
Abstract
During protein biosynthesis, ribosomes bind to messenger (m)RNA, locate its protein-coding information, and translate the nucleotide triplets sequentially as codons into the corresponding sequence of amino acids, forming proteins. Non-coding mRNA features, such as 5′ and 3′ untranslated regions (UTRs), start sites or stop codons of different efficiency, stretches of slower or faster code and nascent polypeptide interactions can alter the translation rates transcript-wise. Most of the homeostatic and signal response pathways of the cells converge on individual mRNA control, as well as alter the global translation output. Among the multitude of approaches to study translational control, one of the most powerful is to infer the locations of translational complexes on mRNA based on the mRNA fragments protected by these complexes from endonucleolytic hydrolysis, or footprints. Translation complex profiling by high-throughput sequencing of the footprints allows to quantify the transcript-wise, as well as global, alterations of translation, and uncover the underlying control mechanisms by attributing footprint locations and sizes to different configurations of the translational complexes. The accuracy of all footprint profiling approaches critically depends on the fidelity of footprint generation and many methods have emerged to preserve certain or multiple configurations of the translational complexes, often in challenging biological material. In this review, a systematic summary of approaches to stabilize translational complexes on mRNA for footprinting is presented and major findings are discussed. Future directions of translation footprint profiling are outlined, focusing on the fidelity and accuracy of inference of the native in vivo translation complex distribution on mRNA.
Introduction
Regulation of messenger (m)RNA translation (decoding) into the amino acid sequences of proteins is one of the major response mechanisms of the live cells to altering environmental and homeostatic stimuli (DeRisi et al. Citation1997; Raser and O'Shea Citation2005; Biology’s Big Bang Citation2007; ENCODE Project Consortium et al. Citation2007). Multiple types of small molecule, ligand, and stress responses including starvation and damage-induced control, immune activation, and viral-host interactions including translational hijacking have been discovered to be integral to the protein biosynthesis regulation. Translational regulation is required to reprogram neuronal connections, control the development of multicellular organisms and is dysfunctional in many diseases, including in cancer, where malignant transformation is often driven or fueled by the dysregulated protein biosynthesis (Hinnebusch Citation2017; Shirokikh and Preiss Citation2018; Teixeira and Lehmann Citation2019; Proud Citation2019; Robichaud et al. Citation2019; Kusnadi et al. Citation2020). The translation is one of the most rapid pathways of a constructive (anabolic; altering the balance of genetic output by synthesis and not degradation or sequestration of biomolecules) cellular response (Advani and Ivanov Citation2019).
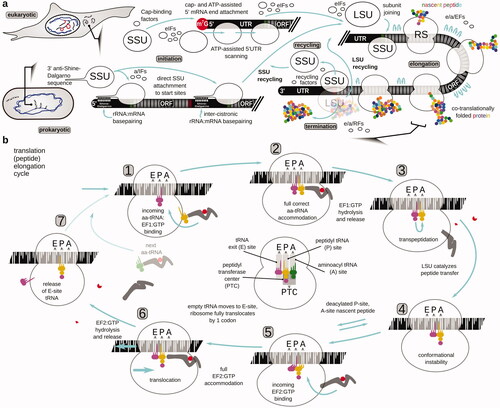
The translation is universally carried out by ancient ribonucleoprotein molecular machines, the ribosomes, which are subdivided into the small (“genetic” and mRNA-binding) and large (“catalytic” and nascent peptide-producing) subunits (the SSU and LSU, respectively), as well as the transfer(t)RNAs charged with respective amino acids, and the protein factors of translation () (Kozak Citation2005; Hinnebusch Citation2017; Shirokikh and Preiss Citation2018; Hershey et al. Citation2019). The mRNA translation cycle most commonly begins with the attachment of specially configured (“initiating”) SSU to mRNA, location of the first coding nucleotide triplet (start codon; collectively peptide synthesis initiation), and then iteration through the coding sequence nucleotide triplets (codons of the open reading frame (ORF) of mRNA) until a nonsense (stop) codon is encountered (peptide elongation). Notably, during peptide elongation, the incoming aminoacyl-tRNA is brought into the respective aminoacyl-tRNA binding site (A-site) of the ribosome (with the help of GTP-dependent tRNA carrier elongation factor e/aEF1A or EF-Tu), and then is shifted into the peptidyl-tRNA binding site (P-site) upon transpeptidation catalyzed by the ribosomal peptidyl transferase center (PTC) and translocation (assisted by the GTP-dependent translocation factor e/aEF2 or EF-G), followed by its shift into the tRNA exit site (E-site) before the detachment from the ribosome (). The A-, P-, and E-sites stretch across both, SSU and LSU, while the PTC is located between the A- and P-sites of the LSU (). Upon reaching the stop codon the ribosomes, most commonly, release the produced peptide (peptide synthesis termination), which is often already folded into the respective protein. Ribosomes are then recycled to start the next round of translation, often through a step-wise disassembly into subunits, with the LSU leaving the mRNA first (). Steps of the translation cycle are facilitated through the alternated binding and release of translation factors (initiation, elongation, termination, and recycling proteins), with the unidirectionality ensured by energy expense in the form of ATP/GTP/NTP hydrolysis. The respective factor and ribosomal structures are diverse enough in minor detail across the taxa so that small molecule inhibitors and antibiotics may have a substantially different activity on the elongation and termination processes, particularly between the higher-order taxonomic groups, such as Eubacteria, Archaea, and Eukarya (Wintermeyer et al. Citation2004; Schmeing and Ramakrishnan Citation2009; Ramakrishnan Citation2014; Frank Citation2017). Ribosomal recycling and translation initiation are evolutionarily more variable, with the largest distinction between the pro- and eukaryotic modes falling on the translation initiation, and, specifically, localization of the start codons. In prokaryotes, translation is co-transcriptional, mRNA is arranged in cistrons (i.e. has several tandem ORFs) and the start site selection is largely defined through the direct interaction of mRNA with SSUs via the Shine-Dalgarno:anti-Shine-Dalgarno sequence basepairing (or interactions with the SSU proteins). The start site can otherwise be located anywhere in the mRNA sequence. In eukaryotic organisms, most start codons are found by the SSUs first binding at the 5′ cap structures of mRNA, then scanning the 5′ untranslated region (UTR) of mRNA in the 3′ direction, and locating commonly the first encountered start site (start codon embedded in the species-specific nucleotide sequence or “context”). Until the start codon recognition step, eukaryotic and prokaryotic initiation are different enough to have a few common mechanisms of control and small molecule inhibitors. All initiation, elongation, termination, and recycling can be regulated in the cells, to various degrees globally and in an mRNA-depended fashion (Hinnebusch Citation2017; Shirokikh and Preiss Citation2018; Hershey et al. Citation2019). Transcript-specific control can be augmented by the covalent nucleotide modification patterns of mRNA and in eukaryotes, splicing-defined alterations including difficultly-discovered nuances, such as microexons (Reixachs-Solé et al. Citation2020).
Figure 1. Overview of the protein biosynthesis pathway (translation). (a) The mRNA translation cycle of eukaryotic and prokaryotic cells, depicting the peptide initiation, elongation and termination, and ribosome recycling phases of the cycle. The focus is brought to the start codon location differences during the initiation phase. For more details about the initiation mechanisms, refer to the respective reviews (e.g. Shirokikh and Preiss Citation2018; Hinnebusch et al. Citation2016; Hinnebusch Citation2017; Hershey et al. Citation2019; Sonenberg and Hinnebusch Citation2009; Gualerzi and Pon Citation2015; Rodnina Citation2018). (b) Steps of translation elongation cycle as ribosome ratchets through the codons of an open reading frame of mRNA and polymerizes the polypeptide. The focus is brought to the events and intermediates that can be specifically targeted and/or can have an impact on the translation complex footprint features and distribution. A taxa-indifferent elongation factor designation is used, with EF1 referring to eEF1A in eukaryotes, aEF1A in archaea and EF-Tu in bacteria, and EF2 referring to eEF2 in eukaryotes, aEF2 in archaea and EF-G in bacteria. In the center of the cycle schematic, the respective aminoacyl (A), peptidyl (P), and exit (E) tRNA localization and functional sites of the SSU and LSU are highlighted (populated with tRNAs from the cycle schematic to provide an example), and the location of the peptidyl transferase center in the LSU (PTC) is schematized.
The classical approaches of protein synthesis investigation comprised early works on “footprinting” of ribosomes on mRNA, where sequences of mRNA protected by ribosomes from a nuclease attack were identified for particular transcripts (Kozak and Shatkin Citation1976; Wolin and Walter Citation1988). The toolset has been substantially expanded with the reverse transcription-based approaches in quantifying mRNA abundance in translated vs. non-translated fractions by polymerase chain reaction (PCR) and ex vivo or in vitro reverse transcription inhibition by translational complexes on mRNA (“toeprinting”) (Pestova et al. Citation1996; Kozak Citation1998; Shirokikh et al. Citation2010). Currently, in vivo high/super-resolution tracking of individual molecules and molecular events is popular (e.g. fluorescence correlation spectroscopy-, Forster resonance energy transfer- or photobleaching-based) (Shashkova and Leake Citation2017; Argüello et al. Citation2018; Boersma et al. Citation2019) (), combined with the large-scale detection screens based on particle sorting and fluid dynamics, as well as computed clustering-assisted cryoelectron microscopy structure determination that is capable of processing complex and heterogeneous samples. Similar to the other fields including DNA and transcription research, high-throughput sequencing methods brought another level of insight into protein biosynthesis studies (). The ability to relate positions of individual translational complexes, ribosomal load of mRNA and transcript-specific, as well as global, metagene information with the models and mechanisms of translation learned from genetic screens, individual biochemical molecular interactions and structures, set translation complex (ribosome) footprint profiling apart as one of the most powerful and versatile suite of approaches () (Jackson and Standart Citation2015; Ingolia et al. Citation2019).
Figure 2. Schematic of popular approaches to study protein biosynthesis. (a) Approaches that provide a high-throughput surveillance of protein and RNA content or report on single-molecule activity of translating ribosomes and poly(ribo)somes. (b) Polysome profiling as one of the most broadly used methods to infer translational involvement across mRNA, including in a high-throughput format when employed together with microarrays or RNA-sequencing.
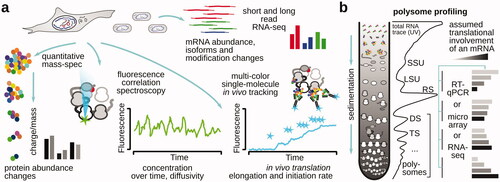
Figure 3. Schematic of nuclease probing methods to study translation. (a) A classical ribosome “footprinting” method using ribosome-protected ribonuclease (RNase)-resistant fragment identification specific to certain mRNAs of choice. (b) Translation (ribosome) footprint profiling, whereby the ribosome-protected RNase-resistant mRNA fragments are isolated based on their association with the ribosomes and then subjected to high-throughput RNA sequencing. Sedimentation-based isolation of the ribosome-associated mRNA fragments is exemplified, although other methods are possible (e.g. Reid et al. Citation2015). In the ribosome profiling, a high-throughput sequencing-derived coverage of a matching intact mRNA often serves as a coverage control and as a way of estimating the level of transnational involvement (“translation efficiency”) of the respective mRNAs (top right plot; gene-wize comparison for all genes). The relative footprint frequency over the respective ORF codons is often used to infer ribosome dwell time and the kinetic landscape of the ORF (bottom right plot; transcript-specific features).
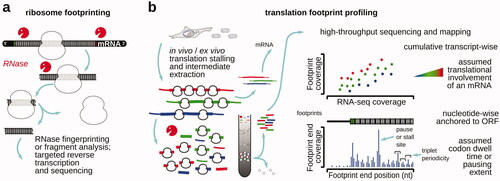
Translation complex (ribosome) footprint profiling has emerged from the combination of polysome profiling () and ribosome footprinting on mRNA () (Ingolia et al. Citation2009). In translation (or polyribosome/polysome) profiling (King and Gerber Citation2016), translational complexes are commonly separated by their sedimentation velocities or size, and the associated mRNA content across polysomes of a different order is analyzed using initially hybridization, then reverse transcription-polymerase chain reaction (RT-PCR) and microarrays, or high-throughput short and long-read sequencing (). In polysome profiling, an assumption is usually made that the higher content of ribosomes per mRNA should be interpreted as its higher propensity to attract ribosomes, particularly when applied to the same mRNA across different conditions in comparison. Ribosome footprinting has been used to identify the position of ribosomal complexes on mRNA, based on the higher ribosomal RNA resistance to certain RNA hydrolysis reactions, such as those catalyzed by single-stranded ribonucleases, due to the compact and mostly structured nature of ribosomal (r)RNA. As a result, only mRNA that is localized inside of the ribosome is left, generating ribosome-protected fragment or “footprint” (), which can be isolated and sequenced. Translation footprint profiling often combines isolation of translated RNA, generation of ribosome footprints, their high-throughput sequencing, and inference of the original ribosomal positions and configurations back in the mRNA varieties of the cell () (Wei and Qian Citation2015; Ingolia Citation2016; Andreev et al. Citation2017; Gobet and Naef Citation2017; McGlincy and Ingolia Citation2017; Ingolia et al. Citation2019). While many methods employ additional footprint purification based on their co-sedimentation with the ribosomal fractions, or co-purification with ribosomes using large size exclusion media, simplified methods based on direct RNA extraction from the nuclease-treated lysates (with subsequent size-selection of the fragments using denaturing gel-electrophoresis) have also been used (Reid et al. Citation2015).
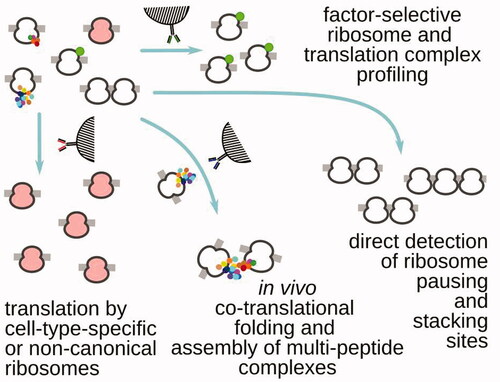
Translation footprint profiling-style experiments have been continuously evolving, bringing capabilities to detect localized translation in the cells (Jan et al. Citation2014; Williams et al. Citation2014; Clamer et al. Citation2018; Padrón et al. Citation2019), tissue and cell type-specific translation (Chen and Dickman Citation2017; Huang et al. Citation2019), translation by distinct ribosome types (Han et al. Citation2014; Ingolia et al. Citation2014; Galmozzi et al. Citation2019), as well as visualize the complete translation cycle (Archer et al. Citation2016; Shirokikh et al. Citation2017; Wagner et al. Citation2020), uncover and glean insight into processes of larger scale in the cell with high association to translation, such as co-translational mRNA-specific protein assembly (Shiber et al. Citation2018; Panasenko et al. Citation2019; Wagner et al. Citation2020) (). Footprint profiling has been used to uncover sequence- and codon-specific alterations in the translation elongation cycle (Li et al. Citation2012; Han et al. Citation2014; Woolstenhulme et al. Citation2015; Buskirk and Green Citation2017; Mohammad et al. Citation2019) and the linked mRNA destabilization mechanisms and quality checkpoints (Pelechano et al. Citation2015; Chen and Tanaka Citation2018; Ikeuchi et al. Citation2019; Han et al. Citation2020; Arpat et al. Citation2020; Wu et al. Citation2020), as well as to suggest models of translation efficiency (Riba et al. Citation2019).
Figure 4. Examples of translation (ribosome) footprint profiling that use additional methods of targeted translation complex isolation, based on the presence of (left to right) specialized ribosomes, in trans interactions with the other translating ribosomes and their nascent peptides, tightly-packed nuclease-resistant stretches of ribosomes (e.g. most commonly, di- and tri-somes) and specific translation factors associated with the respective complexes.
At the core of any ribosome or translation complex profiling experiment are consistency and fidelity of footprint generation, which define the scope of possible interpretations and the accuracy of conclusions made about any characteristic difference in translational dynamics. Critically, footprint patterns depend on the approaches taken to stabilize translational complexes on mRNA, both qualitatively as representing different complex types and locations, and quantitatively, as their abundances can be affected by unwanted destabilization or indirect effects of translation complex extraction from the cells. Enumerating or navigating through all variants of translation footprint profiling with its current diversity might be challenging. The purpose of this review is to systematize known examples of translation profiling based on the targeted stage of the translation cycle and the combination of aids used to stall translation (). Along the way, some of the major findings in the field associated with each of the examples are outlined, important limitations are discussed and avenues for future improvement of the fidelity of translation footprint profiling are presented.
Table 1. Summary of the major approaches employed to arrest or stabilize translational complexes on mRNA for downstream footprinting and footprint profiling.
Translation profiling using antibiotics targeting elongation
Inhibitors of translation elongation have been traditionally used in protein synthesis research (Vazquez Citation1974; Barbacid et al. Citation1975; Huang et al. Citation2011; Garreau de Loubresse et al. Citation2014). The main areas of use have been to study the mechanism of translation (González et al. Citation1981; Wilson Citation2009; Schneider-Poetsch et al. Citation2010; Hobson et al. Citation2020), to use as a co-crystallization aid by making certain ribosomal conformations more homogeneous (Fourmy et al. Citation1996; Pioletti et al. Citation2001; Selmer et al. Citation2006; Ermolenko et al. Citation2007; Borovinskaya et al. Citation2007; Stanley et al. Citation2010; Jenner et al. Citation2013; Svidritskiy et al. Citation2013), and to stabilize translational and polysomal complexes during subcellular fraction separation processes (Hogan and Gross Citation1971; del Prete et al. Citation2007; Vyas et al. Citation2009; Bunnik et al. Citation2013; King and Gerber Citation2016; Chassé et al. Citation2017; Chen et al. Citation2020). The classical variant of ribosome profiling (Ingolia et al. Citation2013; Ingolia Citation2016; Ingolia et al. Citation2019) is not an exception and also primarily utilized data derived from inhibitor-stabilized complexes.
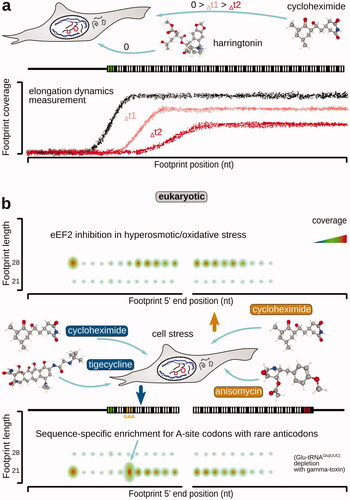
By far, the most popular translation inhibitor to perform ribosome profiling in eukaryotic cells has been cycloheximide (, ; also known as actidione, naramycin A). Cycoleheximide is a biogenic antibiotic from Streptomyces griseus that has been used as a fungicide, despite broad activity against eukaryotic cells and toxicity (Siegel et al. Citation1966; Sisler and Siegel Citation1967; Huang et al. Citation2011; Gupta Citation2018). Its exact mechanism of action has been accurately characterized relatively recently and may still need further detail (Schneider-Poetsch et al. Citation2010; Garreau de Loubresse et al. Citation2014). Cycloheximide has been shown to bind in the LSU E-site at the position usually occupied by the acceptor stem of the outgoing tRNA (Garreau de Loubresse et al. Citation2014). The binding occurs rapidly and competitively, displacing E-site tRNA if it is present (Schneider-Poetsch et al. Citation2010; Garreau de Loubresse et al. Citation2014). Cycloheximide is thus considered to block translocation reaction (Obrig et al. Citation1971) after the transpeptidation has been completed (Schneider-Poetsch et al. Citation2010; Garreau de Loubresse et al. Citation2014). Cycloheximide-protected ribosomal fragments have been found universally within the 27–32 nt range, with a modal peak at 28 nt (Lareau Liana et al. Citation2014; Ingolia Citation2016; Gerashchenko and Gladyshev Citation2017; Ingolia et al. Citation2019). It can be expected that the cycloheximide-stabilized ribosome carries at least peptide-tRNA in the A-site and likely the deacylated P-site tRNA, possibly eEF2:GTP.
Figure 5. Popular specific inhibitors of translation elongation phase used to stabilize translational complexes in the footprint profiling research of eukaryotic cells. (a) Specific translation elongation inhibitors often used in translation footprint profiling research, their chemical structures and schematized mechanism of action (refer to the for more information on the elongation cycle context of the stabilized step). 3D (when available) and 2D structures of inhibitors were downloaded from PubChem (unless otherwise indicated). (b) Typical features of translation footprint patterns associated with the use of respective inhibitors (as indicated by arrows). Shown are schematized commonly observed variants of metagene or transcript-wise footprint distributions, aligned by footprint 5′ or 3′ ends across the sequence of a coding region of mRNA near the respective start (left, green) or stop (right, red) codons (see color version of this figure at www.tandfonline.com/ibmg).
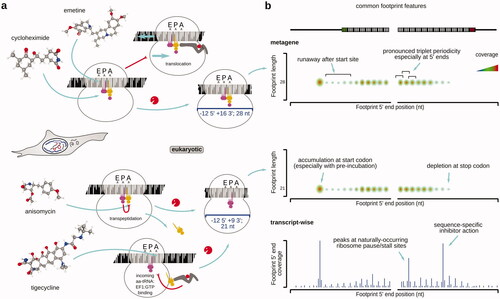
Table 2. Examples of antibiotics and inhibitors used in ribosomal footprinting research to study peptide elongation phase of protein biosynthesis.
Cycloheximide has good cell permeability (Abe and Hiraki Citation2009) and is a relatively potent translation complex binder, with a Kd of 0.14 µM for yeast ribosomes (Garreau de Loubresse et al. Citation2014) [15 µM for mammalian ribosomes (Schneider-Poetsch et al. Citation2010); ]. Nonetheless, much lower dissociation constants are known for many other translation inhibitors, particularly from the macrolide family and alkaloids, such as narciclasine. A noteworthy feature of the cycloheximide-stabilized translational complexes is the partial reversibility of the cycloheximide action and its propensity to dissociate, as well as to allow some “leakage” of the complexes into the subsequent states (Ingolia Citation2016; Mohammad et al. Citation2019; Sharma et al. Citation2019; Koga et al. Citation2021). Thus, many works employ a somewhat related emetine (see more details below), which is characterized by a more “irreversible” action (Ingolia et al. Citation2014; Walczak et al. Citation2019; Hobson et al. Citation2020). The search for the new less reversible analogs continues, e.g. C13-aminobenzoyl cycloheximide has been identified as a more stable and potent ribosome binder (Koga et al. Citation2021). Notably, despite its excellent cell permeability properties which are evolutionary relatable to fighting off fungal competition, cycloheximide induces several known footprint location biases (Gerashchenko and Gladyshev Citation2014; Hussmann et al. Citation2015; Bartholomäus et al. Citation2016; Weinberg et al. Citation2016; Duncan and Mata Citation2017; Gerashchenko et al. Citation2018; Santos et al. Citation2019; Sharma et al. Citation2019). The best-characterized biases are the over-representation of the start codon footprints and under-representation of the footprints derived from ORF 5′-end-proximal ribosomes, partly due to their “leakage” into the downstream codons (Kozak Citation1998; Schneider-Poetsch et al. Citation2010; Weinberg et al. Citation2016) (). More complex and less explored biases may appear in a codon context-dependent manner and when dynamics of translation needs to be investigated (see further in the “Capturing the dynamic state of translational control” section).
The second single-used inhibitor for translation complex footprinting in eukaryotic cells is anisomycin (flagecidin). Anisomycin is slightly more compact than cycloheximide and thus may have better permeability and diffusion features. However, anisomycin has a higher dissociation constant (0.1 mM) and a working concentration range of >1 mM (Kozak Citation1998; Blaha et al. Citation2008). Importantly, anisomycin has a different mechanism of inhibition and acts by binding to the PTC of the LSU, blocking transpeptidation reaction (Garreau de Loubresse et al. Citation2014). The ribosomes are thus stalled in a “rotated” state, with A- and P-sites occupied with the respective tRNAs (except for the stop codon recognition scenario) (Garreau de Loubresse et al. Citation2014). Anisomycin-induced footprints are in the 20–23 nt range with a mode at 21 nt (Lareau Liana et al. Citation2014; Wu et al. Citation2019). The major length difference has been attributed to the openness of the ribosomal A-site as a result of the un-conjugated aminoacyl-tRNA loss from the A-site (Wu et al. Citation2019). Anisomycin is known to allow for a more substantial “leakage” in both, classical toeprinting and footprinting experiments (usually several codons) (Kozak Citation1998; Shirokikh et al. Citation2010; Wu et al. Citation2019), with even a high anisomycin concentration applied to an in vitro system (no permeability barrier) resulting in the transition of the translation complexes into the later states (codons) on mRNA (Kozak Citation1998; Dmitriev et al. Citation2003).
