
Abstract
Background
Blocking Transient Receptor Potential Melastatin 4 (TRPM4) in rodents by our antibody M4P has shown to attenuate cerebral ischaemia-reperfusion injury. Since M4P does not interact with human TRPM4, the therapeutic potential of blocking human TRPM4 remains unclear. We developed a monoclonal antibody M4M that inhibited human TRPM4 in cultured cells. However, M4M has no effect on stroke outcome in wild-type rats. Therefore, M4M needs to be evaluated on animal models expressing human TRPM4.
Methods
We generated a humanised rat model using the CRISPR/Cas technique to knock-in (KI) the human TRPM4 antigen sequence.
Results
In primary neurons from human TRPM4 KI rats, M4M binds to hypoxic neurons, but not normoxic nor wild-type neurons. Electrophysiological studies showed that M4M blocked ATP depletion-induced activation of TRPM4 and inhibited hypoxia-associated cell volume increase. In a stroke model, administration of M4M reduced infarct volume in KI rats. Rotarod test and Neurological deficit score revealed improvement following M4M treatment.
Conclusion
M4M selectively binds and inhibits hypoxia-induced human TRPM4 channel activation in neurons from the humanised rat model, with no effect on healthy neurons. Use of M4M in stroke rats showed functional improvements, suggesting the potential for anti-human TRPM4 antibodies in treating acute ischaemic stroke patients.
Introduction
Transient receptor potential melastatin member 4 (TRPM4) is a voltage-dependent, non-selective monovalent cation channel activated by intracellular Ca2+ and ATP-depletion [Citation1]. TRPM4 can be found in the central nervous system [Citation2], supporting bursts of action potentials of interneurons from the pre-Bötzinger complex [Citation3]. After stroke, aberrant expression and activation of TRPM4 have been implicated in brain tissue damage [Citation4, Citation5]. Therefore, TRPM4 inhibition is considered a potential therapeutic approach to manage stroke. Previous studies using TRPM4-specific siRNA have verified that TRPM4 inhibition could improve stroke outcome in both permanent and transient stroke models [Citation6, Citation7].
To achieve an inhibitory effect, siRNA needs to be administered before TRPM4 protein upregulation. We have shown previously that TRPM4 is upregulated as early as 2 h after stroke onset [Citation6]. Such a narrow time window limits the application of siRNA for stroke management. Therefore, it is critical to develop a direct blocker to inhibit the channel function. However, there are various concerns regarding current available TRPM4 blockers, such as lack of specificity, requiring associated subunits, or toxicity [Citation8]. Recently, we have developed a polyclonal antibody M4P against a 28-amino acid polypeptide of rat TRPM4 channel [Citation9]. This polypeptide lies between the transmembrane segments 5 and 6 of TRPM4, which is close to the channel pore. Our electrophysiological study revealed that the binding of M4P could inhibit TRPM4 activity. Importantly, application of M4P attenuated reperfusion injury in a rat stroke model [Citation9], demonstrating a therapeutic potential for the TRPM4 blocking antibody. By binding directly to TRPM4 channels, M4P has a wider therapeutic time window as compared to siRNA.
M4P was designed to target the rat TRPM4 channel. It was found to have no effect on the human TRPM4 channel because the antigenic sequence of rat TRPM4 is different from that of human TRPM4 [Citation9, Citation10]. Thus, we generated a mouse monoclonal antibody M4M against the human TRPM4 channel, targeting a similar epitope between transmembrane segments 5 and 6 of human TRPM4 [Citation10]. We found that M4M could bind to the human TRPM4 channel on the surface of live cells. In Human Brain Microvascular Endothelial Cells (HBMECs), M4M successfully inhibited human TRPM4 current and ameliorated hypoxia-induced cell swelling [Citation10]. When M4M was tested on wild-type (WT) rats, no functional improvement was observed after stroke induction [Citation10]. To evaluate the in vivo effect of M4M, it is necessary to use animal models carrying human TRPM4 sequences. In this study, we generated a novel transgenic rat model carrying the human TRPM4 sequence that is critical for M4M binding. Our study aims to assess the efficacy of the M4M antibody in inhibiting hypoxia-induced TRPM4 activation and improving functional outcomes in the humanised stroke rat model. Our results provide an insight into the potential clinical application of the TRPM4 blocking antibody for stroke management.
Methods
Generation of TRPM4 humanised rat carrying human TRPM4 sequence
The humanised TRPM4 rat model was produced by Cyagen Biosciences (Suzhou, China) using the In-Fusion cloning technology [Citation11–14]. The rat TRPM4 gene (GenBank accession number: NM_001136229.1; Ensembl: ENSRNOG00000020714) is located on rat chromosome 1. Rat TRPM4 contains 25 exons and Exon 1 was selected as target site for CRISPR/Cas-mediated genome engineering. Following off-target analysis of potential gRNAs, one gRNA with the sequence of AGAGCAGGTATCGCACAGCGCGG was selected. The gRNAs targeting vectors were constructed and confirmed by sequencing. The donor vector which is flanked by homologous arms was constructed too, containing the rat TRPM4 coding sequences (CDS) in which the antibody binding sequence QDRSLPSILRRVFYRPYLQIFGQIPQEEMDVALMNPSNCSAERGSWAHPEGPV was replaced by the corresponding human sequence RDSDFPSILRRVFYRPYLQIFGQIPQEDMDVALMEHSNCSSEPGFWAHPPGAQ. After confirming with DNA sequencing, Cas9 mRNA and gRNA generated by in vitro transcription were co-injected into fertilised eggs with donor vector for knock-in (KI) rat production using Sprague-Dawley (SD) rats. The pups were genotyped using polymerase chain reaction (PCR) followed by sequence analysis.
Genotyping PCR
The rat tail snips were collected and digested using DirectPCR Lysis Reagent (101-T; Viagen Biotech, CA, USA) and Proteinase K (3115828001; Roche, Basel, Switzerland) overnight at 55 °C according to the manufacturer’s protocol. Following digestion, the samples were heated to 85 °C for 1 h. PCR was performed using 5 μL of each digested sample with GoTaq® G2 Green Master Mix (M7823; Promega, WI, USA) following the manufacturer’s instructions. Primers used for PCR-based genotyping were forward primer 5ʹ-AGAGTCTGTCCCTGCTCGCAG-3ʹ; reverse primer 5ʹ- GGTAAGACCCACGGGATTCG-3ʹ to identify the Rat TRPM4-containing allele and forward primer 5ʹ-TTCGAGCACTTCCGTGTCTGTCTC-3ʹ; reverse primer 5ʹ-TGCCTATGGACCCCACTCCTC-3ʹ to identify the Human TRPM4-containing allele. PCR was performed using a standard thermocycler (Biometra T-gradient Thermoblock, Jena, Germany). The PCR conditions were as follows: initial denaturation (95 °C for 3 min), followed by 34 cycles of denaturation (94 °C for 15 s), annealing (60 °C for 15 s) and extension (72 °C for 1 min) and a final extension at 72 °C for 5 min. The PCR products were separated on a 2% agarose gel run at 110 V and imaged on the Chemi XRS Gel Documentation System (Bio-Rad, CA, USA).
