
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Protein lysine methyltransferases (PKMTs) transfer up to three methyl groups to the side chains of lysine residues in proteins and fulfill important regulatory functions by controlling protein stability, localization and protein/protein interactions. The methylation reactions are highly regulated, and aberrant methylation of proteins is associated with several types of diseases including neurologic disorders, cardiovascular diseases, and various types of cancer. This review describes novel insights into the catalytic machinery of various PKMTs achieved by the combined application of biochemical experiments and simulation approaches during the last years, focusing on clinically relevant and well-studied enzymes of this group like DOT1L, SMYD1-3, SET7/9, G9a/GLP, SETD2, SUV420H2, NSD1/2, different MLLs and EZH2. Biochemical experiments have unraveled many mechanistic features of PKMTs concerning their substrate and product specificity, processivity and the effects of somatic mutations observed in PKMTs in cancer cells. Structural data additionally provided information about the substrate recognition, enzyme-substrate complex formation, and allowed for simulations of the substrate peptide interaction and mechanism of PKMTs with atomistic resolution by molecular dynamics and hybrid quantum mechanics/molecular mechanics methods. These simulation technologies uncovered important mechanistic details of the PKMT reaction mechanism including the processes responsible for the deprotonation of the target lysine residue, essential conformational changes of the PKMT upon substrate binding, but also rationalized regulatory principles like PKMT autoinhibition. Further developments are discussed that could bring us closer to a mechanistic understanding of catalysis of this important class of enzymes in the near future. The results described here illustrate the power of the investigation of enzyme mechanisms by the combined application of biochemical experiments and simulation technologies.
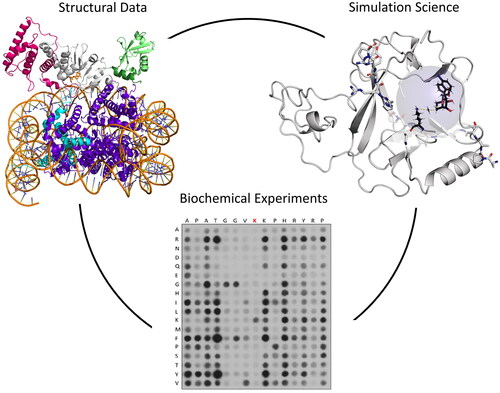
Introduction
The genetic information of every cell is encoded in form of the base pair sequence in the DNA but cellular differentiation is driven by differences in the expression of genes. Epigenetics describes the mechanisms of these often stable but still reversible changes in gene expression patterns that do not involve alterations in the DNA sequence (Allis and Jenuwein Citation2016). Chromatin is the complex of DNA and proteins that compacts the DNA within the nucleus of eukaryotic cells. Nucleosomes are the smallest structural unit of chromatin and consist of a stretch of about 147 base pairs of DNA wrapped around an octamer of histone proteins, containing two copies of the core histones, namely H2A, H2B, H3, and H4. Nucleosomes are connected by linker DNA segments, which vary in length and composition forming a ‘beads-on-a-string’ structure. This linear array of nucleosomes further compacts the chromatin to form higher-order structures, such as chromatin fibers and chromosomes. DNA and protein methylation are among the most important biochemical reactions for influencing gene expression (Jambhekar et al. Citation2019; Chen and Zhang Citation2020; Millán-Zambrano et al. Citation2022). For instance, cytosine bases in DNA can be methylated by DNA methyltransferases (DNMTs) using the cofactor S-adenosyl-L-methionine (SAM) as a methyl group donor. SAM dependent methylation also occurs in proteins or peptides and can be found at side chains of lysine (K), arginine (R), aspartate (D), glutamate (E), histidine (H), asparagine (N), glutamine (Q), and cysteine (C), as well as at N-terminal -amino and C-terminal carboxylate residues (Clarke Citation2013). Histone tails are the flexible ends of the histone proteins sticking out from the histone octamer and are a key target for lysine and arginine methylation and other post translational modifications (PTMs) like acetylation, phosphorylation, ubiquitination, and more (Bannister and Kouzarides Citation2011; Millán-Zambrano et al. Citation2022). The modified amino acids serve as a platform for the recruitment of proteins and protein complexes that interpret and regulate these modifications. Eventually a signaling cascade of downstream effects is triggered, influencing the activity of chromatin remodelers which themselves alter the accessibility of DNA and thus gene transcription.
Enzymes catalyzing the transfer of methyl groups from SAM to histone tails or other proteins are called protein methyltransferases (PMT) if the receiving amino acid is a lysine residue, they are referred to as protein lysine methyltransferases (PKMT). This class of enzymes is going to be the main focus of this review (). For other PMTs with e.g. arginine or histidine as the methylation target, refer to other reviews (Clarke Citation2013; Jarrold and Davies Citation2019; Kwiatkowski and Drozak Citation2020; Jakobsson Citation2021a; Wu et al. Citation2023). The functional diversity of PKMTs is vast, with numerous members belonging to the different families of SET domain and non-SET domain also called seven-beta strand (7BS) PKMTs, each characterized by defined structural features (Qian and Zhou Citation2006; Falnes et al. Citation2023). Many PKMTs methylate lysine residues in histone proteins (often the flexible N-terminal tails of histone H3 and H4) which play important roles in the regulation of chromatin (Husmann and Gozani Citation2019; Millán-Zambrano et al. Citation2022). However, the last decades of research have shown, that lysine methylation is not restricted to histone proteins (Husmann and Gozani Citation2019), but it frequently is observed in many other proteins where it regulates protein stability, localization and protein/protein interactions, often in combination with other post-translational modification (Clarke Citation2013; Biggar and Li Citation2015; Zhang et al. Citation2015; Cornett et al. Citation2019; Di Blasi et al. Citation2021). Some PKMTs are highly specific, targeting only defined lysine residues in one or few substrate proteins, while others exhibit broader substrate recognition capabilities. Additionally, PKMTs can function individually or as components of large protein complexes, adding another layer of complexity to their regulatory mechanisms. Based on their important physiological roles, PKMTs have been implicated in many diseases and they represent important and emerging drug targets (Copeland Citation2018; Bhat et al. Citation2021).
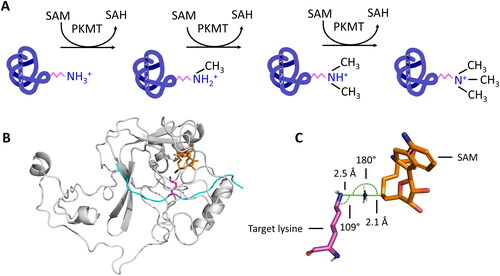
Figure 1. A| Protein lysine methyltransferases (PKMTs) transfer up to three methyl groups to specific lysine residues in proteins. The cofactor S-adenosyl-L-methionine (SAM) provides the methyl group. It is released after the transfer as S-adenosyl-L-homocysteine (SAH). B| The protein substrate (cyan) and SAM (orange, methyl group is colored black) bind at opposing sites of the SET domain (grey) in SET domain PKMTs. The target lysine (pink) is inserted into a narrow tunnel, where the lysine is deprotonated and oriented for the methyl group transfer (image created using simulation results of PDB 6VDB (Schuhmacher et al. Citation2020)). C| The methyl group is transferred using a bimolecular nucleophilic substitution (SN2) mechanism, in which multiple geometric criteria need to be fulfilled to reach the transition state.
In this review, we will specifically summarize studies addressing the catalytic mechanism of PKMTs using simulation technologies mainly including Molecular Dynamics (MD) simulations and Hybrid Quantum Mechanical/Molecular Mechanical (QM/MM) methods. Biochemical experiments matching the simulation experiments were used to complement the simulation approaches and their results. Relevant catalytic properties of PKMTs discussed in this review include: (i) the mechanism of target lysine deprotonation (a necessary precondition for its methylation); (ii) conformational changes upon cofactor and substrate binding; (iii) substrate specificity (the question which target lysine is methylated); (iv) product specificity (the question if mono-, di- or trimethylated lysine is generated); and (v) the assembly of PKMTs with other proteins to regulate their methylation activity or control substrate engagement. One goal of this review is to convey mechanistic principles of PKMT catalyzed methylation reactions of different PKMT subfamilies. Clinically relevant PKMTs like DOT1L, SMYD1-3, SET7/9, G9a/GLP, SETD2, NSD1/2, MLL enzymes and EZH2 were prioritized, but the mechanisms presented are presumably transferable at least to structurally similar PKMTs not mentioned here specifically. Besides investigations addressing mechanistic principles of PKMTs, numerous modelling studies have been conducted to investigate PKMT inhibitors which involved molecular docking, MD simulation, free energy calculation and virtual screening approaches. These studies are extremely valuable especially in a pharmacological context. However, they were excluded from this review, because the approaches and systems used in these studies deal with artificially designed small molecules and/or inhibitors that cause non-natural behaviors, conformational changes and altered PKMTs activities. Hence, the obtained results of inhibition studies do not necessarily provide insights into the natural catalytic mechanisms of the investigated PKMTs. We therefore refer to other excellent reviews covering the interplay of various PKMTs with inhibitors using different simulation and modeling approaches (Luo Citation2015; Schapira Citation2016; Copeland Citation2018; López-López et al. Citation2020; Vougiouklakis et al. Citation2020; Feoli et al. Citation2022).
Simulation technologies
Simulation science is an emerging field of research that combines retrospective analysis and modeling methods with predictive simulations approaches. Modeling of proteins at atomistic resolution can be used to rationalize results of biochemical experiments and give novel insights into the underlying mechanisms like enzyme-substrate interactions, mechanistic consequences of cancer mutants, or conformational changes of flexible protein regions. Moreover, simulations possess an immense predictive capacity, enabling scientists to anticipate the outcomes of experiments and design them accordingly before conducting them physically. By recreating experimental conditions and subsequent systematic changes of parameters, researchers can explore alternative experimental scenarios and test hypotheses in rapid time. This promotes mechanistic understanding, reduces experimental costs, and accelerates the scientific process. In the next two paragraphs, the two main simulation technologies are summarized, which have been applied to PKMTs in recent years.
Molecular dynamics simulations
At present, the Protein Data Bank (PDB) holds more than 206,000 experimentally solved structures, including more than 200,000 proteins (https://www.rcsb.org/ retrieved in June 26, 2023). These structures are extremely valuable for identifying architectures of enzymes, understanding enzyme mechanism and protein interaction with other biomolecules or as templates for homology modeling. However, despite their enormous utility, the structures stored in the PDB provide only a partial view on the 3D structure of proteins, because proteins are flexible entities, and dynamic conformational changes play key roles in their function. This is particularly true for enzymes and their catalytical function. For instance, the autoinhibitory loop of SET domain-containing PKMTs blocks their active sites, but opens up upon binding to the protein substrate (Yang et al. Citation2016; Sato et al. Citation2021; Schnee et al. Citation2022). While at least snapshots of this process are captured in crystal structures (PDB 5LSU for closed, 5V21 for open), the complete process involving the regulatory contacts between enzyme and substrate can only be investigated using simulation technologies. Adding to this, other processes involve regions, which are so flexible that crystallization or cryo-EM techniques are incapable of capturing them. An example is the PKMT SETD2 (aka KMT3A, HYPB, SET2), which was found to have a flexible loop in the post-SET domain. This loop is unstructured in the apo enzyme, but forms a helix and interacts with the core of the enzyme after binding of a substrate (Yang et al. Citation2016; Schnee et al. Citation2022).
Another limitation of resolved structures of macromolecules is the necessity for stable complex formation to enable structural analysis, which is specifically relevant for enzymes, since they catalyze reactions after binding their substrate and then release the product. Bound inhibitors can be used to circumvent this problem, but then critical information about catalysis is lost. For example, SET domain-containing PKMTs use a water channel to deprotonate the target lysine residue in the active site. This water channel is only visible if the cofactor SAM is bound and not if the cofactor product SAH binds in the cofactor binding pocket (Zhang and Bruice Citation2008a). However, in resolved structures mostly SAH is the present cofactor, since it had been used for crystallisation to avoid turnover. Alternatively, a substrate inhibitor, like target lysine K-to-M mutation peptides can be used, which stably bind to SET domain-containing PKMTs, since the methionine cannot be methylated (Fang et al. Citation2016; Yang et al. Citation2016; Zhang et al. Citation2017; Schuhmacher et al. Citation2020). However, important interactions between the missing target lysine and the PKMTs active site are lost in this experimental setup.
Information from resolved structures provide a solid understanding of an enzyme’s 3D structure, but in combination with subsequent modeling approaches a closer approximation to reality is possible. In the modeling methods the resolved structure can be solvated in different buffers, like physiological molarity of sodium chloride, but also in buffers containing different concentrations of detergents or denaturing agents, or the effect of different pH values can be simulated. Missing or flexible amino acids can be rebuilt, or an inhibitor can be exchanged for the real cofactor or substrate. Besides modifying resolved structures, homology modeling with e.g. AlphaFold2 (Jumper et al. Citation2021), PyMOD 3.0 (Janson and Paiardini Citation2021) or SWISS-MODEL (Waterhouse et al. Citation2018) has become a hallmark in modern protein science.
Another limitation of structural models is that they lack the dynamic behavior of proteins. This obstacle can be overcome by employing MD simulations, which apply forces to the atoms in the modelled system based on their interactions with each other. At the start of an MD simulation, each atom is given a random velocity, based on a temperature dependent Maxwell-Boltzmann distribution (Meller Citation2001; Yu and Dalby Citation2020). Subsequently, the forces are applied and by utilizing Newton’s laws of motion, it becomes possible to determine the dynamical behavior and spatial arrangement of all atoms over time. This results in a sequential progression through time, iteratively applying forces to atoms and updating their positions and velocities. Ultimately, the outcome is a dynamic, three-dimensional, atomic-level resolution representation of a protein, e.g. the behavior of a PKMT in the presence of its substrate and SAM (Hospital et al. Citation2015; Hollingsworth and Dror Citation2018).
To calculate the forces acting on each atom, a molecular mechanics force field is employed. This force field is derived and calibrated to fit to the results of quantum mechanical calculations and often incorporates specific experimental parameters (Monticelli and Tieleman Citation2013). The forces can be categorized into two main types: bonded interactions and non-bonded interactions. Bonded interactions describe the chemical bonds between atoms by a bond potential, which encompasses stretching, bending, and torsional rotation. Non-bonded interactions involve can der Waals interactions described by the Lennard-Jones potential accounting for both attractive and repulsive forces, and electrostatic forces between charged atoms which carry full or partial charges modeled by a Coulomb potential and an appropriate dielectric constant (). Electrons are not explicitly treated in classical molecular dynamics force fields. Comparison of simulations with a variety of experimental data indicates that force fields have improved substantially over the years (Lindorff-Larsen et al. Citation2012; Lopes et al. Citation2015; Chmiela et al. Citation2018; Dasetty et al. Citation2019), but the uncertainty introduced by the approximations and small inaccuracies inherent in the force fields should be considered when interpreting simulation results. Moreover, in a classical MD simulation, no covalent bonds are formed or broken meaning that chemical reactions cannot be directly represented. Hybrid quantum mechanics/molecular mechanics (QM/MM) simulations, in which a small part of the system is modeled using quantum mechanical calculations taking electron densities into account, and the remainder of the system is modeled by MD simulation, are frequently employed to study chemical reactions and enzyme catalysis that involve changes in covalent bonds. This method is described in the next chapter.
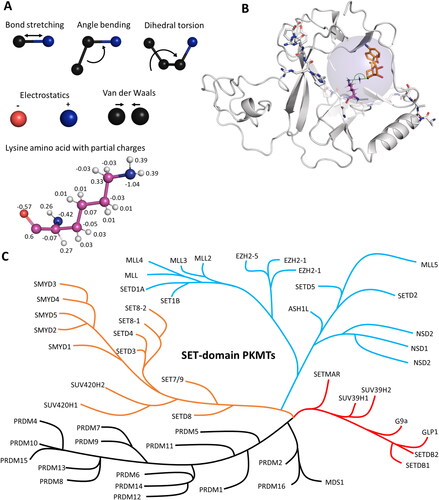
Figure 2. Molecular dynamics (MD) and hybrid quantum mechanics/molecular mechanics (QM/MM) simulations have been applied to investigate protein properties. A| In MD simulations, atoms are connected via spring-like bonds. The bonded forces comprise bond stretching, angle bending and dihedral torsion. Non-bonded interactions consist of electrostatic and van der Waals forces. Electrostatic forces are applied between fully or partially charged atoms. Van der Waals forces are described using the Lennard-Jones potential. B| In QM/MM simulations, the simulated system is divided into a QM region (blue transparent sphere) and a MM region. In the MM region, forces are calculated as for MD simulations using force fields. In the QM region, bonds can be broken or formed, and atoms are treated using quantum mechanics. In applications with PKMTs, the active site has been treated using QM methods to cover the methyl group transfer, the rest of the enzyme and bulk solvent was treated with MM (image created using simulation results of PDB 5V21 (Zhang et al. Citation2017)). C| Phylogenetic tree of SET domain-containing PKMTs. Modified from (Wu et al. Citation2010; Richon et al. Citation2011; Luo Citation2018).
To ensure the stability of the molecular structures during the simulation, the time steps in an MD simulation must be short, typically only a few femtoseconds (10−15 s) each. In contrast, conformational changes of enzymes take place on timescales of nanoseconds, microseconds, up to seconds or even minutes. Hence, billions of time steps are needed to capture the complete dynamics. In each step, all interatomic interactions in the system are calculated, which is computationally expensive. Over time, the continuous and almost exponential advancements in high performance computing power, especially the availability of powerful Graphics Processing Units (GPU), have allowed for more efficient and longer simulations. An example, benchmark simulations with a 159-residue protein (DHFR) in explicit solvent (<24000 atoms) using OpenMM (Eastman and Pande Citation2010; Eastman et al. Citation2017) yielded >1 ms/day on a Nvidia RT2080Ti/A40/V100 system (https://openmm.org/benchmarks). To simulate even longer and larger systems, coarse-grained MD simulations offer a loophole. Here, one artificial particle represents a group of atoms rather than a single atom. Thereby, the resolution decreases but accessible timescales increase by orders of magnitude (Marrink and Tieleman Citation2013). However, these approaches are purely based on empirical force fields which need to be established beforehand and calibrated with experimental data. Recent reviews on this technique can be found in (Kmiecik et al. Citation2016; Joshi and Deshmukh Citation2020).
Due to limited computational resources, it is necessary to restrict the system size in simulation experiments. Therefore, periodic boundary conditions are used, and the system of interest is confined in a box, assuming that the properties of the actual system can be approximated by a virtual infinite system of repeating side-by-side boxes. If a molecule passes through the initial box boundary, it will re-enter the box from the opposite boundary, forming a periodic space (Meller Citation2001; Yu and Dalby Citation2020).
In summary, MD simulations are a helpful tool to reveal molecular processes at an atomistic resolution and can be used to
Assess the flexibility and alternative conformations of various regions of a protein or biomolecule in solution.
Monitor changes in the protein stability in different environments like changing pH, temperature, salt conditions, detergents, or denaturing agents.
Simulate the behavior of intrinsically disordered protein domains in the context of a binding partner.
Perturbate a protein by e.g. deploying, removing, or exchanging a bound ligand and assess how this affects the protein conformation and/or stability.
Find factors controlling ligand binding and dissociation kinetics.
Modify one or more amino acid residues in the protein e.g. by PTMs, altered protonation states of an acidic or basic amino acid, or by mutating residues and simulate the behavior of the modified protein.
Define the structural basis for water and ion binding at the surface and into cavities of proteins.
QM/MM simulations
Hybrid quantum mechanical/molecular mechanical (QM/MM) methods are simulation approaches combining quantum mechanical and molecular mechanical models in one simulation. Therefore, the simulated system is divided into two regions (): the QM region includes the atoms that are treated using quantum mechanics, while the MM region consists of the surrounding environment described by a force field and simulated by MD methods. The QM region is typically small and localized around the specific chemical processes or properties of interest, such as the atoms involved in chemical reactions, or chromophores, if spectroscopic properties are studied. Quantum mechanical methods, such as density functional theory (DFT) are applied to this region to accurately describe the electronic structure and quantum effects. On the other hand, the MM region represents the larger rest of the protein surrounding the QM region, also including the environment like solvent molecules, ions, and other biomolecules. The MM region is treated using force fields, which describe the behavior of atoms based on bonded and non-bonded interactions as described for MD simulations without forming or breaking covalent bonds. The QM and MM regions interact through electrostatic forces, van der Waals interactions, and sometimes covalent bonding. The boundary between the QM and MM regions is typically defined by a set of atoms called the ‘QM/MM interface’ or ‘link atoms’, which bridge the two regions. For a more detailed review focusing also on the physical, mathematical and methodological basis of hybrid QM/MM simulation approaches, we refer to (Senn and Thiel Citation2009; Groenhof Citation2013; Van der Kamp and Mulholland Citation2013).
Drawbacks of QM/MM simulation approaches are additional computational cost for the high accuracy in the QM region. This goes in hand with a limited system size, timescale and sampling of the conformational space. Nevertheless, hybrid QM/MM methods provide a powerful tool for studying complex biological systems, as they combine the accuracy of quantum mechanics for the region of interest with the efficiency of classical mechanics for the surrounding environment. They have been widely applied to investigate chemical reactions, enzyme mechanisms or protein-ligand interactions (Bramley et al. Citation2023). More precisely they can be used to investigate:
Enzymatic reactions by treating the reactive region of the system (e.g. the active site) at the quantum mechanical level, while the surrounding environment (e.g. solvent, and the rest of the protein) is treated with molecular mechanics. This allows for an accurate description of the reaction mechanism and the influence of the surrounding environment on the reaction kinetics.
Protein-ligand interactions, where the ligand can be treated quantum mechanically to accurately describe its electronic structure and reactivity, while the protein environment is described with molecular mechanics. This approach allows for the investigation of binding mechanisms, protein-ligand interactions, and the precise role of specific residues in ligand binding.
Proton transfer reactions in biomolecules, such as enzymatic proton transfers or proton-coupled electron transfer reactions. These simulations can reveal the reaction mechanism for the investigated enzyme, protonation states of key residues, and the influence of the surrounding protein environment on the proton transfer process.
Photoactive proteins, where QM/MM simulations can be used to study the excited-state properties of photoactive proteins. Thereby, the precise mechanisms of light absorption, energy transfer, fluorescence and signal transduction can be uncovered.
Potential mean force calculations
Free energy calculations were frequently used in the studies described in this review to examine enzymatic reaction mechanisms. Their goal is to determine the free energy difference between reactants and products along a predefined reaction coordinate. Free energy calculations for enzyme reactions in QM/MM simulations typically involve the use of enhanced sampling techniques and the calculation of potential of mean force (PMF) profiles. At first, a suitable reaction coordinate needs to be defined that precisely describes the progress of the reaction. This could be an atom to atom distance, an angle, or any other collective variable that captures the essence of the reaction. The reaction coordinate should span from the reactant state to the product state. In the case of a methyl group transfer, the distance of the methyl group C-atom to the donor and acceptor atoms appears suitable to describe the reaction. For PMF calculations, QM/MM is the simulation method of choice, since covalent bonds might be broken or formed. The active site or the amino acids directly involved in the reaction are treated quantum mechanically, while the rest of the enzyme, solvent, and any other surroundings are treated with molecular mechanics. Since enzyme reactions often involve high energy barriers, enhanced sampling techniques are used to explore different regions of the reaction coordinate and enhance the sampling of rare events (Trzesniak et al. Citation2007). Methods like metadynamics, umbrella sampling, or adaptive biasing force are commonly employed to overcome energy barriers and enhance conformational sampling along the reaction coordinate (Park and Schulten Citation2004; You et al. Citation2019; Yoneda et al. Citation2021). By performing simulations at different stages of the reaction along the reaction coordinate and applying biasing potentials or forces, the PMF can be calculated. The PMF represents the free energy profile along the reaction coordinate, providing information about the free energy barriers and the relative stability of different states along the reaction pathway. The free energy difference between the reactant and product states are determined by integrating the PMF profile. This provides insights into the thermodynamics and kinetics of the enzyme reaction, including reaction energetics, transition state (TS) stability, and barriers needed to be overcome. Different substrates, products and enzyme mutants can be compared for their ability to minimize the free energy using their catalytic machinery (You et al. Citation2019). However, the obtained parameters need to be compared to experimental data and carefully used in qualitatively statements by e.g. comparing the differences of two states. An example PMF profile is discussed and visualized in the section Analysis of the product specificity and processivity of PKMTs and in .
Protein lysine methyltransferases
Due to the lone-pair electrons present in the ε-amine of lysine, as well as its preference for localization on the protein surface, lysine residues are highly susceptible to PTMs. Several modifications have been identified at lysine residues in proteins isolated from eukaryotic cells, including methylation, acetylation, ubiquitination, SUMOylation, propionylation, malonylation, butyrylation, succinylation, glutarylation, myristoylation, biotinylation, and neddylation (Kothapalli et al. Citation2005; Luo Citation2012; Choudhary et al. Citation2014; Mattiroli and Sixma Citation2014; Enchev et al. Citation2015; Sabari et al. Citation2017; Seeler and Dejean Citation2017). Protein lysine methylation stands apart from other types of modifications for three reasons. Firstly, the addition of methyl groups to lysine does not affect the overall charge of the residue at physiological pH, unlike acylation modifications that convert the positively charged -amine into a neutral amide. Secondly, lysine methylation represents the smallest PTM, resulting in minimal changes to the size of the side chain when compared to other types of lysine modifications (Luo Citation2018). Thirdly, up to three methyl groups can be transferred to a single target lysine creating monomethyl lysine (Kme1), dimethyl lysine (Kme2) and trimethyl lysine (Kme3) and it even can be combined with other modifications as in the recently discovered acetyl-methyllysine marks (Lu-Culligan et al. Citation2023).
Lysine (K), Kme1 and Kme2 possess lone-pair electrons on their ε-amine groups, which can be methylated. However, due to their high pKa values (10.2 − 10.7), the ε-amines of K, Kme1, and Kme2 predominantly exist in a protonated state under physiological conditions (pH 7.4) making them unreactive as a nucleophile. Therefore, one critical requirement of the lysine methylation reaction catalyzed by PKMT enzymes is to overcome the activation barrier of lysine deprotonation. At the next stage, the deprotonated lysine and SAM must be bound in a conformation that facilitates the subsequent nucleophilic substitution reaction leading to the transfer of the methyl group.
The human genome encodes over 60 characterized PKMTs, which can be categorized into two classes: SET domain-containing PKMTs (class V methyltransferase, denoted as SET due to the genetic phenotypes observed in Drosophila called Suppressor of variegation 3-9, Enhancer of zeste, and Trithorax) and “non-SET” 7BS MTases (class I methyltransferases) () (Copeland et al. Citation2009; Richon et al. Citation2011; Luo Citation2012). The majority of characterized PKMTs belong to the SET domain family (approximately 90%) but several human 7BS PKMTs have already been investigated as well (Luo Citation2018). PKMTs can act in complexes with other proteins or contain domains to achieve binding to certain structures, recognition of specific modifications of the substrate protein or regulation of their own activity. This often includes domains interacting with modified histone tails, like chromodomains, bromodomains, tudor domains, PHD domains or PWWP domains (Patel and Wang Citation2013). For the MLL (Mixed-Lineage Leukemia) complex PKMTs, also known as COMPASS (Complex Proteins Associated with Set1), it is necessary to associate with other proteins and establish important interaction with each other and the nucleosome to stimulate their methylation activity (Shilatifard Citation2012; Basta and Rauchman Citation2015; Park et al. Citation2019). It is therefore crucial to take caution when attributing their biological functions solely to the methyltransferase domain of PKMTs, as these domains may be heavily influenced by interactions partners in their activity and substrate targeting and can have biological functions even independent of their catalytic activities.
SET domain-containing PKMTs
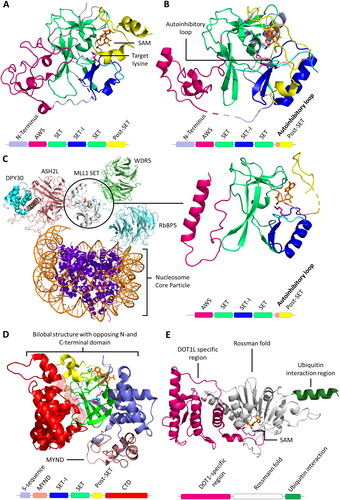
The SET domain is responsible for the methylation activity of this family of PKMTs. It consists of approximately 130 amino acids and is often flanked by so-called pre-SET and post-SET domains (Qian C and Zhou Citation2006). Through phylogenetic analysis of SET domain sequences, human SET domain PKMTs can be further classified into subfamilies, each with its own characteristic structural arrangement (Wu et al. Citation2010). For example, G9a (aka EHMT2, KMT1C), SUV39H1 (aka KMT1A) and SUV39H2 (aka KMT1B) belong to the classical PKMT subfamily, where their SET domains alone are sufficient for catalyzing the methyl group transfer () (Tachibana et al. Citation2001). On the other hand, SETD2, NSD1 (aka KMT3B), NSD2 (aka MMSET, WHSC1), and NSD3 (aka WHSC1L1) reside within a subfamily of PKMTs featuring an autoinhibitory loop () (An et al. Citation2011; Yang et al. Citation2016; Bennett et al. Citation2017). In this case, the apo enzyme of the SET domain is expected to be catalytically inactive (or only weakly active) and needs to undergo conformational changes for substrate binding and enzyme activity. The SET domains of the MLL subfamily PKMTs are inactive on their own, but become catalytically active in the presence of binding partners such as WDR5, RbBP5, ASH2L, and DPY30, collectively referred to as WRAD () (Cao F et al. Citation2014; Borkin et al. Citation2015; Grebien et al. Citation2015). Similar requirements for complex formation have been observed in EZH1 and EZH2 (aka KMT6) (EZH1/2, EED and Suz12 referred as PRC2 complexes) (Kim W et al. Citation2013; He et al. Citation2017). The 17 members of the PRDM subfamily contain a PR domain that is similar to the SET domain and a variable number of Zinc (Zn)-finger repeats (Di Zazzo et al., Citation2013). The 5 member SMYD subfamily is characterized by an insertion of a MYND domain (myeloid translocation protein 8, Nervy and DEAF-1), within their SET domain. The MYND domain is responsible for protein-protein interactions possibly recruiting the enzymes to specific substrate proteins. SMYD enzymes are characterized by a bilobal architecture with the protein substrate in the middle () (Saddic et al. Citation2010; Sirinupong et al. Citation2010; Ferguson et al. Citation2011; Sirinupong et al. Citation2011; Mazur et al. Citation2014; Mzoughi et al. Citation2016).
