
Abstract
Acrylonitrile (ACN) is a known rodent and possible human carcinogen. There have also been concerns as to it causing adverse reproductive health effects. Numerous genotoxicity studies at the somatic level in a variety of test systems have demonstrated ACN’s mutagenicity; its potential to induce mutations in germ cells has also been evaluated. ACN is metabolized to reactive intermediates capable of forming adducts with macromolecules including DNA, a necessary first step in establishing a direct mutagenic mode of action (MOA) for its carcinogenicity. The mutagenicity of ACN has been well demonstrated, however, numerous studies have found no evidence for the capacity of ACN to induce direct DNA lesions that initiate the mutagenic process. Although ACN and its oxidative metabolite (2-cyanoethylene oxide or CNEO) have been shown to bind in vitro with isolated DNA and associated proteins, usually under non-physiological conditions, studies in mammalian cells or in vivo have provided little specification as to an ACN-DNA reaction. Only one early study in rats has shown an ACN/CNEO DNA adduct in liver, a non-target tissue for its carcinogenicity in the rat. By contrast, numerous studies have shown that ACN can act indirectly to induce at least one DNA adduct by forming reactive oxygen species (ROS) in vivo, but it has not been definitively shown that the resulting DNA damage is causative for the induction of mutations. Genotoxicity studies for ACN in somatic and germinal cells are summarized and critically reviewed. Significant data gaps have been identified for bringing together the massive data base that provides the basis of ACN’s current genotoxicity profile.
Introduction
Purpose for this review
Studies of the genotoxicity of acrylonitrile (ACN; CH2=CH–CΞN; CAS No. 107-13-1) have spanned over four decades, fueled by concerns of cancer and/or heritable effects that might result from mutations in critical genetic regions. Virtually all available test systems, including those in vitro in prokaryotic and/or eukaryotic microorganisms or mammalian cells in culture as well as several in vivo in organisms ranging from Drosophila to mammals, including humans, have been employed. Although there is ample evidence that DNA damage occurs, including frank mutations, these investigations have failed to elucidate specific causation. Numerous attempts have been made to characterize the mechanism(s) underlying ACN-induced specific DNA damage, a necessary first step in establishing its mutagenic mode of action. Although ACN itself may react with DNA, the reactivity of 2-cyanoethylene oxide (CNEO), its major oxidative metabolite, is much greater and the most likely toxic intermediate. ACN/CNEO binding to DNA both in vitro or in vivo has been confounded by possible reactions with DNA-associated proteins. Studies of DNA strand breaks, apurinic sites and even DNA repair have demonstrated that DNA damage has occurred, but not how it was initiated. Direct chemical studies of ACN/CNEO with nucleobases or isolated DNA in vitro have identified DNA adducts at several sites but only by employing massive concentrations and under non-physiological conditions. Of central importance, numerous attempts in cells in vitro in culture or in vivo in rodents to identify one or more specific ACN/CNEO DNA adducts that could underlie mutation induction have been largely unsuccessful. Only a single study in vivo in rats has shown a specific adduct, i.e. the non-promutagenic N7 oxyethyl guanine (N7OEG) in liver, a non-target tissue for ACN’s rodent carcinogenicity (Hogy and Guengerich Citation1986). No other ACN/CNEO-specific adducts have been shown in any tissue in any in vivo study. As an alternate possibility to having a direct mutagenic effect, the capacity for ACN to initiate specific pro-mutagenic DNA lesions by forming reactive oxygen species (ROS) has also been investigated. Numerous studies have shown the production of the signature DNA adduct of oxidative DNA damage, i.e. 8oxoguanine, following ACN/CNEO exposures. However, it has not been definitively shown that this oxidative damage is causative for the induction of mutations. The purpose of this review is to examine all currently available reports of ACN’s genotoxicity in a single paper to demonstrate that, at present, no single underlying mutagenic mechanism has been identified, something that must be considered in risk assessment and risk management decisions for ACN.
Sources of human exposure to ACN
ACN is an important high volume industrial chemical. Occupational exposure may occur during its production or use in the manufacture of fibers, resins, polymers, and other chemical intermediates (IARC Citation1999; EC Citation2004, USEPA Citation2011; NTP Citation2021). Major uses of ACN are in the production of acrylic fibers, which find their way into clothing, carpeting and a multitude of other consumer products, in the manufacture of polyacrylonitrile-based carbon fibers and in the production of polymers of ACN that include butadiene and styrene or styrene alone. Consumers of these products may also be exposed to ACN although levels leached from them are quite small (NTP Citation2021; Page and Charbonneau Citation1983, Citation1985). The greatest potential for high to moderate levels of ACN exposure is occupational. At the environmental level, tobacco is an important source of low-level but chronic exposure to a large segment of the population (IARC Citation2004 and references therein; Laugesen and Fowles Citation2005; De Jesús et al. Citation2020, Citation2021; NTP Citation2021). Small amounts of ACN are released during the combustion of plant matter such as biomass and timber. Several studies have quantified emissions of ACN from tropical fires and the burning of biomass (Yokelson et al. Citation2007; Warneke et al. Citation2011).
Methods
Literature searches
Published papers and other reports were identified for this review from a variety of sources including the US National Library of Medicine’s PubMed database and Google Scholar. Searches from these sources used the primary key word “acrylonitrile” and several secondary key words: “genotoxicity,” “mutation,” “cytogenetics,” “DNA damage,” DNA adducts,” “cancer/carcinogenicity,” “germ-cell genotoxicity,” “oxidative DNA damage,” “oxidative stress,” “lipid peroxidation,” “chromosome aberrations,” “aneuploidy” or combinations. Studies were also identified from authoritative reviews in the published literature that focused on carcinogenicity, heritable effects and/or genotoxicity of acrylonitrile, as well from those periodically published by the International Agency for Research on Cancer (IARC) and the United States Environmental Protection Agency (US EPA). Attempts were made to obtain all papers cited in these reviews for evaluation. Papers were also obtained from the literature archives of the Acrylonitrile Group of manufacturers which proved to be a source of older reports and unpublished research reports. The 1985 book, “Evaluation of Short-Term Tests for Carcinogens” (Volume 5, Progress in Mutation Research), also reported on older tests of ACN’s genotoxicity as well as studies in systems not reported elsewhere. Finally, studies of ACN’s genotoxicity conducted by or commissioned by industry were reviewed. Reports of genotoxicity or lack thereof were occasionally included in reports of test performance for one or another test system. When possible, all studies identified from these many sources were obtained for review. The intent was to include all papers relevant to an assessment of ACN’s genotoxicity. The earliest studies identified were from the 1960s.
Background on health effects and toxicokinetics of ACN
Acute toxicity
Acutely, ACN produces irritation of skin, eyes, and mucous membranes, as well as skin sensitization (ECHA REACH). Indirect observations from occupational and animal inhalation studies suggest ACN to be a non-respiratory sensitizing agent (EC RAC Citation2018). At high exposure levels ACN can also produce serious neurological symptoms such as dizziness, headache, confusion or may even lead to loss of consciousness and death (USEPA Citation2011).
Chronic toxicity and carcinogenicity
Several repeated dose toxicity studies of ACN have been reported in rats and mice by oral or inhalation routes of exposure. Dose-response assessments of these studies’ findings identified irritation and neurological effects as critical for establishing no effect levels (NOELs) (Kirman et al. Citation2008; EC RAC Citation2018). Variation in individual exposure and inability to establish accurate exposure levels made dose response difficult to assess in human studies. However, many of the findings seen in animal studies (notably irritant and neurological effects) reflect findings reported in ACN workers (EC RAC Citation2018).
ACN induces tumors at multiple sites in rodents in chronic bioassays following inhalation exposures (Maltoni et al. Citation1977, 1988; Quast, Wade, et al. Citation1980), drinking water exposures (Bigner et al. Citation1986; Gallagher Citation1988; Friedman and Beliles Citation2002; Quast Citation2002; Johannsen and Levinskas Citation2002a, Citation2002b) and gavage (Maltoni et al. Citation1977; Ghanayem et al. Citation2002; Johannsen and Levinskas Citation2002a). Although both rats and mice are sensitive to these effects, in rats there is a predilection for the induction of brain tumors originally reported as astrocytoma, glial tumors or brain tumors-difficult to classify. More recently ACN-induced rat brain tumors from the Quast (Citation2002) and Quast, Wade, et al. (Citation1980) studies were identified as malignant microglial/histiocytic tumors based on robust immunohistochemistry studies (Kolenda-Roberts et al. Citation2013; Moore RR and Hardisty Citation2014). The target cells, microglia, are parenchymal macrophages of the central nervous system. Comparatively oligodendrogliomas, and malignant microglial tumors (all previously diagnosed by H&E staining as astrocytomas), were the most common tumors among twenty-eight spontaneous rat brain tumors chosen from the National Toxicology Program Archives for immunohistochemical staining (Kolenda-Roberts et al. Citation2013). The Kolenda-Roberts et al.’s (Citation2013) report shows that spontaneous rat brain tumors developing at a low incidence in aging rats are not astrocytomas, but are primarily oligodendrogliomas, malignant microgliomas, or perhaps a mixture of both, which supports the susceptibility of tumor development in these two cell types in the rat. Responses of rat astrocytes and microglia to ACN dosing in vitro has been studied (Caito et al. Citation2013, Citation2014, Citation2017). No cytotoxicity was observed at 1 mM. Microglia accumulated less ACN than astrocytes while demonstrating higher levels of the lipid peroxidation by-product F2-isoprostane. Induction of Nrf2, a key transcription factor involved in the response to oxidative stress, was also observed in rat microglia but not in rat or mouse astrocytes or in mouse glial cells. Glutathione (GSH) levels were up-regulated in both rat cell types. These results suggest that rat microglia are more sensitive than rat astrocytes to the oxidative stress effects of ACN (Caito et al. Citation2013), while mouse microglia and astrocytes were found to be resistant to ACN-induced oxidative stress (Caito et al. Citation2017), a species pattern that mirrors tumor formation in these two species.
In addition to the central nervous system, tumors have been reported in rats for the oral cavity, Zymbal’s gland (accessory gland of the rodent ear), forestomach, small intestine and mammary gland. While mice have not shown brain tumors following gavage dosing with ACN (Ghanayem et al. Citation2002), tumors of the forestomach and Harderian gland (an accessory gland of the eye in species with a nictitating membrane) were increased, and equivocal numbers in tumors of the ovary and lung were reported. The evidence relating to key events in ACN rodent brain carcinogenicity and whether the mechanisms of ACN carcinogenicity in rodents are plausible in humans was previously reviewed by Meek et al. (Citation2003). This review concluded that the data available at that time were not sufficient to support a consensus view on a plausible mode of action for ACN-induced rat brain tumors. Prompted by the subsequent finding that the ACN-induced rat brain tumors are microglial/histiocytic in origin (Kolenda-Roberts et al. Citation2013) and the availability of additional mechanistic studies (Caito et al. Citation2013, Citation2014, Citation2017; Williams GM et al. Citation2017; Walker, Walker, et al. Citation2020; Walker, Fennell, et al. Citation2020), a reevaluation of the potential mechanism(s) of action (MOA)s for induction of ACN neoplasia, focused on the brain, forestomach, Zymbal’s gland, Harderian gland, and relevance to humans was recently published by Kobets et al. (Citation2022). Notably, three of these tumor sites are present only in rodents, and the induction of microgliomas in humans appears to be extremely rare (Mathews et al. Citation2016). Kobets et al. concluded that the MOA of ACN carcinogenicity in rodents is consistent with direct and indirect (due to oxidative damage) cytotoxicity, and compensatory cell proliferation, although weak, likely indirect, mutagenicity cannot be ruled out. Overall, Kobets et al. (Citation2022) concluded relevance to humans of findings with ACN in rodent studies is questionable and requires further dose-effect and mechanistic investigation.
Data supporting a nongenotoxic MOA for rodent tumors are limited. A single in vitro study evaluated the effects of ACN on gap junction intercellular communication. Gap junction intercellular communication has been identified as an important factor in regulating cell growth and has been implicated as a potential mechanism for other nongenotoxic carcinogens (Trosko Citation2001). ACN was shown to inhibit gap junction intercellular communication in rat astrocytes (Kamendulis, Jiang, Zhang, et al. Citation1999). This inhibition was reversible upon removal of ACN from the test media and was protected by co-treatment with vitamin E or with a glutathione precursor, suggesting the involvement of oxidative stress. A single in vitro study assessed a potential immunotoxic mode of action for ACN. ACN (20–500 µM) caused damage to lipid raft structures from human T lymphocyte cells, which in turn resulted in Bcl10 protein and lipid raft separation and restrained Ras-Raf-MAPK-extracellular signal-regulated kinase signaling pathways (Li XJ et al. Citation2014).
The animal cancer bioassays of ACN have their counterpart in several human epidemiological studies. Early studies gave inconsistent results on the relationship between ACN exposure and cancer mortality, and in 1999 the International Agency for Research on Cancer (IARC) concluded there was inadequate evidence in humans for the carcinogenicity of acrylonitrile (IARC Citation1999). More recent updates of three previous cohorts showed no clear association between ACN exposure and cancer deaths (Swaen et al. Citation2004; Symons et al. Citation2008; Marsh and Zimmerman Citation2015). The largest and most recent extended mortality study of cancer in ACN-exposed workers reported evidence of an association between ACN exposure and lung cancer death, as well as a possible link between ACN and death from bladder cancer and pneumonitis (Koutros et al. Citation2019). Further analyses of these data were conducted to address potential confounding of standardized mortality ratios by smoking and asbestos using the negative control outcome method of Richardson and sensitivity analyses using Monte Carlo methods (Marsh and Kruchten Citation2023). The authors concluded that their reanalysis provided little evidence to support the National Cancer Institute’s suggestion of associations between ACN exposure and mortality from lung and bladder cancer and pneumonitis.
ACN is classified by IARC as a category 2B carcinogen with sufficient evidence of carcinogenicity in animals but with inadequate evidence in humans (IARC Citation1999) and as “reasonably anticipated to be a human carcinogen” by the NTP (2021). For the purposes of EU harmonized classification and labeling, ACN is considered a 1B carcinogen (EC RAC Citation2018). ACN’s genotoxicity profile, with specific consideration of mechanisms underlying mutagenicity, is a factor in these classifications.
Reproductive and developmental toxicity
Reproductive and developmental effects of ACN have been evaluated in multiple rodent studies (as reviewed in EC RAC Citation2018). Developmental toxicity has been assessed thoroughly in one species (rat). Principal studies were conducted at high dose levels which induced dose-dependent maternal toxicity. No unique fetal susceptibility was identified in any of these studies with effects seen only at high and overtly maternally toxic doses (EC RAC Citation2018).
Malformations, principally an increased incidence of tailless or short-tailed fetuses, were reported in some studies of ACN. However, the most contemporary of the developmental toxicity studies (Saillenfait et al. Citation1993), by the most relevant route of exposure (inhalation) and higher doses, did not show any evidence of exposure-related malformations, even though maternal and fetotoxicity were both evident. In longer-term reproductive toxicity studies of ACN, the overall incidence of tailless pups was too low and sporadic to make a definitive assessment of potential relationship to treatment with ACN (EC RAC Citation2018). Weight-of-evidence evaluation of developmental toxicity and malformations in the ACN animal studies leads to the conclusion that very high, maternally toxic, exposures to ACN result in fetotoxicity, and may result in teratogenicity (Neal et al. Citation2009; EC RAC Citation2018).
If ACN has the potential to be teratogenic at maternally toxic doses, the reported effects do not implicate germ cells. Studies of reproductive outcomes in animals following paternal or maternal administration prior to conception evaluate endpoints of relevance to germ cell genotoxicity. Three such ACN studies have been reported and reviewed in Neal et al. (Citation2009), i.e. a one-generation study of ACN administration in drinking water (TRL Citation1975), a three-generation ACN in drinking water study (Friedman and Beliles Citation2002), and a two-generation ACN by inhalation study (Nemec et al. Citation2008). All studies were conducted in rats. None showed adverse effects such as stillbirths, pre-term deliveries, post-term effects, or maternal mortality. There were no obvious compound-related effects on reproductive success in any of the reproductive toxicity studies, even at exposure levels producing toxicity to the parent animals (EC RAC Citation2018).
Dominant lethal test (DLT) studies of ACN have been reported in mice (Leonard et al. Citation1981; Zhurkov et al. Citation1983) and rats (Working et al. Citation1987). Negative results were reported in each study, demonstrating a lack of male-mediated reproductive toxicity. Details of these studies are described with the genotoxicity data at the germinal level.
Repeated dose toxicity studies may also provide signals pertinent to germ cell genotoxicity and are an important source of information relating to potential germ cell hazards. These studies can indicate both delivery of the agent to male and female germ cells and gonadal tissues, as well cytotoxic effects that may occur following exposure to genotoxicants.
Findings suggestive of effects on sperm quality have been reported in some short-term repeated dose studies of ACN in rats (Abdel-Naim et al. Citation1994 – abstract only; Wang Z et al. Citation1995) and mice (Tandon et al. Citation1988). These findings were not replicated in later longer-term studies (Serota et al. Citation1996; NTP Citation2001; Nemec et al. Citation2008), and no histopathological evidence of testicular toxicity was noted in the various chronic studies of ACN (Neal et al. Citation2009; EC RAC Citation2018).
The only chronic study of ACN in mice (NTP Citation2001) showed an increased incidence of ovarian atrophy in reproductively senescent mice; the biological significance of this finding is unclear. More recently, ovarian follicles in ACN-exposed mice (5–20 mg/kg-day for 28 days) exhibited inflammation, apoptosis, and impaired oocyte development (Luo YS et al. Citation2022). Elevated levels of reactive oxygen species (ROS), early apoptosis, DNA damage, and organelle (mitochondria, endoplasmic reticulum, lysosome) structural and/or functional changes were also reported (Luo YS et al. Citation2022). Transcriptomic data from this study revealed that ACN altered the expression of genes related to apoptosis, oxidative stress, endoplasmic reticulum stress, and autophagy.
Collectively, the available animal reproductive and repeated dose toxicity studies do not support a concern for germ cell toxicity of ACN.
Human reports of potential ACN-mediated reproductive or developmental effects include four epidemiological studies of exposed male and female Chinese workers conducted in the 1990s that observed a variety of adverse reproductive and perinatal outcomes, including spontaneous abortions, still births, birth defects and infertility compared to controls (Wu WK et al. Citation1994; Wu W et al. Citation1995; Dong et al. Citation1996 and reviewed in Wu X and Jin Citation2000; Li Z Citation1996). The reported results among the studies were somewhat consistent. Subsequently two critical reviews of these studies concluded that, although the findings in the Chinese workers were suggestive of an ACN reproductive effect (i.e. hypothesis generating), there were sufficient deficiencies in each that precluded definitively establishing causation (Collins et al. Citation2003; Neal et al. Citation2009). Major among the deficiencies was incomplete exposure assessment that included lack of data on individual workers, timing of exposures relative to reproductive outcomes, and potential industrial co-exposures and other potential lifestyle confounders. The Collins et al. Citation2003 review suggested follow-up studies that never occurred.
Concern for genotoxicity
A concern from animal studies is that, at lower levels and/or chronic exposures, ACN may have additional health effects, the most serious of which are cancer and heritable disorders. These last two outcomes result in whole or in part from toxicity to the genetic material, i.e. genotoxicity to either somatic or germinal cells. Genotoxicity is of particular concern for exposed humans.
Toxicity to the genetic material resulting in mutations in critical genetic regions in somatic cells may initiate events that ultimately result in cancer although this is not an inevitable consequence of mutagenesis and may have other contributory or even primary causes. Mutations at the somatic level therefore are surrogates of deleterious health outcomes, i.e. cancer, and not health outcomes in themselves. By contrast, mutations in germ cells have the potential to be passed to offspring affecting every cell in the body. When occurring in genes necessary for normal function, such mutations in themselves are the cause of a genetic disease. Furthermore, if viable, inherited mutations enter the gene pool and may be passed to subsequent generations. High frequencies of these mutations have an effect for the species. Germ cell mutations per se are adverse health effects.
Mutation research was initially focused on germ cells because of their potential effects on the human species. This focus, however, has gradually shifted to somatic mutations with the realization that genetic events in somatic cells can underlie cancer (Marchetti et al. Citation2020). Technological developments in the early 1970s introduced rapid and simple assays for induced mutations, i.e. specially constructed bacterial tester organisms such as the Ames Assays (Ames Citation1973). As studies in somatic cells developed, it was also concluded that these events were much more frequent than germinal mutations, that mutagenic mechanisms in somatic cells and germinal cells were similar and that protection from environmental agents producing somatic genotoxicity conservatively also protected against germinal genotoxicity (Marchetti et al. Citation2020).
Distribution and metabolism
Exposure to ACN can occur via inhalation, ingestion or, less commonly, the dermal or ocular route. It is rapidly and almost completely absorbed, and widely distributed to all tissues (USEPA Citation2011). As ACN and some of its metabolites are reactive molecules capable of interacting with cellular macromolecules, metabolism is an important determinant of its genotoxicity.
ACN is metabolized by two primary pathways (): (1) conjugation with glutathione (GSH), which can occur either through catalysis with a cytosolic enzyme, glutathione-S-transferase (GST), or nonenzymatically; and (2) oxidation by microsomal enzyme, cytochromes P450 (primarily CYP2E1), forming 2-cyanoethylene oxide (CNEO) (Dahl and Waruszewski Citation1989; Fennell et al. Citation1991; Kedderis, Batra, Koop Citation1993; Burka et al. Citation1994; Gargas et al. Citation1995; Sumner et al. Citation1999). The oxidative pathway can result in the release of cyanide, which has been reported to require CYP2E1 activity (Kedderis, Batra, Koop Citation1993; Wang H et al. Citation2002). However, other enzyme systems may also play a role in ACN oxidation. For example, cytochrome C peroxidase isolated from S. cerevisiae was found to catalyze the oxidation of ACN, as indicated by cyanide release, at a rate that is similar to rat liver microsomal P450 (Chinchilla et al. Citation2014). Lactoperoxidase has also shown activity for oxidation of ACN in vitro (Nasralla et al. Citation2009). Partially purified human lung lipoxygenase has demonstrated an appreciable activity oxidizing ACN to release cyanide in vitro (Roy and Kulkarni Citation1999). Prostaglandin H synthase was reported to oxidize ACN utilizing hydrogen peroxide resulting in the release of cyanide, an activity that was significantly reduced by known prostaglandin H synthase inhibitors (Al-Abbasi et al. Citation2018). These results are supported by studies conducted using structurally similar nitriles, which suggest that other enzyme systems/pathways are involved in their oxidation, including (1) myeloperoxidase oxidation, an activity that may be of particular importance in microglia (Lefkowitz and Lefkowitz Citation2008), of chloroacetonitrile (Abdel-Naim and Mohamadin Citation2004); (2) xanthine oxidase oxidation of dibromoacetonitrile (Mohamadin and Abdel-Naim Citation2003); and (3) non-enzymatic oxidation of dichloroacetonitrile in the presence of reactive oxygen species (peroxides) in vitro (Mohamadin Citation2001).
Figure 1. Metabolism of acrylonitrile. *Reaction can also occur nonenzymatically; P450: Cytochrome P450; GST: Glutathione-S-Transferase; GSH: Reduced glutathione; EH: Epoxide Hydrolase; RH: Rhodanese; Px: Peroxidase; XO: Xanthine oxidase; CHEMA: N-acetyl-S-(1-cyano-2-hydroxyethyl)-L-cysteine; CEMA: N-acetyl-S-(2-cyanoethyl)-L-cysteine; ATCA: 2-aminothiazoline-4-carboxylic acid; CN-: cyanide ion; SCN-: thiocyanate OSCN-: hypothiocyanite.
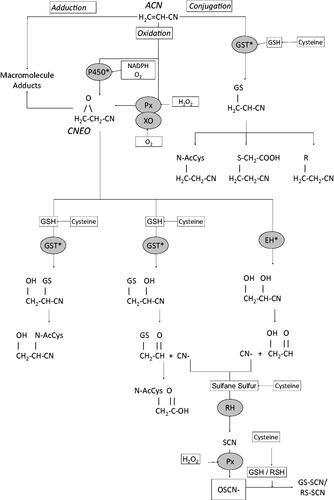
Figure 2. Ratio of ACN urinary metabolites CHEMA:CEMA in humans using NHANES (Citation2015–2016,) data. “X”: data point for individuals with both metabolites detectable in urine. Solid line: linear regression.
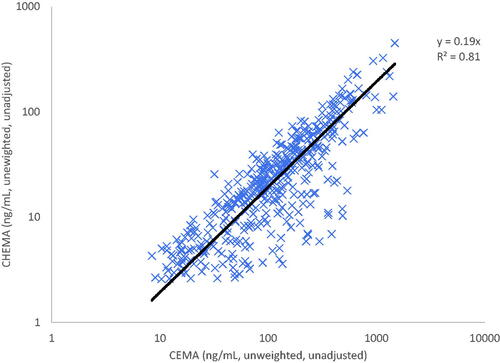
The metabolites of ACN from the oxidative and conjugation pathways are subject to further metabolism. The ACN-GSH conjugate is converted to a mercapturic acid, which is subsequently excreted in urine. CNEO in turn is metabolized by two pathways: (1) conjugation with GSH, either through catalysis by GST or nonenzymatically, forming conjugates on the second or third carbon; and (2) hydrolysis by microsomal enzyme, epoxide hydrolase. The secondary metabolites of CNEO can undergo further metabolism/decomposition. Of toxicological importance, cyanide can be released from the CNEO metabolite generated by the epoxide hydrolase pathway and from the GSH conjugate formed on the third carbon. Cyanide is relatively short-lived in the body and is rapidly metabolized (Ansell and Lewis Citation1970; Hartung Citation1982). Cyanide is primarily detoxified by the mitochondrial enzyme, rhodanese, which uses sulfane sulfur (i.e. thiosulfate) as a cofactor, to form thiocyanate. Thiocyanate was detected in the blood and urine of volunteers following short-term inhalation exposures to ACN (Wilson RH and McCormick Citation1949), in the urine of workers exposed to ACN (Sakurai et al. Citation1978), and has been measured in the blood and brain of rats exposed to ACN by oral gavage (Benz et al. Citation1997; Rao et al. Citation2013). A minor metabolic pathway for cyanide involves its reaction with cystine to form 2-aminothiazoline-4-carboxylic acid (ATCA) (Petrikovics et al. Citation2011), which is excreted in the urine.
In acute exposure scenarios, the formation of thiocyanate from cyanide released from ACN has historically been viewed a detoxification step. However, this may not be the case for some tissues or for long-term exposures to ACN. As a pseuodohalide, the pharmacokinetics of thiocyanate are driven by its active transport and metabolic processes reserved for halides (Br-, Cl-, I-) rather than by tissue partitioning. For this reason, plasma levels of thiocyanate persist considerably longer than either ACN, CNEO, or cyanide (half-life ∼1–6 days in humans; Himwich and Saunders Citation1948; Schulz et al. Citation1979; Junge Citation1985; Lundquist et al. Citation1995). Long-term exposures to thiocyanate are known to produce goiter, due to competition with iodine for uptake by the sodium-iodine symporter into the thyroid (Wolff Citation1998; Tonacchera et al. Citation2004; De Groef et al. Citation2006). Additionally, thiocyanate, as an endogenous antimicrobial agent, is actively transported to external surfaces of the body where its activity is needed, including the oral cavity, gastrointestinal tract, and respiratory tract surface, where thiocyanate levels are generally higher than corresponding plasma levels (Chandler and Day Citation2015). To illustrate this active transport, following an i.v. dose of radiolabeled potassium cyanide administered to rats, approximately 19% of the radiolabel was transported to the GI lumen within 6 h (Crawley and Goddard 1977), presumably in the form of thiocyanate. These data indicate that tissue doses of thiocyanate may vary significantly from one tissue to another depending upon the presence and activity of halide symporters, and may not be readily predicted by blood concentrations. Five minutes after rats received a radiolabeled dose of ACN via i.v. injection, the tissues/media with the highest concentration of radiolabel were the lung, liver, small intestines contents, and spleen (Jacob and Ahmed Citation2003), a distribution pattern that cannot be explained by simple partitioning. Following transport, thiocyanate serves as a substrate for peroxidases (e.g. myeloperoxidase which is active in microglia, lactoperoxidase), which yield hypothiocyanite, an important endogenous antimicrobial agent analogous to hypohalous acids (HOCl, HOBr). However, unlike the hypohalous acids, which react indiscriminately with cellular macromolecules, the antimicrobial activity of hypothiocyanite is attributable to its ability to react almost exclusively with sulfhydryls, a reaction that is largely reversible. Also, unlike hypohalous acids, thiocyanate is capable of diffusing across bi-lipid membranes where it can react with intracellular sulfhydryl groups. As a sulfhydryl reactive agent, hypothiocyanite can deplete levels of reduced GSH (Arlandson et al. Citation2001), inhibit enzyme activities (Arlandson et al. Citation2001; Barrett et al. Citation2012), and oxidize tubulin cysteines, inhibiting microtubule polymerization (Clark et al. Citation2014). While initially considered to be a mild oxidant, there is an increasing body of evidence that the toxicological consequences of hypothiocyanite formation can be significant (Barrett and Hawkins Citation2012; Pattison et al. Citation2012). The role of hypothiocyanite formation by microglial myeloperoxidase has not been evaluated.
The metabolism of ACN is subject to a number of factors that should be considered when interpreting genotoxicity studies, as summarized below:
Species differences – Species differences in the metabolic pathways of ACN have been reported. Clear species differences have been reported for the oxidation of ACN by cytochromes P450. In vitro studies using liver microsomes indicate that mice and rats appear to form CNEO at a greater rate (∼4x and 1.5x, respectively) compared to humans (Roberts et al. Citation1991; Kedderis, Batra, Koop Citation1993). Hydrolysis of CNEO by epoxide hydrolase is significant in humans. It is virtually nondetectable in naive mice and rats (Kedderis et al. Citation1995), but can be induced in both species (Kedderis and Batra Citation1993), as well as in humans (Kroetz et al. Citation1993). With respect to clearance of ACN, GSH conjugates of ACN correspond to approximately 36–43% of urinary metabolites in rats, and 20–28% of urinary metabolites in mice (Fennell et al. 1991; Kedderis, Sumner, et al. Citation1993; Sumner et al. Citation1997). Despite having a higher rate of CNEO formation than rats, mice exhibited circulating levels of CNEO that were notably lower than the levels detected in rats (Roberts et al. Citation1991), suggesting that differences exist between rats and mice with respect to CNEO clearance (e.g. GSH conjugation). Conjugation of CNEO with GSH occurs faster in humans (∼1.5-fold) than in either mice or rats (Kedderis et al. Citation1995). With respect to thiocyanate metabolism, peroxidase activity has been detected in mouse Harderian glands (Strum and Shear Citation1982), which is a target tissue for ACN carcinogenicity, but was not detected in rat Harderian glands (De et al. Citation1987; De Citation1992), which is not a target tissue for ACN carcinogenicity.
Species differences in metabolism can also be assessed by examining the excretion of urinary metabolites and their ratios. At high doses (10 mg/kg), the relative contribution of metabolites from the oxidative pathway [N-acetyl-S-(1-cyano-2-hydroxyethyl)-L-cysteine = CHEMA] is less than that from the direct conjugation pathway [N-acetyl-S-(2-cyanoethyl)-L-cysteine = CEMA], resulting in ratios (CHEMA:CEMA) of 0.3–0.4 in rats and 0.4–0.9 in mice (Fennell et al. 1991; Sumner et al. Citation1997, Citation1999). Kedderis, Sumner, et al. (Citation1993) reported data for the excretion of urinary metabolites in rats and mice exposed to ACN, showing that the ratio of CHEMA:CEMA is highly dose-dependent. At low doses (<0.5 mg/kg), the ratio of CHEMA:CEMA excreted in urine was greater than 3.5 in rats, and greater than 1.5 in mice, suggesting that the oxidative pathway predominates at low doses of ACN. In comparison, Schettgen et al. (Citation2012) reported urinary excretion of the metabolites in humans exposed to ACN in ambient air and/or by smoking, from which CHEMA:CEMA ratios of 0.26 and 0.16 could be calculated for nonsmokers and smokers, respectively. The NHANES biomonitoring data of the US population has included ACN metabolite, CEMA, for multiple sampling periods, and in the more recent data sets (e.g. 2015–2016) also extended to include ACN metabolite, CHEMA (De Jesús et al. Citation2020, 2021). Both biomarkers are notably higher in smokers compared to nonsmokers, and the latter oxidative biomarker is detectable in a small percentage of the sample population (∼15–36%). Based on the sample-weighted geometric mean values, the ratio of CHEMA:CEMA is calculated to be approximately 0.16. A plot of the raw data from NHANES (Citation2015–2016) for the subset of samples (416/2825 or ∼15%) in which both metabolites were detectable yields a slope (CHEMA:CEMA) of 0.19 (). The nondetect samples from this data set were considered to be non-informative for calculating the CHEMA:CEMA ratio, and their inclusion would artificially reduce the slope to a value less than 0.19. The CHEMA:CEMA values based on NHANES are in general agreement with the results of Schettgen et al. (Citation2012). The dose of ACN received by smokers was not specified by the study authors. However, it can be estimated to be less than 0.0075 mg/kg-day, more than an order of magnitude lower than the lowest dose assessed by Kedderis, Sumner, et al. (Citation1993), based upon a maximum cigarette smoking rate of 35/day as reported by the study authors, a maximum ACN content of 15 µg/cigarette (Hoffmann and Hoffmann Citation1997), a body weight of 70 kg, and an assumption of 100% uptake of ACN from cigarettes. Together these data suggest that the oxidative pathway plays a much larger relative role in ACN metabolism in rodents than it does in humans (i.e., CHEMA:CEMA ratios differ by more than an order of magnitude), and that the GSH conjugation pathway plays an important role in ACN metabolism in humans.
Nonlinear Toxicokinetics Due to Sulfhydryl Depletion – An important source of nonlinear toxicokinetics for ACN includes the depletion of cellular sulfhydryls such as GSH, which likely contributes to oxidative stress (Puppel et al. Citation2015). ACN and CNEO both react with GSH, and together are capable of depleting cellular GSH levels. ACN has been shown to be a more effective depletor of tissue GSH levels than several acrylates (Vodicka et al. Citation1990). When administered at oral doses corresponding to the LD50, ACN was more effective than several other nitrile compounds in depleting GSH in rat liver, kidney and brain 1 hr post-exposure (Ahmed et al. Citation1982). GSH depletion has been observed in a number of tissues (brain, lung, liver, kidney, stomach, adrenal gland, erythrocytes) in rats exposed to ACN (Silver and Szabo Citation1982; Cote et al. Citation1984; Gut et al. Citation1985; Vodicka et al. Citation1990; Benz et al. Citation1997). Benz et al. (Citation1997) reported significant GSH depletion in rat tissues at acute doses of approximately 20–50 mg/kg-day. In humans, polymorphisms in GSTT1 may serve to increase variation in susceptibility to GSH depletion (Thier et al. Citation1999, Citation2001). For tissues and cells that have significant peroxidase activity, the formation of hypothiocyanite from thiocyanate creates an additional stressor on GSH levels. In human erythrocytes, GSH was significantly depleted at low concentrations (10 µM) and was completely depleted at 100 µM hypothiocyanite in vitro (Arlandson et al. Citation2001), which are physiologically relevant concentrations in some tissue and fluids. For example, mean thiocyanate and hypothiocyanite concentrations in the saliva young of adults (with no exposure to ACN) were reported to be 1.5 mM and 31 µM, respectively (Jalil, Citation1994). Inspecting the metabolic pathways for ACN (), it is clear that there are multiple steps which are dependent upon maintenance of cysteine levels to support GSH (conjugation reactions with ACN, CNEO, and hypothiocyanite), sulfane sulfur (metabolism of cyanide), and cystine (metabolism of cyanide). For this reason, it is important to consider the magnitude of the ACN exposures used in genotoxicity studies, and the potential role of sulfhydryl depletion as a causative role in producing oxidative stress and subsequent genotoxicity.
