
Abstract
Assessment of genotoxicity is a critical component of mode of action (MOA) analysis and carcinogen risk assessment due to its influence on quantitative risk extrapolation approaches. To date, clear guidance and expert consensus on the determination of a mutagenic MOA remains elusive, resulting in different estimates of carcinogenic risk for the same chemical among different stakeholders. Oral toxicity criteria for hexavalent chromium [Cr(VI)], for example, differ by orders of magnitude due largely to the interpretation of in vivo genotoxicity data. Herein, we review in vivo genotoxicity studies for Cr(VI) to inform the MOA for Cr(VI)-induced tumors observed in a two-year cancer bioassay in mice and rats exposed via drinking water. Overall, genotoxicity results in carcinogenic target tissues (viz., oral cavity and duodenum) are negative. Results in the intestine are consistent with imaging data indicating little to no chromium present in the crypt compartment following oral exposure. Positive genotoxicity results in nontarget tissues have been reported at high doses mostly following nonphysiological routes of exposure. Given the negative genotoxicity results in carcinogenic target organs from oral exposure to Cr(VI), there is scientific justification to support the use of nonlinear low-dose extrapolation methods in the derivation of oral toxicity criteria for Cr(VI). These results highlight important differences between genotoxicity testing for hazard identification purposes and quantitative risk assessment.
1. Introduction
Mode of action (MOA) analysis is a critical component of human health risk assessment of environmental chemicals. Such analyses are most commonly conducted for carcinogens because the MOA directly informs how extrapolation is performed from high doses employed in animal bioassays to the much lower levels humans are typically exposed to in the environment. The 2005 U.S. EPA Guidelines for Carcinogen Risk Assessment characterize mutagenic carcinogens as those that can directly interact with DNA and that can cause gene mutations or structural chromosome aberrations. The same guidelines indicate that nonmutagenic carcinogens can induce indirect DNA effects through mechanisms like mitogenic or cytotoxicity-induced regenerative cell proliferation. Generally, mutagenic carcinogens are assumed to pose some risk at all exposure levels and are therefore extrapolated linearly from a predetermined risk level (e.g. 10% extra risk) to zero exposure. In contrast, carcinogens that induce tumors via nonmutagenic mechanisms can be amenable to nonlinear extrapolation methods such as the application of safety or uncertainty factors (U.S. EPA Citation2005). In practice, however, very few carcinogen risk assessments have employed nonlinear extrapolation approaches due, in part, to unclear guidance on how to assess and interpret genotoxicity data for the purpose of quantitative risk assessment. For example, U.S. EPA (Citation2005) notes that DNA adducts and DNA breakage often lead to gene mutations and chromosomal aberrations, which some risk assessors have interpreted to mean that the former is direct evidence of the latter. It is widely understood among genetic toxicologists, however, that such damage can be eliminated by DNA repair, cell death, or failure of the damaged cell to replicate before mutation fixation or expansion of mutant clones occurs. U.S. EPA (Citation2005) also states that MOA analysis should be performed for each tumor site of interest and that an agent might have different MOAs at different sites, yet this often seems overlooked in practice.
In 2007, the U.S. Environmental Protection Agency (EPA) released a draft document titled “Framework for Determining a Mutagenic Mode of Action for Carcinogenicity” (U.S. EPA Citation2007). The stated purpose of this document was to “help EPA risk assessors…approach the identification of a mutagenic MOA for carcinogenicity in a consistent, objective, transparent, and scientifically sound manner” (U.S. EPA Citation2007). Despite determination of a mutagenic MOA being a critical question for all stakeholders (e.g., government, industry, consumers), the draft was never finalized, and no such guidance appears in the offing. Currently, risk assessors and other stakeholders must approach the determination of a mutagenic MOA largely using their own methodology and interpretation. Others have attempted to fill this guidance vacuum with retrospective analyses of mutagenic MOA determinations by various regulatory bodies to elucidate critical factors in such determinations (Eastmond Citation2012); however, these factors have gained only limited traction (Thompson et al. Citation2017).
A timely example of the disparity in mutagenic MOA determinations can be seen in the risk assessments of the oral carcinogenicity of hexavalent chromium (Cr(VI)), where different risk assessors have proposed linear and nonlinear methods for setting safety standards. Several groups that have reviewed targeted MOA research published over the past decade have developed threshold-based (i.e., nonlinear) toxicity criteria for Cr(VI) that are protective of cancer and noncancer effects (Health Canada Citation2016; TCEQ Citation2016; Thompson et al. Citation2018; FSCJ Citation2019; WHO Citation2019). In contrast, other recent reviews on Cr(VI) have concluded that the MOA and oral genotoxicity of Cr(VI) remain too uncertain to consider nonlinear approaches for safety criteria development. For example, a scientific advisory board tasked to advise the North Carolina Department of Environmental Quality (DEQ) on the most appropriate method to derive an oral safety criteria for Cr(VI) recently recommended that the DEQ conduct linear extrapolation through development an oral cancer slope factor (SSAB Citation2020). The latter recommendation was based, in part, on concerns for the potential of Cr(VI) to cause genotoxicity based largely on data in non-target organs and from non-physiological exposure routes. These disparate approaches to safety criteria development underscore the need for better guidance on evaluating the role of genotoxicity for developing safety criteria for carcinogens.
Evidence for the oral carcinogenicity of Cr(VI) stems primarily from a National Toxicology Program (NTP) 2-year cancer bioassay where B6C3F1 mice and F344 rats were exposed to 5-180 ppm Cr(VI) in drinking water in the form of sodium dichromate dihydrate (SDD) (NTP Citation2008). Mice exhibited cytotoxicity and regenerative cell proliferation (termed diffuse epithelial hyperplasia, DEH) in the proximal small intestine (duodenum and jejunum) at ≥5 ppm and significantly increased incidence of adenomas and carcinomas in the proximal small intestine at ≥30 ppm (). In rats, squamous cell carcinomas of the oral mucosa—primarily originating in the palate adjacent to the upper molar teeth—were significantly increased at 180 ppm (). For context, environmental monitoring data indicate that the median and 95th percentile Cr(VI) concentrations in U.S. drinking water sources are 0.001 and 0.003 ppm, respectively (U.S. EPA Citation2017); a state-by-state analysis of Cr(VI) levels is shown at: cr6study.infoFootnote1. The current maximum contaminant level (MCL) for Cr(VI) in drinking water is 0.1 ppm (U.S. EPA Citation1991). In contrast to Cr(VI), a two-year feed study with trivalent chromium [Cr(III)] in the form of chromium picolinate found no neoplastic effects in ratsFootnote2 or mice at exposures up to 50000 ppm in diet (Stout et al. Citation2009; NTP Citation2010).
Figure 1. Oral carcinogenicity of Cr(VI). (A) Incidence of diffuse epithelial hyperplasia (DEH) in the duodenum and the combined adenoma/carcinoma (A/C) incidence in the duodenum and jejunum of B6CF1 mice exposed to Cr(VI) in drinking water for 2 years. (B) Incidence of oral tumors (squamous cell carcinoma) in F344 rats exposed to Cr(VI) in drinking water for 2 years. The vertical dotted line in each plot represents the current MCL for total chromium (0.1 ppm). The lowest data points in each plot represent the control group assigned 0.003 ppm, the 95th percentile Cr(VI) concentration in U.S. water sources (see text). The red arrows point to the 1E-6 risk concentration of 0.07 ppb (0.00007 ppm).
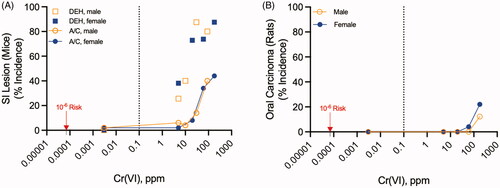
The quantitative differences in safety criteria derived by linear and nonlinear extrapolations approaches and their implications for public health are large. Following the release of the NTP (Citation2008) cancer bioassay, when targeted intestinal genotoxicity data were not available, several agencies developed similar oral cancer slope factors for Cr(VI) based on the small intestine tumors in mice (Stern Citation2010; U.S. EPA Citation2010; OEHHA Citation2011). The cancer slope factor of 0.5 (mg/kg-day)−1 in these assessments result in a 1E-6 extra risk of cancer at 2E-6 mg/kg (1E-6 ÷ 0.5). Assuming a typical 70 kg adult consuming 2 L of water per day, the de minimis risk dose equates to a water concentration of 7E-5 mg/L or 0.00007 ppm (0.07 ppb). This value is 14-fold lower than the mean U.S water concentration of 0.001 ppm and ∼1400-fold lower than the current MCL of 0.1 ppm. The California EPA Office of Environmental Health Hazard Assessment (OEHHA) public health goal (PHG) for Cr(VI) is still lower at 0.02 ppb (OEHHA Citation2011). As a result of linear low-dose extrapolation, such standards imply that the Cr(VI) present in many U.S. drinking water sources (whether natural or anthropogenic) poses an increased cancer risk. The critical question is whether the available scientific data (including genotoxicity studies) support such safety criteria or whether the proposed standards overestimate risk.
As previously mentioned, several groups have more recently developed threshold safety criteria for Cr(VI) (Health Canada Citation2016; TCEQ Citation2016; FSCJ Citation2019; WHO Citation2019). These decisions have been based, in part, on evidence that exposure to Cr(VI) greatly increased the incidence of DEH in the small intestine of mice at both carcinogenic and noncarcinogenic doses (). Rats, in contrast were not noted to have any intestinal hyperplasia or intestinal tumors (NTP Citation2008; Stout et al. Citation2009). It was therefore hypothesized that the intestinal tumors might have been the result of chronic intestinal wounding and regenerative hyperplasia and therefore incompatible with the linear no threshold extrapolation approach (Thompson et al. Citation2011). Over the last decade, more than 30 research and review publications have been published regarding the MOA supporting nonlinear risk assessment approaches for developing Cr(VI) toxicity criteria protective of cancer (see reviews (Thompson et al. Citation2013, Citation2017, Citation2018; Bhat et al. Citation2020)). As described in an adverse outcome pathway (AOP) for small intestinal cancer arising from nongenotoxic mechanisms (Bhat et al. Citation2020), it is important that there be evidence for a nongenotoxic mechanism (e.g. cell proliferation, endocrine disruption, etc.) for intestinal cancer as well as evidence against genotoxicity in the intestine. While the evidence for a nongenotoxic cytotoxicity-induced regenerative cell proliferation MOA has been reviewed elsewhere (Thompson et al. Citation2013, Citation2017; Health Canada Citation2016; Moffat et al. Citation2018; Bhat et al. Citation2020), a formal review of the in vivo genotoxicity of Cr(VI) has not been published.
The purpose of the current review is twofold. The first objective is to collect and summarize the available in vivo genotoxicity data for Cr(VI). Herein, the term genotoxicity is used to encompass gene mutation, chromosomal aberrations (also referred to as chromosomal mutation), as well as DNA breakage. Although the latter is not a direct indicator of mutations as it can be observed in cell populations without replication, some have argued that DNA breakage provides similar information as in vivo mutation assays (Kirkland et al. Citation2019). In vivo genotoxicity was chosen as the focus for this review since these effects are most relevant for informing the MOA and extrapolation approaches for the tumors serving as the basis for the oral carcinogenicity assessment of Cr(VI). Although general reviews of the in vitro and in vivo genotoxicity of Cr(VI) are available (O'Brien et al. Citation2003; McCarroll et al. Citation2010; Zhitkovich Citation2011; Thompson et al. Citation2013, Citation2017), an in-depth treatment of in vivo genotoxicity studies for the specific context of informing human health risk assessment has not yet been published. This current evaluation is limited to in vivo studies that assess (1) small-scale gene mutation (e.g. base pair mutations), (2) large-scale chromosomal aberrations (e.g. micronuclei from clastogens and aneugens and structural and numerical chromosomal aberrations), and (3) DNA breakage (e.g. Comet assay). Adducts and base modifications such as DNA-protein crosslinks and 8-hydroxydeoxyguanosine are not assessed herein, in part because such assays are not generally prescribed in testing batteries for assessing the in vivo genotoxicity of environmental chemicals, food additives, or pharmaceuticals. The second objective of this article is to provide insight into the likely involvement of genotoxicity as an early initiating event in the MOA of the tumors observed in the NTP bioassays so as to potentially inform the oral carcinogenicity risk assessment of Cr(VI).
2. Materials and methods
A comprehensive literature search was conducted to identify all in vivo genotoxicity studies for chromium by any route of exposure. Three independent searches (Supplemental Table S1) were performed in PubMed on November 17, 2020 (and again on September 30, 2021 during peer-review, Table S2) for Cr(III), Cr(VI), and chromium[Mesh] in order to cover all oxidation states of chromium. Resulting hits from the three independent searches were downloaded, unduplicated, and further filtered using data analytics SWIFT Review (Howard et al. Citation2016). Nonresearch articles, including reviews and case studies, in addition to in vitro or studies with no genotoxicity endpoints, were removed using SWIFT title and abstract screening. A total of 86 articles (titles and abstracts) were exported and categorized by chromium oxidation state. Exclusion criteria included those that were not in English, those in nonmammalian species, those that did not include in vivo exposure, and those that did not include standard genotoxicity assays.
A secondary search was conducted in Embase® on September 30, 2021. Three independent searches were performed for Cr(III), Cr(VI), and chromium. Results were filtered to exclude articles from MEDLINE, as MEDLINE articles are captured in PubMed. In addition, to minimize the number of irrelevant results, additional filter syntax was applied to each search (Table S3). Results of this literature search are described in Section 3 below.
Data were extracted and entered into an Excel spreadsheet as reported in the original papers. Critical parameters including test system, route of exposure, dose, units, dose duration, analysis time, tissue, number of animals per group, the group mean and group standard deviation data were collected when possible. Statistical significance was captured as reported and not reanalyzed. With some exceptions, data only presented graphically were reported qualitatively. For a few studies deemed critical based on exposure route and endpoint, data were extracted using WebPlotDigitizer 4.3 and reanalyzed. Summary graphs were also plotted in Prism v9.1.1 (GraphPad Software, LLC).
It should be noted that many Cr(VI) studies use different forms of test article, such as potassium dichromate, sodium dichromate, sodium dichromate dihydrate, and more. Many of the studies are ambiguous as to whether the doses reported are in terms of test article (e.g. potassium dichromate) or chromium ion content (i.e. Cr(VI)). Moreover, very few studies report using analytical dose verification. Therefore, doses are reported herein as reported in the original study unless it is clear that doses are reported as Cr(VI). It is not the intent of this review to evaluate the genotoxic potential of Cr(VI) for the purpose of identifying quantitative endpoints such as no-observable-genotoxic-effect-levels or benchmark doses (Gollapudi et al. Citation2013; Johnson et al. Citation2014).
3. Results
3.1. Literature search results
A targeted literature search for in vivo genotoxicity studies on Cr(VI) resulted in 86 articles, of which 36 were determined to meet criteria for review (see Materials and Methods). The final list was also crosschecked with the U.S. EPA (Citation2010) draft assessment of Cr(VI) and the OEHHA (Citation2011) PHG document, resulting in the addition of a few older studies. During review of this manuscript, the authors became aware of a new publication reviewing occupational exposure to Cr(VI) (den Braver-Sewradj et al. Citation2021); no new genotoxicity studies were identified therein. As such, we believe this to be a thorough representation of the available data for informing the in vivo genotoxicity of Cr(VI). A summary of these data is presented in and summarized in the following sections. Following the PubMed search, a second search was conducted in the proprietary search engine Embase® after removing MEDLINE already captured by PubMed. Nine non-overlapping articles were identified (Table S4), and all were confirmed to not be indexed in PubMed by searching the article titles. Examination of the abstracts indicated that four studies measured MN following intraperitoneal injection, three studies examined chromosomal aberrations following gavage dosing, one abstract provided no experimental details (e.g. dose, route), and one study examined DNA damage in the rat kidney following exposure to 353 ppm Cr(VI) in drinking water for 12 weeks. Except for the latter drinking water study, it was concluded that excluding these articles from the review would not impact the conclusions drawn from the articles found in PubMed and summarized below.