Figure 3. Cartoon representation of SET and 7BS domain PKMT architectures. A| SET domain-containing PKMT G9a complexed with the H3K36 substrate peptide (cyan with the target lysine in pink), and cofactor SAM (orange, PDB 5JIY (Jayaram et al. Citation2016)). SET domain-containing PKMTs incorporate Zn-ions for structural stability in their AWS (Associated with SET, magenta), post-SET (yellow) or MYND (rose) domain depending on the enzyme (Dillon 2005, Wu et al. Citation2011). However, the Zn-ions are not involved in catalysis or conformational changes and they are not shown explicitely in protein structures presented in this review. B| SETD2 complexed with the H3K36 substrate peptide, and cofactor SAM (PDB 5JLB (Yang et al. Citation2016)). The autoinhibitory loop (rose) is in an open position to accommodate the protein substrate. C| MLL1 SET domain (white) associated with WDR5 (green), RbBP5 (light blue), ASH2L (rose) and DPY30 (cyan) bound to a nucleosome core particle (PDB 6PWV (Park et al. Citation2019)). MLL1 SET domain complexed with the H3K4 peptide (cyan, wiht the target K in pink) (PDB 6UH5 (Hsu et al. Citation2019)). D| SMYD2 complexed with the ERα substrate peptide (PDB 4O6F (Jiang et al. Citation2014)) showing the the bilobal or clamshell-like structure and the MYND domain. E| 7BS PKMT DOT1L and cofactor SAM (PDB 1NW3 (Min et al. Citation2003)). The architecture consists of a DOT1L specific region (magenta), a seven-beta sheet rossman fold (white) and a ubiquitin interaction region (green).
Seven beta strand PKMTs
Non-SET domain PKMT and PRMT belong to class I methyltransferases and contain a canonical Rossmann-fold with a seven-stranded β-sheet interconnected by α-helices (7BS PKMTs) (). Among them, certain enzymes have demonstrated PKMT activities (Falnes et al. Citation2016; Luo Citation2018; Falnes et al. Citation2023), one of them, DOT1L, methylates H3K79 (Chandrasekharan et al. Citation2010), others methylate lysine residues in various non-histone proteins, for example METTL13 (Jakobsson et al. Citation2018; Liu et al. Citation2019), METTL20 (Rhein et al. Citation2014; Małecki et al. Citation2015), METTL21A (Jakobsson et al. Citation2013), METTL21B (Hamey et al. Citation2017; Malecki et al. Citation2017), METTL21C (Wiederstein et al. Citation2018; Zoabi et al. Citation2020), METTL21D (Nguyen et al. Citation2023), eEF1A-KMT1 (Jakobsson et al. Citation2017), eEF2-KMT (Davydova et al. Citation2014), and CaM-KMT (Magen et al. Citation2012). Lysine, arginine and histidine residues in proteins serve as substrates for class I methyltransferases, as well as the N-terminal amino groups of proteins, small molecule DNA and RNA are also included in their substrate spectrum (Cheng et al. Citation2005; Richon et al. Citation2011). With approximately 150 candidates encoded by the human genome, many of the 7BS PKMTs have remained uncharacterized (Petrossian and Clarke Citation2011). Unravelling their biochemical properties, catalytic mechanisms and substrates, whether histone or non-histone, is a task for future research.
Simulations of the catalytic mechanism of PKTMs
Key mechanistic features of PKMTs, like the SN2 reaction mechanism and the deprotonation of the lysine substrate, have been investigated by simulations technologies and the results are discussed in the next chapters.
Investigation of the SN2 reaction mechanisms
QM/MM simulations of SET7/9 (aka SETD7, SET7, SET9, KMT7) were first to investigate details of the bimolecular nucleophilic substitution (SN2) mechanism PKMT use to transfer a methyl group from the cofactor SAM to the ε-amino group of a lysine residue (Hu and Zhang Citation2006). The lysine nitrogen (Nε) is first deprotonated (as described later) and then acts as the nucleophile attacking the SAM methyl group, whereas the SAM sulphonium cation (S+) acts as the leaving group converting SAM to SAH. The free electron pair of the deprotonated lysine nitrogen is present in a sp3 orbital at an 109° angle. The SN2 reaction occurs at an aliphatic sp3 carbon center (the C-atom of the transferred methyl group) with the electronegative sulphonium leaving group attached to it. The nucleophile attacks the carbon at a minimal distance of approximately 2.2 Å (Chen et al. Citation2019). Breaking of the C–S bond and the formation of the new bond between the methyl C-atom and the nucleophile occurs instantaneously through a trigonal bipyramidal TS in which the methyl C-atom is sp2 hybridized. The nucleophile attacks the methyl C-atom at a 180° angle to the leaving group, since this provides the best overlap between the nucleophile’s lone pair and the C–S antibonding orbital. The leaving group is then pushed off at the opposite side and the product is formed with inversion of the tetrahedral geometry at the central carbon atom, which is irrelevant in case of the achiral methyl group. The TS structure then collapses and the methyl group covalently binds to the nucleophile, the lysine Nε atom in case of PKMTs, and SAH leaves as product (Copeland et al. Citation2009). Important to note is that the methyl group is transferred rapidly to a deprotonated target lysine once a geometry favorable for the reaction has been achieved due to the high group transfer potential of SAM, thereby preventing a reprotonation. In conclusion, three geometric criteria can be used to describe a state which most accurately describes the TS of the methyl group transfer (Schnee et al. Citation2022; Khella et al. Citation2023):
The distance between the lysine Nε and the SAM methyl C-atom is < 4.6 Å.
The angle between the lysine Nε, the lysine Cδ and the SAM methyl C-atom is close to 109°.
The angle between the lysine Nε, the SAM methyl C-atom and the SAM S-atom is close to 180°.
Later studies based on a combination of computational modeling, QM/MM and kinetic isotope effects showed that PKMT reactions can have different transitions states depending on the enzyme and methylation substrate (Linscott et al. Citation2016; Poulin, Schneck, Matico, McDevitt, et al. Citation2016; Chen et al. Citation2019). For example, in case of SET8 (aka Pr-SET7, SETD8, KMT5A) an early SN2 TS was shown with a short C − S distance (2.0 Å) and a long C − Nε distance (2.4 Å) while a late SN2 TS was observed for NSD2 with a long C − S distance (2.5 Å) and a short C − Nε distance (2.1 Å) (). These are interesting results indicating that although the lysine nucleophile, methyl group transfer and leaving groups are identical, the nature of the TS differs between PKMTs.
Other studies approached the question, how the enzyme-substrate complex reaches a TS conformation with MD simulations. Since these simulations cannot account for bond breakage or formation, TS-like conformations were defined as conformations, which fulfill the geometric SN2 reaction criteria. The frequency of formation of TS-like conformations was then monitored throughout the simulation with different substrates, enzyme mutants or conditions. Thereby, MD simulations can estimate the likelihood of a SN2 reaction without simulating the actual methyl transfer. Of note is that this can only provide qualitative statements and a relation to quantitative biochemical turnover numbers is not recommended (Schnee et al. Citation2022; Khella et al. Citation2023).
A combination of MALDI-TOF MS experiments, NMR analysis, QM/MM and MD simulations revealed that lysine has the optimal length and shape for the SN2 methyl group transfer in the active site of PKMTs. Shorter or longer alkane chains containing a terminal amine (ornithine and homolysine, respectively) lead to a disruption of the SN2 reaction geometry () (Al Temimi et al. Citation2017). In MD simulations of SET7/9, the average distance between the SAM methyl C-atom and the substrate lysine Nε was 3.2 Å and the nucleophile and leaving group were in line fulfilling the geometric 180° criteria for a SN2 reaction. For ornithine placed in the same substrate peptide, the average Nδ to methyl C-atom distance was 4.5 Å and the nucleophile and leaving group were not in line. This difference was also represented in the free energy (PMF) profiles of the two methylation reactions, where exchanging lysine to ornithine raised the free energy barrier from 18.8 to 32 kcal/mol. It was speculated that the lysine nitrogen is stabilized and oriented in the active site in a productive conformation by surrounding residues, namely Y305 in case of SET7/9. The stabilization and orientation were less efficient for the shorter ornithine (Al Temimi et al. Citation2017). Unnatural amine containing N-nucleophiles with the same length as lysine (e.g. azalysine) showed methylation activity, whereas amide/guanidine-containing N-nucleophiles as well as simple O- and C- nucleophiles were not methylated by SET7/9 (Al Temimi et al. Citation2019a). Surprisingly, azalysine had a slightly lower free energy barrier compared to lysine in the SET8 catalyzed methylation reaction (18.0 and 19.4 kcal/mol, respectively). It was assumed that the Nε of azalysine is slightly more acidic than that of lysine and undergoes easier deprotonation in the PKMT active site. The nucleophilic character and the similar conformation of azalysine in the active site compared to lysine may therefore explain the observations that azalysine can in general undergo the PKMT-catalyzed methylation to a similar degree as lysine (Al Temimi et al. Citation2019a). Slightly bulkier substrates like cyclopropyllysine were also accepted by SET7/9 as substrates. However, severely bulkier substrates like meta-amino phenylalanine were not accomodated (Al Temimi et al. Citation2020). Apart from altering the size and shape of lysine mimics, one methylene group in the hydrophobic carbon side chain was substituted with a sulfur resulting in the unnatural amino acid γ-thialysine. Enzyme kinetics and QM/MM free-energy calculations revealed that γ-thialysine and lysine exhibit comparable efficiencies for methyl transfer reactions, indicating that γ-thialysine is a good lysine mimic for PKMT catalysis (Simon et al. Citation2007; Al Temimi et al. Citation2019b).
Figure 4. A| Structure representation of lysine and lysine analogues and their capability to function as PKMT methyl group acceptors. B| Target lysine deprotonation is obligatory for the PKMT catalyzed methyl group transfer. The protonated target lysine (pink, sticks) is oriented by SET7/9 Y335 (white, sticks), the water channel is already present (red spheres, prepared using PDB 1XQH (Chuikov et al. Citation2004)). C| The lysine proton is transferred to the nearby water molecule. D| After lysine deprotonation, the SAM-methyl group is rapidly transferred to the deprotonated target lysine thereby preventing the reprotonation. The excess proton is transferred into the bulk solvent.
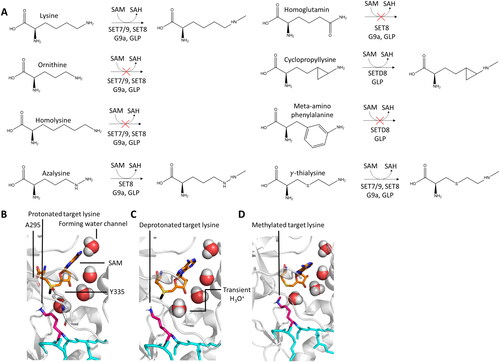
Mechanism of target lysine deprotonation
An essential parameter of lysine methylation is the protonation state of the lysine Nε atom. Whereas under physiological conditions the lysine side chain is protonated, it needs to be deprotonated in the active site of PKMTs to become nucleophilic (Trievel et al. Citation2002). First models trying to explain the deprotonation of the target lysine focused on a conserved tyrosine residue in the SET domain which might deprotonate the target lysine, e.g. Y335 in SET7/9 (Trievel et al. Citation2002; Kwon et al. Citation2003), Y283 in DIM-5 (Zhang et al. Citation2002), Y287 in Rubisco LSMT (Trievel et al. Citation2003), and Y336 in SET8 (Jacobs et al. Citation2002; Xiao et al. Citation2005). This hypothesis was ruled out based on the crystal structures of the PKMT in complex with peptide substrates, which showed that Y335 donates a hydrogen bond to the backbone carbonyl oxygen of A295 and it is too far away from the target lysine to perform the deprotonation function (Wilson et al. Citation2002; Zhang et al. Citation2002; Trievel et al. Citation2003; Xiao et al. Citation2003; Couture et al. Citation2005; Xiao et al. Citation2005). Later, Guo and coworkers suggested a refined model, in which Y335 donates a hydrogen bond to the backbone carboxyl oxygen of A295 when SAM was bound (Guo and Guo Citation2007). In simulations without bound cofactor, they observed that Y355 donates a hydrogen bond to a water molecule suggesting Y355 could be deprotonated in this environment. Subsequently, the deprotonated Y355 was suggested to deprotonate the target lysine. This mechanism was later challenged as the calculated pKa of Y335 (>13) indicated that Y355 cannot take the role as base in this process (Zhang and Bruice Citation2007b). Although Y355 could not be responsible for target lysine deprotonation, it was shown via NMR 1H chemical shift coupled with quantum mechanics calculations that the Y335 hydroxyl group has another critical function in catalysis, because it helps to stabilize the SAM methyl group by a CH-O hydrogen bond and is therefore indispensable for SAM binding and stabilization of the partial positive charge on the methyl group C-atom developing in the TS (Horowitz et al. Citation2011; Horowitz et al. Citation2014).
Despite the lack of identification of a general base in the catalytic center of SET7/9, computational modeling, MD and QM/MM simulations by Zhang and Bruice showcased a conserved mechanism for the deprotonation of the target lysine for multiple SET domain-containing PKMTs (Zhang and Bruice Citation2007b). In this mechanism, the deprotonation of the Nε occurs through the transient formation of dynamic water channels in the enzymes’ active sites. The residues involved in this water channel are G264, G292, A295, Y305, Y335, Y337 in the case of SET7/9 (Hu and Zhang Citation2006; Zhang and Bruice Citation2008b, Citation2008a). A water molecule was frequently discovered in the crystal structures of SET domain-containing PKMTs, however it cannot act as final proton acceptor by itself, since H3O+ is a much stronger acid than Kme1. Instead, the water molecule is suggested to transfer the proton through a chain of water molecules into the aqueous solvent and finally to a buffer molecule (). Adding to this, the electrostatic interactions between the positive charges of the SAM sulfonium moiety and the protonated Nε atom decrease the pKa of the latter from 10.9 to 8.2 (Zhang and Bruice Citation2007b). This could explain the weak methylation activity of PKMTs in acidic and even neutral buffers, as the necessary deprotonation of the target lysine is impeded. In a basic environment, the activity of PKMTs was observed to be increased. Biochemical studies showed that SET7/9 and DIM-5 are active at pH 8 or higher and have a pH optimum of ∼10 (Wilson et al. Citation2002; Zhang et al. Citation2002). Based on this, Bruice and Zhang suggested a stepwise process (Zhang and Bruice Citation2007b), in which 1) the water channel appears, 2) the target lysine is deprotonated, and the proton is transferred into the solvent, 3) the target lysine is methylated using the methyl group provided by SAM. Of note, the steps 2 and 3 can occur in a concerted manner. This concept was based on MD simulation results of several SET domain-containing PKMTs, in which the water channel appearance was tracked and correlated with the experimentally observed product specificity (Zhang and Bruice Citation2007a, Citation2007b, Citation2007c, Citation2008b, Citation2008c, Citation2008a). As an example, a water channel was observed for unmethylated and monomethylated lysine substrates in the Rubisco LSMT, but not for dimethylated lysine, explaining the biochemically observed dimethyltransferase activity of this enzyme. It is important to note, that no water channel could be detected in the absence of cofactor indicating that essential conformational changes occur upon SAM binding.
Exceptions for this model need to be made for 7BS PKMTs like DOT1L (aka KIAA1814, KMT4). Here, the residues located at the target lysine binding pocket seem to be incapable of facilitating a deprotonation or formation of a water channel. It has been speculated that a more hydrophobic active site could reduce the pKa of the target lysine and that the carboxylate of SAM could help in the subsequent deprotonation process (Min et al. Citation2003; Cheng et al. Citation2005; Cortopassi et al. Citation2016).
Analysis of the product specificity and processivity of PKMTs
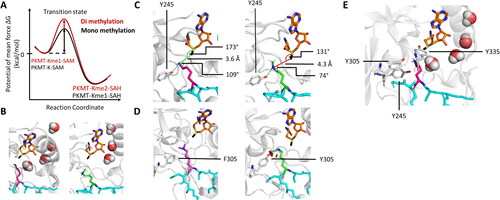
SET7/9 was initially identified as a histone H3 lysine 4 (H3K4) monomethyltransferase and it was one of the first model enzymes to investigate the product specificity of PKMTs (Hu and Zhang Citation2006; Zhang and Bruice Citation2007b; Dhayalan et al. Citation2011). Product specificity refers to the capability of PKMTs to transfer a precise number of methyl groups to its target either only one, or up to two or three methyl groups creating Kme1, Kme2 or Km3, respectively. Despite their similarity in the SET domain, PKMTs exhibit different product specificities. Distinct mechanisms are hence needed to control the number of methylation steps for each individual enzyme. In QM/MM simulation studies, SET7/9 was confirmed as a monomethyltransferase. The free energy barriers determined by PMF for the monomethyl transfer were 22.5 kcal/mol (Wang et al. Citation2007; Hu et al. Citation2008; Zhang and Bruice Citation2008b) or 17–19 kcal/mol (Guo and Guo Citation2007; Zhang and Bruice Citation2007c; Yao et al. Citation2012) depending on the study. Of note, the free energy for a spontaneous methyl transfer in aqueous solution was determined to be 8 kcal/mol higher illustrating the catalytic function of the enzyme (Wang et al. Citation2007). For further methylation of Kme1 substrates to Kme2, an increase in free energy of 3–5 kcal/mol was observed consistent between the studies. For the generation of Kme3 from Kme2, a further increase in free energy of 3 kcal/mol was observed () (Guo and Guo Citation2007; Hu et al. Citation2008; Yao et al. Citation2012). Multiple explanations for the distinct product specificities of PKMTs were proposed, which will be discussed in the following paragraphs.
Figure 5. PKMTs transfer a defined number of methyl groups to their lysine target (pink, sticks). A| Schematic representation of a free energy profile showing a possible first methyl transfer (black line) and an energetically unfavorable second transfer (red line). B| Restricted second methylation because of a disrupted water channel and blocked lysine deprotonation of monomethylated target lysine (green, sticks, PDB 1XQH). C| The SN2 TS cannot be adopted if a monomethyl substrate is present. D| The F/Y-switch position controls the product specificity of certain PKMTs. Phenylalanine (white, sticks) provides more space in the active site allowing to accommodate a dimethyl product (pinks, sticks), while the additional hydroxyl group of a tyrosine leads to sterical clashes preventing formation of the dimethylated product. E| Position of the tyrosine residues 245, 305, 335 in the SET7/9 active site discussed in the main text.
Regulation of PKMT product specificity by deprotonation of the target lysine
PKMTs deprotonate the target lysine prior to the methyl transfer to create a suitable nucleophile. The deprotonation process could therefore be a regulatory step controlling the product specificity. As described above, Zhang and Bruice showcased with multiple PKMTs that a chain of water molecules forms a water tunnel to deprotonate the lysine Nε (Zhang and Bruice Citation2007b). In the context of the product specificity of SET7/9, they proposed that the water channel only forms under defined conditions. In MD simulations of SET7/9, the water channel was only observed in the presence of SAM and unmethylated K4 peptide, but not without cofactor, with SAH or if K4me1 was bound. This could regulate the lysine deprotonation and stop the methylation process after monomethylation. Mechanistically, the methyl group of the monomethylated peptide takes the position of the proton that would be removed through the water channel. Deprotonation and further methylation of Kme1 is therefore impossible () (Zhang and Bruice Citation2008b).
Later, ∼10 times longer MD simulations found that SET7/9 can weakly dimethylate P53-K372, suggesting that there exists a strong preference for monomethylation, but not an absolute limitation to the formation of only Kme1. However, these studies confirmed the proposed mechanism, as a water tunnel was found as well in the complex of monomethylated lysine (Bai et al. Citation2011). Methylation stopped at the dimethylated lysine because this together with freshly bound SAM disrupted the water channel. Free histone methylation experiments indeed showed that SET7/9 can act also as a weak dimethyltransferase (Kwon et al. Citation2003). Furthermore, Dhayalan et al. showed that although SET7/9 was able to transfer two methyl groups to both histone- and non-histone targets in vitro, it catalyzed dimethylation with much lower rates (∼10% of the mono-methylation rate) (Dhayalan et al. Citation2011). While lysine deprotonation might influence the product specificity, QM/MM studies suggest that the deprotonation is not the rate-determining step in the methylation of an unmethylated lysine, as the calculated barrier for the proton transfer is 8.4 kcal/mol, which is more than 10 kcal/mol lower than that of the methyl transfer step (22.5 kcal/mol) (Hu et al. Citation2008).
Regulation of PKMT product specificity by steric constraints
PKMTs transfer one or multiple methyl group(s) from the cofactor SAM to a target lysine in a linear SN2 reaction mechanism (Trievel et al. Citation2002). The above-mentioned geometric requirements are crucial to form the transitions state. As pointed out by Zhang and Bruice in MD and QM/MM simulations of the SET7/9 complexed with the K4 or K4me1 peptide, the distance between the SAM sulfur group and the lysine nitrogen was higher for K4me1 (6.1 Å) than for the unmethylated K4 (5.7 Å) (Zhang and Bruice Citation2007b). A similar observation was made by Hu and Zhang in QM/MM simulations of SET7/9 with a bound peptide substrate, where the SET7/9 Y245 positioned the unmethylated lysine nitrogen for the SN2 reaction by hydrogen bonds (Hu and Zhang Citation2006). In simulations with the H3K4me1 peptide, Y245 precluded the rotation of K4me1, leading to a blockage of the active site, which prevented the adoption of a productive SN2 TS conformation () (Hu and Zhang Citation2006; Yao et al. Citation2012). To support this model, the SET7/9 Y245A mutant was created and tested for product specificity showing that the in-silico activity towards unmethylated lysine decreased whereas di- and trimethylation activity was elevated (Hu and Zhang Citation2006; Yao et al. Citation2012). This was supported by biochemical experiments showing that the Km of SET7/9 WT and SET7/9 Y245A towards the unmethylated TAF10 peptide (K189 as target lysine) were only slightly different (WT 160 17 µM, Y245A 200
35 µM), whereas the kcat differed heavily (WT 17
0.6 min−1, Y245A 0.53
0.04 min−1) (Del Rizzo et al. Citation2010). This indicates that the peptide binding is not altered by Y245A, but the stabilization of the SN2 TS involving Kme0, Kme1 and Kme2.
The space emptied in the active site pocket by the Y-to-A mutation allowed for accommodation and proper orientation of the methylated lysine residues. These simulations are in agreement with results of previous structural and biochemical experiments, which showed that the Y245A mutation converts SET7/9 into a trimethyltransferase with weak monomethyltransferase activity (Xiao et al. Citation2003). Based on this, the Y245A mutation was postulated to function as a switch between mono and up to trimethylation activity, which was later confirmed by crystal structures showing the bound di- and trimethylated TAF10 (TATA box binding protein associated factor) peptide in the SET7/9 Y245A active site (Del Rizzo et al. Citation2010). Additionally, QM/MM simulations of SET7/9 Y245A showed that the free energy barrier was similar for mono-, di- and trimethylation (Yao et al. Citation2012) indicating that the reaction could proceed similarly once a productive conformation is reached.
A similar observation regarding the critical orientation of the target lysine was made for PRDM9 (aka PMF6) Y276F, which structurally overlaps with SET7/9 Y245. Here, QM/MM simulations found that the energy barrier of Y276F increased for the first methyl transfer compared to the WT (Chu et al. Citation2015), which was confirmed by biochemical experiments (Wu et al. Citation2013). This effect is potentially caused by the tyrosine hydroxyl group, which forms hydrogen bonds with the Nε atom of the target lysine residue and helps stabilizing the SN2 TS (Hu and Zhang Citation2006; Yao et al. Citation2012). This coordination is missing for the phenylalanine mutant accounting for lower TS stabilization.
Regulation of PKMT product specificity by F/Y switches
In multiple sequence alignments it had been identified, that PKMTs that possess a tyrosine at a so-called F/Y switch position are limited to catalyzing mono- or dimethylation, whereas enzymes that possess a phenylalanine or another hydrophobic residue at this position display di- or trimethyltransferase activity (Collins et al. Citation2005). This observation was validated and rationalized in a variety of MD, and QM/MM simulations, biochemical and structural experiments. In many examples, the switch of that specific tyrosine or phenylalanine to the other amino acid led to changes in product specificity (Trievel et al. Citation2003; Zhang et al. Citation2003; Collins et al. Citation2005; Qian C and Zhou Citation2006; Wu et al. Citation2010; Cortopassi et al. Citation2016; DiFiore et al. Citation2020). Swaps in product specificity were observed with several SET domain-containing PKMTs, e.g. the trimethyltransferase DIM-5 can be converted into a mono/dimethyltransferase by the F281Y mutation (Zhang et al. Citation2003). The monomethyltransferases SET7/9 and SET8 can be changed to dimethyltransferases through the corresponding Y/F mutation, Y305F for SET7/9 (Zhang et al. Citation2003; Del Rizzo et al. Citation2010) and Y334F for SET8 (Couture et al. Citation2005; Couture et al. Citation2008; Chu et al. Citation2010). In MLL3 (aka KMT2C), the Y4884C mutation which has been observed as somatic mutation in cancer cells was shown to convert the enzyme from a monomethyltransferase with substrate preference for H3K4me0 to a trimethyltransferase with H3K4me1 as the preferred substrate (Weirich et al. Citation2015). Hence Y4884C can be considered a natural mutant using the F/Y switch mechanism for the control of the product specificity of a PKMT. The F/Y switch might not be fixed to a defined position, as more than one position can function in this way. The dimethyltransferase G9a could be turned into a trimethyltransferase by the Y1067F mutation (Wu et al. Citation2010), but into a monomethyltransferase by F1152Y indicating that two susceptible positions exist (Collins et al. Citation2005). Likewise, the dimethyltransferase GLP (G9a-like Protein) could be converted into a monomethyltransferase by F1209Y (Collins et al. Citation2005) and into a trimethyltransferase by Y1124F (Wu et al. Citation2010). However, it is important to note that not every of the tyrosine residues surrounding the active site can be mutated into a phenylalanine to alter the product specificity, since SET7/9 Y245F is catalytically inactive (Xiao et al. Citation2003).
The mechanistic basis of the F/Y-switch solely relies on the presence of one single hydroxyl group. The missing hydroxyl group in the Y-to-F mutants creates additional space in the active site. Thereby, accommodation of water molecules gets easier, leading to an easier deprotonation of the target lysine. In crystal structures of SET7/9 Y305F and the TAF10me1 substrate peptide, water molecules were found in the active site. This was not the case in corresponding structures of WT SET7/9 (Del Rizzo et al. Citation2010). Additionally, MD and QM/MM simulations of SET8 and its F/Y switch mutant Y334F showed a lowered energy barrier for the dimethylation reaction for the Y334F mutant and a connection to the water channel (Chu et al. Citation2010; Chu et al. Citation2012). In contrast, the effect of F-to-Y mutations, which turns trimethyltransferases to mono/dimethyltransferases, could be based on steric effects by the additional hydroxyl group making the active site too narrow to accommodate multiple methyl groups at the lysine Nε () (Hu and Zhang Citation2006; Chu et al. Citation2012). The concept of active site volume dependent product specificity changes has also been illustrated for other mutations like the NSD2 T1150A cancer mutant turning NSD2 from a di- to a trimethyltransferase (Khella et al. Citation2023).
Investigation of the processivity of PKMTs
Enzymes that perform multiple rounds of catalysis on a macromolecular substrate can do so in two different modes of action: In a distributive mechanism, each round of catalysis results in product dissociation and rebinding of a fresh substrate, whereas in a processive mechanism, multiple rounds of catalysis proceed on the same substrate before dissociation of the product. PKMTs using a distributive mechanism dissociate from the substrate protein after methylation of the target lysine residue. Subsequent catalytic turnovers require rebinding of the enzyme to the protein substrate. This also involves dissociation of SAH and rebinding of a new SAM molecule to provide the next methyl group. Each methylation event is generally independent leading to the stochastic generation of Kme1, Kme2 and Kme3, depending on the product specificity of the PKMT. In contrast, processive PKMTs remain associated to their substrate protein and the target lysine residue remains bound in the active site pocket for multiple consecutive catalytic turnovers without dissociation. This allows them to methylate the same lysine residue multiple times without repeated dissociation and binding steps leading to the direct generation of higher methylation states. Still, after the first methyl transfer, SAH is replaced by a fresh SAM, indicating that a SAM/SAH exchange pathway must exist in the presence of the substrate protein.
Biochemical data showed that PKMTs use both mechanisms. Several SET domain-containing PKMTs and also PRMTs were shown to perform multiple rounds of lysine methylation in a processive mechanism (Cheng et al. Citation2005; Smith BC and Denu Citation2009; Van Aller et al. Citation2016). Kinetic studies revealed that G9a catalyzes the transfer of two methyl groups to H3K9 in a processive manner (Poulard et al. Citation2021), which was confirmed in competition methylation experiments (Patnaik et al. Citation2004). A similar behavior was observed for the H3K9 methyltransferase DIM-5 (Zhang et al. Citation2003), SMYD3 (aka ZMYND1) (Van Aller et al. Citation2016) and the H3K36 methyltransferase NSD1 (Khella et al. Citation2023). In MALDI mass spectrometry experiments of NSD1 starting with the unmethylated H3K36 peptide, a clear peak of H3K36me2 product was observed. In contrast, much lower methylation was detectable when the reaction was started under the same conditions with the H3K36me1 substrate indicating that H3K36me0 is the preferred substrate and two methyl groups were transferred in a processive reaction mechanism.