Nonlinear Toxicokinetics Due to Enzyme Induction or Inhibition – Induction of cytochrome P4502E1 (CYP2E1) by ACN does not appear to be an important factor at toxicologically relevant doses. However, enzyme activity for other oxidative pathways is induced by ACN exposure, including stomach myeloperoxidase activity (Hamdy et al. Citation2012) and xanthine oxidase activity (Al-Abbasi Citation2012). These data suggest that for some tissues oxidative metabolism of ACN may be increased at high doses (single oral doses of 25–30 mg/kg). With respect to enzyme inhibition, in human erythrocytes exposed to hypothiocyanite, GST was found to be completely inhibited by 100 µM (Arlandson et al. Citation2001), which, as stated above, is a physiologically relevant concentration for some tissues and fluids. For tissues and cells that have significant peroxidase activity, the formation of hypothiocyanite from thiocyanate could inhibit the conjugation pathways important for ACN and CNEO clearance. Hypothiocyanate has also been shown to reversibly inactivate several enzymes with active site thiol residues (Barrett et al. Citation2012), and so this effect of hypothiocyanite likely extends to multiple enzyme systems.
Local Tissue Metabolism – Studies on the metabolism of ACN have focused upon the liver as the primary site for ACN metabolism, particularly with respect to CYP2E1 and GST activity. The role of local tissue metabolism of ACN, particularly for other enzyme systems (e.g. peroxidases) has not been evaluated. Rodent target tissues for tumor formation (positive species indicated in parentheses) for lifetime exposures to ACN include the following: Brain/microglial (rat); Zymbal’s gland(rat); Forestomach (rat, mouse); Mammary gland (rat); Tongue (rat); Intestines (rat); Nasal turbinate (rat); and Harderian gland (mouse) (Maltoni et al. Citation1977, 1988; Quast, Wade, et al. Citation1980; Quast, Schuetz, et al. Citation1980; NTP Citation2001; Ghanayem et al. Citation2002; Johannsen and Levinskas Citation2002a, Citation2002b). When the list of target tissues is considered within the context of tissues where myeloperoxidase and lactoperoxidase activities are required to support antimicrobial action, there is considerable overlap. At these tissue sites, the formation of hypothiocyanite likely serves as an additional oxidizing stressor to local GSH/sulfhydryl levels (in addition to system-wide stressors contributed by ACN and CNEO metabolism), which in turn may contribute to localized oxidative stress. Recent reports that single doses of ACN inhibit endogenous hydrogen sulfide biosynthesis in rats (Yang B et al. Citation2021) are consistent with the concept of sulfhydryl stress produced by ACN exposure.
Genotoxicity
Genotoxicity is any adverse insult that damages the genetic material. Among these are specific kinds of DNA damage that have the potential to initiate a process resulting in mutations, i.e. heritable structural and/or numerical alterations that irreversibly and permanently alter information content. DNA damage with mutagenic potential includes covalent bonding of a chemical with nucleobases or phosphates producing specific DNA adducts, DNA-protein or DNA-DNA cross-links or actual structural damage in the form of mis-repaired DNA double strand breaks. Numerical changes in chromosome number may also result from DNA damage although these may also result from binding of associated proteins. DNA damage may, in some cases be ignored by a cell or, in others, influence transcription or replication or even be lethal. However, the result of DNA damage that has potential adverse health consequences is the induction of mutations at either the gene or chromosomal level.
For chemical mutagenesis, it is not the chemicals per se that produce mutations; they only produce the DNA alteration leading to mutations. Cells produce mutations, typically by DNA replication on damaged templates or by error prone attempts at DNA repair. The progression of primary DNA damage to mutations is by no means inevitable and may, in fact, be quite rare or not occur at all, depending on the inducing agent, the kinds of interactions between agent and DNA, the efficiency of cellular DNA repair processes and other factors, which may be tissue- or cell-specific.
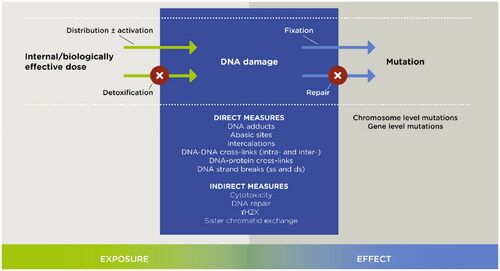
Although it is specific DNA damaging events, i.e. adducts to nucleobases, cross-links and/or double strand breaks, that lead to replication errors and/or mis-repair with fixation of genetic misinformation (; Albertini and Kaden Citation2020), genotoxicity studies of chemicals often measure generic changes to or involving the DNA that, while suggesting a potential for mutation inducing events, fail to identify them. Such changes include uncharacterized co-valent binding of a chemical with DNA, production of single strand breaks/apurinic sites, evidence that past damage has occurred such as its repair as reflected by unscheduled DNA synthesis (UDS), or the formation of sister chromatid exchanges (SCE). Most studies of ACN’s effects on the DNA have focused on generic changes induced in vitro or in vivo. It cannot be determined what portion, if any, of the changes observed reflect the kinds of damage that initiate the mutagenic process. Although not per se informative as to causation, these generic studies indicate that exposure to ACN at least in some way affects the DNA. Relatively fewer studies have focused on specific changes aimed at identification of mutation causing events. The overriding characteristic of DNA damage prior to mutation, excluding cell death, is that it is repairable. Mutations, as fixed changes, are not.
Figure 3. Chemically induced genotoxicity: a continuum that may produce mutation (Albertini and Kaden Citation2020).
Studies of generic changes in DNA are reviewed first, followed by studies of specific changes potentially responsible for initiating the mutagenic process.
Generic changes in DNA ()
Chemical reactivity
Radiolabeled ACN, generally at concentrations in the mM range, showed co-valent binding to isolated DNA, albeit quite slowly, a process considerably accelerated by the addition of rat liver (but not brain) microsomes or a reconstituted CYP450 enzyme system, while radiolabeled CNEO bound rapidly without metabolic activation (Guengerich et al. Citation1981). ACN also bound to proteins without metabolic activation. Of note, incubations with human liver microsomes resulted in no protein and little DNA binding. Peter, Appel, et al. (Citation1983) also showed that ACN at concentrations in the mM range bound slowly to isolated DNA with or without rat microsomes, although their addition significantly accelerated the process. All studies were confounded by the potential for protein contamination. Most recently, Walker, Fennell, et al. (Citation2020) reported that concentrations of radiolabeled CNEO in the µM range bound in a dose-related fashion to DNA isolated from human TK6 cells.
Table 1. ACN/CNEO induced generic DNA damage.
In vivo, radio-labeled CNEO (0.6 mg/kg) administered by i.p. injection to a single F344 rat produced covalent binding to protein in liver and brain, but not to DNA or RNA, one hour later (Hogy and Guengerich Citation1986). Binding to protein was nearly equivalent in the two tissues. Earlier, Peter, Appel, et al. (Citation1983) had injected male Wistar rats i.p. with radiolabeled ACN (1.2 ml of 0.2 mM solution), with sacrifices 14 or 72 h later. Although some radioactivity in liver DNA was associated with nucleotide bases, the peaks observed were too small for identification.
DNA and protein binding of radiolabeled ACN administered as a single oral dose of 46.5 mg/kg (= 0.5 LD50 dose) was also reported in brain, stomach, liver, pulmonary and testicular tissue in Sprague-Dawley rats (Farooqui and Ahmed Citation1983; Ahmed, Abdel-Aziz, et al. 1992; Ahmed, Abdel-Rahman, et al. Citation1992; Abdel-Rahman et al. Citation1994). Again, however, the methods used in these studies for DNA binding were insufficient to differentiate between binding to DNA or associated proteins (Kedderis, Batra, Koop Citation1993).
In the most recent investigation of DNA binding, female F344 and SD rats were administered ACN in drinking water (100 ppm; corresponding to approximately 5 mg/kg bw per day, a dose that is carcinogenic to rats) for 27 days, followed one day later by a single oral gavage dose of labeled ACN (11 mg/kg bw 14C-ACN) with sacrifice 3 h later (Williams GM et al. Citation2017). 14C-benzo[a]pyrene (BP; 5 mg/kg bw single) (BP) was administered by gavage on day 28 to naïve animals as a positive control, with sacrifice 24 h later. There was no association of radiolabeled ACN with brain DNA when determined by liquid scintillation counting. However, when analyzed by accelerator mass spectrometry (AMS), there was significant binding of both ACN and BP in brain tissue although no specific ACN/CNEO-DNA adducts were identified.
DNA damage
Single strand breaks/apurinic sites
DNA strand breaks were reported as early as 1979 in DNA isolated from Syrian Hamster fibroblasts following in vitro exposure to ACN at a lowest effective dose of 200 µg/ml (Parent and Casto Citation1979). Peter, Schwarz, et al. (Citation1983) reported no strand breaks in SV40 DNA exposed to ACN, but did report their induction after treatment with CNEO. Analytic methods are unknown. An in vitro fluorescence based screening assay for DNA damage based on melting and reannealing behavior failed to detect strand breaks in calf thymus DNA incubated for 30 min with 100 mM ACN (Kailasam and Rogers Citation2007).
DNA strand breakage was evaluated in several cell types in a 1985 Collaborative Study on Short Term Tests (CSSTT) sponsored by the International Program on Chemical Safety (IPCS) (Ashby 1985). Bradley (Citation1985) reported induction of single strand breaks in rat hepatocytes at an ACN exposure concentration of 66 µg/ml, which was the lowest concentration tested. Douglas et al. (Citation1985) tested ACN concentrations of 530–5300 µg/ml in CHO cells and found the lowest effective concentration (LEC) for induced DNA strand breaks was 3710 µg/ml, with or without metabolic activation (S9). By contrast, no single strand breaks were reported for CHO cells exposed to ACN, up to a concentration of 5300 µg/ml (Lakhanisky and Hendrickx Citation1985). All three studies utilized either alkaline elution or alkaline sucrose gradient centrifugation methods to identify single strand breaks.
In later studies, Chang et al.(1990) showed an increase in single-strand breaks assessed by alkali elution in human bronchial epithelial cells exposed to ACN at 200 and 500 µg/ml, with toxicity being seen at 600 µg/ml. Yates et al. (Citation1994) reported induction of strand breaks in supercoiled BR322 plasmid DNA by exposure to 50 mM CNEO, again utilizing alkaline sucrose gradient centrifugation. Robbiano et al. (Citation1994) observed single-strand breaks in both human and rat hepatocytes after exposures of 1.0 to 5.6 mM ACN.
The standard alkaline comet assay was employed to measure DNA strand-breaks/alkali-labile sites in rat astrocytes exposed to sub-lethal concentrations of ACN (<1.0 mM) for 24 h (Pu et al. Citation2006; Klaunig and Forney Citation2010). These exposures failed to induce breaks. At the time, astrocytes were selected as the test system in this study (and other studies) since they considered the target cell for carcinogenicity in rats. As noted above, ACN-induced rat brain tumors are now considered to be of microglial origin (Kolenda-Roberts et al. Citation2013).
Numerous studies have also investigated DNA strand breakage in vivo in rodents administered ACN/CNEO. Two of the earliest studies measured single strand breaks by alkaline elution in rat brain and/or liver three hours after i.p. injection of ACN (Hachiya et al. Citation1984, Citation1986). In the first study (Hachiya et al. Citation1984), breaks were reported in liver but not brain. In the second study (Hachiya et al. Citation1986), only liver was studied and an increase in alkali-labile sites but not breaks was observed.
Positive alkaline comet assay results were reported in multiple tissues of rats (in stomach, colon, urinary bladder and lung but not in brain) and mice (in same tissues plus in brain) administered ACN at i.p. doses equivalent to 0.5 X LD50 (Sekihashi et al. Citation2002). Noteworthy are the high doses by a non-physiological route of administration and the positive result in mouse brain, which is not a target species for brain tumor induction, but the negative result in rat brain, which is a target. These results echo those of the simple alkaline elution studies in rats described above (Hachiya et al. Citation1984, Citation1986) where effects were observed in non-target (liver) but not in target tissue (brain).
Most recently, an international validation study of the in vivo standard alkaline comet assay exposed male Sprague-Dawley rats to ACN at 0, 15.7, 31.3, and 62.5 mg/kg/day by oral gavage 48, 24, and 3 h before sacrifice and reported a weak but dose-related positive response significant at the highest dose in liver (Nakagawa et al. Citation2015). Of note in this study – and similar to the findings of tissue specificity noted above (Hachiya et al. Citation1984, Citation1986; Sekihashi et al. Citation2002) – the positive results were observed in liver (non-target tissue) but not in stomach (target tissue). Brain was not studied.
A complex study to define the importance of ACN’s epoxidation for inducing DNA damage as assessed by alkaline comet assays measured effects in several tissues from ACN-exposed B6C3F1 (wild-type = WT) and CYP2E1 knock-out (null) mice exposed to 2.5 (WT only), 10 (WT only), 20 or 60 (null only) mg ACN/kg/day by gavage 5 days/week for 6 weeks with necropsy 24 h post-dosing (Walker, Walker, et al. Citation2020). Assays of target tissues (forestomach and Harderian gland), non-target tissues (glandular stomach and liver) and potential target tissues (lung and ovary) in WT mice under standard electrophoresis conditions were negative for Harderian gland, lung, or glandular stomach cells but positive at the highest dose in cells from forestomach. All assays in somatic cells were negative in the knock-out mice. When the electrophoresis time was extended from 20 to 40 min, significant dose-related increases in DNA damage were detected in forestomach cells of WT mice treated with daily doses of 2.5, 10, or 20 mg ACN/kg, while forestomach cells of null mice given daily doses 60 mg ACN/kg again showed no detectable DNA damage. Significant increases in DNA damage were also found using extended electrophoresis time in liver DNA of WT mice administered daily doses of 10 and 20 mg/kg but not in cells of null mice given 60 mg ACN/kg/day.
These positive reports can be compared with those from the standard alkaline comet studies of Pu et al. (Citation2009, Citation2015) and Williams GM et al. (Citation2017), all of which failed to find positive results in different tissues of rats including lymphocytes or brain at ACN doses of 3, 30, 100, or 200 ppm in drinking water (Pu et al. Citation2009, Citation2015) or Zymbal’s gland at a dose of 100 ppm in drinking water (Williams GM et al. Citation2017) after several days exposure.
Another indicator of DNA strand breakage is fragmentation which was observed in brains of Wistar rats receiving ACN at 100 ppm in drinking water for 14 or 28 days (Mahalakshmi et al. Citation2003). Determinations of fragmentation were made as the ratio of DNA in the supernatant to total DNA in tissue homogenates and were associated with increased levels of lipid peroxidation products (thiobarbituric acid, lipid hydroperoxides) in the brain and plasma.
DNA repair as indicating DNA damage
Unscheduled DNA synthesis (UDS)
Unspecified DNA damage may be inferred by observing DNA repair, which is assessed by measuring unscheduled DNA synthesis (UDS) to reflect the localized synthesis that occurs during nucleotide excision repair (NER), as opposed to the global scheduled DNA synthesis (SDS) that occurs during cell replication. Two methods have been used to measure UDS: (1) liquid scintillation counting of 3H thymidine incorporation in DNA in cells having a hydroxyurea block to eliminate SDS; and (2) direct visualization of incorporation by autoradiography. Of the two, the latter is the more reliable as the newly labeled DNA is visualized as local while even a small amount of SDS that escapes a hydroxyurea block cannot be differentiated from UDS by scintillation counting, potentially producing false positive results (Williams GM et al. Citation1985; Butterworth et al. 1987; Madle et al. 1994; OECD Citation1997).
Perocco et al. (Citation1982) studied UDS in cultured human peripheral blood lymphocytes (PBLs) exposed to ACN concentrations that ranged from 2.5 to 16,500 µg/ml, and observed an increase, particularly at the highest concentration, as determined by liquid scintillation counting. Rizzi et al. (Citation1984) treated HeLa cells to various doses of ACN and observed increases in UDS determined by scintillation counting with or without metabolic activation, with activation producing effects at a lower exposure concentration than corresponding test without activation. Later, four laboratories investigated the ACN-induced UDS response as part of the 1985 IPCS CSSTT venture. Glauert et al. (Citation1985) exposed rat primary hepatocytes to ACN concentrations that ranged from 0.05 to 530.0 µg/ml and reported a positive response at the optimal concentration of 0.5 µg/ml, with toxicity being observed at the higher exposure levels. Martin and Campbell (Citation1985), however, exposed HeLa S3 cells to ACN at unknown concentrations and reported a negative UDS response, even though the method of analysis was liquid scintillation counting. Two additional studies in the IPCS CSSTT series report negative findings. Probst and Hill (Citation1985) and Williams GM et al. (Citation1985) exposed rat hepatocytes to ACN concentrations ranging from 0.03 to 530 and 0.1 to 10,000 µg/ml, respectively, and both reported no increase in UDS. The difference between these two studies and the positive studies is that two of the negative results assayed for UDS by autoradiography.
Rat hepatocytes were again studied for UDS response some years later using the autoradiographic assay (Butterworth et al. Citation1992). The hepatocytes were exposed to either ACN at concentrations ranging from 0.1 to 10 mM, or to CNEO at concentrations ranging from 0.01 to 1.0 mM. No UDS responses were observed in either instance, with toxicity seen at 10 mM and 1.0 mM ACN or CNEO, respectively. This same study exposed human mammary epithelial cells to these same concentrations of ACN or CNEO, with cytotoxicity being observed at the same concentrations as seen for the hepatocytes. As for the hepatocytes, there was no UDS response in the mammary epithelial cells following the ACN exposures. However, there was a positive response in the mammary epithelial cells following CNEO exposures of 0.1 mM, and CNEO exposures of 1.0 mM were found to be toxic.
The ability of ACN to induce UDS has also been studied in vivo. Hogy and Guengerich (Citation1986) reported elevated UDS measured by liquid scintillation counting in liver but not brain two hours following a 50 mg/kg oral dose of ACN to an F344 rat. SDS however, was decreased in brain but not liver. UDS was also measured in the series of experiments described above in which DNA binding was assessed in rats (Ahmed, Abdel-Aziz, et al. 1992; Ahmed, Abdel-Rahman, et al. Citation1992; Abdel-Rahman et al. Citation1994). In all instances, ACN was administered as a 46.5 mg/kg oral dose with increases in UDS and concomitant decreases in SDS reported for lung, testicular and glandular stomach DNA. Again, however, UDS was measured by liquid scintillation counting. In a later study by this group (Ahmed et al. Citation1996), UDS was again measured by liquid scintillation counting in Sprague-Dawley rats administered ACN at 23 or 46 mg/kg orally, and again a positive UDS response was noted in the glandular stomach. This response, however, was partially blocked by the administration of SKF 525 A – a blocker of P450 enzyme activity, taken as evidence that at least part of the ACN UDS response was due to its metabolism to CNEO. The measured UDS response in this study was associated with a significant GSH depletion in the gastric tissue, was increased by the GSH depleting agent diethylmaleate and was inhibited by the administration of sulfhydryl compounds before the ACN administration.
Only a single study measured in vivo UDS by the autoradiographic method. F344 rats administered ACN by gavage as a 75 mg/kg single dose, or at 60 mg/kg daily for five days, failed to show UDS in testes or liver at 2,4, or 12 h following the last dose (Butterworth et al. Citation1992).
Sister chromatid exchanges (SCE)
Sister-chromatid-exchanges (SCEs) are also nonspecific indicators of successful repair of DNA damage (Wilson DM and Thompson Citation2007). As successful repair does not result in changes in DNA information content, SCEs do not have adverse health implications and have no genetic consequences (Bonassi et al. Citation2004)
A positive SCE response was reported in an early study in CHO cells exposed to ACN (unknown concentration), but only with co-incubation with rat hepatocytes (Ved Brat and Williams Citation1982). Similarly, increased frequencies of SCE were induced in phytohemagglutinin stimulated human peripheral blood lymphocytes treated in vitro with ACN 5 × 10−4 M in the presence but not in the absence of metabolic activation; ACN 5 × 10−5 M failed to produce a response (Perocco et al. Citation1982).
Four laboratories investigated the SCE response during the 1985 IPCS CSSTT, employing three different cell types. Gulati et al. (Citation1985) reported a positive response in CHO cells in both the presence and absence of an S9 metabolic activating system. In the presence of S9, an increase in the SCE frequency was seen at an ACN concentration of 50 µg/ml, which was the highest nontoxic concentration (range tested was 1.6 to 160 µg/ml). Seemingly paradoxical, in the absence of S9, a positive SCE response was reported at an ACN concentration of only 16 µg/ml, which was also the highest nontoxic concentration tested in the absence of S9 (range tested was 0.16 to 50 µg/ml). Natarajan et al. (Citation1985) also studied the SCE response in CHO cells exposed to ACN concentrations ranging from 1.0 to 4.0 mM (53 to 212 µg/ml). A positive response was observed at 2.0 mM (106 µg/ml) in the presence of S9 while there was no increase in SCE frequencies in the absence of S9. Priston et al. (Citation1985) measured SCE frequencies in rat liver RL 4 cells exposed to ACN concentrations ranging from 1.25 to 10.0 µg/ml without observing a positive response. Similarly, Obe et al. (Citation1985) exposed human PBLs in culture to ACN concentrations ranging from 1.0 to 10.0 µg/ml, with and without S9, and reported no increases in SCE frequencies in either case. A later study measured SCE frequencies in cultured bronchial epithelial cells exposed to ACN concentrations ranging from 150 to 600 µg/ml (Chang et al. 1990). Increases in SCE frequencies were observed at both the 150 and 300 µg/ml concentrations, with the 600 µg/ml concentration being toxic to the cells.
In an in vivo SCE study in C57BL/6 mice administered ACN at doses up to 60 mg/kg i.p., a weak positive response was seen in bone marrow cells at 45 mg/kg in the single surviving mouse at this dose level (Sharief et al. Citation1986). There was no SCE response at 30 mg/kg or at 60 mg/kg, which killed all of the animals. ACN administered i.p. at 5.0, 7.5, and 10 mg/kg to male mice (strain unknown) induced a significant increase in SCE in bone marrow cells at 10 mg/kg that was, however, considerably weaker than the response induced by mitomycin C in the same experiment (Fahmy Citation1999).
SCE studies in humans are described below.
Unspecified DNA damage
Kawachi et al. (Citation1980) used a B. Subtilis rec assay based on enhanced survival of a DNA recombination-repair-proficient strain of bacteria compared to that of a repair-deficient strain to demonstrate that ACN, after metabolic activation, did induce breaks in that system as the repair-deficient strain failed to rejoin the breaks.
Generic DNA damage in bacteria may be inferred from an increased expression of genes that are up-regulated in response to an adverse exposure, i.e. the SOS response. In a comparative test of 40 chemicals Brams et al. (1987) reported that ACN was negative in a Chromotest that measured induction of afiA gene expression in E. coli PQ37. Similarly, ACN at concentrations up to 2820 µg/ml failed to increase umu gene expression in Salmonella typhimurium (Nakamura et al. Citation1987).
The tumor suppressor proteins p53 and p21WAF1 were employed as indicators of DNA damage occurring in human embryonic fibroblasts in vitro after 24-h exposures to ACN at concentrations ranging from 0.05 to 100 mM (Rössner et al. Citation2002). Earlier studies had suggested this as a biomarker to identify genotoxic carcinogens (Yang J and Duerksen-Hughes Citation1998). Increased production of both was observed in the study by Rössner et al. (Citation2002) at concentrations between 0.3 and 1.0 mM suggesting DNA damage. Changes in cell morphology were seen at ACN concentrations ≥ 0.25 mM.
Specific ACN/CNEO induced adducts ()
Neither uncharacterized DNA binding nor nonspecific generic damage per se identifies an ACN/CNEO chemical reaction that initiates the mutagenic process, i.e. produces a specific DNA adduct or induces a double-strand break.
Adducts are produced at different sites in the DNA because of characteristics of the chemical, structure of the DNA and other factors. Adducts at some sites are pro-mutagenic in that they may cause mutations by their very presence. Adducts that alter DNA structure, prevent replication, or form at coding sites i.e. O6, N1 and N2 of guanine [G] [superscript indicates exo-cyclic oxygen or nitrogen], N1 and N6 of adenine [A], O2, N3 and N4 of cytosine [C] and O4 and N3 of thymine [T] are pro-mutagenic, as are additions of exocyclic carbons to the bases leading to miscoding (Rioux and Delaney Citation2020). However, purine adducts at non-coding cyclic nitrogen sites, i.e. N7G and N3A, usually do not produce mutations unless they distort DNA structure or convert to cyclic or ring-open structures (Singer and Grunberger Citation1983; Boysen et al. Citation2009). N7G and N3A adducts are unstable and usually are removed by spontaneous depurination, leaving behind efficiently repaired apurinic (AP) sites that do not produce mutations unless there is insufficient time for closure prior to DNA synthesis.
In vitro studies with isolated DNA
There have been numerous in vitro studies of the potential for ACN/CNEO to chemically induce specific DNA adducts, with the earliest being in isolated DNA/nucleobases. Guengerich et al. (Citation1981) initially reported formation of the 1,N6 εA etheno adduct following incubation of adenosine with CNEO 100 mM for 40 h at 37 °C, pH7.7. Incubations with the other bases resulted in uncharacterized adducts. It was later shown that ACN itself could react with all four nucleobases in DNA to form specific adducts although at highly non-physiological conditions (Solomon et al. Citation1984, Citation1993). ACN, at 1.4 M, was incubated with calf thymus DNA at 37 °C, pH 7.0, for a total of 40 days. The recovered adducted bases included three of G, i.e. 7 cyanoethyl G (7CNEG); 7,9 bis CNEG and ring opened (iro-) N7, 9 bis 7CNEG; two of A, i.e. 1 carboxyethyl A (1CEA) and N6 CEA; and one each of C (3CEC) and T (3CNET). The adducts were produced in relative amounts of 25.8% (7CNEG), 4.3% (7,9 bis CNEG), 18.9% (iro-7, 9 bis CNEG), 25.9% (1CEA), 7.5% (N6 CEA), 1.5% (3CEC) and 16.3% (3CNET).
Solomon et al. further demonstrated that the reactions of the CNEO metabolite of ACN with DNA were more efficient than the reactions shown in their earlier studies with the parent compound (Solomon et al. Citation1984, Citation1993). CNEO 50 mM incubated with DNA at 37 °C, pH 7.0, for only three hours, produced a single adduct of G, i.e. N7OEG, two of A, i.e. 1, N6 etheno A (1, N6 ε A) and N6 hydroxycarboxyethyl A (N6 HCEA), one of C, which was rapidly converted to uracil (U) by hydrolytic deamination to form 3HCEU, and one of T, i.e. 3OET. These formed in relative amounts of 53.9% (N7OEG), 5.3% (1,N6 ε A), 1.0% (N6 HCEA), 39.2% (3HCEU) and 0.5% (3OET). The most abundant adducts under these conditions were N7OEG and 3HCEU. Adduct profiles following incubations of CNEO with 2′-deoxyribonucleotides were similar, although some additional degradation products have been identified (Yates et al. Citation1993; Solomon et al. Citation1984, Citation1993).
Contemporaneously, Hogy and Guengerich (Citation1986) also identified the N7OEG adduct in vitro following incubation of purified DNA with 5 mM CNEO for 1.0 h at pH 7.4 (Hogy and Guengerich Citation1986). No other adducts following in vitro incubations of DNA were investigated. Manso et al. (Citation2011), however, failed to find covalent binding of ACN to guanosine in vitro under physiological conditions (i.e. pH < 8.0, T = 37° C), although a stable ACN-guanosine adduct formed at higher pH values. Guanosine was present at various concentrations; ACN concentrations varied from 10−3 to 10−4 M and incubations were up to three weeks. Detection was by ultrafast liquid chromatography. By contrast, acrolein-guanosine adducts as positive controls were formed under physiological conditions. Later, Walker, Fennell, et al. (Citation2020) reported that incubation of DNA isolated from rat brain and liver with CNEO concentrations in the µM range resulted in the dose-related formation of N7OEG. It is noteworthy, however, that this adduct was not formed in actively dividing human TK6 lymphoblastoid cells exposed in vitro for two hours to 100 µM CNEO (Walker, Fennell, et al. Citation2020).
In addition to adducts at specific sites on the nucleobases, cyanohydroxyethyl diphosphate adducts were observed after incubations of 2′-deoxynucleotides in vitro with 150 mM CNEO for 3 h, pH 7.0–7.5, 37 °C (Yates et al. Citation1994). It is assumed that such phosphate backbone adducts also occur after incubations with isolated DNA.
In vivo studies
The most convincing evidence that ACN/CNEO-specific DNA adducts initiate the mutagenic process would be discovery of such adducts in ACN exposed cells, in animals or in humans along with the presence of positive mutation results. Although several investigators have attempted to demonstrate induction of ACN/CNEO-specific DNA adducts in vivo, results have yielded no definitive findings.
The first and only report of the specific N7OEG adduct formation in cells or in vivo was in male F344 rat liver (n = 1–3) using a radiometric assay following i.p. injections of ACN (50 mg/kg; 1-14C or 2,3-14C) or unlabeled CNEO (6.0 mg/kg) with sacrifice two hours later (Hogy and Guengerich Citation1986). Adduct levels based on scintillation counting rose from 0.0034 per 106 dG (0.00074 per 106 bases) at baseline to 0.147 and 0.064 per 106 dG (0.032 and 0.014 per 106 bases) following ACN or CNEO administrations, respectively. Despite equal protein binding of these two agents between liver and brain and somewhat higher N7OEG adduct background levels in the latter tissue, no increases in adduct formation following treatments were found in brain. No other adducts were identified. Specifically, no 1,N6 ε A was found (limit of detection 3 pmol adduct/mg DNA) even though this adduct had been detected earlier in vitro (Guengerich et al. Citation1981).
Other in vivo studies, however, failed to identify even the N7OEG ACN/CNEO-specific DNA adduct in various animal tissues. Prior to the study of Hogy and Guengerich (Citation1986), Peter, Appel, et al. (Citation1983) in the binding study described above had injected male Wistar rats i.p. with radiolabeled ACN, with sacrifices 14 or 72 h later. Although some radioactivity in liver DNA was associated with nucleotide bases, the peaks observed were too small for identification. Somewhat later, Prokopczyk et al. (Citation1988) exposed male F344 rats (10 animals per dose) to ACN at 50 or 100 mg/kg by s.c. injection and sacrificed 2 h (50 mg/kg) or 6 h (100 mg/kg) later. DNA was isolated from liver and brain. Assay by HPLC using a method validated to detect 7CNEG at one adduct per 5 × 104 Gs, and O6 CNEG at one adduct per 7 × 104 Gs, failed to find either adduct in liver or brain, although the method was quite insensitive.
No specific DNA adducts were found in brain by 32P post-labeling in the F-344 or SD rats administered ACN at 100 ppm for 27 days in the Williams GM et al. (Citation2017) DNA binding study described above, although interpretation is difficult as no reference standards were used. A single adduct was identified in brain in positive control animals of both strains receiving BP.
A more recently published study in F344 rats administered ACN in drinking water at 300 ppm for up to 105 days (male rats only) or 500 ppm for 15 months (male and female rats) also failed to find significant increases in N7OEG or etheno-adducts adducts in several tissues, i.e. brain, splenic lymphocytes or stomach (Walker, Fennell, et al. Citation2020). Additional adducts were identified but no increases were detected, including N2εG in brain, liver, spleen, or stomach, or N6εA or 3,N4-ethenodeoxycytidine in brain or liver. Failure to find induced adducts in this study cannot simply be attributed to insufficient method sensitivity because concurrent analyses of DNA from liver and other tissues from vinyl chloride-exposed rats found increases in these positive controls (i.e. expected adducts were identified) for the N7OEG and etheno-DNA adduct assays.
It is noteworthy that small but non-significant increases of up to 12 pmol N7OEG per 106 guanines (near the detection limit of 10 pmol per 106 guanines (at a signal to noise ratio of 10:1), were detected in liver of rats exposed to the high-dose levels of ACN in the Walker, Fennell, et al. (Citation2020) study. Other studies have demonstrated that N7OEG can be endogenously produced from lipid peroxidation (Mutlu et al. Citation2012). No such increases were detected in liver or any other tissue from rats exposed to 0, 3, 10, 35, or 100 ppm ACN (Walker, Fennell, et al. Citation2020).
Oxidative DNA damage
Exogenous mutagens damage DNA either directly or indirectly. Indirect mutagenesis frequently results from enhanced production of ubiquitous endogenous mutagens such as reactive oxygen species (ROS) (Hartwig et al. Citation2020). Under background conditions, the DNA in animal and human tissues contains thousands of damaged sites due to endogenous production of reactive chemicals (e.g. formaldehyde, ethylene oxide) and ROS (Swenberg et al. Citation2011). Exposure to exogenous mutagens may increase their production, reduce defenses against them or both.
ACN/CNEO produce depletion of cellular sulfhydryls such as GSH, which likely contributes to oxidative stress (Puppel et al. Citation2015). Both react with GSH, and together are capable of depleting cellular GSH levels. In addition, as discussed above, thiocyanate formed from cyanide released from ACN serves as a substrate for peroxidases (e.g. myeloperoxidase, lactoperoxidase), which yields hypothiocyanite. As a sulfhydryl reactive agent, hypothiocyanite also can deplete levels of reduced GSH (Arlandson et al. Citation2001). In tissues with peroxidase activity, hypothiocyanite may act together with ACN/CNEO to further reduce defenses against oxidative damage.
In addition to producing ROS, exogenous mutagens may indirectly increase the frequencies of mutations by altering cell proliferation, interfering with DNA repair or inhibiting apoptotic cell death, all of which will increase background mutations. Unlike direct DNA reactive genotoxicity, which is likely similar among species, indirect genotoxicity may have species differences that introduce uncertainties in extrapolating among them.