3.2. In vivo mutation studies
The transgenic rodent (TGR) assay is currently the gold standard for assessing in vivo mutagenicity in various tissues of rats or mice. It is the only in vivo genotoxicity assay for assessing gene mutations that has an Organization for Economic Cooperative Development (OECD) test guideline (TG). Per OECD TG 488, a fully compliant study design should have a minimum of five animals per treatment group and a minimum of five treatment groups (including positive and negative controls) with a top dose that is a maximum tolerated dose (MTD) (OECD Citation2020). The recommended study duration is 28 consecutive days of treatment with sampling conducted three days after the final treatment. As will be shown below, several TGR assays for Cr(VI) were identified; however, none were fully OECD compliant. Some were conducted prior to the first OECD TG 488 guidance (2011), and some were conducted with Cr(VI) doses matching those in two-year cancer bioassays as opposed to MTD-based dose spacing. All of these TGR assays were reviewed, with data summarized in .
Table 1. Summary of in vivo mutation assays on Cr(VI).
In addition to the TGR assay, three additional in vivo mutation assays were identified. The coat spot test had an OECD test guideline (OECD Citation1986) that was discontinued in 2014. An eye spot test was identified for which no OECD test guideline has been developed. This assay requires in utero exposure to assess mutation in eye pigment genes and will be discussed in more detail below (Reliene et al. Citation2004; Lambert et al. Citation2005). The third in vivo mutation assay identified, also with no OECD test guideline, is the allele-specific competitive blocker-PCR (ACB-PCR) assay that identifies point mutations in specific codons of target oncogenes of interest (Parsons et al. Citation2010).
3.2.1. Mutation analysis in Cr(VI) oral carcinogenicity target tissues
To date, four in vivo mutation studies have been conducted in tissues (oral cavity and duodenum) that exhibited tumors in the NTP carcinogenicity assay following oral exposure to Cr(VI).
3.2.1.1 Oral cavity
Thompson et al. (Citation2015) conducted a GLP study in male Fisher 344 Big Blue® transgenic rats (TgF344) exposed to 180 ppm Cr(VI) in drinking water in the form of sodium dichromate dihydrate (SDD), the same form as in the NTP 2-year Cr(VI) bioassays, for 28 days with tissue sampling 3 days after the last day of exposure. This study design was not fully OECD 488 compliant, as it was fit for purpose to investigate the MOA at the highest concentration in the NTP (Citation2008) bioassay. Specifically, this study did not determine an MTD and only one concentration was examined (viz., the highest carcinogenic concentration in the NTP bioassay). Two regions of the oral cavity previously qualified with vehicle and the mutagen 4-nitroquinoline-1-oxide (4NQO) were sampled (Thompson et al. Citation2015; Young et al. Citation2015). One region was comprised of the inner gingiva of the upper jaw and the hard palate, which was cut into two halves denoted as the left or right “gingiva/palate”. The other region was comprised of the upper gingiva covering maxillary alveolar process extending from incisors to molars, and the adjacent buccal fold and buccal tissue opposite the molars to the midline of the upper lip. This resulted in left and right “gingiva/buccal” samples (Young et al. Citation2015). Together, these samples comprised the region where oral cavity squamous cell carcinomas (SCC) were thought to originate in F344 rats (NTP Citation2008; Young et al. Citation2015). The mutant frequency (MF) in rats exposed to tap water was 49.8 ± 17.8 × 10−6 and 39.1 ± 7.5 × 10−6 for the gingiva/palate and gingiva/buccal samples, respectively. The MF in rats exposed to 180 ppm Cr(VI) was 57.8 ± 9.1 × 10−6 and 44.4 ± 25.4 × 10−6 for the gingiva/palate and gingiva/buccal samples, respectively. In contrast, exposure to 10 ppm 4NQO increased MF significantly to 1818 ± 362 × 10−6 and 688 ± 250 × 10−6 in the gingiva/palate and gingiva/buccal samples.
The estimated daily dose throughout the study was ∼11 mg/kg Cr(VI), which is comparable to the average daily exposure to male F344 rats exposed to 180 ppm Cr(VI) in the 90-day (∼11.2 mg/kg) and 2-year (5.9 mg/kg) NTP bioassays (NTP Citation2007, Citation2008). The presence of Cr in the gingival/palate and gingival/buccal samples from the side of the mouth not used for mutation analysis was assessed by pooling samples from the vehicle control group and from the Cr(VI) treated group. Using inductively coupled plasma-mass spectrometry (ICP-MS), 0.17 and 0.33 mg/kg Cr was measured in the gingival/buccal and gingival/palate regions of untreated TgF344 rats. These levels are comparable to those previously reported in untreated F344 rats, 0.13 ± 0.16 mg/kg (Kirman et al. Citation2012; Thompson et al. Citation2012). In treated TgF344 rats, 0.66 and 1.0 mg/kg Cr was detected in the gingival/buccal and gingival/palate regions, respectively, demonstrating target tissue dosimetry. However, these values were somewhat lower than the Cr levels detected in the upper palate of F344 rats exposed to 180 ppm Cr(VI) for 90 days (≤6 mg/kg). The latter measurements were taken from rats exposed three times as long and without the 3-day sampling period without Cr(VI) exposure. In summary, Cr(VI) did not significantly increase MF in the rat oral cavity at 180 ppm, the highest tumorigenic concentration in the 2-year NTP cancer bioassay.
3.2.1.2 Duodenum
In 2017, MF was measured in banked and frozen duodenal samples from the above study (Thompson et al. Citation2015). At the time of tissue collection in the original study, duodenal sections from each rat were excised, flushed with saline, cut into approximately 1-inch segments, immediately flash frozen in liquid nitrogen, and stored at −80 °C. At the time of DNA extraction, the duodenum was thawed, cut open, rinsed gently with PBS to remove any intestinal contents and mucus, and then gently scraped to separate intestinal epithelium (including crypts and villi) from the intestinal wall. The intestinal walls were discarded and the intestinal epithelium was further processed for DNA extraction. Because 4NQO may have limited mutagenic action beyond the oral cavity (Young et al. Citation2015), banked and frozen duodenal samples from TgF344 rats previously exposed to 20 mg/kg N-ethyl-N-nitrosourea (ENU) by oral gavage six times (study days 1, 2, 3, 12, 19 and 26) and necropsied on study day 31 were selected to serve as a positive control. The MF in the negative controls and Cr(VI) exposed rats were 23.2 ± 4.8 × 10−6 and 22.7 ± 8.4 × 10−6, respectively, whereas the MF in ENU treated rats was significantly increased to 557 ± 116 × 10−6 (Thompson et al. Citation2017).
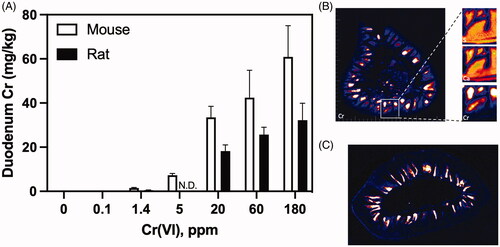
The presence of Cr in the TgF344 rat duodenum was confirmed by ICP-MS. Duodenum Cr levels were significantly higher in treated rats than untreated rats (24.6 ± 3.8 vs 0.96 ± 0.4 mg/kg; p < 0.001), and similar to levels measured in the duodenum of F344 rats exposed to 180 ppm Cr(VI) for 90 days (32 mg/kg) (Kirman et al. Citation2012; Thompson et al. Citation2012). Although Cr(VI) was not carcinogenic to the F344 rat duodenum, the Cr levels in rat duodenum were comparable to levels that were carcinogenic in the mouse duodenum (). Moreover, synchrotron-based X-ray fluorescence (XRF) microscopy revealed localization of Cr to duodenal villi of rats and mice but not the crypt regions (). In summary, 180 ppm Cr(VI) did not significantly increase MF in the rat duodenum, despite unequivocal evidence of tissue exposure () at levels that were carcinogenic to the mouse duodenum.
Figure 2. Chromium dosimetry in the rodent duodenum. (A) Tissue Cr levels in mouse and rat duodenum following exposure to Cr(VI) for 90 days (note: rats were not exposed to 5 ppm Cr(VI)). (B) XRF images from an unstained transverse section of duodenum from a female B6C3F1 mouse exposed to 180 ppm Cr(VI) for 90 days. (Left) full transverse section with chromium (Cr) XRF. (Right) Magnification of the region in the yellow box, with XRF images for calcium (Ca), sulfur (S), and Cr. Signal for Ca and S are contiguous throughout the villus and crypt regions, whereas Cr signal is very high in the villi, but low or absent in the crypt. (C) Chromium XRF image of an unstained 5-µm transverse section from the duodenum of a female F344 rat exposed to 180 ppm Cr(VI) for 90 days. Adapted from Thompson et al. (Citation2011, Citation2015).
Aoki et al. (Citation2019) exposed male gpt delta mice to Cr(VI) in two drinking water studies. Using the OECD TG 488 exposure paradigm (i.e. 28 + 3), MF was measured in the gpt transgene in the duodenum of mice exposed to 30 or 90 ppm Cr(VI) in the form of SDD. Recall that 90 ppm was the highest concentration that male B6C3F1 mice were exposed to in the two-year cancer bioassay (). This study design was not fully OECD 488 compliant, as it was fit for purpose to investigate the MOA at the highest concentration male mice were exposed to in the NTP (Citation2008) bioassay. Specifically, this study did not determine an MTD, and some groups had only four mice, while others had 5–6. There was no significant increase in the MF in the duodenum of mice exposed to Cr(VI) (MF was 0.58 ± 0.31 × 10−5, 0.96 ± 0.69 × 10−5, and 0.91 ± 0.45 × 10−5 for 0, 30 and 90 ppm, respectively). In a separate study, MF was measured in the duodenum of mice exposed to 0, 3, 10, or 30 ppm Cr(VI) in drinking water for 90 days and sampled one day later; again, there was no significant increase in the MF (0.80 ± 0.27 × 10−5, 0.62 ± 0.26 × 10−5, 0.49 ± 0.19 × 10−5, and 0.77 ± 0.28 × 10−5, respectively). Aoki et al. (Citation2019) used potassium bromate (KBrO3) as a positive control, which was administered at 2 g/L in drinking water using the 28 + 3 exposure paradigm. The MF was 0.35 ± 0.19 × 10−5 and 1.03 ± 0.53 × 10−5 in the water and KBrO3-treated animals, respectively. Because the KBrO3 study has its own negative control, it is not clear that it was run concurrently with the aforementioned Cr(VI) study and thus may not be a concurrent positive control. It is important to note that a concurrent positive control is not required by OECD 488 for experienced laboratories (note: 5 animals per group were used in treated animals, and only 4 per group in the negative controls). Notably, Aoki and colleagues argue that the increased MF observed in KBrO3-treated mice was likely secondary to oxidative stress, which suggests that the TGR assay might detect mutations arising from indirect mechanism as well as direct DNA interaction. As such, the negative results for Cr(VI) suggests that Cr(VI) might not be inducing significant oxidative stress in crypts. In summary, ≤90 ppm Cr(VI) did not significantly increase MF in the duodenum of gpt delta mice after 28 or 90 days of exposure.
O’Brien et al. (Citation2013) conducted a non-GLP mutation analysis in the mouse duodenum of wild type B6C3F1 mice using the allele specific competitive PCR (ACB-PCR) method. These mice were cohorts from a larger study conducted at the same contract laboratory that previously conducted the NTP (Citation2008) bioassay (Thompson et al. Citation2011). Mice were exposed to 0, 0.1, 1.4, 5, 20, 60, or 180 ppm Cr(VI) in drinking water for 90 days; at study termination, the duodenum was removed, cut longitudinally, and the intestinal epithelium (containing crypts and villi) were scraped and separated from the outer connective tissues, and stored at −80 °C. Mutation analysis was conducted for a single codon on kras, specifically codon 12 GGT to GAT. The kras oncogene was investigated due to its association with intestinal cancer in humans and apparent sensitivity as a sensitive ‘reporter’ of mutation events (Parsons et al. Citation2010, Citation2012; Rizk and Barker Citation2012; O'Brien et al. Citation2013), as well as evidence that these mutations can lead to increased cell proliferation in the small intestine (Feng et al. Citation2011). Cr(VI) exposure did not significantly increase the kras GGT to GAT MF at any dose, nor was there evidence for a trend in response. Interestingly, the MF of the common tumor driver mutation at codon 12 (exon 2), where a glycine is replaced by aspartic acid (GGT to GAT), in untreated mice was ∼100-fold higher than measured in other tissues such as lung; however, we are unaware of any other studies measuring the MF of kras codon 12 in the B6C3F1 duodenum. Critically, intestinal tumors are relatively rare among tumors induced in NTP cancer bioassays (Chandra et al. Citation2010). It is conceivable that kras mutations frequently arise in daughter cells as they transit through the amplifying portion of the crypt compartment, yet pose little cancer risk due to their eventual differentiation as they reach the villus or due to their sloughing into the lumen within just a few days of their formation. In summary, exposure to ≤180 ppm Cr(VI) for 90 days did not significantly increase MF of kras in the duodenum of B6C3F1 mice.
It is worth noting that a 13-week Cr(VI) bioassay conducted by the NTP included a transgenic strain of mice called PhiX174 (ΦX174) am3-C57BL/6 that harbors the transgene phiX174am3 that can be recovered for scoring of forward and reverse mutations (Malling and Burkhart Citation1989). However, the NTP (Citation2007) 13-week study report indicates that mutation analysis was not conducted due to technical difficulties. A search of the NTP website (https://ntp.niehs.nih.gov)Footnote3 for ‘am3-C57BL/6′ resulted in 13 hits, all of which appeared to relate to Cr(III) or Cr(VI)—suggesting that chromium is the only test article ever tested in this strain by the NTP.
3.2.2. Mutation Analysis in Cr(VI) inhalation carcinogenicity target tissues
Inhalation exposure to Cr(VI) is associated with lung cancer and is therefore discussed here apart from target tissues from oral exposure and nontarget tissues. The increase in lung cancer risk has been observed among workers in chromate production, plating, pigments and ferrochrome production industries where historical exposures to Cr(VI) have been well in excess of 100 µg/m3 (IARC Citation1990; Gibb et al. Citation2000; Luippold et al. Citation2003). Lung tumor incidence is increased in rats exposed to Cr(VI) via inhalation, also around 100 µg/m3 (Glaser et al. Citation1986). No inhalation genotoxicity studies were identified; however, two intratracheal (i.t.) studies conducted by the same group were identified and are summarized below.
Cheng et al. (Citation1998) exposed Big Blue® female mice (4 per group) to potassium dichromate (K2Cr2O7) by intratracheal instillation. Specifically, a surgical cut was made into the ventral neck thereby exposing the trachea followed by injection of test article with a 30.5-gauge needle through the wall of the trachea into the lung. Cr(VI) concentrations tested were 1.7, 3.4, and 6.8 mg/kg Cr(VI). It is assumed that these units are mg/kg bodyweight. The injection volume was not specified and thus the concentration of Cr(VI) in the test article is not known. It is assumed that Cr(VI) was administered once, and it is stated that the mice were sacrificed “1 to 4 weeks” after instillation. The authors report a time-dependent increase in MF in the lung; however, that data were not shown. A dose-dependent increase in MF of the lacI transgene was observed four weeks after instillation of saline, 1.7, 3.4, and 6.8 mg/kg Cr(VI) (MF values of 2.6 ± 0.5 × 10−5, 4.1 ± 0.8 × 10−5, 7.7 ± 2.2 10−5, and 12.5 ± 1.7 × 10−5 respectively) (Cheng et al. Citation1998). Although statistical analyses were not reported, the top two doses appear to be significant by our own Dunnett’s test.