Western blot
Radioimmunoprecipitation assay buffer (150 mM NaCl; 1% IGEPAL CA-630; 0.5% sodium deoxycholate; 0.1% sodium dodecyl sulphate; 50 mM Tris pH8.0; 1 mM EDTA; 1 mM EGTA) containing Mini Protease Inhibitor (04693124001; Roche) was used for homogenisation and lysis of the rat brain tissues for western blot assay as described previously [Citation6, Citation7]. The protein concentration was measured following the protocol from the Pierce BCA Protein Assay Kit (23227; Thermo Fisher). 100 μg of total protein was resolved on 10% SDS-PAGE gels at 80 V, and electrophoretically transferred to Immun-Blot PVDF membranes (1620177; Bio-Rad) at 110 V for 2 h at 4 °C. The membrane was then added with StartingBlock blocking buffer (37538; Thermo Fisher) containing 0.05% Tween®20 for 1 h at room temperature, followed by overnight incubation of the membranes at 4 °C with the mouse M4M antibody at 1:500 dilution and anti-Actin (A1978; Sigma-Aldrich) at 1:5000 dilution. After which, the membranes were washed three times with phosphate buffered saline (PBS) containing 0.1% Tween 20 (PBS-T20) and incubated with the anti-mouse IgG secondary antibody (A4416; Sigma-Aldrich) for 1 h at room temperature. The primary and secondary antibodies were prepared in StartingBlock blocking buffer with 0.05% Tween®20. The blot was washed three times with PBS-T20 prior to detection. The protein bands were detected using the Amersham ECL Western Blotting Analysis System (RPN2109; GE Healthcare, IL, USA) and visualised using the ChemiDocTM MP Imaging System (Bio-Rad, CA, USA).
Middle cerebral artery occlusion (MCAO) model and experimental protocol
All animal procedures were approved by the Institutional Animal Care and Use Committee (protocol no. A19099) of Lee Kong Chian School of Medicine and performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines 2.0 [Citation15]. Allocation of animal treatment was randomised by rolling a dice. All researchers involved in the study were blinded to the intervention. Focal cerebral ischaemia was induced in adult rats by intraluminal middle cerebral artery occlusion (MCAO) as described previously [Citation6, Citation9]. In brief, male transgenic SD rats weighing approximately 250–280 g were anaesthetised with ketamine (75 mg/kg) and xylazine (10 mg/kg) intraperitoneally. The left common carotid artery (CCA), internal carotid artery (ICA), and external carotid artery (ECA) were dissected out, and a 0.37 mm silicon-coated filament (403756PK10; Doccol Corp, CA, USA) was inserted into the left ICA. Cerebral blood flow was monitored by a Laser-Doppler flowmetry (moorVMSPflugers LDF2™, Moor Instruments Inc., DE, USA). Animals with ≥ 70% cerebral blood flow reduction were included for analysis. At 2 h after occlusion, 100 μg of M4M antibody or control mouse IgG was injected intravenously via tail vein at a final volume of 300 µL. At 3 h following occlusion, the filament was removed gently to achieve reperfusion. This 1-h interval was selected to mimic the clinical scenario of reperfusion therapy [Citation8]. During the operation, the heart rate, blood pressure, and rectal temperature were monitored using the PowerLab 4/35 from AD Instruments. The body temperature was maintained at 37 ± 0.5 °C. The total number of rats was 111, of which 16 were excluded from the study due to achieving less than 70% reduction in blood flow (n = 11) or animal death during surgery (n = 5). The overall mortality rate was 13.6% for this study.
Primary culture of cortical neurons
The cortices from embryonic day 15-18 (E15-18) rat pups were dissected and prepared into primary culture of cortical neurons according to the manufacturer’s instructions (LK003150, Worthington Biochemical Corporation, NJ, USA). The tissues were digested for 40 min in Earle’s Balanced Salt Solution containing 20 U/mL papain. The dissociated cells were centrifuged at 300 xg for 5 min, followed by re-suspension of the pellet in albumin-ovomucoid inhibitor/DNase solution. The cell suspension was layered on top of the discontinuous density gradient (albumin-ovomucoid inhibitor), followed by centrifugation at 70 xg for 6 min. The cell pellet was re-suspended in the Neurobasal (NB) culture medium (21103049, Thermo Fisher Scientific) with 2% (v/v) B27 supplement containing 100 IU/mL penicillin, 100 μg/mL streptomycin, and 2 mM glutamax, after which the cells were filtered through a 70 μm cell strainer. The neurons were counted and seeded on 12 mm round glass coverslips that were coated with poly-D-lysine hydrobromide (Sigma Aldrich). The neurons were cultured in NB medium and the was changed every 3 days. The cells were treated with 2 μM cytosine arabinoside from 3-6 days in vitro to limit mitotic cell proliferation and maintained for 10–21 days prior to experimentation.
Surface labelling and immunocytochemistry
Primary cortical neurons were immunostained as described in our previous study [Citation10]. The neurons were fixed for 20 min with 4% (v/v) paraformaldehyde (PFA) prepared in PBS. After washing 3 times with PBS, mouse M4M antibody or control mouse IgG at 30 μg/mL dilution was added on the coverslips and incubated for 1 h at room temperature. The neurons were then washed 3 times with PBS and were then permeabilized with 0.1% (v/v) Triton X-100 in PBS (PBST) for 20 min at room temperature. After washing 3 times with PBS, the slides were blocked with 5% foetal bovine serum (FBS) prepared in PBS (FBS/PBS) for 1 h. The cells were then immunolabeled with rabbit anti-MAP2 antibody (M3696; Sigma-Aldrich) prepared at 1:200 dilution in 5% FBS/PBS for 1 h at room temperature. The coverslips were then washed three times with PBS, and incubated for 1 h at room temperature with the appropriate Alexa Fluor® Dyes secondary antibodies prepared at 1:200 dilution in 5% FBS/PBS. The nuclei were counterstained with DAPI (4ʹ,6-diamidino-2-phenylindole) at 1:3000 dilution for 5 min. The coverslips were mounted onto glass slides using the FluorSave™ reagent (Merck, NJ, USA). Fluorescent images were captured with a laser scanning confocal microscope system (Fluoview FV31S-SW, Olympus, Tokyo, Japan).