ACN/CNEO induction of oxidative stress and lipid peroxidation with production of DNA reactive intermediates (e.g. ROS, malondialdehyde) that damage the genetic material has been repeatedly demonstrated (reviewed in EPA IRIS 2011). These intermediates potentially result in DNA adducts affecting all four nucleobases (reviewed in Wallace Citation2002; Cooke et al. Citation2003; Marnett et al. Citation2003). The “signature” biomarker for oxidative DNA damage is 7,8-dihydro-8-oxo-guanine (8oxoG), the most abundant adduct produced, reflecting reaction at the most readily oxidized site in G, i.e. the eighth position of the imidazolyl ring. Estimates are that a normal cell contain 105 such adducts (Valko et al. Citation2006). This lesion may be transformed, with disruption of the imidazole ring to the 2,6-diamino-4-hydroxy-5-formamidopyrimidine (Fapy-G) lesion. 8oxoG is a promutagenic adduct but the Fapy-G lesion serves as a replication blocker and is lethal in bacteria (Wallace Citation2002). Other potential purine adducts include 7,8-dihydro-8-oxo-adenine (8oxoA), 4,6-diamino-5-formamidopyrimidine (Fapy-A), 2-hydroxyadenine (2 OHA) and α-deoxyadenosine (α dA).
Damaged pyrimidines are also part of the lesion spectrum resulting from oxidative DNA damage. The major cytosine adduct is cytosine glycol, which then is either rapidly deaminated to form uracil glycol or quickly dehydrated to form 5-hydroxycytosine (5OHC). Uracil glycol further dehydrates to form 5-hydroxyuracil (5OHU). Uracil glycol, 5OHC and 5OHU are the stable cytosine adducts produced by oxidative stress. The most abundant oxidized lesion of thymine is the glycol. Others include 5,6-dihydrothymine (DHT) adduct, which may then form two 5-methyl oxidative derivatives, – i.e. 5-hydroxymethyluracil (5HMU) and 5-formyluracil (5fU). Ring-fragmentation or ring opened products of DHT such as urea and β-ureidoisobutyric acid also arise.
Many additional adducts may arise from second-generation reactive intermediates that result from oxygen reactions with cellular components to yield oxidation products. These rearrange to diffusible electrophiles that react with the DNA (Marnett et al. Citation2003). Malondialdehyde, which is the end product of lipid peroxidation, forms the highly mutagenic M1G DNA adduct. A consideration of this large array of lesions, any of which can result in endogenous DNA damage, is beyond the scope of this discussion.
ACN induced ROS DNA adducts
Direct detection of 8oxoG adducts
In vitro studies
Kamendulis, Jiang, Xu, et al. (Citation1999) showed the signature oxidative DNA damage adduct levels, i.e. 8oxoG, to be significantly elevated immediately post-treatment in astrocytes (D1TNC1 derived from Sprague-Dawley rats) but not in hepatocytes in vitro as demonstrated by HPLC, rising from a background level of 3.2 to 3.8, 7.7 and 13.3 adducts per 106 dG at 10, 100, and 1000 µM ACN exposure concentrations and falling significantly 24 h later. Associated markers of oxidative stress such as reductions in GSH content and superoxide dismutase activity and the production of ROS were observed in the ACN-treated astrocytes but not hepatocytes. Lipid peroxidation, catalase activity and glutathione peroxidase were not significantly affected by ACN in either cell type. The oxidative stress/DNA damage induced by ACN was reduced or eliminated by removal of ACN and by 2-oxothiazolidine-4-carboxylic acid (OTC), a GSH precursor, or vitamin E (an antioxidant) co-treatment.
Results similar to those of Kamendulis, Jiang, Xu, et al. (Citation1999) were found in human cultured astrocytes by Jacob and Ahmed (Citation2003) where 8oxoG DNA adduct formation, again as demonstrated by HPLC, was shown in cells incubated at much higher ACN concentrations of 200–400 µM. 8oxoG levels (measured as 8OHdG) rose significantly from ∼1.6 × 106 dG at baseline to ∼2.25 or 5.25 adducts per 106 dG at 200 or 400 µM ACN, respectively. GSH content was significantly reduced, especially at the higher ACN concentrations, while catalase activity, after an initial rise, was significantly reduced at 400 µM ACN. ROS formation was elevated at 200 and 400 µM ACN with TNFα secretion also being elevated at the higher concentration.
In vitro induction of 8oxoG adducts was also measured in Syrian Hamster Embryo (SHE) cell transformation studies by either ACN or cyanide (Zhang et al. Citation2000, Citation2002). Incubation of SHE cells with ACN 75 µg/ml induced 8oxoG adducts at levels to ∼80 to 90% over controls after 2–3 days, but not after 1 or 7 days (Zhang et al. Citation2000). The induction of increased adduct levels was inhibited by antioxidants. Potassium cyanide was also shown to significantly increase 8oxoG adduct levels to ∼50–60% over controls in SHE cells, the latter at 500 µM but not 20 µM, following 1–2 days incubation (Zhang et al. Citation2002). 8oxoG adducts were measured by HPLC and electrodetection in both studies.
A study in D1TNC1 rat astrocytes compared 8oxoG adduct formation by ACN with that formed by methylacrylonitrile (MethACN). While ACN induces brain tumors in rats, MethACN does not. Astrocytes were incubated with either 0.1, 0.5, or 1.0 mM ACN or 0.1, 0.5, 1.0, or 2.5 mM MethACN for 24 h (Klaunig and Forney Citation2010). Analysis by HPLC-electrodetection showed a significant increase in 8oxoG adducts at the two highest ACN concentrations compared to control, i.e. 1.89 and 2.33 per 106 dG respectively, compared to 1.34 per 106 dG for controls. There was no increase over control in the MethACN-treated cells. It must be noted, however, that metabolism of MethACN actually produces more cyanide than does metabolism of ACN (Farooqui and Mumtaz Citation1991).
In vivo studies
ACN induced 8oxoG adduct formation has also been studied in vivo in rodents. Male Sprague-Dawley rats administered ACN at levels of 3, 30, and 300 ppm in drinking water for 21 days with sacrifice on day 22 showed significant elevations of 8oxoG adducts in both brain and liver at the 30 and 300 ppm exposure levels, with the greater amount noted in brain at ACN exposure concentrations as low as 30 ppm (Whysner et al. Citation1998). Brain 8oxoG levels rose from 6.2 per 106 dG at baseline to 8.6, 13.5, and 12.9 adducts per 106 dG at the 3, 30, and 300 ppm exposure levels, respectively. Liver 8oxoG levels rose from 6.7 per 106 dG at baseline to 7.2, 9.5 and 9.6 adducts per 106 dG at the 3, 30, and 300 ppm exposure levels, respectively. 8oxoG levels were also elevated by treatment in stomach DNA but, because of variability, not significantly so. 8oxoG levels in brain and liver appear to plateau above 30 ppm, which may reflect saturation of the oxidative metabolic pathway for ACN. Brain 8oxoG adduct levels were also measured in in a sub-chronic 94-day study F344 rats receiving ACN at 1, 3, 10, 30 or 100 ppm in drinking water for 21 days. Post-treatment adduct levels were again elevated but less so than in Sprague-Dawley rats, being statistically significant only when elevations for the three highest exposure concentrations were combined and compared with the adduct levels at baseline and following 1 ppm exposure. No 8oxoG elevations were seen after administration of methylnitrosourea (MNU) as a control mutagen. GSH, glutathione peroxidase and catalase were not altered by ACN, and no other DNA adducts were monitored.
In a similar study, ACN at doses of 0, 5, 10, 100, or 200 ppm were administered to Sprague-Dawley rats in drinking water with sacrifice of animals and sampling of tissues after 14, 28, or 90 days of continuous treatment (Jiang et al. Citation1998). These exposure levels were chosen to be similar to those that induce brain tumors in long term studies. Significantly elevated levels of 8oxoG as determined by HPLC were found post-exposure in brain but not liver DNA. The baseline levels of 8oxoG adducts in brain remained constant at between 10 and 12 adducts per 106 dG for all time periods while the post-treatment levels rose both as a function of exposure level and time of treatment, reaching levels of 30 to 40 adducts per 106 dG with time and dose. Accompanying the increased levels of 8oxoG was evidence of lipid peroxidation, as reflected in elevations in malondialdehyde, increased levels of ROS, and decreased levels of GSH, catalase activity and SOD activity, all in brain, compared to control animals. Consistent with the failure to find 8oxoG adducts in liver DNA, biomarkers of oxidative stress were not elevated in that tissue.
Somewhat later, 8oxoG adducts were measured by electrodetection in white blood cells (WBCs) of Sprague-Dawley rats receiving ACN at 3, 30, 100, or 200 ppm in drinking water for 28 days (Pu et al. Citation2009). Adduct levels rose from a baseline of approximately 1.2 adducts per 106 dG to significantly elevated levels of approximately 3.8 and 6.8 per 106 dG at the 100 and 200 ppm exposure levels, respectively, but not elevated at the two lower ACN exposure levels. Plasma ROS and reduced GSH in brain were determined as measures of systemic and local oxidative stress, respectively. Brain 8oxoG levels as determined by HPLC had previously been shown to be elevated as a function of ACN dose post-treatment (Jiang et al. Citation1998; Whysner et al. Citation1998). Significant elevations of plasma ROS and reductions in brain reduced GSH were seen, with significant pair-wise correlations between all measures of adducts and oxidative stress, with the exception of GSH levels which were not changed. The oxidative DNA damage and oxidative stress determined in this study were observed at ACN exposure concentrations that significantly correlated with those previously reported to induce rat brain tumors.
These same investigators have recently reported the influence of anti-oxidant diets in reducing the oxidative DNA damage in ACN-treated rats (Pu et al. Citation2015). Female F-344 rats were administered ACN at 100 ppm in drinking water for 28 days (corresponding to a dose of approximately 5 mg/kg-day). 8oxoG levels measured by HPLC-electrodetection in brain rose significantly over control in the treated animals, i.e. approximately 2-fold from 1.5 to 3.0 per 106 dG. The anti-oxidants vitamin E, green tea polyphenols and N-acetyl cysteine in the diet protected against this oxidative DNA damage. Malondialdehyde levels in brain were not increased in the treated animals.
Unlike these several observations in rats, a study in B6C3F1 mice (non-sensitive species for ACN-induced brain tumors) receiving ACN at 0, 2.5, 10, or 20 mg/kg/day in drinking water for 14, 28, or 30 days showed no evidence of increased oxidative DNA damage in either brain or liver and no evidence of increases in oxidative stress indicators (Kamendulis et al. Citation2001).
A recent Taiwanese observational study in humans compared ACN exposure levels determined by measuring urinary CEMA with urinary excretion of the oxidative damage 8oxoG DNA adduct in more than 800 smoking and nonsmoking young adults (Lin et al. 2018). The subjects with the highest 10% of CEMA concentrations in urine were positively and significantly correlated with 8oxoG urine levels, demonstrating in humans also the capacity of ACN to induce oxidative stress/DNA damage.
Detection by modified comet assay
In addition to chemical determinations of 8oxoG adduct formation following ACN treatments, measurements have been made using a modified comet assay. The standard alkaline comet assay can detect both single- and double-strand DNA breaks and alkali-labile sites (abasic sites) in virtually any cell type. After treatment of cells with test agents, cellular membranes are lysed to allow release of coiled DNA and DNA fragments. Electrophoresis at high pH results in migration of fragments in a solid matrix resulting in comet-like structures that gives the assay its name. The DNA strand breaks and alkali-labile sites result in smaller fragments with enhanced migration in the electrophoretic field. Comets are scored as %DNA in the tail, tail length and/or tail moment, all reflecting enhanced migration.
The alkaline comet assay may be modified by the addition of enzymes that cleave the DNA at specific damage (adduct) sites. One such modification employs the enzyme formamidopyrimidine DNA-glycosylase (FPG) to cleave at sites of the 2,6-diamino-4-hydroxy-5-formamidopyrimidine (FPG-G) adduct derived from 8oxoG, thereby serving as an indirect measure of oxidative DNA damage (Collins Citation2009). Unfortunately, FPG-G comets are not totally specific for 8oxoG adducts (Speit et al. Citation2004; Smith et al. Citation2006; Boysen et al. Citation2010). The comet assay modified by addition of human-8-OH-guanine-DNA-glycosylase (hOGG1) enzyme is, however, specific for this adduct (Smith et al. Citation2006).
D1TNC1 rat astrocytes incubated with ACN for 24 h showed a dose dependent increase in modified FPG-G comet tail moments that became significant over control at 1.0 mM concentration while the standard alkaline comet assay did not show such an increase, demonstrating a lack of treatment related direct ACN induced DNA damage (Pu et al. Citation2006). Depletion of intracellular GSH using DL-buthionine –[S,R]-sulfoximine increased the ACN-induced oxidative DNA damage measured in this way while co-treatment with OTC as a GSH precursor reduced it. The oxidative DNA damage was presumed to have been due to ACN’s cyanide metabolite as it was prevented by inhibition of cytochrome P450 activity. Several anti-oxidants also reduced the presumed ACN induced oxidative DNA damage.
Subsequently, the FPG-G comet assay was again used to measure DNA lesions in both rat and human astrocytes incubated with 1.0 mM ACN for 24 h (Pu et al. Citation2009). As before, the rat D1TNC rat astrocytes showed a concentration dependent increase in damage – a result that was again enhanced by buthionine-sulfoximine to deplete GSH but attenuated by treatment with OTC. In contrast, the human astrocytes did not show evidence of increased DNA damage over background by FPG-G comets when incubated with 1.0 mM ACN alone but were sensitized to such damage by co-treatment with buthionine-sulfoximine. The FPG-G comet assay appeared to be less sensitive than HPLC for detecting oxidative DNA damage in that 8oxoG adducts were detected by HPLC in rat astrocytes treated with 10 µM ACN for 4 h, as indicated above, while significant increases in FPG-G comets were not observed until ACN exposures reached 1.0 mM for 24 h (Kamendulis, Jiang, Xu, et al. Citation1999; Pu et al. Citation2006). Similarly, as also noted above, 8oxoG adducts were detected by HPLC in human astrocytes treated with ACN 200–400 µM for 24 h while treatment of these cells with ACN 1.0 mM for a similar duration failed to increase FPG-G comets, requiring GSH depletion to show this effect (Jacob and Ahmed Citation2003). These combined results were interpreted as indicating that human astrocytes have relative protection for ACN induced oxidative DNA damage compared to rat astrocytes because of greater levels of GSH.
D1TNC1 rat astrocytes incubated for 24 h with ACN at 0.1. 0.5, or 1.0 mM concentration or MethACN at 0.1, 0.5., 1.0, or 2.5 mM concentration were also compared for their relative abilities to induce oxidative damage as detected by FPG-G comets (Klaunig and Forney Citation2010). As expected from the chemical determinations described above, ACN at 1.0 mM significantly increased migration in the modified comet assay indicating a significant increase in 8oxoG adducts. There was no increase with MethACN at any concentration. The standard alkaline comet assay was negative for both compounds.
In order to determine if blood cells could be used as surrogates for brain, the FPG-G comet assay was employed to assess presumed oxidative DNA damage in brain and lymphocytes of Sprague-Dawley rats receiving ACN at 3, 30, or 100 ppm in drinking water for 28 days, or up to levels of 200 ppm for 14 or 28 days in a time-course study (Pu et al. Citation2009). Earlier chemical studies had already demonstrated the presence of 8oxoG adducts in brain as chemically determined by HPLC at these exposure levels (Jiang et al. Citation1998; Whysner et al. Citation1998). The more recent study (Pu et al. Citation2009) again found elevated levels over background in brain while also demonstrating elevated levels of this adduct in WBCs as measured by HPLC-electrodetection in both tissues. Furthermore, as expected, both brain and white blood cell 8oxoG levels were found to be elevated as a function of ACN dose and treatment duration when inferred from the elevated FPG-G comet assay responses. Standard alkaline comet assays of lymphocytes from the treated rats failed to show evidence of DNA-strand-breakage/alkali-labile sites. As noted above, the oxidative DNA damage and oxidative stress determined in this study were observed at ACN exposure levels that significantly correlated with those previously reported to induce rat brain tumors.
More recently, these investigators again measured oxidative DNA damage as 8oxoG adducts by HPLC-electrodetection in brain of female F-344 rats administered ACN at 100 ppm in drinking water for 28 days while again determining oxidative DNA damage in WBCs as measured by FPG-G comets in these same animals (Pu et al. Citation2015). The modified comet assay responses in the WBCs (non-target tissue) of the treated animals were elevated in the ACN treated animals, as were the chemically detected 8oxoG levels in brain. The elevated FPG-G responses were also modified by antioxidants. As in their earlier studies, standard alkaline comet assays failed to show increased strand-breakage/alkali-labile sites in the WBCs. ACN treatment also stimulated inflammation responses and growth factors in the treated animals.
Somewhat conflicting results have been reported from the binding study of Williams GM et al. (Citation2017) described earlier when oxidative DNA damage in both brain and Zymbal’s gland tissue was assessed by modified comet assays. In this study, the FPG-G comet assay was positive in brain tissue but negative in Zymbal’s gland. However, and importantly, when the comet assay was modified by substituting the 8oxoG specific hOGG1 for FPG for incision, results were negative for both tissues in the treated animals of both strains. As in earlier studies, standard alkaline comet assays were negative for both tissues in both strains. There were no changes in F2-iso-prostane levels as a measure of oxidative stress/lipid peroxidation in the brains of either strain of ACN-treated rats.
Summary of DNA reactions
Many studies have investigated uncharacterized generic changes in the DNA resulting from exposure to ACN and/or CNEO. Several have focused on the capacity to form co-valent associations with the genetic material, reporting positive results. Studies have also reported the induction of DNA single strand breaks and/or apurinic sites as a function of ACN/CNEO exposures with many claiming positive results. Studies of DNA repair of unspecified ACN/CNEO associated damage as indicated by either UDS or SCE have also yielded positive as well as negative results.
Both ACN and CNEO have been shown to produce specific adducts when reacted in vitro with nucleobases or isolated DNA, with CNEO being the more efficient. For this reason, use of EMA in in vitro test systems increase positive results. In striking contrast, it has been difficult to demonstrate ACN/CNEO induction of specific DNA adducts in living systems, i.e. either in vitro in cultured cells or in vivo in a rodent. By contrast, a large literature has developed that associates ACN with the induction of oxidative DNA damage. The signature 8oxoG DNA adduct has been demonstrated directly by chromatographic methods in cultured cells in vitro and in brain and other tissues in vivo. Additional studies have employed a modified comet assay to demonstrate 8oxoG adducts in relation to ACN/CNEO exposure, with most utilizing FPG-G Comets. Unfortunately, that method is not specific for oxidative DNA damage. In contrast, the human enzyme 8-OH-guanine-DNA glycosylase (hOGG1) modified comet is specific for 8oxodG.
Critique of DNA reactions
Almost none of the studies reporting covalent binding of ACN/CNEO to the genetic material were able to differentiate true DNA binding from binding to associated proteins.
No studies of DNA single strand breaks and/or apurinic sites as a function of ACN/CNEO exposures at the somatic level effects have employed methods that exclusively detect double-strand DNA breakage (Note exception: human study reporting double-strand breaks in sperm of ACN exposed Chinese workers [Xu et al. Citation2003] described below).
Although studies of DNA repair of unspecified ACN/CNEO associated damage as indicated by either UDS or SCE have also yielded positive as well as negative results, positive UDS results were usually obtained using liquid scintillation counting while the negative reports were based autoradiography, which is the more specific indicator of UDS.
Although both ACN and CNEO have been shown to produce specific adducts when reacted in vitro with nucleobases or isolated DNA, these studies were conducted at high concentrations and under non-physiological conditions.
It has been difficult to demonstrate such adducts in living systems with only a single demonstration of a presumptive AN/CNEO specific adduct, i.e., N7OEG s in vivo in rats in non-target (liver) but not brain (target) tissue although several studies have tried and failed to repeat this finding or demonstrate any other ACN/CNEO specific adducts in living systems.
The study that found FPG-G comets positive but hOGG1 comets negative in tissues of ACN treated rats puts in doubt the induction of 8oxoG adducts in that study and raises the question of the significance of the several studies that report induction of this adduct on the basis of FPG-G comet assay alone. The hOGG1 modified comets are the more specific for oxidative DNA damage.
Studies conducted in rat astrocytes (in vitro) and in whole rat brain (in vivo) may not accurately reflect responses in the ultimate target cell identified for ACN-induced rat brain tumors (microglia).
There is at present insufficient information to confidently assign a single simple genotoxic/mutagenic mechanism of action to ACN from studies of its effects on the DNA. The current data base is compatible with multiple mechanisms, the preponderance of which are indirect.
Data gaps
The hypothesis remains that mutation induction is a (the) key event in ACN’s carcinogenic potential. Yet, convincing evidence of an ACN/CNEO induced DNA damaging event such as one or more DNA adducts, either directly induced or resulting indirectly due to oxidative stress, does not exist. To date, there have only been a limited number of DNA adduct studies, and none employing the newest highly sensitive methodologies (e.g. with detection limits of 10−10 to 10−12 normal nucleotides [Swenberg et al. Citation2011] vs. of 10−6–10−7 normal nucleotides. The identification of such adducts would aid in fully assessing human cancer risk due to ACN exposures. Of value would be the identification of specific DNA adducts in cancer target cells such as microglial cells in rats where their detection might be obscured due to tissue dilution when whole tissue (brain) is analyzed. The association of chemical specific and/or oxidative stress related DNA adducts with mutations (either gene or chromosome level) as determined in the same experiment by the same investigators should be determined and results quantified.
Other data gaps include lack of data resolving the ambiguity of origin of some DNA adducts such as N7G as resulting from specific chemical DNA reactions or metabolic enhancements of oxidative stress employing methods using isotope labeled reactants in studies of ACN/CNEO (Swenberg et al. Citation2011). Also, comparisons of mutational spectra in ACN/CNEO induced mutations with those due to known mutagenic carcinogens, already begun, will allow recognition of potential mechanistic similarities that could be followed up for ACN (Walker et al. Citation2020). Finally, there are no data assessing the potential for ACN/CNEO’s protein binding to interfere with metabolism or DNA repair and thereby influence the mutational process. In this regard, follow up of mechanisms by which ACN might interfere with spindle proteins via nitriles would allow better understanding of potential ACN-associated numerical chromosome aberrations.
ACN/CNEO mutations at the somatic level ()
Mutations are the end results of genotoxic insults that have potential deleterious health consequences in somatic cells (discussed in this section) and germ cells (see section below). They are, therefore, the endpoints of most concern. Mutations in DNA that permanently change its information content may be manifest at the gene or chromosome level. The former are submicroscopic changes, while the latter are microscopically observable.
Although, by recent convention, the term mutation has been applied to only gene level changes, and the term chromosome aberration has been applied to chromosome level changes, the extent of an alteration in DNA structure may be a continuum, with overlap between the two. Defining chromosome aberrations in somatic cells as mutations may be questioned because the alterations observed microscopically in cytogenetic assays are not themselves heritable, i.e. many are changes that are cell lethal. There is a similar objection to defining chromosome level changes manifest as micronuclei (MN) as mutations. Both, however, are non-repairable changes in DNA structure or content that unequivocally change genetic information and capture the molecular mechanisms that underlie the cytogenetic changes in malignancies. Furthermore, it is the method of scoring that makes these changes lethal in vitro, i.e. forced mitogen induced DNA synthesis with arrest prior to the first in vitro cell division so that alterations in chromosomes may be observed. Many of these changes in vivo may be mis-repaired to produce reciprocal translocations, peri- and paracentric inversions, interstitial deletions, terminal deletions, interstitial duplications and insertions and/or reintegration of micronuclei, resulting in truly heritable alterations of genetic information (Savage Citation2011; Luijten et al. Citation2018).
Somatic level mutations induced in vitro and/or in vivo by ACN (or CNEO) have been extensively studied in a variety of systems. While in vitro studies are generally designed to yield positive results, induction of mutations in vitro does demonstrate the mutagenic potential of a chemical. In vivo studies, however, are more relevant for assessing human risk. In vitro studies of ACN treated microorganisms or cells frequently employed external metabolic activation (EMA), e.g. liver S9 fractions, to produce the more reactive CNEO metabolite. Mutations at the gene or chromosome level are usually nonspecific as regards causation and may result from either the direct or indirect effects of an inducing agent. Mechanistic insights are occasionally gained from patterns of results in different assays or by analyses of molecular mutational spectra.
Prokaryotes ()
More than sixty specific bacterial strain mutagenicity tests have been conducted over the past forty years for ACN. Those in different E. coli WP2 strains produced mixed results. In an early comparison, strains WP2lexA cells gave negative results in both plate and fluctuation tests following ACN treatments of 53 µg/ml, with or without EMA, while strains WP2, WP2uvrA, and WP2 (PKM101) gave positive results at this same ACN concentration in both plate and fluctuation tests (WP2 only), but only with EMA (Venitt et al. Citation1977). Strain WP2 uvrA, PolA was even more sensitive, giving a positive response in plate assays following 5.3 µg/ml ACN, only with EMA. Subsequent studies in another laboratory, however, in another laboratory failed to find ACN-induced responses in plate assays with either WP2 or WP2 uvrA (McMahon et al. Citation1979). A negative response in E. coli WP2uvrA and WP2uvrA/pKM101 was also reported in a study by the Japanese Ministry of Labor using the pre-incubation method at a variety of ACN concentrations up to 5000 µg/plate with or without EMA (Matsushita and Goto Citation1980). All E. coli WP2 tester strains detect reverse mutations of the trpE gene which require base substitutions at an AT base pair. The strains differ in their DNA repair capacities.
Most bacterial mutagenicity tests of ACN have employed the Salmonella typhimurium histidine reversion assay (Ames test). Overall, twelve strains have been used, with eight being employed in the 1985 IPCS CSSTT series. Most strains are deficient in DNA excision repair and harbor mutations that decrease surface lipopolysaccharides to enhance entry of exogenous agents, especially bulky chemicals (Mortelmans and Zeiger Citation2000). Results here are according to tester strain; studies were performed with and without EMA.
ACN in an early study was negative in eight non-identified tester strains (McMahon et al. Citation1979). Subsequently, results differed according to strain(s). Most using strains TA97 and TA98 were negative in either spot tests, pre-incubation, desiccator or plate assays with ACN concentrations up to 10,000 µg/ml (Florin et al. Citation1980; Kawachi et al. Citation1980; Lijinsky and Andrews Citation1980; Matsushita and Goto Citation1980; Baker and Bonin 1985; Matsushima et al. Citation1985; Rexroat and Probst Citation1985; Zeiger and Haworth Citation1985; Brams et al. 1987). However, bacteria incubated in an atmosphere of 0.2% gaseous ACN produced a weak positive but only with EMA (de Meester et al. Citation1978). Tester strain TA97 has the base sequence CCCCCC in the mutant his D6610 allele and is reverted to wild-type by a single base (C) deletion; TA98 has a deletion in the sequence CGCGCGCG in the mutant his D3052 allele and is reverted by a single base (C or G) addition. Both strains have a deletion of the uvrB gene, which eliminates accurate DNA excision repair (Mortelmans and Zeiger Citation2000).
Studies using strains TA1537, TA1538 and TA1978 have also mostly been negative with only a single definitive and two weak positive results. Studies in TA1537 or TA1538 that used spot or plate assays with or without EMA were negative up to ACN concentrations of 5000 µg/ml (Florin et al. Citation1980; Lijinsky and Andrews Citation1980; Zhurkov et al. Citation1983; Rexroat and Probst Citation1985) as was a study by the Japanese Ministry of Labor using the pre-incubation method (Matsushita and Goto Citation1980). Positive results were reported by a single laboratory for TA1538 and TA1978 (weak) at an ACN concentration of 10,400 ppm or 130 ppm in liquid, respectively, only with EMA (Milvy and Wolff Citation1977). This conclusion, however, was challenged by a reanalysis of the data that was reported in a letter to the editor as a negative response in these two strains (Venitt Citation1978). Notwithstanding, another laboratory reported a weak positive result in strain TA1978 incubated with 0.2% gaseous ACN (de Meester et al. Citation1978). TA1537, TA1538, and TA1978 all contain the hisD3052 mutant allele that is reverted to wild-type by a one base (C or G) addition (Mortelmans and Zeiger Citation2000). TA1537 and TA1538 have the uvrB deletion while TA1978 is wild-type for DNA repair.
In contrast to the above Salmonella tester strains, which revert to wild-type by frame-shift mutations or insertions, strains TA100, TA1530, TA1535 and TA1950 all contain the hisG46 allele, which has been mutated from wild-type by AT→GC transition base-pair substitution. The resultant CCC mutant sequence can revert to wild type by a GC base pair substitution. All also have the uvrB deletion with resultant deficiency in DNA excision repair.
The majority of studies in tester strains containing the hisG46 allele have been reported as positive. Early studies in TA1530 were universally positive in the presence of EMA in desiccator and pre-incubation assays (de Meester et al. Citation1978, Citation1979; Roberfroid et al. Citation1978; Duverger-Van Bogaert et al. Citation1981, Citation1982a, Citation1982b; Zhurkov et al. Citation1983). The lowest ACN concentration that gave a mutagenic response was 2.5 µg/ml (de Meester et al. Citation1978), although most studies employed higher concentrations, and a single study reported a positive response in the absence of EMA (Duverger-Van Bogaert et al. Citation1981). Two additional studies reported mutagenic responses in strain TA1530 exposed to the urine of rodents exposed to ACN (Lambotte-Vandepaer Citation1980, Citation1981).
Responses in strain TA1535 have been almost as consistent in reporting positive results. Six studies have reported positive responses, but only in the presence of EMA, in various assays at ACN concentrations ranging from 0.5 to 167 µg/ml (Milvy and Wolff Citation1977; de Meester et al. Citation1978; Lijinsky and Andrews Citation1980; Matsushita and Goto Citation1980; Cerna et al. Citation1981; Zhurkov et al. Citation1983; Zeiger and Haworth Citation1985). Two laboratories, however, reported negative results in the presence or absence of EMA in spot and plate assays at ACN exposure concentrations up to 5000 µg/plate. An additional study that exposed strain TA1535 to bile from rats exposed to ACN also gave a negative result (Connor et al. Citation1979).
Results of studies in tester strain TA100 have been exceptions to those in other strains carrying the hisG46 mutant allele. In addition to deficiency in DNA excision repair, TA100 harbors a plasmid that increases error-prone recombination repair (Mortelmans and Zeiger Citation2000). Although six studies have reported positive or weakly positive results in the presence of EMA (with two reporting activity in the absence), another six have reported negative results with or without EMA. The positive studies employed a variety of assay methods and lowest effective ACN exposure concentrations (not given in the study by Khudoley et al. Citation1987) ranging from 19 to 806 µg/ml, or 0.2% for vapor exposures (de Meester et al. Citation1978; Cerna et al. Citation1981; Ishidate et al. Citation1981; Zeiger and Haworth Citation1985; Khudoley et al. Citation1987; Hakura et al. Citation2005). The negative studies employed spot, pre-incubation and plate assays and ACN concentrations as high as 10,000 µg/ml in one study (Florin et al. Citation1980; Lijinsky and Andrews Citation1980; Matsushita and Goto Citation1980; Baker and Bonin Citation1985; Matsushima et al. Citation1985; Rexroat and Probst Citation1985; Brams et al. Citation1987).
Two additional Salmonella tester strains have been used in studies of ACN mutagenesis. Strain TA102 was constructed to contain multiple copies of a mutant hisG428 allele on a plasmid – the normal chromosomal gene having been deleted (Mortelmans and Zeiger Citation2000). The mutant codon in hisG428 is the ocher TAA stop codon. Therefore, unlike all other Salmonella tester strains thus far considered, reversion mutations are at T and/or A Strain TA102 does not contain the uvrB deletion and is therefore DNA excision repair competent. It is considered to be particularly sensitive to reverse mutations due to oxidative DNA damage and DNA cross-linking (Mortelmans and Zeiger Citation2000). Three studies (one in three different laboratories) have assessed ACN’s mutagenicity for TA102, with all reporting essentially negative results with or without EMA (Baker and Bonin Citation1985; Matsushima et al. Citation1985; Jung et al. Citation1992), although the Baker and Bonin (Citation1985) study indicated a weak but inconsistent positive result in one test in the absence of EMA. Plate and pre-incubation assays were employed with ACN concentrations at 5000 and 10,000 µg/plate. Later cooperative studies in three independent laboratories were also reported as negative with ACN concentrations up to 5000 µg/plate (Müller et al. Citation1993). However, the most recent study designed to explore discordant results in the bacterial reverse mutation ocher TAA stop codon reported positive results for ACN, again in plate assays with concentrations up to 5000 µg/plate (Stankowski et al. Citation2019).
Salmonella tester strain TM677 is the only strain in this series that measures forward mutations. The target is the XPRT gene and mutants are selected based on their purine analogue resistance (Skopek et al. Citation1978). There are many mutable sites in XPRT with a variety of mutational events being potentially recoverable, making this a “large-target” tester strain. TM677 has the uvrB deletion, making it deficient in DNA excision repair. The single study of ACN mutagenicity reported a positive response, but only in the absence of EMA, at an ACN concentration of 500 µg/ml (Liber Citation1985). Although reproducible, the increase in mutants per surviving cells was observed at only one ACN concentration and there was no net increase in mutants over controls.
As oxidative DNA damage may produce mutagenic DNA adducts (discussed above), results of an early independent study of hypothiocyanite in Salmonella is relevant (White et al. Citation1983). Tester strains TA1535, TA1537 and TA1538 were incubated directly with hypothiocyanite up to toxic concentrations. No increases in mutants over background were observed in any strain. Note that TA1535 gave positive responses when exposed to ACN, suggesting that mutation induction in those studies was not due to oxidative DNA damage resulting from reduction of anti-oxidant defenses.
Eukaryotic microorganisms ()
Due to their higher level of genetic organization compared to bacteria, eukaryotic microorganisms allow assessments of mutations at the gene and chromosomal level. Both forward and reverse point (single gene) mutations are detectable in one or several genes, depending on the fungal strain (Parry Citation1985). Several strains can also detect larger scale genetic events such as intra-chromosomal recombination, manifest as either mitotic crossing-over (generating reciprocal events at mitosis) or gene conversion (generating non-reciprocal events), or numerical changes in chromosomes (aneuploidy) (Parry Citation1985). Large scale mutational events involving several loci are likely to be of greater carcinogenic significance than are point mutations (Schiestl, Gietz, et al. Citation1989; Schiestl, Reynolds, et al Citation1989; Turker et al. Citation1999; Meijer Citation2005; Duesberg et al. Citation2006). A phenomenon termed illegitimate mating, which results from mating-type switching inducible by exposures to mutagens, is also detectible in some fungal strains (Parry Citation1985).
Both Arni (Citation1985) and Parry and Eckardt (Citation1985a) failed to find reverse isoleucine mutations in Saccharomyces cerevisiae strain D7 cells, either in the presence or absence of EMA at ACN concentrations of 5000 and 200 µg/ml, respectively. Similarly, no increases in reverse mutations were found for histidine revertants in Saccharomyces cerevisiae strain RM52 by Mehta and von Borstel (Citation1985) or for adenine frameshift revertants in strains PV2 and PV3 by Inge-Vechtomov et al. (1985), either in the presence or absence of EMA at ACN concentrations of 0.8 and 800 µg/ml, respectively. By contrast, positive responses were reported for adenine and isoleucine reverse gene mutations in Saccharomyces cerevisiae strains D6 and D61-M, either without (D6) or with and without EMA (D61-M) at an ACN concentration of 20 µg/ml (Parry and Eckardt Citation1985b). Similarly, Mehta and von Borstel (Citation1985) reported positive results for isoleucine revertants and either arginine or tryptophan revertants in strains D7-144 and XV185-14C, respectively. The results in D7-144 were seen at an ACN concentration of 8.1 µg/ml and occurred in the presence or absence of EMA, while those in XV185 were seen at 0.8 µg/ml and occurred only without EMA.