In bacterial systems, chloroplasts and mitochondria, chloramphenicol has been broadly used to footprint ribosomal positions on mRNA (; ) (Li et al. Citation2012; Rooijers et al. Citation2013; Woolstenhulme et al. Citation2015; Shell et al. Citation2015; Basu and Yap Citation2016; Wu et al. Citation2019; Chen et al. Citation2020). Chloramphenicol binds at the A-site of the LSU’s PTC, preventing accommodation of the incoming aa-tRNA (Pestka Citation1974; Long and Porse Citation2003; Bulkley et al. Citation2010). The chloramphenicol-stabilized complexes can be expected to be in the post-translocation state with their A-sites empty. Notably, though, the footprint lengths vary broadly in the chloramphenicol-stabilized bacterial translation intermediates and occur in the 15–45 nt range (Woolstenhulme et al. Citation2015; Woolstenhulme et al. Citation2015; Mohammad et al. Citation2016; Buskirk and Green Citation2017; Mohammad et al. Citation2019; Mohammad and Buskirk Citation2019). This variability can be mostly attributed to the 5′-terminal part of the footprints, as their 3′-ends demonstrate good triplet periodicity over the ORFs (Mohammad et al. Citation2019). A short minimal protected length of 15 nt can thus indicate the accessibility of the A-site. The additional protection in the 5′ end direction has been attributed to transient anti-Shine-Dalgarno sequence interactions with the ORF sequence en rote on mRNA, also known to induce ribosome pausing over the ORFs () (Li et al. Citation2012; Mohammad et al. Citation2019). In a similarity to the cycloheximide effects in eukaryotes, chloramphenicol (and other A-site blockers, such as linezolid) induces start codon over-representation and codon/sequence-specific biases () (Marks et al. Citation2016; Mohammad et al. Citation2019). It needs to be mentioned that in addition to the inhibition biases, nucleases suboptimal in the evenness of nucleotide specificity are often used in bacterial systems, as some bacteria efficiently inhibit E. coli RNase I (Gerashchenko and Gladyshev Citation2017; Mohammad et al. Citation2019; Zinshteyn et al. Citation2020). An interesting MetaRibo-Seq approach was used to concentrate RNA material and ribosomal complexes, and stabilize samples derived from difficult culture mixtures, such as those found in gut (Fremin et al. Citation2020). Initial lysis using glass beads into a 1.55 mM chloramphenicol- and RNase-inhibitor-supplemented buffer was directly followed by sodium acetate and ethanol precipitation, and then resuspension into 1.55 mM chloramphenicol-supplemented micrococcal nuclease digestion reaction (Fremin et al. Citation2020). This approach allowed to derive substantial number of footprints from >20 gut microbiome species simultaneously and translationally define >600 novel small proteins.
Figure 6. Same as , but for translation elongation inhibitors commonly used in research of bacterial translation. Erythromycin structure (Fujii et al. Citation2013) was visualized using CCDC JSmol viewer.
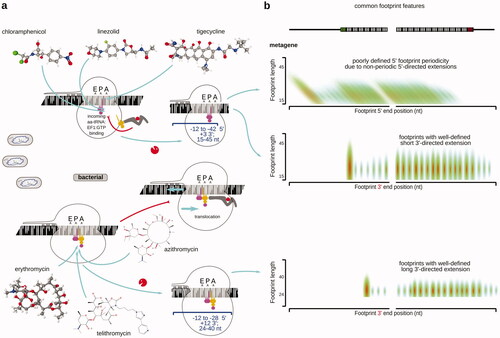
Notably, some bacterial species are “difficult” targets for using chloramphenicol due to extensive resistance mechanisms, such as efflux pumps (Vecchione et al. Citation2009). For several Streptomyces species with resistance (Streptomyces clavuligerus, Streptomyces coelicolor, Streptomyces avermitilis, Streptomyces lividans, Streptomyces venezuelae, and Streptomyces tsukubaensis), thiostrepton was successfully used as a suitable replacement () (Jeong et al. Citation2016; Hwang et al. Citation2019; Kim et al. Citation2020). Thiostrepton is known to bind to the L11/Helix 43/44 protuberance/stalk base (entry, A-proximal side of prokaryotic LSU) and interfere with the binding and function of translation factors, including IF2, EF-Tu, and EF-G (Wilson Citation2009; Polikanov et al. Citation2018). Thiostrepton thus has an ability to suppress initiation as well as accommodation of the incoming aa-tRNA and translocation, likely mimicking the effects of cycloheximide/tigecyclin combination found in eukaryotic cells, but with less bias toward the start codon-associated ribosomes.
Several future directions can be outlined for the use of the full spectrum of antibiotic elongation inhibitors, as only a few elongation inhibitors have been used for genome-wide footprinting (; also references Yonath Citation2005; Tenson and Mankin Citation2006; Wilson Citation2014; Dmitriev et al. Citation2020). In eukaryotic systems, tigecycline (results in post-translocation ribosomes with the empty A-site) (Wu et al. Citation2019) and emetine (results in pre-translocation ribosomes with the occupied A-site) (Ingolia et al. Citation2011; Dunn et al. Citation2013) have been used, each leading to more codon-based elongation cycle insights and different footprint lengths (; ). Tigecycline binds to the SSU in a similarity to tetracycline, occupying A-site and clashing with the A-site tRNA (Schedlbauer et al. Citation2015). Tigecycline has been becoming more popular when there is a requirement to capture post-translocation ribosomes as it allows stabilization of the A-site-unoccupied ribosome. Tigecycline was used to stabilize complexes resulting from non-optimal decoding (such as over CGG, CGA, CUG, and AGU codons) when Carbon Catabolite Repression Negative On TATA-less (CCR4-Not) is binding to the slowed-down ribosome via its Not5 subunit bound to the E-site (and competing with eIF5A), to degrade the slow mRNA and its protein (Buschauer et al. Citation2020). More have been tried in bacteria, such as macrolides erythromycin, telithromycin (Kannan et al. Citation2014), and azithromycin (Davis et al. Citation2014) (result in pre-translocation ribosomes with the occupied A-site), all showing high sequence selectivity of action (; ). While the use of different elongation inhibitors may not lead to better standardization of the profiling experiments (Wei and Qian Citation2015; Weinberg et al. Citation2016; Berg et al. Citation2020), it certainly is of a high utility for gaining more mechanistic insights and/or devising approaches with kinetically or dynamically improved stabilization of the translation intermediates. Inhibitors with a compact structure, better binding and potentially better permeability or kinetics, such as narciclasine (González et al. Citation1981; Garreau de Loubresse et al. Citation2014), remain to be tried in translation profiling research.
Translation profiling using antibiotics targeting initiation
Translation initiation remains one of the primary targets of research due to its role in transcript-specific protein synthesis adjustments and responses. Eukaryotic translation initiation is notoriously complex and is thought to participate in important cellular processes, such as differentiation, synaptic plasticity, immunity and stress response. Bacterial and archaeal translation initiation, despite the apparent simplicity, substantially contributes to the gene expression control with more granularities exposed by recent research (Rudler et al. Citation2019; Saito et al. Citation2020a).
Regular ribosome profiling arresting elongating ribosomes over the ORFs already provides an insight into the initiation site selection. Further, multiple recently developed bioinformatic approaches help to extract parameters characterizing translation initiation efficiency from the global ribo-seq data (Riba et al. Citation2019; Szavits-Nossan and Ciandrini Citation2020). A more direct, biochemical highlighting of initiation sites can still be beneficial. For example, the presence of multiple/short/suboptimal ORFs, upstream (u)ORFs and start sites can hinder start site identification in the total elongating ribosome footprint data. Antibiotics specifically stalling start-codon-recognizing, but not elongating, ribosomes are usually employed to de-noise the data in this case ().
Table 3. Examples of antibiotics and inhibitors used in ribosomal footprinting research to study peptide initiation phase of protein biosynthesis.
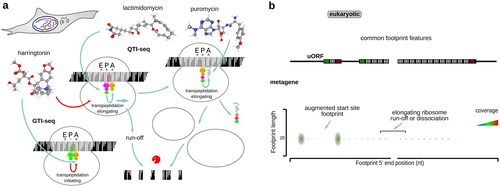
For eukaryotic initiation, lactimidomycin and harringtonin have been used early on (; ) (Ingolia et al. Citation2011; Lee et al. Citation2012; Ingolia et al. Citation2013; Gao et al. Citation2015; Zhang et al. Citation2017). Lactimidomycin has a structure related to cycloheximide and is likewise thought to compete with the E-site tRNA, inhibiting translocation (Garreau de Loubresse et al. Citation2014). Unlike cycloheximide, lactimidomycin does not displace tRNA from the E-site and is less active on any but the start codons, where E-site is unoccupied (Garreau de Loubresse et al. Citation2014). Notably, as lactimidomycin-stabilized start codon ribosomes are insensitive to puromycin, puromycin can further be employed to remove the remaining elongating ribosomes (“QTI-seq”), excluding the necessity of a prolonged run-off incubation and creating a high signal-to-noise start site profile (Gao et al. Citation2015; Aviner Citation2020; Hobson et al. Citation2020). Puromycin alone, due to a lesser sensitivity of initiating ribosomes to it, has been used to produce start site-augmented profiles (Fritsch et al. Citation2012; Aviner Citation2020). Harringtonin was deduced to inhibit the accommodation of the incoming A-site tRNA on the LSU (Fresno et al. Citation1977) and is likely to bind to the A-site of the LSU as the congruently structured homoharringtonine, inhibiting transpeptidation (Garreau de Loubresse et al. Citation2014). Homoharringtonine has been also successfully used to highlight archaeal initiation sites (Gelsinger et al. Citation2020). Harringtonin results in run-off and dissociation of the elongating, but not start codon-associated, ribosomes (“GTI-seq”) (Ingolia et al. Citation2012). Both, lactimidomycin and harringtonin are usually employed with a pre-incubation of the live cells (Yang et al. Citation2015), or post-incubation of the cell lysates, sometimes for as long as 30 min (Lee et al. Citation2012), and thus can result in an exaggerated start site landscape.
Figure 7. Same as , but for translation inhibitors and their combinations commonly used to study eukaryotic translation initiation mechanisms.
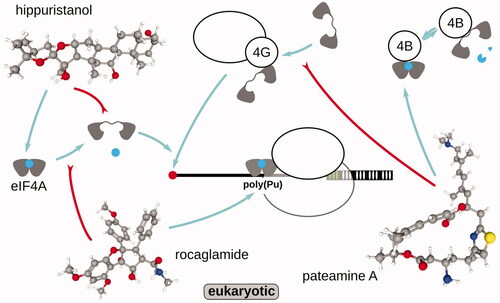
RNA “helicase” eIF4A has long been implicated in the energy coupling for the ATP-depended SSU scanning of mRNA 5′UTRs, a key process of eukaryotic translation initiation. Thus, eIF4A inhibitors, such as hippuristanol, rocaglamide, and pateamine A have been helpful to study the peculiarities of eIF4A-involved initiation (; ). Hippuristanol and related compounds have one of the highest known cytotoxicities against tumor cell lines (Higa et al. Citation1981; González et al. Citation2001) and have been shown to bind to the C-terminal domain of eIF4A (Lindqvist et al. Citation2008; Chang et al. Citation2009; Cencic and Pelletier Citation2016). Hippuristanol binding abolishes eIF4As’ (eIF4AI and eIF4AII) ability to bind RNA and not ATP (Bordeleau et al. Citation2006), but is exceptionally selective and has much diminished activity on related eIF4AIII and other helicases (Cencic and Pelletier Citation2016). Consequently, in the footprinting studies, hippuristanol-sensitive mRNAs were defined as having longer and, potentially, more structured 5′UTRs (Steinberger et al. Citation2020). Rocaglamide and related compounds, such as silvestrol, have been proposed to attach near the RNA-binding pocket and lock eIF4A in the RNA-bound state (without ATP requirement), increasing the helicase activity in low but non-competitively inhibiting eIF4A and scanning in high concentrations (Sadlish et al. Citation2013; Iwasaki et al. Citation2016). In the footprinting studies, rocaglamide has induced translational suppression in a highly sequence-specific manner, attaching to and inhibiting initiation specificity over polypurine sequences (Iwasaki et al. Citation2016). Rocaglamide was then shown to preferentially lock on eIF4A complex with purines due to stacking (Iwasaki et al. Citation2019). Conversely, pateamine A was used to additionally simplify footprint pattern and highlight some (supposedly, less cap-dependent) start sites, while removing most of the signal elsewhere upon 1 µM, 10-min incubation (Popa et al. Citation2016). Pateamine A has been proposed to stabilize eIF4A:eIF4B interaction and ATPase activity, while destabilizing eIF4G:eIF4A interaction (Low et al. Citation2005; Khong et al. Citation2016). It needs to be mentioned that in all these cases, thus far, the readout of the initiation efficiency has been done by measuring the footprint accumulation over start sites (the complete assembled post-initiation ribosome) or ORFs (elongating ribosomes) vs. some normalizing factor, and not the effects to the scanning intermediates or translation initiation rates as such.
Figure 8. Small compounds targeting eukaryotic translation initiation factor and ATP-dependent single-stranded RNA binding protein (RNA helicase) eIF4A and its interactions with the substrates (mRNA, ATP) and other initiation factors and complexes, such as eIF4G:SSU and eIF4B. For more details about scanning and eIF4A action, refer to the specialized reviews (e.g. Shirokikh and Preiss Citation2018; Hinnebusch et al. Citation2016; Hinnebusch Citation2017; Naineni et al. Citation2020). Schematized mechanism of action of each compound is depicted. Note that in the majority of the observations, the footprint signal is found to be concentrated at or nearby the respective ORF start sites, and is observed in the footprints derived from the fraction of translational complexes containing complete ribosomes (as opposed to SSUs).
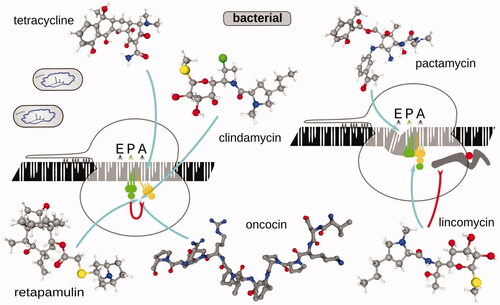
In bacteria, tetracycline, pactamycin, and clindamycin have been used to highlight initiation sites (; ) (Nakahigashi et al. Citation2016; Weaver et al. Citation2019; Meydan et al. Citation2019; Saito et al. Citation2020a). Tetracycline is known to have the primary binding location near the SSU A-site (other sites in SSU and LSU), and while not fully preventing EF-Tu:aminoacyl-tRNA:GTP binding and GTP hydrolysis, is thought to eject the aminoacyl-tRNA before the transpeptidation occurs (Schnappinger and Hillen Citation1996; Brodersen et al. Citation2000; Pioletti et al. Citation2001; Blanchard et al. Citation2004; Jenner et al. Citation2013). Notably, while tetracycline inhibits elongation broadly, it has been observed that under the footprinting conditions over 50% of tetracycline-stabilized complexes sharply localize at start sites, resulting in prominent start site highlighting (Nakahigashi et al. Citation2016). Clindamycin, which binds at the LSU A-site near PTC and blocks aminoacyl-tRNA binding, resulted in a tetracycline-similar start site-augmented profile (Dunkle et al. Citation2010; Ermolenko et al. Citation2013; Matzov et al. Citation2017), as did pactamycin. Similar properties of lyncomicin to stall prokaryotic early elongating ribosomes (up to ∼5 amino acid-long peptides; ribosomes with longer polypeptides are not efficiently stalled) were used to interrogate light-dependent dynamics of elongation in chloroplasts, by following a 10-min (or more) lyncomicin pre-incubation (Chotewutmontri and Barkan Citation2018). Pactamycin is known to bind at the P- and E-sites of the SSU, mimicking and displacing mRNA nucleotides, and thus capable of directly acting on initiating ribosomes (Brodersen et al. Citation2000). Interestingly, the exact mechanism of start site emphasis in the cases of tetracycline and clindamycin remains unclear, but (a) insufficient stabilization away from the just initiated ribosome or (b) less efficient inhibition of elongating ribosomes and run-off due to the competitive nature of the binding could be rationalized. Oncocin Onc112 and retapamulin have also been popular recently (; ) (Weaver et al. Citation2019; Meydan et al. Citation2019; Saito et al. Citation2020a). Retapamulin is a general transpeptidation inhibitor that binds at the LSU PTC mostly at A- and partly P-sites (Poulsen et al. Citation2001; Schlünzen et al. Citation2004; Davidovich et al. Citation2007; Schwarz et al. Citation2016). Its weak action on elongating ribosomes at lower concentrations (e.g. 12.5 µg/ml) allowed to specifically preserve start codon-associated ribosomes by allowing elongating ribosomes to run off in a 5-min incubation, resulting in ∼25–45 nt footrpint range (15 nt from the 3′ end to P-site) (Weaver et al. Citation2019; Meydan et al. Citation2019; Saito et al. Citation2020a). Onc112 is a “proline-rich antimicrobial” peptide that binds at the PTC of the LSU closer to the A-site, and interferes with the positioning of both tRNAs, as well as it blocks the peptide exit tunnel (Roy et al. Citation2015; Seefeldt et al. Citation2015, p. 112; Lomakin et al. Citation2015; Arenz and Wilson Citation2016). Notably, Onc112 is known to bind mRNA- and fMet-tRNAfMet-containing ribosomes (Roy et al. Citation2015), effectively stabilizing start codon-bound initiating intermediates, while it was found not to affect elongating ribosomes much (presumably through the competitive effects of the nascent peptide) (Weaver et al. Citation2019). Onc112 applied with a 10-min pre-incubation at 50 µM, in similarity to retapamulin, also resulted in comparable footprint profiles with sharp start codon-associated peaks, but with a shorter 3′-directed protection of 6–10 nt from the P-site (Weaver et al. Citation2019; Saito et al. Citation2020a).
Figure 9. Same as , but for translation inhibitors commonly used to study bacterial translation initiation mechanisms. Oncocin Onc112 PDB structure 4ZER was visualized using PDB Mol viewer (Seefeldt et al. Citation2015).
Overall, the use of translation initiation inhibitors already has allowed for a deep discovery of nearly all possible start sites across the taxa, and sometimes shed light on the early initiation substeps, such as eIF4A-depended initiation. Clearly, available and new compounds with distinct action on different stages of initiation will allow for even more insightful conclusions. Nonetheless, a more broadly defined translation initiation process that includes earlier events and intermediates of initiation, such as sliding of the bacterial and scanning of the eukaryotic SSUs at the respective UTRs, remains mostly obscured. No specific inhibitors are known that reliably stall and stabilize early translation initiation intermediates on mRNA to obtain their footprints, and any progress in this area will result in vastly improved approaches to investigate translation.
Translation profiling using antibiotics targeting termination and ribosomal recycling
Translation termination, generally, may not be extensively regulated with genetic, evolved natural pathways (Korostelev Citation2011; Nürenberg-Goloub and Tampé Citation2020). A rationalization of this fact may be that cells prefer not to translate a message at all, rather than halt translation when the protein is nearly produced, as it complicates protein folding, ribosome accessibility and interferes with multiple pathways induced by long ribosomal dwell time at a particular site, such as nonsense-mediated mRNA decay (Mühlemann and Karousis Citation2017; Ikeuchi et al. Citation2019; Kim and Maquat Citation2019; Nürenberg-Goloub and Tampé Citation2020). However, the same absence of evolutionary pathways to deal with halted termination or recycling makes them perfect drug targets.
Intriguingly few profiling-style experiments have been performed overall, and none have yet used an inhibitor that would specifically stabilize termination or recycling complexes, somewhat due to the existed inaccessibility of such compounds. Aminoglicosides have been traditionally used to study the effects of termination “inhibition” as they induce codon mismatching including stop codons read-through (Fourmy et al. Citation1996; Borovinskaya et al. Citation2007; Prokhorova et al. Citation2017) (; ). Aminoglicosides commonly bind primarily at the decoding center of the SSU (with multiple secondary binding sites at both SSU and LSU) (Fourmy et al. Citation1996; Borovinskaya et al. Citation2007; Prokhorova et al. Citation2017), and can also inhibit ribosome recycling by reverting ribosome release factor action in bacteria (Borovinskaya et al. Citation2007). Comparisons of geneticin (G418), gentamicin, paromomycin, neomycin, tobramycin, and amikacin 10-min and 24-h treatments by ribosome profiling, with additional stabilization of complexes by adding cycloheximide upon the cell lysis, demonstrated the highest read-through rate induction by geneticin (G418) (Wangen and Green Citation2020). Notably, the read-through depended on the stop codon type (UGA > UAG > UAA) and its context (increased if followed by C) (Wangen and Green Citation2020). Geneticin (G418) also induced less footprints over histone transcripts and more in selenocysteine-coding mRNAs and particularly in S-adenosylmethionine decarboxylase proenzyme (AMD1) mRNA, which is negatively regulated by stalling during its uORF termination (Yordanova et al. Citation2018; Wangen and Green Citation2020). Several compounds, such as mefloquine and CDX5-1 can increase the read-through effect of aminoglycosides, which has not yet been employed in footprint profiling, as well as non-aminoglycoside read-through inducers, such as ataluren, amlexanox, and others [reviewed (Dmitriev et al. Citation2020)].
Figure 10. Specific translation termination inhibitors often used in translation footprint profiling research. (a) Mechanism of action and the stabilized state of the ribosome characteristic to the exemplified compounds. Note that the currently used translation termination inhibitors are as such inducers of read-through and thus, unlike inhibitors used to study the other phases of translation, in fact de-stabilize translation termination complexes. The shown compounds have a broad specificity across taxa and are used in both, eukaryotic and bacterial research. (b) Typical meatgene features of translation footprint patterns associated with the use of respective inhibitors (for designations, refer to ) using an eukaryotic system as example.
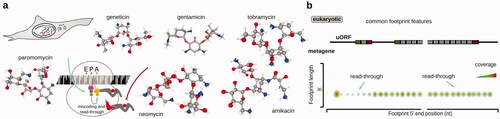
Table 4. Examples of antibiotics and inhibitors used in ribosomal footprinting research to study peptide termination and ribosomal recycling phases of protein biosynthesis.
In spite of the absence of more inhibitor-based studies, depletion or knock-down of Rli1 ribosome recycling factor (yeast ABCE1 ortholog) (Young et al. Citation2015) and SSU recycling factors Tma20 (MCT-1), Tma22 (DENR), and Tma64 (eIF2D) resulted in ribosomal stacking at the stop codon and presence over 3′UTRs, possibly through re-initiation (Young et al. Citation2018). Deletion of Dom34 (Pelota) which may rescue unrecycled ribosomes further increased the 3′UTR footprint accumulation effects (Young et al. Citation2015). Depletion of Hcr1 (eIF3j) which is implicated in mRNA displacement from the SSU also resulted in excessive 3′UTR footprint accumulation (Young and Guydosh Citation2019). A similar effect was observed in bacterial systems upon depletion of Ribosome Recycling Factor (RRF), where ribosomes were observed queuing behind stop codons and leaking into 3′UTRs and intercistronic space (Saito et al. Citation2020b). Depletion of eukaryotic Elongation Factor 3 (eEF3), previously implicated in recycling (Kurata et al. Citation2010), did not reveal major changes by ribosome profiling (Kasari et al. Citation2019).
Importantly, many potent stabilizing inhibitors of termination and recycling have not yet been employed for footprint profiling-style experiments. Blasticidin S was shown to bind near the LSU P-site and aminoacid acceptor stem of P-site tRNA, and in bacteria acts by deforming the P-site tRNA 3′ end toward A-site, which inhibits peptide release by termination factors, but has less detrimental effects on elongation (Svidritskiy et al. Citation2013). It thus has the potential to specifically stall terminating ribosomes [although there is biochemical evidence blasticidin S is less specific toward termination in eukaryotic systems (Lashkevich et al. Citation2020)].
To conclude, with the recent discovery of specific translation termination inhibitors (Ge et al. Citation2019; Li et al. Citation2020), we soon may expect many more insights into the termination and recycling dynamics and their influence on transcript-wise translational control with profiling-style experiments. An interesting and exciting synthetic biology perspective can be outlined for termination inhibitors. As termination implies the presence of a specific nascent peptide in the ribosome in every different mRNA, it may not be impossible to devise mRNA-selective inhibitors, which can have compulsive therapeutic potential.
Translation profiling using a combination of antibiotics targeting different (sub)steps of the translation cycle
A combination of translation inhibitors with different mechanisms of action can be another attractive approach to increase translation complex diversity, improve coverage of translation processes, and possibly, the kinetics of stabilization. The combination of inhibitors has also been useful in trapping multiple configurations of the translational complexes, as well as to assess the dynamics of certain translation steps by chasing one inhibitor with another or assessing the bottleneck stages of translation (Kavčič et al. Citation2020). Further, combinations of antibiotics and inhibitors can be used while stabilizing translation intermediates in difficult target cells and species and in samples with diverse taxonomy.
