citation2015; Soldatow et al. Citation2016; LaRocca et al. Citation2017; Okuda et al. Citation2017a; Peffer et al. Citation2018a; Wiemann et al. Citation2019; Lake et al. Citation2020; Strupp et al. Citation2020). As with PB, these studies have been performed in hepatocyte preparations from male and female donors of various ages and have been performed over a wide range of concentrations including cytotoxic concentrations ().
Table 4. Effect of some CAR activators on RDS in cultured rodent and human hepatocytes.
Overall, many studies have demonstrated that human hepatocytes are refractory to the stimulation of RDS by PB and a number of other nongenotoxic CAR activators. However, as also observed in studies with cultured mouse and rat hepatocytes, the functional viability and responsiveness to mitogenic stimuli of the human hepatocyte preparations used in these various studies was confirmed by significant increases in hepatocyte RDS being observed after treatment with known mitogens, namely EGF and/or hepatocyte growth factor (HGF). The observed species difference that CAR activators produce a stimulation of RDS in rat and mouse, but not in human, hepatocytes has been demonstrated by studies conducted in a number of laboratories employing different procedures for hepatocyte isolation and subsequent culture. Studies with rodent hepatocytes have often been performed using standard liver perfusion procedures employing collagenase (Seglen Citation1976; Lake Citation1997); whereas while some investigations with human hepatocytes have employed cells freshly obtained by liver perfusion techniques (e.g. Parzefall et al. Citation1991), more recent studies (e.g. Yamada et al. Citation2015) have employed cryopreserved cells. Rodent hepatocytes prepared by standard techniques will contain some nonparenchymal cells (Seglen Citation1976) and small amounts of nonparenchymal cells are also observed in preparations of cryopreserved human hepatocytes (unpublished observations). Examination of the literature reveals differences between laboratories in cell culture conditions; including medium hormone (e.g. insulin, glucocorticoid), culture medium and other additions. The observed species difference between rodent and human hepatocytes in the stimulation of RDS by CAR activators has thus been demonstrated by a number of laboratories employing various experimental techniques.
While the above studies were performed with monolayer cultures (i.e. two-dimensional cultures), a study by Plummer et al. (Citation2019) examined the effect of PB on RDS in a three-dimensional culture system where either rat or human hepatocytes were co-cultured in a micro-tissue model containing both primary hepatocytes and non-parenchymal cells (Plummer et al. Citation2019). In this spheroid culture system, the treatment of both rat and human hepatocytes with PB resulted in induction of CYP2B and CYP3A mRNA levels. While PB increased RDS in rat hepatocytes, no such effects were observed in human hepatocytes. However, the functional viability of both the rat and human hepatocyte preparations in this spheroid culture module was confirmed by increases in RDS following treatment with HGF.
Studies in transgenic mice with human CAR or both CAR and PXR
Studies have been performed in mice where mouse CAR has been replaced by hCAR (hCAR mice) and where both the mouse CAR and PXR have been replaced by their human counterparts (hCAR/hPXR mice). However, it should be noted that in these transgenic mouse models the human receptor(s) are operating in a mouse hepatocyte environment. The treatment of hCAR mice with 500 ppm PB in the diet for 1 week resulted in increased relative liver weight, Cyp2b10 mRNA levels and hepatocyte RDS (Huang et al. Citation2005). In a study with hCAR/hPXR mice, treatment with either 80 mg/kg/day PB by intraperitoneal injection or 10 mg/kg/day chlordane by oral gavage for four days resulted in increases in relative liver weight and some CYP enzyme activities (Ross et al. Citation2010). Treatment with PB had no significant effect on hCAR/hPXR mouse hepatocyte RDS, whereas treatment with chlordane produced a small but not statistically significant increase in hepatocyte RDS. However, as subsequent studies with hCAR/hPXR mice have demonstrated significant increases in hepatocyte RDS, the apparent lack of effect in the Ross et al. (Citation2010) study is most likely attributable to the treatment time and/or dose levels of the two CAR activators administered. Haines et al. (Citation2018b) treated C57BL/6J (wild-type) and hCAR/hPXR mice with diets containing 186, 496, 654, and 984 ppm PB for seven days. Due to the absence of mouse hepatic CAR and PXR, terminal plasma levels of PB were significantly increased in hCAR/hPXR mice given 654 and 984 ppm PB to 1.8- and 1.7-fold, respectively, of plasma PB levels in wild-type mice. In the wild-type mice, there was a statistically significant dose-dependent increase in hepatocyte RDS at all PB dose levels examined. However, in the hCAR/hPXR mice, hepatocyte RDS was not significantly increased by treatment with 186 ppm PB, and, although increases in hepatocyte RDS at PB dose levels of 496–984 ppm were statistically significant, the increases were less marked than those observed in wild-type mice. In another investigation, Luisier et al. (Citation2014) demonstrated that administration of 0.05% PB in the drinking water to male hCAR/hPXR mice for 91 days increased relative liver weight, hepatocyte hypertrophy, and induction of Cyp2b10 mRNA levels, together with a transient upregulation of genes associated with DNA replication, cell cycle, and mitosis. These genes were also transiently upregulated in wild-type, but not in CAR KO/PXR KO mice. Finally, in a recent study, the treatment of hCAR/hPXR mice with 1000 ppm PB in the diet for seven days was shown to result in significant increases in relative liver weight, hepatocyte RDS, Cyp2b10, and Cyp3a11 mRNA levels and in mRNA levels of some cell cycling genes, namely Mki67, Mdm2, Pcna, and Gadd45β (Yamada et al. Citation2020). The effects of 1000 ppm PB in hCAR/hPXR mice (terminal plasma PB level 48 μg/mL) on hepatocyte RDS and some of the hepatic mRNA levels measured were somewhat similar to those observed in a previous study in CD-1 mice (terminal plasma PB level 43 μg/mL) given 1500 ppm PB for seven days (Yamada et al. Citation2014), although the stimulation of hepatocyte RDS was less marked in the hCAR/hPXR mice (Yamada et al. Citation2020). Overall, these studies demonstrate that the treatment of either hCAR or hCAR/hPXR mice with PB can result in increased hepatocyte RDS, although for hCAR/hPXR mice a dose–response study (Haines et al. Citation2018b) has shown that the effects of PB treatment are less marked than those observed in wild-type mice, which is consistent with the findings in other studies (Braeuning et al. Citation2014; Yamada et al. Citation2020).
Braeuning et al. (Citation2014) performed an initiation/promotion study where wild-type and hCAR/hPXR mice were given a single dose of the DNA reactive carcinogen DEN followed by treatment with 500 ppm PB in the diet for 40 weeks. At the end of the study, liver levels of PB were significantly higher in hCAR/hPXR mice, being some 1.7-fold greater than in wild-type mice. While tumor incidence assessed as either multiplicity or tumor volume fraction was less marked in the hCAR/hPXR mice, PB promoted DEN-initiated liver tumors in both wild type and hCAR/hPXR mice. Based on these findings, these authors suggested that PB-induced liver tumor formation in rodents could be relevant for humans (Braeuning Citation2014; Braeuning et al. Citation2014; Braeuning and Schwarz Citation2016).
In another study, either mCAR or hCAR receptors were introduced into CAR KO mice (Niu et al. Citation2018). The mCAR transgenic mice were subsequently treated with PB or TCPOBOP, whereas the hCAR transgenic mice were subsequently treated with PB or 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl) oxime (CITCO), the latter being a potent hCAR activator (Omiecinski et al. Citation2011a). Subsequent genome-wide profiling analysis demonstrated that regions exhibiting significantly stronger mCAR binding signals were linked to cell proliferation and apoptosis functions, contrasting to hCAR with higher associations with energy/metabolic functions (Niu et al. Citation2018). However, as with other studies with hCAR and hCAR/hPXR mice described above (Huang et al. Citation2005; Braeuning et al. Citation2014; Luisier et al. Citation2014; Haines et al. Citation2018b; Yamada et al. Citation2020), the mCAR and hCAR receptors in the study of Niu et al. (Citation2018) were operating in a mouse hepatocyte environment, including downstream genes and their activation (Niu et al. Citation2018).
Studies in chimeric mice with human hepatocytes
The chimeric mouse model (Foster et al. Citation2014; Sugahara et al. Citation2020; Tateno and Kojima Citation2020) comprises an in vivo alternative to studies in cultured human hepatocytes, showing nearly normal morphology and expressing most genes at levels similar to those expressed by normal human livers. The chimeric mouse model is therefore useful for relatively longer-term studies compared to in vitro studies. In this model, transplanted human hepatocytes replace most of the host mouse hepatocytes. After the first-generation chimeric human hepatocyte mouse model was established in 2001 (Foster et al. Citation2014; Sugahara et al. Citation2020; Tateno and Kojima Citation2020), this model has been increasingly employed for a variety of applications including efficacy studies on agents for the treatment of hepatitis B or C virus, drug metabolism, and pharmacokinetic studies. We employed a chimeric human hepatocyte mouse model, the PXB-mouse® constructed by PhoenixBio Co., Ltd. (Higashi-Hiroshima, Japan) (Sugahara et al. Citation2020; Tateno and Kojima Citation2020), to investigate the hepatic effects of some nongenotoxic CAR activators. The albumin enhancer/promoter-driven urokinase-type plasminogen activator-transgenic severe combined immunodeficient (uPA/SCID) mouse model was employed for the evaluation of PB (Yamada et al. Citation2014), and in subsequent investigations the cDNA-uPA/SCID mouse model (a novel host strain that has a transgene containing albumin promoter/enhancer-driven urokinase-type plasminogen activator cDNA and has a SCID background) was used in studies with metofluthrin and momfluorothrin (Okuda et al. Citation2017a). The cDNA-uPA/SCID mouse model is an improved model which shows a constant increase of body weight and a constant increase in the human hepatocyte replacement index, as there is no deletion of uPA genes and no kidney disorders in this model (Tateno and Kojima Citation2020). Both the uPA/SCID and cDNA-uPA/SCID mouse models are referred to as the PXB-mouse®. Cryopreserved human hepatocytes from a 2 year old Hispanic female were employed in the study of Yamada et al. (Citation2014), with human hepatocytes from this donor and from two other donors (a 2 year old Caucasian male and a 5 year old African American male) being employed in the study of Okuda et al. (Citation2017a). These donors were selected because cells from young subjects are more responsive to stimulation of hepatocellular proliferation and have better replacement ratios than using hepatocytes from older donors (Masumoto et al. Citation2007).
As described above, studies with cultured human hepatocytes have demonstrated that while hepatocyte RDS can be increased by treatment with mitogenic agents such as EGF or HGF, no chemicals (e.g. drugs or agrochemicals) have been reported to induce RDS in human hepatocytes. Treatment with EGF was also shown to enhance RDS in the human hepatocytes of chimeric mice, thus demonstrating that the transplanted human hepatocytes in chimeric mice can respond to a hepatocyte mitogen (Yamada et al. Citation2014; Okuda et al. Citation2017a). In a study with the PXB-mouse®: cDNA-uPA/SCID mouse model, treatment with KMTR2 (an anti-human TRAIL-R2 monoclonal antibody) was shown to result in hepatotoxicity and apoptosis in human hepatocytes, this being associated with an up regulation of cell cycle related functions likely representing cellular regeneration (Nihira et al. Citation2019).