Results of forward mutations in Saccharomyces cervisiae strain PV1 at a lysine gene were reported as negative, with or without EMA, at an ACN concentration of 800 µg/ml (Inge-Vechtomov et al. 1985). Similarly, increases in forward mutations were not observed for any of six adenine genes in Schizosaccharomyces pombe, either in the presence or absence of EMA, at an ACN concentration of 250 µg/ml (Loprieno et al. 1985). Forward mutations were induced, however, in mitochondrial genes in Saccharomyces cerevisiae strain D5 (“petite” mutations) in the absence of EMA, at an ACN concentration of 30 µg/ml (Ferguson Citation1985).
Rizzi et al. (Citation1984) treated Schizosaccharomyces pombe with ACN at various doses with and without EMA and reported significant increases in forward mutations (locus not specified) at low concentrations, i.e. less than 10 µg/plate, with a greater effect in the presence of EMA.
Compared with the mixed results for gene mutations in fungal studies, those for chromosome level mutations have been generally positive, except in Saccharomyces cervisiae strains PV2 and PV3, where no increases in gene conversion events either with or without EMA were seen at an ACN concentration of 800 µg/ml (Inge-Vechtomov et al. 1985). Increases in gene conversion events were reported, however, for strains JD-1, D7, D7-144, PV4a and PV4b, by Brooks et al. (Citation1985), Parry and Eckardt (Citation1985a), Arni (Citation1985), Mehta and von Borstel (Citation1985) and Inge-Vechtomov et al. (1985), respectively, at ACN concentrations of 250, 20, 25, 0.8 µg/ml and over a range of 1–800 µg/ml, respectively. The positive results in strain JD-1 required EMA while those in strains D7 and D7-144 occurred only in the absence of EMA. The positive results in strains PV4a and PV4b occurred in the presence and absence of EMA.
Mitotic crossing-over leading to mitotic segregation was reported for Saccharomyces cerevisiae strains D61-M by Zimmermann et al. (Citation1985) and strains D6 and D61-M by Parry and Eckardt (Citation1985b) at ACN concentrations of 199 and 20 µg/ml, respectively. Zimmermann et al. (Citation1985) obtained their results in the absence of EMA while Parry and Eckardt (Citation1985b) found theirs with or without EMA.
ACN induced aneuploidy resulting in an abnormal number of chromosomes in a cell was reported for Saccharomyces cerevisiae strain D6, but only in the presence of EMA, following exposure of cells to an ACN concentration of 20 µg/ml (Parry and Eckardt Citation1985b). This same laboratory also reported induction of aneuploidy in strain D61-M, in the presence and absence of EMA, but at a higher ACN concentration of 200 µg/ml (Parry and Eckardt Citation1985b). This finding in D61-M was not replicated, however, by Zimmermann et al. (Citation1985), who failed to find induction of aneuploidy in this strain at an ACN concentration of 199 µg/ml. A later study also reported a negative test for aneuploidy in strain D61M exposed to 2290 µg/ml ACN in the absence of EMA (Whittaker et al. Citation1990). Studies in another fungal species, however, Aspergillus nidulans, did exhibit induced aneuploidy/non-disjunction at an ACN concentration of 806 µg/ml in the absence of EMA (Carere et al. 1985).
Because of the strong association of gross genomic rearrangements with cancer, a strain specially constructed to select for intra-chromosomal recombination events in Saccharomyces cerevisiae was introduced by Schiestl, Gietz, et al. (Citation1989) and Schiestl, Reynolds, et al. (Citation1989). A mutation test based on this strain was termed the deletion (DEL) assay. It was subsequently reported that agents that induce mutations via oxidative damage frequently do so by inducing intra-chromosomal rearrangements and are efficiently detected by the DEL assay (Brennan et al. Citation1994). An evaluation of ACN mutagenicity employing the DEL assay then found both intra-chromosomal recombination events and deletions, with and without EMA, at an ACN exposure concentration of 300 µg/ml (Carls and Schiestl Citation1994).
Cultured mammalian cells ()
ACN’s mutagenicity has been evaluated in studies using cultured mammalian cells, including human cells. As the genetic organization in mammalian cells is more closely related to that in human cells, it is assumed that genotoxicity results will be of greater relevance for risk assessment than are results from studies in lower organisms. However, adaptation of mammalian cell lines to continuous culture conditions has selected for relaxation of normal genetic control mechanisms, e.g. many cell lines are deficient in alkyl guanine transferase DNA repair; L5178Y mouse lymphoma cells carry a mutation in the P53 tumor suppressor gene and are sensitive to oxidative stress; human TK6 cells are relatively deficient in recombination and repair of double strand breaks; the TK6 derived AHH-1 also has a mutation in P53 (Alexander Citation1961; Evans et al. Citation1987; Pegg Citation1990; Morris et al. Citation1996; Clark et al. Citation1998; Bouzyk et al. Citation2000; Szumiel Citation2005). Conditions in vitro may also influence mutagenicity test results such as transient hypoxia, low-pH and high osmolarity which themselves have genotoxic effects (Rice et al. Citation1986; Cifone Citation1987; Galloway et al. Citation1987). Cells cultured in vitro experience ambient oxygen concentrations much higher than those in vivo, increasing the potential for ROS production and DNA damage (Wang CY et al. Citation2013 and references therein).
Most in vitro mammalian gene mutation assays have employed L5178Ymouse lymphoma cells that are heterozygous for the thymidine kinase gene (Tk). Forward mutations of the Tk gene from its heterozygous state (Tk±) in L5178Y tester cells produce mutant homozygous null (Tk-/-) cells that are selected by the pyrimidine analogue trifluorothymidine, which is lethal to cells with proficient TK activity. There are two kinds of Tk-/- mutant colonies, i.e. large, and small or slow-growing, the former thought to signify small or point mutation and the latter large or chromosome level mutations (Moore MM and Doerr Citation1990). Although adequate testing using L5178Y Tk± cells now requires enumeration of both large and small colonies, this was not reported for any of the ACN mutagenicity studies reviewed here.
An early study of ACN’s mutagenicity at the Tk locus in L5178Y cells was reported as negative at concentrations ranging from 0.0005 to 0.01% with or without EMA (Litton Bionetics Citation1976). Later, however, Rudd (Citation1983) reported an increase in the Tk-/- mutant frequency (MF) for L5178Y mouse lymphoma cells incubated with and without EMA at an ACN concentration of 10 µg/ml. Similarly, four groups that studied ACN’s mutagenicity at the Tk locus in L5178Y cells in the IPCS CSSTT exercise (Ashby 1985) reported positive results, with and without EMA (although one group did not add an activating system) (Oberly et al. Citation1984; Amacher and Turner Citation1985; Lee and Webber Citation1985; Myhr et al. Citation1985). These positive responses were elicited at ACN concentrations of 200, 20, 40, and 30 µg/ml, respectively, in the different studies. A fifth study in L5178Y cells in this series reported an inconclusive result at the Tk locus (Styles et al. Citation1985). A final study of ACN at the Tk locus in L5178Y cells as part of an evaluation of two different criteria for positivity for mutagenicity in that assay – i.e. a so-called two-fold rule and a newer ICH guideline – showed that the judgment of ACN’s mutagenicity in this assay differed according to the criteria employed (Oberly et al. Citation1996). ACN was considered positive at 40 µg/ml with EMA by the two-fold criterion. This was a concentration that allowed 10% relative growth of the cells. Of note, ACN would have been considered negative for mutagenicity by the newer proposed criterion.
L5178Y cells were used to detect mutations at the ouabain locus in two studies in the IPCS CCSST series, and both reported negative results with or without EMA at ACN concentrations of 200 and 100 µg/ml (Garner and Campbell Citation1985; Styles et al. Citation1985). A weak positive result was reported in a study of Hprt mutations in these cells, with and without EMA, at an ACN concentration of 200 µg/ml (Garner and Campbell Citation1985).
A study of Hprt mutations in Chinese hamster fibroblast V79 cells was reported as negative with and without EMA at an ACN concentration of 200 µg/ml (Lee and Webber Citation1985). However, Tk -/- mutants were induced by ACN at a concentration of 161 µg/ml in P388 mouse lymphoma cells in the presence, but not in the absence, of EMA (Anderson and Cross Citation1985), while ouabain resistant mutants were induced by ACN 0.76 mM with addition of EMA in Balb/c-3T3 mouse cells (Matthews et al. Citation1985).
The most commonly used human cell line for in vitro mutagenicity testing is TK6, which can detect both forward mutations in the X-chromosomal HPRT gene and at the heterozygous TK locus. Crespi et al. (Citation1985) reported induction of Tk -/- mutants in these cells with but not without EMA at an ACN concentration of 40 µg/ml while, in the same study, HPRT mutations were induced by 25 µg/ml without EMA in the derivative AHH-1 cell line (Crespi et al. Citation1985). Recio and Skopek (Citation1988) used a concentration of 1.4 mM ACN (75 µg/ml) to induce a weak mutagenic response of the TK gene in TK6 cells only with EMA, while CNEO at a concentration of 100 or 150 µM (with and without EMA) produced significant dose-related increases of approximately 5- and 10-fold, respectively, above the spontaneous background TK mutant fraction (Recio and Skopek Citation1988). Later, Recio et al. (Citation1990) also demonstrated a nearly 12-fold increase in the frequency of HPRT mutations in TK6 cells by 150 µM CNEO exposure.
Despite these unambiguous demonstrations of induction of both TK and HPRT mutations in TK6 cells, it is relevant (see above) that there was no increase in N7OEG DNA adducts in TK6 cells at 100 µM CNEO for 2 h (Walker, Fennell, et al. Citation2020).
Recio and Skopek analyzed the TK -/- mutants isolated from the CNEO exposed TK6 cells (mostly induced mutants) and found that most displayed a normal growth rate (Recio and Skopek Citation1988). Molecular analyses of these isolates demonstrated that almost all contained the normal sized Tk 14.8 kb polymorphic fragment, suggesting that they arose from point mutations rather than from large deletions. By contrast, almost all of the slowly growing TK -/- mutant colonies they studied had lost this wild-type band, indicating deletion mutations. However, the conclusion of the study authors, that CNEO induces mostly point mutations, may have been erroneous because the conditions of outgrowth for phenotypic expression by the TK6 cells following their exposures to CNEO in this experiment may have favored overgrowth of normally growing colonies over slowly growing ones. Thus, it may not have been that more normal growth mutants (resulting from point mutations) were induced; it may have been that more normal growth mutants were recovered. This phenomenon is suggested by the increase in the ratio of slowly growing to normally growing cells at the highest concentration of CNEO used in these studies, which was a toxic concentration inhibiting growth of all classes of mutants. A subsequent cytogenetic study of slowly growing TK -/- mutants failed to find a good correlation between cytogenetic abnormalities and the slow-growth status, although some chromosome changes were associated with the CNEO exposures (Kodama et al. Citation1989).
The HPRT mutations induced by CNEO in TK6 cells were later sequenced (Recio et al. Citation1990). Nineteen of 39 mutant isolates studied showed base substitution mutations, 11 in AT base pairs and 8 in GC base pairs. Two −1 frame-shift mutations were seen, both involving GC base pairs. Most of the remaining mutant isolates showed cDNA alterations compatible with splice-site changes, and Southern blot analyses confirmed this in four of five mutants. Two mutants studied for the nature of the splice site change showed point mutations, one transition and one transversion, at sites involving AT base pairs.
ACN induced chromosome level mutations have also been observed in mammalian cells. Early studies of chromosome aberration (CA) induction in vitro in human PBLs or Chinese Hamster DON-6 cells gave a negative result for the PBLs (but testing was only without EMA at an ACN concentration of 5.3 µg/ml) and a weak positive for the DON-6 cells (Sasaki et al. Citation1980; Cerna et al. Citation1981). Chromosome level mutations were repeatedly assessed in the IPCS CCSST 1985 series. MN were induced in CHO cells in the presence and absence of EMA, at an ACN concentration of 1600 µg/ml (Douglas et al. Citation1985). CA also were induced in CHO cells with and without addition of EMA at 1.0 mM ACN (Natarajan et al. Citation1985). Another group of investigators however, found only a weak positive CA response in CHO cells at a much lower exposure concentration only in the presence of EMA, using 100 µg/ml ACN (Gulati et al. Citation1985). There was also induction of CA in Chinese Hamster Lung (CHL) cells, but only with EMA, at ACN concentrations of 18 and 6.2 µg/ml, respectively, in studies by Ishidate et al. (Citation1981) and Ishidate and Sofuni (Citation1985). CA were also evaluated in a study by the Japanese Ministry of Labor in CHL fibroblasts at a range of ACN concentrations (Asakura et al. Citation1994). Significant increases in aberrations were observed at ACN concentrations of 25 µg/ml and above or 20 µg/ml and above following 24 or 48 h incubation in the absence of EMA, respectively, and at 60 µg/ml or 40 µg/ml without or with EMA, respectively, after 6 h incubation. An increased frequency of CA was observed at an ACN concentration of 12.5 µg/ml in a Chinese Hamster liver fibroblast line (CH1-L) in the absence of EMA (Danford Citation1985). However, there were no increases in aberrations in an earlier study that exposed rat liver RL4 cells to 10 µg/ml ACN in the absence of EMA (Priston and Dean Citation1985). All studies of numerical CA (aneuploidy) in cultured mammalian cells have been negative; there was no increase in aneuploidy in CHL cells treated with 25 µg/ml ACN without EMA (Danford Citation1985), no increase in polyploidy in rat liver RL4 cells treated with 10 µg/ml ACN without EMA (Priston and Dean Citation1985) and no increase in spindle damage in CH1-L cells treated with 25 µg/ml ACN, again in the absence of EMA (Parry et al. 1985b).
In vivo in rodents (Table S4)
Studies of mutations at either the gene or chromosomal level arising in vivo are of most relevance for estimating the carcinogenic potential of a genotoxic agent that induces cancer by a mutagenic MOA. Such events demonstrate not only that the agent of concern has penetrated to critical targets but that it and/or its biologically active metabolites have escaped host protective mechanisms to produce the kinds of irreversible genetic effects that alter genetic information content. Studies of in vivo mutations also incorporate effects due to metabolism. In vivo studies have evaluated the mutagenic potential of ACN for both gene and chromosome level events in somatic cells.
In vivo mutations of the Hprt gene have been assessed in both mice and rats exposed by gavage or in drinking water to ACN (Walker, Walker, et al. Citation2020; Walker, Fennell, et al. Citation2020). Male B6C3F1 mice (6 weeks of age) given 1.2, 4.8, or 9.6 mg/kg by gavage 5/days per week for 90 days were studied 24 h after the last day of dosing. There were no significant differences between average Hprt mutant frequencies (MFs) in splenic lymphocytes at any treatment level compared to control background levels. However, in an extension of these studies to investigate the role of epoxidation of ACN via P450 CYP2E1 to CNEO on the mutagenic effects, normal wild-type (WT) female B6C3F1 and CYP2E1 knock-out (deficient in the ability to metabolize ACN to CNEO = null) mice (6 weeks-of-age) were exposed to 0, 2.5 (wild-type mice only), 10 (wild-type mice only), 20, or 60 (knock-out mice only) mg ACN/kg/day by gavage 5 days/week for 6 weeks, with necropsy 24 h post-dosing. In wild-type mice, Hprt MFs significantly increased by pair-wise comparison in splenic lymphocytes only in the 20 mg/kg group but a plot of the daily dose of ACN versus the average induced MFs (induced MF = observed treatment MF minus background MF) showed a significant linear dose-response for mutation induction over the entire dose range. In the CYP2E1 deficient mice, the Hprt MF was also significantly increased by three-fold over background but only at the 60 mg/kg/day dose level. A bolus dose of 60 mg/kg/day was lethal to the WT but not the null mice, suggesting lethality is attributable to an oxidative metabolite(s) of ACN (e.g. CNEO, cyanide).
The dose-response for induction of Hprt and lacZ mutations was also evaluated by these authors in male Muta-Mouse transgenic mice (73–76 days-of-age) exposed for 28 days in drinking water to ACN at 100, 500, or 750 ppm (estimated maximal average daily doses being 8, 58, and 84 mg/kg, respectively, and estimated cumulative doses being 505, 1,620, and 2,350 mg/kg, respectively) (Walker, Walker, et al. Citation2020). Daily doses of 58 and 84 mg ACN/kg were not lethal to the transgenic mice in this study because they were delivered over time as opposed to the bolus delivery used in comet assay study by these same investigators (described below; Walker, Walker, et al. Citation2020). Following a 49-day post-exposure expression period, only 500 or 750 ppm ACN caused significant increases in Hprt MFs in splenic lymphocytes although a plot of average daily ACN doses versus average induced MFs again revealed a significant linear dose-response. LacZ gene mutations showed no significant differences between lacZ MF values in control versus exposed mice at any exposure dose in splenic lymphocytes, bone marrow, brain, lung, or testis (Lambert et al. Citation2005; Walker, Walker, et al. Citation2020).
A dynamic study of Hprt mutation induction, recovery and persistence in T-lymphocytes following ACN drinking water exposures of female F344 rats was conducted to determine the effect of exposure duration on Hprt MFs in T-lymphocytes from thymus and spleen, the effect of time elapsed after ACN exposure on these Hprt MFs and the dose-response for Hprt MFs in splenic T-cells at the time of maximum mutagenic response beginning at 5 weeks-of-age (Walker, Fennell, et al. Citation2020). In rats exposed to 33, 100, or 500 ppm ACN in drinking water, the estimated average daily doses were 8, 21, and 75 mg ACN/kg, respectively. The accumulations of T-cell Hprt mutants in both thymus and spleen in rats necropsied two weeks after 0, 1, 2, 3, or 4 weeks of exposure to 0 or 500 ppm ACN showed significant MF increases that were directly related to the duration of exposure. Hprt MFs in thymus reached their maximum after 2 weeks in animals necropsied 0, 2, 4,6 or 8 weeks after 4 weeks of exposure to 500 ppm ACN (i.e. treated MFs of 6.1 ± 0.5 × 10−6 versus control MFs of 1.9 ± 0.2 × 10−6) and then decreased to background levels. In spleen, MFs were significantly elevated at the end of the 4-week exposure period, increasing in a linear fashion to reach a maximum at 4 weeks post-exposure (i.e. 11.1 ± 2.7 × 10−6 versus control MFs of 2.6 ± 0.4 × 10−6) and then declining to lower but still elevated levels. The dose response for splenic lymphocytes when necropsy at the time post-exposure of maximum mutagenic response for exposures of 0, 33, 100 or 500 ppm was 2.6 ± 0.4 × 10−6, 3.4 ± 0.3 × 10−6 (p = 0.071), 5.1 ± 0.6 × 10−6 (p = 0.036), and 11.1 ± 1.2 × 10−6 (p = 0.018) respectively, demonstrating a significant linear dose-response (p < 0.0001), with the 500 ppm response greater than the 33 or 100 ppm responses.
In addition to this gene level mutagenicity study, several in vivo studies of chromosome level events in rodents were reported between 1978 and 2001. The earliest is unpublished from The Dow Chemical Company in which Sprague Dawley rats were administered ACN by inhalation at concentrations up to 500 ppm for 90 days with no increase in chromosome aberration (CA) in bone marrow of treated animals (Johnston et al. Citation1978 as reported in WHO Environmental Health Criteria [EHC] 28 1983). In a latter study, CA frequencies were not increased over controls in bone marrow cells of Swiss mice receiving ACN orally at doses of 7, 14 and 21 mg/kg/day for 4, 15, or 30 days or following i.p. injections of 10, 15, or 20 mg/kg/day for this same period (Rabello-Gay and Ahmed Citation1980). Similarly, 16 daily oral ACN doses of 40 mg/kg/day failed to increase CA frequencies in bone marrow cells of rats in this same study (Rabello-Gay and Ahmed Citation1980). Leonard et al. (Citation1981) investigated induction of CA in bone marrow cells in NMRI mice following a single i.p. injection of ACN at 20 or 30 mg/kg and found no increases (Leonard et al. Citation1981). Similarly, a study that assessed CA in bone marrow cells in mice exposed to ACN by inhalation at levels up to an equivalent of 100 mg/m3 was reported as negative (Zhurkov et al. Citation1983). Sharief et al. (Citation1986), in the same study that investigated SCE frequencies described above, likewise failed to find increases in CA frequencies in the bone marrow cells of C57BL/6 mice at an ACN exposure level of 30 mg/kg by single i.p. injection. A Russian study in which ACN was administered to BALB/c and C57Bl/6 mice, 5 or 10 mg/kg for single or 20 mg/kg for repeated oral doses, or 10 mg/kg by single or repeated i.p. injections, failed to induce an increase in CA frequencies over controls (Nesterova et al. Citation1999). However, co-administration of the calcium channel blocking agent verapamil with ACN was reported as producing a slight clastogenic effect, but the underlying mechanism(s) is/are unknown. Fahmy (Citation1999) did find dose dependent increases of CA (mostly chromatid type) in bone marrow and spleen cells of Swiss mice receiving ACN doses of 7.75, 15.5, or 31 mg/kg by gavage for 5 days (Fahmy Citation1999).
Chromosome level mutational changes reflected as MN in vivo are frequently assessed in red blood cells (erythrocytes), either those recently released from the bone marrow (polychromatic erythrocytes [PCE] or reticulocytes) or in erythrocytes that have been in the peripheral blood for some time (normochromatic erythrocytes [NCEs]). Both reflect chromosome level mutational effects that have occurred in the bone marrow, the former as acute effects and the latter as chronic. MN in PCE in blood can only be usefully assessed in mice as rats normally clear such cells from the peripheral circulation.
The early report of Leonard et al. (Citation1981) that failed to find induction of CA in NMRI mice receiving ACN i.p. 20 or 30 mg/kg also failed to find induction of MN PCE in bone marrow as assessed at several time points (Leonard et al. Citation1981). Somewhat later, a study from Japan in which ACN 20 mg/kg was administered by a single i.p. injection to mice also failed to find an increase in bone marrow MN PCE (Hachiya Citation1987). Sampling times are unknown in this study.
Two reports from the Collaborative Study Group for the Micronucleus Test, Mammalian Study Group. Environmental Mutagen Society of Japan (CSCMT,MMS,JEMS) describe more inconclusive results for MN PCE induction (Morita et al. Citation1997; Wakata et al. Citation1998). In the first, results were negative in both bone marrow and peripheral blood of CD1 mice treated either orally, i.p. or i.v. with ACN at various doses up to 80% of the LD50 dose but equivocal in bone marrow while negative in peripheral blood of Sprague-Dawley rats at doses of 40 mg/kg i.p. or 98 mg/kg orally (Morita et al. Citation1997, 6th Collaborative study). The second report focused on Sprague Dawley rats only in which MN PCE were induced in bone marrow cells at a dose of 124.8 mg/kg but not in peripheral blood at a dose of 125 mg/kg, both administered i.v. (Wakata et al. Citation1998, 9th Collaborative Study).
The last two reports of chromosome level mutations focused on the potential induction of MN in normochromatic erythrocytes (MN NCEs) or polychromatic erythrocytes (MN PCEs) in male and female B6C3F1 mice. The first study was part of a dose-range finding experiment in preparation for the NTP carcinogenesis studies of ACN in mice, and no increase in the frequencies of MN NCEs was observed in peripheral blood samples from male and female mice administered ACN at doses of 0, 5, 10, 20, or 40 mg/kg/day by gavage, 5 days/week, for 14 weeks (NTP Citation2001). The second study was nested in an epoxidation study described above, which also measured induction of MN (Walker, Walker, et al. Citation2020). Female WT mice given 0 or 20 mg ACN/kg/day and null mice given 0 or 60 mg ACN/kg/day by gavage, 5 days per week for 4 weeks showed no increases in the frequencies of MN NCEs or MN PCEs in either the WT or null mice.
Human studies (Table S5)
In vivo genotoxicity in humans is reflected in epidemiological studies by biomarkers that may identify exposure to an agent of concern, its effect or an unusual susceptibility. A large literature is available describing biomarkers of exposure to ACN. Measurements of the parent compound or its metabolites in blood and/or urine detect acute exposures while ACN/CNEO’s avid binding to proteins has been exploited to develop methods utilizing hemoglobin (Hb) adducts as internal molecular dosimeters reflecting cumulative internal doses.
As human exposures are rarely pure and limited to a single agent, confounding exposures often limit the interpretation of biomarkers of effect, in particular mutations, which are inherently nonspecific as to causation. Inclusions of well validated biomarkers of exposure enhance confidence in interpreting studies of genotoxic effect biomarkers. However, ACN’s biomarkers of exposure are not themselves considered further in this review. Biomarkers of susceptibility such as GST genotypes have shown no consistent relationship to ACN internal doses resulting from external exposures and are likewise not considered here.
ACN’s potential for inducing mutations at either the gene or chromosome level in exposed humans was investigated in Hungarian workers exposed to ACN and dimethylformamide (DMF). Two groups of workers – maintenance and fiber producers, with 13 individuals per group – were studied for HPRT mutations in PBLs at the onset of a new manufacturing technology in the plant, and at seven and 20 months thereafter (Major et al. Citation1998). All exposed workers, however, had three to 10 years previous exposures to unknown concentrations of ACN/DMF. Two control groups were included; a cumulative non-industry control group that consisted of 26 non-exposed individuals and an industry non-exposed group that consisted of six plant workers. Potentially confounding co-exposures, including ionizing radiation were assessed by questionnaires. Smoking status was assessed by questionnaires and by thiocyanate concentrations in serum.
Worker exposures to ACN/DMF were monitored by measuring ambient air and by urine ACN and MMF (monomethylformamide as a measure of DMF) concentrations for individual workers. ACN and DMF concentrations in ambient air ranged from 0 to 17.6 and 0.6 to 23.0 mg/m3 for ACN (0–8.1 ppm) and DMF, respectively. ACN and MMF urine concentrations were also high before and after work shifts, with greater elevations in after shift samples. Urine concentrations of both agents were greater in fiber producers than in maintenance workers. Approximately half of each worker group (control and exposed) smoked cigarettes.
HPRT gene mutations were measured in this study using an autoradiographic assay that scores PBLs that are resistant to 6-thioguanine (6-TG) inhibition of 3H-thymidine incorporation into DNA during first-round synthesis in vitro. Its underlying rationale is that cells that become labeled with 6-TG have lost function of the HPRT enzyme and that this loss is due to mutation of the HPRT gene. In the Major et al.’s (Citation1998) study, mean HPRT variant frequencies (VFs) were reported to be significantly elevated in both the maintenance and fiber producer ACN/DMF worker groups compared to cumulative and industrial controls at the onset of study. Seven months into the study, the HPRT VFs had fallen in the maintenance workers but not in the fiber producers, although mean VFs in both exposed worker groups remained significantly elevated compared to mean values in both control groups. Smoking was associated with increases in HPRT VF values.
There have been two additional studies of biomarker responses possibly related to gene mutations in ACN-exposed workers. Forty-nine workers in the Czech Republic occupationally exposed to ACN 0.05–0.3 mg/m3 (0.02–0.14 ppm) were evaluated for plasma levels of both p53 and p21WAF1 tumor suppressor proteins (Rössner et al. Citation2002). The worker group included males and females, smokers and nonsmokers. No differences were found in concentrations of either protein as compared to a control group of 24 age-, gender-, and smoking-matched non-exposed controls. For comparison, elevated p53 levels in blood were found in other studies of vinyl chloride, PAH or asbestos exposed workers but not in those exposed to uranium (Krajewska et al. Citation1998; Luo JC et al. Citation1999; Schneider et al. Citation1999).
A study in China reported on mitochondrial deletion frequencies in a group of 47 ACN-exposed workers compared to similar deletion frequencies in 47 non-industry age-matched controls (Ding et al. Citation2003). The geometric mean ACN exposure level for the workers was 0.25 mg/m3 (0.12 ppm). Details of exposure assessment are not provided, but this appears to be the same group in the same industry for which MN frequencies (described below) were reported (Fan et al. Citation2006), in which case ACN levels were measured by periodic air sampling between 1997 and 1999. There is no mention of confounders. Mitochondrial deletions were found in eight of the 47 ACN-exposed workers and in none of the controls. For comparison, mitochondrial deletions were found in three of 12 elderly controls not exposed to ACN but in none of 12 young controls, similarly not exposed. The authors conclude that ACN exposures accelerate the mitochondrial deletions associated with aging.
The earliest reported cytogenetic study evaluated 18 ACN-exposed workers with greater than 15 years of exposure (Thiess and Fleig Citation1978). Average ACN workplace air concentration was 1.5 ppm, as measured monthly for approximately two years, but the authors suggested that actual personal exposures may have been greater. Workplace regulations required that respirators be used whenever ACN concentrations in air exceeded 20 ppm. CA frequencies in PBLs were similar for the 18 workers and 18 matched controls leading to the authors’ conclusion that ACN did not induce CA.
Almost 20 years later a second two-part study that measured a variety of biomarkers, including CA and SCEs, in three groups of Portuguese workers arrived at a different conclusion (Borba et al. Citation1996). In a separate report, these authors measured hemoglobin adduct concentrations in nonsmokers in the same three worker groups, i.e. in controls, in maintenance workers and in polymerization workers, as measures of cumulative ACN exposures (Tavares et al. Citation1996). As expected, both worker groups had greater hemoglobin adduct concentrations than did controls while those for the maintenance and polymerization workers showed no real cumulative exposure difference between them. As regards the cytogenetic changes, the mean CA frequency was significantly greater only for the maintenance workers; frequencies for the polymerization workers were not significantly elevated over controls. Although reported as positive for ACN induction of CA, it seems that occupational ACN as reflected by hemoglobin adduct concentrations is insufficient per se to explain these cytogenetic results. As noted below, SCE frequencies did not differ between the worker groups.
It is noteworthy, however, that the inter-group cytogenetic differences in this study correlate with a simultaneously determined biomarker of oxidative stress (Borba et al. Citation1996). Erythrocyte malonaldehyde-complex (MDA) levels were determined to assess lipid peroxidation. The maintenance worker group with elevated CA frequencies showed levels of MDA that were significantly higher than either the control or the polymerization worker group. Additional genotoxic endpoints studied in these workers included urine tests for mutagenicity in the Salmonella TA98 tester strain, using a source of EMA, which were negative for all workers (Borba et al. Citation1996). There was no evidence for ACN induction of hepatic CYP450 enzyme activity as measured by D-glucaric acid in urine (Hunter et al. Citation1971).
The HPRT study of Hungarian workers exposed to a combination of ACN and dimethylformamide (DMF) described above also reported positive results for CA in PBLs (Major et al. Citation1998, Citation1999). Exposure data for the two worker groups (maintenance and fiber production) are given above with the results of the accompanying HPRT studies. An unexpected finding in the cytogenetic portion of the study was that CA mean frequencies for different classes of aberrations were significantly elevated in the non-exposed industry control group compared to means in the cumulative non-industry control group at the onset of study. Worker mean CA frequencies at study onset, however, were higher than those in both control groups – significantly so when compared to the non-industry controls. At the seven- and 20-month study periods, aberration frequencies in both exposed worker groups rose significantly over both control groups, with elevations being greater in the fiber producers compared to maintenance workers, in accord with the relative urine ACN and MMF concentrations in the two worker groups. Many of the CA in this study were of the chromosome type (dicentric and ring chromosomes) with these aberration types being significantly increased in the ACN/DMF exposed workers at 20 months. A somewhat derivative chromosome level study of some of these same workers evaluated the induction of premature centromere division (PCD) by the ACN/DMF exposures (Major et al. Citation1999). This study reported negative results, i.e. no association with ACN/DMF exposures.
In addition to HPRT VF values and CA frequencies in this study, SCEs were significantly increased in industrial controls versus the non-industry cumulative controls at the onset, while both SCEs and high frequency cells (cells with high numbers of SCEs) were significantly increased in both exposed worker groups compared to cumulative controls at seven and 20 months. The lymphocyte proliferation index was significantly greater in both worker groups than in the cumulative non-industry controls at all time points in the study, while UV-stimulated UDS (determined by scintillation counting) was significantly elevated in exposed workers versus cumulative controls at seven months.
A potential confounder in the Hungarian study is clinical illness in several of the exposed workers who showed serious abnormalities of hematological and/or liver function tests (Major et al. Citation1998). Six of the 26 workers in both ACN/DMF exposed groups required hospitalization, and several others reported symptoms. Illnesses were attributed to high DMF inhalation exposures.
Later cytogenetic studies of ACN exposed workers in the Czech Republic measured abnormalities on two occasions in the same sets of workers using both conventional techniques and fluorescence in situ hybridization (FISH) (Srám et al. Citation2004). The first conducted in the year 2000 assessed 45 exposed workers, 23 matched controls living in the same region as the workers (controls I), and 33 controls from Prague (controls II). Stationary monitoring of ambient air in the workplace showed ACN concentrations ranging from 0.05 to 0.30 mg/m3 (0.01–0.14 ppm) in the three-month period preceding blood sampling. Although the authors state that there were no significant differences in mean aberration frequencies among the three subject groups as determined by conventional techniques, in the paper shows a significant increase in mean CA frequencies in control group I individuals, especially among nonsmokers. By comparison, FISH analysis showed a significant increase in mean aberration frequencies in the exposed workers and control group I compared to control group II. Stable CA determined by FISH was significantly correlated with age (positive) and plasma levels of vitamin E (negative). The authors concluded that ACN, at this exposure level, did not induce CA and that the results observed in control I individuals resulted from confounding agents in the region.
Table 2. ACN/CNEO associated specific DNA adducts.
A 2003 study in the same industry evaluated 22 potentially exposed workers and an unspecified number of controls (Beskid et al. Citation2006). It is unclear from these reports if the 22 workers were new additions to the study or if they had also been studied in 2000. In any case, average ACN workplace concentrations were now recorded at 0.05–0.70 mg/m3 (0.02–0.32 ppm). Mean CA frequencies were now significantly elevated in the exposed worker group compared to the control group when determined by conventional techniques while the FISH analysis again failed to observe any such differences. Cytogenetic results were not associated with smoking while aberration frequencies determined by FISH again showed age as a significant confounder.
Patterns of chromosome aberrations as revealed by FISH were analyzed for both Czech studies, i.e. for 39 workers studied in 2000 and 22 workers studied in 2003 (Beskid et al. Citation2006). Although, as reported, there was no significant increase in the overall mean frequencies of cells with aberrant chromosomes as revealed by FISH analysis in the ACN-exposed workers compared to controls, within the category of all FISH aberrations, these workers showed increased frequencies of chromosome translocations compared to industry controls. This finding was, however, attributed to the older age of the workers. Patterns of other CA, and the chromosomes involved in translocations, compared to controls showed some statistical associations with ACN by regression analyses, i.e. an increase in the relative number of reciprocal translocations, of insertions and in the relative number of cells with abnormalities in chromosome 4 but a decrease in the relative number of cells with abnormalities in chromosome 1. The authors suggest that pattern changes may imply an ACN effect. As expected, and in contrast to the Hungarian study, the relative frequency of dicentric chromosomes, as a measure of chromosome type aberrations, was not increased but relatively decreased in the ACN-exposed workers.