For peptide elongation dynamics studies, a combination of harringtonin and cycloheximide was used early on (). Pretreatment of mammalian cells (mouse embryonic stem cells) with 2 µg/ml harringtonin was followed by the addition of 100 µg/ml cycloheximide in 90, 120, and 150 s, which allowed to calculate average elongation speed of 5.6 amino acids per second (Ingolia et al. Citation2011). A similar approach was used for profiling translation in mouse myoblasts and myotubes (de Klerk et al. Citation2015). A combination of harringtonin pretreatment at 2 μg/ml for 5 min to first block initiation, followed by a 1-min cycloheximide pretreatment at 100 μg/ml, was used to discover redirection of initiation to unconventional upstream start sites during tumor initiation (Sendoel et al. Citation2017). A combination of treatments for 15 min with 25 μg/ml azithromycin and 2 min with 100 μg/ml chloramphenicol was used to reveal selective sensitivity of ORFs to macrolide translation inhibition in Staphylococcus aureus and a peptide-specific mechanism of macrolide action (Davis et al. Citation2014). 100 μg/ml chloramphenicol cell pretreatment for 15 min combined with 5-min 100 μg/ml cycloheximide treatment has been used to capture translation by mitoribosomes together with the cytoplasmic ribosomes in primary fibroblasts (Rooijers et al. Citation2013).
Figure 11. Combinations of specific translation inhibitors and their effects to study the dynamics of translation elongation and translation complex identity, as well as codon specificity, of the stalled intermediates in eukaryotic systems. (a) Cycloheximide chase following harringtonin translation initiation arrest to infer the rate of ribosomal progression along mRNA ORFs. Note that the translation initiation is stopped upon the completion of the start codon recognition and the assembly of the elongation-capable ribosome at the respective start sites. (b) Use of tigecycline-cycloheximide and anisomycin-cycloheximide combinations to arrest ribosomes with differently configured A-sites, populated (left, bottom; blocked transpeptidation) and non-populated (right, top; blocked aminoacyl-tRNA accommodation) with the incoming aminoacyl-tRNA:eEF1A:GTP, respectively. Shorter footprints (left, bottom) in this case are reflective of the delays in attracting the respective aminoacyl-tRNA, whereas longer footprints (right, top) are reflective of eEF2 deficiency (see text for more details).
A popular and successful approach to translation stalling has been established by rapid freezing and melting/lysing/homogenizing into a buffer with a cocktail of protein biosynthesis inhibitors. This may remove some of the pre-incubation biases (see more in “Capturing the dynamic state of translational control” section) and also allow for very useful access of cell-impermeable inhibitors, such as non-hydrolyzable nucleotide analogs, as well as high salt, to translational complexes. For example, dynamics of thermal stress response in Arabidopsis thaliana was investigated by thawing and homogenizing plant material in high magnesium ions (35 mM) and 100 μg/ml chloramphenicol, 100 μg/ml cycloheximide, and analyzing chloroplast translation. Co-ordinated transcriptional and translational response was revealed with some mid-course responses solely at the translational level (Lukoszek et al. Citation2016). Drosophila embryos were frozen and lysed into a buffer containing 20 μg/ml emetine and 50 μM GMP-PNP, allowing to observe translating ribosomes in 5′UTRs, as well as transcript-specific levels of read-through past stop codons (Dunn et al. Citation2013). Pulverizing plant (Zea mays leafs) material under liquid nitrogen into buffer containing high magnesium (15 mM) and 100 μg/ml chloramphenicol, 100 μg/ml cycloheximide allowed to observe all, cytoplasmic (bimodal with the prevalence of long fragments; 20–25, 28–33 nt), plastid (equibimodal; 22–32, 33–38 nt) and mitochondrial (bimodal with the prevalence of short fragments; 23–33, 34–39 nt) footprint distributions and characterize plastid elongating ribosome footprint extensions as −30 to −18 nt 5′, +7 nt 3′ ends (Chotewutmontri and Barkan Citation2016). The dynamics of translation underlying proplastid to chloroplast transition have also been investigated (Chotewutmontri and Barkan Citation2016).
Translation inhibitor combinations allowed a more refined outlook at the translation elongation cycle. For example, combining tigecycline to block accommodation of the incoming aminoacyl-tRNA in the A-site or anisomycin to block transpeptidation () with cycloheximide as a general inhibitor of subsequent steps, it was possible to dissect functional effects on different elongation cycle steps () (Wu et al. Citation2019). The shorter, modal 21 nt footprints were enriched in anisomycin/cycloheimide libraries and likely over-represented in ribosomes that have difficulty attracting the corresponding aminoacyl-tRNA. These footprints were indeed correlated with the rarity of tRNA with the respective anticodon and were most sensitive to an artificial depletion of Gly-tRNAGly via glycine starvation, His-tRNAHis via 3-amino-1,2,4-triazole treatment (inhibits histidine production) or Glu-tRNAGlu[UUC] via Kluyveromyces lactis gamma-toxin presence (cleaves tRNAGlu[UUC]), respectively (Wu et al. Citation2019). Conversely, longer 28 nt footprints increased during stress conditions (hyperosmotic and oxidative), where eEF2 is likely inactivated by phosphorylation, resulting in the preferential accumulation of tigecycline/cycloheximide-stalled intermediates (Wu et al. Citation2019). A notable observation has been that using cycloheximide alone just in the cell lysate (no in vivo pretreatment), compared to the combined use with either anisomycin or tigecycline, all resulted in different footprint profiles, suggestive of intense re-configuration of the ribosomes in the lysate and substantial progression of at least some stages of the elongation cycle (Wu et al. Citation2019). These data highlight the necessity of a uniform and rapid stabilization of translational complexes (Tesina et al. Citation2020).
A combination of 100 µg/µl each, cycloheximide and anisomycin, was used to inhibit translation in HEK293 cells and zebrafish embryos to investigate ribosome stalling by the formation of disomes. This combination helped to preserve pre- and post-translocation ribosome configurations and attribute disome-revealed stall sites to the known translation attenuation sites of SEC61B, upstream conserved coding region of AZIN1, and polylysine coding sequence in the middle of ZCRB1 ORFs. Over 600 other sites in zebrafish embryos and HEK 293 cells were characterized, ∼200 of which overlapped on the gene orthologue level, highlighting the conservation of stacking-inducing ribosomal pausing control (Han et al. Citation2020). A distinction in the efficiency of eIF5A to rescue polyproline-induced paused/stacked ribosomes depending on the nascent peptide charge was made, with −8 to −20 (from lead ribosome P-site) positively charged amino acids attenuating the stalling rescue (Han et al. Citation2020). Ribosome-associated quality control (RQC) and its E3 ligases ZNF598 and LTN1 were implicated in triggering ribosome disassembly and peptide degradation (Han et al. Citation2020). Cycloheximide and tigecycline (100 µg/µl each) combination was also used to reveal the reduced A-site occupancy (based on the shorter 21 nt footprints) in several “unfavorable” codon successions. CGA-CCG, CGA-CGA codons, and (AAA)n codons resulted in stacked binding of +4,5 and +4–7 nt of the codon nucleotides to the rRNA, respectively, and non-optimal A-site-decoding-induced stalling of ribosomes with some formation of stacked disomes both in their post-translocation states, which is augmented in the Absence of growth Suppressor of Cyp1 ribosomal protein (Asc1, yeast; Receptor of activated protein C kinase 1, RACK1, higher eukaryotes) (Tesina et al. Citation2020). Overall, the usage of translation stalling “cocktails” will remain a highly productive and popular approach and will be an actively developed direction given the continuously increasing listing of the specific inhibitory compounds.
Translation profiling without immediate high affinity or covalent stabilization of translational complexes on mRNA
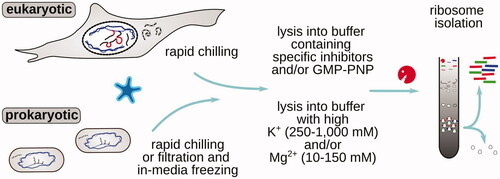
Intriguingly, it has been possible to obtain high-quality footprint datasets with the use of no inhibitor of translation, apart from some stabilization that is offered by rapid chilling and freezing of the samples (; ). Metagene plots of such experiments often appear very sharp, with well-developed triplet periodicity of the footprints over the ORFs. Arguably, the absence of any stabilizing agent removes concern for the stabilization bias it may cause (Ingolia et al. Citation2013; Hussmann et al. Citation2015).
Figure 12. Commonly accepted inhibitor-free approaches to preserve translational complexes on mRNA.
The absence of a translation inhibitor has been historically used as a type of control in ribosome profiling experiments. Although without an inhibitor nearly always less polysomes are observed in sedimentation assays, a relatively clean footprint profile can be recovered with “standard” lysis and polysome buffers in both, eukaryotic (Ingolia et al. Citation2009; Stadler and Fire Citation2011; Lee et al. Citation2012; Gerashchenko and Gladyshev Citation2014; Katz et al. Citation2014; Williams et al. Citation2014; Cenik et al. Citation2015) and prokaryotic (Kannan et al. Citation2014; Balakrishnan et al. Citation2014) cells. The footprint pattern often demonstrates some depletion of the ribosomal density in the 5′-proximal parts of the ORF (Lee et al. Citation2012; Han et al. Citation2014; Gerashchenko and Gladyshev Citation2014; Liu and Qian Citation2016; Stewart et al. Citation2018), possibly reflecting a higher sensitivity of initiation to the cell lysis, while some elongation processes continue. The start codon-associated footprint peak is usually much less pronounced compared to the elongation inhibitor pre-incubations. Even in a buffer with low magnesium (20 mM Tris pH 8.0, 140 mM KCl, 1.5 mM MgCl2, 1% Triton) (Lareau Liana et al. Citation2014), metagene footprint pattern remains well-preserved. Ribosome footprint lengths in these cases exhibit a broadened length spectrum with major 28 nt and minor 21 nt modes, presumably reflecting configurations of ribosome as stabilized with cycloheximide and anisomycin (see above in “Translation profiling using antibiotics targeting elongation”) (Lareau Liana et al. Citation2014). Yet, as many of the inhibitor-omitting studies use biophysical, rather than bioinformatic, footprint length range clipping, some information about complex diversity may have been inadvertently lost.
Most of the currently established techniques either use (1) rapid collection and freezing for initial stabilization, followed by lysis into an inhibitor-containing buffer (Stadler and Fire Citation2011; Weinberg et al. Citation2016); (2) freezing and lysis or immediate lysis into regular polysomal buffers (with 20–200 mM monovalent and 1–10 mM divalent metal ion concentrations) (Ingolia et al. Citation2009; Stadler and Fire Citation2011; Elgamal et al. Citation2014); or (3) freezing and lysis or immediate lysis into buffers containing high monovalent and divalent metal ion concentrations and sometimes additionally specific inhibitors (Ingolia et al. Citation2011).
Well-established examples include rapid yeast filtering and scraping into liquid nitrogen followed by lysis into a cycloheximide-containing buffer to investigate translational control in CAN1 deletion with increased lifespan (Beaupere et al. Citation2017). A similar approach was used to highlight the link between higher A-site codons ribosomal dwell time (“slower elongation”) to the ribosome-induced message destabilization, an application where any biased distribution of ribosomes would be highly detrimental (Hanson et al. Citation2018). Flash freezing to stabilize mammalian translation (zebrafish and HeLa cells) followed by cycloheximide or no inhibitor addition has been used early on (Stadler and Fire Citation2011), and a similar approach was applied to whole animals (Caenorhabditis elegans) (Stadler and Fire Citation2013) and zebrafish developmental course embryos, where additionally high salt (250 mM NaCl, 15 mM MgCl2), as well as 100 μg/ml CHX and 500 μg/ml GMP-PNP were used (Chew et al. Citation2013). Flash-freezing followed by grinding in the extraction buffer is well-suited for plant material and was combined with post-disruption 100 μg/ml chloramphenicol and 100 μg/ml cycloheximide addition to microarray-footprint Zea mays ribosomes (Zoschke et al. Citation2013). A similar approach was used in Glycine max seeds followed by stabilization with 200 mM KCl, 10 mM MgCl2 buffer with no inhibitor (Shamimuzzaman and Vodkin Citation2018). Flash-freezing, tissue disruption, and lysis into 50–100 μg/ml chloramphenicol and 50–100 μg/ml cycloheximide (Juntawong et al. Citation2014) or high-magnesium (35 mM) buffer (Lei et al. Citation2015) was used for footprinting in Arabidopsis thanliana. Rapid organ extraction from relatively large organisms without stabilizing translation, followed by homogenization into cycloheximide-containing buffer, was used to study circadian and temporal dynamics of translation in mouse liver (Atger et al. Citation2015; Janich et al. Citation2015; Janich et al. Citation2016).
No inhibitor or unspecific inhibition combined with rapid freezing is often used in bacterial (Oh et al. Citation2011; Subramaniam et al. Citation2014), other prokaryotic including plastid and mitochondrial (Couvillion and Churchman Citation2017; Rudler et al. Citation2019), and other “difficult” material, such as pathogenic species and parasites (Jensen et al. Citation2014; Vasquez et al. Citation2014). For mitochondrial footprints, an elevated magnesium ions (10 mM) concentration and polysome extraction buffer more compatible with bacterial ribosome preparations (higher pH of 8.0 and ammonium chloride-based formulation) are often used (Couvillion and Churchman Citation2017). The use of standard inhibitors (cycloheximide) can be ineffective for species such as Candida albicans, where rapid cell filtration, freezing, and lysis into buffer containing 10 mg/ml Blasticidin S and 50 mg/ml GMP-PNP has been successfully used (Muzzey et al. Citation2014). 3 mM GMP-PNP has also been used to additionally stabilize bacterial cell lysates before footprinting (Li et al. Citation2012). High potassium and magnesium ions (3.4 M and 100 mM, respectively) were used to stabilize ribosomes in the lysate of Halobacterium salinarum to reveal co-activation of translation in low abundant mRNAs from transcriptionally-activated genes across different growth phases, with ∼20% of regulated genes demonstrating transcriptionally non-correlated translational control (de Lomana et al. Citation2020). Additionally, AMP-PNP was applied for footprinting the ATP-helicase-bound initiating ribosomes in eukaryotic cells, revealing a broad footprint range from 34 to over 45 nt in these conditions (Iwasaki et al. Citation2016). In many instances of prokaryotic footprint preparations, ∼5 mM CaCl2 has been additionally used, although the stabilizing effects need to be better characterized in this case (Chen YX et al. Citation2020; Saito et al. Citation2020a). Interestingly, combinations of antibiotics not affecting translation were applied to investigate difficult specimens, such as in complex or contaminated cultures (Liu et al. Citation2017). Kanamycin (50 μg/ml), carbenicillin (100 μg/ml) and streptomycin sulfate (50 μg/ml) were used to quantify protein syntheses in dynoflagellate Amphidinium carterae usually containing substantial bacterial material. These antibiotics did not interfere with the measurements obtained by using no inhibitor, as well as using cycloheximide, emetine, and harringtonin (Liu et al. Citation2017).
The absence of any specific inhibitors has been used to reveal polyamine-controlled translation based on ribosomal queuing on mRNA. In this case, polyamine-induced slowdown of the elongating ribosomes over Pro-Pro-Trp motif of the antizyme inhibitor 1 (AZIN1) mRNA enhanced initiation of the upstream non-AUG-codon initiated region by the queued SSUs (Ivanov et al. Citation2018). Polyamines were further proposed to counteract eIF5A in facilitating translation through polyproline and other “difficult” sequences (Schuller et al. Citation2017; Ivanov et al. Citation2018).
Further highlighting the advantages and difficulties of non-stabilized ribosome profiling, inhibitor-free methods of translational arrest have been systematically investigated in bacterial cultures (Mohammad et al. Citation2019). The more diverse footprint lengths often attributed to prokaryotic footprints were observed in all, inhibitor-stabilized and non-stabilized cases. 15–45 nt footprint length diversity was confirmed to be linked to the footprint 5′-end heterogeneity, as suggested earlier, likely due to the transient rRNA:mRNA basepairing (O'Connor et al. Citation2013; Mohammad et al. Citation2019). 3′-end of the footprints demonstrated the highest homogeneity, in contrast to the eukaryotic systems, and thus has been mostly exclusively used for the analyzes. Pretreatment of the cells with chloramphenicol induced strong biases regardless of the cell collection method (filtering or centrifugation); and the changes were gene-specific with mostly ORF length-associated bias (the shorter the ORF, the more artificial footprinting density over start sites is influencing the gene score). Codon-wise biases were also detected as chloramphenicol better inhibited (induced accumulation of) the footprints originating from ribosomes with Ala, Gly, and Ser in the E-site (Mohammad et al. Citation2019). Most notably, milder but still substantial biases were observed when cells were rapidly collected without chloramphenicol pretreatment, even using sub-minute rapid filtering and freezing approaches previously considered ideal for the lysate collection. Using lysate translation measurements, these biases were confirmed to originate from two different sources: (1) continued translation in the cell lysate upon cell disruption and chloramphenicol addition, and (2) very rapid cell response to the removal/depletion of some of the nutrients, particularly due to the lower serine and glycine tRNA aminoacylation (Mohammad et al. Citation2019). Interestingly, the least translation in the lysates was observed with high magnesium (50–150 mM) or monovalent salt (1 M NaCl) concentrations. These high-salt conditions, combined with rapid freezing of the whole cell culture (followed by its cryo-disruption and concentration of ribosomes by sedimentation in high-salt conditions), were found to generate footprint profile with acceptably defined triplet periodicity as well as highly sensitive to codon pauses, such as directly induced by aminoacylation of certain tRNAs (e.g. isoleucine tRNA synthase inhibition by mupirocin) (Mohammad et al. Citation2019).
To conclude, unspecific stabilization by rapid freezing and lysis in generally-inhibitive conditions, such as high salt and non-hydrolyzable nucleotide analogs, has been proven to be a robust and broadly applicable method, relieving some of the biases associated with the specific inhibitor use, as well as species-specific matching of the inhibitor efficiency. However, not all complexes are efficiently stabilized as can be evidenced by the absence of reports on the SSU-derived footprints. Further, it can be hypothesized that translational complexes resulting from the more robust, as well as faster processes, would be dominating the footprint patterns in this case. While the absence of translation is critical in preserving the native ribosome footprint distribution profile, ribosomal or protein detachment (likely in high-salt buffers), as well as leakage between different configurations while over the same codon, are still in the need of addressing with methods that do not rely on specific translation complex stabilization.
Translation complex profiling using untargeted covalent stabilization
Stabilization through chemical crosslinking has long been employed to better preserve transient biomolecular complexes. Formaldehyde crosslinking-based stabilization, in particular, has been early used as a general specimen conservation method, as well as in discrete applications, such as in preparing biomolecular complexes for in-cell visualization and ex vivo electron microscopy imaging. The discovery that controlled formaldehyde crosslinking can specifically stabilize direct molecular binders in the background of high concentrations of non-binders opened the way to a multitude of versatile approaches to accurately track molecular interactions, such as in the multiple versions or Chromatin Immunoprecipitation (ChIP) and ChIP-seq, RNA bind-and-sequence (Furey Citation2012; Lambert et al. Citation2014; Lambert et al. Citation2015; Nakato and Sakata Citation2021), and others, where it outperformed any other crosslinker by the sum of the features (Hoffman et al. Citation2015). The versatility of the approach stems from the broad formaldehyde reactivity, stability of the crosslinks in most in vivo conditions (∼0–40 °C), and instability of the crosslinks at slightly higher temperatures (>50 °C), allowing ease of crosslink reversal through hydrolysis, to fully restore the original chemical structures (Hoffman et al. Citation2015). Formaldehyde is thought to first react with a strong nucleophilic group, such as primary amines of lysine, arginine, ornithine, with the formation of methylol adduct, which dehydrates into a highly reactive Schiff base, lending to very fast kinetics of the reaction (). The Schiff base can react with weaker nucleophiles in the vicinity, including amino groups of adenosine, guanosine or cytosine of DNA and RNA, as well as with hydroxyl groups in certain contexts (Hoffman et al. Citation2015) ().
Figure 13. Overview of translation complex footprinting methods based on in vivo stabilization of translation intermediates with covalent crosslinking. (a) Example of a possible formaldehyde-mediated RNA-protein crosslink formation. (b) Schematized TCP-seq, RCP-seq, 40S ribosome profiling and Sel-TCP-seq methods (see text for more details). Characteristic footprint patterns and features for both, SSU- and ribosome-derived footprints are shown, covering the entire translation cycle on mRNA. (c) Factor-selective methods using covalent stabilization allow to obtain additional insight into translation factor involvement along the translation cycle phases (left), and refine start selection mechanisms and dynamics, as well as detect and monitor co-translational complex assembly (right). Designations and structures as in .
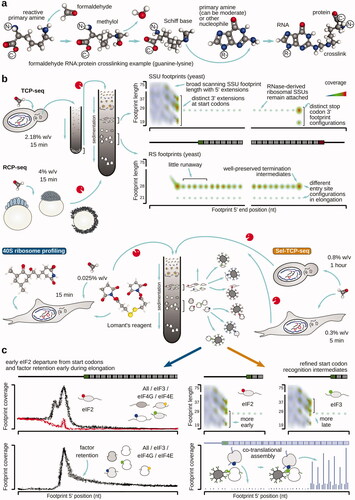
To investigate translation using footprint profiling, formaldehyde-based stabilization of translational complexes was first used in the Translation Complex Profile sequencing (TCP-seq) method in yeast cells (; ). Yeast cells were rapidly chilled by the addition of ice directly into the growth media and simultaneously crosslinked with 2.18% w/v formaldehyde for 10 min; formaldehyde quenching was performed using 250 mM glycine. In TCP-seq, “translated mRNA” (mRNA co-sedimenting with one or more ribosomes) was first isolated, and then disassembled into the monosomal and SSU fractions with RNase I digestion. TCP-seq allowed to visualize a diversity of new translational complexes previously inaccessible to footprinting, such as the SSU scanning complexes, different SSU configurations during start codon recognition, post-termination/recycling SSUs, and SSUs resulting from ribosome disassembly over the coding regions due to the RNase digestion and purification effects. Notably, initiation complexes had their unique length-and-location footprint signatures, which can only be revealed by the broad footprint size selection introduced in TCP-seq. Scanning SSUs were characterized by a footprint length range between 17 and up to ∼75 nt, arguably due to the scanning motor protein interactions in the 5′-direction from the SSU position. A possibility of such an arrangement was further highlighted by cryo-EM structures of the human SSU start codon complex, where eIF4A and eIF4F were located, bound to the eIF3 core, closer to the 5′ cap/exit side of the SSU (Querido et al. Citation2020). Start codon-associated footprints also had distinct modal lengths of 19, 29, and 37 nt and differed in the 3′-direction (+6, +16, and +24 nt 3′ ends, counting from the first nucleotide of the start codon), likely reflecting an initially open A-site and its progressive inaccessibility along with the start codon recognition. Queuing of the SSUs in front of start-codon associated counterparts was seen (−30 nt 5′ end footprint peak), suggestive that at least some mRNAs translate through the cap-severed mechanism. Ranking of mRNAs by the amount of translational control at the scanning step via peak 5′UTR to sum of peak 5′UTR and start codon coverage was suggested as a new metrics of translation, and termination/recycling was shown to complete in a stage-wise manner at least for some mRNAs, with LSU leaving first and the recycling SSUs having a specific footprint signature (29 nt; −12 nt 5′, +16 nt 3′), reminiscent of intermediate start codon recognition step footprints (with the likely closed A-site). Multiple configurations of the elongating ribosomes were recovered, including two major footprint signatures suggestive of inaccessible (31 nt; −13 nt 5′, +18 nt 3′) and more accessible entry/A-site (22 nt; −13 nt 5′, +8 nt 3′), in-line with the other observations obtained either without stabilizing agent or with multiple elongation inhibitors of different elongation stages. Thus, TCP-seq was able to capture translational complexes of any kind. TCP-seq since then has been expanded to include cells and organisms of diverse origin, more comprehensive translation complex capture, and factor-selective footprint investigation, with much room left for further broadening the scope of its applications.