However, attention needs to be paid to details and the general assumption of SET domain PKMTs always acting in a processive manner is incorrect. A distributive methylation mechanism was observed for example for SET domain monomethyltransferases with mutations, which caused them to exceed their monomethylation capability. SET8 Y334F was converted to a dimethyltransferase but exhibited a distributive reaction mechanism (Couture et al. Citation2008). This effect was also observed for MLL1 Y3924F (Patel et al. Citation2009), SET7/9 Y305F and Y245A (Del Rizzo et al. Citation2010). The assumption is plausible, that many canonical di- and trimethyltransferases catalyze multiple methyl group transfers in a processive manner to avoid release of the monomethylated intermediates which in the case of histone methylation often transmit different biological signals than the di- or tri-methylated products. Monomethyltransferases, naturally have to release their substrate after the methyl group transfer. This mechanism seems to be carried over if monomethyltransferases were artificially converted into di- or trimethyltransferases, which then retain the distributive reaction mechanism. One reason for this could be that the active sites of monomethyltransferases have not been evolutionary optimized for multiple turnovers, e.g. by not allowing cofactor exchange while the protein substrate is still bound. The mutation allows for higher methylation levels in these enzymes, but the catalytic mechanism remains unchanged. An exception to this postulation might be the Drosophila Su(var)3–9, which was shown to transfer two methyl groups to H3K9 in a non-processive manner despite it represents the natural enzyme without any mutation (Eskeland et al. Citation2004). In contrast to the SET domain-containing PKMTs, DOT1L has been shown to perform multiple rounds of H3K79 methylation through a distributive mechanism (Frederiks et al. Citation2008).
Rationalizing why a certain PKMT or mutated PKMT operates in processive or distributive manner is still difficult in many cases (Zhao et al. Citation2022). MD simulation appears to be a suitable method to unravel this mechanism. Possible MD approaches to investigate the catalytic mechanism could involve the analysis of conformational changes and active site dynamics. Comparing the active site dynamics and interactions between a processive and distributive a PKMT could give information about conserved mechanisms. Particularly, in a processive mechanism, the active site needs to undergo substantial conformational changes and rearrangements to firstly accommodate the transferred methyl group and subsequently position the target lysine correctly for the next round of catalysis without substrate dissociation. This mechanism also needs to include the requirement for cofactor exchange. These potential rearrangement mechanisms would not be required in distributive PKMTs.
In a distributive mechanism, enzyme, product and cofactor dissociate after catalysis. A repulsive interaction between the product and active site residues after the methyl group transfer could therefore be detected. This can be observed using MD simulation techniques. In an alternative approach, the re-binding of methylated substrates could be probed. Distributive PKMTs need to bind methylated substrates in contrast to processive PKMTs. In such an experiment, adaptive sampling techniques like steered MD simulation (sMD), Monte Carlo sampling or metadynamics could be used to observe differences between the substrate binding of distributive and processive PKMTs. In the sMD approach, external forces are used to guide bimolecular association processes. Thereby, reactions that otherwise would be too slow to be modelled in the timescale accessible to MD simulations are accelerated and at the same time the conformational sampling is concentrated along a specific, predefined reaction coordinate (Yang T et al. Citation2019). Here, it could be studied if a methylated substrate could efficiently be bound by the investigated PKMT. Distributive PKMTs might show mechanisms or conformational changes to productively bind the methylated substrate, whereas processive PKMTs should not. In metadynamics, the sampling of rare events is enhanced by adding bias to the potential energy surface. It builds up a history-dependent bias potential to push the system away from previously visited regions, allowing exploration of new conformational spaces and events (Bussi and Laio Citation2020). For processive PKMTs, the binding of a methylated substrate in the active site will be less likely compared to distributive PKMTs. This would be represented in an energy increase needed to visit these states.
Mechanism of substrate specificity of PKMTs
Given their biological functions, it is essential that PKMTs methylate specific lysine residues on histone tails and other proteins. Mistakes in the substrate choice would lead to aberrant methylation signals which could result in misregulation of chromatin states or protein activity. A highly specific recognition of the protein substrate by PKMTs is therefore indispensable. In this context, multiple research opportunities arise. Approximately 60% of the SET domain- containing proteins in humans have well- documented methylation activity on histone and/or non- histone proteins (Husmann and Gozani Citation2019) while the activity of the remaining ones needs to be discovered. Moreover, in humans approximately 150 7BS proteins exist, which methylate various targets including lysine residues (Bhat et al. Citation2021). Among them, some PKMTs have been discovered, but more enzymes of this class may be identified in future work. As soon as an initial methylation activity of a potential PKMT is detected, deciphering the substrate specificity can directly hint towards its natural methylation targets and the potential biological roles (Kudithipudi and Jeltsch Citation2016). Secondly, different PKMTs might recognize their target amino acid sequence with a variety of recognition processes. Learning about the individual mechanism directly benefits further studies, e.g. specific drugs design, in which molecules can be tailored towards the PKMTs amino acids involved in the substrate recognition.
Analysis of the specificity of PKMTs by SPOT peptide array methylation
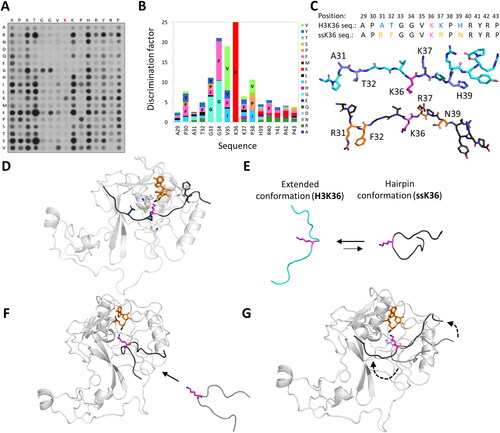
A highly suitable technique to decipher the substrate specificity of PKMTs are experiments investigating the methylation of Celluspots peptide arrays (Bock et al. Citation2011; Weirich and Jeltsch Citation2022). In this technology, peptides are synthesized on a cellulose membrane with solid-phase peptide synthesis. The advantage of this approach is that numerous peptide spots can be synthesized on one membrane, each spot containing an individual peptide sequence (). The membrane is then incubated with the PKMT of interest and radioactively labeled SAM allowing to detect methylation by autoradiography. Then, the signal intensity of the different spots directly indicates which substrate peptides are preferred and which are disfavored by the PKMT. When creating peptide arrays containing all possible single amino acid exchanges of the original substrate sequence, a PKMT-specific substrate specificity profile can be created () (Rathert, Dhayalan, Ma, et al. 2008; Dhayalan et al. Citation2011; Kudithipudi, Kusevic, et al. Citation2014; Kudithipudi, Lungu, et al. Citation2014; Schuhmacher et al. Citation2015; Weirich et al. Citation2016; Kusevic et al. Citation2017). With the obtained specificity profile as a search sequence, novel protein substrate candidates have been identified (Rathert, Dhayalan, Murakami, et al. Citation2008; Dhayalan et al. Citation2011; Schuhmacher et al. Citation2020; Weirich et al. Citation2020). During the investigation of the substrate specificity of the PKMT SETD2, it was found that the canonical H3K36 substrate sequence was not optimal for activity (Schuhmacher et al. Citation2020). Surprisingly, multiple single amino acid mutations caused a higher methylation of the peptide substrates. By combination of the amino acids found to be preferred, a non-natural peptide sequence referred to as ‘super-substrate K36 (ssK36)’ was created. This ssK36 peptide differed at 4 positions from the canonical H3 peptide sequence and was methylated about 100 times more efficiently ().
Figure 6. SETD2 has a ∼ 100-fold higher activity towards an artificially designed ssK36 peptide substrate. A| SPOT peptide array with the 15-residue long H3K36 peptide sequence as starting sequence, incubated with SETD2 and radioactively labeled SAM. Positions are individually mutated to any other amino acid except tryptophan and cysteine. At several positions non-natural amino acids are preferred in the substrate peptide. B| Quantification of the peptide array methylation data generates a PKMT specific specificity profile showing the preference for each position. C| Combination of preferred residues led to the generation of a super-substrate (ssK36) peptide (black, sticks) sequence, which differs at 4 positions (orange, sticks) from the canonical H3K36 peptide sequence (cyan, sticks). D-G| MD and sMD simulation snapshots of SETD2 (white, cartoon) interacting with the ssK36 peptide (black, cartoon). D| Crystal structures show the ssK36 peptide in the SETD2 binding cleft, where special interactions of SETD2 with ssK36 residues are observed (prepared using PDB 6VDB as template (Schuhmacher et al. Citation2020)). E| MD simulation of the H3K36 and ssK36 peptides in solution with subsequent backbone conformation-based clustering show that the ssK36 peptide preferably adopts a hairpin-like conformation with the target lysine facing outwards. The H3K36 peptide (cyan, cartoon) prefers an extended conformation. F| In sMD simulations, hairpin-like shaped peptides docked more often successfully in the SETD2 binding cleft, establishing more TS-like conformations. G| After binding, the hairpin conformations unfold in an extended conformation with contacts spreading gradually from the middle section. A-C| Taken from (Schuhmacher et al. Citation2020; Schnee et al. Citation2022) with permission.
In the case of SUV420H2, specifically methylating H4K20me1 (but not H4K20me0) to H4K20me2/3, MD and QM/MM simulations showed that specific features in the active site are responsible for the particular preference of this enzyme for a monomethylated target (Qian P et al. Citation2017). The relatively low free energy barrier for dimethylation (17.9 kcal/mol) compared to that for monomethylation (23.9 kcal/mol) indicates the more effective TS stabilization with the monomethylated substrate. This is achieved through the strengthening of interactions occuring in the TS such as the CH···O interactions involving the K20me1 methyl group and SUV420H2 residues F160, S161, A179, I181, Y217 and the presence of a cation − π interaction between F191 and the methyl group in the TS of the reaction (Qian P et al. Citation2017). The free energy barrier for trimethylation (23 kcal/mol) is then almost at the level of the monomethylation indicating the preferential generation of H4K20me2.
MD Simulations to investiate the substrate specificity of PKMTs
Complementary to the biochemical approach described in the last chapter, MD simulations can be used to investigate the molecular mechanisms behind the substrate specificity of PKMTs. To do this, MD simulation experiments of PKMTs in complex with preferred and disfavored peptide substrates can be conducted. Potential differences between the two peptide-PKMT complexes regarding contacts or the stabilization of TS-like conformations might be observed. For this, known crystal or cryo-EM structures can be utilized as template even though they were obtained using SAH or target K-to-M mutant substrates, because the active cofactor SAM and the lysine in the substrate peptide can be modelled (). This approach was used to investigate the increased methylation activity of SETD2 towards the super-substrate peptide described above (Schnee et al. Citation2022). Multiple differences in the SETD2-peptide interaction profile were observed for the H3K36 and the ssK36 peptides. This was especially true for the regions surrounding the residues differing between both peptides. Eventually, the altered residues in ssK36 established specific contacts leading to a better stabilization of TS-like conformations. Furthermore, both peptides approached the TS-like conformation in different ways, indicating that a PKMT may employ different TS conformations depending on the amino acid sequence of the substrate (Linscott et al. Citation2016; Poulin, Schneck, Matico, McDevitt, et al. Citation2016; Chen et al. Citation2019). However, the interactions in the SETD2-peptide complex cover only a part of the whole methylation process. This is important, since MD simulations of the free H3K36 and ssK36 peptide in aqueous solution showed that H3K36 in solution adopts an extended conformation, whereas the amino acid changes in ssK36 caused the peptide to preferentially adopt a U-shaped ‘hairpin’ conformation, which was confirmed by FRET experiments () (Schnee et al. Citation2022). In steered molecular dynamics simulations, the hairpin conformation was shown to associate faster into the active site of SETD2 and establish more SN2 TS-like conformations when compared to an extended peptide () (Schnee et al. Citation2022). Upon binding into the binding cleft, the hairpin structure of ssK36 was shown to unfold and extend, gradually establishing contacts with SETD2 towards the peptide ends (). This is in line with FRET experiments and crystal structures of SETD2 presenting the bound peptide in an extended conformation (PDB 5V21, 5JJY, 6VDB (Yang et al. Citation2016; Zhang et al. Citation2017; Schuhmacher et al. Citation2020; Schnee et al. Citation2022)). A similar behavior was observed for SET7/9, as the enzyme showed an increased activity towards an artificially designed peptide with a disulfide bridge, stabilizing a hairpin conformation (Dhayalan et al. Citation2011). Related to these findings is that the lysine demethylase JMJD2A/KDM2A requires a bent peptide conformation for catalytic activity (Ng SS et al. Citation2007). Moreover, substrate conformation might add an additional layer of PKMT regulation. For PKMTs with multiple substrates with similar sequences, discrimination between the substrates could be achieved through the individual substrate conformation, e.g. a hairpin or extended conformation.
SET domain autoinhibition
Autoinhibition refers to a mechanism where the enzyme’s catalytic activity is suppressed or regulated by one of its own structural elements. Upon stimuli, the autoinhibition can be released, leading to the activation of the catalytic activity. The release of autoinhibition can occur through conformational changes induced by substrate binding, binding of regulatory proteins, or post-translational modifications. Autoinhibition thereby provides a control mechanism allowing enzymes to act in a regulated manner, sensible for cellular signals and environmental conditions. In the case of SET domain-containing proteins like ASH1L (aka KMT2H), NSD1-3, SETD2, and SUV39 family member, autoinhibition is mediated through intramolecular interactions between the SET domain and adjacent regions, such as the post-SET domain or the SET-I (SET Insertion or Inhibitory SET) domain. Autoinhibition in SMYD enzymes follows a different mechanism and will be discussed in the SMYD enzyme section.
Autoinhibition by the autoinhibitory loop
First analysis of crystal structures (PDB 3OOI) and MD simulations of NSD1 with bound cofactor SAM, but without bound substrate, showed that an autoinhibitory loop (AL) is placed on top of the substrate binding cleft effectively blocking the entrance of a target peptide () (Trievel et al. Citation2003; Xiao et al. Citation2003; Zhang et al. Citation2003). The AL connects the SET with the post-SET domain, but its sequence is not conserved among SET domain-containing PKMTs (Couture et al. Citation2005; Xiao et al. Citation2005). Mutational studies on the AL of ASH1L showed that stabilizing the closed position of the AL by enforcing hydrophobic interactions (ASH1L K2264L interaction with M2183, C2195, F2197) decreased the methylation activity of ASH1L (Rogawski et al. Citation2015). Besides influencing the general methylation activity, the AL was speculated to regulate the product specificity of PKMTs. For ASH1L, mutational studies found that a Q2265A mutation turns the enzyme from a dimethyltransferase to a trimethyltransferase. Analysis of NMR structures of ASH1L Q2265A showed altered orientations of residues involved in SAM binding and active site shape (An et al. Citation2011; Rogawski et al. Citation2015). This indirect effect could be an explanation for different product specificities observed for PKMTs despite a high sequence similarity in the active site e.g. at the F/Y-switch position. This model was postulated for NSD1, DIM-5 and SETD2, which have differing product specificities (SETD2 and DIM-5 are trimethyltransferases, NSD1 is a dimethyltransferase) and differing AL residues and lengths, but share a high similarity of active site residues (Qiao et al. Citation2011).
Figure 7. In the binary PKMT-SAM structure, the placeholder residue occupies the target lysine channel. A| Alignment of the autoinhibitory loops (AL) of PKMTs with known placeholder residue (bold). B| Cartoon and sticks representation of ASH1L (PDB 4YNM (Rogawski et al. Citation2015)) with S2259 acting as the placeholder residue and the AL in closed position. A target lysine taken from a superposition is shown in fade violet in the target lysine channel to visualize the overlap with the placeholder residue. SAM is shown in orange and the methyl group in black. C| In SETD2, the placeholder residue R1670 (rose, sticks) can adopt multiple conformations. If no peptide is bound, the AL is in a closed position and R1670 occupies the target lysine channel (PDB 4H12 (Zheng et al. Citation2012)). D| In a half-open position, the AL starts to lift and R1670 turns outwards (PDB 5JLE (Yang et al. Citation2016)). E| If a peptide substrate (cyan, target lysine in pink) is bound, the AL is in an open position and R1670 is solvent exposed. The post-SET loop (yellow, cartoon) is closed on top of the bound peptide (PDB 5JLB).
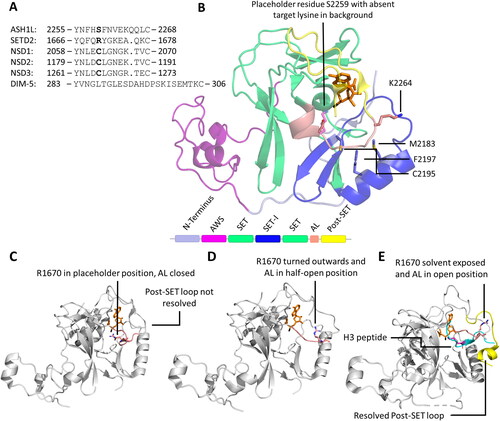
Placeholder residues
Next to the steric hindrance of substrate binding in the closed conformation, the AL was also found to position residues directly in the active site, e.g. C2062 in case of NSD1 (Morishita and di Luccio Citation2011), C1183 in NSD2 (Jaffe et al. Citation2013), R1670 in SETD2 (Yang et al. Citation2016), or S2259 in ASH1L (Yang et al. Citation2016), which in each case occupy the lysine binding tunnel and overlap with the position of the target lysine in a substrate bound state. These residues apparently function as a placeholder stabilizing the closed conformation. This model is supported by the almost abolishment of methylation activity of ASH1L by the S2259M mutation, where the placeholder residue serine was exchanged for methionine, which can establish stronger interactions with the hydrophobic lysine binding tunnel thereby strengthening the closed conformation of the AL (Rogawski et al. Citation2015). Interestingly, methionine was not found to be a placeholder residue in any SET domain PKMT indicating that its binding into the active site pocket may be too strong.
Release of autoinhibition by automethylation
One mechanism to relief autoinhibition and bring the PKMT into an active state was discovered for Clr4, a PKMT of the SUV39H1/2 family found in S. pombe. In this enzyme, automethylation of the placeholder residue K455 by Clr4 led to an increased activity (Iglesias et al. Citation2018). Next to K455, also K472 was shown to be automethylated, which is positioned in the AL of Clr4 as well. Automethylation of both residues led to a conformational switch of the AL away from the active site, resulting in increased enzyme activity towards external substrates. A strong increase in activity towards the H3K9 peptide was also observed for the K455A/K472A double mutant. Here, the AL of Clr4 was brought into a permanently open position, which led to an elimination of the autoinhibition (Iglesias et al. Citation2018). A Clr4 enzyme variant containing a K455M mutation in the AL cannot be automethylated at this site resulting in a strong reduction in methylation activity towards a H3K9 peptide (Khella et al. Citation2020). Mutating the amino acids around K455 to fit the substrate specificity of Clr4 led to a strong increase in automethylation and also increased activity towards the external H3K9 substrate (Khella et al. Citation2020).
Release of autoinhibition by nucleosome interaction
MD simulations, docking experiments and binding isotope effect (BIE) measurements showcased another mechanism to overcome autoinhibition. Interactions with nucleosomes or peptide substrates has been shown to induce conformational changes in PKMTs causing a displacement of the AL and exposition of the substrate binding cleft (Qiao et al. Citation2011; Poulin, Schneck, Matico, McDevitt, et al. Citation2016; Iglesias et al. Citation2018). These studies were stimulated by data showing that the methyltransferase activity of the NSD1–3 and SETD2 enzymes is greatly stimulated when nucleosomes are used as substrates (Li Y et al. Citation2009). Equilibrium BIEs indicated that the methyl group of bound SAM in a binary complex with NSD2 experienced steric constraints, which are resolved in the ternary complex with a bound nucleosome (Poulin, Schneck, Matico, Hou, et al. Citation2016).
For SETD2 a detailed model was developed to describe how this enzyme overcomes autoinhibition in a dynamic manner (Yang et al. Citation2016). In the AL closed state, the placeholder residue R1670 was observed to occupy the lysine binding tunnel, where the target lysine would normally be placed (PDB 4H12) (). However, crystal structures were resolved in which the AL was found in a half-open position (PDB 5JLE). In this state, R1670 was slightly flipped outwards from the active center (). Resolved structures with bound substrate show the AL in a fully open conformation where R1670 is completely flipped outwards and becomes fully solvent exposed. Furthermore, the post-SET loop (Q1676-K1703 for SETD2) was captured on top of the bound peptide substrate hydrophobically interacting with the core enzyme (). This loop is not resolved in crystal structures without bound peptide, indicating a high flexibility in this state. It is suggested that after substrate binding, the post-SET loop closes like a lid and helps to order the peptide in the binding cleft. A working model can be postulated in which at the start of the reaction, the post-SET loop is not binding to the core enzyme and R1670 is in the placeholder position. Upon interactions with a substrate, R1670 flips outwards, the AL is being lifted and the substrate peptide binds in the binding cleft, while the post-SET loop closes behind it. After methylation, the process is reversed (Zheng et al. Citation2012; Yang et al. Citation2016). The complex role of R1670 was highlighted by a strong reduction of SETD2 methylation activity if R1670 is mutated to glycine (Zheng et al. Citation2012), because a “simple” destabilization of the AL by R1670G should have led to a higher catalytic activity. Additionally, alanine mutants of different AL residues (Q1669A and Y1671A) lead to a disruption of the SETD2 methylation activity, emphasizing the importance of the AL in catalysis (Zheng et al. Citation2012). Important to note is that the biochemical, MD simulation and crystallographic results suggesting this model are based on peptides as substrates. For larger substrate structures like proteins or nucleosomes, the post-SET loop might have a different functionality.
This model was later refined, considering that histone tails are extremely flexible structures and adopt a variety of conformations over time (Potoyan and Papoian Citation2011; Ghoneim et al. Citation2021; Huertas et al. Citation2021). Data from sMD simulations suggested an improved association process of peptides in hairpin conformation with the target lysine placed in the loop region of the hairpin (Schnee et al. Citation2022). This could indicate that histone tails could also transiently adopt hairpin conformations and thereby facilitate the association process with PKMTs. Of special interest are the established first contacts between the tip of the hairpin containing the target lysine and the AL, especially the positively charged lysine interacting with solvent exposed residues of the AL. This might not only function as a probing mechanism to recognize the right target lysine, but may also be the start of the conformational changes of the AL needed to overcome autoinhibition (). This model adds to the various roles of the post-SET loop which can also interact with the DNA, thereby weakening contacts with the AL and priming the AL for conformational changes, but further studies are needed for its validation.
Figure 8. Possible mechanism of SETD2 histone tails binding. A| Hypothetical strucutre of the histone tail (modified from PDB 7EA5 (Liu et al. Citation2021)) interacting with the DNA and forming a hairpin-like conformation with the target lysine facing outside. B| First contacts between H3K36 and H3V35 and the SETD2 AL (modified from PDB 6VDB) could trigger conformational changes in the AL and R1670 opening the SETD2 binding cleft.
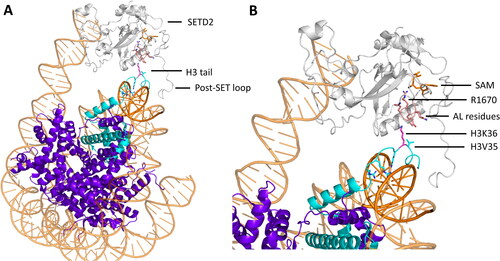
Substrate specificity analysis of SETD2 by Celluspots array methylation showed that residues around the target lysine are crucial for the SETD2 activity (Schuhmacher et al. Citation2020). These residues would be first to interact with the PKMT in a hairpin association. Considering nucleosomes as substrate, a model for the association between PKMTs and histone tails could be envisioned. In this model, the histone tails interact with the nucleosomal DNA, dynamically wrapping around them () (Ghoneim et al. Citation2021). They may gain conformational freedom after partial unwrapping from the DNA and the increased flexibility may stimulate formation of hairpin conformations, exposing the target lysine and nearby residues, which are recognized by the PKMT. This model is in line with cryo-EM structures of MLL1 (aka KMT2A, ALL1, CXXC7, HRX, HTRX, TRX1), MLL3 or yeast Set1 in complex with their associated proteins and their nucleosomal substrate (PDB 6KIX, 6KIU, 6UH5 (Hsu et al. Citation2019; Xue et al. Citation2019)). In these structures, the target lysine and amino acids around the target lysine are resolved (A1-R8 for PDB 6UH5) and positioned correctly in the SET domain of the respective PKMT. However, the H3 tail is not completely resolved, but misses the connecting residues K9-K36. Connecting the resolved K4 region of the H3 tail with the resolved part in close proximity to the nucleosome creates a picture of a flexible histone tail, with one end wrapped around the nucleosomal DNA, forming a hairpin like stucture which exposes the critical K36 and V35 residues to the PKMT (). This model is supported by a large number of positively charged residues in the H3 sequence, which could interact with the negatively charged phosphate sugar backbone (18 arginine, 13 lysine, 2 histidine vs. only 7 glutamate 4 aspartate residues in the H3 sequence (Uniprot P68431)). However, additional experimental and simulation work is needed to further investigate these processes.
Simulation analysis of the effect of disease mutations in PKMTs
NSD2
The NSD2 E1099K mutation has been found frequently in patients with acute lymphoblastic leukemia and other types of cancer (Jaffe et al. Citation2013; Oyer et al. Citation2014). NSD2 T1150A, has been found in mantle cell lymphoma along with the E1099K mutation (Beà et al. Citation2013). Multiple biochemical studies highlighted an aberrant high methylation activity of NSD enzymes bearing these mutations (Li W et al. Citation2021; Sato et al. Citation2021; Khella et al. Citation2023). Li and colleagues solved the cryo-EM structure of the NSD2 E1099K and T1150A mutants bound to a nucleosome (PDB 7CRO), showing a partial unwrapping of the nucleosomal DNA to accommodate NSD2 and the interaction between the AL and the nucleosome facilitating H3 binding (Li W et al. Citation2021). The positively charged lysine residue introduced in the E1099K mutant interacts with the sugar-phosphate backbone of the nucleosomal DNA, which is proposed to be a reason for the increased activity (). This model was supported by the finding that NSD2 exhibits weak and nonspecific lysine methylation activity on histone octamer substrates, whereas it strongly and specifically methylates H3K36 when nucleosomal substrates comprising the histone octamer with bound DNA are used (Li Y et al. Citation2009).
Figure 9. NSD2 cancer mutants E1099K and T1150A result in an increased methylation activity. A| Crystal structure of NSD2 E1099K, T1150A (sticks, red) complexed to a nucleosome binding the H3 tail (cartoon, cyan, PDB 7E8D (Sato et al. Citation2021)). E1099K binds the nucleosome DNA backbone (cartoon, orange), which was suggested to be the reason the increased activity. B-C| Mechanism proposed for the increased activity of T1150A and E1099K by remodeling the H-bonds around the AL (cartoon and sticks, rose) resulting in an easier uplifting of the AL. The placeholder residue C1181 is in an open position. B| H-bond network in NSD2 WT (PDB 5LSU (Tisi et al. Citation2016)). C| H-bond network in NSD2 T1150A, in which the contact with D1182 is lost and the AL is lifted (image created using simulation results as described in Sato et al. (Citation2021)). D| H-bond network in NSD2 E1099K where K1099 forms a novel salt bridge with D1125 which is accompanied by decreased salt bridge formation between D1123 and K1152, since D1123 is in a polar connection with K1124 (image created using simulation results as described in Sato et al. (Citation2021)).
An opposing model was presented by Sato and coworkers, who also solved the NSD2 E1099K and T1150A nucleosome bound structure (Sato et al. Citation2021). MD simulations of the binary NSD2 E1099K or T1150A – SAM complex showed that the AL adopts a more open position in the NSD2 mutant compared to the WT. Hence, E1099K and T1150A were suggested to destabilize the interactions that keep the autoinhibitory loop closed. Thereby, the authors explained the increased activity (). It needs to be noted that the starting conformation of this MD simulation had the AL in a conformation in which the placeholder residue C1183 was already turned outwards. As described earlier, steps before this state are necessary, like the interaction of a substrate with the AL. Still, E1099K/T1150A seemed to destabilize the AL in the closed conformation and support its opening.
Kinetic analysis of both groups revealed that NSD2 E1099K and T1150A have increased catalytic turnover, but no increased nucleosome affinity. A combination of the proposed mechanisms might be necessary to explain the increased activity of the NSD2 cancer mutant, since the destabilizing effect of E1099K and T1150A needs prior conformational changes induced by substrate interactions. These interactions could be enhanced by the improved K1099-DNA contact.
Besides the hyperactivity of NSD2 E1099K and T1150A, it was recently discovered that the product specificity of NSD1 (T2029A) and NSD2 (T1150A) cancer mutants is also altered and that they can trimethylate H3K36 on peptide and protein substrates instead of the mono- or dimethylating activity observed with the NSD1 and NSD2 wildtype enzymes (Khella et al. Citation2023). This phenomenon cannot be explained by the models presented above. To investigate this further, sMD simulations experiments were performed, which found that the binary NSD2-H3K36me2 peptide complex is unable to bind an additional SAM molecule and establish TS-like conformation () (Khella et al. Citation2023). Contrary, the NSD2 cancer mutant T1150A was able to do so (). To rationalize this behavior, MD simulations of the ternary NSD2-SAM-H3K36 peptide complex were conducted, which showed a significantly increased volume of the active site of NSD2 T1150A compared to the NSD2 wildtype (). The mechanistic reason for this was supposed to lie in the contacts established by T1150. An H-bond between the hydroxyl group of T1150 and the backbone amide of Y1092 orients Y1092 and restricts the volume of the active site pocket consequently disfavoring trimethylation. In the case of the T1150A mutant, the contact with Y1092 was much less frequently found, which supported trimethylation. In addition, the truncation of the T1150 side chain in the T1150A mutant creates space in the active site of the PKMT supporting higher product methylation levels. Finally, a hydrophobic contact between the T1150 side chain methyl group and the C-atoms of L1120 additionally restricted the volume in the active site (). Likewise, L1120M was already shown to enhance trimethylation activity in NSD2 (Sato et al. Citation2021), strengthening the important role of L1120 and its interaction with T1150 in the control of the product specificity of NSD2.