In contrast to the effects of EGF on RDS in human hepatocytes of chimeric mice, studies with PB, metofluthrin and momfluorothrin, did not result in any increase in human hepatocyte RDS (Yamada et al. Citation2014; Okuda et al. Citation2017a). The effect of treatment with diets containing 500, 1000, 1500, and 2500 ppm PB for seven days was examined in male CD-1 mice, in male Wistar rats, in chimeric mice with human hepatocytes, and also in male SCID mice administered 1500 ppm PB (Yamada et al. Citation2014). Due to toxicity, the effect of PB in chimeric mice with human hepatocytes could not be examined at a PB dietary level of 2500 ppm, and only two of five animals given 1500 ppm PB in the diet survived. The effect of PB in chimeric mice with human hepatocytes in this study was thus investigated up to the maximum tolerated dose (MTD) level (1000 ppm). Indeed, terminal plasma PB levels in animals treated with 1000 ppm PB were much higher in chimeric mice with human hepatocytes (75 μg/mL) than in the mice (30 μg/mL) and rats (20 μg/mL), which is most likely due to a difference in chemical intake or a more marked induction of enzymes metabolizing PB in the mouse and rat compared to the chimeric mice with human hepatocytes. The 75 μg/mL is much higher than plasma concentrations reported in human subjects given therapeutic doses of 3–6 mg/kg, where plasma levels ranged from 10 to 25 μg/mL (Monro Citation1993). Treatment with PB resulted in significant increases in relative liver weight, hepatocyte RDS, and induction of CYP2B and CYP3A mRNA levels (i.e. mouse Cyp2b10 and Cyp3a11; rat CYP2B1/2 and CYP3A1) in mice and rats. In marked contrast, while treatment of chimeric mice with human hepatocytes with PB resulted in significant increases in CYP2B6 and CYP3A4 mRNA levels, PB treatment did not increase relative liver weight and did not increase hepatocyte RDS. In addition, PB increased hepatic mRNA levels of some cell cycling genes, namely MKI67, PCNA, and GADD45β, in the livers of mice and rats, but had no effect in human hepatocytes of the chimeric mice. PB increased hepatocyte RDS and induction of CYP2B and CYP3A mRNA levels in SCID mice. In another study, the effects of the known nongenotoxic CAR activators metofluthrin and momfluorothrin, which are known to produce liver tumors in the rat (Yamada et al. Citation2009; Okuda et al. Citation2017b; Yamada Citation2018), were investigated in chimeric mice with human hepatocytes from three separate donors (Okuda et al. Citation2017a). Chimeric mice with human hepatocytes were treated with diets containing 1800 ppm metofluthrin and either 1100 or 3000 ppm momfluorothrin for seven days, these dietary levels resulting in intakes either similar to or higher than those which produced liver tumors in the rat bioassays with these two synthetic pyrethroids. The treatment of chimeric mice with human hepatocytes with either metofluthrin or momfluorothrin in some instances increased CYP2B6 mRNA levels, but had no significant effect on relative liver weight and hepatocyte RDS. When the data from the studies with human hepatocytes from three separate donors were combined, in contrast to the effect of treatment with EGF, levels of RDS in chimeric mice with human hepatocytes given either metofluthrin or momfluorothrin were significantly reduced compared to those in control animals (Okuda et al. Citation2017a).
Recently, the effect of transplanting rat hepatocytes into the chimeric mouse model of cDNA-uPA/SCID mice as recipient has been investigated (Yamada et al. Citation2020). Treatment with 1000 ppm PB in the diet for seven days resulted in significant increases in RDS in rat hepatocytes of the chimeric mice, together with a small increase in MKI67 mRNA levels. The stimulation of hepatocyte RDS by PB in rat hepatocytes of the chimeric mice is in agreement with many other previous in vivo and in vitro studies in rats (Whysner et al. Citation1996; Lake Citation2009, Citation2018; Elcombe et al. Citation2014), and demonstrates that the transplanted rat hepatocytes in the chimeric mouse model retained the original character of normal (i.e. non-transplanted) rat hepatocytes by increasing RDS in response to stimulation by a CAR activator (Yamada et al. Citation2020). In addition to PB, the treatment of chimeric mice with rat hepatocytes with EGF also resulted in a significant increase in RDS and MKI67 mRNA levels in rat hepatocytes. As a stimulation of RDS could be readily demonstrated in transplanted rat hepatocytes in the chimeric mouse model, these results strongly support the conclusion that lack of cell proliferation of human hepatocytes in the chimeric mouse model is not due to any functional problems in the mouse liver environment, but rather that human hepatocytes are truly refractory to the mitogenic effects of PB and other rodent CAR activators.
While we conducted short-term studies in the chimeric mouse model, the ultimate test of carcinogenicity assessment in humans using this chimeric model may be a long-term carcinogenicity study. However, to our knowledge, such a study has not been performed and would be technically very difficult. In addition to the very high cost, several preliminary studies would have to be performed such as determining long-term survival rate, suitable dose levels (MTD), etc.
Gene expression studies with PB in mouse and rat experimental models
A number of gene expression studies have been performed with liver samples from animals treated with CAR activators (Deguchi et al. Citation2009; Ross et al. Citation2010; Luisier et al. Citation2014; Yamada et al. Citation2014, Citation2020; Oshida et al. Citation2015a; Ohara et al. Citation2017; Okuda et al. Citation2017b; Peffer et al. Citation2018a; Rooney et al. Citation2019). Studies conducted in our laboratory include investigations with CD-1 mice, hCAR/hPXR mice, chimeric mice with human hepatocytes, and liver hepatocellular adenoma samples from an initiation/promotion study where C3H/He mice were given a single dose of DEN followed by promotion with 500 ppm PB in the diet for 27 weeks (Ohara et al. Citation2017; Yamada et al. Citation2020). In addition, studies in the rat have included investigations with Sprague-Dawley rats, CAR KO rats, and in chimeric mice with rat hepatocytes (Yamada et al. Citation2020).
Several studies have examined gene expression in the Wnt/β-catenin signaling pathway, where mutations in this pathway have been observed in some mouse and human liver tumors (Dong et al. Citation2015; Braeuning and Pavek Citation2020; Shizu and Yoshinari Citation2020). This signaling pathway regulates key aspects of mammalian cell biology (Thompson and Monga Citation2007; Dong et al. Citation2015). Dong et al. (Citation2015) demonstrated that activation of CAR and β-catenin in mouse liver resulted in liver tumor formation. While a high incidence of β-catenin mutations (e.g. 80–90%) are observed in mouse liver tumors from initiation/promotion studies (Strathmann et al. Citation2006; Rignall et al. Citation2011), the incidence of β-catenin mutations was only 40 and 43%, respectively, in chronic studies where mice were given either PB or oxazepam alone without any prior treatment with a genotoxic agent (Devereux et al. Citation1999; Sidaway et al. Citation2020). While chronic treatment with the potent mouse CAR activator TCPOBOP resulted in a 91% incidence (10 of 11 liver tumor samples examined) of β-catenin mutations in liver tumors, treatment with TCPOBOP did not result in increased expression of β-catenin target genes or the nuclear translocation of β-catenin (Mattu et al. Citation2018).
A comprehensive analysis of DNA methylation, hydroxymethylation, and gene expression examined liver samples from several mouse models (Ohara et al. Citation2017). The groups of CD-1 mice and chimeric mice with human hepatocytes had similar mean terminal PB plasma levels, 70 and 75 μg/mL, respectively, whereas the mean terminal plasma PB level of the C3H/He mice given DEN/PB was 15.4 μg/mL (Yamada et al. Citation2014; Ohara et al. Citation2017). Differences in the gene expression profiles were observed between the PB treated chimeric mice with human hepatocytes, the PB treated CD-1 mice, and the hepatocellular adenomas produced by DEN/PB (Ohara et al. Citation2017). Nine cell proliferation/growth-related genes identified as candidate genes responsible for early events in PB-induced liver tumor promotion were increased in the livers of CD-1 mice treated with PB for seven days and also in the hepatocellular adenomas from the DEN/PB study. In contrast, the treatment of chimeric mice with human hepatocytes with PB for seven days had no effect on the expression of these nine genes. Moreover, while the expression of many genes related to the Wnt/β-catenin signaling network were altered in PB treated CD-1 mice and in the hepatocellular adenomas produced by DEN/PB, fewer genes were altered in the human hepatocytes of the chimeric mice (Ohara et al. Citation2017).
The effect of PB treatment on hepatic gene expression associated with Wnt/β-catenin signaling was evaluated in CD-1 mice given 1500 ppm PB in the diet for seven days, hCAR/hPXR mice given 1000 ppm PB for seven days, and chimeric mice with human hepatocytes given either 500 or 1000 ppm PB for seven days (Yamada et al. Citation2020). The groups of CD-1 mice and hCAR/hPXR mice had similar mean PB terminal plasma levels, 43 and 48 μg/mL, respectively, whereas the mean terminal PB plasma levels in the chimeric mice with human hepatocytes given 500 and 1000 ppm PB were 27 and 75 μg/mL, respectively. Comparisons were also made with the hepatocellular adenomas produced by DEN/PB where the mean terminal plasma PB level of the C3H/He mice given DEN/PB was 15.4 μg/mL (Yamada et al. Citation2014, Citation2020; Ohara et al. Citation2017). The hepatic gene expression pattern for Wnt/β-catenin signaling demonstrated that the pattern of the hCAR/hPXR mice clustered most closely with those of hepatocellular adenoma samples, and clustered with those of the CD-1 mice more than with chimeric mice with human hepatocytes (Yamada et al. Citation2020). Different gene clusters were observed for the chimeric mice with human hepatocytes when compared to those of the hCAR/hPXR mice, CD-1 mice and the hepatocellular adenoma tumor samples. Unlike CD-1 mouse hepatocytes, the exposure of human hepatocytes of the chimeric mice to PB (even at a higher terminal plasma PB level) appeared to have little effect on genes associated with the Wnt/β-catenin signaling pathway (Yamada et al. Citation2020).
Gene expression studies were also performed with liver samples from Sprague-Dawley rats, CAR KO rats, and chimeric mice with rat hepatocytes treated with 1000 ppm PB in the diet for seven days (Yamada et al. Citation2020). The mean terminal plasma PB levels were 34, 100, and 15 μg/mL, respectively. Comparisons were also made with liver samples from chimeric mice with human hepatocytes given 500 and 1000 ppm PB in the diet for seven days, where the mean terminal PB plasma levels were 27 and 75 μg/mL, respectively. Several genes related to cell proliferation or cell growth were significantly changed in liver samples from Sprague-Dawley rats and chimeric mice with rat hepatocytes. In contrast, no such changes were detected in liver samples from CAR KO rats and chimeric mice with human hepatocytes, with the exception that cell growth genes were downregulated in CAR KO rats. The hepatic gene expression pattern of the chimeric mice with rat hepatocytes clustered most closely to those of Sprague-Dawley rats. However, the gene expression pattern of both Sprague-Dawley rats and the chimeric mice with rat hepatocytes resulted in different clusters to those observed in chimeric mice with human hepatocytes.
Interpreting the data regarding gene expression changes in tumors, such as adenomas, for relevance to the actual carcinogenic process is uncertain, and relating it to human relevance is problematic. Just because the same genes are mutated in mouse tumors and in human tumors does not signify that the agent causing the change is relevant to human carcinogenesis. Similar gene changes identified in the studies described above are also found in spontaneous tumors arising in untreated controls, as reported in an extensive series from the NTP data base (Riva et al. Citation2020). Furthermore, there generally are not specific gene mutation patterns in tumors for each different chemical. The changes related to cell proliferation in the studies described above with CAR activators are additional markers for the increased cell proliferation occurring in rodent liver in response to these agents as well as in the tumors (which have a proliferation rate higher than normal hepatocytes).
Comparison of the hepatic effects of CAR, PXR, and PPARα activators
As described above, the CAR, PXR, and PPARα nuclear receptors are highly expressed in liver and are involved in xenobiotic and intermediary metabolism (Omiecinski et al. Citation2011a; Corton et al. Citation2014, Citation2018; Yoshinari Citation2019; Daujat-Chavanieu and Gerbal-Chaloin Citation2020). Many studies have demonstrated that both CAR and PPARα activators can stimulate RDS in mouse and rat hepatocytes (Lake Citation2009, Citation2018; Cohen Citation2010; Corton et al. Citation2014; Elcombe et al. Citation2014; Corton et al. Citation2018; Yamada Citation2018). However, whether PXR activators are also mitogenic agents in rodent liver is less clear and no MOA for rodent liver tumor formation by PXR activators has been established (Elcombe et al. Citation2014; Shizu and Yoshinari Citation2020). For example, pregnenolone-16α-carbonitrile (PCN), which is a known rodent PXR activator (Omiecinski et al. Citation2011a; Yoshinari Citation2019), has been shown to stimulate hepatocyte RDS in mouse and rat liver in some studies (Lake et al. Citation1998; Staudinger et al. Citation2001; Haines et al. Citation2018a, Citation2019), but not in other investigations (Thatcher and Caldwell Citation1994; Shizu et al. Citation2013, Citation2021). While PCN has been reported to increase hepatocyte RDS in wild-type mice and rats in some studies, no such effects were observed in PXR KO mice and in both PXR KO and CAR KO/PXR KO rats; although a small increase was observed in PXR KO rats which may be due to crosstalk between the CAR and PXR receptors (Staudinger et al. Citation2001; Haines et al. Citation2018a, Citation2019). It has been suggested that PXR activation alone in either mouse or rat liver does not result in increased RDS, but rather that PXR activation augments RDS produced by other agents including both CAR and PPARα activators (Shizu et al. Citation2013; Yoshinari Citation2019). PCN did not promote mouse liver tumor formation after initiation with DEN, whereas PB produced the expected promotion of liver tumor formation; with the coadministration of PB and PCN not resulting in any enhancement of liver tumor formation over that observed with PB alone (Shizu et al. Citation2021). Overall, the available data suggest that PXR activation by itself does not result in rodent liver tumor formation.