In 2000, Cheng et al. reported more detailed data from Cheng et al. (Citation1998), which they characterized a “preliminary report” in Environment Health Perspectives. The full study additionally reports MF data for kidney and liver, as well as MF data in mice after experimental manipulation of glutathione (GSH) levels. For brevity, we will include results from liver and kidney in this section. Importantly, the i.t. injection volume was reported to be 1 µl/g of bodyweight, and the “chromium dose was calculated as mg Cr(VI) per kg total body weight.” They also report a concurrent dosimetry study with i.t. instillation of 9, 22.5, and 45 mg/kg, and noted that one of three mice died within 8 days of exposure to 9 mg/kg, and all mice died within 2–4 days at higher doses. These data suggest life threatening damage to the lung at i.t. doses ≥9 mg/kg; notably, Cheng et al. characterize 6.8 mg/kg as “well below” the doses that cause lethality. At a typical bodyweight of 25 g, 6.8 mg/kg equates to 0.17 mg Cr(VI) (6.8 mg/kg × 0.025 kg). In an injection volume of 1 µl/g (i.e. 25 μl), the administered solution was 0.0068 mg/µL (0.17 mg ÷ 25 µL), which is equivalent to 6800 mg/L (0.0068 mg/µL × 1E6 µL/L). For reference, the highest concentration of Cr(VI) administered orally in the NTP two-year drinking water study was 180 mg/L, and this dose injures the small intestine after acute exposure (Thompson et al. Citation2011, Citation2015). As such, it seems likely that such concentrations would cause tissue damage and thus potentially lead to increased genotoxicity by secondary mechanisms such as oxidative stress and inflammatory responses. As pointed out by others, Cheng et al. (Citation2000) provided no measures of tissue injury, inflammation etc. following this unusual treatment (Wise and Wise Citation2012).
MF was measured in the lung 1, 2, and 4 weeks after instillation of 6.8 mg/kg, with the negative control (saline only) collected only at week 1 (Cheng et al. Citation2000). MF in Cr(VI)-treated mice increased from 4.6 to 14.6 × 10−5. A dose-dependent increase in MF in the lung was plotted without providing numerical data or statistics; however, MF appears to be significantly elevated at 3.4 mg/kg (∼3400 mg/L) and possibly 1.7 mg/kg (1700 mg/L). MF data were also reported for the liver and kidney without measures of variance. Using the raw data, we calculated the MF in the liver of mice exposed to saline or 6.8 mg/kg Cr(VI) as 2.1 ± 1.0 and 3.8 ± 0.5 × 10−5, respectively. Cheng et al. did not find this difference to be statistically significant. From the raw data, we calculated the MF in the kidney of mice exposed to saline or 6.8 mg/kg Cr(VI) as 1.8 ± 0.4 and 6.6 ± 4.6 × 10−5, which Cheng et al. reported as significant.
Using the same i.t. exposure procedure in wild-type mice, Cheng et al. reported Cr levels in lung and liver to be 41.7 and 4.6 ppm. In oral Cr(VI) studies in B6C3F1 mice exposed to ≤180 ppm Cr(VI) for durations ranging from 90 to 371 days, the liver Cr levels ranged between 40 and 50 mg/kg (ppm) (NTP Citation2008; Kirman et al. Citation2012), yet no tumors were observed in the liver of mice or rats exposed to Cr(VI) for 2 years (NTP Citation2008).
In another set of experiments, MF was measured in the lung of Big Blue® mice exposed to 6.8 mg/kg Cr(VI) by i.t. instillation after receiving either i.p. injection of 8.8 mmol/kg GSH ethyl ester 2 h before Cr(VI) treatment or i.p. injection of 4 mmol/kg buthionine sulfoximine (BSO), an inhibitor of GSH synthesis, twice daily for 3 days prior to Cr(VI) exposure. The study authors investigated effects of GSH levels on MF due to reports suggesting that GSH-mediated Cr(VI) reduction can generate reactive intermediates. Treatment with BSO and GSH ethyl ester were reported to result in lung GSH levels 30% and 170% of “normal”, respectively. The MF in mice treated with GSH ethyl ester (14.1 × 10−5) was similar to the MF in the previous assays at 4 weeks (14.6 and 12.5 1 × 10−5). In contrast, the MF in mice treated with BSO was only 6.9 × 10−5. These data suggest that GSH-mediated reduction of Cr(VI) might be pro-mutagenic; however, others argue that ascorbate-mediated Cr(VI) reduction leads to more mutagenic species than does GSH (Zhitkovich Citation2011).
3.2.3. Mutation analysis in nontarget tissues
Knudsen (Citation1980) investigated the mutagenic potential of Cr(VI) using the mouse spot test OECD TG 484 (OECD Citation1986) that has since been delisted by OECD. In this test, mice heterozygous for several genes involved in coat color are exposed to agents, and mutations that occur in dominant alleles in melanoblasts during gestation lead to coat colors associated with recessive alleles (Lambert et al. Citation2005). Black/grey/brownish spots are generally considered indicative of somatic mutation. Knudsen (Citation1980) exposed pregnant C57/BL6.BOM mice to 0, 10, or 20 mg/kg Cr(VI) on gestational days GD8-10 by intraperitoneal (i.p.) injection. Coat spots were counted at the end of postnatal weeks 2–5. The number (and percentage) of animals with brownish spots were significantly elevated at ≥10 mg/kg Cr(VI) and by 2.5 mg/kg cyclophosphamide (positive control). However, 10 mg/kg cyclophosphamide was embryotoxic (data not shown) and 20 mg/kg Cr(VI) reduced the number of liters, pups, and mutations (Knudsen Citation1980).
Itoh and Shimada (Citation1997) exposed male MutaTM Mice to 40 mg/kg K2CrO4 by i.p. injection for two consecutive days. It should be noted that this study predates OECD guidance on TGR assays, which now caution that i.p. injection “is not recommended since it is not a physiologically relevant route of human exposure” (OECD Citation2020). Three untreated mice served as a negative control, and three mice receiving 100 mg/kg ENU (i.p.) served as a positive control. The injection volume was not specified and thus the concentration of Cr(VI) in the test article is not known. However, a typical i.p. injection volume is 10 ml/kg for mice and rats. Thus, the i.p. injection volume was likely ∼ 0.25 ml for a 0.025 kg mouse. The administered dose is 1 mg (40 mg/kg × 0.025 kg), and therefore, the concentration is 1 mg/0.25 ml or 4 mg/mL, which is equivalent to 4000 mg/L (4000 ppm) K2CrO4 or ∼1000 ppm Cr(VI). MF in the lacZ transgene was measured in the liver and bone marrow 7 days after the last exposure. Only 2–3 mice per group were treated. The MF was determined by dividing the number of confirmed mutant plaques by the total number of plaques examined, that is, confirmed mutant plaques plus blue wild-type plaques (Itoh and Shimada Citation1997). Mean MF in bone marrow in untreated, Cr(VI)-treated, and ENU-treated mice were 28.8, 32.2, and 1821 × 10−6, respectively (note: measures of variance were not reported). In liver, the MF in untreated mice was 35.8 × 10−6; MF increased significantly to 72.3 × 10−6 in Cr(VI)-treated mice and 205 × 10−6 in ENU-treated mice (Itoh and Shimada Citation1997).
In a follow-up study, Itoh and Shimada (Citation1998) exposed male MutaTM Mice to 40 mg/kg K2CrO4 by i.p. injection for a single day. Untreated mice again served as a negative control. The injection volume was not specified (see estimate in previous paragraph). MF was measured in the liver and bone marrow of five mice per group either 1 day or 7 days after exposure. As before, Cr(VI) increased MF in liver at the 7-day sampling time point (51.5 ± 13.7 vs. 104.8 ± 34.5 × 10−6). However, MF was not significantly altered at the 1-day sampling time (51.5 ± 13.7 vs. 43.3 ± 12.4 × 10−6); notably, the same untreated group served as the negative control for both sampling times. In the bone marrow, again Cr(VI) did not increase MF at the 7-day sampling time (46.4 ± 12.6 vs. 39.4 ± 11.0 × 10−6) but did significantly increase MF at the 1-day sampling time to 76.8 ± 14.5 × 10−6. No positive control (e.g. ENU) was reported in this study (Itoh and Shimada Citation1998). In summary, Itoh and Shimada (Citation1997, Citation1998) show that exposure to high mg/kg and highly concentrated Cr(VI) (>1000 ppm) by i.p. injection increases MF in liver and bone marrow in a time dependent manner.
Kirpnick-Sobol et al. (Citation2006) exposed pregnant C57BL/6Jpun/pun mice (4–6 dams per group) to potassium dichromate in drinking water at concentrations of 62.6 ppm and 125 ppm Cr(VI) on postcoitum days 10.5 to 20.5. The C57BL/6Jpun/pun strain of mice carry a pink-eyed unstable mutation (pun), which is a 70 kb tandem duplication. DNA damage leading to homologous recombination leads to deletion of the duplicate sequence in pun and reversion to wild-type p expressed in retinal epithelium precursor cells and subsequent clonal expansion of cells with black pigment against non-pigmented cells. These spots can be counted and used to measure spontaneous reversion frequency as well as reversion frequency in treated animals. Specifically, the eyes are removed, and retinas examined by light microscopy. Each spot corresponds to one pun mutation. This assay is unique in that it is designed to detect deletion mutations.
Kirpnick-Sobol et al. (Citation2006) measured eye spots in offspring at 20 days of age. An average of 5.5 eyespots per retinal pigment epithelium (RPE) were observed in untreated mice, and 27% and 38% more eyespots in mice exposed to 62.6 and 125 ppm Cr(VI), respectively (data and statistics were reported graphically). These averages would therefore be 7/RPE and 7.6/RPE. The eye spot test is not a standard assay so there are relatively few published studies compared to other genotoxicity assays. Further, there was no historical control data reported from this laboratory to determine whether these results were outside of normal variation. Despite the doubling in Cr(VI) concentration in drinking water and the amount of Cr measured in embryos, the reversion rate did not appear to change much in magnitude, although the spots/RPE did differ statistically between 62.5 and 125 ppmFootnote4. shows the data extracted from figures in Kirpnick-Sobol et al. (Citation2006).
Figure 3. Eye spot test (in vivo mutation) results from Kirpnick-Sobol et al. (Citation2006). Data were extracted from in Kirpnick-Sobol et al. (Citation2006) using WebPlotDigitizer v4.3 and replotted in Prism 9. **p-value < 0.01; ****p-value < 0.0001.
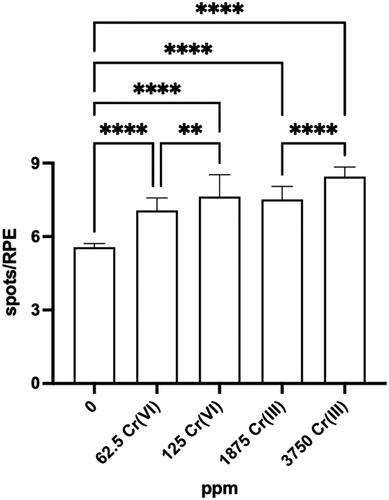
Similar results were observed in mice exposed to 1875 and 3750 ppm Cr(III); doubling the Cr(III) exposure concentrations increased the spots/RPE 36% and 53%, respectively.
3.3. In vivo chromosomal damage studies
The in vivo micronucleus (MN) assay is a standard test for assessing genotoxicity, specifically cytogenetic effects of chromosome damage and numerical chromosome changes. Some agencies view negative in vivo MN assays as dispositive of positive in vitro MN results if adequate bioavailability can be demonstrated. These assays are commonly conducted in peripheral blood or bone marrow for which there is OECD Test Guideline 474. Micronucleus assays can also be conducted in liver, small intestine, and colon, yet there are no guideline studies for these tissues (Goldberg et al. Citation1983; Goldberg and Chidiac Citation1986; Chidiac and Goldberg Citation1987; Morita et al. Citation2011). However, it is acknowledged that the ideal tissues to conduct in vivo genotoxicity tests are those that receive significant dosimetry, are highly proliferative, and that are carcinogenic targets (MacGregor et al. Citation2015). As such, the duodenum is an ideal tissue for assessing the cytogenetic effects of Cr(VI) and the bone marrow and blood less relevant as target tissues. In addition to the MN assay, the chromosomal aberration (CA) assay also detects chromosome damage and numerical chromosome changes, and is described in the OECD Test Guideline 475 (OECD Citation2016a). Only one of the studies described below (Garcia-Rodriguez MC et al. Citation2013) claims to be compliant with either of the aforementioned OECD guidance documents. Data for cytogenetic assays discussed in this section are summarized in .
Table 2. Summary of in vivo chromosomal damage assays on Cr(VI).
3.3.1. Chromosomal Damage in Cr(VI) oral carcinogenicity target tissues
No MN or chromosomal aberration assays were found for the rodent oral mucosa.
3.3.1.1 Duodenum
O’Brien et al. (Citation2013), described in Section 3.2.1.2, also included analyses of MN formation in the duodenum of B6C3F1 mice exposed to Cr(VI). In brief, mice were exposed to 0, 0.1, 1.4, 5, 20, 60, or 180 ppm Cr(VI) in drinking water for 90 days. At study termination, the duodenum was removed, fixed in 10% neutral buffered formalin, embedded in paraffin for transverse sectioning at approximately 5 µm. Paraffin-embedded duodenal sections (3 sections per mouse) were stained for DNA using Feulgen’s stain and analyzed at Experimental Pathology Laboratories, Inc. (EPL; Sterling, VA). MN and KN were assessed by two methods. One method was limited to counting enterocytes with normal nuclei, MN, and karyorrhectic nuclei (KN) in 10 fully intact crypts per animal exposed to Cr(VI) for 90 days using Image-Pro® Plus v7.0 software. Most published MN assays focus on the crypt compartment because cell division is required for MN formation. Identification criteria for aberrant nuclei were consistent with that described previously (Goldberg et al. Citation1983). Specifically, MN consisted of a single dense, ovoid to spherical body that was located adjacent to a normal nucleus within the cytoplasm of the same cell, and karyorrhectic nuclei were fragmented into small, unequally sized, dense spherical bodies, and the cytoplasmic margins of such cells were often indistinct. No treatment-related increases in MN or KN were observed ().
In the second method, KN and MN were counted in both the villus and crypt compartments of three slides obtained from each animal per dose group (4–5 mice in each of 7 dose groups) after 7 or 90 days of exposure. Digital images were randomized prior to the cell counting, and persons performing the counts were unaware of the treatment group status. Aberrant nuclei in digital images were counted by marking each nucleus manually. MN and KN were then tabulated in the crypt and villus regions of these sections (15 per dose group). This was done to increase the thoroughness of the analyses due to concerns for a genotoxic mechanism, even though intestinal carcinogenesis is thought to arise from stem cells in the crypt (Potten and Loeffler Citation1990; Barker et al. Citation2009). No treatment related increases in MN or KN were observed in the crypts of mice in either exposure duration. Because there was no effect and the results were redundant to the analysis with fully intact crypts (above), the crypt results were not published in O’Brien et al. (Citation2013) but are in the original study report (EPL Citation2013) and are included in . MN and KN were significantly increased in the villi (especially tips) of mice exposed to ≥60 ppm for 7 days and ≥20 ppm for 90 days. These results are consistent with the localization of Cr in XRF images in . The biological relevance of MN and KN in villi is uncertain as villus enterocytes are generally differentiated, rarely dividing cells that will slough into the intestinal lumen.
Thompson et al. (Citation2015) conducted a GLP duodenal MN assay in B6C3F1 mice exposed to 1.4, 21, or 180 ppm Cr(VI) in drinking water for 7 days. Positive controls included a single dose of 65 mg/kg dimethyl hydrazine (DMH) by either gavage or i.p., or 50 mg/kg cyclophosphamide by oral gavage. Mice were sacrificed 24 h after dosing on day 7, and intestinal sections were prepared using the Swiss roll technique at BioReliance (Rockville, MD) and shipped to Experimental Pathologies Laboratory (Sterling, VA) for analysis. Sections for MN scoring were stained with Feulgen stain to reveal chromosomal material and images were captured digitally. At least 15 full-length crypts were counted per animal. An American College of Veterinary Pathologists (ACVP) board-certified veterinary pathologist performed counts by differentially marking the cells in each arbitrarily selected crypt using the image analysis software. Cell counts were peer-reviewed by a second ACVP board-certified pathologist at BioReliance. There was no significant increase in MN or KN in Cr(VI) treated animals whereas the positive control cyclophosphamide induced a significant increase in MN and KN in crypt enterocytes (). DMH increased MN by i.p. exposure, albeit not significantly; oral DMH did not increase MN. MN were not assessed in the villi in this study due to their uncertain relevance and the additional analyses described below.