Figure 10. Besides the increasing the methylation activity, the T1150A cancer mutation causes a change in the product specificity of NSD2 from a di- to a trimethyltransferase. A-B| sMD simulations showed that the NSD2-H3K36me2 complex was not able bind SAM in a productive conformation, but the T1150A mutant-H3K36me2 complex was. Shown are representative snapshots from the sMD simulations, where the dashed line indicates the trajectory of the SAM molecule. C| Contacts (dashed red lines) established by T1150 (sticks, white) with Y1092 and L1120 (sticks, green) lead to a compact active site volume (gray spheres) restricting its activity to dimethylation. These contacts are missing for A1150 (sticks, red) resulting in a more spacious active site (blue spheres). Taken and modified from (Khella et al. Citation2023) with permission.
NSD1
Sotos syndrome is a childhood overgrowth syndrome associated with intellectual disability (Kurotaki et al. Citation2002; Douglas et al. Citation2003). More than 80% of patients with Sotos syndrome carry NSD1 mutations mainly in the AWS and SET domains (Tatton-Brown et al. Citation2005; Waggoner et al. Citation2005; Saugier-Veber et al. Citation2007). Biochemical experiments demonstrated that the R1914C, R2005Q and R1952W mutations caused a greatly reduced H3K36 methylation activity in vitro and the NSD1 mutants Y1971, R1984Q and R2017Q/L were catalytically inactive (Qiao et al. Citation2011; Khella et al. Citation2023). Structural analyses and MD simulations suggested that R2017 occupies a central position stabilizing the conformations of three aromatic residues via cation-π interactions, Y1870, Y1977, and F2018, the latter two of them are highly conserved residues in SET domain proteins (Qiao et al. Citation2011). Structural analysis of the NSD1 homologue NSD2 (PDB 7E8D) showed that NSD2 Y1092 (corresponding to NSD1 Y1971) directly contacts the target lysine analog H3K36M side chain together with two more aromatic residues (F1177 and Y1179). Hence, the Y1971C mutation in NSD1 disrupts this aromatic cage which may be needed for positioning and deprotonation of the target lysine explaining the loss of catalytic activity caused by this mutation (Khella et al. Citation2023). Adding to this, R1952, R1984, and R2005 are engaged in intramolecular interactions with several D and E residues. The observed Sotos mutation destroy these interactions and thereby they could lead to protein instability and reduced methylation activity (Qiao et al. Citation2011). In a pure modelling study, the NSD1 I2007F Sotos mutation was investigated by MD simulations showing conformational changes and loss of structural stability due to the bulkier F, which was speculated to lead to loss-of-function of the SET domain (Ha et al. Citation2016). However, a biochemical characterization of this mutant is still missing.
EZH2
Trimethylation of histone H3 on lysine 27 (H3K27me3) is a repressive posttranslational modification introduced by the SET domain-containing PKMT EZH2, which is a subunit of the Polycomb Repressive Complex 2 (PRC2) (Millán-Zambrano et al. Citation2022). Various Y641 mutations (Y641E, Y641F, Y641 N, Y641S, Y641C, and Y641H) in the SET domain of EZH2 have been identified as gain-of-function mutations that promote melanoma and B-cell lymphoma (Morin et al. Citation2010; Sneeringer et al. Citation2010). Kinetic studies showed that EZH2 prefers unmethylated H3K27 as a substrate and it was much less active on substrates with higher methylation states (H3K27me1 and H3K27me2). Conversely, EZH2 Y641 mutations catalyze the conversion of H3K27me2 to H3K27me3 rapidly, but show significantly lower methylation activity for H3K27me0 (Yap et al. Citation2011; McCabe et al. Citation2012; Fioravanti et al. Citation2018). Another EZH2 mutation, A677G, was identified in B-cell lymphoma and also showed increased levels of H3K27me3 and decreased H3K27me1 and H3K27me2 (McCabe et al. Citation2012). Modelling approaches of the SET domain of EZH2 , and its A677G and Y641 mutations uncovered that the mutations alter the lysine binding pocket (McCabe et al. Citation2012). Specifically, Y641 formed a hydrogen bond network with the ε-amino group of K27 and the backbone carbonyl of F665, which supports the interaction with the unmodified K27. This network was weakened by the Y641 mutations possibly causing the shift in substrate preference from unmethylated K27 to mono- and dimethylated K27. Moreover, Y641 mutations create additional space in the active site which further supports generation of higher methylation states as observed for the F/Y-switch position for other SET domain PKMTs. A similar mechanism was observed for A677G, where the missing alanine methyl group created a more spacious active site directly promoting the generation H3K27me2/3 (McCabe et al. Citation2012).
The EED protein is another essential subunit of the PRC2 complex and functions as a methyl lysine reader protein of the WD40 family (Cao Q et al. Citation2014). Through the binding of its aromatic cage to H3K27me3, EED functionally stimulates PRC2 activity. EED I363M is a loss-of-function mutation which has been identified in myeloid disorders such as myelodysplastic syndrome (Suh et al. Citation2019). The PRC2 activator protein JARID2 was found in structural and MD simulation studies to bind EED and EZH2 effectively stabilizing a helix in a region of EZH2, the so called stimulation-responsive motif (SRM, residues 143–153) adjacent to the catalytic SET domain (Justin et al. Citation2016; Lee et al. Citation2018). The SRM helix is supposed to bind to the EZH2 SET-I domain decreasing its occupancy of the substrate-binding channel, thus maintaining EZH2 in its catalytically active conformation (Suh et al. Citation2019). In MD simulations, it was observed that the interactions responsible for this activator function of JARID2 were decreased by the EED I363M mutation (Suh et al. Citation2019). Specifically, the salt bridges between JARID2 R115 and EZH2 D136 and D140 were less frequent in simulations with EED I363M, eventually leading to an unfolding of the SRM helix preventing the allosteric activation of EZH2, which then reduces EZH2 activity and suppresses the propagation of H3K27me3 repressive histone marks (Ueda et al. Citation2012; Suh et al. Citation2019).
Simulations of SMYD enzyme family members
The SMYD enzymes family comprises five PKMTs that are known to target histone and non-histone substrates (Spellmon, Holcomb, et al. Citation2015). All SMYD family members contain a catalytic SET domain with an inserted MYND domain, which is a zinc finger motif known to mediate protein − protein interactions with proline-rich sequences (Boriack-Sjodin and Swinger Citation2016). Crystal structures are known for three of the five family members: SMYD1 (Sirinupong et al. Citation2010), SMYD2 (Ferguson et al. Citation2011; Jiang et al. Citation2011; Wang et al. Citation2011), and SMYD3 (Foreman et al. Citation2011; Sirinupong et al. Citation2011; Xu, Wu, et al. Citation2011), but only for SMYD2 and SMYD3 structures with a bound peptide substrate are known. SMYD1–3 have been reported to methylate H3K4 which promotes active transcription (Hamamoto et al. Citation2004; Abu-Farha et al. Citation2008; Sirinupong et al. Citation2010). However, SMYD enzymes do not have an effect on global H3K4 methylation, but appear to impact selective promoter regions (Cock-Rada et al. Citation2012; Medjkane et al. Citation2012) and they apparently have important roles in the methylation of non-histone protein. SMYD enzymes share a bilobal, clamshell-like architecture. The N-terminal domain (NTD) contains the SET, SET-I, post-SET, and MYND domains, while the α-helical C-terminal domain (CTD) is positioned at the opposing site and shows similarities to TPR (tetratricopeptide repeats) domains despite contrasting sequences () (Spellmon, Holcomb, et al. Citation2015). The MYND domain interacts with the catalytic SET domain but does not participate in substrate or cofactor binding, as its removal does not affect methyltransferase activity (Sirinupong et al. Citation2010; Jiang et al. Citation2011; Sirinupong et al. Citation2011). Rather, it interacts with other proteins containing a similar proline-rich sequence (Liu et al. Citation2007; Abu-Farha et al. Citation2008; Abu-Farha et al. Citation2011) or its positively charged surface contributes to protein-DNA interactions (Hamamoto et al. Citation2004; Spellmon, Holcomb, et al. Citation2015). The SET domain can be divided in SET, SET-I, and post-SET domains, which all directly interact with the cofactor SAM and the protein substrates. Together the interface of the NTD and CTD forms a large, deep binding cleft for protein substrates. In all structures, conserved residues in the NTD form a narrow channel for the lysine substrate (Spellmon, Sun, et al. Citation2015). In the following sections, each member of the SMYD enzyme family will be discussed individually, since different, partially opposing, mechanistic details have been observed in MD simulations. Unfortunately, no structural data or simulation results have been reported for SMYD4.
Figure 11. A| Alignment of the different domains in SMYD enzymes. B| Cartoon representation of SMYD2 complexed with the 10 amino acid long peptide ERα in a U-shape conformation (PDB 4O6F (Jiang et al. Citation2014)). Different domains are colored as in the alignment. C| Cartoon representation of SMYD enzymes 1-5. The color code is as in the domain alignment: SAM is colored orange, and shown as sticks. SMYD1 (PDB 3N71 (Sirinupong et al. Citation2010)) is shown in an open CTD (pink) conformation. SMYD2 (PDB 3RIB (Xu, Zhong, et al. Citation2011)) is shown in an intermediate CTD (magenta) position. SMYD3 (PDB 3PDN (Sirinupong et al. Citation2011)) in a closed CTD (red) conformation. Black arrows indicate the directions the CTD is expected to move based on the starting conformation displayed. SMYD4 (AlphaFold model based on UniProt Q8IYR2) with the characteristic TPR domain (white). SMYD5 (AlphaFold model based on UniProt Q6GMV2) in which part of the CTD is replaced by a poly E-tail (red). D| Results of crystal structures of with SMYD3. Binding of SAM (orange, sticks) into its binding pocket in SMYD3 causes residues F183, S202 and Y239 (green, sticks) to have increased solvent accessible area (PDB 3RU0 (Foreman et al. Citation2011)). F183, S202 and Y239 are part of the target lysine channel. E| SMYD3 shows an increased activity towards the MAP3K2 peptide (cyan) compared to the VEGFR1 peptide (dark green, PDB 5HQ8 (Van Aller et al. Citation2016) and 5EX3 (Fu et al. Citation2016), modified). The molecular mechanism for this focuses on the -2 position of the peptide (in relation to the K260 or K831 target sites in MAP3K2 or VEGFR1, respectively), where MAP3K2 carries F258 (cyan, sticks) which creates a hydrophobic patch (blue circle) with SMYD3 residues (blue, sticks). This effect is reduced for VEGFR1, which has a L258 (dark green, sticks) at this position. Moreover, MAP3K2 T263 and Y264 (cyan, sticks) interact with SMYD3 D332 (red, sticks) causing the peptide to bend into a hairpin conformation while VEGFR1 contains G834 that does not form a contact with SMYD3.
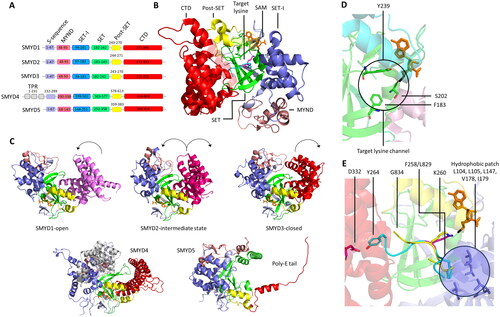
SMYD1
SMYD1 has the largest CTD among the SMYD enzymes and the most exposed substrate binding cleft, based on crystal structure results () (Spellmon, Sun, et al. Citation2015). The structure of the CTD is well conserved among SMYD enzymes, the only difference among them is an extended helix that resembles a ‘handle’ in the SMYD1 structure. The function of the unique SMYD1 C-terminal helix is unknown, but its conserved sequence and involvement in the crystal packing suggests that it may serve as a site for protein–protein interaction (Spellmon, Holcomb, et al. Citation2015). On the other hand, the CTD deletion in SMYD1 resulted in an increased binding and methylation of H3, suggesting that the CTD regulates substrate binding through steric effects (Sirinupong et al. Citation2010; Chandramouli and Chillemi Citation2016). Yet, this assumption is solely based on crystal structures with only the cofactor, but no substrate peptide or protein bound, because to this date no crystal structure of SMYD1 with bound substrate has been published. Homology modeling based on substrate bound SMYD2 or SMYD3 complexes could still give first insights into potential substrate binding modes, due to the high sequence and structural similarity (SMYD1 vs. SMYD2: 30.7% identity, RMSD (backbone atoms) = 3.55 Å based on SMYD1 PDB 3N71 and SMYD2 PDB 3RIB; SMYD1 vs. SMYD3: 31.4% identity, RMSD = 2.48 Å based on SMYD1 PDB 3N71 and SMYD3 PDB 3PDN). However, as seen for other SMYD family members discussed later, the dynamics when interacting with the protein substrate or SAM are crucial for their catalytic mechanism. Homology modeling as a stand-alone approach might therefore not be able to uncover SMYD1 specific mechanisms. Despite the lack of substrate bound structures, it was found that the artificial SMYD1 V214Y mutation leads to an increased binding of H3, potentially due to a tighter lysine channel and additional hydrogen bonds between Y214 and the target lysine (Sirinupong et al. Citation2010).
SMYD2
Structures of SMYD2 with bound P53 (PDB 3S7F) (Ferguson et al. Citation2011; Jiang et al. Citation2011) and ERα (PDB 4O6F (Jiang et al. Citation2014)) peptides have been determined. Both peptides adopt a similar U-shaped conformation in the active site (). Methylation of P53 K370 by SMYD2 reduces the binding efficiency of P53 to promoter genes, thereby repressing the P53 transcriptional activity (Huang et al. Citation2006; Chandramouli and Chillemi Citation2016). The opposite effect was observed when SET7/9 methylates P53 K372, causing an increase in stability and activity of P53 (Marouco et al. Citation2013). A fine tuning of the P53 transcriptional activity, therefore, is obtained via alternative methylation by SMYD2 on K370 and SET7/9 on K372. The mechanism of the interplay between the two lysine residues was unveiled by MD simulation. Methylation of K372 restricted the conformational freedom of the P53 peptide in the SMYD2 active site. This decreased the accessibility of K370 to SAM, effectively hampering SMYD2 methylation of P53 at K370 (Xu, Zhong, et al. Citation2011; Chandramouli et al. Citation2019).
The equilibrium of domain opening and closing can be denoted as ‘protein breathing’, which refers to the inherent dynamic conformational fluctuations within protein ensembles (Klimpel and Fleischman Citation1984; Makowski et al. Citation2008; Mariño Pérez et al. Citation2022). Protein breathing can have significant implications on the enzymatic function, especially in the context of substrate binding and product release. It can regulate the access of substrates to the active sites or contribute to allosteric regulation. Interestingly, SAM binding triggers increased flexibility of the CTD leading SMYD2 to adopt fully open conformations, which then completely expose the substrate binding crevice representing a mechanism to overcome potential autoinhibition (Al-Shar’i and Alnabulsi Citation2016; Chandramouli and Chillemi Citation2016). In the context of a proposed sequential reaction mechanism (Wu et al. Citation2011), SAM binds first and increases the CTD flexibility, thereby allowing the substrate binding site to open fully and facilitating substrate protein binding. The large and flexible substrate binding crevice of SMYD2 may explain its diverse substrate spectrum (Spellmon, Sun, et al. Citation2015; Chandramouli and Chillemi Citation2016). The potentially opening of the substrate binding cleft was further investigated by MD simulations with subsequent dynamical network analysis of SMYD2 with bound SAM but no protein substrate. These studies revealed that SMYD2 exhibits a negative correlation of its inter-lobe motion. This clamshell-like or hinge motion is caused by a twisting movement of the CTD with respect to the NTD () (Spellmon, Sun, et al. Citation2015; Al-Shar’i and Alnabulsi Citation2016). MD simulations of the ternary SMYD2-P53 peptide-SAM or -SAH complex showed that the degree of conformational freedom of the SMYD2 CTD decreases in the presence of the P53 peptide substrate (Chandramouli et al. Citation2019). After catalytic turnover, which is simulated by a replacement of SAM by SAH and using a monomethylated P53 K370 peptide, the CTD exhibited stronger anticorrelation movements with the NTD when compared to the system with unmethylated P53 (Chandramouli et al. Citation2019). This could indicate that the anticorrelated movement of the CTD and NTD is necessary to bind the substrate and to facilitate product release after the methylation reaction.
Biochemical and modeling experiments have addressed the substrate specificity of SMYD2 heading towards the discovery of unknown targets. Lanouette and coworkers used modeled SMYD2-peptide complexes to predict residues necessary for methylation (Lanouette et al. Citation2015). By using the solved SMYD2-P53 peptide structure (PDB 3S7F) as a template, they identified a
motif as the substrate recognition sequence. SPOT peptide array methylation was used to validate the predicted sequence motif, resulting in a
motif, which slightly diverges from the predicted motif. This model was later refined with leucine at the -1 position identified as the most important recognition residue, and aversion of acidic residues at the +1 to +3 site (Weirich et al. Citation2020). Still, the predictive power of the modeling approach was demonstrated, as based on the modeled sequence motif, 4 new SMYD2 substrates were identified (SIX1, SIX2, SIN3B, and DHX15). However, refinements of the method are necessary, because previously known SMYD2 substrates HSP90 and RB would not have been identified. So far, 19 non-histone proteins involved in various biological processes have been identified as cellular substrates of SMYD2 (Lanouette et al. Citation2015; Ahmed et al. Citation2016; Olsen et al. Citation2016) Moreover, by using the substrate specificity profile generated from SPOT peptide arrays, 14 additional protein substrates have been described, six of which were more strongly methylated than P53, the best SMYD2 substrate identified until this point (Weirich et al. Citation2020).
SMYD3
Surprisingly, in contrast to the increased flexibility of the SMYD2 CTD upon SAM binding, simulations of SMYD3 revealed that the CTD of SMYD3 became more rigid after SAM binding (Chandramouli et al. Citation2016; Sun et al. Citation2021). The restricted flexibility of the CTD caused a compaction of the substrate binding cleft, which might have implications on the substrate spectrum (Chandramouli et al. Citation2016). Compared to the large substrate spectrum of SMYD2 with 19 known non-histone targets, SMYD3 has been reported to primarily monomethylate H3K4, H4K5, and H4K20 and only 5 non-histone targets: Vascular Endothelial Growth Factor Receptor 1 (VEGFR1), MAP3 Kinase 2 (MAP3K2), Estrogen Receptor (ER), Human Epidermal Growth Factor Receptor 2 (HER2), and serine/threonine–protein kinase AKT1 (Mazur et al. Citation2014; Van Aller et al. Citation2016; Bottino et al. Citation2020). Hence, SAM binding seems to have opposite effects on the substrate recognition of SMYD2 and SMYD3. Whereas the relaxed CTD with bound SAM in SMYD2 leads to a broad substrate spectrum, the SMYD3 substrate spectrum is narrowed due to the more rigid conformation. However, kinetic experiments showed that the peptide substrates do not discriminate between apo-SMYD3 or binary SMYD3-SAM complexes (Fabini et al. Citation2019; Sun et al. Citation2021). Hence, a random ternary complex mechanism was proposed for SMYD3, in which peptide binding is more frequent before SAM binding (Fabini et al. Citation2019), which is different from the ordered mechanism proposed for SMYD2 as described above. Moreover, Chandramouli et al. performed MD simulations with SMYD3 alone and the binary SMYD3-SAM complex, which showed increased solvent access of residues at the target lysine channel (F183, S202, Y239) after SAM binding () (Chandramouli et al. Citation2016). These residues could potentially have roles in catalysis, since mutagenesis experiments showed that F183A and Y239A led to an abolishment of methylation activity (Xu, Wu, et al. Citation2011).
A combination of crystal structures, biochemical experiments and MD simulations was used to rationalize the substrate preference of SMYD3. In vitro methyltransferase assays showed that SMYD3 has a ∼14-fold higher activity towards the MAP3K2 peptide compared to the VEGFR1 peptide. Methylation of histone substrates (H3K4 and H4K5) was too weak to be detected (Fu et al. Citation2016). This preference on the one hand was shown to be based on the interaction of the F258 residue at the MAP3K2 -2 position (in relation to the K260 target lysine) (Fu et al. Citation2016) which is accommodated in a hydrophobic pocket of SMYD3 (L104, L105, L147, V178, I179) (). VEGFR1 possesses a leucine (L829) at this position, which showed a moderate decrease in binding free energy in 50 ns MD simulations compared to F258. This is speculated to be caused by the weaker hydrophobic interaction and the different shape. Substitution of MAPK3K2 F258 to each of the other 19 natural residues resulted in a decrease of binding free energy in 50 ns MD simulations for each variant (Sun et al. Citation2019). MAP3K2 F258 and VEGFR1 L829 are hydrophobic, whereas a polar arginine residue is found at the corresponding positions in the histone substrates H3K4 and H4K5, explaining the much lower catalytic activity of SMYD3 towards H3K4 and H4K5 (Van Aller et al. Citation2016). The SMYD3 preference for MAP3K2 could be furthermore based on the C-terminal end of the peptides. Structural comparison between the MAP3K2 and VEGFR1 showed that the peptides superimpose well, except at the C-terminal end (PDB 5HQ8 for SMYD3-MAP3K2, PDB 5EX3 for SMYD3-VEGFR1, ). There, the residue in VEGFR1 (G834) differs from the residue in MAP3K2 (Y264) (Fu et al. Citation2016). Y264 of MAP3K2 interacts with SMYD3 D332 which may increase activity, while G834 of VEGFR1 does not interact with D332 but adopts an extended peptide conformation.
SMYD5
The sequence alignment of SMYD1-5 showed, that SMYD5 lacks the TPR-like CTD, which is conserved in all other SMYD enzymes. In SMYD5 this appears to be compensated by a new subdomain, which occupies a position that overlaps with the top part of the CTD domain, thereby contributing to the formation of the substrate binding site. Since the CTD is involved in the formation of the substrate binding site, this structural difference suggests that SMYD5 might use a different mechanism in the recognition of its substrates than the other SMYD enzymes. Reported substrates of SMYD5 include H2K20 (Stender et al. Citation2012), H3K36 and K37 (Aljazi et al. Citation2022) and lysine residues in the HIV Tat protein (Boehm et al. Citation2023).
No crystal structure of SMYD5 has yet been published, nevertheless the protein structure prediction software AlphaFold has provided insights into the structural features of SMYD5, which was assessed by Zhang and coworkers (Zhang et al. Citation2022). The AlphaFold structures were validated by RMSD-based comparisons with other crystal structures and their deviation among each other. Additionally, a novel inter-residue distance map (IRDM)-based metric was developed to validate the predicted AlphaFold models. IRDM comparisons are superposition independent, because each residue is specified by its distance to all other residues and every residue is involved in specifying all other residues. A structural distance network is created precisely describing the overall architecture of the protein. It is claimed, that both metrices (classic RMSD and IRDM) suggest that the AlphaFold model of SMYD5 is reliable.
The overall structure of the AlphaFold SMYD5 model resembles a ‘crab with two large legs’ that are enriched with negatively charged residues (). The body of the crab is made up of four conserved domains, including the SET, MYND, post-SET, and SET-I domains, while the crab legs are formed by the structural features unique to SMYD5. For the legs, the thinner one is formed by the poly-E tract that forms a single helical structure, while the thicker one is formed by two large insertions into the MYND domain (M-insertion) and the SET domain (S-insertion). The region between the poly-E tail and the M- and S-insertions forms a negatively charged deep cleft, potentially binding positively charged protein substrates, which are known to interact with SMYD5 (Stender et al. Citation2012; Zhang et al. Citation2021). The C-terminal poly-glutamic acid tract (poly-E) and a 30-residue long M-insertion have been shown to regulate the structural stability SMYD5 (Zhang et al. Citation2021).
An additional N-terminal sequence is unique to SMYD5 and absent in other SMYD members. This sequence was predicted to be a mitochondrial targeting sequence, but is now claimed to function as a novel non-classical nuclear localization signal (NLS) (Zhang et al. Citation2022). Subcellular localization of SMYD5 was probed by using GFP as a reporter that was tagged to the C-terminus of the protein. It was found that intact SMYD5 is localized to the nucleus in all three cell lines (HEK293, U2OS, and RAW264.7) in an NTD dependent manner (Zhang et al. Citation2022).
Structural organization of MLL PKMTs
The Mixed lineage leukemia (MLL) family of PKMT enzymes, including MLL1-4 (aka KMT2A-2D), SET1A (aka KMT2F) and SET1B (aka KMT2G), represents the major histone H3K4 methyltransferases in mammals (Rao and Dou Citation2015). While monomethylation of H3K4 is located preferentially at active enhancers, trimethylation is a mark found at open and potentially active promoters. Thus, H3K4 methylation is typically associated with gene transcription (Bochyńska et al. Citation2018). Numerous studies were conducted to investigate the interaction between the MLL1/2 enzymes and menin because of its pronounced role in acute myeloid leukemia (AML). Since these studies focus on the development of inhibitors blocking the interaction site between the MLL and menin, they are not included in this review as explained earlier. However, we refer to other publications focusing on this topic (Zhou H et al. Citation2013; Yue et al. Citation2016; Xu et al. Citation2020; Perner et al. Citation2023).
The MLL-WRAD complex
The SET domains of MLL enzymes are slow or even inactive methyltransferases (Luo Citation2018). For their full catalytic activity, the association of binding partners is necessary: WDR5 (Tryptophan-aspartate repeat protein-5), RbBP5 (Retinoblastoma-binding protein-5), ASH2L (Absent, small or homeotic 2-like), and DPY30 (Dumpy-30), forming the so called WRAD complex (Ernst and Vakoc Citation2012). Cryo-EM structures of MLL1 SET-WRAD visualize how the 5 subunits interact and build a complex, capable of H3K4 methylation () (Hsu et al. Citation2019; Park et al. Citation2019; Xue et al. Citation2019; Ayoub et al. Citation2022). An example for the drastic difference between the activities of the MLL SET domain alone and MLL SET domain associated with the WRAD complex was shown for MLL1. Here, the activity towards the H3K4 peptide (residues 1-20) increased by 600-fold if the WRAD complex was present (Patel et al. Citation2009; Del Rizzo and Trievel Citation2011). A mechanism for this effect was proposed by Southall and coworkers, who solved the crystal structure of the MLL1 SET domain complexed with SAH and the H3K4me peptide (PDB 2W5Z). The MLL1 SET structure was then compared to the DIM-5 SET domain complexed with the H3K9 substrate peptide (PDB 1PEG (Southall et al. Citation2009)). It was observed that in DIM-5 the SET-I motif is oriented in close proximity to the Post-SET motif, forming a narrow, well-defined lysine binding channel that constrains the motion of the K9 side chain to promote its methylation. Contrary to this closed conformation, the SET-I motif of MLL1 is shifted away from the Post-SET motif, resulting in an open active site that is unable to optimally orient the K4 side chain for methylation (Southall et al. Citation2009).
Figure 12. Structural organization of the MLL1 SET-WRAD complex. A| Structure of the MLL1 SET domain (white) associated with WDR5 (green), RBBP5 (light blue), ASH2L (rose) and DPY30 (cyan) bound to a nucleosome core particle (NCP, purple) (PDB 6PWV). B| Distinct features of the interaction between RbBP5 and the nucleosome like the quad-R residues, A-loop, I-loop. C| The ASH2L IDR interacts with the NCP thereby facilitating the binding of the complex. D| MLL1 SET-WRAD binds to the NCP (grey) in two distinct binding modes. In the first binding mode, MLL1 SET (dark green) is positioned at the dyad barely interacting with the NCP (PDB 6PWV). Main interaction sites between MLL1 SET-WRAD complex and NCP are established by RbBP5 and ASH2L (WRAD colored green). In the second binding mode, RbBP5 (WRAD colored red) rotates clockwise and MLL1 SET (magenta) is positioned on top of the NCP disk, binding to the H3 histone tail (spheres, cyan, PDB 6KIX (Xue et al. Citation2019)). E| Ubiquitination (yellow) of H2BK120 leads to shift towards the second binding mode of the MLL1 SET (white)-WRAD complex (red, PDB 6KIU (Xue et al. Citation2019)). F| The yeast MLL1 homologue Set1 (white) binds an H2BK120 ubiquitinated NCP in the second binding mode (PDB 6UH5). Histone tail residues H3A1-H3R8 (cyan) are resolved and the target lysine K4 (pink) is correctly bound in the SET domain active site. The H3 tail reaches out from the NCP and is not resolved due to its high flexibility and dynamic behavior. Potentially, it wraps around the nucleosomal DNA to establish a stable binding in the Set1 SET domain.
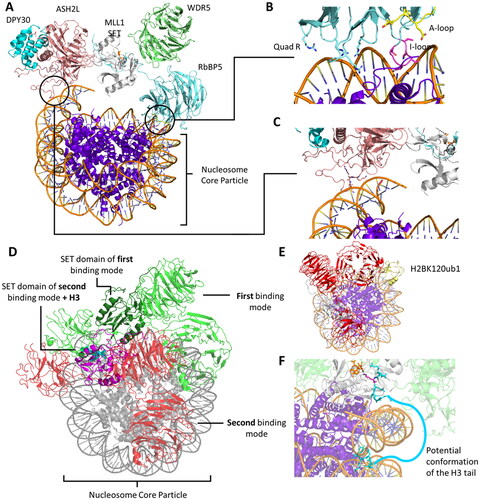
Based on this observation, it was hypothesized that the interaction of the WRAD subunits with the SET-I motif of MLL1 may induce the closure of the active site (Southall et al. Citation2009; Del Rizzo and Trievel Citation2011). Indeed, a combination of MD and QM/MM simulations later confirmed that the association of MLL3 SET with ASH2L and RbBP5 (MLL3 only needs theses subunits for activity (Kwon et al. Citation2020)) impacts the deprotonation and methyl group transfer via interactions with the SET-I motif and the SAM binding site (Miranda-Rojas et al. Citation2021). Salt bridges between MLL3 SET and RbBP5 (R4806-E347, R4812-D353, R4845-E338, R4864/K4867-E341 in PDB 5F6K) restricted the conformational space of two regions in the SET domain. The first region is the interaction network in the active site including Y4800, V4824, Y4825 and Y4884. These residues were shown to be particularly critical for lysine deprotonation (Miranda-Rojas et al. Citation2021). The second restricted conformational space was shown to affect the distance between α-helix B (P4843-Y4846) and the SAM binding site. Both interactions increased the accessibility of active conformations, in which shorter distances between the SAM methyl group and the K4 ε-amino group were observed (Miranda-Rojas et al. Citation2021). Consistent with the simulation results, the Q3867A and R3871A mutations diminished the activity of the MLL1 complex towards H3 peptides, but did not appreciably affect the activity of the isolated MLL1 catalytic domain itself (Southall et al. Citation2009; Del Rizzo and Trievel Citation2011). MLL1 residues Q3867 and R3871 are located in the SET-I domain and interact with RbBP5. Additionally, it was found that the somatic cancer mutants R3864C and R3841W, which interact with ASH2L and WDR5, are not stimulated by WRAD association (Weirich et al. Citation2017). This presumably leads to changes in the cellular MLL1 methylation activity, because the mutant enzymes have lost their activity control by the WRAD complex.