There are many similarities in the effects of nongenotoxic CAR and PPARα activators which produce liver tumors in the mouse and/or rat. For example, in the established MOAs for rodent liver tumor formation by both CAR and PPARα activators, common key events include receptor activation, increased cell proliferation, clonal expansion leading to altered hepatic foci, and liver tumors; whereas common associative events include liver hepatocellular hypertrophy and CYP enzyme induction (Lake Citation2009, Citation2018; Corton et al. Citation2014, Citation2018; Elcombe et al. Citation2014). While both nongenotoxic CAR and PPARα activators stimulate RDS in mice and rats, a number of studies have demonstrated that PPARα activators do not stimulate hepatocyte RDS in the Syrian hamster, guinea pig and primates, and do not produce liver tumors in the Syrian hamster (Tucker and Orton Citation1995; Lake Citation2009; Corton et al. Citation2014, Citation2018). In a study in marmosets, the PPARα activator clofibrate was shown not to produce liver tumors after 6.5 years of treatment, although this is less than the expected life-span of this species (Corton et al. Citation2018). Several studies have demonstrated that rodent PPARα activators do not stimulate RDS in cultured human hepatocytes (Lake Citation2009; Corton et al. Citation2014, Citation2018; Kondo et al. Citation2020). In vivo studies with chimeric mice with human hepatocytes have demonstrated that the PPARα activator fenofibrate does not increase RDS in human hepatocytes and results in downregulation of pathways related to DNA synthesis (Tateno et al. Citation2015; de la Rosa Rodriguez et al. Citation2018) similar to the results with CAR activators in chimeric mice with human hepatocytes. Finally, a number of human epidemiological studies have clearly demonstrated that rodent PPARα activators (e.g. the hypolipidemic drugs bezafibrate, clofibrate, fenofibrate, and gemfibrozil and the insecticide permethrin) do not increase the incidence of liver cancer in humans (Bonovas et al. Citation2012; Corton et al. Citation2014, Citation2018; Boffetta and Desai Citation2018).
For both CAR activators and PPARα activators, human responses in terms of CYP enzyme induction and/or a hypolipidemic effect are similar to those observed in mice and rats. However, the pivotal difference is that unlike mice and rats, a proliferative effect is not observed in human hepatocytes. Since this is a necessary key event for liver tumor formation, this strongly supports the conclusion that neither CAR nor PPARα activators will be carcinogenic in humans.
Discussion
By use of the established IPCS/WHO framework for MOA/human relevance, a robust MOA for PB-induced rodent liver tumor formation has been established (Holsapple et al. Citation2006; Lake Citation2009, Citation2018; Elcombe et al. Citation2014). The findings meet the criteria of temporality and dose response, are consistent between laboratories, and are biologically plausible. The evidence is strong, reproducible, and coherent, meeting all of the Bradford Hill considerations of causality.
This MOA has been shown to apply to a number of other nongenotoxic CAR activators which produce liver tumors in the mouse and/or rat (). The crucial role of CAR activation in this MOA has been clearly demonstrated by studies with animals lacking hepatic CAR where, unlike wild-type animals, effects on the key (e.g. hepatocyte RDS) and associative events are not observed. The MOA for rodent (i.e. mouse and rat) liver tumor formation by PB and other CAR activators is considered to be qualitatively not plausible for humans on the basis that while CAR activators are mitogenic agents in rodent liver, they do not stimulate RDS in human hepatocytes (Lake Citation2009, Citation2018; Cohen Citation2010; Cohen and Arnold Citation2011, Citation2016; Elcombe et al. Citation2014; Yamada Citation2018). While much experimental data has been obtained in studies with PB as the model CAR activator, as shown in , there are now many other nongenotoxic CAR activators which exhibit the same overall MOA for rodent liver tumor formation and lack of human relevance.
The mitogenic effects of a nongenotoxic CAR activator in rodent as against human liver is thus the pivotal species difference in assessing the human relevance of the established MOA for CAR activator-induced rodent liver tumor formation. The data obtained from the three experimental systems described above is assessed below, together with consideration of species differences and a suggested minimal dataset required to establish a CAR-dependent MOA for rodent liver tumor formation.
Assessment of available experimental models employing either human hepatocytes or human hepatic receptors
As described above, many studies from a number of different laboratories have demonstrated a reproducible species difference in that PB and other nongenotoxic CAR activators can stimulate RDS in cultured mouse and/or rat hepatocytes, but do not stimulate RDS in cultured human hepatocytes obtained from both male and female donors from a wide range of ages. It has been suggested that monolayer (i.e. two-dimensional) cultures are not an appropriate model of human liver function and that EGF is not a suitable positive control (Shizu and Yoshinari Citation2020). However, cultured human hepatocytes have been extensively used by many laboratories and are considered the “gold standard” in vitro system for many applications (Soldatow et al. Citation2013). For example, the use of cultured human hepatocytes to reliably screen xenobiotics for induction of human CYP enzymes (e.g. CYP1A, CYP2B, CYP2C, and CYP3A subfamily enzymes) is well established (Parkinson et al. Citation2004; Lake and Price Citation2016). For studies with both rodent and human hepatocytes, either EGF and/or HGF have been employed as markers of functional viability of the rodent and human hepatocyte preparations. The use of a growth factor, rather than a chemical, as a positive control is required because there are no chemicals that have been shown to produce a stimulation of RDS in cultured human hepatocytes. While most studies have been performed with monolayer (i.e. two-dimensional) cultures, a recent study using hepatocyte spheroids (i.e. a three-dimensional culture system including non-parenchymal cells) produced the expected results in that PB stimulated RDS in rat hepatocytes, but no such effect was observed in human hepatocytes (Plummer et al. Citation2019). Overall, it is considered that studies with cultured rodent and human hepatocytes are a validated in vitro model system to permit the evaluation of species differences in the effects of CAR activators on RDS in hepatocytes from experimental animals and humans.
A number of investigations have been performed in transgenic mice where either murine hepatic CAR has been replaced with hCAR (hCAR mice) or both murine CAR and PXR have been replaced with their human counterparts (hCAR/hPXR mice). Studies in both hCAR (Huang et al. Citation2005) and hCAR/hPXR (Luisier et al. Citation2014; Haines et al. Citation2018b; Yamada et al. Citation2020) mice have demonstrated that treatment with PB results in liver hypertrophy, increases in Cyp2b enzymes and a stimulation of hepatocyte RDS. In addition, treatment with PB has also been shown to increase some cell cycling genes in hCAR/hPXR mice (Luisier et al. Citation2014; Yamada et al. Citation2020). One quantitative difference between wild-type mice and hCAR/hPXR mice is that PB produces a more marked stimulation of hepatocyte RDS in wild-type mice as demonstrated in a dose–response study where wild-type and hCAR/hPXR mice were given 186–984 ppm PB in the diet for seven days (Haines et al. Citation2018b). Braeuning et al. (Citation2014) performed an initiation/promotion study with DEN/PB in wild-type and hCAR/hPXR mice. While tumor production assessed as either multiplicity or tumor volume fraction was less marked in the hCAR/hPXR mice, PB promoted DEN-initiated liver tumors in both wild-type and hCAR/hPXR mice. The magnitude of liver tumor formation in this initiation/promotion study thus correlates with the more marked stimulation of hepatocyte RDS by PB in wild-type mice (Haines et al. Citation2018b). Indeed, because of the similar responses of wild-type and hCAR/hPXR mice to PB, a significant increase in liver tumor formation in a DEN/PB initiation/promotion study is to be expected.
The chimeric mouse model offers an in vivo alternative to in vitro cultured hepatocyte studies to examine the effects of chemicals in human hepatocytes. The treatment of chimeric mice with human hepatocytes with PB at up to MTD dose levels and with both metofluthrin and momfluorothrin (at dose levels which produced liver tumors in rats) did not result in a stimulation of RDS in human hepatocytes; whereas the treatment of chimeric mice with EGF did result in significant increases in RDS in human hepatocytes (Yamada et al. Citation2014; Okuda et al. Citation2017a). The reliability and scientific credibility of the chimeric mouse model with donor hepatocytes from other species has been recently confirmed by studies in chimeric mice with rat hepatocytes (Yamada et al. Citation2020). As would be expected from many previous in vivo studies in wild-type rats, the treatment of wild-type rats and chimeric mice with rat hepatocytes with PB resulted in significant increases in hepatocyte RDS; whereas no such effect was observed in CAR KO rats. An increase in hepatocyte RDS was also observed in chimeric mice with rat hepatocytes following treatment with EGF. Gene expression studies demonstrated that several genes related to cell proliferation or cell growth were significantly changed by PB treatment in liver samples from wild-type rats and from chimeric mice with rat hepatocytes; whereas such effects were not observed in liver samples from CAR KO rats.
In terms of species differences between effects of CAR activators in mouse and rat hepatocytes compared to human hepatocytes, the data obtained in in vitro studies with cultured human hepatocytes and in in vivo studies with chimeric mice with human hepatocytes clearly demonstrate that PB and other rodent CAR activators do not stimulate either RDS or some genes associated with cell proliferation in human hepatocytes. Such data are in marked contrast to the results of studies with hCAR and hCAR/hPXR mice where PB has been shown to induce hepatocyte RDS and some genes associated with cell proliferation.
summarizes the differences between wild-type mice, CAR KO mice, transgenic mice with either hCAR or hCAR and PXR receptors and chimeric mice with human hepatocytes. Studies in wild-type mice and in hCAR/hPXR mice clearly demonstrate that PB and other CAR activators can induce endpoints of CAR activation including induction of Cyp2b enzymes and hepatocyte RDS, the latter being associated with increases in some genes associated with cell proliferation. In marked contrast, such effects are not observed in either CAR KO or CAR KO/PXR KO mice thus confirming the need for receptor activation. The pivotal species difference as shown in is that while PB and other CAR activators can induce CYP2B (also CYP3A) enzymes in chimeric mice with human hepatocytes, there is no stimulation of hepatocyte RDS. The lack of effect of PB on RDS in human hepatocytes of chimeric mice is supported by the results of gene array studies, where increases in genes associated with cell proliferation were not observed (Yamada et al. Citation2014, Citation2020). The differences between the effect of PB in wild-type, hCAR/hPXR mice and in chimeric mice with human hepatocytes are not attributable to the dose administered and to subsequent PB exposure as in the chimeric mouse study, PB was administered at up to MTD dose levels, which resulted in greater terminal plasma PB levels than those observed in wild-type mice (Yamada et al. Citation2014).
Figure 1. Effect of CAR activators in different animal models, namely wild-type mice, CAR KO or CAR KO/PXR KO mice, hCAR/hPXR mice and chimeric mice with human hepatocytes. Pathways which do not operate are shown in grey. Thus while Cyp2b enzyme induction and increased cell proliferation are observed in wild-type and hCAR/hPXR mice, these effects are abolished in CAR KO and CAR KO/PXR KO mice due to the absence of the receptor(s). In chimeric mice with human hepatocytes, the human receptors operate through human signaling pathways to produce an induction of CYP2B enzymes but not of cell proliferation.
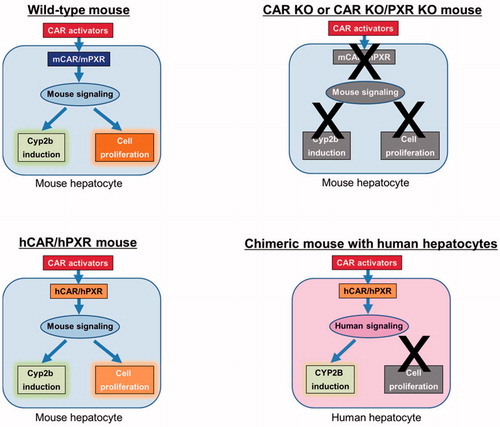
The species difference in PB-induced hepatocyte proliferation could be due to differences in structure/splice variants between the human and mouse CAR proteins and other possible differences, such as: (1) while the DNA binding domains of human and mouse CAR are highly conserved (around 88%), the homology of the ligand binding domains is much weaker (around 70%) (Shizu and Yoshinari Citation2020); (2) a variety of compounds differ with regard to their EC50s/IC50s, and whether they act as agonists, inverse agonists or have no effect when evaluated for effectiveness on hCAR vs. mCAR (Stanley et al. Citation2006); (3) nucleocytoplasmic shuttling sequences differ in rCAR vs. hCAR (Kanno et al. Citation2007); (4) isoform differences mCAR vs. hCAR (Choi et al. Citation1997); and (5) CAR1 and CAR3 reporter assays demonstrate species differences in human, mouse, rat, and dog (Omiecinski et al. Citation2011a). In addition to the differences in the structure/splice variants of CAR, receptor regulation differences could also possibly contribute. From the data obtained, the key difference between the experimental models evaluated is that in cultured human hepatocytes and in chimeric mice with human hepatocytes, hCAR is operating in a human hepatocyte environment, which is not the case for either the hCAR or hCAR/hPXR models. The human receptor(s) in the hCAR and hCAR/hPXR mouse models function in the context of mouse target gene regulatory elements and chromatin structure, and hence gene regulatory protein interactions may differ from those in human hepatocytes (Braeuning Citation2014; Braeuning et al. Citation2014; Yamada Citation2018; Shizu and Yoshinari Citation2020; Yamada et al. Citation2020). hCAR in a mouse hepatocyte environment undergoes post-translational modification(s) that is not the same as in human hepatocytes, possibly due to expression of CAR activators/inhibitors differences, the mouse orthologous genes encoding CAR activators/inhibitors might differ enough from their human orthologs so as to affect CAR differently, and the cis elements in CAR-regulated genes in the mouse might respond differently to hCAR because hCAR is structurally (including a consideration of isoforms) somewhat different from mCAR. Thus, while activation of CAR in a human hepatocyte environment does not result in a proliferative response, the human receptors present in either hCAR or hCAR/hPXR mouse hepatocytes are still able to activate downstream genes leading to the metabolic and proliferative effects expected for a CAR activator like PB, albeit at lower levels of response.