Two additional analyses were conducted in tissue sections from the above study. First, sections were stained for γ-H2AX, which accumulates at sites of DNA double-strand breaks. In all mice, immunoreactivity was observed in the chromatin material of mitotic figures and, to a lesser degree, in goblet cell mucus. Staining of mitotic figures is consistent with increased DNA damage-independent H2AX phosphorylation in mammalian cells during mitosis. In mice exposed to cyclophosphamide, immunoreactivity was additionally observed in cells with KN and MN. In contrast, the crypts of mice exposed to up to 180 ppm Cr(VI) exhibited no difference in immunostaining relative to untreated mice. Unstained sections from mice treated with 180 ppm Cr(VI) were also examined by XRF microscopy. As shown for transverse sections in , Cr fluorescence was localized to intestinal villi in the Swiss roll preps (Thompson et al. Citation2015). Overall, the apparent absence of Cr fluorescence in crypts is consistent with the lack of genotoxic responses in the crypt compartment.
3.3.2. Chromosomal damage in Cr(VI) inhalation carcinogenicity target tissues
Because MN and chromosomal aberration assays are generally conduced in highly proliferating tissues, no such animal studies were found for the lung.
3.3.3. Chromosomal damage in nontarget tissue
In a brief report on 16 chemicals, Wild (1978) exposed NMRI mice (unspecified sex; 4 animals per group) to 0, 12.12, 24.25, or 48.5 mg/kg Cr(VI) as potassium chromate on two consecutive days via i.p. injection. It is important to note that nonrelevant routes (e.g. i.p.) of human exposure are not recommended in OECD TG 474 and require scientific justification. Six hours after the second injection, MN were scored in polychromatic erythrocytes (PCE) of bone smears. The two highest doses significantly increased MN formation ().
Newton and Lilly (Citation1986) exposed Wistar rats (unspecified sex) to Cr(VI) as K2CrO4 by i.p. injection or intravenous (i.v.) exposure. Animals received either one dose 24 h before sacrifice or two doses 24 and 48 h before sacrifice. For two day exposures, rats were exposed to 12, 18, or 21 mg/kg Cr(VI) by i.p. injection, or 24, 30, or 36 mg/kg i.v. For single day exposures, rats were exposed to 21 mg/kg i.p. or 12, 18, 24, or 36 mg/kg i.v. Positive controls included aflatoxin (gavage) and 1-phenyl-3,3 dimethyl triazine (i.v.). Lymphocytes were harvested, stimulated with phytohaemagglutinin, arrested with colcemid, spread on slides and stained with lactoacetic orcein. Bone marrow cells were harvested, spread on slides and stained as described for lymphocytes. Chromosomal aberrations were scored in metaphases with and without gaps. Significant increases in abnormal cells were observed in lymphocytes (two day exposures) at 21 mg/kg i.p. and 36 mg/kg i.v. Significant increases in abnormal bone marrow cells (single day exposures) were observed in all treated groups (Newton and Lilly Citation1986).
In what appears to be the first in vivo oral MN assay with Cr(VI) (as K2CrO4), Shindo et al. (Citation1989) exposed two strains of mice (MS/Ae and CD-1) to Cr(VI) by either i.p. or gavage dosing. Dose selection was based on pilot LD50 studies and pilot MN assays reported graphically (not discussed herein). The LD50 for i.p. and oral routes were respectively 50 and 300 mg/kg for MS/Ae mice and 32 and 180 mg/kg in CD-1 mice. For the main study, both strains were exposed to 0, 10, 20, 40, and 80 mg/kg i.p., and 0, 10, 20, 40, 80, 160, and 320 mg/kg by gavage. The study authors did not mark statistical significance; however, i.p. exposure was reported to increase MN PCEs (%) in both strains, whereas oral exposure had no effect on MN PCEs () (Shindo et al. Citation1989).
Sarkar et al. (Citation1993) exposed male Swiss albino mice to 20 mg/kg Cr(VI) as CrO3 by a single oral gavage. Twenty-four hours after exposure, bone marrow cells were scored for chromosomal abnormalities such as “chromatid and isochromatid gaps and breaks and rearrangements including centric fissions, fusions and dicentrics.” The number of chromosomal aberrations per cell increased from 0.018 ± 0.011 to 0.08 ± 0.0071 following exposure to Cr(VI) (Sarkar et al. Citation1993). Other groups co-exposed to chlorophyllin did not exhibit significant increases in CA (not shown herein); Sarkar et al. speculated that the reduction in chromosomal aberrations was due to either direct scavenging of Cr ions or the anti-oxidant properties of chlorophyllin.
Itoh and Shimada (Citation1996) exposed male Slc:ddY mice to 0, 30, 40, or 50 mg/kg Cr(VI) as K2CrO4 on two consecutive days via i.p. injection. Additional groups of mice were exposed similarly to 62.5, 125, or 250 mg/kg Cr(III) as CrCl3 or 0.5 mg/kg Mitomycin C (positive control). Bone marrow smears collected 24 h after the second dose indicate significant increases in MN PCE (%) in mice exposed to Cr(VI) or Mitomycin C, but not Cr(III). In a separate experiment, mice pretreated with an inducer of metallothionein mitigated the effects of Cr(VI), which the study authors indicated might be due to free radical scavenging. These data might indicate that i.p. injection of highly concentrated Cr(VI) induces DNA damage by indirect mechanisms (Itoh and Shimada Citation1996).
Mirsalis et al. (Citation1996) exposed male and female Swiss-Webster mice to 0, 1, 5, or 20 ppm Cr(VI) by oral gavage or drinking water for two days. Gavage exposure to 300 ppm urethane served as a positive control. After 48 h of exposure via drinking water or 24 h after the final gavage dose, MN PCE (%) were measured. Only mice receiving urethane exhibited significant increases in micronucleated PCEs (). In addition to these studies, a similar set of exposures were used in conjunction with assessment of unscheduled DNA synthesis (UDS) in liver hepatocytes (Mirsalis et al. Citation1996). However, these data are not discussed here because there is general consensus that the UDS is not very predictive of genotoxicity, and it is thought that it might only be useful for agents where the liver is a known or suspected target organ (Kirkland and Speit Citation2008; EFSA Citation2017). Reflecting the decreased use of this assay, the OECD guideline for the in vivo UDS test, OECD 486 1997, was not selected to be updated. Considering that liver tumors were not observed in rats or mice in the NTP (Citation2008) cancer bioassay, there is no particular reason to investigate the liver. Nevertheless, we note that the UDS results with Cr(VI) were negative.
One study reported that green tea mitigated the formation of chromosome aberrations in bone marrow following administration of 20 mg/kg Cr(VI) (potassium dichromate; K2Cr207) by a single oral gavage to male Swiss Albino mice (Mukherjee et al. Citation1997). Twenty-four hours after exposure, the number of CA increased significantly in bone marrow (). Another small study reported increased bone marrow MN PCE (%) 30 h following a single i.p. injection of 20.8 mg/kg K2Cr2O7 in Balb/c mice (Wronska-Nofer et al. Citation1999).
Balansky et al. (Citation2000) studied the effects of Cr(VI) on the genotoxicity of inhaled cigarette smoke in rodents (results were only reported graphically). In one experiment, BDF1 mice were exposed to 50 mg/kg Cr(VI) (as K2Cr2O7) by i.p. injection, with or without prior whole-body exposure to cigarette smoke for 5 days. Additional mice were exposed to smoke alone or served as a negative control. Twenty-four hours after exposure to Cr(VI), MN PCEs (%) isolated from femurs were significantly elevated in Cr(VI)-treated mice relative to the negative control group. In a second experiment, Sprague-Dawley rats were exposed to 0.25 mg/kg Cr(VI) by i.t. instillation (in 100 µl) for 18 days with or without co-exposure to cigarette smoke. Additional rats were exposed to smoke alone or served as a negative control. Twenty-four hours after exposure to Cr(VI), MN were scored in PCEs harvested from femur bone marrow as well as bronchoalveolar lavage cells. Micronucleated PCEs (%) were increased 4-fold relative to controls; however, statistical significance was not indicated. Micronucleated lavage cells were significantly elevated 3.2-fold relative to controls. Conservatively assuming the i.t. dose of 0.25 mg/kg was in terms of potassium dichromate as opposed to Cr(VI) specifically, the concentration is approximately 625 ppm potassium dichromate (equivalent to ∼219 ppm Cr(VI)).
De Flora et al. (Citation2006) reported a series of studies investigating the effects of Cr(VI) on MN formation in blood and bone marrow. In one experiment, adult male BDF1 mice (10 per group) were exposed to 0, 10, or 20 ppm Cr(VI) in the form of K2Cr2O7 in drinking water for 20 days. Two additional sets of 10 mice received a single dose of 50 mg/kg Cr(VI) by either oral gavage or i.p. injection on day 19 of the study. On day 20, the percentage of micronucleated PCE isolated from femur was only significantly elevated compared to control mice in those receiving Cr(VI) via i.p. injection, 1.29 ± 0.36 vs 9.34 ± 1.53 (De Flora et al. Citation2006). In mice receiving Cr(VI) via drinking water for 20 days, the percentage of micronucleated normochromatic erythrocytes (NCE) in blood drawn from the lateral tail vein was not significantly altered at day 5, 12, or 20 of exposure. The percentage of MN PCE were not reported for the single day exposures.
In a second experiment, male and female BDF1 mice (5 animals per group) were exposed to 0, 5, 50, or 500 ppm Cr(VI) in the form of Na2Cr2O7 for in drinking water for 210 days. In addition, separate groups of male and female mice were exposed to 500 ppm Cr(III) as chromic potassium sulfate dodecahydrate in drinking water for 210 days. At study termination (day 210), there were no significant differences in MN PCE (%) in femurs; nor were there any significant differences in MN NCE (%) after 14, 28, 56, and 147 days of exposure.
In a third experiment, pregnant Swiss albino mice were exposed to 0, 5, or 10 ppm Cr(VI) as Na2Cr2O7 in drinking water throughout gestation. Two additional sets of pregnant mice received a single i.p. dose of 50 mg/kg Cr(VI) as either K2Cr2O7 or Na2Cr2O7 on day 17 of pregnancy. At study termination on gestational day 18 (GD18), MN PCE (%) were measured in the femurs of dams, and in the liver and peripheral blood of fetuses. The MN PCE (%) was significantly elevated in dams exposed to both forms of Cr(VI) by i.p. injection. Similarly, the MN PCE (%) was significantly elevated in fetal liver and peripheral blood samples from dams exposed to either form of Cr(VI) via i.p. injection.
In summary, the three experiments in De Flora et al. (Citation2006) indicate no cytogenetic DNA damage in blood and bone marrow cells from mice exposed orally to Cr(VI), whereas i.p. injection consistently increased micronuclei. It is worth noting that i.p. injections were administered in 0.2 ml, indicating concentrations of 6250 mg/L (ppm). Notably, none of the exposures in the three experiments resulted in significant changes in the PCE/NCE ratio.
The NTP conducted a series of studies investigating the effects of Cr(VI) on MN formation in peripheral blood of mice exposed to Cr(VI) (Na2Cr2O7) in drinking water for 90 days (NTP Citation2007). In one experiment, male and female B6C3F1 mice were exposed to 0, 62.5, 125, 250, 500, and 1000 mg/L sodium dichromate dihydrate (SDD; Na2Cr2O7 · H2O)—equivalent to ∼0, 22, 44, 88, 175, 350 ppm Cr(VI). At study termination, no significant increases in micronucleated NCEs were seen in either sex. A decrease in the percentage of PCEs was observed; however, the NTP study authors state that “the changes were small and not well correlated with exposure concentrations” (NTP Citation2007).
In a second experiment, MN NCEs were measured in three strains (B6C3F1, BALB/c, and am3-C57BL/6) of male mice exposed to 0, 62.5, or 125 ppm SDD (0, 22, or 44 ppm Cr(VI)) in drinking water for 90 days. In B6C3F1, there was no statistically significant increase in MN in any Cr(VI) group, yet the NTP study authors considered the results in this second B6C3F1 experiment to be equivocal based on a p-value for trend test being .03 that was close to the criteria for a positive trend of p-value .025. In BALB/c, the NTP study authors concluded there were no increases in MN NCEs relative to controls. In am3-C57BL/6, the two highest dose groups met the necessary cutoff of 0.008 for statistical significance, and there was a significant dose-related trend. Notably, the percentage of PCEs were unaffected in any groups of this second experiment. It is important to note that the values of MN seen in am3 are within normal values of the other mice strains in this study () so the biological relevance is uncertain. Importantly, there may be little or no historical MN data for this strain, as we were unable to find any published MN studies or other NTP reports using am3-C57BL/6 mice.
Overall, with the potential exception of the am3-C57BL/6 strain, there was little evidence for cytogenetic effects in the blood of mice exposed to Cr(VI) in drinking water for extended periods of time (up to 3 months) in the NTP studies.
Garcia-Rodriguez et al. (Citation2013) exposed male CD-1 mice to 20 mg/kg CrO3 by a single i.p. injection with and without pretreatment with 30 mg/kg green tea polyphenol extract via gavage 4 h before Cr(VI) injection. MN were scored 0, 24, 48, and 72 h after exposure (note: the study authors indicate that the study was OECD TG 474 compliant). Exposure to Cr(VI) alone significantly increased MN PCEs from 1.2 ± 0.8 to 13.2 ± 3.8 per 2000 cells at the 48 h time point only. Treatment with green tea extract mitigated but did not abolish the increase in MN observed with Cr(VI) at 48 h (Garcia-Rodriguez MC et al. Citation2013). In a similar study by the same group assessing the protective effects of epigallocatechin-3-gallate (EGCG) on CrO3 genotoxicity in Hsd:ICR mice, i.p. injection of 20 mg/kg CrO3 was again shown to increase MN PCEs at 48 h after injection (from 1.6 ± 0.3 to 10.2 ± 5.6 per 2000 cells), with 10 mg/kg EGCG mitigating the effects of Cr(VI) (Garcia-Rodriguez MC et al. Citation2016) (data not shown herein). In 2021, Garcia-Rodriguez et al. again assessed the antigenotoxic effects of EGCG using i.p. injection of 20 mg/kg CrO3. Consistent with previous studies, Cr(VI) increased MN PCEs at 48 h after injection (from 0.8 ± 5.7 to 11.5 ± 2.04 per 1000 cells), with 8.5 mg/kg EGCG somewhat mitigating the effects of Cr(VI) (Garcia-Rodriguez MDC et al. Citation2021) (data not shown herein).
Elshazly et al. (Citation2016) investigated the ability of radish oil to protect against Cr(VI)-induced DNA damage. Male Sprague-Dawley rats were exposed to 182 ppm Cr(VI) in drinking water for 6 months with and without daily radish oil treatment by oral gavage. The methods state that the radish oil was administered “one hour prior to SDD administration”; however, it is unclear how this was done unless the animals did not have ad libitum access to Cr(VI) drinking water. As such, there are uncertainties in the study design. Elshazly et al. concluded that radish oil provided anti-oxidant protection against Cr(VI)-induced genetic damage, but the number of cells scored in the micronucleus test was inadequate and the differences in frequencies observed among groups were not different at the 0.05 significance level when an appropriate binomial statistical test was applied to the data. The statistical test used is not appropriate for Poisson or binomial distributed data that were based on only 0, 1, or 4 total observed events (micronucleated cells) in the relevant treatment groups, so the claimed differences in micronucleus counts among groups are not supported by the data reported.