A more recent study of 41 workers in a chemical engineering plant, 47 workers in a nitrile fiber plant and 31 healthy, non-industry male controls reported in vivo chromosome level mutations reflected as MN in buccal mucosal and peripheral blood lymphocytes (Fan et al. Citation2006). ACN exposure levels based on air concentrations in the workplace showed a geometric mean of 0.26 mg/m3 (0.11 ppm) for the chemical workers and 2.0 mg/m3 (0.92 ppm) for workers in the fiber plant, thereby classifying the former as a low exposed group and the latter as an intermediate exposed group. Age, smoking and alcohol consumption were similar in the three groups as were years of employment for the two worker groups. The reported buccal mucosal MN frequencies were 2.0 ± 2.2% in controls, 3.7 ± 2.7% in chemical engineering workers and 4.0 ± 2.4% in the fiber workers, with both work group means being significantly different from the controls. The reported lymphocyte MN frequencies were 2.5 ± 1.5%, 2.4 ± 2.1% and 4.2 ± 3.3% for the three groups, respectively, with only the group mean for the fiber workers being significantly different from the control. The authors report that multivariate linear regression indicated that recent ACN exposures, cumulative ACN exposures and cigarette smoking all influenced MN frequencies. Unfortunately, the measurements of workplace air ACN concentrations were made between 1997 and 1999, there were no personal exposure measurements and therefore may not reflect the actual exposures experienced by workers at the time chromosomal endpoints were measured.
Summary of mutations at the somatic level
ACN gene level mutagenicity in prokaryotic microorganisms shows certain patterns. In Salmonella tester strains that require a reverse mutation to occur by a frame shift change, results have been mostly negative. By contrast, most studies in strains carrying his G46 allele have been positive, usually requiring EMA. Reversion of allele G46 in several strains requires a base substitution mutation at G or C. The only Salmonella strain used in the ACN studies that scored for reverse base substitution mutations involving AT base pairs gave mostly negative results. However, studies in E. coli, gave both negative and positive results for ACN mutagenesis. All E. coli WP2 tester strains detect reverse mutations which require base substitutions at an AT base pair. Studies in Salmonella tester strain TA102 which is considered sensitive to oxidative DNA damage gave mostly negative results, although this strain is DNA repair competent compared to most Salmonella strains used which are repair incompetent. Direct incubation of hypothiocyanite with Salmonella tester strain TA1535 failed to induce mutations in this strain carrying the G46 allele while exposure to ACN did.
Both gene and chromosome level mutations induced by ACN have been assessed in eukaryotic microorganisms. Variable results have been reported for gene mutations, employing a variety of loci. These provide few mechanistic clues as the mechanisms underlying the mutations were not known. By contrast, most studies of ACN-induced chromosome level changes in vitro have been reported as positive. Many of these chromosome level mutations involve intra-chromosome recombination and/or gene conversions. An assay in Saccharomyces cerevisiae specifically designed to sensitively detect oxidative damage mutagenesis (DEL assay) has shown positive results for ACN.
Many studies have demonstrated ACN-induced gene level mutations in cultured mammalian cells. Most have used the L5178Y mouse lymphoma assay and have reported positive results, at least for Tk-/-mutations. As noted, L5178Y cells are particularly sensitive to all mutations, in part because they have a mutation in the p53 tumor suppressor gene and in part because they may be especially sensitive to oxidative damage. Also, the Tk locus allows a wide variety of mutational events to be scored because of the presence of an intact allele on the homologous chromosome. Studies of in vitro gene mutations in other cell lines have given variable results, with positive and negative outcomes being reported.
ACN-associated mutations of both the HPRT and TK genes in human TK6 cells have been unambiguously demonstrated. Molecular analyses of both TK-/- and HPRT mutants isolated from these lymphoblastoid cells suggest that point mutations may predominate over deletion mutations. Studies of the HPRT mutations induced by CNEO indicated that base-substitution mutations involving both AT and GC base pairs occur. Despite induction of both TK and HPRT mutations in TK6 cells, there was no increase in N7OEG DNA adducts in TK6 cells at 100 µM CNEO for 2 h.
Many studies of ACN-induced chromosome level mutations in vitro in mammalian cells have been reported as positive, with most requiring EMA. As for the in vitro studies of DNA damage events, all studies of in vitro mutations have employed ACN and/or CNEO exposure concentrations greater than what might be achieved systemically in vivo.
Hprt gene mutations have also been induced in vivo in ACN-treated mice and rats including CYP2E1 deficient knock-out mice while no increases in mutations of the lacZ gene were detected in any tissues.
Although the studies of Hprt mutations suggest that mice are relatively less sensitive than rats for these effects, the mouse studies, unlike those in the rat, were not designed for measuring the peak mutagenic responses.
The demonstration that Hprt mutations could be induced by ACN in CYP2E1 deficient mice suggests that something other than CNEO is the responsible cause, assuming that the CYP2E1 knock-out is complete and that alternative metabolism of ACN to CNEO does not occur.
Human studies have evaluated the occurrence of both gene and chromosome level mutations. The positive Hungarian study of HPRT mutations has a methodological flaw that may invalidate the findings. The autoradiographic assay for HPRT mutations that was used is prone to false positives because PBLs that are in active cell cycle in vivo often are not inhibited by 6-TG sufficiently early in vitro to prevent their incorporating some label into DNA during the first round of DNA synthesis, thus becoming falsely scored as variants (Albertini Citation2001). Cryopreservation is one means used to eliminate or reduce this flaw, but the assay as performed in the study of Major et al. (Citation1998) did not utilize this or any method to eliminate cycling cells. The observation that cell proliferation was increased by ACN exposures in this study would serve to accentuate this phenomenon. The study conclusion that industrial levels of ACN exposure causes HPRT mutations in humans requires confirmation using a more definitive assay conducted in a healthy worker population with a better defined ACN exposure.
The report of ACN induction of mitochondrial deletions cannot be adequately evaluated because the exposure assessment is problematic and the method for detecting mitochondrial deletions is not given in sufficient detail to assess the results, i.e. it is unclear if several deletions were measured or only a single common deletion and the limit of detection is unknown. This study should be repeated with contemporaneous personal ACN exposure measurements, consideration of confounders and specification as to the deletions measured.
ACN-exposed workers in the Czech Republic failed to induce expression of two tumor suppressor genes as manifest by increased serum levels of the respective proteins but the significance of this is unknown.
Results of chromosome level mutations in ACN-exposed humans have been mixed. The Portuguese study was reported as positive but the pattern of CA elevations between exposed worker groups calls into question ACN as the sole causative agent. The elevated CA frequencies in worker groups show the best correlation with evidence of oxidative DNA damage. Interpretation of the CA findings in the Hungarian study is complicated for several reasons; (a) the exposures are complex, consisting of both ACN and DMF, although DMF is not considered to be genotoxic (Antoine et al. Citation1983), (b) there was high level DMF inhalation causing illnesses (c) only mean CA frequencies are given, so the association of aberration frequencies to individual exposure levels is not known and (d) the pattern of CA types is unusual for chemically induced S-phase dependent aberrations, raising again the question of an unknown co-exposure. The studies in Czech workers show results of uncertain significance, i.e. different patterns of CA seen by FISH analysis without an overall increase in aberration frequency and increases in the second (but not first) study of CA determined by standard methods (but not by FISH). A recent study from China does show increases in buccal and PBL MN in workers considered to have a high level of exposure compared to controls and/or workers with lower exposure levels, although there is some ambiguity as to the external exposure assessment. This study does indicate an association with ACN exposures, even though estimates of exposure levels may not have been made contemporaneously with the chromosome studies.
Critique of mutations at the somatic level
Studies of point mutations in prokaryotes indicate that ACN can induce mutations in both GC and AT sites.
Studies in tester strain TA102 and the hypothiocyanite study on strain TA1535 argue against oxidative DNA damage as being responsible for point mutations in prokaryotes.
Studies in eukaryotic microorganisms indicate that ACN preferentially induces chromosomal level mutations in those organisms, with the positive study in Saccharomyces cerevisiae arguing for oxidative DNA damage as the responsible mechanism.
Despite several studies of ACN/CNEO mutations in 5178Y mammalian cells, none have included large and small colony analyses, so the relative contributions of point mutations versus deletion mutations cannot be inferred by this means.
Hprt point mutations induced by CNEO in L5178Y cells in vitro arise at both GC and AT sites.
Induction of both TK and HPRT mutations in vitro in L5178Y cells without an increase in 7OEG DNA adducts suggests that something other than ACN/CNEO adduct formation is responsible for the mutations.
The difference in the sensitivity of the lacZ and Hprt genes in reporting the mutagenic effects of ACN in splenic T-cells of transgenic mice can be explained in part by the high baseline for background mutations in transgene mutation assays, making it difficult to resolve spontaneous and induced MFs, as well as by the restricted target size of the lacZ transgene.
The Hprt MFs measured in ACN-exposed mice likely underestimated their peak mutagenic responses.
Both the mouse and rat mutagenicity studies can be considered in light of potential initiating events. As noted above, oxidative DNA damage in the form of 8oxoG was not found in tissues of ACN exposed mice as opposed to its easy detection in tissues of rats (Kamendulis et al. Citation2001). If this were reproducible, it might suggest that such damage is not the only underlying cause of Hprt mutagenesis. However, more striking is the negative evidence in rats where there is failure to find significant increases in N7OEG or etheno-adducts DNA adducts in several tissues, i.e., brain, splenic lymphocytes or stomach following ACN drinking water exposures at 300 ppm for up to 105 days (males only) or 500 ppm for 15 months (males and females) or to detect N2εG adducts in brain, liver, spleen, or stomach, or N6εA adducts or 3,N4-ethenodeoxycytidine in brains or livers, of rats exposed to 300 or 500 ppm ACN (Walker et al. Citation2020). These negative findings in the same animal species as used for the mutagenicity studies argues against simple direct mutagenicity due to ACN/CNEO specific DNA adducts.
In contrast to the positive studies of gene mutations, the majority of studies assessing chromosome level mutations arising in somatic cells in vivo in mammals (mice or rats) administered ACN by a variety of routes have given negative results.
Although ACN/CNEO is unambiguously clastogenic to mammalian cells in vitro, there is a disparity between the relatively few positive in vivo studies and the more numerous positive in vitro studies.
The report of induction of HPRT mutations in ACN-exposed Hungarian workers is questionable because the frequencies of these mutations in lymphocytes were determined by an autoradiography that is prone to false positives for reasons stated above.
Reports of mitochondrial deletion mutations in AC-exposed humans cannot be fully evaluated for reasons stated above.
Although several studies of chromosomal level mutations in ACN-exposed worker populations can be questioned because of uncertainty in exposure assessment, co-exposures, worker health or data analyses, taken in toto and despite reservations, they do at least suggest that such mutations may result from industrial exposures.
Data gaps
The current data base lacks information of both mutation induction, either in cells or in animals, and the presence of DNA adducts, both chemical specific and oxidative stress related, evaluated in the same study to recognize causative associations. Elimination of oxidative stress by administration of prooxidants or GSH precursors and assessing the effect on mutation induction would further strengthen conclusions as to causation.
Although oxidative stress markers have been assessed in rat microglia (target cell for brain tumors produced by ACN), additional studies would be useful to elucidate they potential role of ACN and its metabolites (CNEO, hypothiocyanite) in causing genotoxicity in this cell type specifically to compare to results obtained to date in rat astrocytes (in vitro) and whole brain (in vivo).
ACN/CNEO induced mutations in several tissues and cell types have been evaluated only once in the Muta-Mouse system. However, a potential deficiency of that system is that it detects only point mutations because larger lesions destroy the transgene contributing to its low sensitivity as demonstrated in the study described above where lacZ mutations were not observed in lymphocytes of the same animals when Hprt mutations were (Walker, Walker, et al. Citation2020; Walker, Fennell, et al. Citation2020). A transgenic rodent system that measures forward mutations in several genes has recently been used for germ cell mutagenicity analysis – the gpt delta transgenic system. The transgene in these animals includes multiple copies of the E. coli gpt gene plus additional genes that inhibit growth in certain selective media, i.e. the λ EG19 construct (Lambert et al. Citation2005). Detection of both point mutations and large deletions detected with this system would be informative as to potential mutagenic carcinogenic mechanisms.
Also, comparisons of mutational spectra in ACN/CNEO inducted mutations with those due to known mutagenic carcinogens, already begun, will allow recognition of potential mechanistic similarities that could be followed up for ACN (Walker, Walker, et al. Citation2020; Walker, Fennell, et al. Citation2020). Finally, there are no data assessing the potential for ACN/CNEO’s protein binding to interfere with metabolism or DNA repair and thereby influence the mutational process. In this regard, follow up of mechanisms by which ACN might interfere with spindle proteins via nitriles would allow better understanding of potential ACN-associated numerical chromosome aberrations.
ACN/CNEO genotoxicity at the germinal level ()
The extensive body of ACN’s genotoxicity literature reviewed above deals almost exclusively with events at the somatic level. Although the steps in the mutagenic process are the same for somatic and germ cells, there are significant differences that can influence this process, especially with respect to induced mutagenicity.
Gametogenesis, which is unique to germ cells, is the developmental process that produces mature germ cells, i.e. ova and spermatozoa in females and males, respectively. The germinal stem cells are oogonia and spermatogonia in females and males, respectively, which mitotically replicate by DNA synthesis to produce two progenies, one to reenter the gonial stage to preserve the stem cell compartment and one to begin the process of meiosis by which chromosome numbers are reduced in two steps to the haploid state of the ova and spermatozoa. The process differs in females and males in that all oogonial cells are produced in the female before birth, with cells entering the first meiotic division to produce primary oocytes and then arresting until puberty, after which the second step with reductive division proceeds at regular cycles. In males, the process of spermatogenesis is continuous throughout life after puberty.
Genotoxicity of agents for germ cells is usually tested by exposing males at all stages of germ cell differentiation, i.e. spermatogonia, spermatocytes (primary and secondary), spermatids and spermatozoa, with DNA replication occurring only in spermatogonia and prior to the first step in meiosis for primary spermatocytes. There is no DNA replication, which is a requisite step in mutation production, in the other germ cell stages. DNA repair is most robust in spermatogonia and spermatocytes, diminishing in early spermatids and formerly considered to be absent in late spermatids and spermatozoa. However, it has recently been found that base-excision repair can persist even in mature spermatozoa (Garcia-Rodriguez et al. Citation2018). These steps are similar in female oogenesis except for the long latent period in completing meiosis. Another complication of testing genotoxins in females is the potential for maternal toxicity affecting results.
The results of ACN’s genotoxic studies in germ cells are considered here according to the genotoxic endpoint(s) measured with specification as to germ cell stages analyzed if available. It is noted that not all studies purporting to have demonstrated genotoxicity in germinal tissues have actually measured the endpoints in germ cells. Germinal tissues of both sexes consist of a framework of somatic cells. Genotoxicity in these cells does not necessarily indicate genotoxicity in germ cells. All studies reviewed here except the first have been in vivo in Drosophila, rodents, or humans. All studies are experimental except the human observational study.
ACN/CNEO DNA damage in germinal tissue
Isolated human and rat testicular cells were treated in vitro (without EMA) with a series of potentially genotoxic chemicals and DNA single strand breaks and/or alkali labile sites were measured by alkaline filter elution (Bjorge et al. Citation1996). Germ cell stages were analyzed by flow cytometry to determine ploidy. Although all stages were apparently in the mixture, the majority of cells were haploid, i.e. secondary spermatocytes, spermatids and spermatozoa. ACN was tested without EMA at 30–1000 µM for human cells and 30–300 µM for rat cells; treatments were for 30 min at 32 °C. The cells were tested shortly after isolation so there was little opportunity for scheduled DNA synthesis or cell replication in the few spermatogonia or primary spermatocytes in the mixture during and after treatment. ACN failed to induce breaks or labile sites at any concentration in either human or rat cells. Five additional chemicals of the 15 evaluated also gave negative results whereas clear positives were obtained for others in cells from one or both species.
Binding to the genetic material of radiolabeled ACN administered as a single oral dose of 46.5 mg/kg (= 0.5 LD50 dose) was studied in Sprague-Dawley rats (Ahmed, Abdel-Rahman, et al. Citation1992). Radioactivity in DNA isolated from testicular tissue was interpreted as covalent binding of ACN to the DNA. It is uncertain the precise cells analyzed in this study, but it appears to be whole testicular tissue. The methods employed could not distinguish protein from DNA binding, with the protein binding being the more likely.
Unscheduled DNA synthesis (UDS) was also measured in the study described above in which DNA binding was assessed in rats (Ahmed, Abdel-Rahman, et al. Citation1992). In all instances, ACN was administered as a 46.5 mg/kg oral dose with increases in UDS and concomitant decreases in SDS being reported for testicular tissues. Again, it is uncertain the precise cells in which an increase in UDS is reported. Regardless of cell type, UDS was measured by liquid scintillation counting, rendering the results unreliable indicators of DNA binding.
By contrast, in vivo UDS measured by autoradiography was studied in F344 rats administered ACN by gavage as a 75 mg/kg single dose, or at 60 mg/kg daily for five days (Working et al.Citation1987; Butterworth et al. Citation1992). At 2, 4, or 12 h following the last dose, animals were sacrificed, testes were dissected, and spermatocytes were isolated and cultured for 24 h in the presence of 3H thymidine. Scoring was by grain counts in mid- to late-stage spermatocytes. ACN did not increase UDS in these studies indicating that these male germ cells were unaffected by treatment. Acrylamide by comparison, deemed to be a germ cell genotoxin by other measures, gave a positive result.
DNA strand breaks/apurinic sites determined by alkaline comet assays were also studied by Walker, Walker, et al. (Citation2020) who administered daily doses of ACN (5 days per week) at 0, 2.5 (wild type [WT] only) 10, 20 or 60 mg/kg bw by gavage for six weeks to female B6C3F1 and CYP2E1 knockout mice with tissues collected for analyses after 24 h as described above for the somatic level analyses. A paradoxical finding was negative results for DNA damage in ovarian tissues from WT animals but positive results in ovarian tissues from knockout mice treated at the highest ACN dose. The increase in DNA damage in the ovaries of knockout but not WT mice did not exhibit a consistent dose-response trend with administered ACN dose (e.g. response was greatest in low-dose animals, and smallest in mid-dose animals). The finding is unexplained, but observation of data ( in the report) shows that values for % migrated DNA, tail length and olive tail movement were considerably lower in the knockout controls compared to the WT controls. Treatment-related increases in CYP2E1 knockout animals are not elevated when compared to the WT control baseline values. Whole ovarian tissues and not germ cells per se were analyzed in these studies so it cannot be determined which cells in the mixture contributed to these findings. The DNA damage in knockout but not WT mice, could conceivably be attributed to increased flux of ACN metabolism via non-oxidative pathways. However, the conjugation pathway for ACN is generally considered to be detoxifying, and therefore these results are more likely are attributed to the unusually low rate in knockout controls, and do not indicate a generalizable germinal tissue effect of ACN.
ACN/CNEO mutations at the germinal level
Germ cell effects in Drosophila following ACN treatments have been studied several times. Although results are not equivalent to those in mammals for estimating risk for heritable mutations, they do allow investigation of underlying mechanisms.
ACN at an inhalation exposure level of 2.7 ppm to test mature oocytes was found to induce sex-chromosome aneuploidy in the offspring of treated females (Osgood et al. Citation1991). The study employed the “ZESTE” assay in which females of a defined genotype are exposed to a test agent and then mated to males, also of a defined genotype, and embryonic progeny are examined for eye color. Different colors other than normal define sex chromosome loss or gain and, depending on timing of examination, the stage of oogenesis affected. ACN-induced sex chromosome loss only (and not gain) in this assay. Timing of analyses indicated that the chromosome loss was manifest only in mature as opposed to immature oocytes.
By contrast to the detection of aneuploidy, two studies of sex-linked recessive lethal (SLRL) gene mutations (approximately 600–800 genes, Abrahamson et al. Citation1980) have been negative. Both point mutations and small deletions may produce SLRLs. An early study administered ACN at 0.1% by intra-abdominal injection to test pre-meiotic cells in female flies without an increase in mutation frequencies (Benesh and Shram Citation1969). Twenty-five years later, Foureman et al. (1994) also reported no increases in SLRL mutation frequencies tested in post-meiotic cells in male flies receiving a single injection of ACN at a dose level of 3500 ppm or by feeding 420 ppm daily for three days.
Gene level lacZ mutations in testes in the transgenic Muta-Mouse system were studied by Walker, Walker, et al. (Citation2020) after 28-day exposures to ACN at 100, 500, or 750 ppm ACN in drinking water with sacrifice 49 days later. There were no significant differences between lacZ MF values in control versus ACN-exposed mice at any exposure dose (Lambert et al. Citation2005; Walker, Walker, et al. Citation2020). As the lacZ mutations were measured in whole testicular tissue, it is not possible to determine which, if any, germ cells were tested. As described above, Hprt mutations were also measured in splenic T-lymphocytes in these same animals with small but significant increases observed, a result that is discordant to the findings in splenocytes for lacZ mutations in these same animals.
Two studies have reported cytogenetic results in male germ cells of ACN treated mice. The first was published in the Russian literature with few details available (Zhurkov et al. Citation1983). Mice (strain unknown) were exposed to 20–100 mg ACN/m3 in air with no induction of chromosome abnormalities reported in spermatogonia at 120 h.
In a second study that analyzed mitotic (M1) primary spermatocytes, statistically significant increases in abnormal metaphases were reported in 9- to 12-week-old male Swiss mice receiving single oral doses of ACN (15.5 or 31.0 mg/kg bw or three to five oral doses of 7.75 mg/kg bw) compared to control (Fahmy Citation1999). The abnormal metaphases increased with dose and duration of treatment. Inspection of in this report shows that most of the abnormal metaphases in the treatment group were due to either sex- or autosomal univalent chromosomes. No increase in aneuploidy was observed in the ACN-treated animals.
Historically the DLT has been the most extensively employed germ cell mutagenicity test in rodents (Yauk et al. Citation2015). The measured endpoint in the DLT is fetal death following mating of treated parents – usually treated males to virgin females – ostensibly caused by induced structural or numerical chromosome changes inherited from exposed parent. However, there have been reports of a decrease in sensitivity in males compared to females for some chemicals (reviewed in Dellarco Citation1993). The DLT may also reflect point mutations. All stages of gametogenesis can be tested by the DLT or a focus can be directed to specific stages depending on timing of post-treatment mating. There is limited sensitivity of this test if only small effects are seen. Current description, interpretation and recommendations are included in OECD Guideline 478.
The earliest DLT reported for ACN-treated eight to ten-week-old male NMRI mice with a single dose of 20 or 30 mg/kg bw administered by i.p. injection before being caged with virgin females of the same strain (Leonard et al. Citation1981). Females were replaced after 7, 14, 21 and 28 days, and analyzed 17 days after mating had begun. IMS and normal saline injections were the positive and negative controls, respectively. All ACN doses and testing intervals were negative. Zhurkov et al. (Citation1983) also assessed DLT mutations in their Russian study mentioned above but with no protocol details other than those noted. However, DLT also was reported as negative. In a final study of DLT mutations, a group of 50 F344 male rats were administered ACN at 60 mg/kg bw/day by gavage for five days (Working et al. Citation1987). Matings were to a single female per week beginning one day after exposure and lasting for ten weeks. Triethylenemelamine by i.p. injection and saline by gavage served as positive and negative controls, respectively. There were no increases in pre- or post-implantation losses in females bred to ACN-treated males i.e. the test was negative. Of interest, the DLT of the structurally related acrylamide conducted simultaneously in this study was positive.
An epidemiological study of germ cell effects of occupational exposure to ACN was conducted in a Chinese industrial facility. Thirty sperm donors, ages ranging from 25 to 30 years, were recruited from a chemical plant where mean exposures to ACN at work sites (presumably in environmental air) were 0.8 ± 0.25 mg/m3 (Xu et al. Citation2003). Additional potential chemical exposures, if any, or work-place features were not reported. Although individual levels or durations of exposure are not stated, the chemical plant had been in production for only 2.8 years so no one had been exposed for a longer time at the plant (but does not preclude exposures from elsewhere prior to employment at this plant). An additional 30 sperm donors, ages ranging from 24 to 35 years, were recruited from the general population as controls. Neither workers nor controls were smokers, heavy alcohol users, had chronic diseases or exposures to chemo- or radiotherapy. DNA double-strand breaks were determined by single-cell electrophoresis at pH 10.4 of all 60 samples (comet assay). Sex chromosome aneuploidy was determined FISH in nine samples from exposed workers and an unknown number of controls. Other measures of sperm quality were made.
Means of comet assay results are presented as group mean values for 9000 sperm from each group, i.e. exposed workers (n = 30) and controls (n = 30), rather than as means of individual values (Xu et al. Citation2003). The mean rate of comet sperm, defined as amount of DNA in the tail, was significantly greater for the 9000 sperm from exposed workers compared to the 9000 sperm from controls (28.7% versus 15.0%) as was tail length (9.8 ± 3.7 versus 4.3 ± 2.3). Also, mean frequencies of sex chromosome aneuploidy, also presented as group means, were significantly greater among 91,015 sperm from exposed workers (n = 9) compared to the 74,679 sperm from controls (n = ???). The authors concluded that exposure to ACN induced both double strand DNA breaks and aneuploidy in sperm. Sperm densities and numbers were also decreased in exposed workers. However, sperm viability and motility were similar in exposed workers and controls and no increase in sperm head abnormalities was observed.
Summary of ACN’s germinal genotoxicity
AN’s genotoxicity profile includes several positive reports of germinal effects. The positive findings are summarized here.
Radio-labeled ACN (2,314C) was administered as a single oral dose (46.5 mg/kg) to Sprague-Dawley rats bound to testicular DNA with maximal effect at 0.5 h and persistence as long as 72 h (Ahmed, Abdel-Aziz, et al. Citation1992). Although protein binding could not be ruled out, this finding does demonstrate that ACN can reach testicular tissue. Decreased DNA synthesis and an increase in DNA repair were reported.
Sex-linked aneuploidy was observed in the offspring of female Drosophila treated with ACN by inhalation at 2.7 ppm (Osgood et al. Citation1991).
ACN 10 mg/kg/bw i.p. produced an increase in SCE in spermatocytes of male mice (Fahmy Citation1999). ACN also showed dose and frequency of treatments related increases in chromosome aberrations in spermatocytes in mice (Fahmy Citation1999).
Workers exposed to ACN (mean concentration in environment 0.54 ppm) for 2.8 years showed an increase in DNA strand breaks in spermatozoa revealed by alkaline comet assays as compared to controls (Xu et al. Citation2003). These workers also showed an increase in aneuploidy in spermatozoa as determined by FISH analyses as compared to controls (Xu et al. Citation2003). A decrease in sperm quantity as manifest by decrease density and numbers was also reported.
ACN’s genotoxicity profile also includes several reports of failures to find germ cell effects.
ACN administered at 0.1% by intra-abdominal injection to female Drosophila failed to increase SLRL mutations (Benesh and Shram Citation1969) while ACN administered as a single injection of 3500 ppm or by feeding at 420 ppm for three days also failed to increase SLRL mutations in Drosophila (Foureman et al. 1994).
F344 rats administered ACN by gavage as a 75 mg/kg single dose or at 60 mg/kg daily for five days failed to show UDS in testes as determined by autoradiography at 2,4 or 12 h following the last dose (Butterworth et al. Citation1992).
Assessment of in vivo lacZ mutations in male germ cells of mice (Mutamouse) administered ACN doses up to ∼2.3 mg/kg) in drinking water for 28 days, followed by a 49-day expression period after the last day of treatment, showed no significant increases (Lambert et al. Citation2005). Treatments of mice by inhalation at ACN concentrations of mg/m3 failed to increase chromosome aberration frequencies in spermatogonial cells (Zhurkov et al. Citation1983) and treatments of NMRI mice by single i.p. injection of 20 or 30 mg/kg/bw also failed to increase dominant lethal mutations at several time points following treatment (Leonard et al. Citation1981). Treatments of mice by inhalation at ACN concentrations of 100 mg/m3 failed to increase dominant lethal mutations (Zhurkov et al. Citation1983). Treatments of F344 rats by oral administration of 60 mg/kg/day for five days also failed to find an increase dominant lethal mutations (Working et al. Citation1987).
Although the majority of studies in AN’s genotoxicity profile failed to demonstrate germ cell effects, some positive studies noted that must be considered. The pattern of results in germinal tissue is similar to that observed in the overall mutation data set, i.e. a tendency to produce chromosome level over single gene effects.
Critique of ACN’s germinal level genotoxicity
There is a relatively small data set for studies of potential genotoxic/mutagenic effects of ACN in germinal tissues. Investigations span a period of over four decades. Many studies were performed before assays were standardized or guidelines issued. Some did not measure effects in germ cells. Critical analyses show that several provide little confidence for decision making as to ACN being a germ cell mutagen. Although the majority of studies of germ cell genotoxicity have been negative, there is a consistency of some positive studies among species which may indicate an indirect effect.
The single in vitro study of isolated human and rat testicular cells, while negative for DNA strand breaks or apurinic sites, does not realistically mimic the in vivo situation. There is no metabolism in this system that may give rise to genotoxic intermediates. Toxicokinetic factors are not considered, nor is repair which is critical for lesion persistence. It can be concluded that ACN did not induce DNA strand breaks or apurinic sites under conditions of the assay, but this does not exonerate it from germ cell genotoxicity at all stages of spermatogenesis or under in vivo conditions.
The studies purporting to demonstrate ACN binding to DNA in testicular tissues of Sprague Dawley rats (Ahmed, Abdel-Rahman, et al. Citation1992) cannot be interpreted as indicating germ cell genotoxicity for two reasons. First, the methods likely measured protein rather than DNA binding and second, there is no reason to believe that what was detected was due to an ACN interaction with macromolecules in germ cells rather than in stromal or other non-germ cells in the tissues.
Similarly, the reports of increased DNA repair in rat testes as manifest by UDS in the same studies is questionable because liquid scintillation counting was used to assess UDS, a method prone to artifact by unanticipated SDS. It is uncertain if the effects reported even arose in germ cells as whole testicular tissue with an abundance of somatic cells was studied. Neither the binding nor the DNA repair aspects of this series of studies are informative as to ACN’s potential germ cell genotoxicity. By contrast, the study that employed autoradiography to detect UDS in specific germ cells and stages of spermatogenesis of ACN-treated F344 rats was clearly negative (Butterworth et al. Citation1992). Although OECD Guideline 486 (OECD Citation1997) is specifically directed at assessing in vivo UDS in liver cells, the methods outlined were followed in the Butterworth et al study (1992) making this a convincing negative report indicating that ACN does not damage germ cell DNA in this system.
The unexpected demonstration of DNA strand breakage/apurinic sites in ovarian tissue from ACN-treated CYP2E1 null mice but not in ovaries of parental wild-type mice is puzzling (Walker, Walker, et al. Citation2020). However, as indicated above, this may simply be a result of unusually low levels of breakage in the control CYP2E1 null animals (<50% of wild-type control levels), as noted by the study authors. Furthermore, as what was measured occurred in whole ovarian tissue cells and not specifically in germ cells, this was not a study of germ cell genotoxicity. In any case, this result does not suggest a generalizable effect of ACN in germ cells.
Although the negative result for ACN induction of lacZ mutations in the testes in the MutaMouse study suggests lack of germinal mutagenicity in this system, it too has deficiencies that render it uninterpretable in terms of germ cell genotoxicity (Walker, Walker, et al. Citation2020). As in the several studies noted above, the study was performed in testicular tissue and not specifically in germ cells. Germ cells may or may not have been in the mix. Also, disconcerting was the observation that ACN did induce Hprt mutations but not lacZ mutations in splenic and thymic lymphocytes at similar exposure doses in these same animals, suggesting a potential sensitivity issue for lacZ assay in this study.
Studies of chromosome aberrations in ACN mice have given mixed results. The earliest was reported as negative (Zhurkov et al. Citation1983). Unfortunately, the report itself is not available and few details are known. These findings cannot be considered as definitive.
A later study evaluated mice treated with multiple ACN doses over multiple time periods and reported induction of aberrations in dose-dependent and time related manners (Fahmy Citation1999). However, as noted, most of the abnormal metaphases in the treatment group were due to either sex- or autosomal univalent chromosomes (i.e., chromosomes that fail to pair during prophase of meiosis, which if leads to malsegregation of chromosomes, it would be a mutational event in germ cells), the significance of which in the production of aneuploidy has been challenged (Allen et al. Citation1986; Liang et al. Citation1986). It is noteworthy that aneuploidy was not increased in the treated animals in this study, indicating that the univalents did not progress and were not indicators of a transmissible numerical chromosome aberration.
Three studies of results of DLTs that measure chromosome level structural or numerical changes induced in tested males have been reported as negative in ACN-treated mice or rats (Leonard et al. Citation1981; Zhurkov et al. Citation1983; Working et al. Citation1987). Detailed reports are available for two of the three allowing confidence in the results. DLTs are time-honored measures of germinal mutations in rodents but may lack the sensitivity of other endpoints. Results of studies in Drosophila provide a mixed picture of ACN’s germ cell genotoxicity in that organism. Sex chromosome aneuploidy was induced in the offspring of treated females, but it was only loss that was observed (Osgood et al. Citation1991). By contrast, two studies of SLRL mutations were negative following ACN treatments of either females or males (Benesh and Shram Citation1969; Foureman et al. 1994). The Drosophila SLRL test, however, does not detect aneuploidy. At face value, these data taken together suggest that ACN may induce numerical chromosome changes in this insect system but not mutations due to damage to the DNA.
Perhaps the most relevant study for assessing ACN’s potential genotoxicity for germ cells is the human study that measured both chromosome breakage and sex chromosome aneuploidy in young men exposed in an industrial setting (Xu et al. Citation2003). An initial concern with this study is in its exposure assessment. A measurement of ACN concentration in air, presumable at worksites, is given. However, there is no personal exposure information. Are there co-exposures? Is the industrial plant producing only ACN or are there productions or processes that employ other chemicals? Importantly, the control group of males is not from the industry. ACN may be one agent in a mix to which study subjects are exposed. Additional information is required to evaluate technical aspects of this study. Although details of the comet assay parameters are well described, it is surprising that there are no measures of cytotoxicity. One potential artifact when interpreting comet assay results is that they may simply be detecting exposure-related cytotoxic effects rather than genotoxic effects because DNA degradation is a part of cell death (Burlinson et al. Citation2007). Measures of low molecular weight DNA, as described above for comet assay results in mouse ovarian tissue, incorporated into the assay allows assessment of cytotoxicity to guard against this artifact and is a critical control (Speit et al. Citation2009; Walker, Walker, et al. Citation2020). The FISH studies of sex chromosome aneuploidy were limited to only nine potentially exposed subjects and an unknown number of controls. The results given in of Xu et al. (Citation2003) are in accord with expectations that first meiotic division non-disjunction produces more aneuploid sperm than second division and that nullisomy (resulting from both chromosome lagging and non-disjunction) is more frequent than disomy. However, there is no consideration as to how the total aneuploid frequencies for both the exposed and control groups compared with reported frequencies for healthy males. A brief comparison with three data sets shows that values for both the controls and exposed in the Xu et al. (Citation2003) study fall within published values for normal males (Williams BJ et al. Citation1993; Luetjens et al. Citation2002; García-Mengual et al. Citation2019). Furthermore, of García-Mengual et al. (Citation2019) shows considerable inter-individual variation among 14 healthy males for sex-chromosome aneuploid frequencies. The finding of inter-individual variability highlights another concern with the Xu et al. (Citation2003) report. Results are pooled for presentation and statistical analysis rather than given as individual values. It cannot be gleaned from these pooled data the extent to which different subjects contributed to the overall picture. The Xu et al. (Citation2003) study might have detected an increase in chromosome breakage and sex-linked aneuploidy in a group of workers compared to the chosen controls, but this cannot be determined with confidence without access to additional data. Even if these abnormalities were found, much more must be known about exposure to assign cause and avoid guilt by association.