Immunohistochemistry
Rats were anaesthetised with ketamine (75 mg/kg) and xylazine (10 mg/kg) and transcardially perfused with PBS followed by fixation with 4% PFA. The brains were harvested and cryosectioned at 30 µm of thickness. The sections were then used for immunofluorescence staining following the standard protocol [Citation6, Citation7] with slight modifications. After washing twice with PBST, the sections were incubated with blocking serum (10% FBS in PBST) for 1 h at room temperature. Following which, the sections were incubated with primary antibodies diluted in 5% FBS prepared in PBST (FBS/PBST). The primary antibodies used in the study were mouse anti-TRPM4 antibody (TA500381; Origene) at 1:100 dilution, mouse M4M antibody at 100 μg/mL dilution, rabbit anti-von Willebrand Factor (vWF) antibody (AB7356; Merck Millipore) at 1:300 dilution, rabbit anti-NeuN antibody (ABN78; Merck Millipore) at 1:300 dilution and rabbit anti-glial fibrillary acidic protein (GFAP) antibody (G9269; Sigma Aldrich) at 1:300 dilution. After overnight incubation at 4 °C, the sections were washed 3 times with PBST and then incubated for 1 h at room temperature with the appropriate Alexa Fluor® Dyes secondary antibodies diluted at 1:200 dilution in 5% FBS/PBST. The sections were washed 3 times with PBS, followed by counterstaining of nuclei by DAPI at 1:3000 dilution for 5 min. Lastly, the slides were mounted with cover glasses using the FluorSave™ and visualised with a laser scanning confocal microscope system.
Infarct volume measurement
2,3,4-triphenyltetrazolium chloride (TTC) staining was used to differentiate between metabolically active and inactive tissues after MCAO [Citation8]. In brief, the rats were sacrificed 24 h after occlusion. The brains were harvested and sectioned into eight slices with 2 mm in thickness. After incubation for 30 min at 37 °C with 0.1% TTC (T4375, Sigma-Aldrich, MI, USA), the brain slices were scanned with an image analyser system (HP Scanjet G3110, HP Inc, CA, USA) and infarction was analysed using ImageJ. Calculation of oedema-corrected infarct volume was performed as described previously [Citation16].
Neurological assessment
Two neurological tests were used to assess motor and behavioural deficits after MCAO. Motor function was evaluated using a rotarod apparatus (Ugo Basile, Gemonio, Italy) as described previously [Citation6, Citation7]. The impairment of motor function was quantified by observing the latency with which the rats fell off the apparatus. Before stroke induction, the rats received three training trials with 15-min intervals each day for five consecutive days. The rotarod was set to accelerate from 4 to 80 rpm within 10 min. One day before operation, the mean duration of time that the animals remained on the device was recorded as an internal baseline control. After surgery, the mean duration of latency was recorded at different time points for comparison among different treatments. The Bederson scale is a global neurological assessment to measure neurological deficit [Citation17]. The assessment includes forelimb flexion, resistance to lateral push and circling behaviour. For each test, the rats were graded on a scale of 0-3 for evaluating the global neurological severity. The Bederson scale was obtained at different time points post-MCAO for comparison.
Electrophysiology
Whole-cell patch clamp was used to evaluate the effect of TRPM4 blocking antibody on primary cultured cortical neurons. Patch electrodes were pulled using a Flaming/Brown micropipette puller (P-1000, Sutter Instrument, CA, USA) and polished with a microforge (MF-200, WPI Inc. FL, USA). Whole-cell currents were recorded using a patch clamp amplifier (Multiclamp 700B equipped with Digidata 1440 A, Molecular Devices, CA, USA). The bath solution contained (in millimole/L): NaCl 140, CaCl2 2, KCl 2, MgCl2 1, glucose 20 and HEPES 20 at pH 7.4. The internal solution contained (in millimole/L): CsCl 156, MgCl2 1, EGTA 10 and HEPES 10 at pH 7.2 adjusted with CsOH. Additional Ca2+ was added in the pipette solution to get 7.4 μM free Ca2+, calculated using WEBMAXC v2.10 [Citation18]. The cells were pre-treated for 30 min with control mouse IgG or mouse monoclonal antibody M4M at a concentration of 10 µg/mL in bath solution before recording. ATP depletion was induced by applying 5 mM NaN3 and 10 mM 2-deoxyglucose (2-DG) into the bath solution continuously through a MicroFil (34 Gauge, WPI Inc. USA) around 10 µm away from the recording cells. The flow rate was kept at 200 µL/min. The current–voltage relations were established by applying voltage ramps from −100 to +100 mV at an interval of 250 ms. Membrane capacitance (Cm) was recorded for 10 min after applying NaN3 and 2-DG by the Membrane Test of Clampex software (Molecular Devices, CA, USA). During the recording, the sampling rate was set at 20 kHz and filtered at 1 KHz. Data were analysed using pClamp10, version 10.2 (Molecular Devices, CA, USA).
Experimental design and Statistical analysis
The data was presented as mean ± SEM and GraphPad Prism version 6.0 was used for the statistical analyses. Comparison of the means of data from three groups was done using one-way analysis of variance (ANOVA) followed by Bonferroni’s post hoc analysis, while two-way ANOVA followed by Bonferroni’s post hoc analysis was used to compare between groups where there are two different factors involved. The data was considered significant if p < 0.05.
Results
Generation and characterisation of the humanised TRPM4 rat model
To generate a novel TRPM4 humanised rat model, a targeting vector was generated and assembled into the rat target allele (). The Rat TRPM4 CDS (coding sequence)-SV40 late polyA (pA) was inserted after Exon 1 of the rat TRPM4, thus the expression of the Rat Trpm4 CDS is controlled by the rat TRPM4 gene regulatory element. The Rat TRPM4 CDS contains a fragment from the human TRPM4 sequence which is responsible for binding to monoclonal antibody M4M (). This sequence also contains the channel pore from human TRPM4. DNA sequencing verified that the human TRPM4 was successfully introduced into the rat genome (). Genotyping by PCR using rat tail tissue shows positive bands (319 bp) of the Rat TRPM4-containing allele in the wild-type (+/+) and heterozygous (+/−) rats (left image). PCR of the Human TRPM4-containing allele shows positive bands (595 bp) in heterozygous (+/−) and homozygous (+/+) rats, which confirms the expression of human TRPM4 in these animals (right image) (). Western blot verified the expression of the targeted human TRPM4 sequence in the heterozygous and homozygous rats (). M4M detected a band of 135 kDa in brain tissues harvested from the heterozygous and homozygous rats, but not in brain tissues from wild-type rats.