3.4. In vivo DNA damage studies
Genotoxicity assays that measure DNA breakage are available using either genomic DNA from tissue (Alkaline Elution Assay or gel electrophoresis) or the more recent in vivo Comet assay using DNA from nuclei from individual cells. Because the Comet assay described in OECD Test Guideline 489 (OECD Citation2016b) was first available in 2016, none of the studies described in this section explicitly claim to be OECD compliant. The Comet assay can be run in most all tissues; however, OECD Test Guideline 489 indicates that it is most commonly run in the liver or sites of first contact (e.g. stomach). The guidance also states that i.p. exposure is not recommended and that of the three measures of DNA damage (% tail DNA, tail length, and tail moment) the % tail DNA is preferred. Although the Comet test is an indicator test, some authoritative groups consider the Comet assay to be a proxy for mutation The European Food Safety Association (EFSA), for example, recommends the in vivo Comet assay as an alternative to the TGR assay for following up on positive in vitro bacteria mutation assays (EFSA Citation2017). Other groups have reported that the Comet and TGR assays have similar predictive capability for carcinogenicity (Zeller et al. Citation2018; Kirkland et al. Citation2019). Limitations of data used for these analyses included relatively few negative or weak mutagenic agents for either assay and many reviewed studies were conducted prior to finalization of the respective OECD TG. This resulted in large number of studies dosed by currently unacceptable routes or durations. Critically, these aforementioned references acknowledge that the TGR assay provides more MOA information than the Comet assay to assess mutagenic hazard. Data for DNA damage studies are discussed below and summarized in .
Table 3. Summary of in vivo DNA damage assays on Cr(VI).
3.4.1. DNA damage in Cr(VI) oral carcinogenicity target tissues
No DNA damage data from either assay were located for oral tumor target tissues.
3.4.2. DNA damage in Cr(VI) inhalation carcinogenicity target tissues
Izzotti et al. (Citation1998) exposed male Sprague-Dawley rats to 0.25 mg/kg Cr(VI) by i.t. instillation in 100 µl for 3 days. DNA fragmentation was determined by gel electrophoresis with densitometric analysis of ethidium bromide staining representing the percentage of DNA fragments with molecular weights less than 6 kb. Twenty-four hours after the last exposure, DNA fragmentation was measured in the lung and liver. In the lung, the percentage of low molecular weight DNA increased significantly from 9.2 ± 4.2 to 30.5 ± 9.7; however, DNA fragmentation was not elevated in the liver. Conservatively assuming the i.t. dose of 0.25 mg/kg was in terms of potassium dichromate as opposed to Cr(VI) specifically, the administered concentration is approximately 625 ppm potassium dichromate (equivalent to ∼219 ppm Cr(VI)).
3.4.3. DNA damage in nontarget tissue
Bagchi, Hassoun, Bagchi, Muldoon et al. (Citation1995) exposed Sprague-Dawley rats to a single oral gavage dose of 25 mg/kg Cr(VI) (sodium dichromate) or 895 mg/kg Cr(III) (chromium chloride) and measured DNA single strand breaks (SSB) by alkaline elution method in hepatocytes 48 h after exposure. Both exposures significantly increased DNA elution constant relative to control animals (Bagchi, Hassoun, Bagchi, Stohs Citation1995).
Bagchi Hassoun, Bagchi, Muldoon et al. (Citation1995) exposed female Sprague-Dawley rats to 10 mg/kg Cr(VI) (sodium dichromate) by oral gavage for 90 consecutive days and measured DNA SSB by alkaline elution method in hepatocytes after 15, 30, 45, 60, 75, and 90 days of exposure. At all time points, the DNA elution constant was significantly elevated compared to time matched untreated animals (Bagchi, Hassoun, Bagchi, Muldoon, et al. Citation1995). With the stated maximum gavage volume of 600 µl, the estimated Cr(VI) concentration is ∼1458 ppm.
Bagchi et al. (Citation1997) exposed Sprague-Dawley rats to 2.5 mg/kg Cr(VI) (sodium dichromate) by oral gavage for up to 120 days. DNA SSB were measured in the liver and brain by the alkaline elution method on days 0, 15, 30, 45, 60, 75 and 120 of exposure. Data were presented graphically and thus not summarized in . The DNA elution constant was significantly increased at multiple timepoints throughout the study duration (Bagchi et al. Citation1997).
Ueno et al. (Citation2001) exposed male albino mice to a single i.p. injection of 20 mg/kg Cr(VI) and DNA damage was measured in the liver, kidney, spleen, lung, and brain 15 min, 3 h, and 24 h after injection by Comet assay. Significant increases in DNA damage were observed in the liver and brain at 15 min but not thereafter () (Ueno et al. Citation2001). No significant increases were observed in the spleen, lung, or brain.
Sekihashi et al. (Citation2001) exposed male ddY mice to an i.p. dose of 120 mg/kg or gavage dose of 320 mg/kg Cr(VI) and sacrificed 3, 8, or 24 h after exposure. The Comet assay was run on stomach, colon, liver, kidney, bladder, lung, brain, and bone marrow. The bone marrow was the only tissue to not exhibit an increase in DNA damage at any time point by either route (data not shown). Other tissues exhibited DNA damage at one or more time points following exposure, but never at 24 h () (Sekihashi et al. Citation2001).
Dana Devi et al. (Citation2001) exposed male Swiss-Albino mice to 0.59, 1.19, 2.38, 4.75, 9.5, 19, 38, or 76 mg/kg Cr(VI) by oral gavage and measured DNA damage in leukocytes via Comet assay at 1, 2, 3, 4, 7, and 14 days after exposure. Based on the gavage volume of 50 µl, the administered concentrations in the lowest and highest doses were ∼103 and 13,300 ppm Cr(VI), respectively. Data were presented graphically without markings of statistical significance. The text indicates that tail length was significantly increased by all Cr(VI) doses at 24-h postexposure. A dose-dependent increase in tail length was observed between 0.59 and 9.5 mg/kg, which decreased between 9.5 and 78 mg/kg. A time-depended increase in tail length was observed between 0 and 48 h, which progressively decreased between 96 h and 14 days. Tail length peaked at ∼22 µm 48 h after exposure to 9.5 mg/kg Cr(VI). A positive control group treated with cyclophosphamide exhibited a significant increase in tail length (from <5µm to 18.4 µm) after 4 h of exposure (Dana Devi et al. Citation2001).
Wang et al. (Citation2006) exposed male Swiss albino mice to 0, 25, 50, or 100 mg/kg Cr(VI) by oral gavage for 1 of 5 consecutive days. Presumably 24 h after the final exposure, lymphocytes were harvested for Comet assay. After 1 day of exposure, the mean tail moment was significantly increased at ≥25 mg/kg. After 5 days of exposure, the mean tail moment was significantly increased at ≥50 mg/kg (Wang et al. Citation2006).
Patlolla et al. (Citation2009) exposed male Sprague-Dawley rats to 0, 2.5, 5, 7.5, and 10 mg/kg Cr(VI) for 5 consecutive days by i.p. injection. The Comet assay was conducted on peripheral blood lymphocytes 24, 48, 72, and 96 h post exposure. The mean tail length was significantly increased at ≥7.5 mg/kg at 24 and 48 h (Patlolla et al. Citation2009).
Kumar et al. (Citation2009) exposed male Swiss albino mice to 10, 20 or 50 ppm potassium dichromate in drinking water (5, 10, or 25 mg/kg) for 3 days. After 3 days of exposure, DNA damage assessed as tail moment, tail DNA %, and tail length was significantly increased in lymphocytes at all doses relative to controls (Kumar et al. Citation2009). In addition, a series of co-exposures to α-lipoic acid was shown to mitigate the genotoxicity of Cr(VI) (not shown here).
In a study by Minigaliyeva et al. (Citation2014), DNA damage was reported in leukocytes following repeat exposure to Cr(VI) by i.p. injection. The DNA damage was reported as the “DNA fragmentation coefficient”, which was described as the ratio of tritium radioactivity in tails and heads of comets; however, there was no mention of how or when tritium was administered in vivo or ex vivo (Minigaliyeva et al. Citation2014). Therefore, these results are not included in .
As described in the previous section, Elshazly et al. (Citation2016) investigated the ability of radish oil to protect against Cr(VI)-induced DNA damage. Using the Comet assay, exposure to Cr(VI) was shown significantly increased DNA damage in liver hepatocytes. Specifically, the tail length increased from 18.88 ± 0.87 in controls to 27.08 ± 1.78 in Cr(VI) treated rats and tail DNA % from 8.67 ± 0.49 to 30.09 ± 2.21. Treatment with both Cr(VI) and radish oil did not significantly increase these markers of DNA damage relative to untreated controls (Elshazly et al. Citation2016).
Orabi and Shawky (Citation2020) exposed male albino rats to 353 ppm Cr(VI) in drinking water for 12 weeks. Other rats received grapefruit seed oil orally daily at 3.7 g/kg with or without exposure to Cr(VI). At study termination, DNA damage was measured in the kidney by Comet analysis. Tail length (µm), tail DNA %, and tail moment all increased significantly in Cr(VI)-treated rats. Rats exposed to Cr(VI) also “showed marked necrosis of the renal tubular epithelium lining with fibrosis of interstitial tissue in the cortex and medulla“ by H&E staining (Orabi and Shawky Citation2020). Notably, 353 ppm Cr(VI) is ∼2-fold higher than the highest concentration administered to rats in the NTP 2-year cancer bioassay (180 ppm), where no nonneoplastic or neoplastic lesions were observed in the kidney. Given that the authors demonstrated DNA damage at the same concentration that frank toxicity was observed in the tissue, these results are not included in .
Fedala et al. (Citation2021) exposed pregnant Wistar rats to a single dose of 10 mg/kg potassium dichromate by subcutaneous injection on day 3 of pregnancy (no justification for this route of exposure was provided). Other rats were co-treated with 0.3 mg/kg Se or 20 mg/kg ZnCl2 by the same route. On the 20th day of gestation, animals were euthanized and thyroids removed. The thyroid gland was analyzed by single-cell Comet assay and scored by a classification scheme based on fluorescence intensity. The results, termed “Thyroid DNA damage (Total score)”, were only presented graphically and therefore not captured in . Cr(VI) exposure was indicated as significantly increasing this score, with the co-treatments ameliorating the effects to various degrees (Fedala et al. Citation2021). A significant uncertainty with this study is that OECD TG 489 indicates that the sampling time from a single dose study should occur at 2-26 h post treatment (OECD Citation2016b).
3.5. Summary of in vivo genotoxicity results
summarizes the in vivo mutation data from drinking water studies in . The only positive results were obtained in the eye spot test (discussed further in the next section). No mutation studies in target tissues for oral carcinogenicity were located that employed other routes of exposure. Mutation studies employing i.p. and i.t. exposure were generally positive, albeit at high doses (). summarizes the in vivo MN results in the duodenum (all drinking water studies; data in ). The only signs of increased MN formation were detected in the villi. These are discussed in detail in Section 4.1 below. No MN studies in target tissues for oral carcinogenicity were located that employed other routes of exposure. presents a summary of the in vivo blood and bone marrow MN studies conducted with drinking water (data in ). Given that very few studies are explicit about whether dose reporting was in terms of Cr(VI) or test article (e.g., Na2Cr2O7), no attempt was made to standardize doses and therefore these results should be viewed as semiquantitative. However, the two studies reporting positive effects clearly indicated concentrations of 60 and 180 ppm Cr(VI). Data for DNA breakage is not shown graphically because there is overwhelming evidence for DNA damage following drinking water and gavage exposure, i.p. administration, and i.t. instillation (). Although no such studies examined target tissues for oral carcinogenicity, one might expect DNA breakage in the villi of the duodenum based on synchrotron imaging ().
Figure 4. Summary of in vivo Cr(VI) mutation assays employing drinking water exposure. Red triangles indicate data points that differed significantly from concurrent controls. For clarity, variance data have been removed. The dotted line marks the current MCL of 0.1 ppm total Cr. Text on each plot designates the tissue examined, duration of exposure, and gene measured. Source: (A, B): Thompson Young et al. (Citation2015); (C) Thompson et al. (Citation2017); (D) O’Brien et al. (Citation2013); (E,F) Aoki et al. (Citation2019); (G) Kirpnick-Sobel et al. (2006).
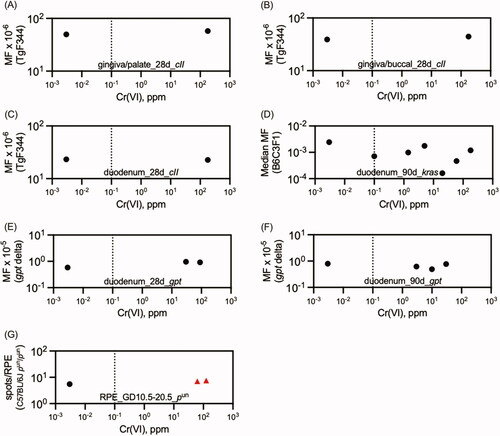
Figure 5. Summary of in vivo Cr(VI) MN assays in the female B6C3F1 mouse duodenum. Red triangles indicate data points that differed significantly form concurrent controls. For clarity, variance data have been removed. The dotted line marks the current MCL of 0.1 ppm total Cr. Text on each plot designates the tissue examined and duration of exposure. Source: (A, C): EPL (Citation2013); (B, D, F) O’Brien et al. (Citation2013); (E) Thompson et al. (Citation2015).
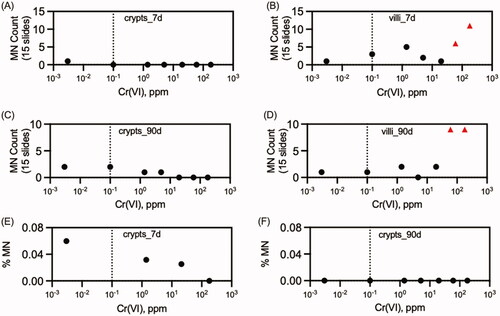
Figure 6. Summary of in vivo Cr(VI) MN assays in blood and bone marrow drinking water studies. Red triangles indicate data points that differed significantly form concurrent controls. For clarity, variance data have been removed. The dotted line marks the current MCL of 0.1 ppm total Cr. Source: (A-B) Mirsalis et al. (Citation1996); (C–F) De Flora et al. (Citation2006); (G–K) NTP (Citation2007); (L) Elshazly et al. (Citation2016).
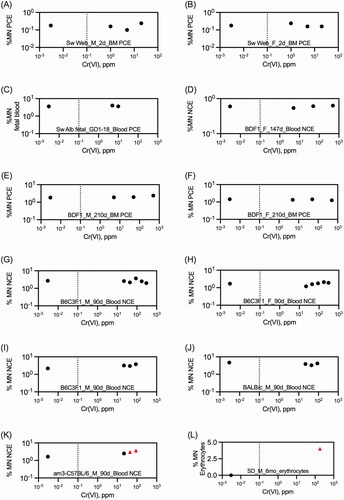
Overall, the data indicate that i.p. and i.v. administration of Cr(VI) can induce DNA damage (e.g., increased Comet tails), cytogenetic effects (e.g. positive MN assays), and mutagenicity (e.g., positive TGR assays). While non-physiological routes of exposure are generally not accepted by relevant OECD test guidelines, these assays inform what is possible from Cr(VI) exposure and can inform our interpretation of results in other assays. In contrast to nonphysiological exposures, oral exposure does not generally appear to cause point mutations or chromosomal damage, although there is potentially DNA damage in villi following exposure to high drinking water concentrations of Cr(VI). While complicated by experimental problems, there is evidence of Cr-DNA adducts in the duodenum following exposure to Cr(VI) (O'Brien et al. Citation2013); subsequent studies with XRF microscopy suggest that any such binding would be predominantly/exclusively limited to the villi (Thompson et al. Citation2015).
Although carcinogenic target tissues provide the most information relevant to assessing the likelihood of genotoxicity in the MOA for Cr(VI)-induced intestinal carcinogenesis, positive indications of genotoxicity in non-target tissues and exposure routes not relevant to human exposure, and possibly by indirect mechanisms, serve to demonstrate that Cr(VI) is capable of inducing various forms of DNA damage (consistent with in vitro genotoxicity studies of Cr(VI)). Therefore, the absence of such in the target tissues dosed by relevant routes of exposure greatly informs the MOA and extrapolation approaches for risk assessment.