Overall, while cultured human hepatocytes represent a rapid in vitro test system to evaluate species differences in the hepatic effects of rodent CAR activators, if required to confirm in vitro data or to evaluate effects of human metabolites of the test chemical, in vivo studies can be conducted in chimeric mice with human hepatocytes. In contrast, studies in either hCAR or hCAR/hPXR mice will not provide useful data for human risk assessment.
Differences between the effects of CAR and PPARα activators in rodent hepatocytes compared to other species
As described above, there are a number of similarities between the MOAs for rodent liver tumor formation by CAR and PPARα activators. For both CAR and PPARα activators, many studies have demonstrated that while these agents can stimulate RDS in mouse and rat hepatocytes, they are not mitogenic agents in human hepatocytes. Similarly, both CAR and PPARα activators have been shown not to induce RDS in the in vivo chimeric mouse model with human hepatocytes. Thus, studies with PPARα activators confirm and extend a basic species difference in response to rodent hepatic mitogenic agents. For CAR activators, limited data are available that PB does not induce hepatocyte RDS in Syrian hamster hepatocytes, and studies with PB, DDT, dieldrin, and toxaphene have been shown not produce liver tumors in the Syrian hamster. PB has also been shown not to increase the incidence of liver tumors in initiation/promotion studies conducted in the Syrian hamster. For PPARα activators, considerable in vitro and in vivo data are available showing that PPARα activators do not stimulate RDS in either the Syrian hamster or guinea pig (Lake Citation2009; Corton et al. Citation2014, Citation2018). Moreover, rodent PPARα activators have also been shown not to stimulate RDS in primate liver and also not to produce liver tumors in the Syrian hamster (Tucker and Orton Citation1995; Corton et al. Citation2018). The data from species differences studies with both CAR and PPARα activators clearly demonstrate that while CAR and PPARα activators are mitogenic agents in mouse and rat liver, they are not mitogenic agents in several other species including the Syrian hamster, guinea pig, primates, and humans.
Overall, while nongenotoxic agents (e.g. rodent CAR and PPARα activators) can stimulate RDS in mouse and rat hepatocytes, several other species, including humans, are refractory to the mitogenic effects of rodent CAR and PPARα activators. Since stimulation of RDS is a required key event for tumorigenesis with these chemicals in rats and mice, its absence in humans means that humans are not susceptible to their tumorigenic effects. This MOA/human relevance evaluation is further supported by the findings of human epidemiology studies described above.
Minimum data required to establish a CAR-activation MOA for rodent liver tumor formation
As described elsewhere (Cohen Citation2010; Elcombe et al. Citation2014; Lake Citation2018; Peffer et al. Citation2018b), data to demonstrate a CAR-dependent MOA for rodent liver tumor formation can be obtained from short-term studies (e.g. 1–4 weeks duration). For established nongenotoxic compounds which have been evaluated in long-term bioassays, short-term MOA studies should be conducted at both carcinogenic and noncarcinogenic dose levels. Required endpoints would include liver hypertrophy (both liver weight and morphology), hepatocyte RDS, and induction of CYP2B enzymes (both enzyme activity and/or mRNA levels). In such studies, CYP2B induction would be an acceptable surrogate marker for CAR receptor activation. To exclude involvement of either AhR, PXR, or PPARα receptors, assays of markers for CYP1A, CYP3A, and CYP4A subfamily enzymes (both enzyme activity and/or mRNA levels) would need to be performed. Data from other available studies (e.g. 13 week studies) can be employed to exclude other MOAs such as cytotoxicity and metal overload (). Gene array studies are now regularly performed in the development of new compounds, and a number of publications have identified gene signatures for activation of AhR, CAR, and PPARα (Deguchi et al. Citation2009; Oshida et al. Citation2015a, Citation2015b, Citation2015c; Okuda et al. Citation2017b; Peffer et al. Citation2018a; Rooney et al. Citation2018; Kondo et al. Citation2019) which may provide an alternative approach to the determination of CYP enzyme and mRNA markers (Peffer et al. Citation2018b). For example, Rooney et al. (Citation2018) have described a novel adverse outcome pathway-driven strategy employing gene array data and other endpoints to identify compounds which can produce liver tumors in rats. A number of reporter gene assays are also available to demonstrate CAR activation (Omiecinski et al. Citation2011a; Currie et al. Citation2014; Pinne et al. Citation2016, Citation2017; Peffer et al. Citation2018a; Lynch et al. Citation2019), but the data obtained need careful evaluation as not all CAR reporter gene assays can detect indirect CAR activators such as PB. Pinne et al. (Citation2017) reported that cell based transactivation assays employing the full-length receptors and native promoters identify both direct and indirect activators of either or both human PXR and CAR.
For new nongenotoxic compounds under development, long-term carcinogenicity studies are currently required by regulatory authorities, although there is movement toward granting waivers to eliminate the need for the long-term bioassay based on MOA considerations (Sistare et al. Citation2011; Craig et al. Citation2019). Potential hepatic effects of chemicals can be screened for in studies of up to 13 weeks duration (Allen et al. Citation2004; Cohen Citation2010), including effects produced by CAR activators. If an effect is detected in this screen, evaluation of MOA can be rapidly investigated as described above. Thus, effects on liver weight and morphology as being due to CAR activation can be confirmed by a 1 week study to evaluate effects on hepatocyte RDS, with liver samples from this investigation also being used to determine effects on CYP enzymes and/or to perform gene profiling studies. Alternative MOAs can be similarly investigated. Since the CAR-dependent MOA for rodent liver tumor formation is not relevant for humans, demonstrating that the MOA for liver proliferative alterations is CAR-dependent in rodents based on data described above, it would not be necessary to perform studies in either cultured human hepatocytes or in chimeric mice with human hepatocytes.
Conclusions
A robust MOA for rodent liver tumor formation by PB and other nongenotoxic CAR activators has been established and includes the key events of CAR activation, increased hepatocellular proliferation, formation of altered hepatic cell foci, and the ultimate development of adenomas and carcinomas. In terms of human relevance, the pivotal species difference is that CAR activators are mitogenic agents in mouse and rat hepatocytes, but they do not stimulate RDS in human hepatocytes. This conclusion is supported by substantial in vitro data with cultured hepatocytes and also by in vivo data from studies with chimeric mice with human hepatocytes. In contrast, data from transgenic mice with either hCAR or hCAR and PXR are not useful because, while human nuclear receptors are present, they function in a mouse hepatocyte environment. Moreover, the data obtained from such models are similar to that obtained from wild-type mice. Examination of the literature reveals many similarities in the hepatic effects and species differences between rodent CAR and PPARα activators. While CAR and PPARα activators can stimulate RDS in mouse and rat hepatocytes, such effects are not observed in other species, including humans. Overall, a critical analysis of the available data demonstrates that the established MOA for rodent liver tumor formation by PB and other CAR activators is qualitatively not plausible for humans. This conclusion is supported by the data from a number of human epidemiology studies showing no increased risk of liver or other tumors.
| Abbreviations | ||
| BRDU | = | 5-bromo-2′-deoxyuridine |
| CAR | = | constitutive androstane receptor |
| CITCO | = | 6-(4-chlorophenyl)imidazo[2,1-b][1,3]thiazole-5-carbaldehyde O-(3,4-dichlorobenzyl)oxime |
| CYP | = | cytochrome P450 |
| DEN | = | diethylnitrosamine |
| DDT | = | 1,1′-(2,2,2-trichloroethylidene)-bis[4-chlorobenzene] |
| ECHA | = | European Chemicals Agency |
| EDU | = | 5-ethynyl-2′-deoxyuridine |
| EGF | = | epidermal growth factor |
| GADD45β | = | growth arrest and DNA-damage-inducible 45 beta |
| HCC | = | hepatocellular carcinoma |
| HGF | = | hepatocyte growth factor |
| hCAR | = | human constitutive androstane receptor |
| hCAR/hPXR | = | human constitutive androstane receptor/human pregnane X receptor |
| IARC | = | International Agency for Research on Cancer |
| ILSI | = | International Life Sciences Institute |
| IPCS | = | International Programme on Chemical Safety |
| KO | = | knockout |
| mCAR | = | mouse constitutive androstane receptor |
| MKI67 | = | marker of proliferation Ki-67 |
| MOA | = | mode of action |
| MTD | = | maximum tolerated dose |
| NaPB | = | sodium phenobarbital |
| PB | = | phenobarbital |
| PCN | = | pregnenolone-16α-carbonitrile |
| PCNA | = | proliferating cell nuclear antigen |
| PPARα | = | peroxisome proliferator-activated receptor alpha |
| PXR | = | pregnane X receptor |
| RDS | = | replicative DNA synthesis |
| RXR | = | retinoid X receptor |
| TCPOBOP | = | 1,4-bis[2-(3,5-dichloropyridyloxy)]benzene |
| uPA/SCID mouse | = | albumin enhancer/promoter-driven urokinase-type plasminogen activator-transgenic severe combined immunodeficient mouse |
| US.EPA | = | US Environmental Protection Agency |
| US.NTP | = | US National Toxicology Program |
| WHO | = | World Health Organization. |
Acknowledgements
The authors gratefully acknowledge Dr. Chris Corton (US.EPA, Research Triangle Park, NC, USA) for discussions on the interpretation of the results of gene array studies for compounds which are CAR activators in rodent liver; Dr. Chise Tateno (PhoenixBio Co., Ltd., Higashi-Hiroshima, Japan) for review and valuable comment on the draft manuscript in particular of characterization and application of chimeric mouse models; members of Environmental Health Science Laboratory, Sumitomo Chemical Company, Ltd. for their encouragement to write this manuscript; and to the external reviewers selected by the Editor and anonymous to the authors whose comments were very valuable in revising and refining the manuscript. All authors gave final approval and agreed to be accountable for all aspects of work in ensuring that questions relating to the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Declaration of interest
Dr. Tomoya Yamada is employed by Sumitomo Chemical Company, Ltd. Prof. Samuel M. Cohen and Prof. Brian G. Lake consult for Sumitomo Chemical Company, Ltd. regarding research on CAR activators as well as other matters. The manuscript was written as part of the authors’ normal employment, but the authors have sole responsibility for the writing and content of this paper. The views presented in this manuscript are those of the authors based on many years of research in the respective areas of investigation reported in this paper. This work was supported by the Sumitomo Chemical Company, Ltd. (https://www.sumitomo-chem.co.jp/english/company/). This company is interested in the human relevance of the CAR-mediated MOA for rodent liver tumor formation, because some company products (e.g. pesticides) have CAR activation activity in rodent liver. Some of the research on CAR activators produced by Sumitomo Chemical Company, Ltd. has been utilized by the company in regulatory submissions to provide MOA information for interpreting the relevance to humans of the liver tumors in rodents. An additional review of the paper for internal approval purposes was conducted by Dr. Yoshitaka Tomigahara, a previous research director of Environmental Health Science Laboratory, Sumitomo Chemical Company, Ltd. No external funding was obtained for manuscript preparation. None of the authors has appeared in any legal or regulatory proceedings related to the contents of this paper. No potential conflict of interest was reported by the authors.
References
- Allen DG, Pearse G, Haseman JK, Maronpot RR. 2004. Prediction of rodent carcinogenesis: an evaluation of prechronic liver lesions as forecasters of liver tumors in NTP carcinogenicity studies. Toxicol Pathol. 32(4):393–401.
- Allen JW, Wolf DC, George MH, Hester SD, Sun G, Thai SF, Delker DA, Moore T, Jones C, Nelson G, et al. 2006. Toxicity profiles in mice treated with hepatotumorigenic and non-hepatotumorigenic triazole conazole fungicides: propiconazole, triadimefon, and myclobutanil. Toxicol Pathol. 34(7):853–862.