Of note, the WRAD complex with a catalytically inactive MLL1 SET mutant can by itself methylate H3K4 peptides. However, the reaction is slow and micromolar concentration of SAM, peptides and the complex were necessary to measure H3K4me1 (Patel et al. Citation2009; Patel et al. Citation2011; Patel et al. Citation2014; Shinsky et al. Citation2014). ASH2L was found to be the responsible subunit to catalyze this weak H3K4 methylation. Because ASH2L has no detectable homology with SET domain or 7BS methyltransferases, it is of great interest to explore the structural features of the cryptic PKMT active site in this enzyme, which can be informative to identify novel PKMTs (Luo Citation2018).
Interactions of the MLL-WRAD complex with nucleosomes
Besides the stimulation of the methylation activity of MLL enzymes upon association with the WRAD complex, MLL activity is further stimulated if nucleosome core particles (NCP) are used as substrates instead of the recombinant H3 protein (Park et al. Citation2019). NCPs consist of the histone protein octamer (H2A, H2B, H3, H4) with typically 147 bp of double stranded DNA wrapped around them. The increased methylation of the NCP substrate indicates that stimulatory interactions must occur between the NCP and the MLL SET-WRAD complex. In a variety of cryo-EM studies, it was found that the MLL1 SET-WRAD complex can bind the NCP in 2 distinct binding modes. In the first binding mode, MLL1 is positioned at the dyad of the NCP () (Park et al. Citation2019). In this conformation, RbBP5 was found to interact with the DNA through four positively charged arginine residues: R220, R251, R272, R294 (denoted as Quad-R), which made electrostatic interactions with the DNA phosphate backbone () (Park et al. Citation2019). In addition, RbBP5 interacts with the NCP in this binding mode with its insertion loop (I-loop, D236-E240) and the anchoring loop (A-loop, T193-T198) (Park et al. Citation2019). The I-loop was placed between the N-terminal tail of histone H4 and the nucleosomal DNA. The A-loop was positioned parallel to the H4 tail, which was placed between the I-/A-loops of RbBP5 and residues L65–D77 of histone H3 (). These interactions were biochemically validated in systematic mutational studies, where R-to-E mutations of the Quad-R residues led to a strong reduction in MLL1 SET methylation activity. Similarly, deletion of the I-loop, and to a lesser extent the A-loop, reduced the activity of the MLL1 SET-WRAD complex. To underline the role of these interactions in NCP methylation, the mutations in Quad-R, the I-loop, and the A-loop had no effects if the free H3 protein was used as substrate (Park et al. Citation2019).
The second major interaction between the MLL1 SET-WRAD complex and the NCP in the first binding mode was mediated by the intrinsically disordered region (IDR) of ASH2L (Park et al. Citation2019). Positively charged residues (K205/R206/K207) in the ASH2L IDR established contacts with the nucleosomal DNA (). Consequently, alanine mutations of K205/R206/K207 reduced the methylation activity of the MLL1 SET-WRAD complex towards the NCP.
In other cryo-EM structures, the MLL1 SET-WRAD complex was found to bind the NCP in a second binding mode (Xue et al. Citation2019; Ayoub et al. Citation2022). In this binding mode, the MLL1 SET-WRAD was not positioned at the NCP dyad, but diagonally across the nucleosome disc (). While ASH2L mostly retained its position, RbBP5 rotated clockwise. The rotation of RbB5 diminished its NCP interactions present in binding mode 1 (Quad-R, I-loop, A-loop). Instead, in this binding mode RbBP5 was found to interact with H2B through a highly conserved amphipathic loop containing L248, V249, N250, R251 (Ayoub et al. Citation2022). In contrast to binding mode 1, in which the MLL1 SET domain is placed above the nucleosome dyad without making significant contacts with the NCP, in binding mode 2 the MLL1 SET domains heavily interacts with the nucleosome disc face (Ayoub et al. Citation2022). In particular, hydrophobic interactions between MLL1 SET (M3812, L3814, M3818) and H2A (N73, L85, L108, P109) as well as a charged interaction (MLL1 SET R3821, H2A D72) were observed (Xue et al. Citation2019; Ayoub et al. Citation2022).
Surprisingly, the MLL1 SET-WRAD complex did not interact with the ‘acidic-patch’ region of the NCP in neither of the binding modes (Park et al. Citation2019; Kwon et al. Citation2020; Worden et al. Citation2020). The acidic patch is a negatively charged region found on the surface of the nucleosome, specifically on the H2A-H2B dimer interface comprinsing H2A: E56, E61, E64, D90, E91, and H2B: E92, E102, E110 (Kalashnikova et al. Citation2013; Park et al. Citation2019; Xue et al. Citation2019). Several other chromatin complexes were observed to engage the acidic-patch region of the NCP through variations of an arginine-finger motif (Barbera et al. Citation2006; Makde et al. Citation2010; Armache et al. Citation2011; Kato et al. Citation2013; Park et al. Citation2019).
Influence of H2B ubiquitination on MLL
The catalytic efficiency of the MLL1 SET-WRAD complex in terms of kcat/Km-values is stimulated 2-fold if the NCP substrate is mono-ubiquitinated on histone H2B lysine 120 (H2BK120ub1) (Xue et al. Citation2019). H2BK120ub1 is a prevalent histone mark that disrupts chromatin compaction and favors open chromatin structures (Kim et al. Citation2009; Fierz et al. Citation2011; Xue et al. Citation2019). Cyro-EM structures of the MLL1-WRAD complex with NCP containing H2BK120ub1 showed that the binding is very similar to binding mode 2 described in the previous chapter (). Besides contacts characteristic of binding mode 2 without H2BK120 monoubiquitination, the WD40 domain of RbBP5 (R228, T232, E238, E240) directly interacts with H2BK120ub1 residues (L8, T9, R42, Q49, H68, R72) (Xue et al. Citation2019). It was proposed that these additional contacts stabilize the complex and lead to an enhanced catalytic efficiency (Xue et al. Citation2019; Kwon et al. Citation2020).
In addition, a recent structural study on the yeast Set1 complex proposed another mechanism to explain how H2BK120ub1 stimulates H3K4 methylation (Hsu et al. Citation2019; Worden et al. Citation2020). In yeast, the human proteins WDR5/RbBP5/ASH2L/DPY30 correspond to Swd3/Swd1/Bre2/Sdc1 (Worden et al. Citation2020). In this complex, the arginine-rich RXXXRR motif of Set1 (R901, R904, R908, R909 in PDB 6VEN (Worden et al. Citation2020)) makes extensive contacts with the acidic patch of histones H2A and H2B. The RXXXRR motif is located on a helix, which directly interacts with ubiquitin regions E34-P37 and L71-G75. Interestingly, this helix is not resolved in cryo-EM structures of Set1 without H2bK120ub1 (PDB 6UGM (Hsu et al. Citation2019)). This could indicate, that H2bK120ub1 stabilizes this part of Set1, potentially leading to the increased activity. In agreement with this model, the RXXXRR motif was previously demonstrated to be critical for the H2Bub-dependent H3K4 methylation activity of the Set1 complex (Kim J et al. Citation2013). Additionally, alanine mutations in the RXXXRR motif resulted in a complete loss of the H3K4 methylation activities of SET1A/B, suggesting an evolutionary conserved process (Kwon et al. Citation2020). However, the RXXXRR motif is conserved only in human SET1A/B but not in MLL1/2/3/4, this mechanism could therefore be specific to SET1A and SET1B complexes (Kwon et al. Citation2020; Worden et al. Citation2020).
Catalytic role of MLL1 binding modes
As described above, the MLL1 SET-WRAD complex binds the NCP in 2 distinct binding modes. In the first binding mode, MLL1 SET is positioned at the NCP dyad anchored by ASH2L and RbBP5 and it does not directly interact with the NCP. In the second binding mode, RbBP5 rotates clockwise and MLL1 SET establishes contacts with the NCP. Multiple studies have been conducted trying to clarify the relation between these MLL1 SET binding modes and catalytic activities (Hsu et al. Citation2019; Xue et al. Citation2019; Worden et al. Citation2020; Ayoub et al. Citation2022; Rahman et al. Citation2022). Indeed, it is a challenging question to find out which binding mode resembles the catalytically ‘active’ conformation and arguments for both models have been provided which are presented shortly in the following paragraph.
As mentioned above, ubiquitination of H2BK120 stimulates the catalytic efficiency of MLL1 (Xue et al. Citation2019; Kwon et al. Citation2020). Cryo-EM structures of MLL1 SET-WRAD and yeast Set1-COMPASS complexed to a H2BK120ub1 NCP have a high similarity with the second binding mode, establishing similar interactions with the NCP, which could hint that the second binding mode is the catalytically active one. Surprisingly, it was found that Set1-COMPASS binds to unmodified nucleosomes and H2BK120ub1 nucleosomes with the same apparent affinity, indicating that the H2BK120ub1 interaction does not lead to an improvement in binding affinity (Worden et al. Citation2020). However, it needs to be noted, that COMPASS engages both the ubiquitinated and non-ubiquitinated nucleosomes in similar fashions, likely because of its pronounced interaction with the H2A/H2B acidic patch (Nakanishi et al. Citation2008; Worden et al. Citation2020). This could point towards a different binding mode of the COMPASS complex than of the human MLL complexes (except SET1A and SET1B).
Moreover, mutagenesis of binding mode 2 specific interactions was used to identify their role and importance in catalysis (Ayoub et al. Citation2022). Hydrophobic interactions between MLL1 SET and H2A are mediated by MLL1 SET A3806-L3814. Their respective alanine mutants reduced H3K4 methylation on the NCP (Ayoub et al. Citation2022). Likewise, mutating MLL1 SET M3812/L3814 to alanine also significantly reduced H3K4 methylation on the NCP (Ayoub et al. Citation2022). However, the described MLL1 SET domain mutants showed similarly reduced methyltransferase activity on recombinant H3, suggesting that they probably function by affecting the intrinsic activity of MLL1 SET instead of specifically disrupting MLL1-NCP interactions. Disruption of other binding mode 2 specific interactions between H2B and the RbbP5 amphipathic loop (L248, N249, N250, R251) by alanine mutations did not affect MLL1 activity on either the NCP or recombinant H3 (Ayoub et al. Citation2022), which may argue against the hypothesis that binding mode 2 is the catalytically relevant conformation.
In the second binding mode with and without H2BK120ub1, one of the flexible H3 tails was found to be resolved and positioned correctly in the MLL1 SET domain (PDB 6KIX without H2BK120ub, PDB 6KIU with H2BK120ub). This is also true for cryo-EM structures of the yeast homologue Set1-COMPASS in complex with H2BK120ub1 NCP (PDB 6VEN, 6UH5) but not for structures in binding mode 1. The second binding mode could therefore represent a state in which the H3 tail is bend around the nucleosomal DNA, binding the SET domain as a hairpin and forming contacts in a zipper-like fashion as described earlier ( and ). This is only possible, because the SET domain is not positioned at the dyad but leans toward one histone tail, suggesting that binding mode 2 is most relevant for activity.
In a working model, one could envision that the MLL1-SET-WRAD complex binds the NCP in the dyad position (binding mode 1) anchoring with RbBP5 and ASH2L. This is supported by the strong effect of the Quad-R deletion on activity. From this position, the complex could shift into the diagonal (binding mode 2) conformation and individually methylate the H3 tails at H3K4, which is suggested by the resolved K4 residues or analogues in the MLL1 SET domain. This conformation is further stabilized by H2BK120ub1 explaining the increased catalytic efficiency (Rahman et al. Citation2022). After methylation, the complex detaches or positions itself again in the dyad conformation, which is consistent with a distributive reaction mechanism (Patel et al. Citation2009). The dynamic switching between the dyad and diagonal conformation could enable locus specific regulation of H3K4 methylation by other chromatin binding proteins (Ayoub et al. Citation2022) like the transcription factor ELL or P-TEFB (Smith E et al. Citation2011) or the chromatin remodeler CHD8 (Subtil-Rodríguez et al. Citation2014). Previous studies have shown that H3K27me3 and H3K4me3 can be deposited asymmetrically at the same nucleosome, but on opposing H3 tails (Voigt et al. Citation2012). This asymmetric, bivalent modification of H3K27 and H3K4 is believed to be associated with maintaining promoters in a poised state during differentiation (Worden et al. Citation2020).
Product specificity of MLL1 and 3
MLL1 primarily acts as a di- and trimethyltransferase, whereas MLL3 is only capable of transferring a single methyl group to the H3K4 target (Shilatifard Citation2012; Rao and Dou Citation2015). Xue and coworkers structurally compared complexes of MLL1 SET-WRAD and MLL3 SET-WRAD bound to an NCP (PDB 6KIU for MLL1, PDB 6KIW for MLL3) to investigate the reason(s) for their differing product specificities (Xue et al. Citation2019). In the cryo-EM structure of MLL1, a hydrophobic patch consisting of MLL1 M3777, F3778, F3780, Y3883, F3904 and RbBP5 W329, F332, F336 directly interacts with the SAM binding pocket, which could facilitate SAM binding. The MLL1-RbBP5 interface is part of the RbBP5 ‘activation segment’ (W329-F336). The RbBP5 activation segment has been previously shown to be a stimulator of MLL activity (Li Y et al. Citation2016). Likewise, deletion of the activation segments led to a 75% loss in activity of MLL1. In contrast, the described hydrophobic patch is weakened in MLL3, since the involved phenylalanine-residues in MLL1 are replaced by tyrosine in MLL3. This could lead to a reduced SAM binding especially at higher methylation stages.
As described earlier for other SET domain-containing PKMTs, tyrosine residues in the active site can modulate the product specificity. Y4884C is a somatic cancer mutation in MLL3. The Y4884 residue is part of an aromatic pocket at the active center of the enzyme. Biochemical data showed that Y4884C has a reduced methylation activity towards recombinant H3 protein and converts MLL3 from a monomethyltransferase with substrate preference for unmethylated H3K4 to a trimethyltransferase with H3K4me1 as preferred substrate (Weirich et al. Citation2015). Moreover, expression of Y4884C has been shown to lead to aberrant H3K4me3 formation in cells (Weirich et al. Citation2015). Adding to this, QM/MM simulation of MLL3 demonstrated that Y4884A is able to di- and trimethylate its target (Blanco-Esperguez et al. Citation2022). However, the phenylalanine mutant Y4884F is inactive, which is also true for Y4800A/F, a tyrosine similarly located in the active site. Like for other SET domain-containing PKMTs, the additional space created by the Y4884A mutant allowed for the accommodation of additional methyl groups, which could explain the altered product specificity of the somatic cancer mutation Y4884C. However, additional biochemical experiments need to confirm the change of product specificity observed with Y4884A in the simulations, although the agreement between biochemical data obtained with Y4884C and simulation data for Y4884A is quite compelling.
Simulations of 7BS PKMTs
Non-SET domain PKMTs belong to the Rossmann-fold-like 7BS family (or class I methyltransferases) with their structural topology featuring a seven-stranded β-sheet connected by α-helices (). Besides proteins, small molecules, DNA and RNA are among the substrates of this group of methyltransferases (Luo Citation2018). SET domain-containing PKMTs and the 7BS methyltransferases use different architectures for their catalytic domain, and little is known about the methyl group transfer mechanism in 7BS PKMTs regarding conformational changes, TS stabilization, substrate- or product specificities. Nonetheless, information from crystal or cryo-EM structures and MD simulations shed first light on the processes involved in catalysis. 7BS PKMTs share structural similarities with PRMTs, which also belong to class I methyltransferases (Richon et al. Citation2011). PRMTs have been investigated in multiple MD simulation studies (Zhang R et al. Citation2013; Zhou R et al. Citation2015; Gathiaka et al. Citation2016). Mechanisms discovered in PRMT, especially conformational changes, could potentially hint towards similar mechanisms in 7BS domain PKMT, given a high sequence and structural similarity, but this conjecture needs to be investigated more closely in the future.
DOT1L
The full-length DOT1L (disruptor of telomeric silencing 1-like) protein transfers up to three methyl groups to H3K79 on nucleosomes marked by H2BK120ub1 (Chandrasekharan et al. Citation2010). DOT1L represents the most intensively studied 7BS PKMT, because of its important role in different forms leukemia (McLean et al. Citation2014; Spangler et al. Citation2022; Yi and Ge Citation2022). AF10 (aka MLLT10) can bind to DOT1L and alter the specificity of DOT1L toward the dimethylated product. Product specificity is thereby regulated through binding partners potentially causing allosteric effects (Ibáñez et al. Citation2010). Notably, fusion proteins can cause leukemia by using their AF region to recruit DOT1L to MLL-targeted genes leading to inappropriate activation of these genes in the affected cells (Feng et al. Citation2002; Sarno et al. Citation2020). MD simulations of the DOT1L-AF10 (PDB 6JN2) complex described the interplay as a coiled-coiled structure and found multiple residues responsible for the interaction of both proteins (Stodola et al. Citation2021). Because of the frequent association with leukemia, multiple studies involving molecular docking and virtual screening approaches have been conducted to develop inhibitors for DOT1L (Yao et al. Citation2011; Basavapathruni et al. Citation2012; Raj et al. Citation2015; Boriack-Sjodin and Swinger Citation2016; Chen et al. Citation2016; Luo et al. Citation2016; Luo Citation2018; Chen and Park Citation2019; Khirsariya et al. Citation2022; Flores-León et al. Citation2023). The presented results show valuable information about conformational changes and altered activities of DOT1L. However, they describe the interaction of artificial molecules with DOT1L and are therefore not included in this review.
Similar to SET domain PKMTs, SAM and the lysine substrate bind at distinct pockets in the active site of the DOT1L enzyme. However, crystal structures of DOT1L already showed differences between SET and 7BS PKMTs in the binding of the SAM cofactor. SET domain PKMTs bind SAM in a conformation, in which the SAM 2’,3’- hydroxyl moieties are solvent exposed and do not engage in essential interactions with protein residues (Luo Citation2018). The comparable 2’,3’ -hydroxyl moieties in DOT1L are deeply embedded in the cofactor-binding pocket () (Luo Citation2018). Molecular dynamics simulations combined with mutagenesis studies showed that especially DOT1L residues D161, G163, G165, E186, F223, and N241 are involved in SAM binding (Stodola et al. Citation2021). The SAM cofactor in SET domain-containing PKMTs is oriented in a relatively packed configuration with the C-C
-S
-C5’ dihedral angle around 120° (e.g. 114° for SET7/9 and 131° for SET8) (Fang et al. Citation2002; Wilson et al. Citation2002; Kwon et al. Citation2003; Xiao et al. Citation2003; Couture et al. Citation2005; Couture et al. Citation2006). In contrast, the SAM cofactor in DOT1L adopts an extended conformation with the C
-C
-S
-C5’ dihedral angle around 180° () (Min et al. Citation2003; Luo Citation2018). The described conformation of SAM in DOT1L overlays well with SAM conformation in PRMTs (Richon et al. Citation2011) and other 7BS methyltransferases (Falnes et al. Citation2016).
Figure 13. Different positioning of SAM in the cofactor binding sites of SET domain and 7BS PKMTs. A| In SET domain-containing PKMTs, SAM is positioned in a bent information, with the sugar ring hydroxyl groups solvent exposed (PDB 3F9W (Couture et al. Citation2008)). B| In the 7BS PKMT DOT1L, SAM is positioned in an extended conformation, with the nicotine amid ring solvent exposed (PDB 1NW3). C| DOT1L requires interaction with H2BK120ub1 (yellow) and the H4 tail (purple) for full catalytic efficiency (PDB 6NQA (Worden et al. Citation2019)). Shown are the residues of DOT1L (green, sticks) interacting with the ubiquitin (yellow, sticks), as well as the residues of DOT1L (white and magenta, stick) interacting with the H4 tail (purple, sticks). The H3 protein with the target K79 is shown as sticks in cyan, whereas the other histone proteins are colored purple.
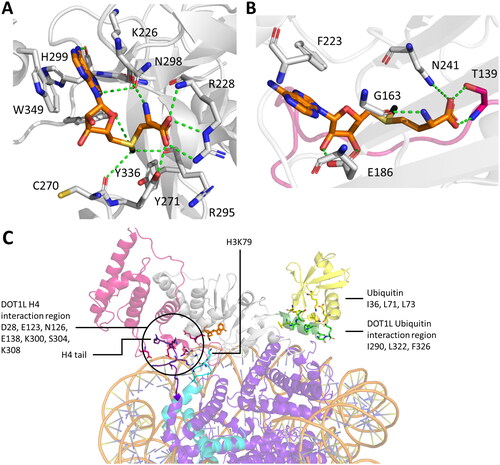
Another difference is found at the target lysine channel, where SET domain PKMTs show well-tuned hydrophobic tunnels to orient the ε-amino group. DOT1L also uses a SN2 reaction mechanism for the methyl group transfer. Yet, no similar aromatic-rich pocket could be mapped for class I PKMTs such as DOT1L. These PKMTs therefore likely adopt different strategies to engage their substrates for catalysis (Min et al. Citation2003; Luo Citation2018). This is especially important regarding the mechanistic requirement for deprotonation of the target lysine. Residues in DOT1L located close to the target lysine are incapable of performing this deprotonation and there is no evidence suggesting the formation of a water channel similar to the one in SET-containing PKMTs (Luo Citation2018). Alternative explanations proposed that the more hydrophobic active site reduces the pKa of the target lysine and that the carboxylate of the SAM plays a role in the lysine deprotonation (Min et al. Citation2003; Cheng et al. Citation2005; Cortopassi et al. Citation2016).
DOT1L methylates H3K79, but biochemical studies showed that histone H3 alone is a poor substrate for DOT1L, suggesting that DOT1L requires activators for full catalytic efficiency (Feng et al. Citation2002; Lacoste et al. Citation2002; Ng, Feng, et al. Citation2002). Cellular and in vitro experiments uncovered that efficient methylation of H3K79 in nucleosomal substrates is only achieved if H2BK120 is ubiquitinated (Briggs et al. Citation2002; Ng, Xu, et al. Citation2002; Kim et al. Citation2005; Valencia-Sánchez et al. Citation2019). However, electrophoretic mobility shift assays and structures of H2BK120ub1 and unmodified nucleosomes suggested, that the binding affinity of DOT1L is not altered by the ubiquitination (Valencia-Sánchez et al. Citation2019). One explanation for the DOT1L activity enhancement by H2BK120ub1 could be the direct interaction of DOT1L with ubiquitin as observed in cryo-EM structures (e.g. PDB 6NQA, 6NN6), where a hydrophobic patch consisting of DOT1L I290, L322 and F326 interacts with ubiquitin I36, L71 and L73 () (Anderson et al. Citation2019; Valencia-Sánchez et al. Citation2019). Similar observations were made for the interaction of MLL1 with H2BK120ub1 as described earlier. Biochemical studies found that disruption of these contacts by I290D, L322D and F326A mutations in DOT1L heavily decreased the in vitro methylation activity of DOT1L (Valencia-Sánchez et al. Citation2019). MD simulations found additional polar interaction between DOT1L residues E323, K330 and ubiquitin (Stodola et al. Citation2021). Previously reported comprehensive mutagenesis studies of the ubiquitin surface identified L71 and L73 as necessary for DOT1L activation (Holt et al. Citation2015). Together these results show that direct interactions between DOT1L and ubiquitin are necessary for DOT1L to deploy its full catalytic potential. However, the exact mechanisms on how these interactions increase the formation of TS conformations remain unclear. Likely conformational changes are most relevant, because the binding affinity is not influenced.
An additional non-catalytic effect of the DOT1L-H2BK120ub1 crosstalk describes the destabilization of the nucleosome. Cryo-EM structures showed a detached DNA from the histone octamer with the degree of the detachment greater for the complex of DOT1L-H2BK120ub1 compared to DOT1L-H2B unmodified. Interestingly, in the structure without H2BK120ub1, the distance between the H3K79 and SAM was 25 Å, whereas it was only 20 Å in the structure with H2BK120ub. While this is still too far for a methylation reaction, it could indicate that H2BK120ub1 changes the binding mode between DOT1L and the nucleosome into a more productive conformation (Jang et al. Citation2019). Single-molecule FRET experiments additionally showed that DOT1L binding to nucleosomes with H2BK120ub1 destabilized the nucleosome structure, potentially leading to conformational changes facilitating methylation of H3K79 (Jang et al. Citation2019).
Besides the interaction with ubiquitin, DOT1L was observed to interact with the basic residues of the H4 tail via D28, E123, N126, E138, K300, S304 and K308 (Valencia-Sánchez et al. Citation2019; Stodola et al. Citation2021). However, the DOT1L-H4 tail interaction surface was relatively small, suggesting that the H4 tail binding might only be transiently facilitating a rapid association and dissociation from the nucleosome (Stodola et al. Citation2021). This model is supported by the observation that not all cryo-EM structures captured DOT1L-H4 tail interactions (Anderson et al. Citation2019). Related to this, DOT1L was shown to be allosterically stimulated by acetylation of H4K16 (Valencia-Sánchez et al. Citation2021). Cryo-EM structures of DOT1L bound to a nucleosome containing H4K16ac showed that the enzyme was bound predominantly in the catalytic conformation while different conformations were observed on a nucleosome lacking H4K16ac (Valencia-Sánchez et al. Citation2021). Based on this it was proposed that acetylation of the H4 tail restricts the conformational sampling space of Dot1L, resulting in a more active complex conformation and increased catalytic activity (Valencia-Sánchez et al. Citation2021). An additional anchor point between DOT1L and the nucleosome is the acidic patch, where DOT1L R278, N280, R282 establish polar contacts with H2B Q47, E113 and H2A E56, E64, N68 (Anderson et al. Citation2019). Hence, multiple interactions between DOT1L and the NCP regulate the methylation activity of DOT1L on H3K79 in a concerted manner.
Experiments with other 7BS PKMTs
Besides DOT1L, several other 7BS PKMTs have demonstrated lysine methylation activities, including METTL10, METTL13, METTL20, METTL21A, METTL21B, METTL21C, METTL21D, METTL22, eEF1A-KMT1, eEF2-KMT, and CaM-KMT (Luo Citation2018; Falnes et al. Citation2023), but so far they were not subjected to MD simulation experiments. However, recently a novel 7BS methyltransferase, denoted Rv2067c, was discovered in Mycobacterium tuberculosis. Rv2067c was found to be secreted into the host macrophages, trimethylating host H3K79 in a non-nucleosomal context (Singh et al. Citation2023). The crystal structure of Rv2067c was solved, revealing that Rv2067c and DOT1L markedly differ in their amino acid sequence, 3D structure, domain composition, architecture, and oligomeric state, as Rv2067c was present as a dimer (Singh et al. Citation2023). Interestingly, in vitro methylation experiments showed that Rv2067c is more active on histone protein substrates compared to nucleosomal substrates, contrary to DOT1L, which is only active on nucleosomal substrates as described above (Singh et al. Citation2023). This difference was rationalized by MD simulations using the solved crystal structure of Rv2067c (Singh et al. Citation2023). Firstly, while the active site of DOT1L is a shallow groove, large enough to easily accommodate SAM and H3K79 on the surface of the large NCP, Rv2067c binds the peptide substrate deeply inside the protein. According to MD simulations and volume calculations, the active site of Rv2067c is able to accommodate the free H3 peptide or the corresponding part of the H3 protein, but not more bulky substrates like the NCP (Singh et al. Citation2023). Secondly, DOT1L engages the H2BK120Ub1 nucleosome multivalently as described earlier but all structural elements involved in these contacts are missing in Rv2067c. The inability of Rv2067c to interact with nucleosomes was further demonstrated in docking experiments attempting to dock Rv2067c onto a NCP from various directions. This rotational scan revealed a minimal number of 211 atoms involved in atomic clashes even in the most favorable Rv2067c-NCP complex model. Moreover, in the corresponding complex, the NCP approaches the Rv2067c active site from a wrong direction, the SAM entry site instead of substrate-binding site, which anyway would not be compatible with catalytic activity.
Finally, the human methyltransferase-like protein 13 (METTL13, aka eEF1A-KNMT and FEAT) is relevant for this review, because of its unique feature of having a double substrate specificity. METTL13 was found to target the α-amino group of the N-terminus (Nt) of proteins and the side chain ε-amino group of K55 in the eukaryotic translation elongation factor 1 alpha (eEF1A) (Jakobsson Citation2021b). This dual reactivity is based on the presence of two independent 7BS domains, one at the N-terminal part (MT13-N) and one at the C-terminal part (MT13-C). In vitro experiments with purified MT13-N and MT13-C domains have demonstrated that MT13-N is responsible for dimethylation of K55 (Jakobsson et al. Citation2018; Liu et al. Citation2019) and MT13-C is responsible for trimethylation of the protein Nt (Jakobsson et al. Citation2018). Methylation of a lysine side chain and the protein Nt are biochemically similar, both occurring on primary amino groups. However, the protein Nt has a pKa close to physiological pH whereas a lysine side chain typically has a pKa above 10 (Grimsley et al. Citation2009). Consequently, the Nt is chemically more reactive under physiological conditions suggesting some mechanistic differences in both reactions which still need to be investigated in more details. Moreover, the functional and biological relevance of combining two active 7BS domains with different specificities in one enzyme in the case of METTL13 needs further investigation.