4. Discussion
No in vivo genotoxicity studies were identified following inhalation exposure to any form of chromium. Only a small number of in vivo genotoxicity studies of the types discussed herein were described for the lung following other routes of exposure in sections 3.2.2, 3.3.2, and 3.4.2. As such, the balance of this review focuses on the oral genotoxicity and carcinogenicity of Cr(VI).
4.1. In vivo genotoxicity of Cr(VI) in oral exposure target tissues
Transgenic rodent (TGR) mutation assays are currently the most definitive assay for assessing in vivo mutagenicity in tissues of rats or mice. The in vivo MN assay and chromosomal aberration assay are the definitive tests for assessing cytogenetic effects in mice or rats. Assays for DNA damage are considered indicator tests and, as such, are given less weight in overall assessments of genotoxicity or MOA analyses. Data from TGR and in vivo cytogenetic tests are key for MOA analysis of carcinogens with emphasis placed on results from tumor target tissues in studies conducted using the same route of exposure as the carcinogenicity study. Cr(VI) is a well-studied agent with both in vivo mutation and in vivo MN assay results at dose levels and exposures relevant to the NTP carcinogenicity study (see Sections 3.2 and 3.3.1):
Negative results for in vivo mutations were observed in two studies in mouse duodenum (O'Brien et al. Citation2013; Aoki et al. Citation2019), one study in the rat duodenum (Thompson et al. Citation2017), and one study in the rat oral mucosa (Thompson et al. Citation2015).
Negative MN results were reported in mouse duodenal crypts in two separate studies (O'Brien et al. Citation2013; Thompson et al. Citation2015). MN and KN increased in mouse villi are consistent with localization of Cr(VI) in villi only in association with localized toxicity resulting in compensatory cell division.
Cell division is required for MN to form; therefore, the increase in KN and MN in villus enterocytes but not crypt enterocytes may be a secondary effect of increased cell turnover in response to toxicity associated with Cr localization in the villi (especially the tips) as visualized by synchrotron-based XRF microcopy (). It is conceivable that increased crypt cell proliferation results in some enterocytes reaching the villus prior to terminal differentiation with the capability of cell division. The biological relevance of cytogenetic effects in villi is uncertain as these cells are expected to be sloughed into the intestinal lumen. As discussed in earlier sections, there is evidence that Cr(VI) DNA damage might involve oxidative stress, and thus, the high levels of Cr detected in villi could cause MN directly and indirectly. Moreover, it has been recently recognized that certain cytoplasmic enzymes can detect self-DNA such as MN thereby leading to immune surveillance mechanisms presumably meant to remove damaged cells; however, such responses can also lead to pro-inflammatory states under certain conditions (Kirsch-Volders et al. Citation2020). Notably, XRF microscopy indicates Cr in intestinal villi (Thompson et al. Citation2015, Citation2015) and H&E staining demonstrates villus histiocytic infiltration in both mice and rats (NTP Citation2008; Thompson et al. Citation2011), perhaps as a result of nuclear damage in villus enterocytes.
4.2. In vivo genotoxicity of Cr(VI) in non-target tissues
Administration of Cr(VI) by i.p. and i.t. routes increased mutations in various non-target tissues (). Only one study analyzed mutations in non-target tissues via an oral route of exposure (Kirpnick-Sobol et al. Citation2006). In this study, a small increase in eye spots following exposure to both Cr(III) or Cr(VI) was observed. For Cr(VI), the total number of eye spots increased only slightly from ∼5.5/RPE in control mice (likely exposed to ∼0.003 ppm) to ∼6.5/RPE in mice exposed to 62.5 ppm (a > 20,000-fold increase in exposure). Doubling the Cr(VI) concentration to 125 ppm approximately doubled the Cr levels in embryos, but only increased eye spots 1.15-fold to ∼7.5/RPE (). Similar results were observed in mice exposed to 1875 and 3750 mg/kg Cr(III). Overall, the eye spot test indicates the potential for high concentrations of Cr(VI) in drinking water to cause large scale DNA deletions; however, the minimal dose-response at such high concentrations weakens the strength of the observations. It is also notable that exposure up to 180 ppm Cr(VI) for 2 years did not result in any tumors beyond the portal of entry in either mice or rats (NTP Citation2008), and exposure up to 5782 mg/kg Cr(III) was not carcinogenic (NTP Citation2010).
Not only are there uncertainties regarding the overall magnitude and dose-response for eye spots reported in Kirpnick-Sobol et al. (Citation2006), but also in the potential mechanism. According to Reliene et al. (Citation2004) there is a high frequency of spontaneous reversion in the pun gene; such reversions result in functional genes leading to pigmented cells (eye spots). In the absence of historical control data from this laboratory, it is not possible to determine if the increase seen with Cr is biologically relevant or within normal variation. Another consideration is that spontaneous reversions in the pun gene are a result of homologous recombination, which might be elevated in the presence of DNA strand breaks observed following Cr(VI) exposure, which itself might be the result of oxidative DNA damage. Indeed, it was previously shown that oral exposure to 300 mg/L titanium dioxide significantly increased eye spots 1.3-fold, which the study authors posited might be secondary to oxidative stress as indicated by increased 8-OHdG in livers from adult male mice exposed to titanium dioxide (Trouiller et al. Citation2009). Neither oxidative mechanisms nor the eye spot data are consistent with a linear dose-response or mutagenic linear no threshold MOA. Given that the eye spot test might be detecting mutations in a specific population of retinal epithelium precursor cells, it is notable that TGR assays have not detected increased mutations in the small intestine—which would arise from either crypt stem cells or some heretofore unidentified stem cell population in villi. While it is conceivable that the negative TGR assays in the intestine are due to their limited ability to detect large scale mutations, other assays (e.g., MN assays) address the potential for such damage in crypt enterocytes (see below).
Ideally, molecular assays for detecting large scale deletions should be tested in the carcinogenic target tissues. New assays have been developed to assess treatment effects on genome integrity, including drug-induced genomic instability test (DiGIT). This assay evaluates structural biomarkers of genomic instability in specific genes such as inter- and intra-chromosomal translocations, deletions, duplications, inversions and copy number variants, and has been proposed for genotoxicity screening (Minocherhomji et al. Citation2020). While these new assays might be used to assess the ability of Cr(VI) to induce larger scale deletions in the intestine, it remains to be determined if they provide additional information for genotoxicity assessment. For example, the DiGIT assay was recently shown to detect structural variations in the rat duodenum following exposure to cyclophosphamide (Minocherhomji et al. Citation2020). As discussed in Section 3.3.1, cyclophosphamide was shown to increase MN formation and λ-H2AX immunopositive aberrant nuclei in the mouse duodenum (Thompson et al. Citation2015) suggesting that cyclophosphamide-induced DiGIT markers occur concomitantly with more traditional cytogenetic markers such as MN. That Cr(VI) did not increase MN formation or λ-H2AX immunopositive aberrant nuclei suggests that it may also fail to increase markers associated with the DiGIT assay.
Chromosomal damage has been reported in blood and bone marrow following i.p., i.v. and high dose oral gavage studies with Cr(VI). Importantly, pharmacokinetic data indicate that a large percentage of Cr(VI) escapes reduction in the stomach at high exposure concentrations such as those employed in the NTP 2-year bioassay (≥5 ppm; ). This results in more Cr(VI) entering the intestinal lumen and damaging the mouse intestinal mucosa than would occur at typical exposure levels in humans. The estimated mg Cr(VI)/kg-day doses in the NTP (Citation2008) bioassay from drinking water exposures of 5-180 ppm ranged from 0.4–9 mg/kg in mice and 0.2–7 mg/kg in rats. Many of the i.p. and p.o. bolus exposures in genotoxicity studies are within or exceed the range of the NTP bioassay, either based on mg/kg dose or test article concentration. While this arguably makes the doses in these studies “relevant”, the internal dosimetry is very different compared to drinking water ingestion and thus positive genotoxicity results from bolus dosing has uncertain relevance to real-world exposure to Cr(VI). In contrast, the genotoxicity results in drinking water studies are uniformly negative for MN induction with the exception of a small increase in MN in am3-C57BL/6 mice (NTP Citation2007) and a questionable effect reported by Elshazly et al. (Citation2016).
4.3. Implications for cancer risk assessment of oral exposure to Cr(VI)
The U.S. EPA Guidelines for Carcinogen Risk Assessment describe the process for deriving safety criteria for carcinogens (U.S. EPA Citation2005). A critical component of these safety criteria is the extrapolation of cancer risk, typically observed in rodents at high doses, to risk in humans at lower environmentally relevant exposure levels. Extrapolation from empirical cancer data can be linear, or nonlinear through the application of uncertainty factors or more rarely by using a biologically based dose-response (BBDR) model that relates exposure over a wide range of doses (e.g. from background to carcinogenic doses) to biological events that increase tumor incidence. The choice between linear and nonlinear risk extrapolation is informed by MOA analysis, which U.S. EPA (Citation2005) explicitly states should be conducted for each tumor of interest. Thus, the MOA is not for the chemical per se but rather the tumor being modeled. Thus, while the data herein indicate that Cr(VI) can be genotoxic in vivo under certain routes of exposure (i.p., i.t.) and at some high exposure situations, the critical question is whether genotoxicity is an early (perhaps initiating) key event in the MOA for the types of exposure and resulting tumor(s) used to derive cancer safety criteria.
4.3.1. Mouse Duodenal tumors
Toxicity and carcinogenicity endpoints assessed in chronic bioassays indicate that drinking water exposure to high levels of Cr(VI) is relatively nontoxic with the important exception of causing proximal intestinal injury (cytotoxicity and regenerative cell proliferation) and ultimately intestinal tumors in mice. As such, the critical question for informing low-dose cancer risk from oral Cr(VI) exposure is whether Cr(VI) causes genotoxicity in the duodenum of the mouse under the exposure conditions employed in the cancer bioassay. In mice, the answer appears to be no, as evidenced by the lack of increased kras codon 12 MF in B6C3F1 mice (O'Brien et al. Citation2013), lack of increased MF in gpt delta mice (Aoki et al. Citation2019), and the lack of increased MN formation in intestinal crypts in B6C3F1 mice (O'Brien et al. Citation2013; Thompson et al. Citation2015). Moreover, Cr(VI) did not increase MF in the duodenum and oral mucosa of TgF344 rats at a tumorigenic concentration (Thompson et al. Citation2015, Citation2017). The absence of genotoxicity in the small intestine is explained, in part, by synchrotron-based XRF microscopy indicating that repeated exposure to 180 ppm Cr(VI) results in Cr fluorescence in villi but not crypts (Thompson et al. Citation2015, ). These findings strongly imply a lack of crypt dosimetry at environmental levels that are ≥60000-fold lower at ≤0.003 ppm. Overall, these data make a compelling case against early or low-dose genotoxicity in the alimentary canal.
In addition to absence of positive genotoxicity results in the duodenum, there is compelling evidence for an alternative MOA involving chronic cytotoxicity induced by regenerative cell proliferation as the MOA for duodenal tumors (Thompson et al. Citation2013; Moffat et al. Citation2018; Bhat et al. Citation2020). Those data are reviewed elsewhere but include evidence for increased cell proliferation after just one week of exposure (Thompson et al. Citation2015). A reanalysis of the NTP duodenal slides indicates similar, albeit milder, noncancer intestinal lesions in rats (Cullen et al. Citation2016). In fact, it has been argued that rats might have developed intestinal tumors if sufficient cytotoxicity and regenerative hyperplasia occurred in the NTP cancer bioassay (Cullen et al. Citation2016; Bhat et al. Citation2020).
4.3.2. Rat oral cavity tumors
The negative TGR assay in rat oral cavity indicates a lack of a mutagenic MOA at concentrations that caused oral tumors to appear late in a two-year cancer bioassay. To date, no obvious non-neoplastic histopathological lesions have been reported in the oral cavity of rats or mice following exposure to Cr(VI). Compared to intestinal tumors in mice, the dose-response for oral cavity tumors in rats is much shallower (). There are concerns about the overall health of the rats at the highest Cr(VI) concentrations that might have contributed to tumorigenesis (De Flora et al. Citation2008; Suh et al. Citation2014). Anemia and iron depletion in the duodenum and other tissues, possibly due to Cr(VI) oxidation of iron in the gut to less absorbable forms, has been demonstrated and might have impacted the health of the rats (Suh et al. Citation2014). De Flora et al. (Citation2008) speculated that irritant and oxidizing effects of Cr(VI) combined with some mechanical stimulus (e.g. water bottle cannula) might have led to oral tumor formation. An independent evaluation of the NTP rat tumor slides confirmed the absence of any preneoplastic alterations, but noted “periodontal pockets surrounding molar teeth [that] contained hair fragments along with feed/bedding material and were associated with low-grade chronic periodontal inflammation and low-grade periodontal epithelial hyperplasia.” (Long Citation2012). While this was observed in both treated and control animals, it is conceivable that any Cr(VI) soaked items (e.g., bedding) might result in prolonged local exposure in this microenvironment. Similarly, foreign items can embed in buccal mucosal folds. Salivary gland atrophy was reported in female rats (NTP Citation2008), which might have affected dosimetry and clearance of Cr(VI) or the overall oral health which might explain the slightly higher tumor incidence in female rats (). Compared to mice, the GSH/GSSG ratios in oral tissue declined more in rats (Thompson et al. Citation2012); however, transcriptomic analyses detected very few gene changes in oral cavity of mice or rats following 7 or 90 days of exposure to ≤180 ppm Cr(VI) (Thompson et al. Citation2016)—possibly indicating a lack of biological response to Cr(VI).
5. Conclusions
Overall, the lack of demonstrable increases in mutant frequency and chromosomal damage in carcinogenic target tissues at exposures comparable to the tumorigenic exposures in the NTP drinking water study suggests that the tumors observed in the NTP (Citation2008) cancer bioassay most likely arose from site of contact toxicity and increased cell proliferation rather than from direct DNA damage. This suggests that a nonlinear approach to cancer risk assessment is scientifically justified. This conclusion does not discount genotoxicity data in non-target tissues as either flawed or irrelevant, but rather considers such studies as providing limited insight into the MOA for the tumors in the NTP bioassay that are being used to derive safety criteria for risk assessment purposes. In fact, the positive indications of genotoxicity in non-target tissues and exposure routes not relevant to human exposure and possibly by indirect mechanisms serve to demonstrate that Cr(VI) is capable of inducing various forms of DNA damage (consistent with in vitro genotoxicity of Cr(VI)) and therefore the absence of such in the target tissues dosed by relevant routes of exposure greatly informs the MOA and extrapolation approaches for risk assessment.
A similar conclusion was drawn by leveraging an analysis that demonstrated that most carcinogens exhibit greater genotoxic potency than tumorigenic potency, i.e., lower benchmark dose values for genotoxic endpoints than carcinogenic endpoints (MacGregor et al. Citation2015; Soeteman-Hernandez et al. Citation2016). However, this relationship did not hold for chloroform and diethanolamine—two carcinogens widely thought to induce tumors via non-mutagenic MOAs. Thompson et al. (Citation2016) recognized that Soeteman-Hernández et al. (2015) included Cr(VI) in their analyses, specifically using benchmark doses for intestinal tumors in B6C3F1 mice and MN induction in am3-C57BL/6 mice. As already discussed, MN results in this strain are highly uncertain. Replacing the genotoxicity benchmark dose with a composite benchmark dose from combined modeling of the other four MN assays in NTP (Citation2007) or even treating the highest no effect levels in the other duodenal genotoxicity assays as benchmark doses, Cr(VI) behaved more like chloroform and diethanolamine, with carcinogenic potency being greater than genotoxic potency (Thompson et al. Citation2016).