Table 3. Summary of ACN induced somatic level mutations in multiple systems.
Table 4. ACN genotoxicity/mutagenicity in germinal tissues.
Data gaps
ACN’s potential for inducing numerical chromosome aberrations, i.e. aneuploidy, is suggested by a single Drosophila study of sex-linked chromosomes. The human study with its several deficiencies also claims to have found sex-linked aneuploidy in ACN-exposed workers. Also, the cytogenetic study that reported an increase in chromosome aberrations in ACN-exposed mice included a class of meiotic changes that theoretically could result in aneuploidy, although no numerical chromosome changes were observed. These findings in the existing data base should all be addressed in a repeat cytogenetic study. Cytogenetic analyses of male germ cells, i.e. spermatogonia, spermatocytes and early spermatids, is a well-established method for detecting both structural and numerical chromosome aberrations in rodents (Allen et al. Citation1986; Adler et al. Citation2012; Yauk et al. Citation2015; OECD Guideline 483). Chromosome painting can add to the precision of the analysis. Micronuclei may also be measured in spermatids (Adler et al. Citation2012). An add-on to the proposed cytogenetic studies that may at least suggest transmission of numerical chromosome aberrations would be FISH analysis of mature sperm using sex-chromosome centromere probes.
The comet assay in widespread use for detecting DNA strand breaks/apurinic sites can also be applied to male germ cells. Although results can give information as to genotoxicity, and may be requested by regulating agencies, the standard comet assay is not yet considered to be fully validated for analyses in sperm (Speit et al. Citation2009; OECD Guideline 489, 2014).
Testing for ACN’s capacity for inducing mutations due to DNA damage in male germ cells can now be accomplished by studies in transgenic rodents. Strict protocols have been developed to focus analyses to specific germ cell stages (Marchetti et al. 2018; OECD Guideline 488, 2019). Studies of mutations in transgenic animals allows for sequencing of mutations to discover specific mutational spectra for identifying causative mutagens. A study in the lacZ Muta-Mouse system is described above. More informative would be a study of germ cell mutations using the gpt delta transgenic system for reasons outlined above in ACN treated animals. Experience from studies in both ENU and acrylamide treated C57BL/6 gpt delta transgenic mice demonstrating that these agents increased mutant frequencies in spermatozoa obtained from the cauda epididymis (Masumura et al. Citation2016, Citation2021; Hagio et al. Citation2021) could guide similar experiments using ACN exposures. Sequencing of mutants obtained from the ENU and acrylamide studies have indicated that causation was due to the treatments in both cases.
Data from these and other newer technologies can potentially inform an answer to the question of ACN’s potential germ cell mutagenicity (Adewoye et al. Citation2015; Beal et al. Citation2019). Two recent studies of germ cell mutations in mutagen treated animals have employed these methods to detect and characterize heritable mutations in tandem repeats (Adewoye et al. Citation2015; Beal et al. Citation2019). The first used ionizing radiation and was performed as a proof of principle study; the second tested benzo(a)pyrene. Both characterized the tandem repeat mutations in the male germ cells and the demonstrated the heritability of these mutations in somatic tissues of progeny, clearly indicating that the mutations occurred in those germ cell stages that were then transmitted.
Beyond information from additional experimental studies, a striking data gap is the lack of human epidemiological evaluations, including follow up on the Chinese reports that was never undertaken (Collins et al. Citation2003) and a repeat of the Xu et al. (Citation2003) study on sex chromosome aneuploidy in sperm that corrects the deficiencies of the reported study.
Discussion
Somatic level effects
The extensive database that describes ACN’s genotoxicity profile provides much information but fails in one critical regard; it has not identified a single event that underlies ACN’s demonstrated mutagenicity. As regards direct DNA reactive mutagenicity, numerous agent specific DNA adducts, many of which are pro-mutagenic, have been induced in isolated DNA in vitro, usually employing massively non-physiological conditions. Radiolabeled ACN or CNEO has also bound to DNA both in vitro in cells and in intact animals, although the possibility of contaminating protein binding is never fully removed. However, with only a single exception, ACN/CNEO-specific adducts have not been identified in living systems, i.e. neither in cells culture nor in whole animals. The adduct that was identified in this single example is N7OEG, which in a non-pro-mutagenic adduct.
Studies that related DNA adduct formation following ACN exposures to the production of mutations have been few. As noted, Walker, Fennell, et al. (Citation2020) failed to find increases in N7OEG adducts in ACN/CNEO treated TK6 cells or of any of several ACN/CNEO-specific adducts in ACN exposed rats while, in both cases, Hprt mutations were induced – by Crespi et al. (Citation1985), Recio and Skopek (Citation1988) and Recio et al. (Citation1990) in TK6 cells in different laboratories and by Walker, Fennell, et al. (Citation2020) in rats in the same laboratory.
The N7 position of guanine is the most susceptible to alkylation and adduct formation (Boysen et al. Citation2009). Even if N7OEG adducts are not usually pro-mutagenic, their presence is a sensitive indicator of DNA alkylation. Clearly, their presence in vivo in brain has been easily demonstrated after exposures to ethylene oxide, an agent that also induces rat brain tumors (Whysner et al. Citation1998 and references therein). The failure to find N7OEG adducts following ACN treatments cannot be simply ascribed to instability or rapid repair of the adduct which also should be the case following ethylene oxide treatments. Similarly, analogous adducts induced by vinyl chloride used as positive controls in the ACN-adduct studies noted above were easily identified (Walker, Fennell, et al. Citation2020). Although ultra-sensitive methods have not been used, the studies that have thus far failed to find an increase in adducts on exposure to ACN did find background adduct levels in the 10−6 range, which is comparable to levels found in studies of many DNA-reactive chemicals.
In comparison to the difficulties in demonstrating ACN/CNEO-specific DNA adducts, oxidative DNA damage has been more easily shown. Several studies in different laboratories have reported increases of the signature 8oxoG adduct, which is pro-mutagenic, following ACN exposures to cells or animals. As described above, ACN-associated increases in 8oxoG adduct levels have always been found in rat brain when looked for. Furthermore, these adduct increases have been associated with other markers of oxidative stress. Several studies reviewed above have indicated that indirect rather than direct effects of ACN may be the more important mediators of genotoxicity/mutagenicity.
The difference between direct and indirect mutagenicity is that, while direct effects imply specific interactions with the genetic material, i.e. ACN → DNA interaction not requiring intermediary cellular processes, the mechanism by which ACN indirectly affects the DNA is multi-step (; Albertini and Kaden Citation2020). A direct reaction with the DNA is the concept that forms the basis of the “one hit” non-threshold model of chemical carcinogenesis for mutagenic carcinogens whereby a single interaction with the DNA minimally raises the probability of cancer, even when undetectable by observation. This concept has been challenged (Heflich et al. Citation2020) but its validity is not the focus of this review. In contrast to direct genotoxicity, an exogenous chemical or its metabolite may indirectly damage the genetic material via a series of interactions with various cellular components leading to alterations of function and/or production of endogenous mutagens and/or reduction of defenses, i.e. exogenous chemical → cellular processes → production of endogenous mutagens → reduction of defenses → ± altered cellular functions. The mechanisms by which exogenous mutagens indirectly damage DNA do not fit the direct model in that there will be a dose effect for chemical interactions with cellular processes that is likely amplified when a series of processes are required before mutations are produced.
Observations supporting ACN-inducted oxidative DNA damage, however, are not without difficulties. Although oxygen stress has usually been attributed to depletion of GSH, this was not universally observed, leaving the mechanism for its production unclear. Although 8oxoG adduct increases have been demonstrated by direct chemical methods and inferred by modified comet assays, there are challenges to both types of existing data. Steps in the preparation methods used for the chemical demonstrations may cause base oxidations giving rise to artifactual elevations in 8oxoG (Loft et al. Citation2008). The modified comet assay used for all save one of the demonstrations of 8oxoG induction has been the FPG-G comet. That particular assay, although sensitive for 8oxoG, is not specific; it detects a variety of DNA alkylations (Smith et al. Citation2008). In the Williams GM et al. (Citation2017) rat study described above, the FPG-G modified comet was positive in brain but not in Zymbal’s gland following ACN oral exposures. However, the comet assay modified by substituting the highly specific hOGG1 instead of FPG gave negative results in both tissues, suggesting that the FPG-G assay was detecting something other than 8oxoG in brain. No studies have been conducted which compare oxidative DNA damage with mutation induction in any system.
Despite the enormity of ACN’s genotoxicity database, no studies have emerged to definitively establish a single mutagenic MOA. Ultrasensitive methods to detect DNA adducts have not been used to look for ACN/CNEO-specific DNA adducts. It is possible that some pro-mutagenic ACN/CNEO-specific adduct remains to be discovered. This possibility should be investigated. Weighing the current evidence suggests that indirect mutagenicity due to oxidative DNA damage will play a role, at least in some tissues, but apparent inconsistencies must be resolved. In any case, the data gaps revealed by this review do indicate what is yet to be investigated. There are answers to the questions posed here; there will be one or more MOAs for ACN’s mutagenicity that, once discovered, will inform its MOA for cancer.
Germinal level effects
The question of ACN being a germ cell mutagen has been addressed over several decades. A series of Chinese human epidemiological studies of ACN-exposed individuals reported an increased frequency of adverse reproductive outcomes (Wu WK et al. Citation1994, 1995; Dong et al. Citation1996 and reviewed in Wu X and Jin Citation2000; Li Z Citation1996). Although such outcomes may have a variety of causes, germ cell mutations may be one. There were deficiencies in design and exposure assessment in these studies and the hope was to repeat them with improved protocols (Collins et al. Citation2003; Neal et al. Citation2009). Unfortunately, this was never accomplished so these results must be view as simply hypothesis generating – the hypothesis being ACN-induced genotoxicity in germ cells.
Several animal reproductive studies have been conducted to test the hypothesis generated by the human epidemiological studies. ACN exposure in rats (up to 90 ppm) did not cause changes to sperm morphology or counts, and slight changes in motility at the highest concentration were not considered to be treatment-related (Nemec et al. Citation2008). Some studies were essentially teratological studies that assess cellular toxicity but not germ cell genotoxicity (Murray et al. Citation1978; Willhite, Ferm, et al. 1981; Willhite, Marin-Padilla, et al. Citation1981; Mehrotra et al. Citation1988; Saillenfait et al. Citation1993; Saillenfait and Sabaté Citation2000 and reviewed in Neal et al. Citation2009). Other studies examined non-genetic toxic manifestations in germ cells (TRL Citation1975; Tandon et al. Citation1988; Wang Z et al. Citation1995; Abdel-Naim et al. as reported in Serota et al. Citation1996; Neal et al. Citation2009; Quast Citation2002; Johannsen and Levinskas Citation2002a, Citation2002b). These too were not assessments of ACN-induced genotoxicity per se. However, three multi-generational studies of adverse pregnancy outcomes and offspring effects in rodents more directly measured heritable effects of ACN exposures (Litton Bionetics Citation1980; Friedman and Beliles Citation2002; Nemec et al. Citation2008), with a high likelihood that any adverse outcomes and effects, if observed, would have a mutagenic basis in germ cells. These studies did not find increased frequencies of heritable effects and, in that sense, were unable to support the hypothesis generated by the human studies (TRL Citation1975; Friedman and Beliles Citation2002; Nemec et al. Citation2008; reviewed in Neal et al. Citation2009).
Despite the importance of the issue, few experimental studies designed to measure genotoxicity in germ cells have been conducted. Three of these studies, i.e. SLRL tests in Drosophila, autoradiographic assessment of DNA repair in rats and DLT in mice and rats were negative for evidence of genotoxicity/mutagenicity, with the SLRL and DLT conducted more than once. The genotoxicity/mutagenicity measured in these studies, if found, would have been due mostly to DNA damage although aneuploidy may give rise to a positive DLT. One study in Drosophila measured sex-chromosome segregation and was clearly positive for an increased frequency of aneuploidy limited to chromosome loss in the ACN-treated flies over control. The kind of mutation measured in this study was numerical chromosome aberration only, which is likely due to damage of protein spindles and not direct DNA interaction damage.
Several of the reported studies of germ cell effects, however, were of questionable relevance to in vivo genotoxicity, did not truly address the question of germ cell genotoxicity or were uninterpretable given the available data. The negative in vitro study of ACN-treated sperm (no EMA) does not address all potentially relevant in vivo conditions. The reports of DNA binding in rodents, UDS determined by scintillation counting in rats, positive comet assay findings in ovarian tissue from ACN-treated knock-out mice and lacZ mutations in ACN-exposed mice all studied effects in germinal tissues but not specifically in germ cells. The first two also had methodological issues that could artifactually elevate results. Two additional studies require more information to allow definitive conclusions. The cytogenetic study that reported an increase in frequency in sperm chromosome aberrations in mice exposed to ACN included a class of changes that are of uncertain significance as to whether they represent a genotoxic effect. The human study of sperm in ACN-exposed male workers requires further information as to exposure, technical procedures and statistical analysis to allow a firm conclusion.
Data gaps
Somatic level effects
The hypothesis remains that mutation induction is the initial key event in ACN’s carcinogenic potential. Yet, convincing evidence of an ACN/CNEO-induced DNA damaging event such as one or more ACN/CNEO-specific DNA adducts, either directly induced, or resulting indirectly due to oxidative stress, does not exist. To date, there have only been a limited number of DNA adduct studies utilizing less sensitive analytical methods (∼10−6–10−7 normal nucleotides), and none employing the newest highly sensitive methodologies (i.e. ∼10−10–10−12 normal nucleotides; Swenberg et al. Citation2011). The identification of such adducts would aid in fully assessing human cancer risk due to ACN exposures. Of value would be the identification of specific DNA adducts in cancer target cells such as microglial cells in rats where their detection might be obscured due to tissue dilution when whole tissue (brain) is analyzed. The current data base also lacks information of both mutation induction, either in mammalian cells or in animals, and the presence of DNA adducts, either chemical specific or oxidative stress related, evaluated in the same study, to recognize causative associations. To date, there have been only four opportunities to compare adduct formation and mutation induction, i.e. (i) lack of ACN/CNEO-specific DNA adducts but positive HPRT mutations in human TK6 cells (Crespi et al. Citation1985; Recio and Skopek Citation1988; Recio et al. Citation1990; Walker, Fennell, et al. Citation2020), (ii) lack of ACN/CNEO-specific DNA adducts but positive Hprt mutations in rats (Walker, Fennell, et al. Citation2020), (iii) presence of 8oxoG DNA adducts and positive Hprt mutations in rats (Pu et al. Citation2009; Walker, Fennell, et al. Citation2020) and (iv) lack of 8oxoG DNA adducts but positive Hprt mutations in mice (Kamendulis et al. Citation2001; Walker, Walker, et al. Citation2020). In the latter case, however, the DNA adduct determination was made in brain and the mutations were induced in lymphocytes not allowing a direct tissue comparison. Furthermore, the studies that are the basis for comparisons were conducted in different laboratories, at different times and using different materials.
ACN/CNEO-induced mutations in several tissues and cell types have been evaluated only once in the transgenic Muta-Mouse system. However, a potential deficiency of that system is that it detects only point mutations because larger lesions destroy the transgene contributing to its low sensitivity as demonstrated in the study described above where lacZ mutations were not observed in lymphocytes of the same animals when Hprt mutations were (Walker, Walker, et al. Citation2020). A transgenic rodent system that measures forward mutations in several genes has recently been used for germ cell mutagenicity analysis – the gpt delta transgenic system. The transgene in these animals includes multiple copies of the E. coli gpt gene plus additional genes that inhibit growth in certain selective media, i.e. the λ EG19 construct (Lambert et al. Citation2005). Detection of both point mutations and large deletions detected with this system, along with markers of DNA damage (direct adducts and indirect oxidative) would be informative as to potential mutagenic carcinogenic mechanisms.
Because many studies on ACN genetic damage have been conducted in rat astrocytes (in vitro) or whole rat brain (in vivo), the results may not accurately reflect the effects of ACN on the more recently identified target cell in rat brain (microglia). Future in vitro studies that assess the effects of ACN, CNEO, and hypothiocyanite separately and in combination (at physiologically relevant concentrations) on DNA damage (direct and oxidative) in rat microglia (quiescent and activated) could provide valuable insight into potential MOAs for ACN carcinogenicity. Similarly, in vivo studies in rat that are able to isolate the potential effects of ACN (and thereby in combination with its metabolites) on microglial cell populations, if technically feasible, could prove to be insightful as well.
Finally, there are no data assessing the potential for ACN/CNEO’s protein binding to interfere with metabolism or DNA repair, thereby potentially influencing the mutational process. In this regard, follow-up of mechanisms by which ACN might interfere with spindle proteins via nitriles would allow better understanding of potential ACN-associated numerical chromosome aberrations.
Germinal level effects
ACN’s potential for inducing numerical chromosome aberrations, i.e. aneuploidy, is suggested by a single Drosophila study of sex-linked chromosomes. The human study with its several deficiencies also claims to have found sex-linked aneuploidy in ACN-exposed workers. Also, the cytogenetic study that reported an increase in chromosome aberrations in ACN-exposed mice included a class of meiotic changes that theoretically could result in aneuploidy, although no numerical chromosome changes were observed. These findings in the existing data base could all be addressed in a repeat cytogenetic study. Cytogenetic analyses of male germ cells, i.e. spermatogonia, spermatocytes and early spermatids, is a well-established method for detecting both structural and numerical chromosome aberrations in rodents (Allen et al. Citation1986; Adler et al. Citation2012; Yauk et al. Citation2015; OECD Guideline 483, 2016). Chromosome painting can add to the precision of the analysis. Micronuclei may also be measured in spermatids (Adler et al. Citation2012). An add-on to the proposed cytogenetic studies that may at least suggest transmission of numerical chromosome aberrations would be FISH analysis of mature sperm using sex-chromosome centromere probes.
The comet assay in widespread use for detecting DNA strand breaks/apurinic sites can also be applied to male germ cells. Although results can give information as to genotoxicity, and may be requested by regulating agencies, the standard comet assay is not yet considered to be fully validated for analyses in sperm (Speit et al. Citation2009; OECD Guideline 489, 2014).
Testing for ACN’s capacity for inducing mutations due to DNA damage in male germ cells can now be accomplished by studies in transgenic rodents. Strict protocols have been developed to focus analyses to specific germ cell stages (Marchetti et al. 2018; OECD Guideline 488, 2019). Studies of mutations in transgenic animals allow for sequencing of mutations to discover specific mutational spectra for identifying causative mutagens. A negative study of lacZ mutations in the transgenic Muta-Mouse system was described above. More informative would be a study of germ cell mutations in ACN-treated mice using the gpt delta transgenic system for reasons outlined above. Much has been gleaned from studies in both ENU and acrylamide-treated C57BL/6 gpt delta transgenic mice demonstrating that these agents increased mutant frequencies in spermatozoa obtained from the cauda epididymis (Masumura et al. Citation2016, Citation2021; Hagio et al. Citation2021). Sequencing of mutants indicated that causation was due to the treatments in both cases.
Data from these and other newer technologies can potentially inform an answer to the question of ACN’s potential germ cell mutagenicity.
Beyond information from additional experimental studies, a striking data gap is the lack of human epidemiological evaluations, including follow up on the Chinese reports that was never undertaken (Collins et al. Citation2003) and a repeat of the Xu et al. (Citation2003) study on sex chromosome aneuploidy with a more thorough characterization and analysis of worker exposure histories.
Conclusion
Despite the enormity of ACN’s genotoxicity data base, no studies have emerged to definitively establish its mutagenic MOA. Ultrasensitive methods to detect DNA adducts have not been used to look for ACN/CNEO-specific DNA adducts. It is possible that some pro-mutagenic ACN/CNEO-specific adduct remains to be discovered. This possibility should be investigated. Weighing the current evidence suggests that indirect mutagenicity due to oxidative DNA damage will play a role, at least in some tissues, but apparent contradictions must be resolved. In any case, the data gaps revealed by this review do indicate what remains to be investigated. There is one or more underlying mechanism for ACN’s mutagenicity that, once discovered, will inform its MOA for cancer.
| Abbreviations | ||
| ACN | = | acrylonitrile |
| AP | = | apurinic |
| ATCA | = | 2-aminothiazoline-4-carboxylic acid |
| αdA | = | α-deoxyadenosine |
| CA | = | chromosomal aberration |
| 1CEA | = | 1-carboxyethyl A |
| 7CNEG | = | 7-cyanoethyl guanine |
| CEMA | = | N-acetyl-S-(2-cyanoethyl)-L-cysteine |
| CHEMA | = | N-acetyl-S-(1-cyano-2-hydroxyethyl)-L-cysteine |
| CNEO | = | 2-cyanoethylene oxide |
| CHO | = | Chinese hamster ovary |
| DHT | = | 5,6-dihydrothymine |
| DLT | = | dominant lethal test |
| DMF | = | dimethylformamide |
| DNA | = | deoxyribonucleic acid |
| EH | = | epoxide hydrolase |
| EMA | = | external metabolic activation |
| Fapy-G | = | 2,6-diamino-4-hydroxy-5-formamidopyrimidine |
| FISH | = | fluorescence in situ hybridization |
| FPG | = | formamidopyrimidine DNA-glycosylase |
| 5FU | = | 5-formyluracil |
| GSH | = | glutathione |
| 5HMU | = | 5-hydroxymethyluracil |
| hOGG1 | = | human-8-OH-guanine-DNA-glycolase |
| HPLC | = | high performance liquid chromatography |
| KCN | = | potassium cyanide |
| LD50 | = | lethal dose, 50% |
| MethACN | = | methylacrylonitrile |
| MDA | = | malonaldehyde-complex |
| MF | = | mutation frequency |
| MN | = | micronuclei |
| MNU | = | methylnitrosourea |
| MOA | = | mode of action |
| NCE | = | normochromatic erythrocytes |
| N7OEG | = | N7 oxyethyl guanine |
| NER | = | nucleotide excision repair |
| 5OHC | = | 5-hydroxycytosine |
| 5OHU | = | 5-hydroxyuracil |
| 2OHA | = | 2-hydroxyadenine |
| 8oxoG | = | 7,8-dihydro-8-oxo-guanine |
| OTC | = | 2-oxothiazolidine-4-carboxylic acid |
| PBL | = | peripheral blood lymphocyte |
| PCD | = | premature centromere division |
| PCE | = | polychromate erythrocytes |
| PHS | = | prostaglandin H synthase |
| ROS | = | reactive oxygen species |
| SCE | = | sister chromatid exchange |
| SDS | = | scheduled DNA synthesis |
| SLRL | = | sex-linked recessive lethal |
| SOD | = | superoxide dismutase |
| UDS | = | unscheduled DNA synthesis |
| VF | = | variant frequency |
| WBC | = | white blood cells |
| WT | = | wild type |
Supplemental Material
Download MS Word (87.6 KB)Acknowledgments
The authors would like to thank the anonymous peer reviewers selected by the journal Editor. The insightful comments by two external reviewers lead to rewrites and additions that added to clarity, consistency, and completeness for readers leading to a substantially improved manuscript. We would also like to thank Dr. Michael Aschner (Albert Einstein College of Medicine) for his thoughtful comments on early drafts of the metabolism and mechanistic portions of the manuscript.
Declaration of interest
The conclusions of this review are those of the authors. RJA is an independent consultant (retired, University of Vermont). CRK is an independent consultant and the owner of Summit Toxicology. DES is an independent consultant and owner of ToxSolve. This project was funded by the Acrylonitrile (AN) Group, whose members consist of companies that manufacture or use acrylonitrile. Each author received funding separately via direct contract with the AN Group. Member company representatives were given the opportunity to review the draft manuscript for completeness and clarity purposes, but authors retained full control over the review content. None of the authors has any financial interest in the conclusions reached in this paper.
References
- Abdel-Naim AB, Mohamadin AM. 2004. Myeloperoxidase-catalyzed oxidation of chloroacetonitrile to cyanide. Toxicol Lett. 146(3):249–257.
- Abdel-Naim AB, Hamada F, Abdel Aziz AH, Ahmed AE. 1994. Acrylonitrile (VCN)-induced testicular toxicity in the rat. Toxicologist. 14(1):268.
- Abdel-Rahman SZ, Nouraldeen AM, Ahmed AE. 1994. Molecular interaction of [2,3-14C]acrylonitrile with DNA in gastric tissue of rat. J Biochem Toxicol. 9(4):191–198.
- Abrahamson S, Würgler FE, DeJongh C, Meyer HU. 1980. How many loci on the X-chromosome of Drosophila melanogaster can mutate to recessive lethals? Environ Mutagen. 2(4):447–453.
- Adewoye AB, Lindsay SJ, Dubrova YE, Hurles ME. 2015. The genome-wide effects of ionizing radiation on mutation induction in the mammalian germline. Nat Commun. 6:6684.
- Adler ID, Pacchierotti F, Russo A. 2012. The measurement of induced genetic change in mammalian germ cells. Methods Mol Biol. 817:335–375.
- Ahmed AE, Farooqui YH, Upreti RK, El-Shabrawy O. 1982. Distribution and covalent interactions of [1-14C]acrylonitrile in the rat. Toxicology. 23(2–3):159–175.
- Ahmed AE, Abdel-Aziz AH, Abdel-Rahman SZ, Haque AK, Nouraldeen AM, Shouman SA. 1992. Pulmonary toxicity of acrylonitrile: covalent interaction and effect on replicative and unscheduled DNA synthesis in the lung. Toxicology. 76(1):1–14.
- Ahmed AE, Abdel-Rahman SZ, Nour-Al Deen AM. 1992. Acrylonitrile interaction with testicular DNA in rats. J Biochem Toxicol. 7(1):5–11.
- Ahmed AE, Nouraldeen AM, Abdel-Rahman SZ, Rajaraman S. 1996. Role of glutathione modulation in acrylonitrile induced gastric DNA damage in rats. Arch Toxicol. 70(10):620–627.
- Al-Abbasi FA, Esmat A, Mohamadin AM, Abdel-Naim AB. 2018. Report-role of prostaglandin H synthase in activation of acrylonitrile to cyanide. Pak J Pharm Sci. 31(4):1431–1435.
- Al-Abbasi FA. 2012. Acrylonitrile-induced gastric toxicity in rats: the role of xanthine oxidase. Med Sci Monit. 18(6):BR208–14.
- Albertini RJ, Kaden DA. 2020. Mutagenicity monitoring in humans: global versus specific origin of mutations. Mutat Res Rev Mutat Res. 786:108341.
- Albertini RJ. 2001. Validated biomarker responses influence medical surveillance of individuals exposed to genotoxic agents. Radiat Prot Dosimetry. 97(1):47–54.
- Alexander P. 1961. Mouse lymphoma cells with different radiosensitivities. Nature. 192:572–573.
- Allen JW, Liang JC, Carrano AV, Preston RJ. 1986. Review of literature on chemical-induced aneuploidy in mammalian male germ cells. Mutat Res. 167(1–2):123–137.
- Amacher E, Turner G. 1985. Tests for gene mutational activity in the L5158Y/TK assay system. Prog Mutat Res. 5:487–496.
- Ames BN, Lee FD, Durston WE. 1973. An improved bacterial test system for the detection and classification of mutagens and carcinogens. Proc Natl Acad Sci U S A. 70(3):782–786.
- Anderson D, Cross MF. 1985. Suitability of the P388F mouse lymphoma system for detecting potential carcinogens and mutagens. Food Chem Toxicol. 23(1):115–118.
- Ansell M, Lewis FA. 1970. A review of cyanide concentrations found in human organs. A survey of literature concerning cyanide metabolism, 'normal’, non-fatal, and fatal body cyanide levels. J Forensic Med. 17(4):148–155.
- Antoine JL, Arany J, Léonard A, Henrotte J, Jenar-Dubuisson G, Decat G. 1983. Lack of mutagenic activity of dimethylformamide. Toxicology. 26(3–4):207–212.
- Arlandson M, Decker T, Roongta VA, Bonilla L, Mayo KH, MacPherson JC, Hazen SL, Slungaard A. 2001. Eosinophil peroxidase oxidation of thiocyanate. Characterization of major reaction products and a potential sulfhydryl-targeted cytotoxicity system. J Biol Chem. 276(1):215–224.
- Arni P. 1985. Induction of various genetic effects in the yeast Saccharomvces cerevisiae strain D7. In: Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH, Matter BE, Shelby MD, editors. Progress in mutation research: evaluation of short-term tests for carcinogens; report of the international programme on chemical safety’s collaborative study on in vitro assays. Vol. 5. Amsterdam: Elsevier Science Publishers; p. 217–224.
- Asakura M, Noguchi T, Sugiyama Y, Inoue M, Satake H. 1994. Research study on evaluation of mutagenicity for existing chemical substances. Research study on industrial safety and health in 1994-3. In vitro chromosomal aberration test on cultured mammalian cells. Tokyo: Testing Laboratory: Japan Industrial Safety and Health Association: Japan Bioassay Research Center. Owner company: Japan Industrial Safety and Health Association.
- Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH, Matter BE, Shelby MD, editors. Progress in mutation research: evaluation of short-term tests for carcinogens; report of the international programme on chemical safety’s collaborative study on in vitro assays. Vol. 5. Amsterdam: Elsevier Science Publishers; p. 1–752.
- Singer B, Grunberger D. 1983. Molecular biology of mutagens and carcinogens. New York (NY): Springer.
- Baker RU, Bonin AM. 1985. Tests with the Salmonella plate incorporation assay. In: Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH, Matter BE, Shelby MD, editors. Progress in mutation research: evaluation of short-term tests for carcinogens; report of the international programme on chemical safety’s collaborative study on in vitro assays. Vol. 5. Amsterdam: Elsevier Science Publishers; p. 177–180.
- Barrett TJ, Hawkins CL. 2012. Hypothiocyanous acid: benign or deadly? Chem Res Toxicol. 25(2):263–273.
- Barrett TJ, Pattison DI, Leonard SE, Carroll KS, Davies MJ, Hawkins CL. 2012. Inactivation of thiol-dependent enzymes by hypothiocyanous acid: role of sulfenyl thiocyanate and sulfenic acid intermediates. Free Radic Biol Med. 52(6):1075–1085.
- Beal MA, Meier MJ, Williams A, Rowan-Carroll A, Gagné R, Lindsay SJ, Fitzgerald T, Hurles ME, Marchetti F, Yauk CL. 2019. Paternal exposure to benzo(a)pyrene induces genome-wide mutations in mouse offspring. Commun Biol. 2:228.
- Benesh V, Shram R. 1969. Mutagenic activity of some pesticides in Drosophila melanogaster. Ind Med. 38:442–444.
- Benz FW, Nerland DE, Li J, Corbett D. 1997. Dose dependence of covalent binding of acrylonitrile to tissue protein and globin in rats. Fundam Appl Toxicol. 36(2):149–156.
- Beskid O, Dusek Z, Solanský I, Srám RJ. 2006. The effects of exposure to different clastogens on the pattern of chromosomal aberrations detected by FISH whole chromosome painting in occupationally exposed individuals. Mutat Res. 594(1–2):20–29.
- Bigner DD, Bigner SH, Burger PC, Shelburne JD, Friedman HS. 1986. Primary brain tumours in F344 rats chronically exposed to acrylonitrile in their drinking water. Food Chem Toxicol. 24(2):129–137.
- Bjorge C, Brunborg G, Wiger R, Holme JA, Scholz T, Dybing E, Søderlund EJ. 1996. A comparative study of chemically induced DNA damage in isolated human and rat testicular cells. Reprod Toxicol. 10(6):509–519.
- Bonassi S, Lando C, Ceppi M, Landi S, Rossi AM, Barale R. 2004. No association between increased levels of high-frequency sister chromatid exchange cells (HFCs) and the risk of cancer in healthy individuals. Environ Mol Mutagen. 43(2):134–136.
- Borba H, Monteiro M, Proença MJ, Chaveca T, Pereira V, Lynce N, Rueff J. 1996. Evaluation of some biomonitoring markers in occupationally exposed populations to acrylonitrile. Teratog Carcinog Mutagen. 16(4):205–218.
- Bouzyk E, Gradzka I, Iwaneńko T, Kruszewski M, Sochanowicz B, Szumiel I. 2000. The response of L5178Y lymphoma sublines to oxidative stress: antioxidant defence, iron content and nuclear translocation of the p65 subunit of NF-kappaB. Acta Biochim Pol. 47(4):881–888.
- Boysen G, Collins LB, Liao S, Luke AM, Pachkowski BF, Watters JL, Swenberg JA. 2010. Analysis of 8-oxo-7,8-dihydro-2'-deoxyguanosine by ultra high pressure liquid chromatography-heat assisted electrospray ionization-tandem mass spectrometry. J Chromatogr B Analyt Technol Biomed Life Sci. 878(3–4):375–380.
- Boysen G, Pachkowski BF, Nakamura J, Swenberg JA. 2009. The formation and biological significance of N7-guanine adducts. Mutat Res. 678(2):76–94.
- Bradley MO. 1985. Measurement of DNA single-strand breaks by alkaline elution in rat hepatocytes. Prog Mutat Res. 5:353–357.
- Brams A, Buchet JP, Crutzen-Fayt MC, De Meester C, Lauwerys R, Léonard A. 1987. A comparative study, with 40 chemicals, of the efficiency of the Salmonella assay and the SOS chromotest (kit procedure). Toxicol Lett. 38(1–2):123–133.
- Brennan RJ, Swoboda BE, Schiestl RH. 1994. Oxidative mutagens induce intrachromosomal recombination in yeast. Mutat Res. 308(2):159–167.
- Brooks TM, Gonzalez LP, Calvert R, Parry JM. 1985. The induction of mitotic gene conversion in the yeast Saccharomyces cerevisiae strain JD1. Prog Mutat Res. 5:225–228.
- Burka LT, Sanchez IM, Ahmed AE, Ghanayem BI. 1994. Comparative metabolism and disposition of acrylonitrile and methacrylonitrile in rats. Arch Toxicol. 68(10):611–618.
- Burlinson B, Tice RR, Speit G, Agurell E, Brendler-Schwaab SY, Collins AR, Escobar P, Honma M, Kumaravel TS, Nakajima M, et al. 2007. Fourth International workgroup on genotoxicity testing: results of the in vivo comet assay workgroup. Mutat Res. 627(1):31–35.
- Butterworth BE, Ashby J, Bermudez E, Casciano D, Mirsalis J, Probst G, Williams G. 1987. A protocol and guide for the in vitro rat hepatocyte DNA-repair assay. Mutat Res. 189(2):113–121.