- Andersen ME, Preston RJ, Maier A, Willis AM, Patterson J. 2014. Dose–response approaches for nuclear receptor-mediated modes of action for liver carcinogenicity: results of a workshop. Crit Rev Toxicol. 44(1):50–63.
- Boffetta P, Desai V. 2018. Exposure to permethrin and cancer risk: a systematic review. Crit Rev Toxicol. 48(6):433–442.
- Bogen KT. 2018. Biphasic hCAR inhibition-activation by two aminoazo liver carcinogens. Nucl Recept Res. 5:12.
- Bonovas S, Nikolopoulos GK, Bagos PG. 2012. Use of fibrates and cancer risk: a systematic review and meta-analysis of 17 long-term randomized placebo-controlled trials. PLoS One. 7(9):e45259.
- Boobis AR, Cohen SM, Dellarco V, McGregor D, Meek ME, Vickers C, Willcocks D, Farland W. 2006. IPCS framework for analyzing the relevance of a cancer mode of action for humans. Crit Rev Toxicol. 36(10):781–792.
- Boobis AR, Doe JE, Heinrich-Hirsch B, Meek ME, Munn S, Ruchirawat M, Schlatter J, Seed J, Vickers C. 2008. IPCS framework for analyzing the relevance of a noncancer mode of action for humans. Crit Rev Toxicol. 38(2):87–96.
- Braeuning A. 2014. Liver cell proliferation and tumor promotion by phenobarbital: relevance for humans? Arch Toxicol. 88(10):1771–1772.
- Braeuning A, Gavrilov A, Brown S, Wolf CR, Henderson CJ, Schwarz M. 2014. Phenobarbital-mediated tumor promotion in transgenic mice with humanized CAR and PXR. Toxicol Sci. 140(2):259–270.
- Braeuning A, Pavek P. 2020. β-Catenin signaling, the constitutive androstane receptor and their mutual interactions. Arch Toxicol. 94(12):3983–3991.
- Braeuning A, Schwarz M. 2016. Is the question of phenobarbital as potential liver cancer risk factor for humans really resolved? Arch Toxicol. 90(6):1525–1526.
- Bucher JR, Shackelford CC, Haseman JK, Johnson JD, Kurtz PJ, Persing RL. 1994. Carcinogenicity studies of oxazepam in mice. Fundam Appl Toxicol. 23(2):280–297.
- Butler WH, Gabriel KL, Osimitz TG, Preiss FJ. 1998. Oncogenicity studies of piperonyl butoxide in rats and mice. Hum Exp Toxicol. 17(6):323–330.
- Cabral JRP, Hall RK, Bronczyk SA, Shubik P. 1979. A carcinogenicity study of the pesticide dieldrin in hamsters. Cancer Lett. 6(4–5):241–246.
- Cabral JR, Hall RK, Rossi L, Bronczyk SA, Shubik P. 1982. Lack of carcinogenicity of DDT in hamsters. Tumori. 68(1):5–10.
- Carthew P, Edwards RE, Nolan BM. 1998. The quantitative distinction of hyperplasia from hypertrophy in hepatomegaly induced in the rat liver by phenobarbital. Toxicol Sci. 44(1):46–51.
- Choi HS, Chung M, Tzameli I, Simha D, Lee YK, Seol W, Moore DD. 1997. Differential transactivation by two isoforms of the orphan nuclear hormone receptor CAR. J Biol Chem. 272(38):23565–23571.
- Choi CJ, Rushton EK, Vardy A, Higgins L, Augello A, Parod RJ. 2017. Mode of action and human relevance of THF-induced mouse liver tumors. Toxicol Lett. 276:138–143.
- Cohen SM. 2004. Human carcinogenic risk evaluation: an alternative approach to the two-year rodent bioassay. Toxicol Sci. 80(2):225–229.
- Cohen SM. 2010. Evaluation of possible carcinogenic risk to humans based on liver tumors in rodent assays: the two-year bioassay is no longer necessary. Toxicol Pathol. 38(3):487–501.
- Cohen SM. 2017. The relevance of experimental carcinogenicity studies to human safety. Curr Opin Toxicol. 3:6–11.
- Cohen SM, Arnold LL. 2011. Chemical carcinogenesis. Toxicol Sci. 120(Suppl. 1):S76–S92.
- Cohen SM, Arnold LL. 2016. Critical role of toxicologic pathology in a short-term screen for carcinogenicity. J Toxicol Pathol. 29(4):215–227.
- Cohen SM, Boobis AR, Dellarco VL, Doe JE, Fenner-Crisp PA, Moretto A, Pastoor TP, Schoeny RS, Seed JG, Wolf DC. 2019. Chemical carcinogenicity revisited 3: risk assessment of carcinogenic potential based on the current state of knowledge of carcinogenesis in humans. Regul Toxicol Pharmacol. 103:100–105.
- Cohen SM, Ellwein LB. 1990. Cell proliferation in carcinogenesis. Science. 249(4972):1007–1011.
- Cohen SM, Klaunig J, Meek ME, Hill RN, Pastoor T, Lehman-McKeeman L, Bucher J, Longfellow DG, Seed J, Dellarco V, et al. 2004. Evaluating the human relevance of chemically induced animal tumors. Toxicol Sci. 78(2):181–186.
- Corton JC, Cunningham ML, Hummer BT, Lau C, Meek B, Peters JM, Popp JA, Rhomberg L, Seed J, Klaunig JE. 2014. Mode of action framework analysis for receptor-mediated toxicity: the peroxisome proliferator-activated receptor alpha (PPARα) as a case study. Crit Rev Toxicol. 44(1):1–49.
- Corton JC, Peters JM, Klaunig JE. 2018. The PPARα-dependent rodent liver tumor response is not relevant to humans: addressing misconceptions. Arch Toxicol. 92(1):83–119.
- Craig E, Lowe K, Akerman G, Dawson J, May B, Reaves E, Lowit A. 2019. Reducing the need for animal testing while increasing efficiency in a pesticide regulatory setting: lessons from the EPA Office of Pesticide Programs' Hazard and Science Policy Council. Regul Toxicol Pharmacol. 108:104481.
- Cui X, Thomas A, Han Y, Palamanda J, Montgomery D, White RE, Morrison RA, Cheng KC. 2005. Quantitative PCR assay for cytochromes P450 2B and 3A induction in rat precision-cut liver slices: correlation study with induction in vivo. J Pharmacol Toxicol Methods. 52(2):234–243.
- Cunningham ML, Maronpot RR, Thompson M, Bucher JR. 1994. Early responses of the liver of B6C3F1 mice to the hepatocarcinogen oxazepam. Toxicol Appl Pharmacol. 124(1):31–38.
- Currie RA, Peffer RC, Goetz AK, Omiecinski CJ, Goodman JI. 2014. Phenobarbital and propiconazole toxicogenomic profiles in mice show major similarities consistent with the key role that constitutive androstane receptor (CAR) activation plays in their mode of action. Toxicology. 321:80–88.
- Daujat-Chavanieu M, Gerbal-Chaloin S. 2020. Regulation of CAR and PXR expression in health and disease. Cells. 9(11):2395.
- de la Iglesia FA, Barsoum N, Gough A, Mitchell L, Martin RA, Di Fonzo C, McGuire EJ. 1981. Carcinogenesis bioassay of prazepam (Verstran) in rats and mice. Toxicol Appl Pharmacol. 57(1):39–54.
- de la Rosa Rodriguez MA, Sugahara G, Hooiveld G, Ishida Y, Tateno C, Kersten S. 2018. The whole transcriptome effects of the PPARalpha agonist fenofibrate on livers of hepatocyte humanized mice. BMC Genomics. 19(1):443.
- Deguchi Y, Yamada T, Hirose Y, Nagahori H, Kushida M, Sumida K, Sukata T, Tomigahara Y, Nishioka K, Uwagawa S, et al. 2009. Mode of action analysis for the synthetic pyrethroid metofluthrin-induced rat liver tumors: evidence for hepatic CYP2B induction and hepatocyte proliferation. Toxicol Sci. 108(1):69–80.
- Dekant W. 2019. Tetrahydrofuran-induced tumors in rodents are not relevant to humans: quantitative weight of evidence analysis of mode of action information does not support classification of tetrahydrofuran as a possible human carcinogen. Regul Toxicol Pharmacol. 109:104499.
- Devereux TR, Anna CH, Foley JF, White CM, Sills RC, Barrett JC. 1999. Mutation of beta-catenin is an early event in chemically induced mouse hepatocellular carcinogenesis. Oncogene. 18(33):4726–4733.
- Diwan BA, Ward JM, Anderson LM, Hagiwara A, Rice JM. 1986. Lack of effect of phenobarbital on hepatocellular carcinogenesis initiated by N-nitrosodiethylamine or methylazoxymethanol acetate in male Syrian golden hamsters. Toxicol Appl Pharmacol. 86(2):298–307.
- Doe JE, Boobis AR, Dellarco V, Fenner-Crisp PA, Moretto A, Pastoor TP, Schoeny RS, Seed JG, Wolf DC. 2019. Chemical carcinogenicity revisited 2: current knowledge of carcinogenesis shows that categorization as a carcinogen or non-carcinogen is not scientifically credible. Regul Toxicol Pharmacol. 103:124–129.
- Dong B, Lee JS, Park YY, Yang F, Xu G, Huang W, Finegold MJ, Moore DD. 2015. Activating CAR and β-catenin induces uncontrolled liver growth and tumorigenesis. Nat Commun. 6:5944.
- Elcombe CR, Peffer RC, Wolf DC, Bailey J, Bars R, Bell D, Cattley RC, Ferguson SS, Geter D, Goetz A, et al. 2014. Mode of action and human relevance analysis for nuclear receptor-mediated liver toxicity: a case study with phenobarbital as a model constitutive androstane receptor (CAR) activator. Crit Rev Toxicol. 44(1):64–82.
- Ellwein LB, Cohen SM. 1988. A cellular dynamics model of experimental bladder cancer: analysis of the effect of sodium saccharin in the rat. Risk Anal. 8(2):215–221.
- Faucette SR, Zhang TC, Moore R, Sueyoshi T, Omiecinski CJ, LeCluyse EL, Negishi M, Wang H. 2007. Relative activation of human pregnane X receptor versus constitutive androstane receptor defines distinct classes of CYP2B6 and CYP3A4 inducers. J Pharmacol Exp Ther. 320(1):72–80.
- Foster JR, Lund G, Sapelnikova S, Tyrrell DL, Kneteman NM. 2014. Chimeric rodents with humanized liver: bridging the preclinical/clinical trial gap in ADME/toxicity studies. Xenobiotica. 44(2):109–122.
- Franzosa JA, Bonzo JA, Jack J, Baker NC, Kothiya P, Witek RP, Hurban P, Siferd S, Hester S, Shah I, et al. 2021. High-throughput toxicogenomic screening of chemicals in the environment using metabolically competent hepatic cell cultures. NPJ Syst Biol Appl. 7(1):7.
- Friedman GD, Jiang S-F, Udaltsova N, Quesenberry CP, Chan J, Habel LA. 2009. Epidemiologic evaluation of pharmaceuticals with limited evidence of carcinogenicity. Int J Cancer. 125(9):2173–2178.
- Gamer AO, Jaeckh R, Leibold E, Kaufmann W, Gembardt C, Bahnemann R, van Ravenzwaay B. 2002. Investigations on cell proliferation and enzyme induction in male rat kidney and female mouse liver caused by tetrahydrofuran. Toxicol Sci. 70(1):140–149.
- Goettel M, Fegert I, Honarvar N, Vardy A, Haines C, Chatham LR, Lake BG. 2020. Comparative studies on the effects of sodium phenobarbital and two other constitutive androstane receptor (CAR) activators on induction of cytochrome P450 enzymes and replicative DNA synthesis in cultured hepatocytes from wild type and CAR knockout rats. Toxicology. 433–434:152394.
- Gold LS, Manley NB, Slone TH, Ward JM. 2001. Compendium of chemical carcinogens by target organ: results of chronic bioassays in rats, mice, hamsters, dogs, and monkeys. Toxicol Pathol. 29(6):639–652.
- Goodman JI. 2018. Goodbye to the bioassay. Toxicol Res (Camb). 7(4):558–564.
- Goodman JI, Brusick DJ, Busey WM, Cohen SM, Lamb JC, Starr TB. 2000. Reevaluation of the cancer potency factor of toxaphene: recommendations from a peer review panel. Toxicol Sci. 55(1):3–16.