In the Ribosome Complex Profiling (RCP-seq) variation (), stage-matched zebrafish embryos were fixed with 4% de-polymerized paraformaldehyde for 15 min on ice, followed by 250 mM glycine quenching for 5 min (Giess et al. Citation2020). mRNAs were not separated from the clarified lysate based on co-sedimentation with ribosomes, and RNase I footprinting was performed directly in the clarified lysate, thus possibly containing footprints derived from non-translated mRNA (Giess et al. Citation2020). Despite these and library construction approach differences to TCP-seq, the metagene ribosome and SSU distributions largely resembled that of the TCP-seq, most notably with the prominent 5′-directed extensions of the start codon-associated SSUs, and the broad range of footprint lengths, also up to ∼75 nt, of the scanning SSUs (Giess et al. Citation2020). The pattern of the increase of the SSU footprint length upon footprint 5′ end alignment over transcript start and moving into 3′ direction was interpreted in RCP-seq as a result of mRNA threading through SSU (Giess et al. Citation2020). It should be noted though, that while these patterns are not inconsistent with mRNA SSU threading in general, the diversity of the SSU footprint lengths over 5′UTRs is observed in all TCP-seq-like works across the entire length of the 5′UTRs except nearby start codons, where a more complex patter is present. Thus, the range of scanning SSU-protected lengths, starting from the smallest accurately mappable length of 17–19 nt, is a feature of any nucleotide position in the 5′UTR and not just the 5′ termini of the transcript. The apparent length increase while moving downstream away from the 5′ end can be an effect of the absence of transcript in the 5′ direction. RCP-seq was used to compare the SSU footprint presences before and after uORFs with certain stop codons, to demonstrate that UGA (a weaker stop codon) results in significantly lower scanning resumption than UAA (a stronger stop codon), possibly due to the leakage of the elongation downstream (Giess et al. Citation2020), although a probability of some kinetic effects, such as a slower termination (higher ribosome to SSU ratio was observed for UGA) should not be dismissed.
Methods further derived from TCP-seq have been developed for use in model mammalian cell lines, such as HEK 293, HeLa, and NIH 3T3. In HeLa and NIH 3T3 cells in the selective 40S footprinting method (), the complex stabilization approach was substantially different and included a pre-wash of the cells with chilled 800 µM cycloheximide in magnesium-supplemented phosphate-buffered saline, combined with 2 mg/ml harringtonin pretreatment at growth conditions for some of the experiments (Bohlen, Fenzl, et al. Citation2020). The crosslinking was performed in magnesium-supplemented phosphate-buffered saline containing 400 µM cycloheximide, 0.025% de-polymerized paraformaldehyde, and 0.5 mM Lomant's reagent (di(N-succinimidyl)-3,3′-dithiodipropionate) at room temperature for 15 min (Bohlen, Fenzl, et al. Citation2020). Formaldehyde and Lomant's reagent quenching was performed with 300 mM glycine in cold and all buffers contained cycloheximide. Lomant's reagent is “homobifunctional” and mostly active as a crosslinker for highly-reactive primary amines (e.g. lysines of proteins), and has a spacer length of ∼12 Å, ensuring the broader spatial reach of the crosslinks and usually capturing more proteins than formaldehyde-based methods (Tian et al. Citation2012). A sequential application of Lomant's reagent followed by formaldehyde has been known from ChIP-style DNA crosslinking (e.g. XchIP) (Tian et al. Citation2012), however, simultaneous application of the inhibitor-based (cycloheximide has one potentially active hydroxyl group), Lomant's reagent (which is bi-functionalized via disulfide bridge and formaldehyde is known to react highly with, e.g. cysteines and shift disulfide balance) (Kamps et al. Citation2019) and formaldehyde may lead to very complex chemistry and kinetics of the stabilization, while arguably capturing some of the intermediates more comprehensively. Nonetheless, TCP-seq-like scanning and start codon footprint signature were prominently present in the HeLa cell data, including all three major SSU footprint length groups found over the start codons (also differing by 3′-end extensions), 5′-directed scanning and start codon SSU extensions, and the broad scanning SSU protection range (Bohlen, Fenzl, et al. Citation2020). Importantly, using antibodies to eIF3b, eIF2α, eIF4E, and eIF4GI, it was possible to trace ribosomal and SSU footprints enriched for the presence of these initiation factors. While eIF2α was depleted beginning from the start codon and into the ORF body even for the SSU fraction, eIF3b, eIF4E, and eIF4GI all showed gradually decreasing presence for up to 200–300 nt of the start codons, indicating these factors may stay associated with the ribosomes as previously suggested () (such distances are likely impossible for ribosomes to “leak” into given combination of the stabilizing agents used, although the capability of Lomant's reagent to induce longer crosslinks and facilitate some of the association cannot be dismissed). Further, using additional stalling of the initiating ribosomes with harringtonin pretreatment, it was possible to observe the difference in the accumulation of the queued SSUs before start codons, markedly more in NIH 3T3 compared to HeLa cells (Bohlen, Fenzl, et al. Citation2020). While some queuing could have also been induced by the use of cycloheximide in the background control (and thus reduce the overall difference), it allows making an important conclusion that different cell types may have different levels of cap-tethered or cap-severed modes of scanning. The even coverage of all initiation factor-enriched footprints over 5′UTRs, including eIF4E, is not inconsistent with the cap tethered hypothesis, but should not be used as the absolute proof of exclusively cap-tethered initiation until it remains unknown where the chain of cap:eIF4E:eIF4G:eIF3:SSU interactions is most frequently broken first and that eIF4E cannot re-bind to the scanning complex from solution if the detachment between cap:eIF4E and eIF4G took place in the initiation round.
Sel-TCP-seq, based on profiling of pure formaldehyde-fixed, eIF-selective footprints, has been developed and applied in both, yeast and human (HEK 293 T) cells () (Wagner et al. Citation2020). In Sel-TCP-seq, yeast cells were fixed upon chilling with ice and formaldehyde (0.8% w/v) addition directly to the cell media for 1 h on ice (subsequent quenching with 94 mM glycine). Despite these differences and the substantially altered library preparation method, metagene footprint signatures were nearly identical to the previous TCP-seq results, confirming the robustness of the approach. In HEK 293 T cells, a lower formaldehyde concentration of 0.3% w/v was used by adding directly to the cell media at room temperature with subsequent 5-min incubation in cold (quenching with 75 mM glycine for 5 min). HEK 293 T footprint patterns resembled many features of the yeast footprints and were consistent across works with other higher eukaryote cells. Native eIF3a antibody and anti-FLAG antibody for yeast FLAG-tagged Sui3p (eIF2β), Tif32p (eIF3a), and Nip1p (eIF3c) were used to enrich complexes upon RNase I disassembly of the polysomes; no pre-isolation of “translated RNA” was used. Sel-TCP-seq allowed to further clarify several important questions of the dynamics of start codon recognition, and critically, to demonstrate a co-assembly of multisubunit protein complexes (Wagner et al. Citation2020). All tested eIFs produced SSU footprint enrichment along the entire length of 5′UTRs, as expected for the cap-dependent scanning of mRNA. In-line with the former predictions and in parallel to the results in HeLa cells and other recent observations of eIF3 association with ribosomes, an increased presence of the footprints derived from the elongating ribosomal complexes enriched by eIF3b for HEK 293 T and eIF3a for yeast was observed within the first 200 nt after the main start codons. Using eIF2 and eIF3-enriched SSUs, it was further possible to sharpen the interpretation of footprint extensions over the start codons in yeast cells: longer (+18 nt 3′) footprints were associated with eIF3-, while shorter (+8, +16 nt 3′) with eIF2-enriched SSUs, further underpinning the observation of the more progressively inaccessible entry (A) site of the SSU in the course of start codon recognition. In eIF2-enriched SSUs of Sel-TCP-seq, +16 nt 3′ end footprint group could further be split into +15 nt and +18 nt 3′ end peaks, likely reflecting fine staging of the entry site closed configuration into POUT and PIN (deeper tRNAi positioning in the P-site) sub-stages (Hinnebusch Citation2017; Shirokikh and Preiss Citation2018; Thakur et al. Citation2020). With Sel-TCP-seq in yeast, much more detail became available for the uORFs of GCN4 mRNA. First, uORFs were shown to hold large amounts of SSUs with eIF2 and eIF3, possibly to provide a source of re-initiating SSUs to translate this mRNA in an event of GCN2-induced downregulation of translation, as scanning SSUs may not bind efficiently to mRNA in the absence of active eIF2 in this case (Wagner et al. Citation2020). Highly importantly, using the eIF3 subunits and footprint-level enrichment alteration across its potential interactors, Sel-TCP-seq has revealed eIF3c as the nucleation core of the eIF3 assembly, as well as the co-translational attachment of other factors () (Wagner et al. Citation2020). Using Sel-TCP-seq, it was further possible to clarify the likely sequentialities of the assembly events in vivo (Wagner et al. Citation2016; Zeman et al. Citation2019), leading to the eIF3c→eIF3a (also eIF1, eIF5)→eIF3b (also eIF2β)→eIF2γ (likely eIF2α)→eIF3i,g assembly sequence (Wagner et al. Citation2020).
Formaldehyde crosslinking has further been used to clarify details of factor-specific translation termination and recycling, as it allows ease of terminating ribosome and post-termination SSU stabilization on mRNA. Each of the MCT-1 (MCTS1):DENR ribosomal recycling factors have been knocked out from HeLa cells to observe increased ribosome dwell time at stop codons by regular cycloheximide-based footprinting; most of the additional density accumulated immediately before the footprint corresponding to the last (terminating) ribosome, suggesting impediment by another particle. Intriguingly, translation of mRNAs with complex 5′UTRs [such as uORFs-containing transcripts of ATF4, a-Raf (ARAF), c-Raf (RAF1), PI3K, Drosha, and MAP2K6] was inhibited in the knockouts, synergistic to the siRNA suppression of eIF2D, which mimics MCT-1:DENR. Using conditions similar to the selective 40S footprinting (but lysis into very high-salt, high-inhibitor buffer with 50 mM MgCl2, 1 M KCl, 1,000 μM cycloheximide) (Bohlen, Fenzl, et al. Citation2020), it was shown that the absence of DENR directly suppresses detachment of the SSUs, although contributions of ribosome disassembly may need to be taken into account (as follows from the retention of triplet periodicity of the SSU signal over ORFs) (Bohlen, Harbrecht, et al. Citation2020). Furthermore, SSU footprinting allowed to discover codon-specific retention of the post-termination intermediates, with a strong preference for CTGLeu, CTTLeu, CTCLeu, ATGMet, GCGAla, and AGGArg in the penultimate positions of main and upstream ORFs, explaining the MCT-1:DENR/eIF2D-driven re-initiation dependence (Bohlen, Harbrecht, et al. Citation2020). Highly similar findings were reported in a study that utilized a fixation approach similar to Sel-TCP-seq (0.8% formaldehyde on ice for 1 h, glycine quenching, and lysis into 140 mM KCl, 1.5 mM MgCl2 buffer) in yeast Tma20/Tma22 (MCT-1:DENR) and Tma64 (eIF2D) deletion mutants (Young et al. Citation2021). Notably, Tma64 (eIF2D) deletion had only minor effects on recycling in yeast, but possibly had different codon specificity (Young et al. Citation2021). It is noteworthy that while the footprint density from ribosomes stalled with the initiation inhibitor lactimidomycin and formaldehyde-crosslinked start codon SSU footprints showed a high correlation (Young et al. Citation2021), in both studies, the SSU-associated footprint signal has been nearly completely absent in the ribosomal libraries prepared with standard inhibitors (cycloheximide, lactimidomycin) (Bohlen, Harbrecht, et al. Citation2020; Young et al. Citation2021), bolstering the utility of the untargeted covalent stabilization.
Overall, formaldehyde-based stabilization of translational complexes remains an extremely attractive option for many reasons. It has now been established as a robust approach leading to highly similar observations even in the background of vastly different material sources, crosslinking handling, and library preparation approach. There is a potential for expansion into prokaryotic and plant applications as it is known that plant translational complexes can be efficiently stabilized with formaldehyde (Firmino et al. Citation2020). It is a universal and flexible method, arguably the only one allowing to capture the full diversity of translational complexes. Due to its process-agnostic nature and rapidity (see more below in the “Capturing the dynamic state of translational control” section), there is an expectation of suppression of many of the metabolic processes and thus lesser susceptibility to a post-stabilization bias. It is a method amenable to compatibility with co-purification of large (and fragile) complexes and electron microscopy approaches, as well as co-analysis of other cellular processes (such as transcription) (Kiernan Citation2000; Rajala et al. Citation2015; Gavrilov et al. Citation2015; Hoffman et al. Citation2015; Tayri-Wilk et al. Citation2020; Wagner et al. Citation2020). The main caveats remain in the domain of crosslinking optimization (as subtle changes in cell or material type may lead to the necessity to re-optimize crosslinking), possible inconvenience of formaldehyde handling in certain settings, and much more complex data that can be difficult to analyze. For example, the co-translational assembly, as shown in Sel-TCP-seq, is uniquely captured by this method due to the stabilization of protein:protein interactions, allowing to use the progression of ribosomes over mRNA as “molecular clock” for the assembly event timings. Nonetheless, these data may be difficult to extract if co-translational interactions are of low stoichiometry and/or very diverse.
To fully empower translational analysis with formaldehyde- and other potential covalent-based stabilization assays, some improvements are necessary. Surprisingly, the complete spectrum of formaldehyde and formaldehyde-initiated adducts, particularly those in nucleic acids, remains to be carefully systematized (Gavrilov et al. Citation2015; Hoffman et al. Citation2015; Kamps et al. Citation2019; Tayri-Wilk et al. Citation2020). Thus, the chemistry and biases of the subsequent reactions are not fully accessible. While the majority of the adducts and, particularly, crosslinks, can be expected to be mediated by monomeric formaldehyde residues due to the instability of longer paraformaldehyde chains in water environments (and thus be highly precise and localized), some of the crosslinks (particularly, made with the high formaldehyde concentration) can result from short formaldehyde oligomers and be less sensitive to the distance between the crosslinked entities. These and other dependencies, such as the possible higher loss of ribosomes attached to larger cellular structures in crosslinked material from subsequent analyses, need to be investigated to allow for more accurate modeling. This will open opportunities for the development of more comprehensive and sophisticated computational methods, the inclusion of more of the factor-specific datasets, and accurate, species-specific inference of complex identity from the footprint features. Based on these anticipated advances, it will be possible to decipher co-localization of all co-translational interactions, define “protein complex assembly factories”, build networks of interactions and define the diffusional boundaries of each of the polysome types in the cell.
Capturing the dynamic state of translational control
The largest challenge, but also a great field for potential expansion of the protection-based translation profiling-style experiments, remains in the high dynamic nature of translational control. As the ultimate goal is to understand the mechanisms that drive the change of translational response, capturing accurate snapshots of translation in the exact moment of time is paramount. Several major hurdles to accurate translational snapshots have been already identified and are being successfully addressed, while others still require more detailed characterization and attention.
Biases in library preparation methods that can affect the accuracy of footprint identification and quantification
The accuracy of ribosome profiling greatly depends on the footprint generation, although this can be application-specific. For understanding global gene- and in some cases transcript-level alterations of footprint counts between experiments, footprint accuracy is not critically important (Artieri and Fraser Citation2014; Hussmann et al. Citation2015; Bartholomäus et al. Citation2016; O'Connor et al. Citation2016; Zinshteyn et al. Citation2020). Conversely, any other application that attempts to categorize footprint types and address mRNA location or complex type-specific differences in the footprints relies on the assumption that the mapped footprint 5′ and 3′ ends are a function of the position and configuration of the respective translational complexes. Thus, a less nucleotide-specific, frequently RNA-hydrolyzing technique would be favored, qualities for which RNase I has been popular in footprint profiling-style experiments, including the first works (Ingolia et al. Citation2009; Ingolia et al. Citation2012; Gerashchenko and Gladyshev Citation2017).
In a systematic overview of different RNases, RNase I generally outperformed others by the sum of its features (Zinshteyn et al. Citation2020). The most homogeneous and sharp protection length groups from cycloheximide-stalled ribosomes have been observed with RNase I (narrow 28 nt), compared to RNases A (broader 29 nt; cleaves after pyrimidines), T1 (broader 27 nt; cleaves after guanine nucleotides), and micrococcal (RNase S7) nuclease (broader 28 nt; cleaves mostly before pyrimidines)(Gerashchenko and Gladyshev Citation2017). RNase I was also excellently preserving the apparent integrity of yeast ribosomes, as measured by the homogeneous and unshifted monosome sedimentation velocity, indicating that in these conditions RNase I is indeed highly specific to single-stranded RNA, as most of rRNA is structured (Gerashchenko and Gladyshev Citation2017). However, RNase I was early noted for its inability to digest many prokaryotic polysomes, including those of E. coli, apparently due to the RNase-inhibiting activity of the ribosomes themselves (Kitahara and Miyazaki Citation2011; Glaub et al. Citation2019; Mohammad et al. Citation2019). Thus, alternatives must be used for prokaryotic polysomes and while the most popular is a micrococcal nuclease, it fails to generate an accurate triplet periodicity pattern, as compared to the bacterial toxin and endoribouclease RelE, which binds to and specifically cleaves bacterial A-site-located mRNA (Griffin et al. Citation2013; Hwang and Buskirk Citation2017; Mohammad et al. Citation2019). Clearly, a combination of different RNase treatments is also possible (including exoribonucleases, and albeit this requires effort in optimizing the digestion), e.g. RNase I, micrococcal nuclease, XRN-1, RNase R, and RNase T cocktail has been successfully used (Hücker et al. Citation2017). Importantly, different nucleases resulted in vastly different transcript-wise footprint coverage profiles (with RNase I giving the most even coverage) (Gerashchenko and Gladyshev Citation2017), let alone footprint end identities, highlighting the necessity of nuclease treatment improvement and further bias characterization, as many modern algorithms rely on local transcript-wise footprint abundance changes to infer ribosomal dwell time and codon-level dynamics of elongation. Even more unexpectedly, polysomes from different organisms and tissues exhibited different sensitivity to RNase type, and their ribosomes also non-uniformly resisted the treatment (Gerashchenko and Gladyshev Citation2017). Mus musculus, C. elegans, and particularly, Drosophila melanogaster ribosomes were highly sensitive to RNases I, A, and micrococcal nuclease, but not RNase T1 (Gerashchenko and Gladyshev Citation2017). Further, lysates from organs, such as spleen, contained high intrinsic nuclease activity, suppressible with high heparin concentrations (Gerashchenko and Gladyshev Citation2017). Combined with the finding that RNase I can have a calcium-ion-dependent double-stranded RNA cleaving activity (Grünberg et al. Citation2021), these data suggest that extreme care needs to be taken for footprint generation, which must be done in highly controlled environments, such as in isolated polysomal fraction or large-molecule-size-excluded cell lysates, to remove any intrinsic nucleases, ions and small molecules. Given such sensitivity to nuclease type, we can expect that different ribosomal configurations as stabilized by various inhibitors will have nuclease-dependent footprint protection differences as well. Formaldehyde-based stabilization, in this case, stands out as formaldehyde modifies amines of the bases involved in Watson-Crick structure formation (and specific single-stranded RNA:protein interactions). Thus, mild formaldehyde treatment as in TCP-seq may induce “crosslink-or-leave” type of modification where any unstable secondary/tertiary structure or protein interactions would be further destabilized, unless they are immediately crosslinked. At high formaldehyde concentrations, this effect has been traditionally used to denature DNA and RNA (Rio Citation2015). Therefore, formaldehyde fixation may counter-intuitively improve RNA accessibility within unstable structures, and thus footprint evenness and end resolution, effects that prompt further systematic investigation for finding the best fixation and digestion combinations. Overall, while the biases of different RNase application and limitations are becoming better understood, less bias-prone methods, such as short radiation-induced pulses of hydroxyl radical cleavage, which can be used directly in frozen lysates (Adilakshmi et al. Citation2006), need to be considered, as well as the broader employment of exoribonucleases for accurate footprint end trimming.
Another area where improvement is clearly required is the efficient and unbiased (or one with a well-characterized bias) fragment library integration and RNA depletion techniques. Circular, 5′, and 3′ adapter ligation and template switching [reviewed (Boone et al. Citation2018)] have been mostly used with different claims of efficiency and potential nucleotide bias, but no systematic comparison is still available. Some fragment integration biases were clearly identified, such as the depletion footprints terminating at certain sites, such as AUG start codons (Archer et al. Citation2016; Shirokikh et al. Citation2017; Wagner et al. Citation2020). This depletion is present across different footprint extension techniques [3′ poly(A) addition by E. coli poly(A) polymerase (Archer et al. Citation2016; Shirokikh et al. Citation2017) or e.g. T4 RNA Ligase 1 linker ligation (Wagner et al. Citation2020)] and is likely produced by the cDNA circularization preferences of CircLigase. While rRNA fragments are efficiently reduced in the more classical ribosome profiling experiments solely based on the very narrow footprint selection range, more translation complex-comprehensive methods, such as TCP-seq and derivatives, di- and multi-some profiling and profiling of prokaryotic ribosomes which footprints stretch over to ∼70 nt, require more efficient rRNA depletion. Promising progress has been made with oligonucleotide-assisted technology, such as RNase H-facilitated depletion, which can result in very high footprint enrichment (Huang et al. Citation2020). However, such approaches also have been shown to be prone to substantial off-target effects and alterations of the footprint end identities (Zinshteyn et al. Citation2020). A more specific approach using duplex-specific nuclease [particularly, at the cDNA stage when RNA fragment ends remain unchanged (Archer et al. Citation2014; Archer et al. Citation2015; Chung et al. Citation2015)] was not yet assessed, as well as CRISPR-based depletion techniques, both of which can promise higher fidelity (Hardigan et al. Citation2019; Prezza et al. Citation2020).
Difficulty stabilizing all translation intermediates simultaneously or with sufficient agility
The fidelity of footprinting depends on the evenness and speed of complex stabilization. Footprinting fidelity can have a different weight in selecting the translation stabilization approach, such as it can be extremely important for the understanding mechanistic differences across complex types as the translation cycle progresses, in the discovery of unexpected translation complex configurations, and less crucial if a comparative “bulk” ribosomal load across mRNA is in question.
The average elongation speed (including that measured by ribosomal footprinting itself) is usually quoted in the range of 2–20 amino acids per second (aa/s) (∼5 aa/s as a realistic average) (Ingolia et al. Citation2011; Riba et al. Citation2019). Because average ribosomal spacing is estimated to be in the range of 70–300 nt (∼50 codons) (Viero et al. Citation2015; Chassé et al. Citation2017; Andreeva et al. Citation2018; Samatova et al. Citation2021), we can expect an initiation and termination event every 10 s for an mRNA molecule. Any sub-steps of the translation cycle, such as mRNA cap-binding, scanning, start/stop codon recognition and recycling, consequently, will have even shorter characteristic times. Therefore few, likely none, inhibitors will be able to capture a native translation complex snapshot in vivo. This stems from the fact that the sub-processes of the translation cycle not only are fast, but also have different speeds and are differentially affected by the inhibitors, whether due to the type of translational complex, sequence and codon context, or intracellular accessibility and permeability of the location of the translated mRNA to the stabilizing compound.
Well-established consequences of uneven stabilization include the inhibitor-induced (best characterized for cycloheximide) artificial piling up/queuing/accumulation of ribosomes over initiation sites (as a result of continued initiation in the background of slowed down elongation) and severe bias toward stabilizing a particular configuration of the translating ribosome, such as characterized for the pre- or post-translocated ribosomes with open or closed entry A-sites (Gerashchenko and Gladyshev Citation2014; Lareau Liana et al. Citation2014; Weinberg et al. Citation2016; Wu et al. Citation2019). These effects can be extremely useful as analytical tools and sometimes are nearly irreplaceable with any other type of analysis. For example, the combined use of harringtonin and cycloheximide administered intravenously with incremental delay allowed to accurately characterize global dynamics of elongation in different organs and tissues of live mice, highlighting age-dependent translational slowdown (Gerashchenko et al. Citation2021). However, native complex distribution remains inaccessible with these approaches. Insufficiently rapid stabilization is exemplified by run-away effects, whereby in the case of elongation a region adjacent to start codons becomes artificially depleted of footprints resulting from continued elongation due to the reversibility of the inhibitor action and/or its slow intracellular concentration build-up (Gerashchenko and Gladyshev Citation2014). It is well-established that regular stabilization approaches act poorly on prokaryotic and mitoribosomes (Rooijers et al. Citation2013). In addition to the inhibitor-inaccessible eukaryotic steps, such as scanning, translation initiation in bacteria was also shown to be able to proceed without Shine-Dalgarno sequence even for species normally relying on Shine-Dalgarno:anti-Shine-Dalgarno interactions (Saito et al. Citation2020a). Instead, A-rich sequences devoid of the canonical structure were identified to be promotive of this type of initiation (Saito et al. Citation2020a). Thus, some form of “passive” scanning might still take place during bacterial initiation (although this has been somewhat challenged by an observation of the lack of dependence of downstream cistron translation on ribosomal recycling) (Saito et al. Citation2020b), and the dynamics of this process requires the development of special inhibitors or an unbiased stabilization approach. Even more sophisticated and less predictable biases can occur; for example, cycloheximide was proposed not to stabilize ribosomes remaining in the 3′-portions of transcripts upon nonsense-medicated decay cleavage (Serdar et al. Citation2020). With the popularity of locating ribosomal pause sites by the regions of di- or multisome accumulation (Tesina et al. Citation2020; Wu et al. Citation2020), any elongation inhibitor should be used with extreme caution, as stacking of ribosomes may prevent transition through the steps of elongation cycle for the trailing particles and thus defeat their stabilization.
