
Abstract
The main molecular mechanisms explaining the well-established antioxidant and reducing activity of N-acetylcysteine (NAC), the N-acetyl derivative of the natural amino acid l-cysteine, are summarised and critically reviewed. The antioxidant effect is due to the ability of NAC to act as a reduced glutathione (GSH) precursor; GSH is a well-known direct antioxidant and a substrate of several antioxidant enzymes. Moreover, in some conditions where a significant depletion of endogenous Cys and GSH occurs, NAC can act as a direct antioxidant for some oxidant species such as NO2 and HOX. The antioxidant activity of NAC could also be due to its effect in breaking thiolated proteins, thus releasing free thiols as well as reduced proteins, which in some cases, such as for mercaptoalbumin, have important direct antioxidant activity. As well as being involved in the antioxidant mechanism, the disulphide breaking activity of NAC also explains its mucolytic activity which is due to its effect in reducing heavily cross-linked mucus glycoproteins. Chemical features explaining the efficient disulphide breaking activity of NAC are also explained.
Introduction
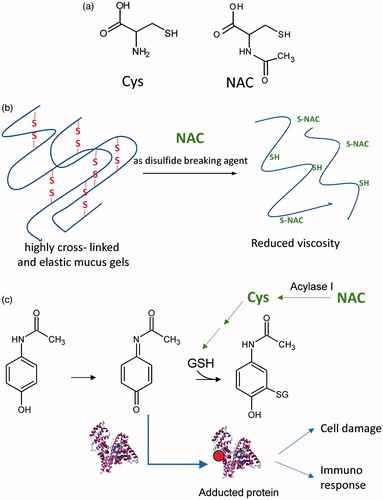
Since the 1960s, N-acetylcysteine (NAC), the N-acetyl derivative of the natural amino acid l-cysteine (), has been widely prescribed as a mucolytic agent [Citation1] and, since the 1970s, has been used for the treatment of acetaminophen poisoning [Citation2]. The molecular mechanisms underlying these two therapeutic effects are well known. In particular, the mucolytic action is due to the ability of NAC to break the disulphide bridges in the high-molecular-weight glycoproteins of mucus, resulting in reduced viscosity [Citation3] (). The effect of NAC on acetaminophen poisoning is a result of the action of NAC in replenishing hepatic reduced glutathione (GSH), which is the main endogenous nucleophilic peptide that reacts with and neutralises electrophilic and hence damaging molecules such as N-acetyl-p-benzoquinone imine (NAPQI), the electrophilic metabolite of acetaminophen [Citation4] (). Since the 1980s, NAC has also been proposed for the treatment of diseases in which oxidative stress is considered to be involved in the onset and progression of the disease state [Citation5].
Figure 1. (a) Chemical structures of Cys and of NAC. (b) The mucolytic action of NAC is due to its ability to break the disulphide bridges of the high-molecular-weight glycoproteins in the mucus, resulting in reduced viscosity. (c) NAC for the treatment of acetaminophen poisoning. NAC acts by replenishing hepatic GSH, which is the main endogenous nucleophilic peptide able to neutralise N-acetyl-p-benzoquinone imine (NAPQI), the electrophilic metabolite of acetaminophen. Paracetamol is metabolised forming the electrophilic metabolite (NAPQI) which is detoxified by GSH. In the case of GSH depletion, NAPQI reacts with proteins forming adducts which can induce cell damage or immune response. NAC acts by replenishing the GSH pool as a precursor of Cys, the building block and the rate-limiting step in glutathione synthesis. NAC is converted to Cys through a deacetylation reaction catalysed by acylase.
Several in vitro studies have reported efficient antioxidant activity of NAC using different oxidants, substrates, and methods to assess the oxidative processes [Citation6–10]. The wide application of NAC is not only because of its well-described antioxidant and radical scavenging activity but also because, as a thiol molecule, it is quite stable and commercially available at a low price. Based on these facts and considering its bioavailability and safety, NAC has also been used as an antioxidant in several in vivo studies [Citation5]. In vivo, NAC treatment was found to prevent and/or inhibit the oxidative process as measured by different biomarkers of oxidative stress [Citation11–16]. The in vivo antioxidant activity can be explained by considering the antioxidant and radical scavenging mechanism of NAC as observed in in vitro conditions. However, this is a mere simplification because the antioxidant behaviour of a compound tested in in vitro conditions is not the same as that in vivo. In the former, the antioxidant under test is in a simplified environment represented by the radical species and the substrate, whereas in vivo, the situation is more complex because competing reactions with other enzymatic and non-enzymatic antioxidants, as well as substrates, occur.