- Graillot C, Gak JC, Lancret C, Truhaut R. 1975. Research on the modalities and mechanisms of toxic action of organochlorine insecticides. II. Long term toxicity effects of DDT in hamsters. Eur J Toxicol Environ Hyg. 8(6):353–359.
- Greenfield RE, Ellwein LB, Cohen SM. 1984. A general probabilistic model of carcinogenesis: analysis of experimental urinary bladder cancer. Carcinogenesis. 5(4):437–445.
- Griffin RJ, Burka LT, Cunningham ML. 1995. Activity of hepatic drug metabolizing enzymes following oxazepam-dosed feed treatment in B6C3F1 mice. Toxicol Lett. 76(3):251–256.
- Griffin RJ, Dudley CN, Cunningham ML. 1996. Biochemical effects of the mouse hepatocarcinogen oxazepam: similarities to phenobarbital. Fundam Appl Toxicol. 29(1):147–154.
- Haines C, Chatham LR, Vardy A, Elcombe CR, Foster JR, Lake BG. 2018a. Comparison of the hepatic and thyroid gland effects of sodium phenobarbital in wild type and constitutive androstane receptor (CAR) knockout rats and pregnenolone-16α-carbonitrile in wild type and pregnane X receptor (PXR) knockout rats. Toxicology. 400–401:20–27.
- Haines C, Chatham LR, Vardy A, Elcombe CR, Foster JR, Lake BG. 2019. Comparison of the hepatic and thyroid gland effects of sodium phenobarbital and pregnenolone-16α-carbonitrile in wild-type and constitutive androstane receptor (CAR)/pregnane X receptor (PXR) knockout rats. Xenobiotica. 49(2):227–238.
- Haines C, Elcombe BM, Chatham LR, Vardy A, Higgins LG, Elcombe CR, Lake BG. 2018b. Comparison of the effects of sodium phenobarbital in wild type and humanized constitutive androstane receptor (CAR)/pregnane X receptor (PXR) mice and in cultured mouse, rat and human hepatocytes. Toxicology. 396–397:23–32.
- Harada T, Takeda M, Kojima S, Tomiyama N. 2016. Toxicity and carcinogenicity of dichlorodiphenyltrichloroethane (DDT). Toxicol Res. 32(1):21–33.
- Hester S, Moore T, Padgett WT, Murphy L, Wood CE, Nesnow S. 2012. The hepatocarcinogenic conazoles: cyproconazole, epoxiconazole, and propiconazole induce a common set of toxicological and transcriptional responses. Toxicol Sci. 127(1):54–65.
- Heusinkveld H, Braakhuis H, Gommans R, Botham P, Corvaro M, van der Laan JW, Lewis D, Madia F, Manou I, Schorsch F, et al. 2020. Towards a mechanism-based approach for the prediction of nongenotoxic carcinogenic potential of agrochemicals. Crit Rev Toxicol. 50(9):725–739.
- Hirose Y, Nagahori H, Yamada T, Deguchi Y, Tomigahara Y, Nishioka K, Uwagawa S, Kawamura S, Isobe N, Lake BG, et al. 2009. Comparison of the effects of the synthetic pyrethroid metofluthrin and phenobarbital on CYP2B form induction and replicative DNA synthesis in cultured rat and human hepatocytes. Toxicology. 258(1):64–69.
- Hoflack JC, Mueller L, Fowler S, Braendli-Baiocco A, Flint N, Kuhlmann O, Singer T, Roth A. 2012. Monitoring Cyp2b10 mRNA expression at cessation of 2-year carcinogenesis bioassay in mouse liver provides evidence for a carcinogenic mechanism devoid of human relevance: the dalcetrapib experience. Toxicol Appl Pharmacol. 259(3):355–365.
- Holsapple MP, Pitot HC, Cohen SM, Cohen SH, Boobis AR, Klaunig JE, Pastoor T, Dellarco VL, Dragan YP. 2006. Mode of action in relevance of rodent liver tumors to human cancer risk. Toxicol Sci. 89(1):51–56.
- Huang W, Zhang J, Washington M, Liu J, Parant JM, Lozano G, Moore DD. 2005. Xenobiotic stress induces hepatomegaly and liver tumors via the nuclear receptor constitutive androstane receptor. Mol Endocrinol. 19(6):1646–1653.
- Huff J, Cirvello J, Haseman J, Bucher J. 1991. Chemicals associated with site-specific neoplasia in 1394 long-term carcinogenesis experiments in laboratory rodents. Environ Health Perspect. 93:247–270.
- Hughes BJ, Thomas J, Lynch AM, Borghoff SJ, Green S, Mensing T, Sarang SS, LeBaron MJ. 2016. Methyl isobutyl ketone-induced hepatocellular carcinogenesis in B6C3F1 mice: a constitutive androstane receptor (CAR)-mediated mode of action. Regul Toxicol Pharmacol. 81:421–429.
- IARC. 1996. IARC monographs on the evaluation of carcinogenic risks to humans. Vol. 66, some pharmaceutical drugs. Lyon, France: IARC Press; p. 37–96.
- IARC. 2001. IARC monographs on the evaluation of carcinogenic risks to humans. Vol. 79, some thyrotropic agents: phenobarbital and its sodium salt. Lyon, France: IARC Press; p. 161–288.
- IARC. 2018. IARC monographs on the evaluation of carcinogenic risks to humans. Vol. 113. DDT, lindane, and 2,4-D. Lyon, France: IARC Press; p. 37–266.
- IPCS. 1999. CARBAMAZEPINE International Programme on Chemical Safety poisons information monograph 100 Pharmaceutical. Updated by Dr M.O. Rambourg-Schepens & Dr L. Murray; [accessed 2021 May 14]. http://www.inchem.org/documents/pims/pharm/pim100.htm#SectionTitle:7.3%20%20Carcinogenicity.
- Iqbal U, Nguyen PA, Syed-Abdul S, Yang HC, Huang CW, Jian WS, Hsu MH, Yen Y, Li YC. 2015. Is long-term use of benzodiazepine a risk for cancer? Medicine (Baltimore). 94(6):e483.
- James NH, Roberts RA. 1996. Species differences in response to peroxisome proliferators correlate in vitro with induction of DNA synthesis rather than suppression of apoptosis. Carcinogenesis. 17(8):1623–1632.
- Jones HB, Orton TC, Lake BG. 2009. Effect of chronic phenobarbitone administration on liver tumour formation in the C57BL/10J mouse. Food Chem Toxicol. 47(6):1333–1340.
- Kanno Y, Suzuki M, Miyazaki Y, Matsuzaki M, Nakahama T, Kurose K, Sawada J, Inouye Y. 2007. Difference in nucleocytoplasmic shuttling sequences of rat and human constitutive active/androstane receptor. Biochim Biophys Acta. 1773(6):934–944.
- Kazantseva YA, Yarushkin AA, Pustylnyak VO. 2013. Dichlorodiphenyltrichloroethane technical mixture regulates cell cycle and apoptosis genes through the activation of CAR and ERα in mouse livers. Toxicol Appl Pharmacol. 271(2):137–143.
- Khasawinah AM, Grutsch JF. 1989. Chlordane: thirty-month tumorigenicity and chronic toxicity test in rats. Regul Toxicol Pharmacol. 10(2):95–109.
- Kiyosawa N, Kwekel JC, Burgoon LD, Williams KJ, Tashiro C, Chittim B, Zacharewski TR. 2008. o,p′-DDT elicits PXR/CAR-, not ER-, mediated responses in the immature ovariectomized rat liver. Toxicol Sci. 101(2):350–363.
- Kolaja KL, Stevenson DE, Johnson JT, Walborg EF Jr, Klaunig JE. 1996. Subchronic effects of dieldrin and phenobarbital on hepatic DNA synthesis in mice and rats. Fundam Appl Toxicol. 29(2):219–228.
- Kondo M, Kikumoto H, Osimitz TG, Cohen SM, Lake BG, Yamada T. 2020. An evaluation of the human relevance of the liver tumors observed in female mice treated with permethrin based on mode of action. Toxicol Sci. 175(1):50–63.
- Kondo M, Miyata K, Nagahori H, Sumida K, Osimitz TG, Cohen SM, Lake BG, Yamada T. 2019. Involvement of peroxisome proliferator-activated receptor-alpha in liver tumor production by permethrin in the female mouse. Toxicol Sci. 168(2):572–696.
- Kostka G, Kopeć-Szlezak J, Palut D. 1996. Early hepatic changes induced in rats by two hepatocarcinogenic organohalogen pesticides: bromopropylate and DDT. Carcinogenesis. 17(3):407–412.
- La Vecchia C, Negri E. 2014. A review of epidemiological data on epilepsy, phenobarbital, and risk of liver cancer. Eur J Cancer Prev. 23(1):1–7.
- Lake BG. 1997. In vitro methods. In: McCuskey RS, Earnest DL, editors. Comprehensive toxicology. Volume 9. Hepatic and gastrointestinal toxicology. Oxford (UK): Elsevier Science Ltd; p. 233–249.
- Lake BG. 2009. Species differences in the hepatic effects of inducers of CYP2B and CYP4A subfamily forms: relationship to rodent liver tumour formation. Xenobiotica. 39(8):582–596.
- Lake BG. 2018. Human relevance of rodent liver tumour formation by constitutive androstane receptor (CAR) activators. Toxicol Res (Camb). 7(4):697–717.
- Lake BG, Price RJ. 2016. Induction of hepatic cytochrome P450 enzymes: importance in drug development and toxicity. In: Wilson AGE, editor. New horizons in predictive drug metabolism and pharmacokinetics. Cambridge (UK): RSC Publishing; p. 309–332.
- Lake BG, Price RJ, Scott MP, Chatham LR, Vardy A, Osimitz TG. 2020. Piperonyl butoxide: mode of action analysis for mouse liver tumour formation and human relevance. Toxicology. 439:152465.
- Lake BG, Renwick AB, Cunninghame ME, Price RJ, Surry D, Evans DC. 1998. Comparison of the effects of some CYP3A and other enzyme inducers on replicative DNA synthesis and cytochrome P450 isoforms in rat liver. Toxicology. 131(1):9–20.
- LaRocca JL, Rasoulpour RJ, Gollapudi BB, Eisenbrandt DL, Murphy LA, LeBaron MJ. 2017. Integration of novel approaches demonstrates simultaneous metabolic inactivation and CAR-mediated hepatocarcinogenesis of a nitrification inhibitor. Toxicol Rep. 4:586–597.
- LeBaron MJ, Gollapudi BB, Terry C, Billington R, Rasoulpour RJ. 2014a. Human relevance framework for rodent liver tumors induced by the insecticide sulfoxaflor. Crit Rev Toxicol. 44(Suppl. 2):15–24.
- LeBaron MJ, Rasoulpour RJ, Gollapudi BB, Sura R, Kan HL, Schisler MR, Pottenger LH, Papineni S, Eisenbrandt DL. 2014b. Characterization of nuclear receptor-mediated murine hepatocarcinogenesis of the herbicide pronamide and its human relevance. Toxicol Sci. 142(1):74–92.
- LeBaron MJ, Geter DR, Rasoulpour RJ, Gollapudi BB, Thomas J, Murray J, Kan HL, Wood AJ, Elcombe C, Vardy A, et al. 2013. An integrated approach for prospectively investigating a mode-of-action for rodent liver effects. Toxicol Appl Pharmacol. 270(2):164–173.
- Luijten M, Corvi R, Mehta J, Corvaro M, Delrue N, Felter S, Haas B, Hewitt NJ, Hilton G, Holmes T, et al. 2020. A comprehensive view on mechanistic approaches for cancer risk assessment of non-genotoxic agrochemicals. Regul Toxicol Pharmacol. 118:104789.
- Luisier R, Lempiäinen H, Scherbichler N, Braeuning A, Geissler M, Dubost V, Müller A, Scheer N, Chibout S-D, Hara H, et al. 2014. Phenobarbital induces cell cycle transcriptional responses in mouse liver humanized for constitutive androstane and pregnane x receptors. Toxicol Sci. 139(2):501–511.
- Lynch C, Mackowiak B, Huang R, Li L, Heyward S, Sakamuru S, Wang H, Xia M. 2019. Identification of modulators that activate the constitutive androstane receptor from the Tox21 10k compound library. Toxicol Sci. 167(1):282–292.
- Maeda J, Inoue K, Ichimura R, Takahashi M, Kodama Y, Saito N, Yoshida M. 2015. Essential role of constitutive androstane receptor in Ginkgo biloba extract induced liver hypertrophy and hepatocarcinogenesis. Food Chem Toxicol. 83:201–209.
- Maglich JM, Stoltz CM, Goodwin B, Hawkins-Brown D, Moore JT, Kliewer SA. 2002. Nuclear pregnane x receptor and constitutive androstane receptor regulate overlapping but distinct sets of genes involved in xenobiotic detoxification. Mol Pharmacol. 62(3):638–646.