- Butterworth BE, Eldridge SR, Sprankle CS, Working PK, Bentley KS, Hurtt ME. 1992. Tissue-specific genotoxic effects of acrylamide and acrylonitrile. Environ Mol Mutagen. 20(3):148–155.
- Caito S, Park M, Aschner M. 2017. Resistance of mouse primary microglia and astrocytes to acrylonitrile-induced oxidative stress. Neurotoxicology. 63:120–125.
- Caito S, Yu Y, Aschner M. 2013. Differential response to acrylonitrile toxicity in rat primary astrocytes and microglia. Neurotoxicology. 37:93–99.
- Caito SW, Yu Y, Aschner M. 2014. Differential inflammatory response to acrylonitrile in rat primary astrocytes and microglia. Neurotoxicology. 42:1–7.
- Carere A, Conti G, Conti L, Crebelli R. 1985. Assays in Aspergillus nidulans for the induction of forward-mutation in haploid strain 35 and for mitotic nondisjunction, haploidization and crossing-over in diploid strain P1. In: Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH, Matter BE, Shelby MD, editors. Progress in mutation research: evaluation of short-term tests for carcinogens; report of the international programme on chemical safety’s collaborative study on in vitro assays. Vol. 5. Amsterdam: Elsevier Science Publishers.
- Carls N, Schiestl RH. 1994. Evaluation of the yeast DEL assay with 10 compounds selected by the International Program on Chemical Safety for the evaluation of short-term tests for carcinogens. Mutat Res. 320(4):293–303.
- Cerna M, Kocisova J, Kodytkova J, Kopecky J, Sram RJ. 1981. Mutagenic activity of oxiranecarbonitrile (glycidonitrile). In: Gut I, Cikrt M, Plaa GL, editors. Industrial and environmental xenobiotics. Berlin Heidelberg New York: Springer; p. 251–254.
- Chandler JD, Day BJ. 2015. Biochemical mechanisms and therapeutic potential of pseudohalide thiocyanate in human health. Free Radic Res. 49(6):695–710.
- Chang CM, Hsia MT, Stoner GD, Hsu IC. 1990. Acrylonitrile-induced sister-chromatid exchanges and DNA single strand breaks in adult human bronchial epithelial cells. Mutat Res. 241(4):355–360.
- Chinchilla D, Kilheeney H, Vitello LB, Erman JE. 2014. Kinetic and equilibrium studies of acrylonitrile binding to cytochrome c peroxidase and oxidation of acrylonitrile by cytochrome c peroxidase compound I. Biochem Biophys Res Commun. 443(1):200–204.
- Cifone MA, Myhr B, Eiche A, Bolcsfoldi G. 1987. Effect of pH shifts on the mutant frequency at the thymidine kinase locus in mouse lymphoma L5178Y TK+/- cells. Mutat Res. 189(1):39–46.
- Clark HM, Hagedorn TD, Landino LM. 2014. Hypothiocyanous acid oxidation of tubulin cysteines inhibits microtubule polymerization. Arch Biochem Biophys. 541:67–73.
- Clark LS, Hart DW, Vojta PJ, Harrington-Brock K, Barrett JC, Moore MM, Tindall KR. 1998. Identification and chromosomal assignment of two heterozygous mutations in the Trp53 gene in L5178Y/Tk(+/-)-3.7.2C mouse lymphoma cells. Mutagenesis. 13(5):427–434.
- Collins AR. 2009. Investigating oxidative DNA damage and its repair using the comet assay. Mutat Res. 681(1):24–32.
- Collins JJ, Cheng R, Buck GM, Zhang J, Klebanoff M, Schisterman EF, Scheffers T, Ohta H, Takaya K, Miyauchi H, et al. 2003. The feasibility of conducting a reproductive outcome study of Chinese acrylonitrile worker. J Environ Occup Med. 1:29–32.
- Connor TH, Meyne J, Molina L, Legator MS. 1979. A combined testing protocol approach for mutagenicity testing. Mutat Res. 64(1):19–26.
- Cooke MS, Evans MD, Dizdaroglu M, Lunec J. 2003. Oxidative DNA damage: mechanisms, mutation, and disease. FASEB J. 17(10):1195–1214.
- Cote IL, Bowers A, Jaeger RJ. 1984. Effects of acrylonitrile on tissue glutathione concentrations in rat, mouse, and hamster. Res Commun Chem Pathol Pharmacol. 43(3):507–510.
- Crawley FE, Goddard EA. 1977. Internal dose from carbon-14 labelled compounds. The metabolism of carbon-14 labelled potassium cyanide in the rat. Health Phys. 32(3):135–142.
- Crespi CL, Ryan CG, Seixas GM, Turner TR, Penman BW. 1985. Tests for mutagenic activity using mutation assays at two loci in the human lymphoblast cell lines YK6 and AHH-1. Prog Mutat Res. 5:497–516.
- Dahl AR, Waruszewski BA. 1989. Metabolism of organonitriles to cyanide by rat nasal tissue enzymes. Xenobiotica. 19(11):1201–1205.
- Danford N. 1985. Tests for chromosomal aberrations and aneuploidy in the Chinese hamster fibroblast cell line CH1-L. Prog Mutat Res. 5:397–411.
- De Groef B, Decallonne BR, Van der Geyten S, Darras VM, Bouillon R. 2006. Perchlorate versus other environmental sodium/iodide symporter inhibitors: potential thyroid-related health effects. Eur J Endocrinol. 155(1):17–25.
- De Jesús VR, Bhandari D, Zhang L, Reese C, Capella K, Tevis D, Zhu W, Del Valle-Pinero AY, Lagaud G, Chang JT, et al. 2020. Urinary biomarkers of exposure to volatile organic compounds from the population assessment of tobacco and health study wave 1 (2013-2014). Int J Environ Res Public Health. 17(15):5408.
- De Jesús VR, Zhang L, Bhandari D, Zhu W, Chang JT, Blount BC. 2021. Characterization of acrylonitrile exposure in the United States based on urinary n-acetyl-S-(2-cyanoethyl)-L-cysteine (2CYEMA): NHANES 2011-2016. J Expo Sci Environ Epidemiol. 31(2):377–385.
- de Meester C, Bogaert MD-V, Lambotte-Vandepaer M, Roberfroid M, Poncelet F, Mercier M. 1979. Liver extract mediated mutagenicity of acrylonitrile. Toxicology. 13(1):7–15.
- de Meester C, Poncelet F, Roberfroid M, Mercier M. 1978. Mutagenicity of acrylonitrile. Toxicology. 11(1):19–27.
- De PK, Roy A, Banerjee RK. 1987. Immunological characterization of soluble peroxidases from rat tissues including preputial gland. Mol Cell Biochem. 77(2):127–134.
- De PK. 1992. Tissue distribution of constitutive and induced soluble peroxidase in rat. Purification and characterization from lacrimal gland. Eur J Biochem. 206(1):59–67.
- Dellarco VL. 1993. Genetic anomalies in mammalian germ cells and their significance for human reproductive and developmental risk. Environ Health Perspect. 101(Suppl 2):5–11.
- Ding S, Lai-Ji MA, Fan W, Zhu RJ, Ying Q, Zhou YL, Jin FS. 2003. Study on mitochondrial DNA damage in peripheral blood nucleate cells of the workers exposed to acrylonitrile. Chinese J Indust Hyg Occup Dis. 21:99–101. Chinese).
- Dong D, Wang D, Ai X, Zhang H. 1996. Unpublished study of acrylonitrile hazardous effects on workers’ reproductive system (as reported in Collins et al., 2003).
- Douglas GR, Blakey DH, Liu-Lee VW, Bell RDL, Bayley JM. 1985. Alkaline sucrose sedimentation, sister chromatid exchange and micronucleus assays in CHO cells. Prog Mutat Res. 5:359–366.
- Duesberg P, Li R, Fabarius A, Hehlmann R. 2006. Aneuploidy and cancer: from correlation to causation. Contrib Microbiol. 13:16–44.
- Duverger-Van Bogaert M, Lambotte-Vandepaer M, De Meester C, Mercier M, Poncelet F. 1982a. Role of glutathione in liver-mediated mutagenicity of acrylonitrile. Toxicol Lett. 11(3–4):305–311.
- Duverger-Van Bogaert M, Lambotte-Vandepaer M, De Meester C, Mercier M, Poncelet F. 1982b. Vinyl chloride and acrylonitrile: activation mechanism and mutagenicity. Toxicol Eur Res. 4(1):35–37.
- Duverger-Van Bogaert M, Lambotte-Vandepaer M, de Meester C, Rollmann B, Poncelet F, Mercier M. 1981. Effect of several factors on the liver extract mediated mutagenicity of acrylonitrile and identification of four new in vitro metabolites. Toxicol Lett. 7(4–5):311–319.
- EC [European Commission]. 2004. European union risk assessment report for acrylonitrile. Final report. European Chemicals Bureau (Existing Substances, 1st Priority List Volume 32). European Commission, JRC, 2004.
- EC [European Commission] RAC [Risk Assessment Committee]. 2018. ANNEX 1. Background document in support of the Committee for Risk Assessment (RAC) evaluation of limit values for acrylonitrile in the workplace. ECHA/RAC/O-0000001412-86-188/F. 9 March 2018.
- ECHA REACH. 2022. Acrylonitrile Registration Dossier: https://echa.europa.eu/registration-dossier/-/registered-dossier/15561.
- Evans HH, Ricanati M, Horng MF. 1987. Deficiency in DNA repair in mouse lymphoma strain L5178Y-S. Proc Natl Acad Sci U S A. 84(21):7562–7566.
- Fahmy MA. 1999. Evaluation of the genotoxicity of acrylonitrile in different tissues of male mice. Cytologia. 64(1):1–9.
- Fan W, Wang WL, Ding S, Zhou YL, Jin FS. 2006. Application of micronucleus test of buccal mucosal cells in assessing the genetic damage of workers exposed to acrylonitrile. Zhonghua Lao Dong Wei Sheng Zhi Ye Bing Za Zhi. 24(2):106–108. [Chinese]
- Farooqui MY, Mumtaz MM. 1991. Toxicology of methacrylonitrile. Toxicology. 65(3):239–250.
- Farooqui MY, Ahmed AE. 1983. In vivo interactions of acrylonitrile with macromolecules in rats. Chem Biol Interact. 47(3):363–371.
- Fennell TR, Kedderis GL, Sumner SCJ. 1991. Urinary metabolites of [1,2,3-13C]acrylonitrile in rats and mice detected by carbon-13 nuclear magnetic resonance spectroscopy. Chem Res Toxicol. 4(6):678–687.
- Ferguson LR. 1985. Petite mutagenesis in Saccharomyces cerevisiae strain D5. In: Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH, Matter BE, Shelby MD, editors. Progress in mutation research: evaluation of short-term tests for carcinogens; report of the international programme on chemical safety’s collaborative study on in vitro assays. Vol. 5. Amsterdam: Elsevier Science Publishers; p. 229–234.
- Florin I, Rutberg L, Curvall M, Enzell CR. 1980. Screening of tobacco smoke constituents for mutagenicity using the Ames’ test. Toxicology. 15(3):219–232.
- Foureman P, Mason JM, Valencia R, Zimmering S. 1994. Chemical mutagenesis testing in Drosophila. X. Results of 70 coded chemicals tested for the National Toxicology Program. Environ Mol Mutagen. 23(3):208–227. I don’t see ACN in this paper. Should this be: Foureman P, Mason JM, Valencia R, Zimmering S. Chemical mutagenesis testing in Drosophila. IX. Results of 50 coded compounds tested for the National Toxicology Program. Environ Mol Mutagen. 1994;23(1):51–63.
- Friedman MA, Beliles RP. 2002. Three-generation reproduction study of rats receiving acrylonitrile in drinking water. Toxicol Lett. 132(3):249–261.
- Gallagher GT, Maull EA, Kovacs K, Szabo S. 1988. Neoplasms in rats ingesting acrylonitrile for two years. J Am Coll Toxicol. 7(5):603–615.
- Galloway SM, Deasy DA, Bean CL, Kraynak AR, Armstrong MJ, Bradley MO. 1987. Effects of high osmotic strength on chromosome aberrations, sister-chromatid exchanges and DNA strand breaks, and the relation to toxicity. Mutat Res. 189(1):15–25.
- García-Mengual E, Triviño JC, Sáez-Cuevas A, Bataller J, Ruíz-Jorro M, Vendrell X. 2019. Male infertility: establishing sperm aneuploidy thresholds in the laboratory. J Assist Reprod Genet. 36(3):371–381.
- Garcia-Rodriguez A, de la Casa M, Serrano M, Gosálvez J, Roy Barcelona R. 2018. Impact of polymorphism in DNA repair genes OGG1 and XRCC1 on seminal parameters and human male infertility. Andrologia. 50(10):e13115.
- Gargas ML, Andersen ME, Teo SK, Batra R, Fennell TR, Kedderis GL. 1995. A physiologically based dosimetry description of acrylonitrile and cyanoethylene oxide in the rat. Toxicol Appl Pharmacol. 134(2):185–194.
- Garner RC, Campbell J. 1985. Tests for the induction of mutations to ouabain or 6-thioguanine resistance in mouse lymphoma L5178Y cells. Prog Mutat Res. 5:525–529.
- Ghanayem BI, Nyska A, Haseman JK, Bucher JR. 2002. Acrylonitrile is a multisite carcinogen in male and female B6C3F1 mice. Toxicol Sci. 68(1):59–68.
- Glauert HP, Kennan WS, Sattler GL, Pitot HC. 1985. Assays to measure theinduction of unscheduled DNA synthesis in cultured hepatocytes. In: Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH, Matter BE, Shelby MD, editors. Progress in mutation research: evaluation of short-term tests for carcinogens; report of the international programme on chemical safety’s collaborative study on in vitro assays. Vol. 5. Amsterdam: Elsevier Science Publishers; p. 371–373.
- Guengerich FP, Geiger LE, Hogy LL, Wright PL. 1981. In vitro metabolism of acrylonitrile to 2-cyanoethylene oxide, reaction with glutathione, and irreversible binding to proteins and nucleic acids. Cancer Res. 41(12 Pt 1):4925–4933.
- Gulati DK, Sabharwal PS, Shelby MD. 1985. Tests for the induction of chromosomal aberrations and sister chromatid exchanges in cultured Chinese hamster ovary (CHO) cells. In: Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH, Matter BE, Shelby MD, editors. Progress in mutation research: evaluation of short-term tests for carcinogens; report of the international programme on chemical safety’s collaborative study on in vitro assays. Vol. 5. Amsterdam: Elsevier Science Publishers; p. 413–426.
- Gut I, Nerudová J, Stiborová A, Kopecký J, Frantík E. 1985. Acrylonitrile inhalation in rats: II. Excretion of thioethers and thiocyanate in urine. J Hyg Epidemiol Microbiol Immunol. 29(1):9–13.
- Hachiya N. 1987. Evaluation of chemical genotoxicity by a series of short-term tests. Akita Med. 14:269–292.
- Hachiya N, Sato M, Takizawa Y. 1984. Detection of DNA damage in mutagen-treated mammalian tissues by alkaline elution assay. Mutat Res. 130(5):363.
- Hachiya N, Tanaka N, Takizawa Y. 1986. DNA damages in mammalian tissues, II. DNA single-strand breaks and alkali-liable sites detected by alkaline elution assay. Mutat Res. 164(4):266.
- Hagio S, Tsuji N, Furukawa S, Takeuchi K, Hayashi S, Kuroda Y, Honma M, Masumura K. 2021. Effect of sampling time on somatic and germ cell mutations induced by acrylamide in gpt delta mice. Genes Environ. 43(1):4.
- Hakura A, Shimada H, Nakajima M, Sui H, Kitamoto S, Suzuki S, Satoh T. 2005. Salmonella/human S9 mutagenicity test: a collaborative study with 58 compounds. Mutagenesis. 20(3):217–228.
- Hamdy NM, Al-Abbasi FA, Alghamdi HA, Tolba MF, Esmat A, Abdel-Naim AB. 2012. Role of neutrophils in acrylonitrile-induced gastric mucosal damage. Toxicol Lett. 208(2):108–114.
- Hartung R. 1982. Cyanides and nitriles. In: Clayton GD, Clayton E, editors. Patty’s industrial hygiene and toxicology. Vol. 2C. New York (NY): John Wiley & Sons; p. 4845–4900.
- Hartwig A, Arand M, Epe B, Guth S, Jahnke G, Lampen A, Martus HJ, Monien B, Rietjens IMCM, Schmitz-Spanke S, et al. 2020. Mode of action-based risk assessment of genotoxic carcinogens. Arch Toxicol. 94(6):1787–1877. Erratum in: arch Toxicol. 2020 Sep;94(9):3347.
- Heflich RH, Johnson GE, Zeller A, Marchetti F, Douglas GR, Witt KL, Gollapudi BB, White PA. 2020. Mutation as a toxicological endpoint for regulatory decision-making. Environ Mol Mutagen. 61(1):34–41.
- Himwich WA, Saunders JP. 1948. Enzymatic conversion of cyanide to thiocyanate. Am J Physiol. 153(2):348–354.
- Hoffmann D, Hoffmann I. 1997. The changing cigarette, 1950-1995. J Toxicol Environ Health. 50(4):307–364.
- Hogy LL, Guengerich FP. 1986. In vivo interaction of acrylonitrile and 2-cyanoethylene oxide with DNA in rats. Cancer Res. 46(8):3932–3938.
- Hunter J, Maxwell JD, Carrella M, Stewart DA, Williams R. 1971. Urinary d-glucaric acid excretion as a measure of hepatic enzyme induction in man. Clin Sci. 40(3):10P.
- IARC. 2004. Tobacco smoke and involuntary smoking. IARC Monogr Eval Carcinog Risks Hum. 83:1–1438.
- IARC (International Agency for Research on Cancer). 1999. Predictive value of rodent forestomach and gastric neuroendocrine tumours in evaluating carcinogenic risks to humans, views and expert opinions of an IARC Working Group. Technical Publication No. 39. Lyon (France): International Agency for Research on Cancer.
- Inge-Vechtomov SG, Pavlov YI, Noskov VN, Repnevskaya MV, Karpova TS, Khromov-Borisov NN, Chekuolene J, Chitavichus D. 1985. Tests for genetic activity in the yeast Saccharomyces cerevisiae: study of forward and reverse mutation, mitotic recombination and illegitimate mating induction. In: Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH, Matter BE, Shelby MD, editors. Progress in mutation research: evaluation of short-term tests for carcinogens; report of the international programme on chemical safety’s collaborative study on in vitro assays. Vol. 5. Amsterdam: Elsevier Science Publishers; p. 243–255.
- Ishidate M, Jr, Sofuni T. 1985. The in vitro chromosomal aberration test using Chinese hamster lung (CHL) fibroblast cells in culture. Prog Mutat Res. 5:427–432.
- Ishidate M, Jr, Sofuni T, Yoshikawa K. 1981. Chromosomal aberration tests in vitro as a primary screening tool for environmental mutagens and/or carcinogens. Gann. 27:95–108.
- Jacob S, Ahmed AE. 2003. Acrylonitrile-induced neurotoxicity in normal human astrocytes: oxidative stress and 8-hydroxy-2'-deoxyguanosine formation. Toxicol Mech Methods. 13(3):169–179.
- Jalil RA. 1994. Concentrations of thiocyanate and hypothiocyanite in the saliva of young adults. J Nihon Univ Sch Dent. 36(4):254–260.
- Jiang J, Xu Y, Klaunig JE. 1998. Induction of oxidative stress in rat brain by acrylonitrile (ACN). Toxicol Sci. 46(2):333–341.
- Johannsen FR, Levinskas GJ. 2002a. Comparative chronic toxicity and carcinogenicity of acrylonitrile by drinking water and oral intubation to Spartan Sprague-Dawley rats. Toxicol Lett. 132(3):197–219.
- Johannsen FR, Levinskas GJ. 2002b. Chronic toxicity and oncogenic dose-response effects of lifetime oral acrylonitrile exposure to F344 rats. Toxicol Lett. 132(3):221–247.
- Johnston RV, Schwetz BA, Middleton J, Humiston JCG, Lisowe RW. 1978. Unpublished report from the Dow Chemical Company (as reported in WHO, 1983)
- Jung R, Engelhart G, Herbolt B, Jäckh R, Müller W. 1992. Collaborative study of mutagenicity with Salmonella typhimurium TA102. Mutat Res. 278(4):265–270.
- Junge B. 1985. Changes in serum thiocyanate concentration on stopping smoking. Br Med J (Clin Res Ed). 291(6487):22.
- Kailasam S, Rogers KR. 2007. A fluorescence-based screening assay for DNA damage induced by genotoxic industrial chemicals. Chemosphere. 66(1):165–171.
- Kamendulis LM, Jiang J, Zhang H, deFeijter-Rupp H, Trosko JE, Klaunig JE. 1999. The effect of acrylonitrile on gap junctional intercellular communication in rat astrocytes. Cell Biol Toxicol. 15(3):173–183.
- Kamendulis LM, Jiang J, Xu Y, Klaunig JE. 1999. Induction of oxidative stress and oxidative damage in rat glial cells by acrylonitrile. Carcinogenesis. 20(8):1555–1560.
- Kawachi T, Yahagi T, Kada T, Tazima Y, Ishidate M, Sasaki M, Sugiyama T. 1980. Cooperative programme on short-term assays for carcinogenicity in Japan. IARC Sci Publ. (27):323–330.
- Kedderis GL, Batra R. 1993. Species differences in the hydrolysis of 2-cyanoethylene oxide, the epoxide metabolite of acrylonitrile. Carcinogenesis. 14(4):685–689.
- Kedderis GL, Batra R, Koop DR. 1993. Epoxidation of acrylonitrile by rat and human cytochromes P450. Chem Res Toxicol. 6(6):866–871.
- Kedderis GL, Batra R, Turner MJ. Jr. 1995. Conjugation of acrylonitrile and 2-cyanoethylene oxide with hepatic glutathione. Toxicol Appl Pharmacol. 135(1):9–17.
- Kedderis GL, Sumner SC, Held SD, Batra R, Turner MJ, Roberts AE, Fennell TR. 1993. Dose-dependent urinary excretion of acrylonitrile metabolites by rats and mice. Toxicol Appl Pharmacol. 120(2):288–297.
- Khudoley VV, Mizgireuv I, Pliss GB. 1987. The study of mutagenic activity of carcinogens and other chemical agents with Salmonella typhimurium assays: testing of 126 compounds. Arch Geschwulstforsch. 57:453–462.
- Kirman CR, Sweeney LM, Gargas ML, Strother DE, Collins JJ, Deskin R. 2008. Derivation of noncancer reference values for acrylonitrile. Risk Anal. 28(5):1375–1394.
- Klaunig JE, Forney RB. 2010. Nongenotoxic Mechanisms of Acrylonitrile Carcinogenicity Study I: Antioxidants screening [unpublished]. Oakland (CA): Testing Laboratory: Center for Environmental Health, USA. Owner company: The AN group Inc.
- Kobets T, Iatropoulos MJ, Williams GM. 2022. Acrylonitrile induction of rodent neoplasia: potential mechanism of action and relevance to humans. Toxicol Res Appl. 6:239784732110553.
- Kodama Y, Boreiko CJ, Skopek TR, Recio L. 1989. Cytogenetic analysis of spontaneous and 2-cyanoethylene oxideinduced tk-/- mutants in TK6 human lymphoblastoid cultures. Environ Mol Mutagen. 14(3):149–154.
- Kolenda-Roberts HM, Harris N, Singletary E, Hardisty JF. 2013. Immunohistochemical characterization of spontaneous and acrylonitrile-induced brain tumors in the rat. Toxicol Pathol. 41(1):98–108.
- Koutros S, Lubin JH, Graubard BI, Blair A, Stewart PA, Beane Freeman LE, Silverman DT. 2019. Extended mortality follow-up of a cohort of 25,460 workers exposed to acrylonitrile. Am J Epidemiol. 188(8):1484–1492.
- Krajewska B, Lutz W, Piłacik B. 1998. Determination of blood serum oncoprotein NEU and antioncoprotein p-53–molecular biomarkers in various types of occupational exposure. Int J Occup Med Environ Health. 11(4):343–348.
- Kroetz DL, Kerr BM, McFarland LV, Loiseau P, Wilensky AJ, Levy RH. 1993. Measurement of in vivo microsomal epoxide hydrolase activity in white subjects. Clin Pharmacol Ther. 53(3):306–315.
- Kamendulis LM, Siesky AM, Park JJ, Ren B, Xu Y, Klaunig JE. 2001. Examination of DNA synthesis and oxidative stress following treatment with acrylonitrile in the mouse. Toxicologist. Abstract 1366.
- Lakhanisky T, Hendrickx B. 1985. Induction of DNA single-strand breaks in CHO cells in culture. Prog Mutat Res. 5:367–370.
- Lambert IB, Singer TM, Boucher SE, Douglas GR. 2005. Detailed review of transgenic rodent mutation assays. Mutat Res. 590(1–3):1–280.
- Lambotte-Vandepaer M, Duverger-van Bogaert M, de Meester C, Poncelet F, Mercier M. 1980. Mutagenicity of urine from rats and mice treated with acrylonitrile. Toxicology. 16(1):67–71.
- Lambotte-Vandepaer M, Bogaert M-V, de Meester C, Rollmann B, Poncelet F, Mercier M. 1981. Identification of two urinary metabolites of rats treated with acrylonitrile; influence of several inhibitors on the mutagenicity of those urines. Toxicol Lett. 7(4–5):321–327.
- Laugesen M, Fowles J. 2005. Scope for regulation of cigarette smoke toxicity according to brand differences in published toxicant emissions. N Z Med J. 118(1213):U1401.
- Lee CG, Webber TD. 1985. The induction of gene mutations in the mouse lymphoma L5178Y/TK+/- assay and the Chinese hamster V79/HGPRT assay. Prog Mutat Res. 5:547–554.
- Lefkowitz DL, Lefkowitz SS. 2008. Microglia and myeloperoxidase: a deadly partnership in neurodegenerative disease. Free Radic Biol Med. 45(5):726–731.
- Leonard A, Garny V, Poncelet F, Mercier M. 1981. Mutagenicity of acrylonitrile in mouse. Toxicol Lett. 7(4–5):329–334.
- Li XJ, Li B, Huang JS, Shi JM, Wang P, Fan W, Zhou YL. 2014. Effects of acrylonitrile on lymphocyte lipid rafts and RAS/RAF/MAPK/ERK signaling pathways. Genet Mol Res. 13(3):7747–7756.
- Li Z. 1996. Survey of reproductive system of female workers exposed to acrylonitrile. China Ind Med Mag. 9:33–39.
- Liang JC, Sherron DA, Johnston D. 1986. Lack of correlation between mutagen-induced chromosomal univalency and aneuploidy in mouse spermatocytes. Mutat Res. 163(3):285–297.
- Liber HL. Jr, 1985. Mutation tests with salmonella using 8-azaguanine resistance as the genetic marker. In: Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH, Matter BE, Shelby MD, editors. Progress in mutation research: evaluation of short-term tests for carcinogens; report of the international programme on chemical safety’s collaborative study on in vitro assays. Vol. 5. Amsterdam: Elsevier Science Publishers; p. 213–216.
- Lijinsky W, Andrews AW. 1980. Mutagenicity of vinyl compounds in Salmonella typhimurium. Teratog Carcinog Mutagen. 1(3):259–267.
- Lin CY, Lee HL, Sung FC, Su TC. 2018. Investigating the association between urinary levels of acrylonitrile metabolite N-acetyl-S-(2-cyanoethyl)-L-cysteine and the oxidative stress product 8-hydroxydeoxyguanosine in adolescents and young adults. Environ Pollut. 239:493–498.
- Litton Bionetics. 1976. Mutagenicity evaluation of MCA 730. Final report, LBI Project No. 2548, prepared for the Manufacturing Chemists Association. Kensington (MD): Litton Bionetics.
- Litton Bionetics. 1980. Three-generation reproduction study of rats receiving acrylonitrile in drinking water. Unpublished report. Kensington (MD): Litton Bionetics.
- Loft S, Høgh Danielsen P, Mikkelsen L, Risom L, Forchhammer L, Møller P. 2008. Biomarkers of oxidative damage to DNA and repair. Biochem Soc Trans. 36(Pt 5):1071–1076.
- Loprieno N, Boncristiani G, Forster R, Goldstein B, Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH. 1985. Assays for forward mutation in Schizosaccharomyces pombe strain P1. In: Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH, Matter BE, Shelby MD, editors. Progress in mutation research: evaluation of short-term tests for carcinogens; report of the international programme on chemical safety’s collaborative study on in vitro assays. Vol. 5. Amsterdam: Elsevier Science Publishers; p. 297–306.
- Luetjens CM, Rolf C, Gassner P, Werny JE, Nieschlag E. 2002. Sperm aneuploidy rates in younger and older men. Hum Reprod. 17(7):1826–1832.
- Luijten MNH, Lee JXT, Crasta KC. 2018. Mutational game changer: chromothripsis and its emerging relevance to cancer. Mutat Res Rev Mutat Res. 777:29–51.
- Lundquist P, Kågedal B, Nilsson L. 1995. An improved method for determination of thiocyanate in plasma and urine. Eur J Clin Chem Clin Biochem. 33(6):343–349.
- Luo JC, Liu HT, Cheng TJ, Du CL, Wang JD. 1999. Plasma p53 protein and anti-p53 antibody expression in vinyl chloride monomer workers in Taiwan. J Occup Environ Med. 41(6):521–526.
- Luo YS, He QK, Sun MX, Qiao FX, Liu YC, Xu CL, Xu ZR, Zhao SC, Wang HL, Qi ZQ, et al. 2022. Acrylonitrile exposure triggers ovarian inflammation and decreases oocyte quality probably via mitochondrial dysfunction induced apoptosis in mice. Chem Biol Interact. 360:109934.
- Madle S, Dean SW, Andrae U, Brambilla G, Burlinson B, Doolittle DJ, Furihata C, Hertner T, McQueen CA, Mori H. 1994. Recommendations for the performance of UDS tests in vitro and in vivo. Mutat Res. 312(3):263–285. 10.1016/0165-1161(94)00013-1.
- Mahalakshmi K, Pushpakiran G, Anuradha CV. 2003. Taurine prevents acrylonitrile-induced oxidative stress in rat brain. Pol J Pharmacol. 55:1037–1043.
- Major J, Hudák A, Kiss G, Jakab MG, Szaniszló J, Náray M, Nagy I, Tompa A. 1998. Follow-up biological and genotoxicological monitoring of acrylonitrile- and dimethylformamide-exposed viscose rayon plant workers. Environ Mol Mutagen. 31(4):301–310.
- Major J, Jakab MG, Tompa A. 1999. The frequency of induced premature centromere division in human populations occupationally exposed to genotoxic chemicals. Mutat Res. 445(2):241–249.
- Maltoni C, Ciliberti A, Cotti G, Perino G. 1988. Long-term carcinogenicity bioassays on acrylonitrile administered by inhalation and by ingestion to Sprague-Dawley rats. Ann N Y Acad Sci. 534:179–202.
- Maltoni C, Ciliberti A, Di MV. 1977. Carcinogenicity bioassays on rats of acrylonitrile administered by inhalation and by ingestion. Med Lav. 68(6):401–411.
- Manso JA, Camacho IFC, Calle E, Casado J. 2011. Potential of α,β-unsaturated compounds. Org Biomol Chem. 9(18):6226–6233.
- Marchetti F, Aardema MJ, Beevers C, van Benthem J, Godschalk R, Williams A, Yauk CL, Young R, Douglas GR. 2018. Identifying germ cell mutagens using OECD test guideline 488 (transgenic rodent somatic and germ cell gene mutation assays) and integration with somatic cell testing. Mutat Res Genet Toxicol Environ Mutagen. 832–833:7–18. Erratum in: Mutat Res Genet Toxicol Environ Mutagen. 2019;844:70–71.
- Marchetti P, Fovez Q, Germain N, Khamari R, Kluza J. 2020. Mitochondrial spare respiratory capacity: mechanisms, regulation, and significance in non-transformed and cancer cells. FASEB J. 34(10):13106–13124.
- Marnett LJ, Riggins JN, West JD. 2003. Endogenous generation of reactive oxidants and electrophiles and their reactions with DNA and protein. J Clin Invest. 111(5):583–593.
- Marsh GM, Kruchten AT. 2023. A reevaluation of selected mortality risks in the updated NCI/NIOSH acrylonitrile cohort study. Front Public Health. 11:1122346.
- Marsh GM, Zimmerman SD. 2015. Mortality among chemical plant workers exposed to acrylonitrile: 2011 follow-up. J Occup Environ Med. 57(2):134–145.
- Martin CN, Campbell J. 1985. Tests for the induction of unscheduled DNA repair synthesis in HeLa cells. Prog Mutat Res. 5:375–379.
- Masumura K, Ando T, Toyoda-Hokaiwado N, Ukai A, Nohmi T, Honma M. 2021. Comparison of the frequencies of ENU-induced point mutations in male germ cells and inherited germline mutations in their offspring. Genes Environ. 43(1):43.
- Masumura K, Toyoda-Hokaiwado N, Ukai A, Gondo Y, Honma M, Nohmi T. 2016. Estimation of the frequency of inherited germline mutations by whole exome sequencing in ethyl nitrosourea-treated and untreated gpt delta mice. Genes Environ. 38:10.
- Mathews A, Ohsawa K, Buckland ME, Kesavadas C, Ratheesan K, Kusumakumary P, Burger PC, Kohsaka S, Graeber MB. 2016. Microglioma in a child – a further case in support of the microglioma entity and distinction from histiocytic sarcoma. Clin Neuropathol. 35(5):302–313.
- Matsushima T, Muramatsu M, Haresaku M. 1985. Mutation tests on Salmonella tvohimurium by the preincubation method. In: Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH, Matter BE, Shelby MD, editors. Progress in mutation research: evaluation of short-term tests for carcinogens; report of the international programme on chemical safety’s collaborative study on in vitro assays. Vol. 5. Amsterdam: Elsevier Science Publishers; p. 181–186.
- Matsushita H, Goto S. 1980. Study for the screening technology and development of carcinogen focused on mutagenicity. Contract research on occupational health in 1980: 3) mutation assays of 30 chemical substances on Salmonella typhimurium and Escherichia coli. Tokyo (Japan): Testing laboratory: The Institute of Public Health. Owner company: Ministry of Labor.
- Matthews EJ, DelBalzo T, Rundell JO. 1985. Assays for morphological transformation and mutation to ouabain resistance of Balb/c-3T3 cells in culture. Prog Mutat Res. 5:639–650.
- McMahon RE, Cline JC, Thompson CZ. 1979. Assay of 855 test chemicals in ten tester strains using a new modification of the Ames test for bacterial mutagens. Cancer Res. 39(3):682–693.
- Meek ME, Bucher JR, Cohen SM, Dellarco V, Hill RN, Lehman-McKeeman LD, Longfellow DG, Pastoor T, Seed J, Patton DE. 2003. A framework for human relevance analysis of information on carcinogenic modes of action. Crit Rev Toxicol. 33(6):591–653.
- Mehrotra J, Khanna VK, Husain R, Seth PK. 1988. Biochemical and developmental effects in rats following in utero exposure to acrylonitrile: a preliminary report. Ind Health. 26(4):251–255.