Conclusions
Protein lysine methyltransferases have essential regulatory roles in biology. Understanding their cellular functions critically depends on understanding the underlying mechanistic principles of their catalytic machinery. This includes substrate and product specificity, processivity, autoinhibition and other regulatory principles including the formation of complexes with other proteins. Major achievements have been made in understanding these prosses, facilitated by a combination of biochemical, structural and molecular simulation studies (MD and QM/MM). As described in many examples in this review, simulation science benefits from an increasing amount of structural data and computational power. Conversely, biochemists can use simulation predictions to design new experiments leading to a powerful synergy. Moreover, simulations can allow to identify dynamic conformations at an atomic resolution which is not possible by any other technology. The resulting interdisciplinary approach considers multiple scales, conditions and is capable of precisely describing how PKMTs transfer methyl groups to their targets and thereby influence the cellular metabolism. This methodological interplay provides a promising avenue for the mechanistic understanding of PKMTs and other families of related enzymes.
Author contributions
P.S. and A.J. prepared the draft manuscript. P.S. prepared the figures. All authors contributed to the final manuscript preparation. All authors approved the final version of the manuscript.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Abu-Farha M, Lambert JP, Al-Madhoun AS, Elisma F, Skerjanc IS, Figeys D. 2008. The tale of two domains: proteomics and genomics analysis of SMYD2, a new histone methyltransferase. Mol Cell Proteomics. 7(3):560–572. doi: 10.1074/mcp.M700271-MCP200.
- Abu-Farha M, Lanouette S, Elisma F, Tremblay V, Butson J, Figeys D, Couture JF. 2011. Proteomic analyses of the SMYD family interactomes identify HSP90 as a novel target for SMYD2. J Mol Cell Biol. 3(5):301–308. doi: 10.1093/jmcb/mjr025.
- Ahmed H, Duan S, Arrowsmith CH, Barsyte-Lovejoy D, Schapira M. 2016. An Integrative Proteomic Approach Identifies Novel Cellular SMYD2 Substrates. J Proteome Res. 15(6):2052–2059. doi: 10.1021/acs.jproteome.6b00220.
- Al-Shar’i NA, Alnabulsi SM. 2016. Explaining the autoinhibition of the SMYD enzyme family: A theoretical study. J Mol Graph Model. 68:147–157. doi: 10.1016/j.jmgm.2016.07.001.
- Al Temimi AHK, Reddy YV, White PB, Guo H, Qian P, Mecinović J. 2017. Lysine Possesses the Optimal Chain Length for Histone Lysine Methyltransferase Catalysis. Sci Rep. 7(1):16148. doi: 10.1038/s41598-017-16128-4.
- Al Temimi AHK, Tran V, Teeuwen RS, Altunc AJ, Amatdjais-Groenen HIV, White PB, Lenstra DC, Proietti G, Wang Y, Wegert A, et al. 2020. Examining sterically demanding lysine analogs for histone lysine methyltransferase catalysis. Sci Rep. 10(1):3671. doi: 10.1038/s41598-020-60337-3.
- Al Temimi AHK, Amatdjais-Groenen HIV, Reddy YV, Blaauw RH, Guo H, Qian P, Mecinović J. 2019a. The nucleophilic amino group of lysine is central for histone lysine methyltransferase catalysis. Commun Chem. 2(1) doi: 10.1038/s42004-019-0210-8.
- Al Temimi AHK, van der Wekken-de Bruijne R, Proietti G, Guo H, Qian P, Mecinović J. 2019b. Gamma-Thialysine versus Lysine: An Insight into the Epigenetic Methylation of Histones. Bioconjug Chem. 30(6):1798–1804. doi: 10.1021/acs.bioconjchem.9b00313.
- Aljazi MB, Gao Y, Wu Y, He J. 2022. SMYD5 is a histone H3-specific methyltransferase mediating mono-methylation of histone H3 lysine 36 and 37. Biochem Biophys Res Commun. 599:142–147. doi: 10.1016/j.bbrc.2022.02.043.
- Allis CD, Jenuwein T. 2016. The molecular hallmarks of epigenetic control. Nat Rev Genet. 17(8):487–500. doi: 10.1038/nrg.2016.59.
- An S, Yeo KJ, Jeon YH, Song JJ. 2011. Crystal structure of the human histone methyltransferase ASH1L catalytic domain and its implications for the regulatory mechanism. J Biol Chem. 286(10):8369–8374. doi: 10.1074/jbc.M110.203380.
- Anderson CJ, Baird MR, Hsu A, Barbour EH, Koyama Y, Borgnia MJ, McGinty RK. 2019. Structural Basis for Recognition of Ubiquitylated Nucleosome by Dot1L Methyltransferase. Cell Rep. 26(7):1681–1690.e5. e1685. doi: 10.1016/j.celrep.2019.01.058.
- Armache KJ, Garlick JD, Canzio D, Narlikar GJ, Kingston RE. 2011. Structural basis of silencing: Sir3 BAH domain in complex with a nucleosome at 3.0 Å resolution. Science. 334(6058):977–982. doi: 10.1126/science.1210915.
- Ayoub A, Park SH, Lee YT, Cho US, Dou Y. 2022. Regulation of MLL1 Methyltransferase Activity in Two Distinct Nucleosome Binding Modes. Biochemistry. 61(1):1–9. doi: 10.1021/acs.biochem.1c00603.
- Bai Q, Shen Y, Yao X, Wang F, Du Y, Wang Q, Jin N, Hai J, Hu T, Yang J. 2011. Modeling a new water channel that allows SET9 to dimethylate p53. PLoS One. 6(5):e19856. doi: 10.1371/journal.pone.0019856.
- Bannister AJ, Kouzarides T. 2011. Regulation of chromatin by histone modifications. Cell Res. 21(3):381–395. doi: 10.1038/cr.2011.22.
- Barbera AJ, Chodaparambil JV, Kelley-Clarke B, Joukov V, Walter JC, Luger K, Kaye KM. 2006. The nucleosomal surface as a docking station for Kaposi’s sarcoma herpesvirus LANA. Science. 311(5762):856–861. doi: 10.1126/science.1120541.
- Basavapathruni A, Jin L, Daigle SR, Majer CRA, Therkelsen CA, Wigle TJ, Kuntz KW, Chesworth R, Pollock RM, Scott MP, et al. 2012. Conformational adaptation drives potent, selective and durable inhibition of the human protein methyltransferase DOT1L. Chem Biol Drug Des. 80(6):971–980. doi: 10.1111/cbdd.12050.
- Basta J, Rauchman M. 2015. The nucleosome remodeling and deacetylase complex in development and disease. Transl Res. 165(1):36–47. doi: 10.1016/j.trsl.2014.05.003.
- Beà S, Valdés-Mas R, Navarro A, Salaverria I, Martín-Garcia D, Jares P, Giné E, Pinyol M, Royo C, Nadeu F, et al. 2013. Landscape of somatic mutations and clonal evolution in mantle cell lymphoma. Proc Natl Acad Sci U S A. 110(45):18250–18255. doi: 10.1073/pnas.1314608110.
- Bennett RL, Swaroop A, Troche C, Licht JD. 2017. The Role of Nuclear Receptor-Binding SET Domain Family Histone Lysine Methyltransferases in Cancer. Cold Spring Harb Perspect Med. 7(6):a026708. doi: 10.1101/cshperspect.a026708.
- Bhat KP, Ümit Kaniskan H, Jin J, Gozani O. 2021. Epigenetics and beyond: targeting writers of protein lysine methylation to treat disease. Nat Rev Drug Discov. 20(4):265–286. doi: 10.1038/s41573-020-00108-x.
- Biggar KK, Li SSC. 2015. Non-histone protein methylation as a regulator of cellular signalling and function. Nat Rev Mol Cell Biol. 16(1):5–17. doi: 10.1038/nrm3915.
- Blanco-Esperguez K, Tunon I, Kastner J, Mendizabal F, Miranda-Rojas S. 2022. Unraveling the Role of the Tyrosine Tetrad from the Binding Site of the Epigenetic Writer MLL3 in the Catalytic Mechanism and Methylation Multiplicity. Int J Mol Sci. 23(18)
- Bochyńska A, Lüscher-Firzlaff J, Lüscher B. 2018. Modes of Interaction of KMT2 Histone H3 Lysine 4 Methyltransferase/COMPASS Complexes with Chromatin. Cells. 7(3):17. doi: 10.3390/cells7030017.
- Bock I, Kudithipudi S, Tamas R, Kungulovski G, Dhayalan A, Jeltsch A. 2011. Application of Celluspots peptide arrays for the analysis of the binding specificity of epigenetic reading domains to modified histone tails. BMC Biochem. 12(1):48. doi: 10.1186/1471-2091-12-48.
- Boehm D, Lam V, Schnolzer M, Ott M. 2023. The lysine methyltransferase SMYD5 amplifies HIV-1 transcription and is post-transcriptionally upregulated by Tat and USP11. Cell Rep. 42(3):112234. doi: 10.1016/j.celrep.2023.112234.
- Boriack-Sjodin PA, Swinger KK. 2016. Protein Methyltransferases: A Distinct, Diverse, and Dynamic Family of Enzymes. Biochemistry. 55(11):1557–1569. doi: 10.1021/acs.biochem.5b01129.
- Borkin D, He S, Miao H, Kempinska K, Pollock J, Chase J, Purohit T, Malik B, Zhao T, Wang J, et al. 2015. Pharmacologic inhibition of the Menin-MLL interaction blocks progression of MLL leukemia in vivo. Cancer Cell. 27(4):589–602. doi: 10.1016/j.ccell.2015.02.016.
- Bottino C, Peserico A, Simone C, Caretti G. 2020. SMYD3: An Oncogenic Driver Targeting Epigenetic Regulation and Signaling Pathways. Cancers (Basel). 12(1):142. doi: 10.3390/cancers12010142.
- Bramley GA, Beynon OT, Stishenko PV, Logsdail AJ. 2023. The application of QM/MM simulations in heterogeneous catalysis. Phys Chem Chem Phys. 25(9):6562–6585. doi: 10.1039/d2cp04537k.
- Briggs SD, Xiao T, Sun ZW, Caldwell JA, Shabanowitz J, Hunt DF, Allis CD, Strahl BD. 2002. Gene silencing: trans-histone regulatory pathway in chromatin. Nature. 418(6897):498–498. doi: 10.1038/nature00970.
- Bussi G, Laio A. 2020. Using metadynamics to explore complex free-energy landscapes. Nat Rev Phys. 2(4):200–212. doi: 10.1038/s42254-020-0153-0.
- Cao F, Townsend EC, Karatas H, Xu J, Li L, Lee S, Liu L, Chen Y, Ouillette P, Zhu J, et al. 2014. Targeting MLL1 H3K4 methyltransferase activity in mixed-lineage leukemia. Mol Cell. 53(2):247–261. doi: 10.1016/j.molcel.2013.12.001.
- Cao Q, Wang X, Zhao M, Yang R, Malik R, Qiao Y, Poliakov A, Yocum AK, Li Y, Chen W, et al. 2014. The central role of EED in the orchestration of polycomb group complexes. Nat Commun. 5(1):3127. doi: 10.1038/ncomms4127.
- Chandramouli B, Chillemi G. 2016. Conformational Dynamics of Lysine Methyltransferase Smyd2. Insights into the Different Substrate Crevice Characteristics of Smyd2 and Smyd3. J Chem Inf Model. 56(12):2467–2475. doi: 10.1021/acs.jcim.6b00652.
- Chandramouli B, Melino G, Chillemi G. 2019. Smyd2 conformational changes in response to p53 binding: role of the C-terminal domain. Mol Oncol. 13(6):1450–1461. doi: 10.1002/1878-0261.12502.
- Chandramouli B, Silvestri V, Scarno M, Ottini L, Chillemi G. 2016. Smyd3 open & closed lock mechanism for substrate recruitment: The hinge motion of C-terminal domain inferred from mu-second molecular dynamics simulations. Biochim Biophys Acta. 1860(7):1466–1474. doi: 10.1016/j.bbagen.2016.04.006.
- Chandrasekharan MB, Huang F, Sun ZW. 2010. Histone H2B ubiquitination and beyond: Regulation of nucleosome stability, chromatin dynamics and the trans-histone H3 methylation. Epigenetics. 5(6):460–468. doi: 10.4161/epi.5.6.12314.
- Chen J, Park HJ. 2019. Computer-Aided Discovery of Massonianoside B as a Novel Selective DOT1L Inhibitor. ACS Chem Biol. 14(5):873–881. doi: 10.1021/acschembio.8b00933.
- Chen S, Kapilashrami K, Senevirathne C, Wang Z, Wang J, Linscott JA, Luo M. 2019. Substrate-Differentiated Transition States of SET7/9-Catalyzed Lysine Methylation. J Am Chem Soc. 141(20):8064–8067. doi: 10.1021/jacs.9b02553.
- Chen S, Li L, Chen Y, Hu J, Liu J, Liu YC, Liu R, Zhang Y, Meng F, Zhu K, et al. 2016. Identification of Novel Disruptor of Telomeric Silencing 1-like (DOT1L) Inhibitors through Structure-Based Virtual Screening and Biological Assays. J Chem Inf Model. 56(3):527–534. doi: 10.1021/acs.jcim.5b00738.
- Chen Z, Zhang Y. 2020. Role of Mammalian DNA Methyltransferases in Development. Annu Rev Biochem. 89(1):135–158. doi: 10.1146/annurev-biochem-103019-102815.
- Cheng X, Collins RE, Zhang X. 2005. Structural and sequence motifs of protein (histone) methylation enzymes. Annu Rev Biophys Biomol Struct. 34(1):267–294. doi: 10.1146/annurev.biophys.34.040204.144452.
- Chmiela S, Sauceda HE, Müller K-R, Tkatchenko A. 2018. Towards exact molecular dynamics simulations with machine-learned force fields. Nat Commun. 9(1):3887. doi: 10.1038/s41467-018-06169-2.
- Choudhary C, Weinert BT, Nishida Y, Verdin E, Mann M. 2014. The growing landscape of lysine acetylation links metabolism and cell signalling. Nat Rev Mol Cell Biol. 15(8):536–550. doi: 10.1038/nrm3841.
- Chu Y, Sun L, Zhong S. 2015. How Y357F, Y276F mutants affect the methylation activity of PRDM9: QM/MM MD and free energy simulations. J Mol Model. 21(5):125. doi: 10.1007/s00894-015-2673-6.
- Chu Y, Xu Q, Guo H. 2010. Understanding Energetic Origins of Product Specificity of SET8 from QM/MM Free Energy Simulations: What Causes the Stop of Methyl Addition during Histone Lysine Methylation? J Chem Theory Comput. 6(4):1380–1389. doi: 10.1021/ct9006458.
- Chu Y, Yao J, Guo H. 2012. QM/MM MD and free energy simulations of G9a-like protein (GLP) and its mutants: understanding the factors that determine the product specificity. PLoS One. 7(5):e37674. doi: 10.1371/journal.pone.0037674.
- Chuikov S, Kurash JK, Wilson JR, Xiao B, Justin N, Ivanov GS, McKinney K, Tempst P, Prives C, Gamblin SJ, et al. 2004. Regulation of p53 activity through lysine methylation. Nature. 432(7015):353–360. doi: 10.1038/nature03117.
- Clarke SG. 2013. Protein methylation at the surface and buried deep: thinking outside the histone box. Trends Biochem Sci. 38(5):243–252. doi: 10.1016/j.tibs.2013.02.004.
- Cock-Rada AM, Medjkane S, Janski N, Yousfi N, Perichon M, Chaussepied M, Chluba J, Langsley G, Weitzman JB. 2012. SMYD3 promotes cancer invasion by epigenetic upregulation of the metalloproteinase MMP-9. Cancer Res. 72(3):810–820. doi: 10.1158/0008-5472.CAN-11-1052.
- Collins RE, Tachibana M, Tamaru H, Smith KM, Jia D, Zhang X, Selker EU, Shinkai Y, Cheng X. 2005. In vitro and in vivo analyses of a Phe/Tyr switch controlling product specificity of histone lysine methyltransferases. J Biol Chem. 280(7):5563–5570. doi: 10.1074/jbc.M410483200.
- Copeland RA. 2018. Protein methyltransferase inhibitors as precision cancer therapeutics: a decade of discovery. Philos Trans R Soc Lond B Biol Sci. 373(1748):20170080. doi: 10.1098/rstb.2017.0080.
- Copeland RA, Solomon ME, Richon VM. 2009. Protein methyltransferases as a target class for drug discovery. Nat Rev Drug Discov. 8(9):724–732. doi: 10.1038/nrd2974.
- Cornett EM, Ferry L, Defossez PA, Rothbart SB. 2019. Lysine Methylation Regulators Moonlighting outside the Epigenome. Mol Cell. 75(6):1092–1101. doi: 10.1016/j.molcel.2019.08.026.
- Cortopassi WA, Kumar K, Duarte F, Pimentel AS, Paton RS. 2016. Mechanisms of histone lysine-modifying enzymes: A computational perspective on the role of the protein environment. J Mol Graph Model. 67:69–84. doi: 10.1016/j.jmgm.2016.04.011.
- Couture JF, Collazo E, Brunzelle JS, Trievel RC. 2005. Structural and functional analysis of SET8, a histone H4 Lys-20 methyltransferase. Genes Dev. 19(12):1455–1465. doi: 10.1101/gad.1318405.
- Couture JF, Collazo E, Hauk G, Trievel RC. 2006. Structural basis for the methylation site specificity of SET7/9. Nat Struct Mol Biol. 13(2):140–146. doi: 10.1038/nsmb1045.
- Couture JF, Dirk LM, Brunzelle JS, Houtz RL, Trievel RC. 2008. Structural origins for the product specificity of SET domain protein methyltransferases. Proc Natl Acad Sci U S A. 105(52):20659–20664. doi: 10.1073/pnas.0806712105.
- Dasetty S, Meza-Morales PJ, Getman RB, Sarupria S. 2019. Simulations of interfacial processes: recent advances in force field development. Curr Opin Chem Eng. 23:138–145. doi: 10.1016/j.coche.2019.04.003.
- Davydova E, Ho AYY, Malecki J, Moen A, Enserink JM, Jakobsson ME, Loenarz C, Falnes PO. 2014. Identification and characterization of a novel evolutionarily conserved lysine-specific methyltransferase targeting eukaryotic translation elongation factor 2 (eEF2). J Biol Chem. 289(44):30499–30510. doi: 10.1074/jbc.M114.601658.
- Del Rizzo PA, Couture JF, Dirk LM, Strunk BS, Roiko MS, Brunzelle JS, Houtz RL, Trievel RC. 2010. SET7/9 catalytic mutants reveal the role of active site water molecules in lysine multiple methylation. J Biol Chem. 285(41):31849–31858. doi: 10.1074/jbc.M110.114587.
- Del Rizzo PA, Trievel RC. 2011. Substrate and product specificities of SET domain methyltransferases. Epigenetics. 6(9):1059–1067. doi: 10.4161/epi.6.9.16069.
- Dhayalan A, Kudithipudi S, Rathert P, Jeltsch A. 2011. Specificity analysis-based identification of new methylation targets of the SET7/9 protein lysine methyltransferase. Chem Biol. 18(1):111–120. doi: 10.1016/j.chembiol.2010.11.014.
- Di Blasi R, Blyuss O, Timms JF, Conole D, Ceroni F, Whitwell HJ. 2021. Non-Histone Protein Methylation: Biological Significance and Bioengineering Potential. ACS Chem Biol. 16(2):238–250. doi: 10.1021/acschembio.0c00771.
- Di Zazzo E, De Rosa C, Abbondanza C, Moncharmont B. 2013. PRDM Proteins: Molecular Mechanisms in Signal Transduction and Transcriptional Regulation. Biology. 2(1):107–141. doi: 10.3390/biology2010107.
- DiFiore JV, Ptacek TS, Wang Y, Li B, Simon JM, Strahl BD. 2020. Unique and Shared Roles for Histone H3K36 Methylation States in Transcription Regulation Functions. Cell Rep. 31(10):107751. doi: 10.1016/j.celrep.2020.107751.
- Douglas J, Hanks S, Temple IK, Davies S, Murray A, Upadhyaya M, Tomkins S, Hughes HE, Cole TR, Rahman N. 2003. NSD1 mutations are the major cause of Sotos syndrome and occur in some cases of Weaver syndrome but are rare in other overgrowth phenotypes. Am J Hum Genet. 72(1):132–143. doi: 10.1086/345647.
- Eastman P, Pande VS. 2010. OpenMM: A Hardware Independent Framework for Molecular Simulations. Comput Sci Eng. 12(4):34–39. doi: 10.1109/MCSE.2010.27.
- Eastman P, Swails J, Chodera JD, McGibbon RT, Zhao Y, Beauchamp KA, Wang LP, Simmonett AC, Harrigan MP, Stern CD, et al. 2017. OpenMM 7: Rapid development of high performance algorithms for molecular dynamics. PLoS Comput Biol. 13(7):e1005659. doi: 10.1371/journal.pcbi.1005659.
- Enchev RI, Schulman BA, Peter M. 2015. Protein neddylation: beyond cullin-RING ligases. Nat Rev Mol Cell Biol. 16(1):30–44. doi: 10.1038/nrm3919.
- Ernst P, Vakoc CR. 2012. WRAD: enabler of the SET1-family of H3K4 methyltransferases. Brief Funct Genomics. 11(3):217–226. doi: 10.1093/bfgp/els017.
- Eskeland R, Czermin B, Boeke J, Bonaldi T, Regula JT, Imhof A. 2004. The N-Terminus of Drosophila SU(VAR)3 − 9 Mediates Dimerization and Regulates Its Methyltransferase Activity. Biochemistry. 43(12):3740–3749. doi: 10.1021/bi035964s.
- Fabini E, Talibov VO, Mihalic F, Naldi M, Bartolini M, Bertucci C, Del Rio A, Danielson UH. 2019. Unveiling the Biochemistry of the Epigenetic Regulator SMYD3. Biochemistry. 58(35):3634–3645. doi: 10.1021/acs.biochem.9b00420.
- Falnes PØ, Jakobsson ME, Davydova E, Ho A, Małecki J. 2016. Protein lysine methylation by seven-β-strand methyltransferases. Biochem J. 473(14):1995–2009. doi: 10.1042/BCJ20160117.
- Falnes PØ, Małecki JM, Herrera MC, Bengtsen M, Davydova E. 2023. Human seven-beta-strand (METTL) methyltransferases - conquering the universe of protein lysine methylation. J Biol Chem. 299(6):104661. doi: 10.1016/j.jbc.2023.104661.
- Fang D, Gan H, Lee J-H, Han J, Wang Z, Riester SM, Jin L, Chen J, Zhou H, Wang J, et al. 2016. The histone H3.3K36M mutation reprograms the epigenome of chondroblastomas. Science. 352(6291):1344–1348. doi: 10.1126/science.aae0065.
- Fang J, Feng Q, Ketel CS, Wang H, Cao R, Xia L, Erdjument-Bromage H, Tempst P, Simon JA, Zhang Y. 2002. Purification and Functional Characterization of SET8, a Nucleosomal Histone H4-Lysine 20-Specific Methyltransferase. Curr Biol. 12(13):1086–1099. doi: 10.1016/s0960-9822(02)00924-7.
- Feng Q, Wang H, Ng HH, Erdjument-Bromage H, Tempst P, Struhl K, Zhang Y. 2002. Methylation of H3-Lysine 79 Is Mediated by a New Family of HMTases without a SET Domain. Curr Biol. 12(12):1052–1058. doi: 10.1016/s0960-9822(02)00901-6.
- Feoli A, Viviano M, Cipriano A, Milite C, Castellano S, Sbardella G. 2022. Lysine methyltransferase inhibitors: where we are now. RSC Chem Biol. 3(4):359–406. doi: 10.1039/d1cb00196e.
- Ferguson AD, Larsen NA, Howard T, Pollard H, Green I, Grande C, Cheung T, Garcia-Arenas R, Cowen S, Wu J, et al. 2011. Structural basis of substrate methylation and inhibition of SMYD2. Structure. 19(9):1262–1273. doi: 10.1016/j.str.2011.06.011.
- Fierz B, Chatterjee C, McGinty RK, Bar-Dagan M, Raleigh DP, Muir TW. 2011. Histone H2B ubiquitylation disrupts local and higher-order chromatin compaction. Nat Chem Biol. 7(2):113–119. doi: 10.1038/nchembio.501.
- Fioravanti R, Stazi G, Zwergel C, Valente S, Mai A. 2018. Six Years (2012-2018) of Researches on Catalytic EZH2 Inhibitors: The Boom of the 2-Pyridone Compounds. Chem Rec. 18(12):1818–1832. doi: 10.1002/tcr.201800091.
- Flores-León CD, Colorado-Pablo LF, Santos-Contreras MÁ, Aguayo-Ortiz R. 2023. Determination of nucleoside DOT1L inhibitors’ residence times by τRAMD simulations. Front Drug Discov. 2 doi: 10.3389/fddsv.2022.1083198.
- Foreman KW, Brown M, Park F, Emtage S, Harriss J, Das C, Zhu L, Crew A, Arnold L, Shaaban S, et al. 2011. Structural and functional profiling of the human histone methyltransferase SMYD3. PLoS One. 6(7):e22290. doi: 10.1371/journal.pone.0022290.
- Frederiks F, Tzouros M, Oudgenoeg G, van Welsem T, Fornerod M, Krijgsveld J, van Leeuwen F. 2008. Nonprocessive methylation by Dot1 leads to functional redundancy of histone H3K79 methylation states. Nat Struct Mol Biol. 15(6):550–557. doi: 10.1038/nsmb.1432.
- Fu W, Liu N, Qiao Q, Wang M, Min J, Zhu B, Xu RM, Yang N. 2016. Structural Basis for Substrate Preference of SMYD3, a SET Domain-containing Protein Lysine Methyltransferase. J Biol Chem. 291(17):9173–9180. doi: 10.1074/jbc.M115.709832.
- Gathiaka S, Boykin B, Cáceres T, Hevel JM, Acevedo O. 2016. Understanding protein arginine methyltransferase 1 (PRMT1) product specificity from molecular dynamics. Bioorg Med Chem. 24(20):4949–4960. doi: 10.1016/j.bmc.2016.08.009.
- Ghoneim M, Fuchs HA, Musselman CA. 2021. Histone Tail Conformations: A Fuzzy Affair with DNA. Trends Biochem Sci. 46(7):564–578. doi: 10.1016/j.tibs.2020.12.012.
- Grebien F, Vedadi M, Getlik M, Giambruno R, Grover A, Avellino R, Skucha A, Vittori S, Kuznetsova E, Smil D, et al. 2015. Pharmacological targeting of the Wdr5-MLL interaction in C/EBPalpha N-terminal leukemia. Nat Chem Biol. 11(8):571–578. doi: 10.1038/nchembio.1859.
- Grimsley GR, Scholtz JM, Pace CN. 2009. A summary of the measured pK values of the ionizable groups in folded proteins. Protein Sci. 18(1):247–251. doi: 10.1002/pro.19.
- Groenhof G. 2013. Introduction to QM/MM simulations. Methods Mol Biol. 924:43–66.
- Guo H, Guo H. 2007. Mechanism of histone methylation catalyzed by protein lysine methyltransferase SET7/9 and origin of product specificity. Proc Natl Acad Sci U S A. 104(21):8797–8802. doi: 10.1073/pnas.0702981104.
- Ha K, Anand P, Lee JA, Jones JR, Kim CA, Bertola DR, Labonne JD, Layman LC, Wenzel W, Kim HG. 2016. Steric Clash in the SET Domain of Histone Methyltransferase NSD1 as a Cause of Sotos Syndrome and Its Genetic Heterogeneity in a Brazilian Cohort. Genes (Basel).). 7(11):96. doi: 10.3390/genes7110096.
- Hamamoto R, Furukawa Y, Morita M, Iimura Y, Silva FP, Li M, Yagyu R, Nakamura Y. 2004. SMYD3 encodes a histone methyltransferase involved in the proliferation of cancer cells. Nat Cell Biol. 6(8):731–740. doi: 10.1038/ncb1151.
- Hamey JJ, Wienert B, Quinlan KGR, Wilkins MR. 2017. METTL21B Is a Novel Human Lysine Methyltransferase of Translation Elongation Factor 1A: Discovery by CRISPR/Cas9 Knockout. Mol Cell Proteomics. 16(12):2229–2242. doi: 10.1074/mcp.M116.066308.
- He Y, Selvaraju S, Curtin ML, Jakob CG, Zhu H, Comess KM, Shaw B, The J, Lima-Fernandes E, Szewczyk MM, et al. 2017. The EED protein–protein interaction inhibitor A-395 inactivates the PRC2 complex. Nat Chem Biol. 13(4):389–395. doi: 10.1038/nchembio.2306.
- Hollingsworth SA, Dror RO. 2018. Molecular Dynamics Simulation for All. Neuron. 99(6):1129–1143. doi: 10.1016/j.neuron.2018.08.011.
- Holt MT, David Y, Pollock S, Tang Z, Jeon J, Kim J, Roeder RG, Muir TW. 2015. Identification of a functional hotspot on ubiquitin required for stimulation of methyltransferase activity on chromatin. Proc Natl Acad Sci U S A. 112(33):10365–10370. doi: 10.1073/pnas.1504483112.
- Horowitz S, Adhikari U, Dirk LM, Del Rizzo PA, Mehl RA, Houtz RL, Al-Hashimi HM, Scheiner S, Trievel RC. 2014. Manipulating unconventional CH-based hydrogen bonding in a methyltransferase via noncanonical amino acid mutagenesis. ACS Chem Biol. 9(8):1692–1697. doi: 10.1021/cb5001185.
- Horowitz S, Yesselman JD, Al-Hashimi HM, Trievel RC. 2011. Direct evidence for methyl group coordination by carbon-oxygen hydrogen bonds in the lysine methyltransferase SET7/9. J Biol Chem. 286(21):18658–18663. doi: 10.1074/jbc.M111.232876.
- Hospital A, Goñi JR, Orozco M, Gelpí JL. 2015. Molecular dynamics simulations: advances and applications. Adv Appl Bioinform Chem. 8:37–47. doi: 10.2147/AABC.S70333.
- Hsu PL, Shi H, Leonen C, Kang J, Chatterjee C, Zheng N. 2019. Structural Basis of H2B Ubiquitination-Dependent H3K4 Methylation by COMPASS. Mol Cell. 76(5):712–723.e4. e714. doi: 10.1016/j.molcel.2019.10.013.