- Martignoni M, Groothuis GMM, de Kanter R. 2006. Species differences between mouse, rat, dog, monkey and human CYP-mediated drug metabolism, inhibition and induction. Expert Opin Drug Metab Toxicol. 2(6):875–894.
- Marx-Stoelting P, Knebel C, Braeuning A. 2020. The connection of azole fungicides with xeno-sensing nuclear receptors, drug metabolism and hepatotoxicity. Cells. 9:1192.
- Masumoto N, Tateno C, Tachibana A, Utoh R, Morikawa Y, Shimada T, Momisako H, Itamoto T, Asahara T, Yoshizato K. 2007. GH enhances proliferation of human hepatocytes grafted into immunodeficient mice with damaged liver. J Endocrinol. 194(3):529–537.
- Mattu S, Saliba C, Sulas P, Zavattari P, Perra A, Kowalik MA, Monga SP, Columbano A. 2018. High frequency of β-catenin mutations in mouse hepatocellular carcinomas induced by a nongenotoxic constitutive androstane receptor agonist. Am J Pathol. 188(11):2497–2507.
- Meek ME, Boobis A, Cote I, Dellarco V, Fotakis G, Munn S, Seed J, Vickers C. 2014a. New developments in the evolution and application of the WHO/IPCS framework on mode of action/species concordance analysis. J Appl Toxicol. 34(1):1–18.
- Meek ME, Bucher JR, Cohen SM, Dellarco V, Hill RN, Lehman-McKeeman LD, Longfellow DG, Pastoor T, Seed J, Patton DE. 2003. A framework for human relevance analysis of information on carcinogenic modes of action. Crit Rev Toxicol. 33(6):591–653.
- Meek ME, Palermo CM, Bachman AN, North CM, Jeffrey Lewis R. 2014b. Mode of action human relevance (species concordance) framework: evolution of the Bradford Hill considerations and comparative analysis of weight of evidence. J Appl Toxicol. 34(6):595–606.
- Monro A. 1993. The paradoxical lack of interspecies correlation between plasma concentrations and chemical carcinogenicity. Regul Toxicol Pharmacol. 18(1):115–135.
- Moolgavkar SH, Knudson AG. 1981. Mutation and cancer: a model for human carcinogenesis. J Natl Cancer Inst. 66(6):1037–1052.
- Negishi M, Kobayashi K, Sakuma T, Sueyoshi T. 2020. Nuclear receptor phosphorylation in xenobiotic signal transduction. J Biol Chem. 295(45):15210–15225.
- Nihira K, Nan-Ya KI, Kakuni M, Ono Y, Yoshikawa Y, Ota T, Hiura M, Yoshinari K. 2019. Chimeric mice with humanized livers demonstrate human-specific hepatotoxicity caused by a therapeutic antibody against TRAIL-receptor 2/death receptor 5. Toxicol Sci. 167(1):190–201.
- Niu B, Coslo DM, Bataille AR, Albert I, Pugh BF, Omiecinski CJ. 2018. In vivo genome-wide binding interactions of mouse and human constitutive androstane receptors reveal novel gene targets. Nucleic Acids Res. 46(16):8385–8403.
- Ohara A, Takahashi Y, Kondo M, Okuda Y, Takeda S, Kushida M, Kobayashi K, Sumida K, Yamada T. 2017. Candidate genes responsible for early key events of phenobarbital-promoted mouse hepatocellular tumorigenesis based on differentiation of regulating genes between wild type mice and humanized chimeric mice. Toxicol Res (Camb). 6(6):795–813.
- Okuda Y, Kushida M, Kikumoto H, Nakamura Y, Higuchi H, Kawamura S, Cohen SM, Lake BG, Yamada T. 2017a. Evaluation of the human relevance of the constitutive androstane receptor-mediated mode of action for rat hepatocellular tumor formation by the synthetic pyrethroid momfluorothrin. J Toxicol Sci. 42(6):773–788.
- Okuda Y, Kushida M, Sumida K, Nagahori H, Nakamura Y, Higuchi H, Kawamura S, Lake BG, Cohen SM, Yamada T. 2017b. Editor's highlight: mode of action analysis for rat hepatocellular tumors produced by the synthetic pyrethroid momfluorothrin: evidence for activation of the constitutive androstane receptor and mitogenicity in rat hepatocytes. Toxicol Sci. 158(2):412–430.
- Omiecinski CJ, Coslo DM, Chen T, Laurenzana EM, Peffer RC. 2011a. Multi-species analyses of direct activators of the constitutive androstane receptor. Toxicol Sci. 123(2):550–562.
- Omiecinski CJ, Vanden Heuvel JP, Perdew GH, Peters JM. 2011b. Xenobiotic metabolism, disposition, and regulation by receptors: from biochemical phenomenon to predictors of major toxicities. Toxicol Sci. 120(Suppl. 1):S49–S75.
- Oshida K, Vasani N, Jones C, Moore T, Hester S, Nesnow S, Auerbach S, Geter DR, Aleksunes LM, Thomas RS, et al. 2015a. Identification of chemical modulators of the constitutive activated receptor (CAR) in a gene expression compendium. Nucl Recept Signal. 13:e002.
- Oshida K, Vasani N, Thomas RS, Applegate D, Gonzalez FJ, Aleksunes LM, Klaassen CD, Corton JC. 2015b. Screening a mouse liver gene expression compendium identifies modulators of the aryl hydrocarbon receptor (AhR). Toxicology. 336:99–112.
- Oshida K, Vasani N, Thomas RS, Applegate D, Rosen M, Abbott B, Lau C, Guo G, Aleksunes LM, Klaassen C, et al. 2015c. Identification of modulators of the nuclear receptor peroxisome proliferator-activated receptor α (PPARα) in a mouse liver gene expression compendium. PLoS One. 10(2):e0112655.
- Osimitz TG, Droege W, Boobis AR, Lake BG. 2013. Evaluation of the utility of the lifetime mouse bioassay in the identification of cancer hazards for humans. Food Chem Toxicol. 60:550–562.
- Osimitz TG, Lake BG. 2009. Mode-of-action analysis for induction of rat liver tumors by pyrethrins: relevance to human cancer risk. Crit Rev Toxicol. 39(6):501–511.
- Parkinson A, Leonard N, Draper A, Ogilvie BW. 2006. On the mechanism of hepatocarcinogenesis of benzodiazepines: evidence that diazepam and oxazepam are CYP2B inducers in rats, and both CYP2B and CYP4A inducers in mice. Drug Metab Rev. 38(1–2):235–259.
- Parkinson A, Mudra DR, Johnson C, Dwyer A, Carroll KM. 2004. The effects of gender, age, ethnicity, and liver cirrhosis on cytochrome P450 enzyme activity in human liver microsomes and inducibility in cultured human hepatocytes. Toxicol Appl Pharmacol. 199(3):193–209.
- Parzefall W, Erber E, Sedivy R, Schulte-Hermann R. 1991. Testing for induction of DNA synthesis in human hepatocyte primary cultures by rat liver tumor promoters. Cancer Res. 51(4):1143–1147.
- Peffer RC, Cowie DE, Currie RA, Minnema DJ. 2018a. Sedaxane-use of nuclear receptor transactivation assays, toxicogenomics, and toxicokinetics as part of a mode of action framework for rodent liver tumors. Toxicol Sci. 162(2):582–598.
- Peffer RC, LeBaron MJ, Battalora M, Bomann WH, Werner C, Aggarwal M, Rowe RR, Tinwell H. 2018b. Minimum datasets to establish a CAR-mediated mode of action for rodent liver tumors. Regul Toxicol Pharmacol. 96:106–120.
- Peffer RC, Moggs JG, Pastoor T, Currie RA, Wright J, Milburn G, Waechter F, Rusyn I. 2007. Mouse liver effects of cyproconazole, a triazole fungicide: role of the constitutive androstane receptor. Toxicol Sci. 99(1):315–325.
- Pinne M, Ponce E, Raucy JL. 2016. Transactivation assays to assess canine and rodent pregnane x receptor (PXR) and constitutive androstane receptor (CAR) activation. PLoS One. 11(10):e0164642.
- Pinne M, Ponce E, Raucy JL. 2017. Transactivation assays that identify indirect and direct activators of human pregnane x receptor (PXR, NR1I2) and constitutive androstane receptor (CAR, NR1I3). Drug Metab Lett. 11(2):128–137.
- Plummer S, Beaumont B, Wallace S, Ball G, Wright J, McInnes L, Currie R, Peffer R, Cowie D. 2019. Cross-species comparison of CAR-mediated procarcinogenic key events in a 3D liver microtissue model. Toxicol Rep. 6:998–1005.
- Price RJ, Giddings AM, Scott MP, Walters DG, Capen CC, Osimitz TG, Lake BG. 2008. Effect of pyrethrins on cytochrome P450 forms in cultured rat and human hepatocytes. Toxicology. 243(1–2):84–95.
- Price RJ, Walters DG, Finch JM, Gabriel KL, Capen CC, Osimitz TG, Lake BG. 2007. A mode of action for induction of liver tumors by pyrethrins in the rat. Toxicol Appl Pharmacol. 218(2):186–195.
- Rignall B, Braeuning A, Buchmann A, Schwarz M. 2011. Tumor formation in liver of conditional β-catenin-deficient mice exposed to a diethylnitrosamine/phenobarbital tumor promotion regimen. Carcinogenesis. 32(1):52–57.
- Riva L, Pandiri AR, Li YR, Droop A, Hewinson J, Quail MA, Iyer V, Shepherd R, Herbert RA, Campbell PJ, et al. 2020. The mutational signature profile of known and suspected human carcinogens in mice. Nat Genet. 52(11):1189–1197.
- Rooney J, Hill T 3rd, Qin C, Sistare FD, Christopher Corton J. 2018. Adverse outcome pathway-driven identification of rat liver tumorigens in short-term assays. Toxicol Appl Pharmacol. 356:99–113.
- Rooney JP, Oshida K, Kumar R, Baldwin WS, Corton JC. 2019. Chemical activation of the constitutive androstane receptor leads to activation of oxidant-induced Nrf2. Toxicol Sci. 167(1):172–189.
- Ross J, Plummer SM, Rode A, Scheer N, Bower CC, Vogel O, Henderson CJ, Wolf CR, Elcombe CR. 2010. Human constitutive androstane receptor (CAR) and pregnane X receptor (PXR) support the hypertrophic but not the hyperplastic response to the murine nongenotoxic hepatocarcinogens phenobarbital and chlordane in vivo. Toxicol Sci. 116(2):452–466.
- Rossi L, Barbieri O, Sanguineti M, Cabral JR, Bruzzi P, Santi L. 1983. Carcinogenicity study with technical-grade dichlorodiphenyltrichloroethane and 1,1-dichloro-2,2-bis(p-chlorophenyl)ethylene in hamsters. Cancer Res. 43(2):776–781.
- Sakamoto Y, Inoue K, Takahashi M, Taketa Y, Kodama Y, Nemoto K, Degawa M, Gamou T, Ozawa S, Nishikawa A, et al. 2013. Different pathways of constitutive androstane receptor-mediated liver hypertrophy and hepatocarcinogenesis in mice treated with piperonyl butoxide or decabromodiphenyl ether. Toxicol Pathol. 41(8):1078–1092.
- Sakamoto Y, Yoshida M, Tamura K, Takahashi M, Kodama Y, Inoue K. 2015. Dose-dependent difference of nuclear receptors involved in murine liver hypertrophy by piperonyl butoxide. J Toxicol Sci. 40(6):787–796.
- Scheer N, Ross J, Rode A, Zevnik B, Niehaves S, Faust N, Wolf CR. 2008. A novel panel of mouse models to evaluate the role of human pregnane X receptor and constitutive androstane receptor in drug response. J Clin Invest. 118(9):3228–3239.
- Seed J, Carney EW, Corley RA, Crofton KM, DeSesso JM, Foster PM, Kavlock R, Kimmel G, Klaunig J, Meek ME, et al. 2005. Overview: using mode of action and life stage information to evaluate the human relevance of animal toxicity data. Crit Rev Toxicol. 35(8–9):664–672.
- Seglen PO. 1976. Preparation of isolated rat liver cells. Methods Cell Biol. 13:29–83.
- Shizu R, Benoki S, Numakura Y, Kodama S, Miyata M, Yamazoe Y, Yoshinari K. 2013. Xenobiotic-induced hepatocyte proliferation associated with constitutive active/androstane receptor (CAR) or peroxisome proliferator-activated receptor α (PPARα) is enhanced by pregnane X receptor (PXR) activation in mice. PLoS One. 8(4):e61802.