Global translational responses are thought to be very rapid, such as starvation response in eukaryotes and prokaryotes is observed over minutes, likely sub-minute scale (Karlsen et al. Citation2018). Considering the typical diffusional measurements for cycloheximide (as an example of well-permeating and compact inhibitor) of 700 µm2/s (Dykstra et al. Citation1988) and simple approximation of the Fick’s law to 5 µm diffusional barrier (such as half of the typical cultured mammalian eukaryotic cell thickness), slightly over 1 s is required to reach 90% concentration in the cells (excluding any membrane permeability slow-downs), which should be sufficient for fairly fast measurements (). However, increasing the distance to just 50 µm (which can be encountered in cell aggregates and homogenized tissues, as well as in certain cell locations in organisms) results in ∼2 min wait time, which is unacceptable for measurements of translational response (). Apparently, larger or less lipophilic inhibitors would have even more undesirable kinetics.
Figure 14. Formaldehyde-based covalent stabilization of translational complexes is a versatile approach compatible with rapid protein biosynthesis dynamics. (a) Estimates of formaldehyde vs. cycloheximide characteristic diffusion times to a given % of their initial concentration (blue line demarks 90%) over a typical barrier (5 µm for attached cell thickness or 50 µm for a group of cells or tissue fragment). Diffusion coefficients in water and a 2-term Taylor series approximation of the Fick’s law were used; calculations and plotting was done in GeoGebra (https://www.geogebra.org). (b,c) Mapping of HEK 293 formaldehyde-fixed TCP-seq-style footprints demonstrating the presence of footprints from nuclease-resistant disome fraction (b), specific signals from mitochondrial SSU and ribosomes in the respective fractions, and mitochondrial gene coverage in all, SSU, ribosome and disome fractions (c). These results suggest universal and taxonomically indifferent applicability of the formaldehyde-based covalent stabilization of translational complexes (see color version of this figure at www.tandfonline.com/ibmg).
Ex vivo stabilization has been becoming popular as a workaround to distances, cell membrane, and other diffusion barriers. Indeed, rapid freezing followed by homogenization in a frozen state with buffers containing several translation inhibitors directed at diverse complex types may be one of the attractive and successfully applied solutions. Possible limitations, apart from difficulty applying to large organisms, and liquid cell cultures with requirements for plentiful starting material, are in the uncertainties of inhibition action upon grindate thawing as faster processes may still be able to disproportionally complete, and transient complexes destabilized, as a result of the freeze-thaw process.
Covalent stabilization, such as with formaldehyde appears, as most suited for rapid and unbiased complex capture. Formaldehyde offers high diffusivity (∼2000 µm2/s) (GSI Chemical Database Citation2021) and can cross water environment up to 90% of the starting concentration for 5 µm in <0.4 s, 50 µm in <40 s, which makes it more suitable for the stabilization of difficult material (). Formaldehyde crosslinking preserves mitochondrial footprints efficiently even upon gentle cell lysis with detergents (). It is conceivable that formaldehyde crosslinking can also be coupled with rapid freezing and mixing with formaldehyde buffer in a frozen state or fixation directly in the frozen state by vapor diffusion. Additional crosslinking aids can be used to further stabilize complexes upon cell disruption. For example, protein-protein crosslinking agents with limited nucleic acid reactivity can further help preserving translational complexes during footprint generation to yield higher fidelity of end definition and less ribosomal degradation, while not affecting footprint liberation yield. Crosslinkers like glyoxal and genipin have been highly successfully used in this role for other applications (Zhang et al. Citation2014; Bussolati et al. Citation2017; Festuccia et al. Citation2019; Channathodiyil and Houseley Citation2021). While kinetically highly promising, however, currently fixation dynamics of translational complexes with formaldehyde is poorly characterized and we are yet to uncover the limitations of its applicability to rapid processes, complete understanding on the effects of RNase action and biases associated with the loss of heavy and fast-sedimenting crosslinked material.
Translation-dependent biases of approaches to stall translational complexes
Adding to the complexity of cell responses, inhibitor-induced translational stalling can result in some unexpected translational alterations, usually via mechanisms embedded into the naturally evolved pathways of translational control. As these regulatory pathways often include signal multiplication steps, such as through activation of certain protein kinases, the resultant effect can be seen as disproportionally acute.
A clear indication of the translation-dependent effects has been elucidated in studying rapid yeast response to starvation in the background of cycloheximide addition. It was proposed that under starvation, cycloheximide depresses TORC1 signaling which is normally shut down to reduce the production of growth homeostasis genes, such as ribosome biogenesis components (Santos et al. Citation2019). As TORC1 signaling, in this case, relies on the rapid shuffling of transcription factors, such as Dot6, Stb3, and Tod6 (repressors of ribosomal biogenesis genes) between nucleus and cytoplasm and thus is translationally insensitive, the ribosome biogenesis genes are induced back in the background of cycloheximide-inhibited translation (Santos et al. Citation2019). This creates a pool of new transcripts that appear as translationally-regulated (repressed) due to their low “translation efficiency”, as commonly calculated in the form of ribosome footprint coverage normalized to RNA-seq coverage (Santos et al. Citation2019). Importantly, although the data did not contain absolute mRNA level measurements, in the addition to cycloheximide-specific effects of cell homeostasis, they highlight the inability of the commonly employed “translation efficiency” to distinguish different pools of mRNA, for example newly produced and even nuclear RNA fractions, as well as many other scenarios of principal inaccessibility of mRNA to translation due to any form of compartmentalization (condensation, inclusion in higher-order structures, etc.).
Further complicating investigation of any translational dynamics with translation inhibitors, intermediate (but not high) doses of elongation inhibitors, such as anisomycin and emetine (1–100 and 0.048–48 µg/ml, respectively) were shown to induce the formation of stacked ribosomes, presumably due to the differential action on some of the codon combinations and concentrations still permissive of elongation (Wu et al. Citation2020). Ribosomes stacked in this way in HEK 293 and HeLa cells were shown to trigger Stress-Activated Protein Kinases (SAPKs; p38 and c-Jun N-terminal kinase JNK) and ribotoxic stress response through mitogen-activated protein kinase kinase kinase ZAK (Wu et al. Citation2020). ZAK was shown to self-activate by autophosporylation once bound to disomes, leading to programmed cell death activation, as well as likely GCN1-dependent GCN2 activation and translation initiation suppression as a survival response, in the case of a more moderate stalling (Wu et al. Citation2020). In a more physiological context, similar responses were observed in a UV- or amino acid starvation-induced stress responses and by overwhelming translation machinery with messages containing “difficult” and slow code, such as poly(A) stretches. Notably, most of the ribosome stacking in the case of externally UV-irradiated mRNA occurred before sequential pyrimidines, likely due to their dimerization and failed decoding with the unoccupied ribosomal A-site (short footprints) (Wu et al. Citation2020). Thus, while once again highlighting the power of the use of selective translation inhibitors, these results demonstrate activation of cell responses and re-distribution of mRNAa and nascent peptides that extends far beyond the direct action of the translation inhibitors. Just the use of an array of the inhibitors (harringtonine, cycloheximide, lactimidomycin, ricin, as well as anisomycin and emetine) but in a different range of concentrations (0.1–100 µM) also revealed ZAK-dependent p38 activation in U2OS cells, but led to a different conclusion of elongation slowdown and stalling—and not ribosomal stacking—as the driving force of the response (Vind et al. Citation2020).
Other translation-dependent biases have been identified. For example, translation elongation inhibitors can skew initiation rates toward recognition of the non-AUG codons (Kearse et al. Citation2019). Used in their commonly applied and relatively high concentrations, cycoheximide (100 μg/ml), anisomycin (0.5 μg/ml), didemnin B (1 μM), and bouvardin (1 μM) were shown to unexpectedly promote translation initiation from near-cognate/alternative codons, such as GUG/ACG/CUG/AUU/UUG. Didemnin B is a pluripotent antibiotic and anti-cancer compound, with direct effects on stabilizing aa-tRNA:eEF1ɑ1:GTP in the ribosomal A-site and binding and inhibiting palmitoyl-protein thioesterase 1 (Potts et al. Citation2015). Conversely, bouvardin has been proposed to stabilize eEF2 binding to the ribosomes, resulting in effects that somewhat mimic eEF2 kinase-dependent translational control (Stickel et al. Citation2015). All these inhibitors can stall elongating ribosomes amidst the coding sequence and allow some degree of elongation “leakage”, likely combined with differential dwell time over particular sequences while having little to no effect on translation initiation. Therefore, it was proposed that the elongation slowdown has allowed for more initiating and scanning ribosomes to queue up before the slowed down elongating ribosomes, augmenting the known mechanisms of kinetic preference of the start codon selection, where longer dwell times of the scanning complexes eventually allow recognizing a triplet as start codon even in suboptimal contexts (Kozak Citation1990; Kearse et al. Citation2019). It can be confidently suggested that such “miscoding” or “misinitiation” can induce multiple secondary effects through the appearance of aberrant peptides in the cell.
To exclude or account for the translation-dependent biases of footprinting, it can be recommended to perform complex stalling with a set of translation inhibitors with diverse action concurrently, or supplement data with footprinting in the conditions of covalent or non-covalent translation stabilization.
Influence and dependence of the footprint snapshots on cell systems at the levels other than translation
The translation is arguably one of the most rapid means of the cells to actively change gene expression, but other responses and pathways can be involved concomitantly, too. Translation-independent or effects secondary to translation suppression at the levels of transcription, splicing, nuclear export/import, stability, condensation status of mRNA, and other translational components, usually accompany purely translational responses. Most of the footprint profiling-type experiments to date do not account for non-translational alterations and can lead to misinterpreted cell responses.
Translational inhibitors in vivo are known to cause pleiotropic effects. Cycloheximide has long been known to inhibit DNA synthesis, likely through suppressed replicon initiation and ribosomal RNA maturation (Venkatesan Citation1977). Cycloheximide has been implicated in the activation of proteasome and in fact, much of cell sensitivity (e.g. in yeast) has been attributed to this “secondary” effect, as proteasome protein mutants confer resistance to the lower range of cycloheximide concentrations (Gerlinger et al. Citation1997). Cycoheximide is known to initiate apoptosis through the Mcl-1 abundance downregulation and Bax/Bak pathway (Goodall et al. Citation2016) and can have a strong inhibitory effect on transcription when used in just 1 µg/ml range, likely as a secondary effect of reducing transcription factor supply, but apparent enough (∼50% at 20 min post-incubation for ribosomal RNA in lymphosarcoma cells) to substantially alter any measurements (Gokal et al. Citation1986). In a similarity, anisomycin is known to affect DNA replication processes. As anisomycin may induce quality control ribosome dissociation of stalled post-translocation ribosomes by the ribosome-mediated quality control (RQC) and RQC-trigger complex and activate ERK1/2, JNK, and p38 (Matsuo et al. Citation2017), it also transcriptionally induces higher transcription factor early growth response-1 protein production and elevated activity of immediate-early growth response genes in a 2-h timeframe (Shin et al. Citation2006). Oncocin and bactenecin were shown to bind DnaK (prokaryotic “chaperone”) and inhibit its ATPase activity (albeit in the higher concentration range than their ribosomal targets), which can result in unexpected translational effects during co-translational folding and accumulation of unfolded polypeptides (Kragol et al. Citation2001; Krizsan et al. Citation2014; Mardirossian et al. Citation2014; Lomakin et al. Citation2015). Strong elongation inhibitors, such as erythromycin and chloramphenicol, have been also known to induce ribosome assembly defects (Siibak et al. Citation2009). Translation inhibitor effects on ribosome assembly were shown to be very rapid (5 min in prokaryotes) and likely resultant from the reduced protein supply as no distinct assembly intermediate was favored (Siibak et al. Citation2009). Remarkably, formaldehyde crosslinking was shown to exhibit bias on transcript abundance in vivo, mostly affecting oxidative stress, mitochondrial and bioenergetic pathways (Wehmas et al. Citation2020). The formaldehyde fixation bias in large (bulky) samples was linked to the gradual increase of its concentration combined with longer required incubation time, allowing the cells to respond to the crosslink-induced stress and accumulate detectable changes (Gavrilov et al. Citation2015; Wehmas et al. Citation2020). Overall, prolonged incubation with any inhibitor (including untargeted covalent stabilization) can be deemed as strongly bias-inducing. Shorter incubation times with quick use of high doses of stabilizing agents immediately before or during cell disruption can address many of the pleiotropic biases (Sharma et al. Citation2019; Glaub et al. Citation2020) but are difficult to implement for large organisms in vivo.
Dynamic translational response occurring in the background of other processes can lead to unexpected results during the rapid change of conditions. Accurate modeling of mRNA abundance accounting for mRNA maturation delays and possible effects of different constant degradation rates (such as mRNA half-life within 2–64 min) revealed the appearance of peak abundance of newly-produced mRNA in just 60–100 min (inferred from RNA-seq and ChIP-seq data from MCF-7 breast cancer cells responding to estrogen receptor α activation) (Honkela et al. Citation2015). Approximately 50% of the peak transcriptional response can be modeled within the first 30–40 min, time duration not uncommon in translation profiling-style works (Honkela et al. Citation2015). Very similar estimates were obtained upon the stimulation of NIH 3T3 cells with serum or TGF-β1 and monitoring for short-lived luciferase activity in vivo (Molina et al. Citation2013). Transcript abundance alterations have been reliably shown in yeast in response to a variety of environmental signals (nutrients, osmotic shock, temperature, oxidation) within as early as 10–15 min, sometimes 5 min (Novo et al. Citation2007; Strassburg et al. Citation2010; Taymaz-Nikerel et al. Citation2016). Rapid transcription-level reactions accompanied with mRNA degradation (30 min timeframe) have been reported in plants as well (Cookson et al. Citation2016). Transcription-independent alterations of mRNA availability, such as rapid degradation and inclusion into condensates and autophagy (Roy and Chanfreau Citation2014; Janapala et al. Citation2019; Ivanov et al. Citation2019; Moon et al. Citation2020; Makino et al. Citation2021), can arguably occur even faster and kinetically compete with translational responses. The coupled transcription-translation of prokaryotes may appear somewhat simpler in respect to the subcellular localization and availability of mRNA with its almost instantaneous transcription-driven response (Chen et al. Citation2018; Jayaraman et al. Citation2018; Uluşeker et al. Citation2019), but nearly all respective calculations of footprint frequencies are compounded by the usually high cell division rate of bacteria.
Intracellular mRNA dynamics can be controlled by or depend on translation, further complicating the understanding of the translational response. For example, dengue virus has been shown (by using pulse-inhibition with 180 µM cycloheximde for 30 s and disrupting plasma and endoplasmic reticulum membranes with digitonin and n-Dodecyl-B-D-Maltoside and lysis into the stabilizing 15 mM MgCl2, and 4 mM CaCl2 buffer) to specifically suppress host endoplasmic reticulum-associated translation and as a result, endoplasmic reticulum-mediated stress responses, while only mildly disrupting the cytosolic translation (Reid et al. Citation2018). Short footprints (15–34 nt) were used to observe signatures of exosome-independent, translation stalling-induced endonucleolytic degradation of mRNA (Guydosh and Green Citation2017). Translation deceleration sites (measured as increased footprint density; see more below) have been linked with the localization of protein transmembrane domains and membrane-localized translation, e.g. in E. coli and chloroplasts (Fluman et al. Citation2014; Zoschke and Barkan Citation2015; Gawroński et al. Citation2018; Samatova et al. Citation2021). Overall, dynamics of mRNA translation in subcellular locations remains notoriously difficult to accurately estimate as some of the best existing tools, such as puromycin-assisted in vivo/in situ visualization of active translation sites combined with inhibitor-stabilized elongation, have been shown to be insufficiently accurate (Hobson et al. Citation2020). Thus, the greatest remaining challenges in the footprint-based profiling are associated with the current poor distinction between the actual availability of an mRNA to translation and its competitive efficiency as the ability to sustain a higher flux of translating ribosomes over time. For example, mRNA can be still located in the nucleus, or be selectively condensed or diverted into degradation, dependent on its localization and translation status, simultaneously affecting the abundance measurements by RNA-seq. The majority of existing approaches are based on the “translation efficiency” (or metrics derived from it) as calculated by normalizing the relative footprint counts to relative mRNA abundance. Thus, especially during a rapid response while away from the steady-state, any mRNA abundance-derived measurement may be inaccurate. Another critical assumption of the existing approaches is that a higher relative footprint count per relative mRNA abundance per mRNA (more accurately: ORF) length is indicative of its specific translation “power”. Clearly, this assumption is contradictory to the other simultaneously used dogma that higher footprint counts are associated with a higher translation complex dwell time and thus slower processes (Darnell et al. Citation2018; Arpat et al. Citation2020). Even further, without localized slowdowns, known evidence points that at least some mRNAs, while highly densely occupied by the ribosomes, may become less translationally efficient. Inefficient translation in polysomes of higher order sometimes is attributed to specialized mechanisms, such as stacking-induced destabilization, but also to the limiting initiation rate by blocking start site and possibly, sterical limitations of the tightly packed structure (Dai et al. Citation2016; Andreeva et al. Citation2018; Reid Citation2020). Critically, additional or new measurements are in an acute need of development to account for the translation-independent dynamics and the availability of mRNA to translational components in live cells.
Perspectives
The repertoire of translation footprinting approaches has matured to an impressive biochemical toolset within the last 5 years, capable of interrogating nearly any question of dynamic control of protein biosynthesis. Localized translation, translational responses to diverse stimuli, dynamics of the use of many translation-related protein and RNA factors, and the complete set of protein biosynthesis intermediates have been addressed. Approaches to tackling multiple variants of ribosomal stalling and stacking and investigating the function of specific ribosome configurations have emerged (Chen et al. Citation2020). In a perspective, we can expect more broad integration of the translation footprinting data with the in vivo structural, isofomal, and modification patterns of RNA and its interactors, such as RNA-binding proteins. An area of critically needed progress remains in understanding the impacts of differential cell responses in dividing cells, as mostly non-synchronized cultures have been investigated. Further, mRNA abundance response magnitudes have been shown to vary across different cells in a population (Schwabe and Bruggeman Citation2014), prompting the use of single-cell translation complex profiling approaches. Complementing advances in biochemistry, within the last 5 years there has been a burst in different ribosome profiling metrics and tools, many of which focus on footprint triplet periodicity and length analysis, ORF and UTR classification and discovery, elongating ribosomes reading frame and metagene footprint distributions (Ingolia Citation2016; Berg et al. Citation2020). More comprehensive, as well as user-friendly bioinformatic packages begin to emerge, and this trend will likely continue. Packages, such as RiboToolkit, begin to offer simultaneous analysis of codon usage, stall sites and translation efficiency, and some ORF detection capabilities (Liu et al. Citation2020), along with the many other extremely useful tools, such as RiboGalaxy, deltaTE, GWIPs-viz, RiboVIEW, and others (Michel et al. Citation2014; Michel et al. Citation2016; Chothani et al. Citation2019; Legrand and Tuorto Citation2020). We can expect that the deconvolution into translational vs. RNA abundance cell response, and means of tracking the dynamic alterations of translation in the background of concomitant changes of transcription, modification, localization, and degradation of RNA, will constitute the future progress.
To summarize, high-throughput sequencing-based approaches continue to develop as some of the most productive discovery methods in gene expression research. Protein biosynthesis profiling by tracking ribosome-protected fragments of RNA (footprints) has become a de-facto standard of translation interrogation.
Mechanistic insights into translation are becoming accessible through a variety of translation complex stalling approaches, including identification of footprints from diverse initiation, elongation, termination, and recycling intermediates, as well as elongation stall site detection footprints reflective of ribosomal stacking into compactly arranged di- and multi-somes. Integrated computational tools begin to emerge capable of deciphering main and upstream/downstream open reading frames, codon-specific dynamics, and isoform-resolved footprint frequency.
Improvements to translation footprint fidelity and methods to preserve footprint diversity and distribution best reflecting native translation complex state of the cells can be anticipated. Computational methods allowing to identify the dynamics of translational response in the background of concomitant DNA transcription and RNA modification, mRNA localization and degradation, and provide experimental data-driven integrative modeling of these processes, will be developing.
Acknowledgments
The author is very grateful for the critical scientific discussions with all members of the Division of Genome Sciences and Cancer of The John Curtin School of Medical Research, The Australian National University.
Disclosure statement
The author declares that there are no competing interests associated with the manuscript.
Additional information
Funding
References
- Abe F, Hiraki T. 2009. Mechanistic role of ergosterol in membrane rigidity and cycloheximide resistance in Saccharomyces cerevisiae. Biochim Biophys Acta. 1788(3):743–752.
- Adilakshmi T, Lease RA, Woodson SA. 2006. Hydroxyl radical footprinting in vivo: mapping macromolecular structures with synchrotron radiation. Nucleic Acids Res. 34(8):e64.
- Advani VM, Ivanov P. 2019. Translational control under stress: reshaping the translatome. BioEssays. 41(5):1900009.
- Andreev DE, O'Connor PBF, Loughran G, Dmitriev SE, Baranov PV, Shatsky IN. 2017. Insights into the mechanisms of eukaryotic translation gained with ribosome profiling. Nucleic Acids Res. 45(2):513–526.
- Andreeva I, Belardinelli R, Rodnina MV. 2018. Translation initiation in bacterial polysomes through ribosome loading on a standby site on a highly translated mRNA. Proc Natl Acad Sci USA. 115(17):4411–4416.
- Archer SK, Shirokikh NE, Beilharz TH, Preiss T. 2016. Dynamics of ribosome scanning and recycling revealed by translation complex profiling. Nature. 535(7613):570–574.
- Archer SK, Shirokikh NE, Preiss T. 2014. Selective and flexible depletion of problematic sequences from RNA-seq libraries at the cDNA stage. BMC Genomics. 15(1):401.
- Archer SK, Shirokikh NE, Preiss T. 2015. Probe‐directed degradation (PDD) for flexible removal of unwanted cDNA sequences from RNA‐seq libraries. Curr Protoc Hum Genet. 85(1):11.15.1–11.15.36.
- Arenz S, Wilson DN. 2016. Blast from the past: reassessing forgotten translation inhibitors, antibiotic selectivity, and resistance mechanisms to aid drug development. Mol Cell. 61(1):3–14.
- Argüello RJ, Reverendo M, Mendes A, Camosseto V, Torres AG, Sa van de Gatti E, Pierre P. 2018. SunRiSE – measuring translation elongation at single-cell resolution by means of flow cytometry. J Cell Sci. 131(10):jcs214346.
- Arpat AB, Liechti A, Matos MD, Dreos R, Janich P, Gatfield D. 2020. Transcriptome-wide sites of collided ribosomes reveal principles of translational pausing. Genome Res. 30(7):985–999.
- Artieri CG, Fraser HB. 2014. Accounting for biases in riboprofiling data indicates a major role for proline in stalling translation. Genome Res. 24(12):2011–2021.
- Atger F, Gobet C, Marquis J, Martin E, Wang J, Weger B, Lefebvre G, Descombes P, Naef F, Gachon F. 2015. Circadian and feeding rhythms differentially affect rhythmic mRNA transcription and translation in mouse liver. Proc Natl Acad Sci USA. 112(47):E6579–E6588.
- Aviner R. 2020. The science of puromycin: from studies of ribosome function to applications in biotechnology. Comput Struct Biotechnol J. 18:1074–1083.
- Balakrishnan R, Oman K, Shoji S, Bundschuh R, Fredrick K. 2014. The conserved GTPase LepA contributes mainly to translation initiation in Escherichia coli. Nucleic Acids Res. 42(21):13370–13383.
- Barbacid M, Fresno M, Vazquez D. 1975. Inhibitors of polypeptide elongation on yeast polysomes. J Antibiot. 28(6):453–462.
- Barbacid M, Vazquez D. 1974. [3H]anisomycin binding to eukaryotic ribosomes. J Mol Biol. 84(4):603–623.
- Bartholomäus A, Campo CD, Ignatova Z. 2016. Mapping the non-standardized biases of ribosome profiling. Biol Chem. 397(1):23–35.
- Basu A, Yap M-NF. 2016. Ribosome hibernation factor promotes Staphylococcal survival and differentially represses translation. Nucleic Acids Res. 44(10):4881–4893.
- Beaupere C, Wasko BM, Lorusso J, Kennedy BK, Kaeberlein M, Labunskyy VM. 2017. CAN1 arginine permease deficiency extends yeast replicative lifespan via translational activation of stress response genes. Cell Rep. 18(8):1884–1892.
- Belousoff MJ, Eyal Z, Radjainia M, Ahmed T, Bamert RS, Matzov D, Bashan A, Zimmerman E, Mishra S, Cameron D, et al. 2017. Structural basis for linezolid binding site rearrangement in the Staphylococcus aureus ribosome. mBio. 8(3):e00395–e00417.
- Berg JA, Belyeu JR, Morgan JT, Ouyang Y, Bott AJ, Quinlan AR, Gertz J, Rutter J. 2020. XPRESSyourself: enhancing, standardizing, and automating ribosome profiling computational analyses yields improved insight into data. PLOS Comput Biol. 16(1):e1007625.