Figure 1. Generation and characterisation of the TRPM4 humanised KI rat model. (A) Schematic strategy for generating the KI rat model. (B) KI rat contains a human TRPM4 polypeptide to replace the corresponding rat sequences. The amino acid sequences used for generating M4M were circled with boxes. Filled grey box indicates the channel pore region. M4M binding epitope EPGF were highlighted in red. (C) DNA sequencing results on homozygous KI rats. (D) Genotyping of the WT (+/+), heterozygous (+/−) and homozygous (−/−) rats. (E) Western blot on brains from WT (+/+), heterozygous (+/−) and homozygous (−/−) rats using monoclonal antibody M4M that is specific for human TRPM4 [Citation10]. The Immunoblot was probed with anti-Actin antibody as the housekeeping protein.
![Figure 1. Generation and characterisation of the TRPM4 humanised KI rat model. (A) Schematic strategy for generating the KI rat model. (B) KI rat contains a human TRPM4 polypeptide to replace the corresponding rat sequences. The amino acid sequences used for generating M4M were circled with boxes. Filled grey box indicates the channel pore region. M4M binding epitope EPGF were highlighted in red. (C) DNA sequencing results on homozygous KI rats. (D) Genotyping of the WT (+/+), heterozygous (+/−) and homozygous (−/−) rats. (E) Western blot on brains from WT (+/+), heterozygous (+/−) and homozygous (−/−) rats using monoclonal antibody M4M that is specific for human TRPM4 [Citation10]. The Immunoblot was probed with anti-Actin antibody as the housekeeping protein.](/cms/asset/9b70331d-9836-4c00-acc4-31b5d2aa7f2c/idrt_a_2313522_f0001_c.jpg)
Characterisation of M4M in primary cultured neurons
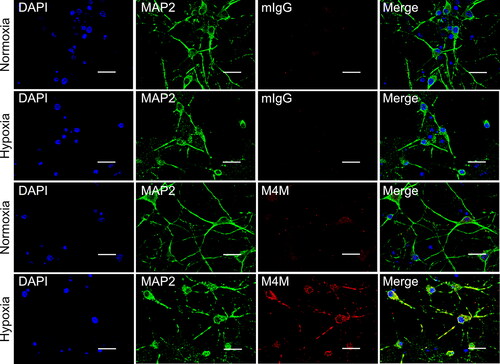
To examine the in vitro role of M4M in TRPM4 humanised rats, primary cortical neurons were cultured from embryonic WT and homozygous rat brains. Neurons were cultured under normoxic or hypoxic conditions for 24 h prior to experiments. Cell surface staining was achieved by incubating the neurons with M4M or control mouse IgG prior to permeabilization with Triton X-100. For comparison, the coverslips were processed together with similar experimental conditions including the same solutions for processing and the same exposure time during image capture. shows the staining of homozygous neurons. Under normoxic conditions, M4M demonstrates a very weak staining signal. In contrast, a strong staining was observed under hypoxic neurons. For control mouse IgG, no staining signal was detected under both normoxia and hypoxia. In WT neurons, neither M4M nor mouse IgG stained the neurons under normoxia and hypoxia (Supplementary Figure 1). These results indicate that hypoxia induces TRPM4 expression in neurons, similar as reported previously [Citation6, Citation9]. Importantly, M4M is specific for homozygous neurons expressing the human TRPM4 sequence and does not cross-react with rat TRPM4 in WT neurons.
Figure 2. Immunofluorescent staining of primary cultured neurons from embryonic homozygous rat brains. Neurons were treated under normoxia or hypoxia for 24 h prior to fixation. Control mouse IgG or M4M (red) were used to stain neurons that were labelled with neuronal marker MAP2 (green). Nuclei were counterstained with DAPI (blue). Scale bars: 20 µm.
Next, we employed whole-cell patch-clamp recordings to compare the electrophysiological properties of M4M on primary cultured cortical neurons from WT (+/+) and homozygous (−/−) rats. Activation of TRPM4 channels was induced by applying 5 mM sodium azide and 10 mM 2-deoxyglucose (2-DG) for 7 min to deplete ATP [Citation19]. In both WT and homozygous neurons treated with vehicle, ATP depletion resulted in a significant increase of current (). Treatment with mouse IgG did not change the current increase in both WT and homozygous neurons (). In primary cultured homozygous neurons, application of M4M completely inhibited the TRPM4 current induced by ATP depletion (). No current increase was observed during the 7-min hypoxia treatment. In contrast, M4M exhibited no effect on suppressing the TRPM4 current in WT neurons under hypoxia (). It should be noted that at 0 min, no difference in baseline current was observed among the 3 treatments in both WT and homozygous neurons, suggesting that M4M or the other two treatments has no effect on TRPM4 activity under normoxia.
Figure 3. Functional characterisation of M4M on primary cultured neurons from TRPM4 humanised rats. (A) Current voltage relationships of primary cultured neurons from wild-type (+/+) rats before (0 min) and after 7-min of hypoxic induction under the treatment of vehicle, 10 µg/mL control mouse IgG (mIgG) or 10 µg/mL M4M. Ramp protocols were applied from − 80 to + 80 mV with a holding potential at 0 mV. (B) Summary of the currents at + 80 and -80 mV. (C) current voltage relationships of primary cultured neurons from homozygous rats (−/−) expressing human TRPM4 sequences. (D) Summary of the currents at + 80 and -80 mV from C. (E) Sample images of WT (+/+) neurons before (0 min) and after 10 min of ATP depletion by 5 mM NaN3 and 10 mM 2-DG. Scale bars: 10 µm. (F) time course of membrane capacitance (Cm) during 10 min of ATP depletion in WT neurons. The Cm is normalised to baseline at 0 min. (G) Sample images of homozygous (−/−) neurons before and after 10 min of ATP depletion. Scale bars: 10 µm. (H) Time course of Cm changes during 10 min of ATP depletion in homozygous (−/−) neurons. Statistical analysis was performed by two-way ANOVA test with post hoc Bonferroni’s analysis. *p < 0.05, **p < 0.01, ***p < 0.0001, #p < 0.0001.