- Hu P, Wang S, Zhang Y. 2008. How Do SET-Domain Protein Lysine Methyltransferases Achieve the Methylation State Specificity? Revisited by Ab Initio QM/MM Molecular Dynamics Simulations. J Am Chem Soc. 130(12):3806–3813. doi: 10.1021/ja075896n.
- Hu P, Zhang Y. 2006. Catalytic Mechanism and Product Specificity of the Histone Lysine Methyltransferase SET7/9: An ab Initio QM/MM-FE Study with Multiple Initial Structures. J Am Chem Soc. 128(4):1272–1278. doi: 10.1021/ja056153+.
- Huang J, Perez-Burgos L, Placek BJ, Sengupta R, Richter M, Dorsey JA, Kubicek S, Opravil S, Jenuwein T, Berger SL. 2006. Repression of p53 activity by Smyd2-mediated methylation. Nature. 444(7119):629–632. doi: 10.1038/nature05287.
- Huertas J, Schöler HR, Cojocaru V. 2021. Histone tails cooperate to control the breathing of genomic nucleosomes. PLoS Comput Biol. 17(6):e1009013. doi: 10.1371/journal.pcbi.1009013.
- Husmann D, Gozani O. 2019. Histone lysine methyltransferases in biology and disease. Nat Struct Mol Biol. 26(10):880–889. doi: 10.1038/s41594-019-0298-7.
- Ibáñez G, McBean JL, Astudillo YM, Luo M. 2010. An enzyme-coupled ultrasensitive luminescence assay for protein methyltransferases. Anal Biochem. 401(2):203–210. doi: 10.1016/j.ab.2010.03.010.
- Iglesias N, Currie MA, Jih G, Paulo JA, Siuti N, Kalocsay M, Gygi SP, Moazed D. 2018. Automethylation-induced conformational switch in Clr4 (Suv39h) maintains epigenetic stability. Nature. 560(7719):504–508. doi: 10.1038/s41586-018-0398-2.
- Jacobs SA, Harp JM, Devarakonda S, Kim Y, Rastinejad F, Khorasanizadeh S. 2002. The active site of the SET domain is constructed on a knot. Nat Struct Biol. 9(11):833–838. doi: 10.1038/nsb861.
- Jaffe JD, Wang Y, Chan HM, Zhang J, Huether R, Kryukov GV, Bhang H-eC, Taylor JE, Hu M, Englund NP, et al. 2013. Global chromatin profiling reveals NSD2 mutations in pediatric acute lymphoblastic leukemia. Nat Genet. 45(11):1386–1391. doi: 10.1038/ng.2777.
- Jakobsson ME. 2021a. Enzymology and significance of protein histidine methylation. J Biol Chem. 297(4):101130. doi: 10.1016/j.jbc.2021.101130.
- Jakobsson ME. 2021b. Structure, Activity and Function of the Dual Protein Lysine and Protein N-Terminal Methyltransferase METTL13. Life (Basel). 11(11)doi: 10.3390/life11111121.
- Jakobsson ME, Malecki J, Nilges BS, Moen A, Leidel SA, Falnes PO. 2017. Methylation of human eukaryotic elongation factor alpha (eEF1A) by a member of a novel protein lysine methyltransferase family modulates mRNA translation. Nucleic Acids Res. 45(14):8239–8254. doi: 10.1093/nar/gkx432.
- Jakobsson ME, Małecki JM, Halabelian L, Nilges BS, Pinto R, Kudithipudi S, Munk S, Davydova E, Zuhairi FR, Arrowsmith CH, et al. 2018. The dual methyltransferase METTL13 targets N terminus and Lys55 of eEF1A and modulates codon-specific translation rates. Nat Commun. 9(1):3411. doi: 10.1038/s41467-018-05646-y.
- Jakobsson ME, Moen A, Bousset L, Egge-Jacobsen W, Kernstock S, Melki R, Falnes PO. 2013. Identification and characterization of a novel human methyltransferase modulating Hsp70 protein function through lysine methylation. J Biol Chem. 288(39):27752–27763. doi: 10.1074/jbc.M113.483248.
- Jambhekar A, Dhall A, Shi Y. 2019. Roles and regulation of histone methylation in animal development. Nat Rev Mol Cell Biol. 20(10):625–641. doi: 10.1038/s41580-019-0151-1.
- Jang S, Kang C, Yang HS, Jung T, Hebert H, Chung KY, Kim SJ, Hohng S, Song JJ. 2019. Structural basis of recognition and destabilization of the histone H2B ubiquitinated nucleosome by the DOT1L histone H3 Lys79 methyltransferase. Genes Dev. 33(11-12):620–625. doi: 10.1101/gad.323790.118.
- Janson G, Paiardini A. 2021. PyMod 3: a complete suite for structural bioinformatics in PyMOL. Bioinformatics. 37(10):1471–1472. doi: 10.1093/bioinformatics/btaa849.
- Jarrold J, Davies CC. 2019. PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends Mol Med. 25(11):993–1009. doi: 10.1016/j.molmed.2019.05.007.
- Jayaram H, Hoelper D, Jain SU, Cantone N, Lundgren SM, Poy F, Allis CD, Cummings R, Bellon S, Lewis PW. 2016. S-adenosyl methionine is necessary for inhibition of the methyltransferase G9a by the lysine 9 to methionine mutation on histone H3. Proc Natl Acad Sci U S A. 113(22):6182–6187. doi: 10.1073/pnas.1605523113.
- Jiang Y, Sirinupong N, Brunzelle J, Yang Z. 2011. Crystal structures of histone and p53 methyltransferase SmyD2 reveal a conformational flexibility of the autoinhibitory C-terminal domain. PLoS One. 6(6):e21640. doi: 10.1371/journal.pone.0021640.
- Jiang Y, Trescott L, Holcomb J, Zhang X, Brunzelle J, Sirinupong N, Shi X, Yang Z. 2014. Structural insights into estrogen receptor alpha methylation by histone methyltransferase SMYD2, a cellular event implicated in estrogen signaling regulation. J Mol Biol. 426(20):3413–3425. doi: 10.1016/j.jmb.2014.02.019.
- Joshi SY, Deshmukh SA. 2020. A review of advancements in coarse-grained molecular dynamics simulations. Mol Simul. 47(10-11):786–803. doi: 10.1080/08927022.2020.1828583.
- Jumper J, Evans R, Pritzel A, Green T, Figurnov M, Ronneberger O, Tunyasuvunakool K, Bates R, Žídek A, Potapenko A, et al. 2021. Highly accurate protein structure prediction with AlphaFold. Nature. 596(7873):583–589. doi: 10.1038/s41586-021-03819-2.
- Justin N, Zhang Y, Tarricone C, Martin SR, Chen S, Underwood E, De Marco V, Haire LF, Walker PA, Reinberg D, et al. 2016. Structural basis of oncogenic histone H3K27M inhibition of human polycomb repressive complex 2. Nat Commun. 7(1):11316. doi: 10.1038/ncomms11316.
- Kalashnikova AA, Porter-Goff ME, Muthurajan UM, Luger K, Hansen JC. 2013. The role of the nucleosome acidic patch in modulating higher order chromatin structure. J R Soc Interface. 10(82):20121022. doi: 10.1098/rsif.2012.1022.
- Kato H, Jiang J, Zhou BR, Rozendaal M, Feng H, Ghirlando R, Xiao TS, Straight AF, Bai Y. 2013. A conserved mechanism for centromeric nucleosome recognition by centromere protein CENP-C. Science. 340(6136):1110–1113. doi: 10.1126/science.1235532.
- Khella MS, Brohm A, Weirich S, Jeltsch A. 2020. Mechanistic Insights into the Allosteric Regulation of the Clr4 Protein Lysine Methyltransferase by Autoinhibition and Automethylation. Int J Mol Sci. 21(22)
- Khella MS, Schnee P, Weirich S, Bui T, Bröhm A, Bashtrykov P, Pleiss J, Jeltsch A. 2023. The T1150A cancer mutant of the protein lysine dimethyltransferase NSD2 can introduce H3K36 trimethylation. J Biol Chem. 299(6):104796. doi: 10.1016/j.jbc.2023.104796.
- Khirsariya P, Pospíšil P, Maier L, Boudný M, Babáš M, Kroutil O, Mráz M, Vácha R, Paruch K. 2022. Synthesis and Profiling of Highly Selective Inhibitors of Methyltransferase DOT1L Based on Carbocyclic C-Nucleosides. J Med Chem. 65(7):5701–5723. doi: 10.1021/acs.jmedchem.1c02228.
- Kim J, Guermah M, McGinty RK, Lee JS, Tang Z, Milne TA, Shilatifard A, Muir TW, Roeder RG. 2009. RAD6-Mediated transcription-coupled H2B ubiquitylation directly stimulates H3K4 methylation in human cells. Cell. 137(3):459–471. doi: 10.1016/j.cell.2009.02.027.
- Kim J, Hake SB, Roeder RG. 2005. The human homolog of yeast BRE1 functions as a transcriptional coactivator through direct activator interactions. Mol Cell. 20(5):759–770. doi: 10.1016/j.molcel.2005.11.012.
- Kim J, Kim JA, McGinty RK, Nguyen UT, Muir TW, Allis CD, Roeder RG. 2013. The n-SET domain of Set1 regulates H2B ubiquitylation-dependent H3K4 methylation. Mol Cell. 49(6):1121–1133. doi: 10.1016/j.molcel.2013.01.034.
- Kim W, Bird GH, Neff T, Guo G, Kerenyi MA, Walensky LD, Orkin SH. 2013. Targeted disruption of the EZH2-EED complex inhibits EZH2-dependent cancer. Nat Chem Biol. 9(10):643–650. doi: 10.1038/nchembio.1331.
- Klimpel GR, Fleischman WR. 1984. Spectrographic Representation of Globular Protein Breathing Motions. Science. 223(4632):181–182.
- Kmiecik S, Gront D, Kolinski M, Wieteska L, Dawid AE, Kolinski A. 2016. Coarse-Grained Protein Models and Their Applications. Chem Rev. 116(14):7898–7936. doi: 10.1021/acs.chemrev.6b00163.
- Kothapalli N, Camporeale G, Kueh A, Chew YC, Oommen AM, Griffin JB, Zempleni J. 2005. Biological functions of biotinylated histones. J Nutr Biochem. 16(7):446–448. doi: 10.1016/j.jnutbio.2005.03.025.
- Kudithipudi S, Jeltsch A. 2016. Approaches and Guidelines for the Identification of Novel Substrates of Protein Lysine Methyltransferases. Cell Chem Biol. 23(9):1049–1055. doi: 10.1016/j.chembiol.2016.07.013.
- Kudithipudi S, Kusevic D, Weirich S, Jeltsch A. 2014. Specificity analysis of protein lysine methyltransferases using SPOT peptide arrays. J Vis Exp. (93):e52203. doi: 10.3791/52203.
- Kudithipudi S, Lungu C, Rathert P, Happel N, Jeltsch A. 2014. Substrate specificity analysis and novel substrates of the protein lysine methyltransferase NSD1. Chem Biol. 21(2):226–237. doi: 10.1016/j.chembiol.2013.10.016.
- Kurotaki N, Imaizumi K, Harada N, Masuno M, Kondoh T, Nagai T, Ohashi H, Naritomi K, Tsukahara M, Makita Y, et al. 2002. Haploinsufficiency of NSD1 causes Sotos syndrome. Nat Genet. 30(4):365–366. doi: 10.1038/ng863.
- Kusevic D, Kudithipudi S, Iglesias N, Moazed D, Jeltsch A. 2017. Clr4 specificity and catalytic activity beyond H3K9 methylation. Biochimie. 135:83–88. doi: 10.1016/j.biochi.2017.01.013.
- Kwiatkowski S, Drozak J. 2020. Protein Histidine Methylation. Curr Protein Pept Sci. 21(7):675–689. doi: 10.2174/1389203721666200318161330.
- Kwon M, Park K, Hyun K, Lee JH, Zhou L, Cho YW, Ge K, Skalnik DG, Muir TW, Kim J. 2020. H2B ubiquitylation enhances H3K4 methylation activities of human KMT2 family complexes. Nucleic Acids Res. 48(10):5442–5456. doi: 10.1093/nar/gkaa317.
- Kwon T, Chang JH, Kwak E, Lee CW, Joachimiak A, Kim YC, Lee J, Cho Y. 2003. Mechanism of histone lysine methyl transfer revealed by the structure of SET7/9-AdoMet. Embo J. 22(2):292–303. doi: 10.1093/emboj/cdg025.
- Lacoste N, Utley RT, Hunter JM, Poirier GG, Côte J. 2002. Disruptor of telomeric silencing-1 is a chromatin-specific histone H3 methyltransferase. J Biol Chem. 277(34):30421–30424. doi: 10.1074/jbc.C200366200.
- Lanouette S, Davey JA, Elisma F, Ning Z, Figeys D, Chica RA, Couture JF. 2015. Discovery of substrates for a SET domain lysine methyltransferase predicted by multistate computational protein design. Structure. 23(1):206–215. doi: 10.1016/j.str.2014.11.004.
- Lee CH, Yu JR, Kumar S, Jin Y, LeRoy G, Bhanu N, Kaneko S, Garcia BA, Hamilton AD, Reinberg D. 2018. Allosteric Activation Dictates PRC2 Activity Independent of Its Recruitment to Chromatin. Mol Cell. 70(3):422–434.e6. e426. doi: 10.1016/j.molcel.2018.03.020.
- Li W, Tian W, Yuan G, Deng P, Sengupta D, Cheng Z, Cao Y, Ren J, Qin Y, Zhou Y, et al. 2021. Molecular basis of nucleosomal H3K36 methylation by NSD methyltransferases. Nature. 590(7846):498–503. doi: 10.1038/s41586-020-03069-8.
- Li Y, Han J, Zhang Y, Cao F, Liu Z, Li S, Wu J, Hu C, Wang Y, Shuai J, et al. 2016. Structural basis for activity regulation of MLL family methyltransferases. Nature. 530(7591):447–452. doi: 10.1038/nature16952.
- Li Y, Trojer P, Xu C-F, Cheung P, Kuo A, Drury WJ, Qiao Q, Neubert TA, Xu R-M, Gozani O, et al. 2009. The target of the NSD family of histone lysine methyltransferases depends on the nature of the substrate. J Biol Chem. 284(49):34283–34295. doi: 10.1074/jbc.M109.034462.
- Lindorff-Larsen K, Maragakis P, Piana S, Eastwood MP, Dror RO, Shaw DE. 2012. Systematic validation of protein force fields against experimental data. PLoS One. 7(2):e32131. doi: 10.1371/journal.pone.0032131.
- Linscott JA, Kapilashrami K, Wang Z, Senevirathne C, Bothwell IR, Blum G, Luo M. 2016. Kinetic isotope effects reveal early transition state of protein lysine methyltransferase SET8. Proc Natl Acad Sci U S A. 113(52):E8369–E8378.
- Liu S, Hausmann S, Carlson SM, Fuentes ME, Francis JW, Pillai R, Lofgren SM, Hulea L, Tandoc K, Lu J, et al. 2019. METTL13 Methylation of eEF1A Increases Translational Output to Promote Tumorigenesis. Cell. 176(3):491–504.e21. e421. doi: 10.1016/j.cell.2018.11.038.
- Liu Y, Chen W, Gaudet J, Cheney MD, Roudaia L, Cierpicki T, Klet RC, Hartman K, Laue TM, Speck NA, et al. 2007. Structural basis for recognition of SMRT/N-CoR by the MYND domain and its contribution to AML1/ETO’s activity. Cancer Cell. 11(6):483–497. doi: 10.1016/j.ccr.2007.04.010.
- Liu Y, Zhang Y, Xue H, Cao M, Bai G, Mu Z, Yao Y, Sun S, Fang D, Huang J. 2021. Cryo-EM structure of SETD2/Set2 methyltransferase bound to a nucleosome containing oncohistone mutations. Cell Discov. 7(1):32. doi: 10.1038/s41421-021-00261-6.
- Lopes PEM, Guvench O, MacKerell AD. 2015. Current Status of Protein Force Fields for Molecular Dynamics Simulations. In: Kukol A, editor. Molecular Modeling of Proteins. New York, NY: Springer New York; p. 47–71.
- López-López E, Rabal O, Oyarzabal J, Medina-Franco JL. 2020. Towards the understanding of the activity of G9a inhibitors: an activity landscape and molecular modeling approach. J Comput Aided Mol Des. 34(6):659–669. doi: 10.1007/s10822-020-00298-x.
- Lu-Culligan WJ, Connor LJ, Xie Y, Ekundayo BE, Rose BT, Machyna M, Pintado-Urbanc AP, Zimmer JT, Vock IW, Bhanu NV, et al. 2023. Acetyl-methyllysine marks chromatin at active transcription start sites. Nature. 622(7981):173–179. doi: 10.1038/s41586-023-06565-9.
- Luo M. 2012. Current chemical biology approaches to interrogate protein methyltransferases. ACS Chem Biol. 7(3):443–463. doi: 10.1021/cb200519y.
- Luo M. 2015. Inhibitors of protein methyltransferases as chemical tools. Epigenomics. 7(8):1327–1338. doi: 10.2217/epi.15.87.
- Luo M. 2018. Chemical and Biochemical Perspectives of Protein Lysine Methylation. Chem Rev. 118(14):6656–6705. doi: 10.1021/acs.chemrev.8b00008.
- Luo M, Wang H, Zou Y, Zhang S, Xiao J, Jiang G, Zhang Y, Lai Y. 2016. Identification of phenoxyacetamide derivatives as novel DOT1L inhibitors via docking screening and molecular dynamics simulation. J Mol Graph Model. 68:128–139. doi: 10.1016/j.jmgm.2016.06.011.
- Magen S, Magnani R, Haziza S, Hershkovitz E, Houtz R, Cambi F, Parvari R. 2012. Human calmodulin methyltransferase: expression, activity on calmodulin, and Hsp90 dependence. PLoS One. 7(12):e52425. doi: 10.1371/journal.pone.0052425.
- Makde RD, England JR, Yennawar HP, Tan S. 2010. Structure of RCC1 chromatin factor bound to the nucleosome core particle. Nature. 467(7315):562–566. doi: 10.1038/nature09321.
- Makowski L, Rodi DJ, Mandava S, Minh DD, Gore DB, Fischetti RF. 2008. Molecular crowding inhibits intramolecular breathing motions in proteins. J Mol Biol. 375(2):529–546. doi: 10.1016/j.jmb.2007.07.075.
- Malecki J, Aileni VK, Ho AYY, Schwarz J, Moen A, Sorensen V, Nilges BS, Jakobsson ME, Leidel SA, Falnes PO. 2017. The novel lysine specific methyltransferase METTL21B affects mRNA translation through inducible and dynamic methylation of Lys-165 in human eukaryotic elongation factor 1 alpha (eEF1A). Nucleic Acids Res. 45(8):4370–4389.
- Małecki J, Ho AYY, Moen A, Dahl H-A, Falnes PØ. 2015. Human METTL20 is a mitochondrial lysine methyltransferase that targets the beta subunit of electron transfer flavoprotein (ETFbeta) and modulates its activity. J Biol Chem. 290(1):423–434. doi: 10.1074/jbc.M114.614115.
- Mariño Pérez L, Ielasi FS, Bessa LM, Maurin D, Kragelj J, Blackledge M, Salvi N, Bouvignies G, Palencia A, Jensen MR. 2022. Visualizing protein breathing motions associated with aromatic ring flipping. Nature. 602(7898):695–700. doi: 10.1038/s41586-022-04417-6.
- Marouco D, Garabadgiu A, Melino G, Barlev NA. 2013. Lysine-specific modifications of p53: a matter of life and death? Oncotarget. 4(10):1556–1571. doi: 10.18632/oncotarget.1436.
- Marrink SJ, Tieleman DP. 2013. Perspective on the Martini model. Chem Soc Rev. 42(16):6801–6822. doi: 10.1039/c3cs60093a.
- Mattiroli F, Sixma TK. 2014. Lysine-targeting specificity in ubiquitin and ubiquitin-like modification pathways. Nat Struct Mol Biol. 21(4):308–316. doi: 10.1038/nsmb.2792.
- Mazur PK, Reynoird N, Khatri P, Jansen PWTC, Wilkinson AW, Liu S, Barbash O, Van Aller GS, Huddleston M, Dhanak D, et al. 2014. SMYD3 links lysine methylation of MAP3K2 to Ras-driven cancer. Nature. 510(7504):283–287. doi: 10.1038/nature13320.
- McCabe MT, Graves AP, Ganji G, Diaz E, Halsey WS, Jiang Y, Smitheman KN, Ott HM, Pappalardi MB, Allen KE, et al. 2012. Mutation of A677 in histone methyltransferase EZH2 in human B-cell lymphoma promotes hypertrimethylation of histone H3 on lysine 27 (H3K27). Proc Natl Acad Sci U S A. 109(8):2989–2994. doi: 10.1073/pnas.1116418109.
- McLean CM, Karemaker ID, van Leeuwen F. 2014. The emerging roles of DOT1L in leukemia and normal development. Leukemia. 28(11):2131–2138. doi: 10.1038/leu.2014.169.
- Medjkane S, Cock-Rada A, Weitzman JB. 2012. Role of the SMYD3 histone methyltransferase in tumorigenesis: local or global effects? Cell Cycle. 11(10):1865–1865. doi: 10.4161/cc.20415.
- Meller J. 2001. Molecular Dynamics. Encyclopedia of Life Sciences. Nature Publishing Group.
- Millán-Zambrano G, Burton A, Bannister AJ, Schneider R. 2022. Histone post-translational modifications—cause and consequence of genome function. Nat Rev Genet. 23(9):563–580. doi: 10.1038/s41576-022-00468-7.
- Min J, Feng Q, Li Z, Zhang Y, Xu RM. 2003. Structure of the Catalytic Domain of Human DOT1L, a Non-SET Domain Nucleosomal Histone Methyltransferase. Cell. 112(5):711–723. doi: 10.1016/s0092-8674(03)00114-4.
- Miranda-Rojas S, Blanco-Esperguez K, Tuñón I, Kästner J, Mendizábal F. 2021. Exploration of the Activation Mechanism of the Epigenetic Regulator MLL3: A QM/MM Study. Biomolecules. 11(7):1051. doi: 10.3390/biom11071051.
- Monticelli L, Tieleman DP. 2013. Force Fields for Classical Molecular Dynamics. In: Monticelli L, Salonen E, editors. Biomolecular Simulations: Methods and Protocols. Totowa, NJ: Humana Press; p. 197–213.
- Morin RD, Johnson NA, Severson TM, Mungall AJ, An J, Goya R, Paul JE, Boyle M, Woolcock BW, Kuchenbauer F, et al. 2010. Somatic mutations altering EZH2 (Tyr641) in follicular and diffuse large B-cell lymphomas of germinal-center origin. Nat Genet. 42(2):181–185. doi: 10.1038/ng.518.
- Morishita M, di Luccio E. 2011. Structural insights into the regulation and the recognition of histone marks by the SET domain of NSD1. Biochem Biophys Res Commun. 412(2):214–219. doi: 10.1016/j.bbrc.2011.07.061.
- Mzoughi S, Tan YX, Low D, Guccione E. 2016. The role of PRDMs in cancer: one family, two sides. Curr Opin Genet Dev. 36:83–91. doi: 10.1016/j.gde.2016.03.009.
- Nakanishi S, Sanderson BW, Delventhal KM, Bradford WD, Staehling-Hampton K, Shilatifard A. 2008. A comprehensive library of histone mutants identifies nucleosomal residues required for H3K4 methylation. Nat Struct Mol Biol. 15(8):881–888. doi: 10.1038/nsmb.1454.
- Ng HH, Feng Q, Wang H, Erdjument-Bromage H, Tempst P, Zhang Y, Struhl K. 2002. Lysine methylation within the globular domain of histone H3 by Dot1 is important for telomeric silencing and Sir protein association. Genes Dev. 16(12):1518–1527. doi: 10.1101/gad.1001502.
- Ng HH, Xu RM, Zhang Y, Struhl K. 2002. Ubiquitination of histone H2B by Rad6 is required for efficient Dot1-mediated methylation of histone H3 lysine 79. J Biol Chem. 277(38):34655–34657. doi: 10.1074/jbc.C200433200.
- Ng SS, Kavanagh KL, McDonough MA, Butler D, Pilka ES, Lienard BM, Bray JE, Savitsky P, Gileadi O, von Delft F, et al. 2007. Crystal structures of histone demethylase JMJD2A reveal basis for substrate specificity. Nature. 448(7149):87–91. doi: 10.1038/nature05971.
- Nguyen TQ, Koh S, Kwon J, Jang S, Kang W, Yang JK. 2023. Structural basis for recognition and methylation of p97 by METTL21D, a valosin-containing protein lysine methyltransferase. iScience. 26(7):107222. doi: 10.1016/j.isci.2023.107222.
- Olsen JB, Cao XJ, Han B, Chen LH, Horvath A, Richardson TI, Campbell RM, Garcia BA, Nguyen H. 2016. Quantitative Profiling of the Activity of Protein Lysine Methyltransferase SMYD2 Using SILAC-Based Proteomics. Mol Cell Proteomics. 15(3):892–905. doi: 10.1074/mcp.M115.053280.
- Oyer JA, Huang X, Zheng Y, Shim J, Ezponda T, Carpenter Z, Allegretta M, Okot-Kotber CI, Patel JP, Melnick A, et al. 2014. Point mutation E1099K in MMSET/NSD2 enhances its methyltranferase activity and leads to altered global chromatin methylation in lymphoid malignancies. Leukemia. 28(1):198–201. doi: 10.1038/leu.2013.204.
- Park S, Schulten K. 2004. Calculating potentials of mean force from steered molecular dynamics simulations. J Chem Phys. 120(13):5946–5961. doi: 10.1063/1.1651473.
- Park SH, Ayoub A, Lee YT, Xu J, Kim H, Zheng W, Zhang B, Sha L, An S, Zhang Y, et al. 2019. Cryo-EM structure of the human MLL1 core complex bound to the nucleosome. Nat Commun. 10(1):5540. doi: 10.1038/s41467-019-13550-2.
- Patel A, Dharmarajan V, Vought VE, Cosgrove MS. 2009. On the mechanism of multiple lysine methylation by the human mixed lineage leukemia protein-1 (MLL1) core complex. J Biol Chem. 284(36):24242–24256. doi: 10.1074/jbc.M109.014498.
- Patel A, Vought VE, Dharmarajan V, Cosgrove MS. 2011. A novel non-SET domain multi-subunit methyltransferase required for sequential nucleosomal histone H3 methylation by the mixed lineage leukemia protein-1 (MLL1) core complex. J Biol Chem. 286(5):3359–3369. doi: 10.1074/jbc.M110.174524.
- Patel A, Vought VE, Swatkoski S, Viggiano S, Howard B, Dharmarajan V, Monteith KE, Kupakuwana G, Namitz KE, Shinsky SA, et al. 2014. Automethylation activities within the mixed lineage leukemia-1 (MLL1) core complex reveal evidence supporting a "two-active site" model for multiple histone H3 lysine 4 methylation. J Biol Chem. 289(2):868–884. doi: 10.1074/jbc.M113.501064.
- Patel DJ, Wang Z. 2013. Readout of epigenetic modifications. Annu Rev Biochem. 82(1):81–118. doi: 10.1146/annurev-biochem-072711-165700.
- Patnaik D, Chin HG, Estève P-O, Benner J, Jacobsen SE, Pradhan S. 2004. Substrate specificity and kinetic mechanism of mammalian G9a histone H3 methyltransferase. J Biol Chem. 279(51):53248–53258. doi: 10.1074/jbc.M409604200.
- Perner F, Stein EM, Wenge DV, Singh S, Kim J, Apazidis A, Rahnamoun H, Anand D, Marinaccio C, Hatton C, et al. 2023. MEN1 mutations mediate clinical resistance to menin inhibition. Nature. 615(7954):913–919. doi: 10.1038/s41586-023-05755-9.
- Petrossian TC, Clarke SG. 2011. Uncovering the human methyltransferasome. Mol Cell Proteomics. 10(1):M110. 000976.
- Potoyan DA, Papoian GA. 2011. Energy landscape analyses of disordered histone tails reveal special organization of their conformational dynamics. J Am Chem Soc. 133(19):7405–7415. doi: 10.1021/ja1111964.
- Poulard C, Noureddine LM, Pruvost L, Le Romancer M. 2021. Structure, Activity, and Function of the Protein Lysine Methyltransferase G9a. Life (Basel). 11(10) doi: 10.3390/life11101082.
- Poulin MB, Schneck JL, Matico RE, Hou W, McDevitt PJ, Holbert M, Schramm VL. 2016. Nucleosome Binding Alters the Substrate Bonding Environment of Histone H3 Lysine 36 Methyltransferase NSD2. J Am Chem Soc. 138(21):6699–6702. doi: 10.1021/jacs.6b01612.
- Poulin MB, Schneck JL, Matico RE, McDevitt PJ, Huddleston MJ, Hou W, Johnson NW, Thrall SH, Meek TD, Schramm VL. 2016. Transition state for the NSD2-catalyzed methylation of histone H3 lysine 36. Proc Natl Acad Sci U S A. 113(5):1197–1201. doi: 10.1073/pnas.1521036113.
- Qian C, Zhou MM. 2006. SET domain protein lysine methyltransferases: Structure, specificity and catalysis. Cell Mol Life Sci. 63(23):2755–2763. doi: 10.1007/s00018-006-6274-5.
- Qian P, Guo H, Wang L, Guo H. 2017. QM/MM Investigation of Substrate and Product Specificities of Suv4-20h2: How Does This Enzyme Generate Dimethylated H4K20 from Monomethylated Substrate? J Chem Theory Comput. 13(6):2977–2986. doi: 10.1021/acs.jctc.7b00069.
- Qiao Q, Li Y, Chen Z, Wang M, Reinberg D, Xu RM. 2011. The structure of NSD1 reveals an autoregulatory mechanism underlying histone H3K36 methylation. J Biol Chem. 286(10):8361–8368. doi: 10.1074/jbc.M110.204115.