- Biology’s Big Bang. 2007. The Economist. https://www.economist.com/leaders/2007/06/14/biologys-big-bang
- Blaha G, Gurel G, Schroeder SJ, Moore PB, Steitz TA. 2008. Mutations outside the anisomycin-binding site can make ribosomes drug-resistant. J Mol Biol. 379(3):505–519.
- Blanchard SC, Gonzalez RL, Kim HD, Chu S, Puglisi JD. 2004. tRNA selection and kinetic proofreading in translation. Nat Struct Mol Biol. 11(10):1008–1014.
- Boersma S, Khuperkar D, Verhagen BMP, Sonneveld S, Grimm JB, Lavis LD, Tanenbaum ME. 2019. Multi-color single-molecule imaging uncovers extensive heterogeneity in mRNA decoding. Cell. 178(2):458–472.e19.
- Bohlen J, Fenzl K, Kramer G, Bukau B, Teleman AA. 2020. Selective 40S footprinting reveals cap-tethered ribosome scanning in human cells. Mol Cell. 79(4):561–574.e5.
- Bohlen J, Harbrecht L, Blanco S, Clemm von Hohenberg K, Fenzl K, Kramer G, Bukau B, Teleman AA. 2020. DENR promotes translation reinitiation via ribosome recycling to drive expression of oncogenes including ATF4. Nat Commun. 11(1):4676.
- Boone M, De Koker A, Callewaert N. 2018. Capturing the ‘ome’: the expanding molecular toolbox for RNA and DNA library construction. Nucleic Acids Res. 46(6):2701–2721.
- Bordeleau M-E, Mori A, Oberer M, Lindqvist L, Chard LS, Higa T, Belsham GJ, Wagner G, Tanaka J, Pelletier J. 2006. Functional characterization of IRESes by an inhibitor of the RNA helicase eIF4A. Nat Chem Biol. 2(4):213–220.
- Borovinskaya MA, Pai RD, Zhang W, Schuwirth BS, Holton JM, Hirokawa G, Kaji H, Kaji A, Cate JHD. 2007. Structural basis for aminoglycoside inhibition of bacterial ribosome recycling. Nat Struct Mol Biol. 14(8):727–732.
- Brodersen DE, Clemons WM, Carter AP, Morgan-Warren RJ, Wimberly BT, Ramakrishnan V. 2000. The structural basis for the action of the antibiotics tetracycline, pactamycin, and hygromycin B on the 30S ribosomal subunit. Cell. 103(7):1143–1154.
- Bulkley D, Innis CA, Blaha G, Steitz TA. 2010. Revisiting the structures of several antibiotics bound to the bacterial ribosome. Proc Natl Acad Sci USA. 107(40):17158–17163.
- Bunnik EM, Chung D-WD, Hamilton M, Ponts N, Saraf A, Prudhomme J, Florens L, Le Roch KG. 2013. Polysome profiling reveals translational control of gene expression in the human malaria parasite Plasmodium falciparum. Genome Biol. 14(11):R128.
- Buschauer R, Matsuo Y, Sugiyama T, Chen Y-H, Alhusaini N, Sweet T, Ikeuchi K, Cheng J, Matsuki Y, Nobuta R, et al. 2020. The Ccr4-not complex monitors the translating ribosome for codon optimality. Science. 368(6488):eaay6912.
- Buskirk AR, Green R. 2017. Ribosome pausing, arrest and rescue in bacteria and eukaryotes. Phil Trans R Soc B. 372(1716):20160183.
- Bussolati G, Annaratone L, Berrino E, Miglio U, Panero M, Cupo M, Gugliotta P, Venesio T, Sapino A, Marchiò C. 2017. Acid-free glyoxal as a substitute of formalin for structural and molecular preservation in tissue samples. PLOS One. 12(8):e0182965.
- Carocci M, Yang PL. 2016. Lactimidomycin is a broad-spectrum inhibitor of dengue and other RNA viruses. Antiviral Res. 128:57–62.
- Cencic R, Pelletier J. 2016. Hippuristanol – a potent steroid inhibitor of eukaryotic initiation factor 4A. Translation. 4(1):e1137381.
- Cenik C, Cenik ES, Byeon GW, Grubert F, Candille SI, Spacek D, Alsallakh B, Tilgner H, Araya CL, Tang H, et al. 2015. Integrative analysis of RNA, translation, and protein levels reveals distinct regulatory variation across humans. Genome Res. 25(11):1610–1621.
- Chang JH, Cho YH, Sohn SY, Choi JM, Kim A, Kim YC, Jang SK, Cho Y. 2009. Crystal structure of the eIF4A–PDCD4 complex. Proc Natl Acad Sci USA. 106(9):3148–3153.
- Channathodiyil P, Houseley J. 2021. Glyoxal fixation facilitates transcriptome analysis after antigen staining and cell sorting by flow cytometry. PLOS One. 16(1):e0240769.
- Chassé H, Boulben S, Costache V, Cormier P, Morales J. 2017. Analysis of translation using polysome profiling. Nucleic Acids Res. 45(3):e15.
- Chen C-W, Tanaka M. 2018. Genome-wide translation profiling by ribosome-bound tRNA capture. Cell Rep. 23(2):608–621.
- Chen M, Asanuma M, Takahashi M, Shichino Y, Mito M, Fujiwara K, Saito H, Floor SN, Ingolia NT, Sodeoka M, et al. 2020. Dual targeting of DDX3 and eIF4A by the translation inhibitor rocaglamide A. Cell Chem Biol. 28(4):475–486.e8.
- Chen X, Dickman D. 2017. Development of a tissue-specific ribosome profiling approach in Drosophila enables genome-wide evaluation of translational adaptations. PLOS Genet. 13(12):e1007117.
- Chen Y, Ho JML, Shis DL, Gupta C, Long J, Wagner DS, Ott W, Josić K, Bennett MR. 2018. Tuning the dynamic range of bacterial promoters regulated by ligand-inducible transcription factors. Nat Commun. 9(1):64.
- Chen Y-X, Xu Z, Ge X, Hong J-Y, Sanyal S, Lu ZJ, Javid B. 2020. Selective translation by alternative bacterial ribosomes. Proc Natl Acad Sci USA. 117(32):19487–19496.
- Chew G-L, Pauli A, Rinn JL, Regev A, Schier AF, Valen E. 2013. Ribosome profiling reveals resemblance between long non-coding RNAs and 5′ leaders of coding RNAs. Development. 140(13):2828–2834.
- Chotewutmontri P, Barkan A. 2016. Dynamics of chloroplast translation during chloroplast differentiation in maize. PLoS Genet. 12(7):e1006106.
- Chotewutmontri P, Barkan A. 2018. Multilevel effects of light on ribosome dynamics in chloroplasts program genome-wide and psbA-specific changes in translation. PLoS Genet. 14(8):e1007555.
- Chothani S, Adami E, Ouyang JF, Viswanathan S, Hubner N, Cook SA, Schafer S, Rackham OJL. 2019. deltaTE: Detection of translationally regulated genes by integrative analysis of Ribo-seq and RNA-seq data. Curr Protoc Mol Biol. 129(1):e108.
- Chung BY, Hardcastle TJ, Jones JD, Irigoyen N, Firth AE, Baulcombe DC, Brierley I. 2015. The use of duplex-specific nuclease in ribosome profiling and a user-friendly software package for Ribo-seq data analysis. RNA. 21(10):1731–1745.
- Clamer M, Tebaldi T, Lauria F, Bernabò P, Gómez-Biagi RF, Marchioretto M, Kandala DT, Minati L, Perenthaler E, Gubert D, et al. 2018. Active ribosome profiling with RiboLace. Cell Rep. 25(4):1097–1108.e5.
- Colón-Ramos DA, Shenvi CL, Weitzel DH, Gan EC, Matts R, Cate J, Kornbluth S. 2006. Direct ribosomal binding by a cellular inhibitor of translation. Nat Struct Mol Biol. 13(2):103–111.
- Cookson SJ, Yadav UP, Klie S, Morcuende R, Usadel B, Lunn JE, Stitt M. 2016. Temporal kinetics of the transcriptional response to carbon depletion and sucrose readdition in Arabidopsis seedlings. Plant Cell Environ. 39(4):768–786.
- Couvillion MT, Churchman LS. 2017. Mitochondrial ribosome (mitoribosome) profiling for monitoring mitochondrial translation in vivo. Curr Protoc Mol Biol. 119:4.28.1–4.28.25.
- Dai X, Zhu M, Warren M, Balakrishnan R, Patsalo V, Okano H, Williamson JR, Fredrick K, Wang Y-P, Hwa T. 2016. Reduction of translating ribosomes enables Escherichia coli to maintain elongation rates during slow growth. Nat Microbiol. 2(2):1–9.
- Darnell AM, Subramaniam AR, O'Shea EK. 2018. Translational control through differential ribosome pausing during amino acid limitation in mammalian cells. Mol Cell. 71(2):229–243.e11.
- Davidovich C, Bashan A, Auerbach-Nevo T, Yaggie RD, Gontarek RR, Yonath A. 2007. Induced-fit tightens pleuromutilins binding to ribosomes and remote interactions enable their selectivity. Proc Natl Acad Sci USA. 104(11):4291–4296.
- Davis AR, Gohara DW, Yap M-NF. 2014. Sequence selectivity of macrolide-induced translational attenuation. Proc Natl Acad Sci USA. 111(43):15379–15384.
- de Klerk E, Fokkema IFAC, Thiadens KAMH, Goeman JJ, Palmblad M, den Dunnen JT, von Lindern M, ’T Hoen PAC. 2015. Assessing the translational landscape of myogenic differentiation by ribosome profiling. Nucleic Acids Res. 43(9):4408–4428.
- de Lomana ALG, Kusebauch U, Raman AV, Pan M, Turkarslan S, Lorenzetti APR, Moritz RL, Baliga NS. 2020. Selective translation of low abundance and upregulated transcripts in Halobacterium salinarum. mSystems. 5(4):e00329–20.
- del Prete MJ, Vernal R, Dolznig H, Müllner EW, Garcia-Sanz JA. 2007. Isolation of polysome-bound mRNA from solid tissues amenable for RT-PCR and profiling experiments. RNA. 13(3):414–421.
- DeRisi JL, Iyer VR, Brown PO. 1997. Exploring the metabolic and genetic control of gene expression on a genomic scale. Science. 278(5338):680–686.
- Dmitriev SE, Pisarev AV, Rubtsova MP, Dunaevsky YE, Shatsky IN. 2003. Conversion of 48S translation preinitiation complexes into 80S initiation complexes as revealed by toeprinting. FEBS Lett. 533(1–3):99–104.
- Dmitriev SE, Vladimirov DO, Lashkevich KA. 2020. A quick guide to small-molecule inhibitors of eukaryotic protein synthesis. Biochemistry. 85(11):1389–1421.
- Duncan CDS, Mata J. 2017. Effects of cycloheximide on the interpretation of ribosome profiling experiments in Schizosaccharomyces pombe. Sci Rep. 7(1):10331.
- Dunkle JA, Xiong L, Mankin AS, Cate JHD. 2010. Structures of the Escherichia coli ribosome with antibiotics bound near the peptidyl transferase center explain spectra of drug action. Proc Natl Acad Sci USA. 107(40):17152–17157.
- Dunn JG, Foo CK, Belletier NG, Gavis ER, Weissman JS. 2013. Ribosome profiling reveals pervasive and regulated stop codon readthrough in Drosophila melanogaster. eLife. 2:e01179.
- Dykstra KH, Li X-M, Wang HY. 1988. Computer modeling of antibiotic fermentation with on-line product removal. Biotechnol Bioeng. 32(3):356–362.
- Elgamal S, Katz A, Hersch SJ, Newsom D, White P, Navarre WW, Ibba M. 2014. EF-P dependent pauses integrate proximal and distal signals during translation. PLoS Genet. 10(8):e1004553.
- ENCODE Project Consortium, Birney E, Stamatoyannopoulos JA, Dutta A, Guigó R, Gingeras TR, Margulies EH, Weng Z, Snyder M, Dermitzakis ET, et al. 2007. Identification and analysis of functional elements in 1% of the human genome by the ENCODE pilot project. Nature. 447(7146):799–816.
- Ermolenko DN, Cornish PV, Ha T, Noller HF. 2013. Antibiotics that bind to the A site of the large ribosomal subunit can induce mRNA translocation. RNA. 19(2):158–166.
- Ermolenko DN, Spiegel PC, Majumdar ZK, Hickerson RP, Clegg RM, Noller HF. 2007. The antibiotic viomycin traps the ribosome in an intermediate state of translocation. Nat Struct Mol Biol. 14(6):493–497.
- Eustice DC, Wilhelm JM. 1984. Mechanisms of action of aminoglycoside antibiotics in eucaryotic protein synthesis. Antimicrob Agents Chemother. 26(1):53–60.
- Festuccia N, Owens N, Papadopoulou T, Gonzalez I, Tachtsidi A, Vandoermel-Pournin S, Gallego E, Gutierrez N, Dubois A, Cohen-Tannoudji M, et al. 2019. Transcription factor activity and nucleosome organization in mitosis. Genome Res. 29(2):250–260.
- Firmino AAP, Gorka M, Graf A, Skirycz A, Martinez-Seidel F, Zander K, Kopka J, Beine-Golovchuk O. 2020. Separation and paired proteome profiling of plant chloroplast and cytoplasmic ribosomes. Plants. 9(7):892.
- Fluman N, Navon S, Bibi E, Pilpel Y. 2014. mRNA-programmed translation pauses in the targeting of E. coli membrane proteins. eLife. 3:e03440.
- Fourmy D, Recht MI, Blanchard SC, Puglisi JD. 1996. Structure of the A site of Escherichia coli 16S ribosomal RNA complexed with an aminoglycoside antibiotic. Science. 274(5291):1367–1371.
- Frank J. 2017. The translation elongation cycle—capturing multiple states by cryo-electron microscopy. Phil Trans R Soc B. 372(1716):20160180.
- Fremin BJ, Sberro H, Bhatt AS. 2020. MetaRibo-seq measures translation in microbiomes. Nat Commun. 11(1):3268.
- Fresno M, Jiménez A, Vázquez D. 1977. Inhibition of translation in eukaryotic systems by harringtonine. Eur J Biochem. 72(2):323–330.
- Fritsch C, Herrmann A, Nothnagel M, Szafranski K, Huse K, Schumann F, Schreiber S, Platzer M, Krawczak M, Hampe J, et al. 2012. Genome-wide search for novel human uORFs and N-terminal protein extensions using ribosomal footprinting. Genome Res. 22(11):2208–2218.
- Fujii K, Aoki M, Uekusa H. 2013. Solid-state hydration/dehydration of erythromycin A investigated by ab initio powder X-ray diffraction analysis: stoichiometric and nonstoichiometric dehydrated hydrate. Crystal Growth Des. 13(5):2060–2066.
- Fujita T, Kurihara Y, Iwasaki S. 2019. The plant translatome surveyed by ribosome profiling. Plant Cell Physiol. 60(9):1917–1926.
- Furey TS. 2012. ChIP-seq and beyond: new and improved methodologies to detect and characterize protein-DNA interactions. Nat Rev Genet. 13(12):840–852.
- Galmozzi CV, Merker D, Friedrich UA, Döring K, Kramer G. 2019. Selective ribosome profiling to study interactions of translating ribosomes in yeast. Nat Protoc. 14(8):2279–2317.
- Gao X, Wan J, Liu B, Ma M, Shen B, Qian S-B. 2015. Quantitative profiling of initiating ribosomes in vivo. Nat Methods. 12(2):147–153.
- Garreau de Loubresse N, Prokhorova I, Holtkamp W, Rodnina MV, Yusupova G, Yusupov M. 2014. Structural basis for the inhibition of the eukaryotic ribosome. Nature. 513(7519):517–522.
- Garza-Ramos G, Xiong L, Zhong P, Mankin A. 2001. Binding site of macrolide antibiotics on the ribosome: new resistance mutation identifies a specific interaction of ketolides with rRNA. J Bacteriol. 183(23):6898–6907.
- Gavrilov A, Razin SV, Cavalli G. 2015. In vivo formaldehyde cross-linking: it is time for black box analysis. Brief Funct Genomics. 14(2):163–165.
- Gawroński P, Jensen PE, Karpiński S, Leister D, Scharff LB. 2018. Pausing of chloroplast ribosomes is induced by multiple features and is linked to the assembly of photosynthetic complexes. Plant Physiol. 176(3):2557–2569.
- Ge X, Oliveira A, Hjort K, Bergfors T, Gutiérrez-de-Terán H, Andersson DI, Sanyal S, Åqvist J. 2019. Inhibition of translation termination by small molecules targeting ribosomal release factors. Sci Rep. 9(1):15424.
- Gelsinger DR, Dallon E, Reddy R, Mohammad F, Buskirk AR, DiRuggiero J. 2020. Ribosome profiling in archaea reveals leaderless translation, novel translational initiation sites, and ribosome pausing at single codon resolution. Nucleic Acids Res. 48(10):5201–5216.
- Gerashchenko MV, Gladyshev VN. 2014. Translation inhibitors cause abnormalities in ribosome profiling experiments. Nucleic Acids Res. 42(17):e134.
- Gerashchenko MV, Gladyshev VN. 2017. Ribonuclease selection for ribosome profiling. Nucleic Acids Res. 45(2):e6.
- Gerashchenko MV, Peterfi Z, Gladyshev VN. 2018. Organ-specific translation elongation rates measured by in vivo ribosome profiling. bioRxiv. 279257.
- Gerashchenko MV, Peterfi Z, Yim SH, Gladyshev VN. 2021. Translation elongation rate varies among organs and decreases with age. Nucleic Acids Res. 49(2):e9.
- Gerlinger U-M, Gückel R, Hoffmann M, Wolf DH, Hilt W. 1997. Yeast cycloheximide-resistant CRL mutants are proteasome mutants defective in protein degradation. Mol Biol Cell. 8(12):2487–2499.
- Giess A, Torres Cleuren YN, Tjeldnes H, Krause M, Bizuayehu TT, Hiensch S, Okon A, Wagner CR, Valen E. 2020. Profiling of small ribosomal subunits reveals modes and regulation of translation initiation. Cell Rep. 31(3):107534.
- Glaub A, Huptas C, Neuhaus K, Ardern Z. 2019. Improving bacterial ribosome profiling data quality. bioRxiv. 863266.
- Glaub A, Huptas C, Neuhaus K, Ardern Z. 2020. Recommendations for bacterial ribosome profiling experiments based on bioinformatic evaluation of published data. J Biol Chem. 295(27):8999–9011.
- Gobet C, Naef F. 2017. Ribosome profiling and dynamic regulation of translation in mammals. Curr Opin Genet Dev. 43:120–127.
- Gokal PK, Cavanaugh AH, Thompson EA. 1986. The effects of cycloheximide upon transcription of rRNA, 5S RNA, and tRNA genes. J Biol Chem. 261(6):2536–2541.
- González A, Santamaría F, Vázquez D, Jiménez A. 1981. A mutation of Saccharomyces cerevisiae leading to resistance to some inhibitors of peptide bond formation. Mol Gen Genet. 181(1):140–146.
- González N, Barral MA, Rodrı́guez J, Jiménez C. 2001. New cytotoxic steroids from the gorgonian Isis hippuris. Structure–activity studies. Tetrahedron. 57(16):3487–3497.
- Goodall KJ, Finch-Edmondson ML, van Vuuren J, Yeoh GC, Gentle IE, Vince JE, Ekert PG, Vaux DL, Callus BA. 2016. Cycloheximide can induce Bax/Bak dependent myeloid cell death independently of multiple BH3-only proteins. PLoS One. 11(11):e0164003.
- Griffin MA, Davis JH, Strobel SA. 2013. Bacterial Toxin RelE: a highly efficient ribonuclease with exquisite substrate specificity using atypical catalytic residues. Biochemistry. 52(48):8633–8642.
- Grünberg S, Coxam B, Chen T-H, Dai N, Saleh L, Corrêa IR, Jr, Nichols NM, Yigit E. 2021. E. coli RNase I exhibits a strong Ca2+-dependent inherent double-stranded RNase activity. Nucleic Acids Res. 49(9):5265–5277.
- GSI Chemical Database. 2021. GSI Environmental Inc. https://www.gsi-net.com/http://www.gsi-net.com/en/publications/gsi-chemical-database/single/288-CAS-50000.html
- Gualerzi CO, Pon CL. 2015. Initiation of mRNA translation in bacteria: structural and dynamic aspects. Cell Mol Life Sci. 72(22):4341–4367.
- Gupta PK. 2018. Chapter 45 – toxicity of fungicides. In: Gupta RC, editor. Veterinary toxicology. 3rd ed. Elsevier Science Publishing Co Inc, United States: Academic Press; p. 569–580.
- Guydosh NR, Green R. 2017. Translation of poly(A) tails leads to precise mRNA cleavage. RNA. 23(5):749–761.
- Han P, Shichino Y, Schneider-Poetsch T, Mito M, Hashimoto S, Udagawa T, Kohno K, Yoshida M, Mishima Y, Inada T, et al. 2020. Genome-wide survey of ribosome collision. Cell Rep. 31(5):107610.
- Han Y, Gao X, Liu B, Wan J, Zhang X, Qian S-B. 2014. Ribosome profiling reveals sequence-independent post-initiation pausing as a signature of translation. Cell Res. 24(7):842–851.
- Hanson G, Alhusaini N, Morris N, Sweet T, Coller J. 2018. Translation elongation and mRNA stability are coupled through the ribosomal A-site. RNA. 24(10):1377–1389.
- Hardigan AA, Roberts BS, Moore DE, Ramaker RC, Jones AL, Myers RM. 2019. CRISPR/Cas9-targeted removal of unwanted sequences from small-RNA sequencing libraries. Nucleic Acids Res. 47(14):e84.
- Hershey JWB, Sonenberg N, Mathews MB. 2019. Principles of translational control. Cold Spring Harb Perspect Biol. 11(9):a032607.
- Higa T, Tanaka J, Tsukitani Y, Kikuchi H. 1981. Hippuristanols, cytotoxic polyoxygenated steroids from the gorgonian Isis hippuris. Chem Lett. 10(11):1647–1650.
- Hinnebusch AG, Ivanov IP, Sonenberg N. 2016. Translational control by 5′-untranslated regions of eukaryotic mRNAs. Science. 352(6292):1413–1416.
- Hinnebusch AG. 2017. Structural insights into the mechanism of scanning and start codon recognition in eukaryotic translation initiation. Trends Biochem Sci. 42(8):589–611.
- Hobson BD, Kong L, Hartwick EW, Gonzalez RL, Sims PA. 2020. Elongation inhibitors do not prevent the release of puromycylated nascent polypeptide chains from ribosomes. eLife. 9:e60048.
- Hoffman EA, Frey BL, Smith LM, Auble DT. 2015. Formaldehyde crosslinking: a tool for the study of chromatin complexes. J Biol Chem. 290(44):26404–26411.
- Hogan B, Gross PR. 1971. The effect of protein synthesis inhibition on the entry of messenger RNA into the cytoplasm of sea urchin embryos. J Cell Biol. 49(3):692–701.
- Honkela A, Peltonen J, Topa H, Charapitsa I, Matarese F, Grote K, Stunnenberg HG, Reid G, Lawrence ND, Rattray M. 2015. Genome-wide modeling of transcription kinetics reveals patterns of RNA production delays. Proc Natl Acad Sci USA. 112(42):13115–13120.
- Huang K, Chen W, Zhu F, Li PW-L, Kapahi P, Bai H. 2019. RiboTag translatomic profiling of Drosophila oenocytes under aging and induced oxidative stress. BMC Genomics. 20(1):50.
- Huang S-X, Yu Z, Robert F, Zhao L-X, Jiang Y, Duan Y, Pelletier J, Shen B. 2011. Cycloheximide and congeners as inhibitors of eukaryotic protein synthesis from endophytic actinomycetes Streptomyces sps. YIM56132 and YIM56141. J Antibiot. 64(1):163–166.
- Huang Y, Sheth RU, Kaufman A, Wang HH. 2020. Scalable and cost-effective ribonuclease-based rRNA depletion for transcriptomics. Nucleic Acids Res. 48(4):e20.
- Hücker SM, Ardern Z, Goldberg T, Schafferhans A, Bernhofer M, Vestergaard G, Nelson CW, Schloter M, Rost B, Scherer S, et al. 2017. Discovery of numerous novel small genes in the intergenic regions of the Escherichia coli O157:H7 Sakai genome. PLoS One. 12(9):e0184119.
- Hussmann JA, Patchett S, Johnson A, Sawyer S, Press WH. 2015. Understanding biases in ribosome profiling experiments reveals signatures of translation dynamics in yeast. PLoS Genet. 11(12):e1005732.