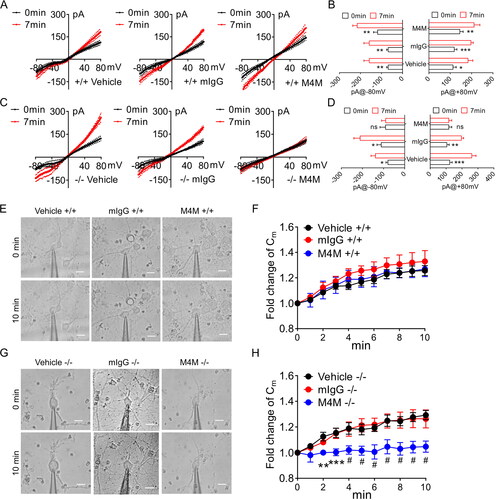
It has been previously reported that TRPM4 blocking antibodies could inhibit oncotic cell death induced by hypoxia [Citation9, Citation19]. To study the effect of M4M on cell volume change, the neurons were incubated with a bath solution containing 5 mM NaN3 and 10 mM 2-DG to deplete ATP for 10 min. Cell membrane capacitance (Cm) was recorded every minute as a proxy for cell volume [Citation20]. In WT neurons, 10 min of ATP depletion caused a change in cell morphology, exhibiting cell swelling in all three treatments: vehicle, mouse IgG, and M4M (). It is evidenced that during the 10-min interval, ATP depletion continued to increase the Cm of the WT neurons in all three groups (). At the end of 10 min, Cm is increased by around 20–25%. There is no difference among the three treatments at any time point. In homozygous neurons, ATP depletion induced similar cell swelling in vehicle and mouse IgG groups. Whereas cell morphology remained unchanged in M4M treated neurons (). Recordings of Cm showed the same increase continued in vehicle and mouse IgG groups during the 10 min ATP depletion period. In sharp contrast, M4M treatment significantly inhibited Cm increase from the 2nd minute onwards (H).
TRPM4 expression in humanised rat brains after stroke
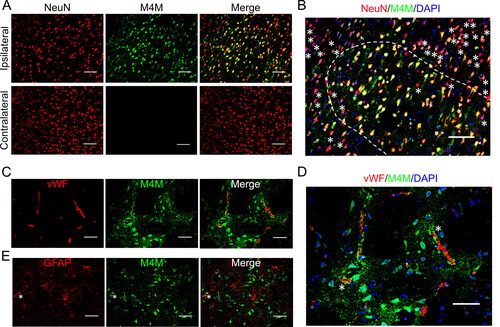
To investigate the role of M4M in stroke, a transient stroke model was created on the wild-type and homozygous humanised rats. In this model, the left middle cerebral artery (MCA) was occluded for 3 h, and then the filament was withdrawn to recanalize the MCA. One day post-operation, the rats were sacrificed, and the brains were collected for immunostaining. The neurons were first stained by M4M and neuronal marker NeuN. Consistent with previous reports [Citation6, Citation7, Citation9], TRPM4 upregulation was identified in neurons within the ipsilateral hemisphere close to the infarct core in homozygous rat brains, but not in contralateral hemisphere (). Corresponding areas within the contralateral hemisphere were negatively stained by M4M, suggesting that the expression of TRPM4 is low in healthy brain regions. In the enlarged image, the staining of neurons by M4M is heterogeneous (), consistent with previous report showing that the effects of hypoxia can vary across different cells and locations [Citation6]. The neurons with more positive TRPM4 staining may suffer from more severe hypoxia. Next, we used M4M to stain wild-type rats. No positive staining of M4M was found on both ipsilateral and contralateral hemispheres (Supplementary Figure 2), indicating that M4M does not recognise rodent TRPM4 channel.
Figure 4. Immunofluorescent staining of TRPM4 using M4M in homozygous rats. Left MCAO was created for 3 h followed by 24-h reperfusion. (A) M4M (green) and neuronal marker, anti-NeuN (red) were used to co-stain neurons within the ipsilateral and contralateral hemispheres. (B) Co-localisation of anti-NeuN, M4M and DAPI (blue) within the ipsilateral hemisphere from A. Asterisks: M4M negative neurons. Majority of the neurons within the dotted line were positive for both NeuN and M4M. Scale bars for A–B: 100 µm. (C) Double staining of M4M (green) and endothelial marker vWF (red) on the ipsilateral hemisphere. (D) Co-localisation of vWF, M4M and DAPI (blue) within the ipsilateral hemisphere from C. Asterisks: partly colocalization of M4M and anti-vWF. (E) Double staining of M4M (green) and astrocyte marker GFAP (red) on the ipsilateral hemisphere. Asterisks: partly colocalization of M4M and GFAP. Scale bars for C–E: 50 µm.
A commercial anti-TRPM4 antibody (Origene) that recognises rodent TRPM4 was used to validate the results from M4M (Supplementary Figure 3). The staining pattern of the commercial antibody was similar to that of M4M, with negative staining of neurons from the contralateral hemisphere (Supplementary Figure 3A) and positive staining of neurons from the ipsilateral hemisphere (Supplementary Figure 3B). The commercial antibody again demonstrated heterogeneous staining of wild-type neurons within the ipsilateral hemisphere (Supplementary Figure 3C).
Possible localisation of M4M to vascular endothelial cells and astrocytes was examined by co-staining with endothelial marker vWF and astrocyte marker GFAP in homozygous and wild-type brains (C–E and Supplementary Figure 2). As shown in , very few endothelial cells and astrocytes within the ipsilateral hemisphere of the homozygous rats were positively stained by M4M. In the wild-type brains, M4M showed no positive staining on the ipsilateral brain tissues when co-stained with vWF and GFAP (Supplementary Figure 3B). In contrast, the commercial anti-TRPM4 antibody demonstrated a similar staining pattern in both homozygous and wild-type rat brains (Supplementary Figure 3B and Supplementary Figure 4B). TRPM4 co-localized partly or rarely with vWF and GFAP in the ipsilateral hemisphere.
M4M improves stroke outcome in humanised rats
To examine the in vivo effect of M4M in stroke, an ischaemia reperfusion model was established in rats in which middle cerebral arteries were transiently occluded for 3 h followed by recanalization. M4M or control mouse IgG (mIgG) was applied intravenously 1 h before recanalization (). This experimental design is to mimic the clinical scenario in which the antibody can be delivered together with the reperfusion drug tPA [Citation8]. Infarct formation was assessed 1 day after operation in wild-type (+/+), heterozygous (+/−), and homozygous (−/−) rats to compare the effect of M4M with mIgG (). Summary of infarct volume shows that compared to mIgG, M4M treatment significantly reduced infarct formation in homozygous rats, but not in heterozygous or wild-type rats (). The absence of therapeutic effect of M4M in wild-type rats is consistent with our previous study [Citation10]. When we compared the percentage of infarct volume of M4M treatment in all three groups of animals (), the homozygous group is notably lower than the wild-type group. The average value of percentage infarct of the heterozygous group is higher than that of the homozygous group, and lower than the wild-type group. However, there is no significant difference between the heterozygous group and either two groups.