Although several in vivo studies have found that NAC significantly prevents or inhibits oxidative stress under certain conditions, a clear molecular mechanism through which NAC exerts this activity is still not known. The present review focuses on the molecular mechanisms through which NAC regulates oxidative stress in in vivo conditions.
The in vivo antioxidant activity of NAC can be related to at least three different mechanisms and in particular:
A direct antioxidant effect toward certain oxidant species.
An indirect antioxidant effect as a result of the ability of NAC to act as a precursor of Cys, which is a building block and the rate-limiting step in glutathione synthesis, GSH being a well-known direct antioxidant and a substrate of several antioxidant enzymes.
A breaking effect on disulphides and the ability to restore thiol pools, which in turn regulate the redox state.
Understanding the antioxidant and reducing mechanisms of NAC
Direct antioxidant activity of NAC
As a thiol compound, NAC can react in the test tube with most typical radical and non-radical oxidants. However, to act as an antioxidant in a biological matrix, the reaction rate of any antioxidant, including NAC, toward oxidants, should be higher than that of the endogenous antioxidants and clearly much higher than that of the substrates. The reaction rate is the product of the molecule concentration at the site where the oxidants are produced and the reaction rate constant of the antioxidant toward specific oxidants. To understand the potential activity of NAC as an antioxidant in a certain situation, some parameters must be considered, and in particular the reaction rate of NAC and the endogenous antioxidants and substrates toward the oxidants formed in that situation, together with the concentrations of each of the above-mentioned reactants. The reaction rate constants for NAC and enzymatic and non-enzymatic antioxidants have already been determined and summarised [Citation17], and the concentrations for endogenous antioxidants and for NAC, at least in plasma, and after different administration regimens, are available in the literature. Taking into account the values summarised in , it is quite evident that for some oxidants such as H2O2 and O2•−, the reaction rate of NAC, as well as of other thiols such as Cys and GSH, is negligible compared with that of antioxidant enzymes. For example, hydrogen peroxide reacts with NAC and with GSH peroxidase 3 with a reaction rate constant of 0.16 M−1 s−1 and 1.8 × 108 M−1 s−1, respectively. By considering a NAC plasma concentration of 100 µM, which is the steady-state concentration reached after four doses of 2 g of NAC injected intravenously [Citation18], and that of GSH peroxidase 3, which is present at a concentration of 0.5–0.8 µM, then the reaction rate constant × concentration is 0.016 × 10−3 s−1 and 90 s−1 for NAC and the peroxidase, respectively. Hence, the reaction between H2O2 and the enzyme would be 5.625 × 106 times faster than the reaction with NAC. This difference increases 1000-fold in cells where the GSH concentration reaches the millimolar concentration range.
Table 1. Reaction rate constants of NAC, Cys, GSH, and endogenous enzymatic antioxidants toward the main oxidant species.
Superoxide anion (O2•−) is also untargeted by NAC and in general by thiol-containing compounds, irrespective of their concentration. There is a general consensus that the reaction between O2•− and GSH is relatively slow for a radical reaction, with a rate constant of 200 M−1 s−1, whereas that for manganese-containing superoxide dismutase is 2.3 × 109 M−1 s−1 [Citation19]. The reaction rate for NAC, 68 M−1 s−1, is even lower than that of GSH [Citation19]. In addition, the reaction rate of NAC with peroxynitrite (ONOOH) (rate constant, 415 M−1 s−1) is limited in in vivo conditions by considering the reaction rates of GSH and Cys [Citation20], which are in the order of 103 M−1 s−1. Moreover, rate constants in the order of 106–107 M−1 s−1 have been reported for the reaction of peroxynitrite with very reactive thiols in proteins, such as those present in peroxiredoxins (Prx), which constitute an efficient key detoxification system of this oxidant species [Citation21].