In the AOP for small intestinal cancer arising from nongenotoxic mechanisms described in Bhat et al. (Citation2020), two conditions were proposed for concluding that an intestinal carcinogen acts through a nongenotoxic MOA. The first condition is consistent evidence against genotoxicity in the intestine, ideally with corroborating dosimetry data in crypts and villi. The second condition is clear and consistent evidence for a plausible MOA containing key events that increase the chance of intestinal tumor formation such as, but not limited to, chronic regenerative hyperplasia. Evidence for the latter has been reviewed in several places (Thompson et al. Citation2013, Citation2017; Health Canada Citation2016; Moffat et al. Citation2018; Bhat et al. Citation2020), whereas this is the first such review of the in vivo oral genotoxicity of Cr(VI). Taken together, the MOA and genotoxicity data strongly support exploration and use of nonlinear risk assessment approaches for developing oral toxicity criteria for Cr(VI). Several organizations have already done so (Health Canada Citation2016; TCEQ Citation2016; FSCJ Citation2019; WHO Citation2019). Importantly, as new or improved genotoxicity assays and/or new guidance become available, information from future testing can be used to update and revise Cr(VI) risk assessments as warranted.
Acknowledgments
The authors acknowledge the valuable peer-review comments from reviewers selected by the Editor and anonymous to the authors. The authors also thank Dr. Deborah Barsotti (Emilcott) for review and comment on an earlier version of this manuscript.
Declaration of interest
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by The Hexavalent Chromium Panel of the American Chemistry Council.
The authors’ employment affiliations are shown in the title block above. ToxStrategies is a private consulting firm providing services to private and public organizations on toxicology and risk assessment issues. Drs. Aardema and MacGregor and Mr. Robert Young are private consultants. This idea for this work was conceived and presented by ToxStrategies to the Hexavalent Chromium Science Panel of the American Chemistry Council (ACC) (https://www.americanchemistry.com) in 2020, as it represented a data gap in the science and risk assessment of hexavalent chromium [Cr(VI)]. The ACC represents more than 170 companies engaged in the production, manufacture and use of chemicals. Members of the Hexavalent Chromium Science Panel of ACC, under the direction and coordination of Ms. Eileen Conneely, were given the opportunity to review the draft manuscript to provide feedback to the authors regarding the clarity of the science presented but not on the interpretation of research findings. The contents of this manuscript reflect solely the view of the authors. Those providing comments are listed in the Acknowledgements section. The project was funded through contracts between ACC and ToxStrategies. ToxStrategies is currently contracted with multiple chemical panels at ACC to provide scientific consulting support. ToxStrategies is also currently contracted by ACC on other projects involving the evaluation of the available science and a risk assessment for Cr(VI). All the scientists at ToxStrategies (CT, MH) involved in the development of the current manuscript were provided salary compensation as part of their normal employment as scientific consultants. Authors MA, JM and RY were subcontracted by ToxStrategies.
There are no conflicts of interest for any of the authors to disclose related to the submission of this manuscript. None of the authors are currently engaged to testify as experts on behalf of the sponsors in litigation related to Cr(VI). CT has participated in meetings with the EPA and other agencies, on behalf of ACC, to discuss the current state of the science for Cr(VI) and the need to consider mode of action data in a risk assessment for Cr(VI). BY has coauthored two TGR manuscripts on Cr(VI) on behalf of the Electric Power Research Institute. JM is a coauthor of a previous review of the genotoxic risks from Cr(III), an essential trace element that is widely used as a food and feed supplement, that was partially funded by Zinpro Corporation (Critical Reviews in Toxicology 38:173-190, 2008).
Notes
1 This website is administered by ToxStrategies and is sponsored by the American Chemistry Council. It contains information related to Cr(VI) studies funded by the ACC as well as other related information on Cr(VI).
2 Equivocal evidence of preputial gland neoplasms were observed in male rats.
3 Accessed 11/18/2020.
4 Based on our analysis after digitally extracting data from plots in Kirpnick-Sobol et al. (Citation2006).
References
- Aoki Y, Matsumoto M, Matsumoto M, Masumura K, Nohmi T. 2019. Mutant frequency is not increased in mice orally exposed to sodium dichromate. Food Saf (Tokyo)). 7(1):2–10.
- Bagchi D, Hassoun EA, Bagchi M, Muldoon DF, Stohs SJ. 1995. Oxidative stress induced by chronic administration of sodium dichromate [Cr(VI)] to rats. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol. 110(3):281–287.
- Bagchi D, Hassoun EA, Bagchi M, Stohs SJ. 1995. Chromium-induced excretion of urinary lipid metabolites, DNA damage, nitric oxide production, and generation of reactive oxygen species in Sprague-Dawley rats. Comp Biochem Physiol C Pharmacol Toxicol Endocrinol. 110(2):177–187.
- Bagchi D, Vuchetich PJ, Bagchi M, Hassoun EA, Tran MX, Tang L, Stohs SJ. 1997. Induction of oxidative stress by chronic administration of sodium dichromate [chromium VI] and cadmium chloride [cadmium II] to rats. Free Radic Biol Med. 22(3):471–478.
- Balansky RM, D'Agostini F, Izzotti A, De Flora S. 2000. Less than additive interaction between cigarette smoke and chromium(VI) in inducing clastogenic damage in rodents. Carcinogenesis. 21:1677–1682.
- Barker N, Ridgway RA, van Es JH, van de Wetering M, Begthel H, van den Born M, Danenberg E, Clarke AR, Sansom OJ, Clevers H. 2009. Crypt stem cells as the cells-of-origin of intestinal cancer [Research Support, Non-U.S. Gov't]. Nature. 457(7229):608–611.
- Bhat VS, Cohen SM, Gordon EB, Wood CE, Cullen JM, Harris MA, Proctor DM, Thompson CM. 2020. An adverse outcome pathway for small intestinal tumors in mice involving chronic cytotoxicity and regenerative hyperplasia: a case study with hexavalent chromium, captan, and folpet. Crit Rev Toxicol. 50(8):685–706.
- Chandra SA, Nolan MW, Malarkey DE. 2010. Chemical carcinogenesis of the gastrointestinal tract in rodents: an overview with emphasis on NTP carcinogenesis bioassays. Toxicol Pathol. 38(1):188–197.
- Cheng L, Liu S, Dixon K. 1998. Analysis of repair and mutagenesis of chromium-induced DNA damage in yeast, mammalian cells, and transgenic mice. Environ Health Perspect. 106 Suppl 4(Suppl 4):1027–1032.
- Cheng L, Sonntag DM, de Boer J, Dixon K. 2000. Chromium(VI)-induced mutagenesis in the lungs of big blue transgenic mice. J Environ Pathol Toxicol Oncol. 19(3):239–249.
- Chidiac P, Goldberg MT. 1987. Lack of induction of nuclear aberrations by captan in mouse duodenum [Research Support, Non-U.S. Environ Mutagen. 9(3):297–306.
- Cullen JM, Ward JM, Thompson CM. 2016. Reevaluation and classification of duodenal lesions in B6C3F1 mice and F344 rats from 4 studies of hexavalent chromium in drinking water. Toxicol Pathol. 44(2):279–289.
- Dana Devi K, Rozati R, Saleha Banu B, Jamil K, Grover P. 2001. In vivo genotoxic effect of potassium dichromate in mice leukocytes using comet assay. Food Chem Toxicol. 39(8):859–865.
- De Flora S, D'Agostini F, Balansky R, Micale R, Baluce B, Izzotti A. 2008. Lack of genotoxic effects in hematopoietic and gastrointestinal cells of mice receiving chromium(VI) with the drinking water. Mutat Res. 659(1-2):60–67.
- De Flora S, Iltcheva M, Balansky RM. 2006. Oral chromium(VI) does not affect the frequency of micronuclei in hematopoietic cells of adult mice and of transplacentally exposed fetuses. Mutat Res. 610(1-2):38–47.
- den Braver-Sewradj SP, van Benthem J, Staal YCM, Ezendam J, Piersma AH, Hessel EVS. 2021. Occupational exposure to hexavalent chromium. Part II. Hazard assessment of carcinogenic effects. Regul Toxicol Pharmacol. 126:105045.
- Eastmond DA. 2012. Factors influencing mutagenic mode of action determinations of regulatory and advisory agencies. Mutat Res. 751(1):49–63.
- EFSA. 2017. Clarification of some aspects related to genotoxicity assessment. Efsa J. 15(12):5113.
- Elshazly MO, Morgan AM, Ali ME, Abdel-Mawla E, Abd El-Rahman SS. 2016. The mitigative effect of Raphanus sativus oil on chromium-induced geno- and hepatotoxicity in male rats. J Adv Res. 7(3):413–421.
- EPL. 2013. Quantitative histopathologic evaluations of mouse duodenum specimens. Sterling, VA. 928-002 and 928-003.
- Fedala A, Adjroud O, Abid-Essefi S, Timoumi R. 2021. Protective effects of selenium and zinc against potassium dichromate-induced thyroid disruption, oxidative stress, and DNA damage in pregnant Wistar rats. Environ Sci Pollut Res Int. 28(18):22563–22576.
- Feng Y, Bommer GT, Zhao J, Green M, Sands E, Zhai Y, Brown K, Burberry A, Cho KR, Fearon ER. 2011. Mutant KRAS promotes hyperplasia and alters differentiation in the colon epithelium but does not expand the presumptive stem cell pool. Gastroenterology. 141(3):1003–1013. e1001-1010.
- FSCJ. 2019. Risk Assessment Report Hexavalent Chromium (Beverages). Food Safety Commission of Japan. 7(2):56–57.
- Garcia-Rodriguez MC, Carvente-Juarez MM, Altamirano-Lozano MA. 2013. Antigenotoxic and apoptotic activity of green tea polyphenol extracts on hexavalent chromium-induced DNA damage in peripheral blood of CD-1 mice: analysis with differential acridine orange/ethidium bromide staining. Oxid Med Cell Longev. 2013:486419.
- Garcia-Rodriguez MC, Montano-Rodriguez AR, Altamirano-Lozano MA. 2016. Modulation of hexavalent chromium-induced genotoxic damage in peripheral blood of mice by epigallocatechin-3-gallate (EGCG) and its relationship to the apoptotic activity. J Toxicol Environ Health A. 79(1):28–38.
- Garcia-Rodriguez MDC, Serrano-Reyes G, Hernandez-Cortes LM, Altamirano-Lozano M. 2021. Antigenotoxic effects of (-)-epigallocatechin-3-gallate (EGCG) and its relationship with the endogenous antioxidant system, 8-hydroxydeoxyguanosine adduct repair (8-OHdG), and apoptosis in mice exposed to chromium(VI). J Toxicol Environ Health A. 84(8):331–344.
- Gibb HJ, Lees PS, Pinsky PF, Rooney BC. 2000. Clinical findings of irritation among chromium chemical production workers. Am J Ind Med. 38(2):127–131.
- Glaser U, Hochrainer D, Kloppel H, Oldiges H. 1986. Carcinogenicity of sodium dichromate and chromium (VI/III) oxide aerosols inhaled by male Wistar rats. Toxicology. 42(2-3):219–232. eng.
- Goldberg MT, Blakey DH, Bruce WR. 1983. Comparison of the effects of 1,2-dimethylhydrazine and cyclophosphamide on micronucleus incidence in bone marrow and colon. Mutat Res. 109(1):91–98.
- Goldberg MT, Chidiac P. 1986. An in vivo assay for small intestine genotoxicity. Mutat Res. 164(4):209–215.
- Gollapudi BB, Johnson GE, Hernandez LG, Pottenger LH, Dearfield KL, Jeffrey AM, Julien E, Kim JH, Lovell DP, Macgregor JT, et al. 2013. Quantitative approaches for assessing dose-response relationships in genetic toxicology studies. Environ Mol Mutagen. 54(1):8–18.
- Health Canada. 2016. Guidelines for Canadian drinking water quality: guideline technical document — chromium. In: Water and Air Quality Bureau HEaCSB, editor. Ottawa, Ontario.: Health Canada.
- Howard BE, Phillips J, Miller K, Tandon A, Mav D, Shah MR, Holmgren S, Pelch KE, Walker V, Rooney AA, et al. 2016. SWIFT-Review: a text-mining workbench for systematic review. Syst Rev. 5:87.
- IARC. 1990. Chromium, nickel and welding, IARC Monogr. Eval. Carcinog. Risks Hum. 49:1–648.
- Itoh S, Shimada H. 1996. Micronucleus induction by chromium and selenium, and suppression by metallothionein inducer. Mutat Res. 367(4):233–236.
- Itoh S, Shimada H. 1997. Clastogenicity and mutagenicity of hexavalent chromium in lacZ transgenic mice. Toxicol Lett. 91(3):229–233.
- Itoh S, Shimada H. 1998. Bone marrow and liver mutagenesis in lacZ transgenic mice treated with hexavalent chromium. Mutat Res. 412(1):63–67.
- Izzotti A, Bagnasco M, Camoirano A, Orlando M, De Flora S. 1998. DNA fragmentation, DNA-protein crosslinks, postlabeled nucleotidic modifications, and 8-hydroxy-2'-deoxyguanosine in the lung but not in the liver of rats receiving intratracheal instillations of chromium(VI). Chemoprevention by oral N-acetylcysteine. Mutat Res. 400:233–244.
- Johnson GE, Soeteman-Hernandez LG, Gollapudi BB, Bodger OG, Dearfield KL, Heflich RH, Hixon JG, Lovell DP, MacGregor JT, Pottenger LH, et al. 2014. Derivation of point of departure (PoD) estimates in genetic toxicology studies and their potential applications in risk assessment. Environ Mol Mutagen. 55(8):609–623.
- Kirkland D, Levy DD, LeBaron MJ, Aardema MJ, Beevers C, Bhalli J, Douglas GR, Escobar PA, Farabaugh CS, Guerard M, et al. 2019. A comparison of transgenic rodent mutation and in vivo comet assay responses for 91 chemicals. Mutat Res Genet Toxicol Environ Mutagen. 839:21–35.
- Kirkland D, Speit G. 2008. Evaluation of the ability of a battery of three in vitro genotoxicity tests to discriminate rodent carcinogens and non-carcinogens III. Appropriate follow-up testing in vivo. Mutat Res. 654(2):114–132.
- Kirman CR, Hays SM, Aylward LL, Suh M, Harris MA, Thompson CM, Haws LC, Proctor DM. 2012. Physiologically based pharmacokinetic model for rats and mice orally exposed to chromium. Chem Biol Interact. 200(1):45–64.
- Kirpnick-Sobol Z, Reliene R, Schiestl RH. 2006. Carcinogenic Cr(VI) and the nutritional supplement Cr(III) induce DNA deletions in yeast and mice. Cancer Res. 66(7):3480–3484.
- Kirsch-Volders M, Bolognesi C, Ceppi M, Bruzzone M, Fenech M. 2020. Micronuclei, inflammation and auto-immune disease. Mutat Res Rev Mutat Res. 786:108335.
- Knudsen I. 1980. The mammalian spot test and its use for the testing of potential carcinogenicity of welding fume particles and hexavalent chromium. Acta Pharmacol Toxicol (Copenh)). 47(1):66–70.
- Kumar S, Budhwar R, Nigam A, Priya S. 2009. Cytoprotection against Cr(6+)-induced DNA damage by alpha-lipoic acid: implications in reducing occupational cancer risk. Mutagenesis. 24(6):495–500.
- Lambert IB, Singer TM, Boucher SE, Douglas GR. 2005. Detailed review of transgenic rodent mutation assays. Mutat Res. 590(1-3):1–280.
- Long P. 2012. Review of Carcinogenesis Study of Sodium Dichromate Dihydrate (CAS NO. 7789-12-0) in F344 rats (drinking water study). Anatomic Pathology Expert Report by Vet Path Services.
- Luippold RS, Mundt KA, Austin RP, Liebig E, Panko J, Crump C, Crump K, Proctor D. 2003. Lung cancer mortality among chromate production workers [Research Support, Non-U.S. Gov't]. Occup Environ Med. 60(6):451–457.
- MacGregor JT, Frotschl R, White PA, Crump KS, Eastmond DA, Fukushima S, Guerard M, Hayashi M, Soeteman-Hernandez LG, Johnson GE, et al. 2015. IWGT report on quantitative approaches to genotoxicity risk assessment II. Use of point-of-departure (PoD) metrics in defining acceptable exposure limits and assessing human risk. Mutat Res Genet Toxicol Environ Mutagen. 783:66–78.