- Rahman S, Hoffmann NA, Worden EJ, Smith ML, Namitz KEW, Knutson BA, Cosgrove MS, Wolberger C. 2022. Multistate structures of the MLL1-WRAD complex bound to H2B-ubiquitinated nucleosome. Proc Natl Acad Sci U S A. 119(38):e2205691119.
- Raj U, Kumar H, Gupta S, Varadwaj PK. 2015. Novel DOT1L ReceptorNatural Inhibitors Involved in Mixed Lineage Leukemia: a Virtual Screening, Molecular Docking and Dynamics Simulation Study. Asian Pac J Cancer Prev. 16(9):3817–3825. doi: 10.7314/apjcp.2015.16.9.3817.
- Rao RC, Dou Y. 2015. Hijacked in cancer: the KMT2 (MLL) family of methyltransferases. Nat Rev Cancer. 15(6):334–346. doi: 10.1038/nrc3929.
- Rathert P, Dhayalan A, Ma H, Jeltsch A. 2008. Specificity of protein lysine methyltransferases and methods for detection of lysine methylation of non-histone proteins. Mol Biosyst. 4(12):1186–1190. doi: 10.1039/b811673c.
- Rathert P, Dhayalan A, Murakami M, Zhang X, Tamas R, Jurkowska R, Komatsu Y, Shinkai Y, Cheng X, Jeltsch A. 2008. Protein lysine methyltransferase G9a acts on non-histone targets. Nat Chem Biol. 4(6):344–346. doi: 10.1038/nchembio.88.
- Rhein VF, Carroll J, He J, Ding S, Fearnley IM, Walker JE. 2014. Human METTL20 methylates lysine residues adjacent to the recognition loop of the electron transfer flavoprotein in mitochondria. J Biol Chem. 289(35):24640–24651. doi: 10.1074/jbc.M114.580464.
- Richon VM, Johnston D, Sneeringer CJ, Jin L, Majer CR, Elliston K, Jerva LF, Scott MP, Copeland RA. 2011. Chemogenetic analysis of human protein methyltransferases. Chem Biol Drug Des. 78(2):199–210. doi: 10.1111/j.1747-0285.2011.01135.x.
- Rogawski DS, Ndoj J, Cho HJ, Maillard I, Grembecka J, Cierpicki T. 2015. Two Loops Undergoing Concerted Dynamics Regulate the Activity of the ASH1L Histone Methyltransferase. Biochemistry. 54(35):5401–5413. doi: 10.1021/acs.biochem.5b00697.
- Sabari BR, Zhang D, Allis CD, Zhao Y. 2017. Metabolic regulation of gene expression through histone acylations. Nat Rev Mol Cell Biol. 18(2):90–101. doi: 10.1038/nrm.2016.140.
- Saddic LA, West LE, Aslanian A, Yates JR, Rubin SM, Gozani O, Sage J. 2010. Methylation of the retinoblastoma tumor suppressor by SMYD2. J Biol Chem. 285(48):37733–37740. doi: 10.1074/jbc.M110.137612.
- Sarno F, Nebbioso A, Altucci L. 2020. DOT1L: a key target in normal chromatin remodelling and in mixed-lineage leukaemia treatment. Epigenetics. 15(5):439–453. doi: 10.1080/15592294.2019.1699991.
- Sato K, Kumar A, Hamada K, Okada C, Oguni A, Machiyama A, Sakuraba S, Nishizawa T, Nureki O, Kono H, et al. 2021. Structural basis of the regulation of the normal and oncogenic methylation of nucleosomal histone H3 Lys36 by NSD2. Nat Commun. 12(1):6605. doi: 10.1038/s41467-021-26913-5.
- Saugier-Veber P, Bonnet C, Afenjar A, Drouin-Garraud V, Coubes C, Fehrenbach S, Holder-Espinasse M, Roume J, Malan V, Portnoi MF, et al. 2007. Heterogeneity of NSD1 alterations in 116 patients with Sotos syndrome. Hum Mutat. 28(11):1098–1107. doi: 10.1002/humu.20568.
- Schapira M. 2016. Chemical Inhibition of Protein Methyltransferases. Cell Chem Biol. 23(9):1067–1076. doi: 10.1016/j.chembiol.2016.07.014.
- Schnee P, Choudalakis M, Weirich S, Khella MS, Carvalho H, Pleiss J, Jeltsch A. 2022. Mechanistic basis of the increased methylation activity of the SETD2 protein lysine methyltransferase towards a designed super-substrate peptide. Commun Chem. 5(1):139. doi: 10.1038/s42004-022-00753-w.
- Schuhmacher MK, Beldar S, Khella MS, Bröhm A, Ludwig J, Tempel W, Weirich S, Min J, Jeltsch A. 2020. Sequence specificity analysis of the SETD2 protein lysine methyltransferase and discovery of a SETD2 super-substrate. Commun Biol. 3(1):511. doi: 10.1038/s42003-020-01223-6.
- Schuhmacher MK, Kudithipudi S, Kusevic D, Weirich S, Jeltsch A. 2015. Activity and specificity of the human SUV39H2 protein lysine methyltransferase. Biochim Biophys Acta. 1849(1):55–63. doi: 10.1016/j.bbagrm.2014.11.005.
- Seeler JS, Dejean A. 2017. SUMO and the robustness of cancer. Nat Rev Cancer. 17(3):184–197. doi: 10.1038/nrc.2016.143.
- Senn HM, Thiel W. 2009. QM/MM methods for biomolecular systems. Angew Chem Int Ed Engl. 48(7):1198–1229. doi: 10.1002/anie.200802019.
- Shilatifard A. 2012. The COMPASS family of histone H3K4 methylases: mechanisms of regulation in development and disease pathogenesis. Annu Rev Biochem. 81(1):65–95. doi: 10.1146/annurev-biochem-051710-134100.
- Shinsky SA, Hu M, Vought VE, Ng SB, Bamshad MJ, Shendure J, Cosgrove MS. 2014. A non-active-site SET domain surface crucial for the interaction of MLL1 and the RbBP5/Ash2L heterodimer within MLL family core complexes. J Mol Biol. 426(12):2283–2299. doi: 10.1016/j.jmb.2014.03.011.
- Simon MD, Chu F, Racki LR, de la Cruz CC, Burlingame AL, Panning B, Narlikar GJ, Shokat KM. 2007. The site-specific installation of methyl-lysine analogs into recombinant histones. Cell. 128(5):1003–1012. doi: 10.1016/j.cell.2006.12.041.
- Singh PR, Dadireddy V, Udupa S, Kalladi SM, Shee S, Khosla S, Rajmani RS, Singh A, Ramakumar S, Nagaraja V. 2023. The Mycobacterium tuberculosis methyltransferase Rv2067c manipulates host epigenetic programming to promote its own survival. Nat Commun. 14(1):8497. doi: 10.1038/s41467-023-43940-6.
- Sirinupong N, Brunzelle J, Doko E, Yang Z. 2011. Structural insights into the autoinhibition and posttranslational activation of histone methyltransferase SmyD3. J Mol Biol. 406(1):149–159. doi: 10.1016/j.jmb.2010.12.014.
- Sirinupong N, Brunzelle J, Ye J, Pirzada A, Nico L, Yang Z. 2010. Crystal structure of cardiac-specific histone methyltransferase SmyD1 reveals unusual active site architecture. J Biol Chem. 285(52):40635–40644. doi: 10.1074/jbc.M110.168187.
- Smith BC, Denu JM. 2009. Chemical mechanisms of histone lysine and arginine modifications. Biochim Biophys Acta. 1789(1):45–57. doi: 10.1016/j.bbagrm.2008.06.005.
- Smith E, Lin C, Shilatifard A. 2011. The super elongation complex (SEC) and MLL in development and disease. Genes Dev. 25(7):661–672. doi: 10.1101/gad.2015411.
- Sneeringer CJ, Scott MP, Kuntz KW, Knutson SK, Pollock RM, Richon VM, Copeland RA. 2010. Coordinated activities of wild-type plus mutant EZH2 drive tumor-associated hypertrimethylation of lysine 27 on histone H3 (H3K27) in human B-cell lymphomas. Proc Natl Acad Sci U S A. 107(49):20980–20985. doi: 10.1073/pnas.1012525107.
- Southall SM, Wong PS, Odho Z, Roe SM, Wilson JR. 2009. Structural basis for the requirement of additional factors for MLL1 SET domain activity and recognition of epigenetic marks. Mol Cell. 33(2):181–191. doi: 10.1016/j.molcel.2008.12.029.
- Spangler CJ, Yadav SP, Li D, Geil CN, Smith CB, Wang GG, Lee TH, McGinty RK. 2022. DOT1L activity in leukemia cells requires interaction with ubiquitylated H2B that promotes productive nucleosome binding. Cell Rep. 38(7):110369. doi: 10.1016/j.celrep.2022.110369.
- Spellmon N, Holcomb J, Trescott L, Sirinupong N, Yang Z. 2015. Structure and function of SET and MYND domain-containing proteins. Int J Mol Sci. 16(1):1406–1428. doi: 10.3390/ijms16011406.
- Spellmon N, Sun X, Sirinupong N, Edwards B, Li C, Yang Z. 2015. Molecular Dynamics Simulation Reveals Correlated Inter-Lobe Motion in Protein Lysine Methyltransferase SMYD2. PLoS One. 10(12):e0145758. doi: 10.1371/journal.pone.0145758.
- Stender JD, Pascual G, Liu W, Kaikkonen MU, Do K, Spann NJ, Boutros M, Perrimon N, Rosenfeld MG, Glass CK. 2012. Control of proinflammatory gene programs by regulated trimethylation and demethylation of histone H4K20. Mol Cell. 48(1):28–38. doi: 10.1016/j.molcel.2012.07.020.
- Stodola TJ, Chi Y-I, De Assuncao TM, Leverence EN, Tripathi S, Dsouza NR, Mathison AJ, Volkman BF, Smith BC, Lomberk G, et al. 2021. Computational modeling reveals key molecular properties and dynamic behavior of disruptor of telomeric silencing 1-like (DOT1L) and partnering complexes involved in leukemogenesis. Proteins. 90(1):282–298. doi: 10.1002/prot.26219.
- Subtil-Rodríguez A, Vázquez-Chávez E, Ceballos-Chávez M, Rodríguez-Paredes M, Martín-Subero JI, Esteller M, Reyes JC. 2014. The chromatin remodeller CHD8 is required for E2F-dependent transcription activation of S-phase genes. Nucleic Acids Res. 42(4):2185–2196. doi: 10.1093/nar/gkt1161.
- Suh JL, Barnash KD, Abramyan TM, Li F, The J, Engelberg IA, Vedadi M, Brown PJ, Kireev DB, Arrowsmith CH, et al. 2019. Discovery of selective activators of PRC2 mutant EED-I363M. Sci Rep. 9(1):6524. doi: 10.1038/s41598-019-43005-z.
- Sun J, Li Z, Yang N. 2021. Mechanism of the Conformational Change of the Protein Methyltransferase SMYD3: A Molecular Dynamics Simulation Study. Int J Mol Sci. 22(13)
- Sun J, Shi F, Yang N. 2019. Exploration of the Substrate Preference of Lysine Methyltransferase SMYD3 by Molecular Dynamics Simulations. ACS Omega. 4(22):19573–19581. doi: 10.1021/acsomega.9b01842.
- Tachibana M, Sugimoto K, Fukushima T, Shinkai Y. 2001. Set domain-containing protein, G9a, is a novel lysine-preferring mammalian histone methyltransferase with hyperactivity and specific selectivity to lysines 9 and 27 of histone H3. J Biol Chem. 276(27):25309–25317. doi: 10.1074/jbc.M101914200.
- Tatton-Brown K, Douglas J, Coleman K, Baujat G, Cole TR, Das S, Horn D, Hughes HE, Temple IK, Faravelli F, et al. 2005. Genotype-phenotype associations in Sotos syndrome: an analysis of 266 individuals with NSD1 aberrations. Am J Hum Genet. 77(2):193–204. doi: 10.1086/432082.
- Tisi D, Chiarparin E, Tamanini E, Pathuri P, Coyle JE, Hold A, Holding FP, Amin N, Martin ACL, Rich SJ, et al. 2016. Structure of the Epigenetic Oncogene MMSET and Inhibition by N-Alkyl Sinefungin Derivatives. ACS Chem Biol. 11(11):3093–3105. doi: 10.1021/acschembio.6b00308.
- Trievel RC, Beach BM, Dirk LMA, Houtz RL, Hurley JH. 2002. Structure and Catalytic Mechanism of a SET Domain Protein Methyltransferase. Cell. 111(1):91–103. doi: 10.1016/s0092-8674(02)01000-0.
- Trievel RC, Flynn EM, Houtz RL, Hurley JH. 2003. Mechanism of multiple lysine methylation by the SET domain enzyme Rubisco LSMT. Nat Struct Biol. 10(7):545–552. doi: 10.1038/nsb946.
- Trzesniak D, Kunz AP, van Gunsteren WF. 2007. A comparison of methods to compute the potential of mean force. Chemphyschem. 8(1):162–169. doi: 10.1002/cphc.200600527.
- Ueda T, Sanada M, Matsui H, Yamasaki N, Honda ZI, Shih LY, Mori H, Inaba T, Ogawa S, Honda H. 2012. EED mutants impair polycomb repressive complex 2 in myelodysplastic syndrome and related neoplasms. Leukemia. 26(12):2557–2560. doi: 10.1038/leu.2012.146.
- Valencia-Sánchez MI, De Ioannes P, Wang M, Truong DM, Lee R, Armache J-P, Boeke JD, Armache K-J. 2021. Regulation of the Dot1 histone H3K79 methyltransferase by histone H4K16 acetylation. Science. 371(6527):eabc6663. doi: 10.1126/science.abc6663.
- Valencia-Sánchez MI, De Ioannes P, Wang M, Vasilyev N, Chen R, Nudler E, Armache J-P, Armache K-J. 2019. Structural Basis of Dot1L Stimulation by Histone H2B Lysine 120 Ubiquitination. Mol Cell. 74(5):1010–1019.e6. e1016. doi: 10.1016/j.molcel.2019.03.029.
- Van Aller GS, Graves AP, Elkins PA, Bonnette WG, McDevitt PJ, Zappacosta F, Annan RS, Dean TW, Su D-S, Carpenter CL, et al. 2016. Structure-Based Design of a Novel SMYD3 Inhibitor that Bridges the SAM-and MEKK2-Binding Pockets. Structure. 24(5):774–781. doi: 10.1016/j.str.2016.03.010.
- Van der Kamp MW, Mulholland AJ. 2013. Combined quantum mechanics/molecular mechanics (QM/MM) methods in computational enzymology. Biochemistry. 52(16):2708–2728. doi: 10.1021/bi400215w.
- Voigt P, LeRoy G, Drury WJ, Zee BM, Son J, Beck DB, Young NL, Garcia BA, Reinberg D. 2012. Asymmetrically modified nucleosomes. Cell. 151(1):181–193. doi: 10.1016/j.cell.2012.09.002.
- Vougiouklakis T, Bernard BJ, Nigam N, Burkitt K, Nakamura Y, Saloura V. 2020. Clinicopathologic significance of protein lysine methyltransferases in cancer. Clin Epigenetics. 12(1):146. doi: 10.1186/s13148-020-00897-3.
- Waggoner DJ, Raca G, Welch K, Dempsey M, Anderes E, Ostrovnaya I, Alkhateeb A, Kamimura J, Matsumoto N, Schaeffer GB, et al. 2005. NSD1 analysis for Sotos syndrome: insights and perspectives from the clinical laboratory. Genet Med. 7(8):524–533. doi: 10.1097/01.GIM.0000178503.15559.d3.
- Wang L, Li L, Zhang H, Luo X, Dai J, Zhou S, Gu J, Zhu J, Atadja P, Lu C, et al. 2011. Structure of human SMYD2 protein reveals the basis of p53 tumor suppressor methylation. J Biol Chem. 286(44):38725–38737. doi: 10.1074/jbc.M111.262410.
- Wang S, Hu P, Zhang Y. 2007. Ab Initio Quantum Mechanical/Molecular Mechanical Molecular Dynamics Simulation of Enzyme Catalysis: The Case of Histone Lysine Methyltransferase SET7/9. J Phys Chem B. 111(14):3758–3764. doi: 10.1021/jp067147i.
- Waterhouse A, Bertoni M, Bienert S, Studer G, Tauriello G, Gumienny R, Heer FT, de Beer TAP, Rempfer C, Bordoli L, et al. 2018. SWISS-MODEL: homology modelling of protein structures and complexes. Nucleic Acids Res. 46(W1):W296–W303. doi: 10.1093/nar/gky427.
- Weirich S, Jeltsch A. 2022. Specificity Analysis of Protein Methyltransferases and Discovery of Novel Substrates Using SPOT Peptide Arrays. In: Margueron R, Holoch D, editors. Histone Methyltransferases: Methods and Protocols. New York, NY: Springer US; p. 313–325.
- Weirich S, Kudithipudi S, Jeltsch A. 2016. Specificity of the SUV4-20H1 and SUV4-20H2 protein lysine methyltransferases and methylation of novel substrates. J Mol Biol. 428(11):2344–2358. doi: 10.1016/j.jmb.2016.04.015.
- Weirich S, Kudithipudi S, Jeltsch A. 2017. Somatic cancer mutations in the MLL1 histone methyltransferase modulate its enzymatic activity and dependence on the WDR5/RBBP5/ASH2L complex. Mol Oncol. 11(4):373–387. doi: 10.1002/1878-0261.12041.
- Weirich S, Kudithipudi S, Kycia I, Jeltsch A. 2015. Somatic cancer mutations in the MLL3-SET domain alter the catalytic properties of the enzyme. Clin Epigenetics. 7(1):36. doi: 10.1186/s13148-015-0075-3.
- Weirich S, Schuhmacher MK, Kudithipudi S, Lungu C, Ferguson AD, Jeltsch A. 2020. Analysis of the Substrate Specificity of the SMYD2 Protein Lysine Methyltransferase and Discovery of Novel Non-Histone Substrates. Chembiochem. 21(1-2):256–264. doi: 10.1002/cbic.201900582.
- Wiederstein JL, Nolte H, Günther S, Piller T, Baraldo M, Kostin S, Bloch W, Schindler N, Sandri M, Blaauw B, et al. 2018. Skeletal Muscle-Specific Methyltransferase METTL21C Trimethylates p97 and Regulates Autophagy-Associated Protein Breakdown. Cell Rep. 23(5):1342–1356. doi: 10.1016/j.celrep.2018.03.136.
- Wilson JR, Jing C, Walker PA, Martin SR, Howell SA, Blackburn GM, Gamblin SJ, Xiao B. 2002. Crystal Structure and Functional Analysis of the Histone Methyltransferase SET7/9. Cell. 111(1):105–115. doi: 10.1016/s0092-8674(02)00964-9.
- Worden EJ, Hoffmann NA, Hicks CW, Wolberger C. 2019. Mechanism of Cross-talk between H2B Ubiquitination and H3 Methylation by Dot1L. Cell. 176(6):1490–1501.e12. e1412. doi: 10.1016/j.cell.2019.02.002.
- Worden EJ, Zhang X, Wolberger C. 2020. Structural basis for COMPASS recognition of an H2B-ubiquitinated nucleosome. Elife. 9 doi: 10.7554/eLife.53199.
- Wu H, Mathioudakis N, Diagouraga B, Dong A, Dombrovski L, Baudat F, Cusack S, de Massy B, Kadlec J. 2013. Molecular basis for the regulation of the H3K4 methyltransferase activity of PRDM9. Cell Rep. 5(1):13–20. doi: 10.1016/j.celrep.2013.08.035.
- Wu H, Min J, Lunin VV, Antoshenko T, Dombrovski L, Zeng H, Allali-Hassani A, Campagna-Slater V, Vedadi M, Arrowsmith CH, et al. 2010. Structural biology of human H3K9 methyltransferases. PLoS One. 5(1):e8570. doi: 10.1371/journal.pone.0008570.
- Wu J, Cheung T, Grande C, Ferguson AD, Zhu X, Theriault K, Code E, Birr C, Keen N, Chen H. 2011. Biochemical characterization of human SET and MYND domain-containing protein 2 methyltransferase. Biochemistry. 50(29):6488–6497. doi: 10.1021/bi200725p.
- Wu K, Niu C, Liu H, Fu L. 2023. Research progress on PRMTs involved in epigenetic modification and tumour signalling pathway regulation (Review). Int J Oncol. 62(5) doi: 10.3892/ijo.2023.5510.
- Xiao B, Jing C, Kelly G, Walker PA, Muskett FW, Frenkiel TA, Martin SR, Sarma K, Reinberg D, Gamblin SJ, et al. 2005. Specificity and mechanism of the histone methyltransferase Pr-Set7. Genes Dev. 19(12):1444–1454. doi: 10.1101/gad.1315905.
- Xiao B, Jing C, Wilson JR, Walker PA, Vasisht N, Kelly G, Howell S, Taylor IA, Blackburn GM, Gamblin SJ. 2003. Structure and catalytic mechanism of the human histone methyltransferase SET7/9. Nature. 421(6923):652–656. doi: 10.1038/nature01378.
- Xu S, Aguilar A, Huang L, Xu T, Zheng K, McEachern D, Przybranowski S, Foster C, Zawacki K, Liu Z, et al. 2020. Discovery of M-808 as a Highly Potent, Covalent, Small-Molecule Inhibitor of the Menin-MLL Interaction with Strong In Vivo Antitumor Activity. J Med Chem. 63(9):4997–5010. doi: 10.1021/acs.jmedchem.0c00547.
- Xu S, Wu J, Sun B, Zhong C, Ding J. 2011. Structural and biochemical studies of human lysine methyltransferase Smyd3 reveal the important functional roles of its post-SET and TPR domains and the regulation of its activity by DNA binding. Nucleic Acids Res. 39(10):4438–4449. doi: 10.1093/nar/gkr019.
- Xu S, Zhong C, Zhang T, Ding J. 2011. Structure of human lysine methyltransferase Smyd2 reveals insights into the substrate divergence in Smyd proteins. J Mol Cell Biol. 3(5):293–300. doi: 10.1093/jmcb/mjr015.
- Xue H, Yao T, Cao M, Zhu G, Li Y, Yuan G, Chen Y, Lei M, Huang J. 2019. Structural basis of nucleosome recognition and modification by MLL methyltransferases. Nature. 573(7774):445–449. doi: 10.1038/s41586-019-1528-1.
- Yang S, Zheng X, Lu C, Li GM, Allis CD, Li H. 2016. Molecular basis for oncohistone H3 recognition by SETD2 methyltransferase. Genes Dev. 30(14):1611–1616. doi: 10.1101/gad.284323.116.
- Yang T, Zhang W, Cheng J, Nie Y, Xin Q, Yuan S, Dou Y. 2019. Formation Mechanism of Ion Channel in Channelrhodopsin-2: Molecular Dynamics Simulation and Steering Molecular Dynamics Simulations. Int J Mol Sci. 20(15)
- Yao J, Chu Y, An R, Guo H. 2012. Understanding product specificity of protein lysine methyltransferases from QM/MM molecular dynamics and free energy simulations: the effects of mutation on SET7/9 beyond the Tyr/Phe switch. J Chem Inf Model. 52(2):449–456. doi: 10.1021/ci200364m.
- Yao Y, Chen P, Diao J, Cheng G, Deng L, Anglin JL, Prasad BV, Song Y. 2011. Selective inhibitors of histone methyltransferase DOT1L: design, synthesis, and crystallographic studies. J Am Chem Soc. 133(42):16746–16749. doi: 10.1021/ja206312b.
- Yap DB, Chu J, Berg T, Schapira M, Cheng SW, Moradian A, Morin RD, Mungall AJ, Meissner B, Boyle M, et al. 2011. Somatic mutations at EZH2 Y641 act dominantly through a mechanism of selectively altered PRC2 catalytic activity, to increase H3K27 trimethylation. Blood. 117(8):2451–2459. doi: 10.1182/blood-2010-11-321208.
- Yi Y, Ge S. 2022. Targeting the histone H3 lysine 79 methyltransferase DOT1L in MLL-rearranged leukemias. J Hematol Oncol. 15(1):35. doi: 10.1186/s13045-022-01251-1.
- Yoneda S, Saito T, Nakajima D, Watanabe G. 2021. Potential of mean force and umbrella sampling simulation for the transport of 5-oxazolidinone in heterotetrameric sarcosine oxidase. Proteins. 89(7):811–818. doi: 10.1002/prot.26060.
- You W, Tang Z, Chang CA. 2019. Potential Mean Force from Umbrella Sampling Simulations: What Can We Learn and What Is Missed? J Chem Theory Comput. 15(4):2433–2443. doi: 10.1021/acs.jctc.8b01142.
- Yu H, Dalby PA. 2020. A beginner’s guide to molecular dynamics simulations and the identification of cross-correlation networks for enzyme engineering. Methods Enzymol. 643:15–49. doi: 10.1016/bs.mie.2020.04.020.
- Yue L, Du J, Ye F, Chen Z, Li L, Lian F, Zhang B, Zhang Y, Jiang H, Chen K, et al. 2016. Identification of novel small-molecule inhibitors targeting menin-MLL interaction, repurposing the antidiarrheal loperamide. Org Biomol Chem. 14(36):8503–8519. doi: 10.1039/c6ob01248e.
- Zhang R, Li X, Liang Z, Zhu K, Lu J, Kong X, Ouyang S, Li L, Zheng YG, Luo C. 2013. Theoretical insights into catalytic mechanism of protein arginine methyltransferase 1. PLoS One. 8(8):e72424. doi: 10.1371/journal.pone.0072424.
- Zhang X, Bruice T. 2007a. Catalytic Mechanism and Product Specificity of Rubisco Large Subunit Methyltransferase: QM/MM and MD Investigations. Biochemistry. 46(18):5505–5514. doi: 10.1021/bi700119p.
- Zhang X, Bruice T. 2007b. Histone Lysine Methyltransferase SET7/9: Formation of a Water Channel Precedes Each Methyl Transfer. Biochemsitry. 46(51):14838–14844. doi: 10.1021/bi7014579.
- Zhang X, Bruice T. 2007c. A Quantum Mechanics/Molecular Mechanics Study of the Catalytic Mechanism and Product Specificity of Viral Histone Lysine Methyltransferase. Biochemistry. 46(34):9743–9751. doi: 10.1021/bi700515q.
- Zhang X, Bruice T. 2008a. Enzymatic mechanism and product specificity of SET-domain protein lysine methyltransferases. PNAS. 105
- Zhang X, Bruice T. 2008b. Mechanism of Product Specificity of AdoMet Methylation Catalyzed by Lysine Methyltransferases: Transcriptional Factor p53 Methylation by Histone Lysine Methyltransferase SET7/9. Biochemistry. 47(9):2743–2748. doi: 10.1021/bi702370p.
- Zhang X, Bruice T. 2008c. Product Specificity and Mechanism of Protein Lysine Methyltransferases: Insights from the Histone Lysine Methyltransferase SET8. Biochemistry. 47(25):6671–6677. doi: 10.1021/bi800244s.
- Zhang X, Huang Y, Shi X. 2015. Emerging roles of lysine methylation on non-histone proteins. Cell Mol Life Sci. 72(22):4257–4272. doi: 10.1007/s00018-015-2001-4.
- Zhang X, Tamaru H, Khan SI, Horton JR, Keefe LJ, Selker EU, Chen X. 2002. Structure of the Neurospora SET domain protein DIM-5, a histone H3 lysine methyltransferase. Cell. 111(1):117–127. doi: 10.1016/s0092-8674(02)00999-6.
- Zhang X, Yang Z, Khan SI, Horton JR, Tamaru H, Selker EU, Cheng X. 2003. Structural basis for the product specificity of histone lysine methyltransferases. Mol Cell. 12(1):177–185. doi: 10.1016/s1097-2765(03)00224-7.
- Zhang Y, Alshammari E, Sobota J, Yang A, Li C, Yang Z. 2022. Unique SMYD5 Structure Revealed by AlphaFold Correlates with Its Functional Divergence. Biomolecules. 12(6) doi: 10.3390/biom12060783.
- Zhang Y, Hayden S, Spellmon N, Xue W, Martin K, Muzzarelli K, Kovari L, Yang Z. 2021. Sperm chromatin-condensing protamine enhances SMYD5 thermal stability. Biochem Biophys Res Commun. 550:1–7. doi: 10.1016/j.bbrc.2021.02.073.
- Zhang Y, Shan CM, Wang J, Bao K, Tong L, Jia S. 2017. Molecular basis for the role of oncogenic histone mutations in modulating H3K36 methylation. Sci Rep. 7(1):43906. doi: 10.1038/srep43906.
- Zhao YY, Deng H, Rahman A, Xu XL, Qian P, Guo H. 2022. Computational Study of Methionine Methylation Process Catalyzed by SETD3. Interdiscip Sci. 14(4):929–936. doi: 10.1007/s12539-022-00516-0.
- Zheng W, Ibáñez G, Wu H, Blum G, Zeng H, Dong A, Li F, Hajian T, Allali-Hassani A, Amaya MF, et al. 2012. Sinefungin derivatives as inhibitors and structure probes of protein lysine methyltransferase SETD2. J Am Chem Soc. 134(43):18004–18014. doi: 10.1021/ja307060p.
- Zhou H, Liu L, Huang J, Bernard D, Karatas H, Navarro A, Lei M, Wang S. 2013. Structure-based design of high-affinity macrocyclic peptidomimetics to block the menin-mixed lineage leukemia 1 (MLL1) protein-protein interaction. J Med Chem. 56(3):1113–1123. doi: 10.1021/jm3015298.
- Zhou R, Xie Y, Hu H, Hu G, Patel VS, Zhang J, Yu K, Huang Y, Jiang H, Liang Z, et al. 2015. Molecular Mechanism underlying PRMT1 Dimerization for SAM Binding and Methylase Activity. J Chem Inf Model. 55(12):2623–2632. doi: 10.1021/acs.jcim.5b00454.
- Zoabi M, Zhang L, Li TM, Elias JE, Carlson SM, Gozani O. 2020. Methyltransferase-like 21C (METTL21C) methylates alanine tRNA synthetase at Lys-943 in muscle tissue. J Biol Chem. 295(33):11822–11832. doi: 10.1074/jbc.RA120.014505.