Figure 5. Evaluating the in vivo role of M4M in a stroke reperfusion animal model. (A) Diagram showing experimental protocol for 3-h transient MCAO. M4M (100 µg) or control mouse IgG (mIgG, 100 µg) was injected intravenously (i.v. Ab) 1 h before recanalization. (B) Representative images of TTC-stained brains from homozygous (−/−), heterozygous (+/−), and wild-type (+/+) rats at 1 day after transient MCAO induction that were treated with mIgG or M4M. C, Summary of infarct volume formation of rat brains at 1 day after transient MCAO induction. n = 6 rats for M4M and mIgG treatments in wild-type and homozygous rats except for treatments in heterozygous rats (n = 7). (D) Comparison of percentage of infarct volume normalised to the whole brain region after M4M treatment among homozygous, heterozygous, and wild-type rats. (E) Assessment of motor functions by Rotarod test in homozygous rats after the treatment of M4M (n = 7), mIgG (n = 6), or vehicle (n = 6). (F) Comparison of the neurological severity scores in homozygous rats treated with vehicle (n = 6), mIgG (n = 6), and M4M (n = 7). In C, E, and F, statistical analysis was performed by two-way ANOVA with Bonferroni post hoc test, and in D by one-way ANOVA with Bonferroni post hoc test. *p < 0.05, **p < 0.01. In E and F, significance was observed in M4M versus vehicle or mIgG at day 1. After day 1, significance was found only in M4M versus mIgG.
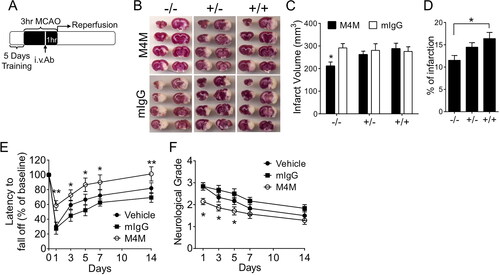
To verify that the infarct volume reduction by M4M in homozygous rats is associated with functional improvement, we first performed the Rotarod test to assess the motor function in homozygous rats (). One day after operation, the performance of the M4M group was significantly better than that of the mIgG group or the vehicle group. On days 3, 5, 7, and 10, M4M group demonstrated significantly better motor functions compared to the mIgG group, but not to the vehicle group. Neurological deficit was further examined using a neurological severity score system [Citation21]. The highest score was observed at day 1 after operation, and the score gradually decreased in the following days (). Similar to the Rotarod test, the neurological scores of the M4M group were significantly lower than those of both mIgG and vehicle groups at day 1. At days 3 and 5, the neurological scores of the M4M group were significantly lower than that of the mIgG group, but not the vehicle group.
Discussion
Mouse monoclonal antibody M4M was first designed to block human TRPM4 channel [Citation10]. The antigenic sequence for generating M4M is located extracellularly, close to the channel pore. Epitope scanning revealed that a four amino-acid peptide EPGF from human TRPM4 is critical for binding. Any amino acid change to EPGF significantly disrupts antibody binding [Citation10, Citation22]. In rat and mouse TRPM4, the corresponding sequence is ERGS. Therefore, M4M is specific to human TRPM4, demonstrating no effect on rodent TRPM4 [Citation10]. Prior to M4M, we have generated a polyclonal antibody M4P, targeting a similar extracellular domain of rat TRPM4 [Citation9]. As the homology between the rat antigenic sequence and the human antigenic sequence is 58.3%, M4P does not cross-react with human TRPM4 [Citation9, Citation10]. Characterisation of M4M thus requires testing in animal models expressing human TRPM4.
In the present study, we created a humanised rat model expressing human TRPM4 sequence. This TRPM4 humanised rat model expresses the antigen-containing sequence, that was used to generate M4M, leaving most of the rat TRPM4 sequence unchanged. This design could minimise potential side effects arising from replacing the whole rat TRPM4 with human TRPM4 sequence. It should be noted that the channel pore is located between the two antigenic fragments [Citation10]. Therefore, this humanised rat model contains the human TRPM4 channel pore. Of the putative 21-amino acid channel pore sequence, the human TRPM4 channel differs from the rat TRPM4 by three amino acids [Citation10]. When we examined the electrophysiological properties of TRPM4 in the primary cultured neurons from the homozygous rats, typical outward rectifying TRPM4 currents were identified when the neurons were exposed to ATP depletion. This result suggests that the humanised animals express functional TRPM4 channel. It should be noted that majority of the baseline current is not from TRPM4 as the channel activity under normoxia remains low [Citation2].
Compared to normoxic neurons, M4M was revealed to bind preferentially to the homozygous neurons upon ATP depletion (). This result is consistent with our previous report showing that M4M could bind to the TRPM4 channel in HEK 293 cells with human TRPM4 overexpression [Citation10]. Functionally, application of M4M completely abolished hypoxia-induced activation of TRPM4 in primary cultured neurons from homozygous rats. Accordingly, cell volume increase associated with ATP depletion was inhibited by M4M treatment. These results verify our previous study on cultured Human Brain Microvascular Endothelial Cells (HBMECs) [Citation10]. The lack of inhibitory effect on wild-type neurons further indicates the specificity of M4M for the human TRPM4 channel.
The binding epitope of M4M, EPGF, is located extracellularly [Citation10, Citation22]. Post-translational modifications to the antigenic domain may potentially affect antibody binding. It has been reported that TRPM4 can be glycosylated [Citation23]. This N-glycosylation site is located within the antigenic sequence of rodent TRPM4 for binding with polyclonal antibody M4P [Citation9]. Previous in vitro and in vivo characterisation of M4P revealed that the N-glycosylation does not affect M4P function on rodent TRPM4 [Citation9]. As the N-glycosylation site is conserved across rodents and humans, it is necessary to examine whether the glycosylation (N992 in human TRPM4) affects anti-human TRPM4 antibody M4M. Previous characterisation of M4M using HBMECs or HEK 293 cells transfected with human TRPM4 found that M4M successfully inhibited human TRPM4 current and downregulated its surface expression [Citation10]. Here, the efficacy of M4M was verified using the humanised rat model suggesting that the N-glycosylation does not affect in vivo functions of M4M. This may be because the glycosylation site N992 is very close to the channel pore, with a distance away from the M4M binding epitope EPGF (996-999), which is located to the outermost of the channel, forming the tip of the extracellular turret [Citation22]. Thus, potential N992 glycosylation does not interact with M4M binding.