- Hwang J-Y, Buskirk AR. 2017. A ribosome profiling study of mRNA cleavage by the endonuclease RelE. Nucleic Acids Res. 45(1):327–336.
- Hwang S, Lee N, Jeong Y, Lee Y, Kim W, Cho S, Palsson BO, Cho B-K. 2019. Primary transcriptome and translatome analysis determines transcriptional and translational regulatory elements encoded in the Streptomyces clavuligerus genome. Nucleic Acids Res. 47(12):6114–6129.
- Ikeuchi K, Tesina P, Matsuo Y, Sugiyama T, Cheng J, Saeki Y, Tanaka K, Becker T, Beckmann R, Inada T. 2019. Collided ribosomes form a unique structural interface to induce Hel2-driven quality control pathways. EMBO J. 38(5):e100276.
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. 2012. The ribosome profiling strategy for monitoring translation in vivo by deep sequencing of ribosome-protected mRNA fragments. Nat Protoc. 7(8):1534–1550.
- Ingolia NT, Brar GA, Rouskin S, McGeachy AM, Weissman JS. 2013. Genome-wide annotation and quantitation of translation by ribosome profiling. Curr Protoc Mol Biol. 103(1):4.18.1–4.18.19.
- Ingolia NT, Brar GA, Stern-Ginossar N, Harris MS, Talhouarne GJS, Jackson SE, Wills MR, Weissman JS. 2014. Ribosome profiling reveals pervasive translation outside of annotated protein-coding genes. Cell Rep. 8(5):1365–1379.
- Ingolia NT, Ghaemmaghami S, Newman JRS, Weissman JS. 2009. Genome-wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science. 324(5924):218–223.
- Ingolia NT, Hussmann JA, Weissman JS. 2019. Ribosome profiling: global views of translation. Cold Spring Harb Perspect Biol. 11(5):a032698.
- Ingolia NT, Lareau LF, Weissman JS. 2011. Ribosome profiling of mouse embryonic stem cells reveals the complexity and dynamics of mammalian proteomes. Cell. 147(4):789–802.
- Ingolia NT. 2016. Ribosome footprint profiling of translation throughout the genome. Cell. 165(1):22–33.
- Ivanov IP, Shin B-S, Loughran G, Tzani I, Young-Baird SK, Cao C, Atkins JF, Dever TE. 2018. Polyamine control of translation elongation regulates start site selection on antizyme inhibitor mRNA via ribosome queuing. Mol Cell. 70(2):254–264.e6.
- Ivanov P, Kedersha N, Anderson P. 2019. Stress granules and processing bodies in translational control. Cold Spring Harb Perspect Biol. 11(5):a032813.
- Iwasaki S, Floor SN, Ingolia NT. 2016. Rocaglates convert DEAD-box protein eIF4A into a sequence-selective translational repressor. Nature. 534(7608):558–561.
- Iwasaki S, Iwasaki W, Takahashi M, Sakamoto A, Watanabe C, Shichino Y, Floor SN, Fujiwara K, Mito M, Dodo K, et al. 2019. The translation inhibitor rocaglamide targets a bimolecular cavity between eIF4A and polypurine RNA. Mol Cell. 73(4):738–748.e9.
- Jackson R, Standart N. 2015. The awesome power of ribosome profiling. RNA. 21(4):652–654.
- Jan CH, Williams CC, Weissman JS. 2014. Principles of ER cotranslational translocation revealed by proximity-specific ribosome profiling. Science. 346(6210):1257521.
- Janapala Y, Preiss T, Shirokikh NE. 2019. Control of translation at the initiation phase during glucose starvation in yeast. IJMS. 20(16):4043.
- Janich P, Arpat AB, Castelo-Szekely V, Gatfield D. 2016. Analyzing the temporal regulation of translation efficiency in mouse liver. Genom Data. 8:41–44.
- Janich P, Arpat AB, Castelo-Szekely V, Lopes M, Gatfield D. 2015. Ribosome profiling reveals the rhythmic liver translatome and circadian clock regulation by upstream open reading frames. Genome Res. 25(12):1848–1859.
- Jayaraman P, Yeoh JW, Zhang J, Poh CL. 2018. Programming the dynamic control of bacterial gene expression with a chimeric ligand- and light-based promoter system. ACS Synth Biol. 7(11):2627–2639.
- Jelić D, Antolović R. 2016. From erythromycin to azithromycin and new potential ribosome-binding antimicrobials. Antibiotics. 5(3):29.
- Jenner L, Starosta AL, Terry DS, Mikolajka A, Filonava L, Yusupov M, Blanchard SC, Wilson DN, Yusupova G. 2013. Structural basis for potent inhibitory activity of the antibiotic tigecycline during protein synthesis. Proc Natl Acad Sci USA. 110(10):3812–3816.
- Jensen BC, Ramasamy G, Vasconcelos EJR, Ingolia NT, Myler PJ, Parsons M. 2014. Extensive stage-regulation of translation revealed by ribosome profiling of Trypanosoma brucei. BMC Genomics. 15(1):911.
- Jeong Y, Kim J-N, Kim MW, Bucca G, Cho S, Yoon YJ, Kim B-G, Roe J-H, Kim SC, Smith CP, et al. 2016. The dynamic transcriptional and translational landscape of the model antibiotic producer Streptomyces coelicolor A3(2). Nat Commun. 7(1):11605.
- Jonker HRA, Baumann S, Wolf A, Schoof S, Hiller F, Schulte KW, Kirschner KN, Schwalbe H, Arndt H-D. 2011. NMR structures of thiostrepton derivatives for characterization of the ribosomal binding site. Angew Chem Int Ed Engl. 50(14):3308–3312.
- Juntawong P, Girke T, Bazin J, Bailey-Serres J. 2014. Translational dynamics revealed by genome-wide profiling of ribosome footprints in Arabidopsis. Proc Natl Acad Sci USA. 111(1):E203–E212.
- Kamps JJAG, Hopkinson RJ, Schofield CJ, Claridge TDW. 2019. How formaldehyde reacts with amino acids. Commun Chem. 2(1):1–14.
- Kannan K, Kanabar P, Schryer D, Florin T, Oh E, Bahroos N, Tenson T, Weissman JS, Mankin AS. 2014. The general mode of translation inhibition by macrolide antibiotics. Proc Natl Acad Sci USA. 111(45):15958–15963.
- Kannan K, Vázquez-Laslop N, Mankin AS. 2012. Selective protein synthesis by ribosomes with a drug-obstructed exit tunnel. Cell. 151(3):508–520.
- Karlsen J, Asplund-Samuelsson J, Thomas Q, Jahn M, Hudson EP. 2018. Ribosome profiling of synechocystis reveals altered ribosome allocation at carbon starvation. mSystems. 3(5):e00126–18.
- Kasari V, Margus T, Atkinson GC, Johansson MJO, Hauryliuk V. 2019. Ribosome profiling analysis of eEF3-depleted Saccharomyces cerevisiae. Sci Rep. 9(1):3037.
- Katz Y, Li F, Lambert NJ, Sokol ES, Tam W-L, Cheng AW, Airoldi EM, Lengner CJ, Gupta PB, Yu Z, et al. 2014. Musashi proteins are post-transcriptional regulators of the epithelial-luminal cell state. Elife. 3:e03915.
- Kavčič B, Tkačik G, Bollenbach T. 2020. Mechanisms of drug interactions between translation-inhibiting antibiotics. Nat Commun. 11(1):4013.
- Kearse MG, Goldman DH, Choi J, Nwaezeapu C, Liang D, Green KM, Goldstrohm AC, Todd PK, Green R, Wilusz JE. 2019. Ribosome queuing enables non-AUG translation to be resistant to multiple protein synthesis inhibitors. Genes Dev. 33(13–14):871–885.
- Khong A, Bonderoff JM, Spriggs RV, Tammpere E, Kerr CH, Jackson TJ, Willis AE, Jan E. 2016. Temporal regulation of distinct internal ribosome entry sites of the Dicistroviridae cricket paralysis virus. Viruses. 8(1):25.
- Kiernan JA. 2000. Formaldehyde, formalin, paraformaldehyde and glutaraldehyde: what they are and what they do. Micros Today. 8(1):8–13.
- Kim W, Hwang S, Lee N, Lee Y, Cho S, Palsson B, Cho B-K. 2020. Transcriptome and translatome profiles of Streptomyces species in different growth phases. Sci Data. 7(1):138.
- Kim YK, Maquat LE. 2019. UPFront and center in RNA decay: UPF1 in nonsense-mediated mRNA decay and beyond. RNA. 25(4):407–422.
- King HA, Gerber AP. 2016. Translatome profiling: methods for genome-scale analysis of mRNA translation. Brief Funct Genomics. 15(1):22–31.
- Kitahara K, Miyazaki K. 2011. Specific inhibition of bacterial RNase T2 by helix 41 of 16S ribosomal RNA. Nat Commun. 2:549.
- Koga Y, Hoang EM, Park Y, Keszei AFA, Murray J, Shao S, Liau BB. 2021. Discovery of C13-aminobenzoyl cycloheximide derivatives that potently inhibit translation elongation. J Am Chem Soc. 143(34):13473–13477.
- Korostelev AA. 2011. Structural aspects of translation termination on the ribosome. RNA. 17(8):1409–1421.
- Kouvela EC, Petropoulos AD, Kalpaxis DL. 2006. Unraveling new features of clindamycin interaction with functional ribosomes and dependence of the drug potency on polyamines. J Biol Chem. 281(32):23103–23110.
- Kozak M. 1990. Downstream secondary structure facilitates recognition of initiator codons by eukaryotic ribosomes. Proc Natl Acad Sci USA. 87(21):8301–8305.
- Kozak M. 1998. Primer extension analysis of eukaryotic ribosome-mRNA complexes. Nucleic Acids Res. 26(21):4853–4859.
- Kozak M. 2005. Regulation of translation via mRNA structure in prokaryotes and eukaryotes. Gene. 361:13–37.
- Kragol G, Lovas S, Varadi G, Condie BA, Hoffmann R, Otvos L. 2001. The antibacterial peptide pyrrhocoricin inhibits the ATPase actions of DnaK and prevents chaperone-assisted protein folding. Biochemistry. 40(10):3016–3026.
- Krause KM, Serio AW, Kane TR, Connolly LE. 2016. Aminoglycosides: an overview. Cold Spring Harb Perspect Med. 6(6):a027029.
- Krizsan A, Volke D, Weinert S, Sträter N, Knappe D, Hoffmann R. 2014. Insect-derived proline-rich antimicrobial peptides kill bacteria by inhibiting bacterial protein translation at the 70S ribosome. Angew Chem Int Ed. 53(45):12236–12239.
- Kurata S, Nielsen KH, Mitchell SF, Lorsch JR, Kaji A, Kaji H. 2010. Ribosome recycling step in yeast cytoplasmic protein synthesis is catalyzed by eEF3 and ATP. Proc Natl Acad Sci USA. 107(24):10854–10859.
- Kusnadi EP, Trigos AS, Cullinane C, Goode DL, Larsson O, Devlin JR, Chan KT, De Souza DP, McConville MJ, McArthur GA, et al. 2020. Reprogrammed mRNA translation drives resistance to therapeutic targeting of ribosome biogenesis. EMBO J. 39(21):e105111.
- Lambert N, Robertson A, Jangi M, McGeary S, Sharp PA, Burge CB. 2014. RNA Bind-n-Seq: quantitative assessment of the sequence and structural binding specificity of RNA binding proteins. Mol Cell. 54(5):887–900.
- Lambert NJ, Robertson AD, Burge CB. 2015. Chapter 15 – RNA Bind-n-Seq: measuring the binding affinity landscape of RNA-binding proteins. In: Woodson SA, Allain FHT, editors. Methods in Enzymology. Vol. 558. Elsevier Science Publishing Co Inc, United States: Academic Press; p. 465–493.
- Lareau LF, Hite DH, Hogan GJ, Brown PO. 2014. Distinct stages of the translation elongation cycle revealed by sequencing ribosome-protected mRNA fragments. eLife. 3:e01257.
- Lashkevich KA, Shlyk VI, Kushchenko AS, Gladyshev VN, Alkalaeva EZ, Dmitriev SE. 2020. CTELS: a cell-free system for the analysis of translation termination rate. Biomolecules. 10(6):911.
- Lee S, Liu B, Lee S, Huang S-X, Shen B, Qian S-B. 2012. Global mapping of translation initiation sites in mammalian cells at single-nucleotide resolution. Proc Natl Acad Sci USA. 109(37):E2424–E2432.
- Legrand C, Tuorto F. 2020. RiboVIEW: a computational framework for visualization, quality control and statistical analysis of ribosome profiling data. Nucleic Acids Res. 48(2):e7.
- Lei L, Shi J, Chen J, Zhang M, Sun S, Xie S, Li X, Zeng B, Peng L, Hauck A, et al. 2015. Ribosome profiling reveals dynamic translational landscape in maize seedlings under drought stress. Plant J. 84(6):1206–1218.
- Li G-W, Oh E, Weissman JS. 2012. The anti-Shine-Dalgarno sequence drives translational pausing and codon choice in bacteria. Nature. 484(7395):538–541.
- Li W, Chang ST-L, Ward FR, Cate JHD. 2020. Selective inhibition of human translation termination by a drug-like compound. Nat Commun. 11(1):4941.
- Lindqvist L, Oberer M, Reibarkh M, Cencic R, Bordeleau M-E, Vogt E, Marintchev A, Tanaka J, Fagotto F, Altmann M, et al. 2008. Selective pharmacological targeting of a DEAD box RNA helicase. PLoS One. 3(2):e0001583.
- Liu B, Qian S-B. 2016. Characterizing inactive ribosomes in translational profiling. Translation. 4(1):e1138018.
- Liu C-L, Place AR, Jagus R. 2017. Use of antibiotics for maintenance of axenic cultures of Amphidinium carterae for the analysis of translation. Mar Drugs. 15(8):242.
- Liu Q, Shvarts T, Sliz P, Gregory RI. 2020. RiboToolkit: an integrated platform for analysis and annotation of ribosome profiling data to decode mRNA translation at codon resolution. Nucleic Acids Res. 48(W1):W218–W229.
- Lomakin IB, Gagnon MG, Steitz TA. 2015. Antimicrobial peptides targeting bacterial ribosome. Oncotarget. 6(22):18744–18745.
- Long KS, Porse BT. 2003. A conserved chloramphenicol binding site at the entrance to the ribosomal peptide exit tunnel. Nucleic Acids Res. 31(24):7208–7215.
- Low W-K, Dang Y, Schneider-Poetsch T, Shi Z, Choi NS, Merrick WC, Romo D, Liu JO. 2005. Inhibition of eukaryotic translation initiation by the marine natural product pateamine A. Mol Cell. 20(5):709–722.
- Lu W, Roongsawang N, Mahmud T. 2011. Biosynthetic studies and genetic engineering of pactamycin analogs with improved selectivity toward malarial parasites. Chem Biol. 18(4):425–431.
- Lukoszek R, Feist P, Ignatova Z. 2016. Insights into the adaptive response of Arabidopsis thaliana to prolonged thermal stress by ribosomal profiling and RNA-Seq. BMC Plant Biol. 16(1):221.
- Makino S, Kawamata T, Iwasaki S, Ohsumi Y. 2021. Selectivity of mRNA degradation by autophagy in yeast. Nat Commun. 12(1):2316.
- Mardirossian M, Grzela R, Giglione C, Meinnel T, Gennaro R, Mergaert P, Scocchi M. 2014. The host antimicrobial peptide Bac71-35 binds to bacterial ribosomal proteins and inhibits protein synthesis. Chem Biol. 21(12):1639–1647.
- Kozak M, Shatkin AJ. 1976. Characterization of ribosome-protected fragments from reovirus messenger RNA. J Biol Chem. 251(14):4259–4266.
- Marks J, Kannan K, Roncase EJ, Klepacki D, Kefi A, Orelle C, Vázquez-Laslop N, Mankin AS. 2016. Context-specific inhibition of translation by ribosomal antibiotics targeting the peptidyl transferase center. Proc Natl Acad Sci USA. 113(43):12150–12155.
- Matsuo Y, Ikeuchi K, Saeki Y, Iwasaki S, Schmidt C, Udagawa T, Sato F, Tsuchiya H, Becker T, Tanaka K, et al. 2017. Ubiquitination of stalled ribosome triggers ribosome-associated quality control. Nat Commun. 8(1):159.
- Matzov D, Eyal Z, Benhamou RI, Shalev-Benami M, Halfon Y, Krupkin M, Zimmerman E, Rozenberg H, Bashan A, Fridman M, et al. 2017. Structural insights of lincosamides targeting the ribosome of Staphylococcus aureus. Nucleic Acids Res. 45(17):10284–10292.
- Meydan S, Marks J, Klepacki D, Sharma V, Baranov PV, Firth AE, Margus T, Kefi A, Vázquez-Laslop N, Mankin AS. 2019. Retapamulin-assisted ribosome profiling reveals the alternative bacterial proteome. Mol Cell. 74(3):481–493.e6.
- McGlincy NJ, Ingolia NT. 2017. Transcriptome-wide measurement of translation by ribosome profiling. Methods. 126:112–129.
- Michel AM, Fox G, M Kiran A, De Bo C, O'Connor PBF, Heaphy SM, Mullan JPA, Donohue CA, Higgins DG, Baranov PV. 2014. GWIPS-viz: development of a Ribo-seq genome browser. Nucleic Acids Res. 42(Database issue):D859–D864.
- Michel AM, Mullan JPA, Velayudhan V, O'Connor PBF, Donohue CA, Baranov PV. 2016. RiboGalaxy: a browser based platform for the alignment, analysis and visualization of ribosome profiling data. RNA Biol. 13(3):316–319.
- Mohammad F, Buskirk AR. 2019. Protocol for ribosome profiling in bacteria. Bio Protoc. 9(24):e3468.
- Mohammad F, Green R, Buskirk AR. 2019. A systematically-revised ribosome profiling method for bacteria reveals pauses at single-codon resolution. eLife. 8:e42591.
- Mohammad F, Woolstenhulme CJ, Green R, Buskirk AR. 2016. Clarifying the translational pausing landscape in bacteria by ribosome profiling. Cell Rep. 14(4):686–694.
- Molina N, Suter DM, Cannavo R, Zoller B, Gotic I, Naef F. 2013. Stimulus-induced modulation of transcriptional bursting in a single mammalian gene. Proc Natl Acad Sci USA. 110(51):20563–20568.
- Moon SL, Morisaki T, Stasevich TJ, Parker R. 2020. Coupling of translation quality control and mRNA targeting to stress granules. J Cell Biol. 219:e202004120.
- Mühlemann O, Karousis ED. 2017. New functions in translation termination uncovered for NMD factor UPF3B. EMBO J. 36(20):2928–2930.
- Muzzey D, Sherlock G, Weissman JS. 2014. Extensive and coordinated control of allele-specific expression by both transcription and translation in Candida albicans. Genome Res. 24(6):963–973.
- Naineni SK, Maiga RI, Cencic R, Putnam AA, Amador LA, Rodriguez AD, Jankowsky E, Pelletier J. 2020. A comparative study of small molecules targeting eIF4A. RNA. 26(5):541–549.
- Nakahigashi K, Takai Y, Kimura M, Abe N, Nakayashiki T, Shiwa Y, Yoshikawa H, Wanner BL, Ishihama Y, Mori H. 2016. Comprehensive identification of translation start sites by tetracycline-inhibited ribosome profiling. DNA Res. 23(3):193–201.
- Nakato R, Sakata T. 2021. Methods for ChIP-seq analysis: a practical workflow and advanced applications. Methods. 187:44–53.
- Novo M, Beltran G, Rozes N, Guillamon J-M, Sokol S, Leberre V, François J, Mas A. 2007. Early transcriptional response of wine yeast after rehydration: osmotic shock and metabolic activation. FEMS Yeast Res. 7(2):304–316.
- Nürenberg-Goloub E, Tampé R. 2020. Ribosome recycling in mRNA translation, quality control, and homeostasis. Biol Chem. 401(1):47–61.
- O'Connor PBF, Andreev DE, Baranov PV. 2016. Comparative survey of the relative impact of mRNA features on local ribosome profiling read density. Nat Commun. 7(1):12915.
- O'Connor PBF, Li G-W, Weissman JS, Atkins JF, Baranov PV. 2013. rRNA:mRNA pairing alters the length and the symmetry of mRNA-protected fragments in ribosome profiling experiments. Bioinformatics. 29(12):1488–1491.
- Obrig TG, Culp WJ, McKeehan WL, Hardesty B. 1971. The mechanism by which cycloheximide and related glutarimide antibiotics inhibit peptide synthesis on reticulocyte ribosomes. J Biol Chem. 246(1):174–181.
- Oh E, Becker AH, Sandikci A, Huber D, Chaba R, Gloge F, Nichols RJ, Typas A, Gross CA, Kramer G, et al. 2011. Selective ribosome profiling reveals the cotranslational chaperone action of trigger factor in vivo. Cell. 147(6):1295–1308.
- Padrón A, Iwasaki S, Ingolia NT. 2019. Proximity RNA labeling by APEX-seq reveals the organization of translation initiation complexes and repressive RNA granules. Mol Cell. 75(4):875–887.e5.
- Panasenko OO, Somasekharan SP, Villanyi Z, Zagatti M, Bezrukov F, Rashpa R, Cornut J, Iqbal J, Longis M, Carl SH, et al. 2019. Co-translational assembly of proteasome subunits in NOT1-containing assemblysomes. Nat Struct Mol Biol. 26(2):110–120.
- Pelechano V, Wei W, Steinmetz LM. 2015. Widespread co-translational RNA decay reveals ribosome dynamics. Cell. 161(6):1400–1412.
- Pestka S. 1974. Antibiotics as probes of ribosome structure: binding of chloramphenicol and erythromycin to polyribosomes; effect of other antibiotics. Antimicrob Agents Chemother. 5(3):255–267.
- Pestova TV, Hellen CU, Shatsky IN. 1996. Canonical eukaryotic initiation factors determine initiation of translation by internal ribosomal entry. Mol Cell Biol. 16(12):6859–6869.
- Pestova TV, Kolupaeva VG. 2002. The roles of individual eukaryotic translation initiation factors in ribosomal scanning and initiation codon selection. Genes Dev. 16(22):2906–2922.
- Pioletti M, Schlünzen F, Harms J, Zarivach R, Glühmann M, Avila H, Bashan A, Bartels H, Auerbach T, Jacobi C, et al. 2001. Crystal structures of complexes of the small ribosomal subunit with tetracycline, edeine and IF3. Embo J. 20(8):1829–1839.
- Polikanov YS, Aleksashin NA, Beckert B, Wilson DN. 2018. The mechanisms of action of ribosome-targeting peptide antibiotics. Front Mol Biosci. 5:48.
- Popa A, Lebrigand K, Barbry P, Waldmann R. 2016. Pateamine A-sensitive ribosome profiling reveals the scope of translation in mouse embryonic stem cells. BMC Genomics. 17(1):52.
- Potts MB, McMillan EA, Rosales TI, Kim HS, Ou Y-H, Toombs JE, Brekken RA, Minden MD, MacMillan JB, White MA. 2015. Mode of action and pharmacogenomic biomarkers for exceptional responders to didemnin B. Nat Chem Biol. 11(6):401–408.
- Poulsen SM, Karlsson M, Johansson LB, Vester B. 2001. The pleuromutilin drugs tiamulin and valnemulin bind to the RNA at the peptidyl transferase centre on the ribosome. Molecular Microbiology. 41(5):1091–1099.
- Prezza G, Heckel T, Dietrich S, Homberger C, Westermann AJ, Vogel J. 2020. Improved bacterial RNA-seq by Cas9-based depletion of ribosomal RNA reads. RNA. 26(8):1069–1078.
- Prokhorova I, Altman RB, Djumagulov M, Shrestha JP, Urzhumtsev A, Ferguson A, Chang C-WT, Yusupov M, Blanchard SC, Yusupova G. 2017. Aminoglycoside interactions and impacts on the eukaryotic ribosome. Proc Natl Acad Sci USA. 114(51):E10899–E10908.
- Proud CG. 2019. Phosphorylation and signal transduction pathways in translational control. Cold Spring Harb Perspect Biol. 11(7):a033050.
- Querido JB, Sokabe M, Kraatz S, Gordiyenko Y, Skehel JM, Fraser CS, Ramakrishnan V. 2020. Structure of a human 48S translational initiation complex. Science. 369(6508):1220–1227.
- Rajala N, Hensen F, Wessels HJCT, Ives D, Gloerich J, Spelbrink JN. 2015. Whole cell formaldehyde cross-linking simplifies purification of mitochondrial nucleoids and associated proteins involved in mitochondrial gene expression. PLoS One. 10(2):e0116726.
- Ramakrishnan V. 2014. The ribosome emerges from a black box. Cell. 159(5):979–984.
- Ramirez MS, Tolmasky ME. 2017. Amikacin: uses, resistance, and prospects for inhibition. Molecules. 22(12):2267.