- Mehta RD Jr., von Borstel RC. 1985. Tests for genetic activity in the yeast Saccharomyces cerevisiae using strains D7-144, XV185-14C, and RM52. In: Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH, Matter BE, Shelby MD, editors. Progress in mutation research: evaluation of short-term tests for carcinogens; report of the international programme on chemical safety’s collaborative study on in vitro assays. Vol. 5. Amsterdam: Elsevier Science Publishers; p. 271–284.
- Meijer GA. 2005. Chromosomes and cancer, Boveri revisited. Cell Oncol. 27(5–6):273–275.
- Milvy P, Wolff M. 1977. Mutagenic studies with acrylonitrile. Mutat Res. 48(3–4):271–278.
- Mohamadin AM, Abdel-Naim AB. 2003. In vitro activation of dibromoacetonitrile to cyanide: role of xanthine oxidase. Arch Toxicol. 77(2):86–93.
- Mohamadin AM. 2001. Possible role of hydroxyl radicals in the oxidation of dichloroacetonitrile by Fenton-like reaction. J Inorg Biochem. 84(1–2):97–105.
- Moore MM, Doerr CL. 1990. Comparison of chromosome aberration frequency and small-colony TK-deficient mutant frequency in L5178Y/TK(+/-)-3.7.2C mouse lymphoma cells. Mutagenesis. 5(6):609–614.
- Moore RR, Hardisty JF. 2014. Immunohistochemical characterization of brain tumors: a two-year toxicity and oncogenicity study with acrylonitrile following inhalation exposure in rats (Quast et al. 1980). EPL Project No. 694-004. Final Pathology Report, November 4, 2014.
- Morita T, Asano N, Awogi T, Sasaki YF, Sato S, Shimada H, Sutou S, Suzuki T, Wakata A, Sofuni T, et al. 1997. Evaluation of the rodent micronucleus assay in the screening of IARC carcinogens (groups 1,2A and 2B): the summary report of the 6th collaborative study by CSGMT/JEMS MMS. Collaborative study of the micronucleus group test. Mammalian Mutagenicity Study Group. Mutat Res. 389(1):3–122.
- Morris SM, Manjanatha MG, Shelton SD, Domon OE, McGarrity LJ, Casciano DA. 1996. A mutation in the p53 tumor suppressor gene of AHH-1 tk+/- human lymphoblastoid cells. Mutat Res. 356(2):129–134.
- Mortelmans K, Zeiger E. 2000. The Ames Salmonella/microsome mutagenicity assay. Mutat Res. 455(1–2):29–60.
- Müller W, Engelhart G, Herbold B, Jäckh R, Jung R. 1993. Evaluation of mutagenicity testing with Salmonella typhimurium TA102 in three different laboratories. Environ Health Perspect. 101(Suppl 3):33–36.
- Murray FJ, Schwetz BA, Nitschke KD, John JA, Norris JM, Gehring PJ. 1978. Teratogenicity of acrylonitrile given to rats by gavage or by inhalation. Food Cosmet Toxicol. 16(6):547–551.
- Mutlu E, Jeong YC, Collins LB, Ham AJ, Upton PB, Hatch G, Winsett D, Evansky P, Swenberg JA. 2012. A new LC-MS/MS method for the quantification of endogenous and vinyl chloride-induced 7-(2-Oxoethyl)guanine in sprague-dawley rats. Chem Res Toxicol. 25(2):391–399.
- Myhr B, Bowers L, Kaspary WJ. 1985. Assays for the induction of gene mutations at the thymidine kinase locus in L5178Y mouse lymphoma cells in culture. Prog Mutat Res. 5:555–568.
- Nakagawa Y, Toyoizumi T, Sui H, Ohta R, Kumagai F, Usumi K, Saito Y, Yamakage K. 2015. In vivo comet assay of acrylonitrile, 9-aminoacridine hydrochloride monohydrate and ethanol in rats. Mutat Res Genet Toxicol Environ Mutagen. 786–788:104–113.
- Nakamura SI, Oda Y, Shimada T, Oki I, Sugimoto K. 1987. SOS-inducing activity of chemical carcinogens and mutagens in Salmonella typhimurium TA1535-psk-1002: examination with 151 chemicals. Mutat Res. 192(4):239–246.
- Nasralla SN, Ghoneim AI, Khalifa AE, Gad MZ, Abdel-Naim AB. 2009. Lactoperoxidase catalyzes in vitro activation of acrylonitrile to cyanide. Toxicol Lett. 191(2–3):347–352.
- Natarajan AT, Bussmann CJM, van Kesteren-van Leeuwen AC, Meijers M, Van Rijn JLS. 1985. Tests for chromosomal aberrations and sister chromatid exchanges in cultured Chinese hamster ovary (CHO) cells. Prog Mutat Res. 5:433–437.
- Neal BH, Collins JJ, Strother DE, Lamb JC. 2009. Weight-of-the-evidence review of acrylonitrile reproductive and developmental toxicity studies. Crit Rev Toxicol. 39(7):589–612.
- Nemec MD, Kirkpatrick DT, Sherman J, Van Miller JP, Pershing ML, Strother DE. 2008. Two-generation reproductive toxicity study of inhaled acrylonitrile vapors in Crl: CD(SD) rats. Int J Toxicol. 27(1):11–29.
- Nesterova EV, Durnev AD, Seredenin SB. 1999. Tsitogeneticheskie éffekty akrilamida, akrilonitrila i ikh kombinatsii s verapamilom in vivo [Cytogenetic effects of acrylamide, acrylonitrile and their combination with verapamil in vivo]. Biull Eksp Biol Med. 128(12):684–689.
- NHANES. 2015–2016. Laboratory data. [accessed 2022 Aug 1]. https://wwwn.cdc.gov/nchs/nhanes/Default.aspx.
- NTP (National Toxicology Program). 2001. Toxicology and carcinogenesis studies of acrylonitrile (CAS No. 107- 13-1) in B6C3F1 mice (gavage studies). Public Health Service, U.S. Department of Health and Human Services; NTP TR506. Available from the National Institute of Environmental Health Sciences, Research Triangle Park, NC. [accessed 2009 Apr 21]. http://ntp.niehs.nih.gov/ntpweb/index.cfm?objectid=D16D6C59-F1F6-975E-7D23D1519B8CD7A5#tr500.
- NTP [National Toxicology Program]. 2021. 15th ROC [report on carcinogens] acrylonitrile. https://ntp.niehs.nih.gov/whatwestudy/assessments/cancer/roc/index.html
- Obe G, Hille A, Jonas R, Schmidt S, Thenhaus U. 1985. Tests for the induction ofsister-chromatid exchanges in human peripheral lymphocytes in culture. In: Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH, Matter BE, Shelby MD, editors. Progress in mutation research: evaluation of short-term tests for carcinogens; report of the international programme on chemical safety’s collaborative study on in vitro assays. Vol. 5. Amsterdam: Elsevier Science Publishers; p. 439–442.
- Oberly TJ, Bewsey BJ, Probst GS. 1984. An evaluation of the L5178Y TK+/- mouse lymphoma forward mutation assay using 42 chemicals. Mutat Res. 125(2):291–306.
- Oberly TJ, Hoffman WP, Garriott ML. 1996. An evaluation of the twofold rule for assessing a positive response in the L5178Y TK(+/-) mouse lymphoma assay. Mutat Res. 369(3–4):221–232.
- OECD. 1997. Test no. 486: unscheduled DNA synthesis (UDS) test with mammalian liver cells in vivo. OECD guidelines for the testing of chemicals, Section 4. Paris: OECD Publishing.
- Osgood C, Bloomfield M, Zimmering S. 1991. Aneuploidy in drosophila. IV. Inhalation studies on the induction of aneuploidy by nitriles. Mutat Res. 259(2):165–176.
- Page BD, Charbonneau CF. 1983. Determination of acrylonitrile in foods by headspace gas-liquid chromatography with nitrogen-phosphorus detection. J Assoc off Anal Chem. 66(5):1096–1105. 6630123.
- Page BD, Charbonneau CF. 1985. Improved procedure for determination of acrylonitrile in foods and its application to meat. J Assoc off Anal Chem. 68(3):606–608.
- Parent RA, Casto BC. 1979. Effect of acrylonitrile on primary Syrian golden hamster embryo cells in culture: transformation and DNA fragmentation. J Natl Cancer Inst. 62(4):1025–1029.
- Parry JM. 1985. Summary report on the performance of the yeast and Aspergillus assays. In: Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH, Matter BE, Shelby MD, editors. Progress in mutation research: evaluation of short-term tests for carcinogens; report of the international programme on chemical safety’s collaborative study on in vitro assays. Vol. 5. Amsterdam: Elsevier Science Publishers; p. 25–46.
- Parry JM, Eckardt F. 1985a. The detection of mitotic gene conversion, point mutation and mitotic segregation using the yeast Saccharomyces cerevisiae strain D7. In: Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH, Matter BE, Shelby MD, editors. Progress in mutation research: evaluation of short-term tests for carcinogens; report of the international programme on chemical safety’s collaborative study on in vitro assays. Vol. 5. New York: Elsevier; p. 261–269.
- Parry JM, Eckardt F. 1985b. The induction of mitotic aneuploidy, point mutation and mitotic crossing-over in the yeast Saccharomyces ceretqsiae D61.M and D6. In: Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH, Matter BE, Shelby MD, editors. Progress in mutation research: evaluation of short-term tests for carcinogens; report of the international programme on chemical safety’s collaborative study on in vitro assays. Vol. 5. Amsterdam: Elsevier Science Publishers; p. 285–295.
- Pattison DI, Davies MJ, Hawkins CL. 2012. Reactions and reactivity of myeloperoxidase-derived oxidants: differential biological effects of hypochlorous and hypothiocyanous acids. Free Radic Res. 46(8):975–995.
- Pegg AE. 1990. Properties of mammalian O6-alkylguanine-DNA transferases. Mutat Res. 233(1–2):165–175.
- Perocco P, Pane G, Bolognesi S, Zannotti M. 1982. Increase of sister chromatid exchange and unscheduled synthesis of deoxyribonucleic acid by acrylonitrile in human lymphocytes in vitro. Scand J Work Environ Health. 8(4):290–293.
- Peter H, Appel KE, Berg R, Bolt HM. 1983. Irreversible binding of acrylonitrile to nucleic acids. Xenobiotica. 13(1):19–25.
- Peter H, Schwarz M, Mathiasch B, Appel KE, Bolt HM. 1983. A note on synthesis and reactivity towards DNA of glycidonitrile, the epoxide of acrylonitrile. Carcinogenesis. 4(2):235–237.
- Petrikovics I, Thompson DE, Rockwood GA, Logue BA, Martin S, Jayanna P, Yu JC. 2011. Organ-distribution of the metabolite 2-aminothiazoline-4-carboxylic acid in a rat model following cyanide exposure. Biomarkers. 16(8):686–690.
- Priston RJ, Dean BJ, et al. 1985. Tests for the induction of chromosomeaberrations, polyploidy and sister chromatid exchanges in rat liver (RL) cells. In: Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH, Matter BE, Shelby MD, editors. Progress in mutation research: evaluation of short-term tests for carcinogens; report of the international programme on chemical safety’s collaborative study on in vitro assays. Vol. 5. Amsterdam: Elsevier Science Publishers; p. 387–395.
- Priston RAJ, Dean BJ. 1985. Tests for the induction of chromosomal aberrations, polyploidy and sister chromatid exchanges in rat liver (RL4) cells. Prog Mutat Res. 5:387–395.
- Probst GS, Hill LE. 1985. Tests for the induction of DNA-repair synthesis in primary cultures of adult rat hepatocytes. Prog Mutat Res. 5:381–386.
- Prokopczyk B, Bertinato P, Hoffman D. 1988. Cyanoethylation of DNA in vivo in 3-(methylnitrosamino)-propionitrile, an Areca-derived carcinogen. Cancer Res. 48(23):6780–6784.
- Pu X, Wang Z, Klaunig JE. 2015. Alkaline comet assay for assessing DNA damage in individual cells. Curr Protoc Toxicol. 65:3.12.1–3.12.11.
- Pu X, Kamendulis LM, Klaunig JE. 2009. Acrylonitrile-induced oxidative stress and oxidative DNA damage in male Sprague-Dawley rats. Toxicol Sci. 111(1):64–71.
- Pu X, Kamendulis LM, Klaunig JE. 2006. Acrylonitrile-induced oxidative DNA damage in rat astrocytes. Environ Mol Mutagen. 47(8):631–638.
- Puppel K, Kapusta A, Kuczyńska B. 2015. The etiology of oxidative stress in the various species of animals, a review. J Sci Food Agric. 95(11):2179–2184.
- Quast JF. 2002. Two-year toxicity and oncogenicity study with acrylonitrile incorporated in the drinking water of rats. Toxicol Lett. 132(3):153–196.
- Quast JF, Schuetz DJ, Balmer MF, Gushow TS, Park CN, McKenna MJ. 1980. A two-year toxicity and oncogenicity study with acrylonitrile following inhalation exposure of rats. Midland (MI): Dow Chemical Co., Toxicology Research Laboratory.
- Quast JF, Wade C, Humiston C, Gushow TS, Park CN, McKenna MJ. 1980. Two-year toxicity and oncogenicity study with acrylonitrile incorporated in the drinking water of rats. Midland (MI): Dow Chemical Co., Toxicology Research Laboratory.
- Rabello-Gay MN, Ahmed AE. 1980. Acrylonitrile: in vivo cytogenetic studies in mice and rats. Mutat Res. 79(3):249–255.
- Rao P, Singh P, Yadav SK, Gujar NL, Bhattacharya R. 2013. Acute toxicity of some synthetic cyanogens in rats: time-dependent cyanide generation and cytochrome oxidase inhibition in soft tissues after sub-lethal oral intoxication. Food Chem Toxicol. 59:595–609.
- Recio L, Simpson D, Cochrane J, Liber H, Skopek TR. 1990. Molecular analysis of hprt mutants induced by 2-cyanoethylene oxide in human lymphoblastoid cells. Mutat Res. 242(3):195–208.
- Recio L, Skopek TR. 1988. The cellular and molcular analysis of acrylonitrile-induced mutations in human cells. CIIT Activities. 8:1–6.
- Rexroat MA, Probst GS, et al. 1985. Mutation tests with Salmonella using the plate-incorporation assay. In: Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH, Matter BE, Shelby MD, editors. Progress in mutation research: evaluation of short-term tests for carcinogens; report of the international programme on chemical safety’s collaborative study on in vitro assays. Vol. 5. Amsterdam: Elsevier Science Publishers; p. 201–212.
- Rice GC, Hoy C, Schimke RT. 1986. Transient hypoxia enhances the frequency of dihydrofolate reductase gene amplification in Chinese hamster ovary cells. Proc Natl Acad Sci U S A. 83(16):5978–5982.
- Rioux KL, Delaney S. 2020. 1,N6-ethenoadenine: from molecular to biological consequences. Chem Res Toxicol. 33(11):2688–2698.
- Rizzi R, Chiesara E, Cova D, Mattioli M, Di Lernia R. 1984. Acrylonitrile: mutagenicity in yeasts and genotoxicity in HeLa cells. Mut Res Environ Mutagen Relat Subj. 130(3):223.
- Robbiano L, Allavena A, Bagarolo C, Martelli A, Brambilla G. 1994. Comparison in human and rat hepatocytes of the DNA-damaging activity of five chemicals probably carcinogenic to humans. Toxicol in Vitro. 8(1):131–137.
- Roberfroid M, Poncelet F, Lambotte-Vandepaer M, Duverger-Van Bogaert M, de Meester C, Mercier M. 1978. Acute biotoxic effect of styrene on rat liver. Correlation with enzyme-mediated mutagenicity of benzpyrene and acrylonitrile. Scand J Work Environ Health. 4(2):163–168.
- Roberts AE, Kedderis GL, Turner MJ, Rickert DE, Swenberg JA. 1991. Species comparison of acrylonitrile epoxidation by microsomes from mice, rats and humans: relationship to epoxide concentrations in mouse and rat blood. Carcinogenesis. 12(3):401–404.
- Rössner P, Jr, Binková B, Chvátalová I, Srám RJ. 2002. Acrylonitrile exposure: the effect on p53 and p21(WAF1) protein levels in the blood plasma of occupationally exposed workers and in vitro in human diploid lung fibroblasts. Mutat Res. 517(1–2):239–250.
- Roy P, Kulkarni AP. 1999. Co-oxidation of acrylonitrile by soybean lipoxygenase and partially purified human lung lipoxygenase. Xenobiotica. 29(5):511–531.
- Rudd CJ. 1983. L5178Y tk+/? mouse lymphoma forward mutation assay of acrylonitrile. Prepared by SRI International, Menlo Park, CA, for the Environmental Research Center, U.S. Environmental Protection Agency, Research Triangle Park, NC; SRI Project LSU-3447, EPA Contract No. 68-02-3703.
- Saillenfait A, Bonnet P, Guenier J, Deceaurriz J. 1993. Relative developmental toxicities of inhaled aliphatic mononitriles in rats. Fundam Appl Toxicol. 20(3):365–375.
- Saillenfait AM, Sabaté JP. 2000. Comparative development toxicities of aliphatic nitriles: in vivo and in vitro observations. Toxicol Appl Pharmacol. 163(2):149–163.
- Sakurai H, Onodera M, Utsunomiya T. 1978. Health effects of acrylonitrile in acrylic fibre factories. Br J Ind Med. 35(3):219–225.
- Sasaki M, Sugimura K, Yoshida MA, et al. 1980. Cytogenetic effects of 60 chemicals on cultured human and Chinese hamster cells. La Kromosomo II. 20:574–584.
- Savage JRK. 2011. An introduction to chromosomal aberrations. Atlas of genetics and cytogenetics in oncology and haematology. https://atlasgeneticsoncology.org/deep-insight/20001/an-introduction-to-chromosomal-aberrations/
- Schettgen T, Bertram J, Kraus T. 2012. Accurate quantification of the mercapturic acids of acrylonitrile and its genotoxic metabolite cyanoethylene-epoxide in human urine by isotope-dilution LC-ESI/MS/MS. Talanta. 98:211–219.
- Schiestl RH, Gietz RD, Mehta RD, Hastings PJ. 1989. Carcinogens induce intrachromosomal recombination in yeast. Carcinogenesis. 10(8):1445–1455.
- Schiestl RH, Reynolds P, Prakash S, Prakash L. 1989. Cloning and sequence analysis of the Saccharomyces cerevisiae RAD9 gene and further evidence that its product is required for cell cycle arrest induced by DNA damage. Mol Cell Biol. 9(5):1882–1896.
- Schneider J, Presek P, Braun A, Woitowitz HJ. 1999. Serum levels of pantropic p53 protein and EGF-receptor, and detection of anti-p53 antibodies in former uranium miners (SDAG Wismut). Am J Ind Med. 36(6):602–609.
- Schulz V, Bonn R, Kindler J. 1979. Kinetics of elimination of thiocyanate in 7 healthy subjects and in 8 subjects with renal failure. Klin Wochenschr. 57(5):243–247.
- Sekihashi K, Yamamoto A, Matsumura Y, Ueno S, Watanabe-Akanuma M, Kassie F, Knasmüller S, Tsuda S, Sasaki YF. 2002. Comparative investigation of multiple organs of mice and rats in the comet assay. Mutat Res. 517(1–2):53–75.
- Serota DG, Giles HD, Coyne JM, Hogan DB. 1996. Sub-chronic toxicity study in B6C3F1 mice. Birmingham (AL): Testing Laboratory: Southern Research Institute. Report no: SRI-LIF-95- 593-8618-I.
- Sharief Y, Brown AM, Backer LC, Campbell JA, Westbrook-Collins B, Stead AG, Allen JW. 1986. Sister chromatid exchange and chromosome aberration analyses in mice after in vivo exposure to acrylonitrile, styrene, or butadiene monoxide. Environ Mutagen. 8(3):439–448.
- Silver EH, Szabo S. 1982. Possible role of lipid peroxidation in the actions of acrylonitrile on the adrenals, liver and gastrointestinal tract. Res Commun Chem Pathol Pharmacol. 36:33–43.
- Skopek TR, Liber HL, Kaden DA, Thilly WG. 1978. Relative sensitivities of forward and reverse mutation assays in Salmonella typhimurium. Proc Natl Acad Sci U S A. 75(9):4465–4469.
- Smith CC, Adkins DJ, Martin EA, O'Donovan MR. 2008. Recommendations for design of the rat comet assay. Mutagenesis. 23(3):233–240. Erratum in: Mutagenesis. 2009;24(1):95. Dosage error in article text.
- Smith CC, O'Donovan MR, Martin EA. 2006. hOGG1 recognizes oxidative damage using the comet assay with greater specificity than FPG or ENDOIII. Mutagenesis. 21(3):185–190.
- Solomon JJ, Cote IL, Wortman M, Decker K, Segal A. 1984. In vitro alkylation of calf thymus DNA by acrylonitrile. Isolation of cyanoethyl-adducts of guanine and thymidine and carboxyethyl-adducts of adenine and cytosine. Chem Biol Interact. 51(2):167–190.
- Solomon JJ, Singh US, Segal A. 1993. In vitro reactions of 2-cyanoethylene oxide with calf thymus DNA. Chem Biol Interact. 88(2–3):115–135.
- Speit G, Vasquez M, Hartmann A. 2009. The comet assay as an indicator test for germ cell genotoxicity. Mutat Res. 681(1):3–12.
- Speit G, Schütz P, Bonzheim I, Trenz K, Hoffmann H. 2004. Sensitivity of the FPG protein towards alkylation damage in the comet assay. Toxicol Lett. 146(2):151–158.
- Srám RJ, Beskid O, Binkova B. 2004. Cytogenetic analysis using fluorescence in situ hybridization (FISH) to evaluate occupational exposure to carcinogens. Toxicol Lett. 149:335–344.
- Stankowski LF, Jr, Lallier B, Tydrick C, Baba R, Tincher S, Callupe J, Kwok VK. 2019. Re-evaluation of discordant results in related OECD TG741 tester strains. https://www.criver.com/sites/default/files/resource-files/SP-EMGS-18-re-evaluation-of-discordant-results-in-related-OECD-TG471-tester-strains.pdf.
- Strum JM, Shear CR. 1982. Harderian glands in mice: fluorescence, peroxidase activity and fine structure. Tissue Cell. 198214(1):135–148.
- Styles JA, Clay P, Cross MF. 1985. Assays for the induction of gene mutations at the thymidine kinase and the Na+/K + ATPase loci in two different mouse lymphoma cell lines in culture. Prog Mutat Res. 5:587–596.
- Sumner SC, Selvaraj L, Nauhaus SK, Fennell TR. 1997. Urinary metabolites from F344 rats and B6C3F1 mice coadministered acrylamide and acrylonitrile for 1 or 5 days. Chem Res Toxicol. 10(10):1152–1160.
- Sumner SC, Fennell TR, Moore TA, Chanas B, Gonzalez F, Ghanayem BI. 1999. Role of cytochrome P450 2E1 in the metabolism of acrylamide and acrylonitrile in mice. Chem Res Toxicol. 12(11):1110–1116.
- Swaen GMH, Bloemen LJN, Twisk J, Scheffers T, Slangen JJM, Collins JJ, ten Berge WFJP. 2004. Mortality update of workers exposed to acrylonitrile in the Netherlands. J Occup Environ Med. 46(7):691–698.
- Swenberg JA, Lu K, Moeller BC, Gao L, Upton PB, Nakamura J, Starr TB. 2011. Endogenous versus exogenous DNA adducts: their role in carcinogenesis, epidemiology, and risk assessment. Toxicol Sci. 120(Suppl 1):S130–S145.
- Symons JM, Kreckmann KH, Sakr CJ, Kaplan AM, Leonard RC. 2008. Mortality among workers exposed to acrylonitrile in fiber production: an update. J Occup Environ Med. 50(5):550–560.
- Szumiel I. 2005. L5178Y sublines: a look back from 40 years. Part 1: general characteristics. Int J Radiat Biol. 81(5):339–352.
- Tandon R, Saxena DK, Chandra SV, Seth PK, Srivastava SP. 1988. Testicular effects of acrylonitrile in mice. Toxicol Lett. 42(1):55–63.
- Tavares R, Borba H, Monteiro M, Proença MJ, Lynce N, Rueff J, Bailey E, Sweetman GM, Lawrence RM, Farmer PB, et al. 1996. Monitoring of exposure to acrylonitrile by determination of N-(2-cyanoethyl)valine at the N-terminal position of haemoglobin. Carcinogenesis. 17(12):2655–2660.
- Thier R, Balkenhol H, Lewalter J, Selinski S, Dommermuth A, Bolt HM. 2001. Influence of polymorphisms of the human glutathione transferases and cytochrome P450 2E1 enzyme on the metabolism and toxicity of ethylene oxide and acrylonitrile. Mutat Res. 482(1–2):41–46. 6.
- Thier R, Lewalter J, Kempkes M, Selinski S, Brüning T, Bolt HM. 1999. Haemoglobin adducts of acrylonitrile and ethylene oxide in acrylonitrile workers, dependent on polymorphisms of the glutathione transferases GSTT1 and GSTM1. Arch Toxicol. 73(4–5):197–202.
- Thiess AM, Fleig I. 1978. Analysis of chromosomes of workers exposed to acrylonitrile. Arch Toxicol. 41(2):149–152.
- Tonacchera M, Pinchera A, Dimida A, Ferrarini E, Agretti P, Vitti P, Santini F, Crump K, Gibbs J. 2004. Relative potencies and additivity of perchlorate, thiocyanate, nitrate, and iodide on the inhibition of radioactive iodide uptake by the human sodium iodide symporter. Thyroid. 14(12):1012–1019.
- TRL (Toxicological Research Laboratory). 1975. Results of a reproduction study in rats maintained on drinking water containing acrylonitrile. Unpublished report. Midland (MI): Dow Chemical Company, Health and Environmental Research.
- Trosko JE. 2001. Commentary: Is the concept of “tumor promotion” a useful paradigm? Mol Carcinog. 30(3):131–137.
- Turker MS, Gage BM, Rose JA, Elroy D, Ponomareva ON, Stambrook PJ, Tischfield JA. 1999. A novel signature mutation for oxidative damage resembles a mutational pattern found commonly in human cancers. Cancer Res. 59(8):1837–1839.
- USEPA. 2011. Toxicological review of acrylonitrile (external review draft). Archived.
- Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. 2006. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact. 160(1):1–40.
- Ved Brat S, Williams GM. 1982. Hepatocyte-mediated production of sister chromatid exchange in co-cultured cells by acrylonitrile: evidence for extra cellular transport of a stable reactive intermediate. Cancer Lett. 17(2):213–216.
- Venitt S. 1978. Letter on acrylonitrile mutagenicity. Mutat Res. 57(1):107–113.
- Venitt S, Bushell CT, Osborne M. 1977. Mutagenicity of acrylonitrile (cyanoethylene) in Escherichia coli. Mutat Res. 45(2):283–288.
- Vodicka P, Gut I, Frantík E. 1990. Effects of inhaled acrylic acid derivatives in rats. Toxicology. 65(1–2):209–221.
- Wakata A, Miyamae Y, Sato S-i, Suzuki T, Morita T, Asano N, Awogi T, Kondo K, Hayashi M. 1998. Evaluation of the rat micronucleus test with bone marrow and peripheral blood: summary of the 9th collaborative study by CSGMT/JEMS.MMS. Environ Mol Mutagen. 32(1):84–100.
- Walker VE, Fennell TR, Walker DM, Bauer MJ, Upton PB, Douglas GR, Swenberg JA. 2020. Analysis of DNA adducts and mutagenic potency and specificity in rats exposed to acrylonitrile. Chem Res Toxicol. 33(7):1609–1622.
- Walker VE, Walker DM, Ghanayem BI, Douglas GR. 2020. Analysis of biomarkers of DNA Damage and mutagenicity in mice exposed to acrylonitrile. Chem Res Toxicol. 33(7):1623–1632.
- Wallace SS. 2002. Biological consequences of free radical-damaged DNA bases. Free Radic Biol Med. 33(1):1–14.
- Wang CY, Liu LN, Zhao ZB. 2013. The role of ROS toxicity in spontaneous aneuploidy in cultured cells. Tissue Cell. 45(1):47–53.
- Wang Z, Li Z, Wei X. 1995. Study on acrylonitrile to teratogenesis of rat sperm. J Lanzhou Med Coll. 21:3.
- Wang H, Chanas B, Ghanayem BI. 2002. Cytochrome P450 2E1 (CYP2E1) is essential for acrylonitrile metabolism to cyanide: comparative studies using CYP2E1-null and wild-type mice. Drug Metab Dispos. 30(8):911–917.
- Warneke C, Roberts JM, Veres P, Gilman J, Kuster WC, Burling I, Yokelson R, de Gouw JA. 2011. VOC identification and inter-comparison from laboratory biomass burning using PTR-MS and PIT-MS. Int J Mass Spectrom. 303(1):6–14.
- White WE, Jr, Pruitt KM, Mansson-Rahemtulla B. 1983. Peroxidase-thiocyanate-peroxide antibacterial system does not damage DNA. Antimicrob Agents Chemother. 23(2):267–272.
- Whittaker SG, Zimmermann FK, Dicus B, Piegorsch WW, Resnick MA, Fogel S. 1990. Detection of induced mitotic chromosome loss in Saccharomyces cerevisiae–an interlaboratory assessment of 12 chemicals. Mutat Res. 241(3):225–242.
- Whysner J, Steward RE, Chen D, Conaway CC, Verna LK, Richie JP, Ali N, Williams GM. 1998. Formation of 8-oxodeoxyguanosine in brain DNA of rats exposed to acrylonitrile. Arch Toxicol. 72(7):429–438.
- Willhite CC, Ferm VH, Smith RP. 1981a. Teratogenic effects of aliphatic nitriles. Teratology. 23(3):317–323.
- Willhite CC, Marin-Padilla M, Ferm VH, Smith RP. 1981. Morphogenesis of axial skeletal (dysraphic) disorders induced by aliphatic nitriles. Teratology. 23(3):325–333.
- Williams BJ, Ballenger CA, Malter HE, Bishop F, Tucker M, Zwingman TA, Hassold TJ. 1993. Non-disjunction in human sperm: results of fluorescence in situ hybridization studies using two and three probes. Hum Mol Genet. 2(11):1929–1936.
- Williams GM, Kobets T, Duan JD, Iatropoulos MJ. 2017. Assessment of DNA binding and oxidative DNA damage by acrylonitrile in two rat target tissues of carcinogenicity: implications for the mechanism of action. Chem Res Toxicol. 30(7):1470–1480.
- Williams GM, Tong CV, Brat S. 1985. Tests with the rat hepatocyte primary culture/DNA-repair test. Prog Mutat Res. 5:341–345.
- Wilson DM, Thompson LH. 2007. Molecular mechanisms of sister-chromatid exchange. Mutat Res. 616(1–2):11–23.
- Wilson RH, McCormick WE. 1949. Acrylonitrile; its physiology and toxicology. Ind Med Surg. 18(6):243–245.
- Wolff J. 1998. Perchlorate and the thyroid gland. Pharmacol Rev. 50(1):89–105.
- Working PK, Bentley KS, Hurtt ME, Mohr KL. 1987. Comparison of the dominant lethal effects of acrylonitrile and acrylamide in male F344 rats. Mutagenesis. 2(3):215–220.
- Wu WK, Su J, Huang MY. 1994. Epidemiological investigation of reproductive outcomes in acrylonitrile-exposed male workers. J Clio Ind Hygiene Occup Med. 12:261–263.
- Wu X, Jin T. 2000. An overview of the study of acrylonitrile on reproductive toxicology. J Labour Med. 17(1):51–52.
- Wu W, Su J, Huang M. 1995. An epidemiological study on reproductive effects in female workers exposed to acrylonitrile. Zhonghua Yu Fang Yi Xue Za Zhi (China Prevent Med Mag). 29:83–85. (Chinese).
- Xu D-X, Zhu Q-X, Zheng L-K, Wang Q-N, Shen H-M, Deng L-X, Ong C-N. 2003. Exposure to acrylonitrile induced DNA strand breakage and sex chromosome aneuploidy in human spermatozoa. Mutat Res. 537(1):93–100.
- Yang B, Zhao W, Yin C, Bai Y, Wang S, Xing G, Li F, Bian J, Aschner M, Cai J, et al. 2021. Acute acrylonitrile exposure inhibits endogenous H2S biosynthesis in rat brain and liver: the role of CBS/3-MPST-H2S pathway in its astrocytic toxicity. Toxicology. 451:152685.
- Yang J, Duerksen-Hughes P. 1998. A new approach to identifying genotoxic carcinogens: p53 induction as an indicator of genotoxic damage. Carcinogenesis. 19(6):1117–1125.
- Yates JM, Fennell TR, Turner MJ, Recio L, Sumner SC. Jr 1994. Characterization of phosphodiester adducts produced by the reaction of cyanoethylene oxide with nucleotides. Carcinogenesis. 15(2):277–283.,
- Yates JM, Summer SC, Turner MJ, Recio L, Fennell TR. 1993. Characterization of an adduct and its degradation product produced by the reaction of cyanoethylene oxide with deoxythymidine and DNA. Carcinogenesis. 14(7):1363–1369.
- Yauk CL, Aardema MJ, van Benthem J, Bishop JB, Dearfield KL, DeMarini DM, Dubrova YE, Honma M, Lupski JR, Marchetti F, et al. 2015. Approaches for identifying germ cell mutagens: report of the 2013 IWGT workshop on germ cell assays(⋆). Mutat Res Genet Toxicol Environ Mutagen. 783:36–54.
- Yokelson RJ, Karl T, Artaxo P, Blake DR, Christian TJ, Griffith David WT, Guenther A, Hao WM. 2007. The tropical forest and fire emissions experiment: overview and airborne fire emission factor measurements. Chemistry and Biochemistry Faculty Publications. 3. https://scholarworks.umt.edu/chem_pubs/3.
- Zeiger E, Haworth S. 1985. Tests with a preincubation modification ofthe Salmonella/microsome assay. In: Ashby J, de Serres FJ, Draper M, Ishidate M, Jr, Margolin BH, Matter BE, Shelby MD, editors. Progress in mutation research: evaluation of short-term tests for carcinogens; report of the international programme on chemical safety’s collaborative study on in vitro assays. Vol. 5. Amsterdam: Elsevier Science Publishers; p. 187–199.
- Zhang H, Kamendulis LM, Klaunig JE. 2002. Mechanisms for the induction of oxidative stress in Syrian hamster embryo cells by acrylonitrile. Toxicol Sci. 67(2):247–255.
- Zhang H, Kamendulis LM, Jiang J, Xu Y, Klaunig JE. 2000. Acrylonitrile-induced morphological transformation in Syrian hamster embryo cells. Carcinogenesis. 21(4):727–733.
- Zhurkov VS, Shram RY, Dugan AM. 1983. Analysis of the mutagenic activity of acrylonitrile. Gig Sanit. 1:71–72.
- Zimmermann FK, Mayer VW, Scheel I, Resnick MA. 1985. Acetone, methyl ethyl ketone, ethyl acetate, acetonitrile and other polar aprotic solvents are strong inducers of aneuploidy in Saccharomyces cerevisiae. Mutat Res. 149(3):339–351.