The in vivo role of M4M was examined on an ischaemia reperfusion model created in wild-type and homozygous rats. Surviving neurons close to the infarct core, in homozygous rats, were positively stained by M4M. TRPM4 upregulation differs among neurons, suggesting possible differential hypoxic conditions in different brain areas close to the infarct. Neurons in the contralateral hemisphere were negatively stained by M4M. This result is consistent with staining using a commercial TRPM4 antibody, and our previous study using M4P on wild-type animals [Citation6]. These results indicate that hypoxia induces TRPM4 expression in neurons following stroke [Citation6, Citation24]. In vascular endothelial cells and astrocytes, M4M demonstrated little staining signal. The staining pattern of astrocytes supports our previous study showing that blocking TRPM4 has no functional impact on astrocytes during acute ATP depletion [Citation19]. However, the staining pattern of vascular endothelium is different from our previous study on the permanent stroke model where TRPM4 expression was prominent in vascular endothelial cells one day after stroke induction [Citation7]. One possible explanation is that the blood vessels in this current reperfusion model have been recanalized for 24 h and the hypoxic impact on endothelium was ameliorated.
The therapeutic potential of M4M was evaluated by examining the motor function and the neurological severity scores. As M4M is a mouse monoclonal antibody, we used mouse IgG as a control, in addition to vehicle control. M4M treatment improved functional outcome in both Rotarod test and neurological scores (). However, there is a slight difference between the control mouse IgG and the vehicle groups. M4M group performed significantly better at multiple timepoints when compared to mouse IgG, whereas it only showed significant improvement compared to vehicle at day 1 post-operation. Although there is no difference between the mouse IgG group and the vehicle group at all-time points, the vehicle group tends to behave better than the mouse IgG group. As the in vivo study was performed in rats, mouse IgG might elicit an adverse effect due to the species difference. Further research is needed to test this hypothesis.
Potential of possible side effects might be a concern when the TRPM4 antibody is used to block human TRPM4 channel. TRPM4 is expressed in certain areas of the central nervous system such as the pre-Bötzinger complex, supporting bursts of action potentials of interneurons [Citation3]. TRPM4-specific short hairpin RNA (shRNA) progressively decreased the tidal volume of breaths and increased breathing frequency, leading to gasping and even fatal respiratory failure. However, TRPM4 knockout mice are viable and fertile without noteworthy abnormalities [Citation25]. In our previous studies to block TRPM4 in animal models [Citation6–10, Citation26, Citation27], no significantly higher mortality rate was observed even after 2 weeks to 2 months treatment. Blocking TRPM4 consistently showed better therapeutic outcomes than control treatments. An advantage of using antibody therapy is that antibodies enter the brain when the blood-brain barrier (BBB) is disrupted during stroke. In healthy brain areas, the large molecular weights of antibodies prevent them from crossing the BBB.
Although M4M has shown excellent therapeutic potential in animal models of stroke, it cannot be used in humans due to its murine nature. Humanisation of M4M is the next step towards clinical application. To our best knowledge, this is the first time that a blocking antibody against an ion channel has been developed for therapeutic purpose. Antibodies against various drug targets have been proposed to treat stroke [Citation28]. The use of monoclonal antibodies in combination therapy with other therapeutic modalities has been proposed in various other studies to further propel clinical success [Citation29]. Therefore the TRPM4 blocking antibody can be a novel therapy to reduce reperfusion injury and potentially extend the current limited time window for reperfusion therapy to provide better clinical outcome for patients [Citation8].
Ethics approval and consent to participate
All animal procedures were approved by the Institutional Animal Care and Use Committee (protocol no. A19099) of Lee Kong Chian School of Medicine and performed in accordance with the NIH Guide for the Care and Use of Laboratory Animals and the Animal Research: Reporting of In Vivo Experiments (ARRIVE) guidelines 2.0.
Authors’ contributions
P.L. conceived and directed the project. C.P.P., S.W., B.C., S.W.L., J.T.S.Q., A.T-H.L, B.N. and P.L. conceived, analysed data, and wrote the paper. C.P.P., S.W., B.C. and S.W.L. performed experiments and analysed data. All authors have read and agreed to the published version of the manuscript.
Availability of data and material
All data generated or analysed during this study are included in this published article and its supplementary.
| Abbreviations | ||
| 2-DG | = | 2-deoxyglucose |
| ANOVA | = | analysis of variance |
| ATP | = | adenosine triphosphate |
| CCA | = | common carotid artery |
| CDS | = | coding sequences |
| Cm | = | membrane capacitance |
| DAPI | = | 4′,6-diamidino-2-phenylindole |
| ECA | = | external carotid artery |
| FBS | = | foetal bovine serum |
| GFAP | = | glial fibrillary acidic protein |
| HBMECs | = | Human Brain Microvascular Endothelial Cells |
| ICA | = | internal carotid artery |
| KI | = | knock-in |
| MCAO | = | Middle Cerebral Artery Occlusion |
| NB | = | Neurobasal |
| PBS | = | phosphate buffered saline |
| PCR | = | polymerase chain reaction |
| PFA | = | paraformaldehyde |
| SD | = | Sprague-Dawley |
| siRNA | = | small interfering RNA |
| TRPM4 | = | Transient Receptor Potential Melastatin 4 |
| TTC | = | 2,3,4-triphenyltetrazolium chloride |
| vWF | = | von Willebrand Factor |
| WT | = | wild-type |
Supplementary Material
Download Zip (12.2 MB)Disclosure statement
P.L. has a patent WO-2014209239-A1 (Trpm4 Channel Inhibitors for Stroke) for the TRPM4 blocking antibody. The authors declare that they have no other competing interests.
Additional information
Funding
References
- Nilius B, Prenen J, Droogmans G, et al. Voltage dependence of the Ca2+-activated cation channel TRPM4. J Biol Chem. 2003;278(33):30813–30820. doi: 10.1074/jbc.M305127200.
- Mathar I, et al. Trpm4. Handb Exp Pharmacol. 2014;222:461–487.
- Picardo MCD, Sugimura YK, Dorst KE, et al. Trpm4 ion channels in pre-Botzinger complex interneurons are essential for breathing motor pattern but not rhythm. PLoS Biol. 2019;17(2):e2006094. doi: 10.1371/journal.pbio.2006094.