- Raser JM, O'Shea EK. 2005. Noise in gene expression: origins, consequences, and control. Science. 309(5743):2010–2013.
- Reid D, Bicknell A, Presnyak V, Licata M, Donovan S, Cheng J, Jones A, Moore M, Köhrer C. 2020. High ribosome density is associated with rapid mRNA decay in mammalian cells. In: RNA 2020 – online! Abstracts (Concurrent Session 1: Regulation of Translation): 314. https://app.oxfordabstracts.com/events/1385/program-app/submission/182603
- Reid DW, Campos RK, Child JR, Zheng T, Chan KWK, Bradrick SS, Vasudevan SG, Garcia-Blanco MA, Nicchitta CV. 2018. Dengue virus selectively annexes endoplasmic reticulum-associated translation machinery as a strategy for co-opting host cell protein synthesis. J Virol. 92(7):e01766–17.
- Reid DW, Shenolikar S, Nicchitta CV. 2015. Simple and inexpensive ribosome profiling analysis of mRNA translation. Methods. 91:69–74.
- Reixachs-Solé M, Ruiz-Orera J, Albà MM, Eyras E. 2020. Ribosome profiling at isoform level reveals evolutionary conserved impacts of differential splicing on the proteome. Nat Commun. 11(1):1768.
- Riba A, Nanni ND, Mittal N, Arhné E, Schmidt A, Zavolan M. 2019. Protein synthesis rates and ribosome occupancies reveal determinants of translation elongation rates. Proc Natl Acad Sci USA. 116(30):15023–15032.
- Rio DC. 2015. Denaturation and electrophoresis of RNA with formaldehyde. Cold Spring Harb Protoc. 2015(2):219–222.
- Robichaud N, Sonenberg N, Ruggero D, Schneider RJ. 2019. Translational control in cancer. Cold Spring Harb Perspect Biol. 11(7):a032896.
- Rodnina MV. 2018. Translation in prokaryotes. Cold Spring Harb Perspect Biol. 10(9):a032664.
- Rooijers K, Loayza-Puch F, Nijtmans LG, Agami R. 2013. Ribosome profiling reveals features of normal and disease-associated mitochondrial translation. Nat Commun. 4(1):2886.
- Roy K, Chanfreau G. 2014. Stress-induced nuclear RNA degradation pathways regulate yeast bromodomain factor 2 to promote cell survival. PLoS Genet. 10(9):e1004661.
- Roy RN, Lomakin IB, Gagnon MG, Steitz TA. 2015. The mechanism of inhibition of protein synthesis by the proline-rich peptide oncocin. Nat Struct Mol Biol. 22(6):466–469.
- Rubio CA, Weisburd B, Holderfield M, Arias C, Fang E, DeRisi JL, Fanidi A. 2014. Transcriptome-wide characterization of the eIF4A signature highlights plasticity in translation regulation. Genome Biol. 15(10):476.
- Rudler DL, Hughes LA, Perks KL, Richman TR, Kuznetsova I, Ermer JA, Abudulai LN, Shearwood A-MJ, Viola HM, Hool LC, et al. 2019. Fidelity of translation initiation is required for coordinated respiratory complex assembly. Sci Adv. 5(12):eaay2118.
- Sadlish H, Galicia-Vazquez G, Paris CG, Aust T, Bhullar B, Chang L, Helliwell SB, Hoepfner D, Knapp B, Riedl R, et al. 2013. Evidence for a functionally relevant rocaglamide binding site on the eIF4A-RNA complex. ACS Chem Biol. 8(7):1519–1527.
- Saito K, Green R, Buskirk AR. 2020a. Translational initiation in E. coli occurs at the correct sites genome-wide in the absence of mRNA-rRNA base-pairing. eLife. 9:e55002.
- Saito K, Green R, Buskirk AR. 2020b. Ribosome recycling is not critical for translational coupling in Escherichia coli. eLife. 9:e59974.
- Sako H, Yada K, Suzuki K. 2016. Genome-wide analysis of acute endurance exercise-induced translational regulation in mouse skeletal muscle. PLoS One. 11(2):e0148311.
- Salian S, Matt T, Akbergenov R, Harish S, Meyer M, Duscha S, Shcherbakov D, Bernet BB, Vasella A, Westhof E, et al. 2012. Structure-activity relationships among the Kanamycin aminoglycosides: role of ring I hydroxyl and amino groups. Antimicrob Agents Chemother. 56(12):6104–6108.
- Samatova E, Daberger J, Liutkute M, Rodnina MV. 2021. Translational control by ribosome pausing in bacteria: how a non-uniform pace of translation affects protein production and folding. Front Microbiol. 11:619430.
- Santos DA, Shi L, Tu BP, Weissman JS. 2019. Cycloheximide can distort measurements of mRNA levels and translation efficiency. Nucleic Acids Res. 47(10):4974–4985.
- Scaiola A, Leibundgut M, Boehringer D, Caspers P, Bur D, Locher HH, Rueedi G, Ritz D. 2019. Structural basis of translation inhibition by cadazolid, a novel quinoxolidinone antibiotic. Sci Rep. 9(1):5634.
- Schedlbauer A, Kaminishi T, Ochoa-Lizarralde B, Dhimole N, Zhou S, López-Alonso JP, Connell SR, Fucini P. 2015. Structural characterization of an alternative mode of tigecycline binding to the bacterial ribosome. Antimicrob Agents Chemother. 59(5):2849–2854.
- Schlünzen F, Pyetan E, Fucini P, Yonath A, Harms JM. 2004. Inhibition of peptide bond formation by pleuromutilins: the structure of the 50S ribosomal subunit from Deinococcus radiodurans in complex with tiamulin. Mol Microbiol. 54(5):1287–1294.
- Schmeing TM, Ramakrishnan V. 2009. What recent ribosome structures have revealed about the mechanism of translation. Nature. 461(7268):1234–1242.
- Schnappinger D, Hillen W. 1996. Tetracyclines: antibiotic action, uptake, and resistance mechanisms. Arch Microbiol. 165(6):359–369.
- Schneider-Poetsch T, Ju J, Eyler DE, Dang Y, Bhat S, Merrick WC, Green R, Shen B, Liu JO. 2010. Inhibition of eukaryotic translation elongation by cycloheximide and lactimidomycin. Nat Chem Biol. 6(3):209–217.
- Schuller AP, Wu CC-C, Dever TE, Buskirk AR, Green R. 2017. eIF5A functions globally in translation elongation and termination. Mol Cell. 66(2):194–205.e5.
- Schwabe A, Bruggeman FJ. 2014. Single yeast cells vary in transcription activity not in delay time after a metabolic shift. Nat Commun. 5(1):4798.
- Schwarz S, Shen J, Kadlec K, Wang Y, Michael GB, Feßler AT, Vester B. 2016. Lincosamides, streptogramins, phenicols, and pleuromutilins: mode of action and mechanisms of resistance. Cold Spring Harb Perspect Med. 6(11):a027037.
- Seefeldt AC, Nguyen F, Antunes S, Pérébaskine N, Graf M, Arenz S, Inampudi KK, Douat C, Guichard G, Wilson DN, et al. 2015. The proline-rich antimicrobial peptide Onc112 inhibits translation by blocking and destabilizing the initiation complex. Nat Struct Mol Biol. 22(6):470–475.
- Selmer M, Dunham CM, Murphy FV, Weixlbaumer A, Petry S, Kelley AC, Weir JR, Ramakrishnan V. 2006. Structure of the 70S ribosome complexed with mRNA and tRNA. Science. 313(5795):1935–1942.
- Sendoel A, Dunn JG, Rodriguez EH, Naik S, Gomez NC, Hurwitz B, Levorse J, Dill BD, Schramek D, Molina H, et al. 2017. Translation from unconventional 5′ start sites drives tumour initiation. Nature. 541(7638):494–499.
- Serdar LD, Whiteside DL, Nock SL, McGrath D, Baker KE. 2020. Inhibition of post-termination ribosome recycling at premature termination codons in UPF1 ATPase mutants. eLife. 9:e57834.
- Shamimuzzaman M, Vodkin L. 2018. Ribosome profiling reveals changes in translational status of soybean transcripts during immature cotyledon development. PLoS One. 13(3):e0194596.
- Sharma P, Nilges BS, Wu J, Leidel SA. 2019. The translation inhibitor cycloheximide affects ribosome profiling data in a species-specific manner. bioRxiv. 746255.
- Shashkova S, Leake MC. 2017. Single-molecule fluorescence microscopy review: shedding new light on old problems. Biosci Rep. 37(4):BSR20170031.
- Shell SS, Wang J, Lapierre P, Mir M, Chase MR, Pyle MM, Gawande R, Ahmad R, Sarracino DA, Ioerger TR, et al. 2015. Leaderless transcripts and small proteins are common features of the mycobacterial translational landscape. PLoS Genet. 11(11):e1005641.
- Shiber A, Döring K, Friedrich U, Klann K, Merker D, Zedan M, Tippmann F, Kramer G, Bukau B. 2018. Cotranslational assembly of protein complexes in eukaryotes revealed by ribosome profiling. Nature. 561(7722):268–272.
- Shin SY, Lee JH, Min BW, Lee YH. 2006. The translation inhibitor anisomycin induces Elk-1-mediated transcriptional activation of egr-1 through multiple mitogen-activated protein kinase pathways. Exp Mol Med. 38(6):677–685.
- Shirokikh NE, Alkalaeva EZ, Vassilenko KS, Afonina ZA, Alekhina OM, Kisselev LL, Spirin AS. 2010. Quantitative analysis of ribosome-mRNA complexes at different translation stages. Nucleic Acids Res. 38(3):e15.
- Shirokikh NE, Archer SK, Beilharz TH, Powell D, Preiss T. 2017. Translation complex profile sequencing to study the in vivo dynamics of mRNA–ribosome interactions during translation initiation, elongation and termination. Nat Protoc. 12(4):697–731.
- Shirokikh NE, Dutikova YS, Staroverova MA, Hannan RD, Preiss T. 2019. Migration of small ribosomal subunits on the 5′ untranslated regions of capped messenger RNA. IJMS. 20(18):4464.
- Shirokikh NE, Preiss T. 2018. Translation initiation by cap-dependent ribosome recruitment: Recent insights and open questions. Wiley Interdiscip Rev RNA. 9(4):e1473.
- Shirokikh NE, Spirin AS. 2008. Poly(A) leader of eukaryotic mRNA bypasses the dependence of translation on initiation factors. Proc Natl Acad Sci USA. 105(31):10738–10743.
- Siegel MR, Sisler HD, Johnson F. 1966. Relationship of structure to fungitoxicity of cycloheximide and related glutarimide derivatives. Biochem Pharmacol. 15(8):1213–1223.
- Siibak T, Peil L, Xiong L, Mankin A, Remme J, Tenson T. 2009. Erythromycin- and chloramphenicol-induced ribosomal assembly defects are secondary effects of protein synthesis inhibition. Antimicrob Agents Chemother. 53(2):563–571.
- Sisler HD, Siegel MR. 1967. Cycloheximide and other glutarimide antibiotics. In: Gottlieb D, Shaw PD, editors. Mechanism of action. Berlin; Heidelberg: Springer; p. 283–307.
- Sonenberg N, Hinnebusch AG. 2009. Regulation of translation initiation in eukaryotes: mechanisms and biological targets. Cell. 136(4):731–745.
- Stadler M, Fire A. 2011. Wobble base-pairing slows in vivo translation elongation in metazoans. RNA. 17(12):2063–2073.
- Stadler M, Fire A. 2013. Conserved translatome remodeling in nematode species executing a shared developmental transition. PLoS Genet. 9(10):e1003739.
- Stanley RE, Blaha G, Grodzicki RL, Strickler MD, Steitz TA. 2010. The structures of the anti-tuberculosis antibiotics viomycin and capreomycin bound to the 70S ribosome. Nat Struct Mol Biol. 17(3):289–293.
- Steinberger J, Shen L, J, Kiniry S, Naineni SK, Cencic R, Amiri M, Aboushawareb SAE, Chu J, Maïga RI, Yachnin BJ, et al. 2020. Identification and characterization of hippuristanol-resistant mutants reveals eIF4A1 dependencies within mRNA 5′ leader regions. Nucleic Acids Res. 48(17):9521–9537.
- Stewart H, Brown K, Dinan AM, Irigoyen N, Snijder EJ, Firth AE. 2018. Transcriptional and translational landscape of equine torovirus. J Virol. 92(17):e00589–18.
- Stickel SA, Gomes NP, Frederick B, Raben D, Su TT. 2015. Bouvardin is a radiation modulator with a novel mechanism of action. Radiat Res. 184(4):392–403.
- Strassburg K, Walther D, Takahashi H, Kanaya S, Kopka J. 2010. Dynamic transcriptional and metabolic responses in yeast adapting to temperature stress. Omics. 14(3):249–259.
- Subramaniam AR, Zid BM, O'Shea EK. 2014. An integrated approach reveals regulatory controls on bacterial translation elongation. Cell. 159(5):1200–1211.
- Sun Y, Atas E, Lindqvist LM, Sonenberg N, Pelletier J, Meller A. 2014. Single-molecule kinetics of the eukaryotic initiation factor 4AI upon RNA unwinding. Structure. 22(7):941–948.
- Svidritskiy E, Ling C, Ermolenko DN, Korostelev AA. 2013. Blasticidin S inhibits translation by trapping deformed tRNA on the ribosome. Proc Natl Acad Sci USA. 110(30):12283–12288.
- Szavits-Nossan J, Ciandrini L. 2020. Inferring efficiency of translation initiation and elongation from ribosome profiling. Nucleic Acids Res. 48(17):9478–9490.
- Taymaz-Nikerel H, Cankorur-Cetinkaya A, Kirdar B. 2016. Genome-wide transcriptional response of Saccharomyces cerevisiae to stress-induced perturbations. Front Bioeng Biotechnol. 4:17.
- Tayri-Wilk T, Slavin M, Zamel J, Blass A, Cohen S, Motzik A, Sun X, Shalev DE, Ram O, Kalisman N. 2020. Mass spectrometry reveals the chemistry of formaldehyde cross-linking in structured proteins. Nat Commun. 11(1):3128.
- Teixeira FK, Lehmann R. 2019. Translational control during developmental transitions. Cold Spring Harb Perspect Biol. 11(6):a032987.
- Tenson T, Mankin A. 2006. Antibiotics and the ribosome. Mol Microbiol. 59(6):1664–1677.
- Tereshchenkov AG, Dobosz-Bartoszek M, Osterman IA, Marks J, Sergeeva VA, Kasatsky P, Komarova ES, Stavrianidi AN, Rodin IA, Konevega AL, et al. 2018. Binding and action of amino-acid analogues of chloramphenicol upon the bacterial ribosome. J Mol Biol. 430(6):842–852.
- Tesina P, Lessen LN, Buschauer R, Cheng J, Wu CC-C, Berninghausen O, Buskirk AR, Becker T, Beckmann R, Green R. 2020. Molecular mechanism of translational stalling by inhibitory codon combinations and poly(A) tracts. EMBO J. 39(3):e103365.
- Thakur A, Gaikwad S, Vijjamarri AK, Hinnebusch AG. 2020. eIF2α interactions with mRNA control accurate start codon selection by the translation preinitiation complex. Nucleic Acids Res. 48(18):10280–10296.
- Tian B, Yang J, Brasier AR. 2012. Two-step crosslinking for analysis of protein-chromatin interactions. Methods Mol Biol. 809:105–120.
- Tu D, Blaha G, Moore PB, Steitz TA. 2005. Structures of MLSBK antibiotics bound to mutated large ribosomal subunits provide a structural explanation for resistance. Cell. 121(2):257–270.
- Uluşeker C, Torres-Bacete J, García JL, Hanczyc MM, Nogales J, Kahramanoğulları O. 2019. Quantifying dynamic mechanisms of auto-regulation in Escherichia coli with synthetic promoter in response to varying external phosphate levels. Sci Rep. 9(1):2076.
- Vasquez J-J, Hon C-C, Vanselow JT, Schlosser A, Siegel TN. 2014. Comparative ribosome profiling reveals extensive translational complexity in different Trypanosoma brucei life cycle stages. Nucleic Acids Res. 42(6):3623–3637.
- Vazquez D. 1974. Inhibitors of protein synthesis. FEBS Lett. 40(S1):S48–S62.
- Vecchione JJ, Alexander B, Sello JK. 2009. Two distinct major facilitator superfamily drug efflux pumps mediate chloramphenicol resistance in Streptomyces coelicolor. Antimicrob Agents Chemother. 53(11):4673–4677.
- Venkatesan N. 1977. Mechanism of inhibition of DNA synthesis by cycloheximide in Balb3T3 cells. Biochim Biophys Acta. 478(4):437–453.
- Viero G, Lunelli L, Passerini A, Bianchini P, Gilbert RJ, Bernabò P, Tebaldi T, Diaspro A, Pederzolli C, Quattrone A. 2015. Three distinct ribosome assemblies modulated by translation are the building blocks of polysomes. J Cell Biol. 208(5):581–596.
- Vind AC, Snieckute G, Blasius M, Tiedje C, Krogh N, Bekker-Jensen DB, Andersen KL, Nordgaard C, Tollenaere MAX, Lund AH, et al. 2020. ZAKα recognizes stalled ribosomes through partially redundant sensor domains. Mol Cell. 78(4):700–713.
- Vyas K, Chaudhuri S, Leaman DW, Komar AA, Musiyenko A, Barik S, Mazumder B. 2009. Genome-wide polysome profiling reveals an inflammation-responsive posttranscriptional operon in gamma interferon-activated monocytes. Mol Cell Biol. 29(2):458–470.
- Wagner S, Herrmannová A, Hronová V, Gunišová S, Sen ND, Hannan RD, Hinnebusch AG, Shirokikh NE, Preiss T, Valášek LS. 2020. Selective translation complex profiling reveals staged initiation and co-translational assembly of initiation factor complexes. Mol Cell. 79(4):546–560.e7.
- Wagner S, Herrmannová A, Šikrová D, Valášek LS. 2016. Human eIF3b and eIF3a serve as the nucleation core for the assembly of eIF3 into two interconnected modules: the yeast-like core and the octamer. Nucleic Acids Res. 44(22):10772–10788.
- Walczak CP, Leto DE, Zhang L, Riepe C, Muller RY, DaRosa PA, Ingolia NT, Elias JE, Kopito RR. 2019. Ribosomal protein RPL26 is the principal target of UFMylation. Proc Natl Acad Sci USA. 116(4):1299–1308.
- Wangen JR, Green R. 2020. Stop codon context influences genome-wide stimulation of termination codon readthrough by aminoglycosides. eLife. 9:e52611.
- Weaver J, Mohammad F, Buskirk AR, Storz G. 2019. Identifying Small Proteins by Ribosome Profiling with Stalled Initiation Complexes. mBio. 10(2):e02819–18.
- Wehmas LC, Hester SD, Wood CE. 2020. Direct formalin fixation induces widespread transcriptomic effects in archival tissue samples. Sci Rep. 10(1):14497.
- Wei S, Qian S-B. 2015. Ribosome profiling: principles and variations. In: eLS. Chichester, UK: John Wiley & Sons, Ltd. DOI:https://doi.org/10.1002/9780470015902.a0025984
- Weinberg DE, Shah P, Eichhorn SW, Hussmann JA, Plotkin JB, Bartel DP. 2016. Improved ribosome-footprint and mRNA measurements provide insights into dynamics and regulation of yeast translation. Cell Rep. 14(7):1787–1799.
- Williams CC, Jan CH, Weissman JS. 2014. Targeting and plasticity of mitochondrial proteins revealed by proximity-specific ribosome profiling. Science. 346(6210):748–751.
- Wilson DN. 2009. The A-Z of bacterial translation inhibitors. Crit Rev Biochem Mol Biol. 44(6):393–433.
- Wilson DN. 2014. Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat Rev Microbiol. 12(1):35–48.
- Wilson JE, Pestova TV, Hellen CUT, Sarnow P. 2000. Initiation of protein synthesis from the A site of the ribosome. Cell. 102(4):511–520.
- Wintermeyer W, Peske F, Beringer M, Gromadski KB, Savelsbergh A, Rodnina MV. 2004. Mechanisms of elongation on the ribosome: dynamics of a macromolecular machine. Biochem Soc Trans. 32(Pt 5):733–737.
- Wolin SL, Walter P. 1988. Ribosome pausing and stacking during translation of a eukaryotic mRNA. EMBO J. 7(11):3559–3569.
- Wong W, Bai X, Brown A, Fernandez IS, Hanssen E, Condron M, Tan YH, Baum J, Scheres SH. 2014. Cryo-EM structure of the Plasmodium falciparum 80S ribosome bound to the anti-protozoan drug emetine. eLife. 3:e03080.
- Woolstenhulme CJ, Guydosh NR, Green R, Buskirk AR. 2015. High-precision analysis of translational pausing by ribosome profiling in bacteria lacking EFP. Cell Rep. 11(1):13–21.
- Wu CC-C, Peterson A, Zinshteyn B, Regot S, Green R. 2020. Ribosome collisions trigger general stress responses to regulate cell fate. Cell. 182(2):404–416.e14.
- Wu CC-C, Zinshteyn B, Wehner KA, Green R. 2019. High-resolution ribosome profiling defines discrete ribosome elongation states and translational regulation during cellular stress. Mol Cell. 73(5):959–970.e5.
- Yan K, Madden L, Choudhry AE, Voigt CS, Copeland RA, Gontarek RR. 2006. Biochemical characterization of the interactions of the novel pleuromutilin derivative retapamulin with bacterial ribosomes. Antimicrob Agents Chemother. 50(11):3875–3881.
- Yang Z, Cao S, Martens CA, Porcella SF, Xie Z, Ma M, Shen B, Moss B. 2015. Deciphering poxvirus gene expression by RNA sequencing and ribosome profiling. J Virol. 89(13):6874–6886.
- Yonath A. 2005. Antibiotics targeting ribosomes: resistance, selectivity, synergism and cellular regulation. Annu Rev Biochem. 74(1):649–679.
- Yordanova MM, Loughran G, Zhdanov AV, Mariotti M, Kiniry SJ, O'Connor PBF, Andreev DE, Tzani I, Saffert P, Michel AM, et al. 2018. AMD1 mRNA employs ribosome stalling as a mechanism for molecular memory formation. Nature. 553(7688):356–360.
- Young DJ, Guydosh NR, Zhang F, Hinnebusch AG, Green R. 2015. Rli1/ABCE1 recycles terminating ribosomes and controls translation reinitiation in 3′UTRs in vivo. Cell. 162(4):872–884.
- Young DJ, Guydosh NR. 2019. Hcr1/eIF3j is a 60S ribosomal subunit recycling accessory factor in vivo. Cell Rep. 28(1):39–50.e4.
- Young DJ, Makeeva DS, Zhang F, Anisimova AS, Stolboushkina EA, Ghobakhlou F, Shatsky IN, Dmitriev SE, Hinnebusch AG, Guydosh NR. 2018. Tma64/eIF2D, Tma20/MCT-1, and Tma22/DENR recycle post-termination 40S subunits in vivo. Mol Cell. 71(5):761–774.e5.
- Young DJ, Meydan S, Guydosh NR. 2021. 40S ribosome profiling reveals distinct roles for Tma20/Tma22 (MCT-1/DENR) and Tma64 (eIF2D) in 40S subunit recycling. Nat Commun. 12(1):2976.
- Zeman J, Itoh Y, Kukačka Z, Rosůlek M, Kavan D, Kouba T, Jansen ME, Mohammad MP, Novák P, Valášek LS. 2019. Binding of eIF3 in complex with eIF5 and eIF1 to the 40S ribosomal subunit is accompanied by dramatic structural changes. Nucleic Acids Res. 47(15):8282–8300.
- Zhang P, He D, Xu Y, Hou J, Pan B-F, Wang Y, Liu T, Davis CM, Ehli EA, Tan L, et al. 2017. Genome-wide identification and differential analysis of translational initiation. Nat Commun. 8(1):1749.
- Zhang X, Chen X, Yang T, Zhang N, Dong L, Ma S, Liu X, Zhou M, Li B. 2014. The effects of different crossing-linking conditions of genipin on type I collagen scaffolds: an in vitro evaluation. Cell Tissue Bank. 15(4):531–541.
- Zinshteyn B, Wangen JR, Hua B, Green R. 2020. Nuclease-mediated depletion biases in ribosome footprint profiling libraries. RNA. 26(10):1481–1488.
- Zoschke R, Barkan A. 2015. Genome-wide analysis of thylakoid-bound ribosomes in maize reveals principles of cotranslational targeting to the thylakoid membrane. Proc Natl Acad Sci USA. 112(13):E1678–E1687.
- Zoschke R, Watkins KP, Barkan A. 2013. A rapid ribosome profiling method elucidates chloroplast ribosome behavior in vivo. Plant Cell. 25(6):2265–2275.