Let us now consider another example of an untargeted oxidant species and in particular the reaction of HO•. As reported in , the reaction rate constant between NAC and HO• is high, as are the values of other thiols, but this is due to the high reactivity of HO•, which makes the reaction rate of other molecules, including substrates, high. By considering that the concentrations of substrates are much higher than the concentration of NAC, we can easily rule out a possible antioxidant action of NAC toward HO• species.
Hence, based on the reaction rates and concentrations, the antioxidant activity of NAC should be ruled out for some oxidant species such as H2O2, O2•−, OHNOO, and HO•, but for others, including NO2 and hypohalous acids, HOX, it could be more plausible [Citation17] as explained below.
Hypochlorous acid (HOCl) and related species (hypobromous acid, HOBr; hypothiocyanous acid, HOSCN) are oxidants produced by activated neutrophils and monocytes through the activity of myeloperoxidase (MPO). MPO catalyses the reaction between hydrogen peroxide (H2O2) formed by dismutation of O2•− and halides (Cl−, I−, Br−, or SCN–) to produce the corresponding hypohalous acids (HOX). The reaction occurs preferentially with Cl− forming hypochlorous acid (HOCl) because of its high concentration in body fluids compared with other halides [Citation22]. These oxidant species are potent bactericides and disinfectants and play a role in the human response to invading pathogens. However, HOX, due to their high reactivity, are not specific oxidants and also react with many biologically important molecules, thus inducing a cytotoxic effect and the development of a number of diseases such as atherosclerosis, cancer, and promyelocytic leukaemia, as well as neurodegenerative diseases, including Alzheimer’s disease and multiple sclerosis [Citation22]. MPO and HOX are also involved in the pathophysiology of some lung diseases. O’Donnell et al. [Citation23] reported the presence of 3-chlorotyrosine (3Cl−Tyr), a reaction product of HOCl with protein tyrosine, in the sputum of patients with chronic obstructive pulmonary disease (COPD), which correlated well with MPO activity in sputum, suggesting that an active process related to MPO may play a role in the pathophysiology of this disease. High levels of MPO protein and activity, together with a significant increase of halogenated proteins, protein oxidative cross-links, and disulphide bonds, was reported in the airway mucus from patients with cystic fibrosis (CF), suggesting that oxidation arising from airway inflammation contributes to pathologic mucus gel formation in the lungs of patients with CF [Citation24].
HOX is quite reactive toward thiols and, based on the reaction rate constant, it is obvious that NAC can potentially act when its relative concentration is higher than that of GSH and that of free or protein Cys. This situation can potentially occur in some situations and pathologic conditions, such as in lung fluids exposed to an inflammatory/oxidative process, as already demonstrated for some pathologies [Citation25]. Under these conditions, a significant decrease in the pool SH occurs and hence NAC, given at a dose of 300 mg by aerosol or by endotracheal–bronchial administration, could be a direct neutraliser of HOX species. Similarly, NAC can have a potential trapping effect for nitrogen oxide (NO2), which is classically known as a major component of both indoor and outdoor air pollution and is involved in epithelial injury in the lung. NO2 is a toxic free radical gas produced by several exogenous sources, including motor vehicles, burning fuel, cigarette smoke, and cooking gas. NO2 can also be formed during inflammation by the decomposition of ONOO- or through peroxidase-catalysed reactions [Citation26]. Rate constants for reaction of NO2 with a spectrum of potential targets have been estimated, and the most relevant compounds are thiols, reduced purines such as uric acid, ascorbic acid, and phenols such as tyrosine [Citation27]. Reaction constants at pH 7.4 for Cys, GSH, and NAC are reported in . NAC as a trapping agent of NO2 was then tested in in vivo conditions. In vivo administration of NAC to NO2-inhaling rats protected bronchoalveolar lavage (BAL) parameters and the physiology of type II pneumocytes from impairment [Citation28].
The reaction rate constants of NAC and of the endogenous thiol-containing compounds toward oxidant species follows the general order: Cys > GSH > NAC. Hence, assuming the same concentrations, NAC is the weakest antioxidant, and this is explained by considering that the antioxidant activity of SH is due to the thiolate anion, the relative concentration of which is regulated by the acidity of thiol. In other words, the acidity (Ka) of the thiol group regulates the equilibrium and hence the relative amount of S− with respect to SH. Accordingly, the pKa of the above-mentioned thiols follows the same order: cysteine (pKa 8.30) > GSH (8.83) > NAC (9.52). At pH 7.4, for each 100 SH molecules in the SH state, 12, 3.7, and 0.7 are in a thiolate form for Cys, GSH, and NAC, respectively.