- Walcott BP, Kahle KT, Simard JM. Novel treatment targets for cerebral edema. Neurotherapeutics. 2012;9(1):65–72. doi: 10.1007/s13311-011-0087-4.
- Loh KY, Wang Z, Liao P. Oncotic cell death in stroke. Rev Physiol Biochem Pharmacol. 2019;176:37–64.
- Chen B, Ng G, Gao Y, et al. Non-Invasive multimodality imaging directly shows TRPM4 inhibition ameliorates stroke reperfusion injury. Transl Stroke Res. 2019;10(1):91–103. doi: 10.1007/s12975-018-0621-3.
- Loh KP, Ng G, Yu CY, et al. TRPM4 inhibition promotes angiogenesis after ischemic stroke. Pflugers Arch. 2014;466(3):563–576. doi: 10.1007/s00424-013-1347-4.
- Chen B, Wei S, Low SW, et al. TRPM4 blocking antibody protects cerebral vasculature in delayed stroke reperfusion. Biomedicines. 2023;11(5):1480. doi: 10.3390/biomedicines11051480.
- Chen B, Gao Y, Wei S, et al. TRPM4-specific blocking antibody attenuates reperfusion injury in a rat model of stroke. Pflugers Arch. 2019;471(11-12):1455–1466. doi: 10.1007/s00424-019-02326-8.
- Low SW, Gao Y, Wei S, et al. Development and characterization of a monoclonal antibody blocking human TRPM4 channel. Sci Rep. 2021;11(1):10411. doi: 10.1038/s41598-021-89935-5.
- VanRyzin JW, Arambula SE, Ashton SE, et al. Generation of an Iba1-EGFP transgenic rat for the study of microglia in an outbred rodent strain. eNeuro. 2021;8(5):ENEURO.0026-21.2021. doi: 10.1523/ENEURO.0026-21.2021.
- Pettibone JR, Yu JY, Derman RC, et al. Knock-In rat lines with cre recombinase at the dopamine D1 and adenosine 2a receptor loci. eNeuro. 2019;6(5):ENEURO.0163-19.2019. doi: 10.1523/ENEURO.0163-19.2019.
- Bäck S, Necarsulmer J, Whitaker LR, et al. Neuron-Specific genome modification in the adult rat brain using CRISPR-Cas9 transgenic rats. Neuron. 2019;102(1):105–119.e8. doi: 10.1016/j.neuron.2019.01.035.
- Ma Y, Yu L, Pan S, et al. CRISPR/Cas9-mediated targeting of the Rosa26 locus produces cre reporter rat strains for monitoring cre-loxP-mediated lineage tracing. Febs J. 2017;284(19):3262–3277. doi: 10.1111/febs.14188.
- Percie Du Sert N, Hurst V, Ahluwalia A, et al. The ARRIVE guidelines 2.0: updated guidelines for reporting animal research. Exp Physiol. 2020;105(9):1459–1466. doi: 10.1113/EP088870.
- Walberer M, Stolz E, Müller C, et al. Experimental stroke: ischaemic lesion volume and oedema formation differ among rat strains (a comparison between wistar and Sprague-Dawley rats using MRI). p. Lab Anim. 2006;40(1):1–8. doi: 10.1258/002367706775404426.
- Bederson JB, Pitts LH, Tsuji M, et al. Rat Middle cerebral artery occlusion: evaluation of the model and development of a neurologic examination. Stroke. 1986;17(3):472–476. doi: 10.1161/01.str.17.3.472.
- Schattling B, Steinbach K, Thies E, et al. TRPM4 cation channel mediates axonal and neuronal degeneration in experimental autoimmune encephalomyelitis and multiple sclerosis. Nat Med. 2012;18(12):1805–1811. doi: 10.1038/nm.3015.
- Wei S, Low SW, Poore CP, et al. Comparison of anti-oncotic effect of TRPM4 blocking antibody in neuron, astrocyte and vascular endothelial cell under hypoxia. Front Cell Dev Biol. 2020;8:562584. doi: 10.3389/fcell.2020.562584.
- Satoh H, Delbridge LM, Blatter LA, et al. Surface:volume relationship in cardiac myocytes studied with confocal microscopy and membrane capacitance measurements: species-dependence and developmental effects. Biophys J. 1996;70(3):1494–1504. doi: 10.1016/S0006-3495(96)79711-4.
- Wei S, Chen B, Low SW, et al. SLC26A11 inhibition reduces oncotic neuronal death and attenuates stroke reperfusion injury. Mol Neurobiol. 2023;60(10):5931–5943. doi: 10.1007/s12035-023-03453-1.
- Wei S, Behn J, Poore CP, et al. Binding epitope for recognition of human TRPM4 channel by monoclonal antibody M4M. Sci Rep. 2022;12(1):19562. doi: 10.1038/s41598-022-22077-4.
- Syam N, Rougier JS, Abriel H. Glycosylation of TRPM4 and TRPM5 channels: molecular determinants and functional aspects. Front Cell Neurosci. 2014;8:52. doi: 10.3389/fncel.2014.00052.
- Mehta RI, Tosun C, Ivanova S, et al. Sur1-Trpm4 cation channel expression in human cerebral infarcts. J Neuropathol Exp Neurol. 2015;74(8):835–849. doi: 10.1097/NEN.0000000000000223.
- Barbet G, Demion M, Moura IC, et al. The calcium-activated nonselective cation channel TRPM4 is essential for the migration but not the maturation of dendritic cells. Nat Immunol. 2008;9(10):1148–1156. doi: 10.1038/ni.1648.
- Liao P, Loh KP. TRPM4 channel inhibitors for stroke treatment. US Patent. 2019;10,221,239.
- Hazalin N, Liao P, Hassan Z. TRPM4 inhibition improves spatial memory impairment and hippocampal long-term potentiation deficit in chronic cerebral hypoperfused rats. Behav Brain Res. 2020;393:112781. doi: 10.1016/j.bbr.2020.112781.
- Yu CY, Ng G, Liao P. Therapeutic antibodies in stroke. Transl Stroke Res. 2013;4(5):477–483. doi: 10.1007/s12975-013-0281-2.
- Aghanejad A, Bonab SF, Sepehri M, et al. A review on targeting tumor microenvironment: the main paradigm shift in the mAb-based immunotherapy of solid tumors. Int J Biol Macromol. 2022;207:592–610. doi: 10.1016/j.ijbiomac.2022.03.057.