- Malling HV, Burkhart JG. 1989. Use of phi X174 as a shuttle vector for the study of in vivo mammalian mutagenesis. Mutat Res. 212(1):11–21.
- McCarroll N, Keshava N, Chen J, Akerman G, Kligerman A, Rinde E. 2010. An evaluation of the mode of action framework for mutagenic carcinogens case study II: chromium (VI). Environ Mol Mutagen. 51(2):89–111.
- Minigaliyeva IA, Katsnelson BA, Privalova LI, Gurvich VB, Panov VG, Varaksin AN, Makeyev OH, Sutunkova MP, Loginova NV, Kireyeva EP, et al. 2014. Toxicodynamic and toxicokinetic descriptors of combined chromium (VI) and nickel toxicity. Int J Toxicol. 33(6):498–505.
- Minocherhomji S, Liu Y, He YD, Fielden MR. 2020. Biomarkers of genome instability in normal mammalian genomes following drug-induced replication stress. Environ Mol Mutagen. 61(8):770–785.
- Mirsalis JC, Hamilton CM, O'Loughlin KG, Paustenbach DJ, Kerger BD, Patierno S. 1996. Chromium (VI) at plausible drinking water concentrations is not genotoxic in the in vivo bone marrow micronucleus or liver unscheduled DNA synthesis assays. Environ Mol Mutagen. 28(1):60–63.
- Moffat I, Martinova N, Seidel C, Thompson CM. 2018. Hexavalent chromium in drinking water. Journal AWWA. 110(5):E22–E35.
- Morita T, MacGregor JT, Hayashi M. 2011. Micronucleus assays in rodent tissues other than bone marrow [Research Support, Non-U.S. Gov't Review]. Mutagenesis. 26(1):223–230.
- Mukherjee P, Sarkar D, Sharma A. 1997. Effects of dietary consumption of black tea infusion alone and in combination with known clastogens on mouse bone marrow chromosomes in vivo. Food Chem Toxicol. 35(7):657–661.
- Newton MF, Lilly LJ. 1986. Tissue-specific clastogenic effects of chromium and selenium salts in vivo. Mutat Res. 169(1-2):61–69.
- NTP. 2007. National Toxicology Program technical report on the toxicity studies of sodium dichromate dihydrate (CAS No. 7789-12-0) administered in drinking water to male and female F344/N rats and B6C3F1 mice and male BALB/c and am3-C57BL/6 mice. NTP Toxicity Report Series Number 72, NIH Publication No 07-5964.
- NTP. 2008. National Toxicology Program technical report on the toxicology and carcinogenesis studies of sodium dichromate dihydrate (CAS No. 7789-12-0) in F344/N rats and B6C3F1 mice (drinking water studies), NTP TR 546. NIH Publication No 08-5887.
- NTP. 2010. National Toxicology Program technical report on the toxicology and carcinogenesis studies of chromium picolinate monohydrate (CAS NO. 27882-76-4) in F344/N rats and B6C3F1 mice (feed studies). NIH Publication No 08-5897.
- O'Brien TJ, Ceryak S, Patierno SR. 2003. Complexities of chromium carcinogenesis: role of cellular response, repair and recovery mechanisms. Mutat Res. 533(1-2):3–36.
- O'Brien TJ, Ding H, Suh M, Thompson CM, Parsons BL, Harris MA, Winkelman WA, Wolf JC, Hixon JG, Schwartz AM, et al. 2013. Assessment of K-Ras mutant frequency and micronucleus incidence in the mouse duodenum following 90-days of exposure to Cr(VI) in drinking water [Research Support, Non-U.S. Mutat Res. 754(1-2):15–21.
- OECD. 1986. OECD Guideline Test No. 484: Genetic Toxicology: Mouse Spott Test (deleted April 2, 2014).
- OECD. 2016a. Test Guideline No. 475 - Mammalian Bone Marrow Chromosomal Aberration Test.
- OECD. 2016b. Test Guideline No. 489 - In Vivo Mammalian Alkaline Comet Assay.
- OECD. 2020. Test Guideline No. 488, - Transgenic Rodent Somatic and Germ Cell Gene Mutation Assays. OECD, Paris, France.
- OEHHA. 2011. Final Technical Support Document on Public Health Goal for Hexavalent Chromium in Drinking Water. Pesticide and Environmental Toxicology Branch, Office of Environmental Health Hazard Assessment, California Environmental Protection Agency.
- Orabi SH, Shawky SM. 2020. Ameliorative effects of grape seed oil on chromium-induced nephrotoxicity and oxidative stress in rats. SVR. 57(3):123–131.
- Parsons BL, Manjanath MB, Myers MB, McKim KL, Wang Y, Gollapudi BB, Moore N, Haber Lt, Moore MM. 2012. Induction of cII and K-Ras mutation in lung DNA of Big Blue mice exposed to ethylene oxide by inhalation,. 43rd Annual Meeting Environmental Mutagen Society; Bellevue, Washington.
- Parsons BL, Marchant-Miros KE, Delongchamp RR, Verkler TL, Patterson TA, McKinzie PB, Kim LT. 2010. ACB-PCR quantification of K-RAS codon 12 GAT and GTT mutant fraction in colon tumor and non-tumor tissue. Cancer Invest. 28(4):364–375.
- Parsons BL, Myers MB, Meng F, Wang Y, McKinzie PB. 2010. Oncomutations as biomarkers of cancer risk [Review. Environ Mol Mutagen. 51(8-9):836–850.
- Patlolla AK, Barnes C, Yedjou C, Velma VR, Tchounwou PB. 2009. Oxidative stress, DNA damage, and antioxidant enzyme activity induced by hexavalent chromium in Sprague-Dawley rats. Environ Toxicol. 24(1):66–73.
- Potten CS, Loeffler M. 1990. Stem cells: attributes, cycles, spirals, pitfalls and uncertainties. Lessons for and from the crypt [Research Support, Non-U.S. Gov't Review]. Development. 110(4):1001–1020.
- Reliene R, Bishop AJ, Aubrecht J, Schiestl RH. 2004. In vivo DNA deletion assay to detect environmental and genetic predisposition to cancer. Methods Mol Biol. 262:125–139.
- Rizk P, Barker N. 2012. Gut stem cells in tissue renewal and disease: methods, markers, and myths. Wiley Interdiscip Rev Syst Biol Med. 4(5):475–496.
- Sarkar D, Sharma A, Talukder G. 1993. Differential protection of chlorophyllin against clastogenic effects of chromium and chlordane in mouse bone marrow in vivo. Mutat Res. 301(1):33–38.
- Sekihashi K, Sasaki T, Yamamoto A, Kawamura K, Ikka T, Tsuda S, Sasaki YF. 2001. A comparison of intraperitoneal and oral gavage administration in comet assay in mouse eight organs. Mutat Res. 493(1-2):39–54.
- Shindo Y, Toyoda Y, Kawamura K, Kurebe M, Shimada H, Hattori C, Satake S. 1989. Micronucleus test with potassium chromate(VI) administered intraperitoneally and orally to mice. Mutat Res. 223(4):403–406.
- Soeteman-Hernandez LG, Johnson GE, Slob W. 2016. Estimating the carcinogenic potency of chemicals from the in vivo micronucleus test. Mutagenesis. 31(3):347–358.
- SSAB. 2020. Memorandum: T. Augspurger (Chair, Secretaries' Science Advisory Board) to S. Holman (NC Department of Environmental Quality), Secretaries' Science Advisory Board response to inquiry on hexavalent chromium.
- Stern AH. 2010. A quantitative assessment of the carcinogenicity of hexavalent chromium by the oral route and its relevance to human exposure. Environ Res. 110(8):798–807.
- Stout MD, Herbert RA, Kissling GE, Collins BJ, Travlos GS, Witt KL, Melnick RL, Abdo KM, Malarkey DE, Hooth MJ. 2009. Hexavalent chromium is carcinogenic to F344/N rats and B6C3F1 mice after chronic oral exposure. Environ Health Perspect. 117(5):716–722.
- Stout MD, Nyska A, Collins BJ, Witt KL, Kissling GE, Malarkey DE, Hooth MJ. 2009. Chronic toxicity and carcinogenicity studies of chromium picolinate monohydrate administered in feed to F344/N rats and B6C3F1 mice for 2 years. Food Chem Toxicol. 47(4):729–733.
- Suh M, Thompson CM, Kirman CR, Carakostas MC, Haws LC, Harris MA, Proctor DM. 2014. High concentrations of hexavalent chromium in drinking water alter iron homeostasis in F344 rats and B6C3F1 mice [Research Support, Non-U.S. Gov't]. Food Chem Toxicol. 65:381–388.
- TCEQ 2016. Hexavalent Chromium Oral Reference Dose: Development Support Document (Final).
- Thompson CM, Bichteler A, Rager JE, Suh M, Proctor DM, Haws LC, Harris MA. 2016. Comparison of in vivo genotoxic and carcinogenic potency to augment mode of action analysis: case study with hexavalent chromium. Mutat Res Genet Toxicol Environ Mutagen. 800-801:28–34.
- Thompson CM, Haws LC, Harris MA, Gatto NM, Proctor DM. 2011. Application of the U.S. EPA mode of action Framework for purposes of guiding future research: a case study involving the oral carcinogenicity of hexavalent chromium [Research Support, Non-U.S. Gov't Review]. Toxicol Sci. 119(1):20–40.
- Thompson CM, Kirman CR, Hays SM, Suh M, Harvey SE, Proctor DM, Rager JE, Haws LC, Harris MA. 2018. Integration of mechanistic and pharmacokinetic information to derive oral reference dose and margin-of-exposure values for hexavalent chromium. J Appl Toxicol. 38(3):351–365.
- Thompson CM, Proctor DM, Haws LC, Hebert CD, Grimes SD, Shertzer HG, Kopec AK, Hixon JG, Zacharewski TR, Harris MA. 2011. Investigation of the mode of action underlying the tumorigenic response induced in B6C3F1 mice exposed orally to hexavalent chromium [Research Support, Non-U.S. Gov't]. Toxicol Sci. 123(1):58–70.
- Thompson CM, Proctor DM, Suh M, Haws LC, Hebert CD, Mann JF, Shertzer HG, Hixon JG, Harris MA. 2012. Comparison of the effects of hexavalent chromium in the alimentary canal of F344 rats and B6C3F1 mice following exposure in drinking water: implications for carcinogenic modes of action [Research Support, Non-U.S. Gov't]. Toxicol Sci. 125(1):79–90.
- Thompson CM, Proctor DM, Suh M, Haws LC, Kirman CR, Harris MA. 2013. Assessment of the mode of action underlying development of rodent small intestinal tumors following oral exposure to hexavalent chromium and relevance to humans. Crit Rev Toxicol. 43(3):244–274.
- Thompson CM, Rager JE, Suh M, Ring CL, Proctor DM, Haws LC, Fry RC, Harris MA. 2016. Transcriptomic responses in the oral cavity of F344 rats and B6C3F1 mice following exposure to Cr(VI): implications for risk assessment. Environ Mol Mutagen. 57(9):706–716.
- Thompson CM, Seiter J, Chappell MA, Tappero RV, Proctor DM, Suh M, Wolf JC, Haws LC, Vitale R, Mittal L, et al. 2015. Synchrotron-Based Imaging of Chromium and γ-H2AX immunostaining in the duodenum following repeated exposure to Cr(VI) in drinking water . Toxicol Sci. 143(1):16–25.
- Thompson CM, Suh M, Proctor DM, Haws LC, Harris MA. 2017. Ten factors for considering the mode of action of Cr(VI)-induced gastrointestinal tumors in rodents. Mutat Res Genet Toxicol Environ Mutagen. 823:45–57.
- Thompson CM, Wolf JC, Elbekai RH, Paranjpe MG, Seiter JM, Chappell MA, Tappero RV, Suh M, Proctor DM, Bichteler A, et al. 2015. Duodenal crypt health following exposure to Cr(VI): Micronucleus scoring, γ-H2AX immunostaining, and synchrotron X-ray fluorescence microscopy . Mutat Res Genet Toxicol Environ Mutagen. 789-790:61–66.
- Thompson CM, Young RR, Dinesdurage H, Suh M, Harris MA, Rohr AC, Proctor DM. 2017. Assessment of the mutagenic potential of hexavalent chromium in the duodenum of big blue® rats. Toxicol Appl Pharmacol. 330:48–52.
- Thompson CM, Young RR, Suh M, Dinesdurage HR, Elbekai RH, Harris MA, Rohr AC, Proctor DM. 2015. Assessment of the mutagenic potential of Cr(VI) in the oral mucosa of Big Blue® transgenic F344 rats. Environ Mol Mutagen. 56(7):621–628.
- Trouiller B, Reliene R, Westbrook A, Solaimani P, Schiestl RH. 2009. Titanium dioxide nanoparticles induce DNA damage and genetic instability in vivo in mice. Cancer Res. 69(22):8784–8789.
- U.S. EPA. 1991. National primary drinking water regulations-synthetic organic chemicals and inorganic chemicals; monitoring for unregulated contaminants; national primary drinking water regulations implementation; national secondary drinking water regulations. Final rule. Fed Regist. 56:3526–3597.
- U.S. EPA. 2005. Guidelines for carcinogen risk assessment, EPA/630/P-03/001F. Risk Assessment Forum: US Environmental Protection Agency, Washington, DC.
- U.S. EPA. 2007. Framework for Determining a Mutagenic Mode of Action for Carcinogenicity: Using EPA’s 2005 Cancer Guidelines and Supplemental Guidance for Assessing Susceptibility from Early-Life Exposure to Carcinogens. US Environmental Protection Agency, Washinton, DC. (EPA 120/R-07/002-A).
- U.S. EPA. 2010. Toxicological review of hexavalent chromium in support of summary information on the integrated risk information system (IRIS). External Review DRAFT. US Environmental Protection Agency, Washington, DC.
- U.S. EPA. 2017. Data Summary of The Third Unregulated Contaminant Monitoring Rule (UCMR3). EPA 815-S-17-001.(Data accessed on January 15, 2017).
- Ueno S, Kashimoto T, Susa N, Furukawa Y, Ishii M, Yokoi K, Yasuno M, Sasaki YF, Ueda J, Nishimura Y, et al. 2001. Detection of dichromate (VI)-induced DNA strand breaks and formation of paramagnetic chromium in multiple mouse organs. Toxicol Appl Pharmacol. 170(1):56–62.
- Wang XF, Xing ML, Shen Y, Zhu X, Xu LH. 2006. Oral administration of Cr(VI) induced oxidative stress, DNA damage and apoptotic cell death in mice. Toxicology. 228(1):16–23. eng.
- WHO. 2019. Chromium in Drinking Water: Draft Background Document for Development of WHO Guidelines for Drinking-water quality.
- Wise SS, Wise JP. Sr. 2012. Chromium and genomic stability [Research Support, N.I.H., Extramural Research Support, Non-U.S. Gov't]. Mutat Res. 733(1-2):78–82.
- Wronska-Nofer T, Wisniewska-Knypl J, Dziubaltowska E, Wyszynska K. 1999. Prooxidative and genotoxic effect of transition metals (cadmium, nickel,.chromium, and vanadium) in mice. Trace Elem Electrolytes. 16(2):87–92.
- Young RR, Thompson CM, Dinesdurage HR, Elbekai RH, Suh M, Rohr AC, Proctor DM. 2015. A robust method for assessing chemically induced mutagenic effects in the oral cavity of transgenic Big Blue® rats. Environ Mol Mutagen. 56(7):629–636.
- Zeller A, Pfuhler S, Albertini S, Bringezu F, Czich A, Dietz Y, Fautz R, Hewitt NJ, Kirst A, Kasper P. 2018. A critical appraisal of the sensitivity of in vivo genotoxicity assays in detecting human carcinogens. Mutagenesis. 33(2):179–193.
- Zhitkovich A. 2011. Chromium in drinking water: sources, metabolism, and cancer risks [Research Support, N.I.H., Extramural. Chem Res Toxicol. 24(10):1617–1629].