- Shizu R, Ishimura M, Nobusawa S, Hosaka T, Sasaki T, Kakizaki S, Yoshinari K. 2021. The influence of the long-term chemical activation of the nuclear receptor pregnane X receptor (PXR) on liver carcinogenesis in mice. Arch Toxicol. 95(3):1089–1102.
- Shizu R, Yoshinari K. 2020. Nuclear receptor CAR-mediated liver cancer and its species differences. Expert Opin Drug Metab Toxicol. 16(4):343–351.
- Sidaway JE, Orton TC, Kalaitzi K, Jones HB, Foster A, Lake BG. 2020. Analysis of β-catenin gene mutations and gene expression in liver tumours of C57BL/10J mice produced by chronic administration of sodium phenobarbital. Toxicology. 430:152343.
- Sistare FD, Morton D, Alden C, Christensen J, Keller D, Jonghe SD, Storer RD, Reddy MV, Kraynak A, Trela B, et al. 2011. An analysis of pharmaceutical experience with decades of rat carcinogenicity testing: support for a proposal to modify current regulatory guidelines. Toxicol Pathol. 39(4):716–744.
- Skoda J, Dusek J, Drastik M, Stefela A, Dohnalova K, Chalupsky K, Smutny T, Micuda S, Gerbal-Chaloin S, Pavek P. 2020. Diazepam promotes translocation of human constitutive androstane receptor (CAR) via direct interaction with the ligand-binding domain. Cells. 9(12):2532.
- Soldatow VY, LeCluyse EL, Griffith LG, Rusyn I. 2013. In vitro models for liver toxicity testing. Toxicol Res (Camb). 2(1):23–39.
- Soldatow V, Peffer RC, Trask OJ, Cowie DE, Andersen ME, LeCluyse E, Deisenroth C. 2016. Development of an in vitro high content imaging assay for quantitative assessment of CAR-dependent mouse, rat, and human primary hepatocyte proliferation. Toxicol In Vitro. 36:224–237.
- Sonich-Mullin C, Fielder R, Wiltse J, Baetcke K, Dempsey J, Fenner-Crisp P, Grant D, Hartley M, Knaap A, Kroese D, et al. 2001. IPCS conceptual framework for evaluating a mode of action for chemical carcinogenesis. Regul Toxicol Pharmacol. 34(2):146–152.
- Stanley LA, Horsburgh BC, Ross J, Scheer N, Wolf CR. 2006. PXR and CAR: nuclear receptors which play a pivotal role in drug disposition and chemical toxicity. Drug Metab Rev. 38(3):515–597.
- Staudinger J, Liu Y, Madan A, Habeebu S, Klaassen CD. 2001. Coordinate regulation of xenobiotic and bile acid homeostasis by pregnane X receptor. Drug Metab Dispos. 29(11):1467–1472.
- Stenbäck F, Mori H, Furuya K, Williams GM. 1986. Pathogenesis of dimethylnitrosamine-induced hepatocellular cancer in hamster liver and lack of enhancement by phenobarbital. J Natl Cancer Inst. 76(2):327–333.
- Stevenson DE, Walborg EF, North DW, Sielken RL, Ross CE, Wright AS, Xu Y, Kamendulis LM, Klaunig JE. 1999. Monograph: reassessment of human cancer risk of aldrin/dieldrin. Toxicol Lett. 109(3):123–186.
- Strathmann J, Schwarz M, Tharappel JC, Glauert HP, Spear BT, Robertson LW, Appel KE, Buchmann A. 2006. PCB 153, a non-dioxin-like tumor promoter, selects for beta-catenin (Catnb)-mutated mouse liver tumors. Toxicol Sci. 93(1):34–40.
- Stritzelberger J, Lang JD, Mueller TM, Reindl C, Westermayer V, Kostev K, Hamer HM. 2021. Anti-seizure medication is not associated with an increased risk to develop cancer in epilepsy patients. J Neurol. 268(6):2185–2191.
- Strupp C, Quesnot N, Richert L, Moore J, Bomann WH, Singh P. 2020. Weight of evidence and human relevance evaluation of the benfluralin mode of action in rodents (Part I): liver carcinogenesis. Regul Toxicol Pharmacol. 117:104758.
- Sugahara G, Ishida Y, Sun J, Tateno C, Saito T. 2020. Art of making artificial liver: depicting human liver biology and diseases in mice. Semin Liver Dis. 40(2):189–212.
- Takayama S, Sieber SM, Dalgard DW, Thorgeirsson UP, Adamson RH. 1999. Effects of long-term oral administration of DDT on nonhuman primates. J Cancer Res Clin Oncol. 125(3–4):219–225.
- Tamura K, Inoue K, Takahashi M, Matsuo S, Irie K, Kodama Y, Gamo T, Ozawa S, Yoshida M. 2015. Involvement of constitutive androstane receptor in liver hypertrophy and liver tumor development induced by triazole fungicides. Food Chem Toxicol. 78:86–95.
- Tamura K, Inoue K, Takahashi M, Matsuo S, Irie K, Kodama Y, Ozawa S, Nishikawa A, Yoshida M. 2013. Dose–response involvement of constitutive androstane receptor in mouse liver hypertrophy induced by triazole fungicides. Toxicol Lett. 221(1):47–56.
- Tanaka T, Mori H, Williams GM. 1987. Enhancement of dimethylnitrosamine-initiated hepatocarcinogenesis in hamsters by subsequent administration of carbon tetrachloride but not phenobarbital or p,p′-dichlorodiphenyltrichloroethane. Carcinogenesis. 8(9):1171–1178.
- Tateishi T, Asoh M, Nakura H, Watanabe M, Tanaka M, Kumai T, Kobayashi S. 1999. Carbamazepine induces multiple cytochrome P450 subfamilies in rats. Chem Biol Interact. 117(3):257–268.
- Tateno C, Kojima Y. 2020. Characterization and applications of chimeric mice with humanized livers for preclinical drug development. Lab Anim Res. 36:2.
- Tateno C, Yamamoto T, Utoh R, Yamasaki C, Ishida Y, Myoken Y, Oofusa K, Okada M, Tsutsui N, Yoshizato K. 2015. Chimeric mice with hepatocyte-humanized liver as an appropriate model to study human peroxisome proliferator-activated receptor-α. Toxicol Pathol. 43(2):233–248.
- Thatcher NJ, Caldwell J. 1994. Origins of hepatomegaly produced by dexamethasone (DEX), pregnenolone 16 alpha-carbonitrile (PCN) and phenobarbitone (PB) in female Sprague-Dawley rats. Biochem Soc Trans. 22(2):132S.
- Thompson MD, Monga SP. 2007. WNT/beta-catenin signaling in liver health and disease. Hepatology. 45(5):1298–1305.
- Thoolen B, Ten Kate FJ, van Diest PJ, Malarkey DE, Elmore SA, Maronpot RR. 2012. Comparative histomorphological review of rat and human hepatocellular proliferative lesions. J Toxicol Pathol. 25(3):189–199.
- Thoolen B, Maronpot RR, Harada T, Nyska A, Rousseaux C, Nolte T, Malarkey DE, Kaufmann W, Küttler K, Deschl U, et al. 2010. Proliferative and nonproliferative lesions of the rat and mouse hepatobiliary system. Toxicol Pathol. 38(7 Suppl.):5S–81S.
- Tinwell H, Rouquie D, Schorsch F, Geter D, Wason S, Bars R. 2014. Liver tumor formation in female rat induced by fluopyram is mediated by CAR/PXR nuclear receptor activation. Regul Toxicol Pharmacol. 70(3):648–658.
- Tucker MJ, Orton TC. 1995. Comparative toxicology of hypolipidaemic fibrates. London: Taylor & Francis.
- Wang Z, Neal BH, Lamb JC, Klaunig JE. 2015. Mechanistic investigation of toxaphene induced mouse liver tumors. Toxicol Sci. 147(2):549–561.
- Wang Z, Wu Q, Li X, Klaunig JE. 2020. Constitutive androstane receptor (CAR) mediates dieldrin-induced liver tumorigenesis in mouse. Arch Toxicol. 94(8):2873–2884.
- Wang Z, Li X, Wu Q, Lamb JC, Klaunig JE. 2017. Toxaphene-induced mouse liver tumorigenesis is mediated by the constitutive androstane receptor. J Appl Toxicol. 37(8):967–975.
- Ward WO, Delker DA, Hester SD, Thai SF, Wolf DC, Allen JW, Nesnow S. 2006. Transcriptional profiles in liver from mice treated with hepatotumorigenic and nonhepatotumorigenic triazole conazole fungicides: propiconazole, triadimefon, and myclobutanil. Toxicol Pathol. 34(7):863–878.
- Wei P, Zhang J, Egan-Hafley M, Liang S, Moore DD. 2000. The nuclear receptor CAR mediates specific xenobiotic induction of drug metabolism. Nature. 407(6806):920–923.
- Whysner J, Ross PM, Williams GM. 1996. Phenobarbital mechanistic data and risk assessment: enzyme induction, enhanced cell proliferation, and tumor promotion. Pharmacol Ther. 71(1–2):153–191.
- Wiemann C, Goettel M, Vardy A, Elcombe BM, Elcombe CR, Chatham LR, Wang H, Li L, Buesen R, Honarvar N, et al. 2019. Metazachlor: mode of action analysis for rat liver tumour formation and human relevance. Toxicology. 426:152282.
- Wolf DC, Cohen SM, Boobis AR, Dellarco VL, Fenner-Crisp PA, Moretto A, Pastoor TP, Schoeny RS, Seed JG, Doe JE. 2019. Chemical carcinogenicity revisited 1: a unified theory of carcinogenicity based on contemporary knowledge. Regul Toxicol Pharmacol. 103(10386):86–92.
- Wood CE, Hukkanen RR, Sura R, Jacobson-Kram D, Nolte T, Odin M, Cohen SM. 2015. Scientific and regulatory policy committee (SRPC) review: interpretation and use of cell proliferation data in cancer risk assessment. Toxicol Pathol. 43(6):760–775.
- Yamada T. 2018. Case examples of an evaluation of the human relevance of the pyrethroids/pyrethrins-induced liver tumours in rodents based on the mode of action. Toxicol Res (Camb). 7(4):681–696.
- Yamada T, Kikumoto H, Lake BG, Kawamura S. 2015. Lack of effect of metofluthrin and sodium phenobarbital on replicative DNA synthesis and Ki-67 mRNA expression in cultured human hepatocytes. Toxicol Res. 4(4):901–913.
- Yamada T, Ohara A, Ozawa N, Maeda K, Kondo M, Okuda Y, Abe J, Cohen SM, Lake BG. 2020. Comparison of the hepatic effects of phenobarbital in chimeric mice containing either rat or human hepatocytes with humanized constitutive androstane receptor and pregnane X receptor mice. Toxicol Sci. 177(2):362–376.
- Yamada T, Okuda Y, Kushida M, Sumida K, Takeuchi H, Nagahori H, Fukuda T, Lake BG, Cohen SM, Kawamura S. 2014. Human hepatocytes support the hypertrophic but not the hyperplastic response to the murine nongenotoxic hepatocarcinogen sodium phenobarbital in an in vivo study using a chimeric mouse with humanized liver. Toxicol Sci. 142(1):137–157.
- Yamada T, Uwagawa S, Okuno Y, Cohen SM, Kaneko H. 2009. Case study: an evaluation of the human relevance of the synthetic pyrethroid metofluthrin-induced liver tumors in rats based on mode of action. Toxicol Sci. 108(1):59–68.
- Yamamoto Y, Moore R, Goldsworthy TL, Negishi M, Maronpot RR. 2004. The orphan nuclear receptor constitutive active/androstane receptor is essential for liver tumor promotion by phenobarbital in mice. Cancer Res. 64(20):7197–7200.
- Yang H, Wang H. 2014. Signaling control of the constitutive androstane receptor (CAR). Protein Cell. 5(2):113–123.
- Yano BL, Hardisty JF, Seely JC, Butterworth BE, McConnell EE, Swenberg JA, Williams GM, Stebbins KE, Golllapudi BB, Eisenbrandt DL. 2008. Nitrapyrin: a scientific advisory group review of the mode of action and carcinogenicity in B6C3F1 mice. Regul Toxicol Pharmacol. 51(1):53–65.
- Yoshinari K. 2019. Role of nuclear receptors PXR and CAR in xenobiotic-induced hepatocyte proliferation and chemical carcinogenesis. Biol Pharm Bull. 42(8):1243–1252.
- Yoshinari K, Ueda R, Kusano K, Yoshimura T, Nagata K, Yamazoe Y. 2008. Omeprazole transactivates human CYP1A1 and CYP1A2 expression through the common regulatory region containing multiple xenobiotic-responsive elements. Biochem Pharmacol. 76(1):139–145.