If, on the one hand, NAC is the weakest antioxidant among endogenous thiol compounds, on the other hand, it is more stable in aqueous solution. NAC was subjected to stability studies for 24 h at 4-h intervals, and the results were obtained in terms of percentage degradation. The results suggest that there was a degradation of 0.89% and 0.48% in the solution stored at room temperature and in refrigerated conditions, respectively [Citation29].
In addition to a direct antioxidant activity, a direct pro-oxidant effect of thiols, including NAC should also be considered. It is well established that any antioxidant under certain conditions can act as a pro-oxidant. As an example ascorbic acid in the presence of transition metals such as Fe2+ [Citation30] or copper [Citation31] results in the generation of ROS. Moreover, the well-established lipid antioxidant alpha-tocoferol, can, in an oxidising milieu, form the corresponding radical species (tocopheroxyl radical) which, if not recycled to the corresponding non-radical species, can act as pro-oxidant [Citation32]. Also thiols such as Cys and GSH as well as NAC, besides acting as antioxidants, can in some case can also act as pro-oxidants through the formation of HO° and thyil radicals [Citation33–35]. Regarding NAC, some cellular and in vitro studies reported that NAC, when in combination with some compounds and in certain conditions, shows a pro-oxidant effect. For instance, NAC enhances fisetin-induced cytotoxicity via induction of ROS-independent apoptosis in human colonic cancer cells, an effect which was considered of interest in the treatment of colonic cancer [Citation36]. NAC in the presence of transition metal ions such as Cu2+ [Citation37] or of vitamin B12 [Citation38] was shown to exert a pro-oxidant effect. However, it should be pointed out that the pro-oxidant action of NAC is unlikely to occur in in vivo conditions because it was found to happen under certain reaction conditions, such as the presence of free transition metal ions, high NAC concentrations, and simplified matrices which do not occur in in vivo conditions.
Indirect antioxidant activity of NAC
Glutathione is a tripeptide (γ-l-glutamyl-l-cysteinylglycine, GSH) synthesised and maintained at high (mM) concentrations in cells [Citation39]. The γ-glutamylcysteine intermediate is first synthesised from l-glutamate and cysteine via the enzyme γ-glutamylcysteine synthetase (glutamate cysteine ligase). This reaction is the rate-limiting step in glutathione synthesis. In a subsequent synthetic step, l-glycine is added to the C-terminus of γ-glutamylcysteine via the enzyme glutathione synthetase.
In addition to directly reacting with radicals/oxidants and electrophiles, forming GSSG and GSH Michael adducts, GSH serves as a substrate or cofactor of a large number of detoxifying cellular enzymes, including glutathione reductase, glutaredoxin (Grx), glutathione peroxidase, peroxiredoxin (Prx), glyoxalases 1 and 2, glutathione transferase, and MAPEG (membrane-associated proteins in eicosanoid and glutathione metabolism), as recently reviewed by Deponte [Citation40]. In particular, GSH is involved in the reduction of intra- or intermolecular disulphides of proteins and low-molecular-weight compounds, reactions that can occur either non-enzymatically or enzymatically with the help of Grx, protein disulphide isomerase, and some glutathione S-transferase (GST) isoforms. GSH is widely involved in the cellular removal of H2O2 and of other hydroperoxides, a reaction catalysed by a variety of enzymes, including specialised GPx, Prx, GST, and a few Grx isoforms. GSH is then involved in the detoxification of 2-oxoaldehydes such as glyoxal (GO) and methylglyoxal (MGO), a reaction that is catalysed by the isomerase Glo1 and the thioesterase Glo2. In addition to glycolysis, MGO, GO, and other 2-oxoaldehydes (2-OA) are also formed during lipid peroxidation as well as by the metabolism of acetone, glycerol, and threonine [Citation40]. These reactive carbonyl species can condense with the nucleophilic sites of proteins, lipids, and nucleic acids, thereby yielding the so-called advanced glycation end products, which are involved in the inflammatory and profibrotic response so that their removal by a detoxification system is beneficial [Citation41–43].
In the case of severe and prolonged oxidative stress, GSH depletion can occur for several reasons: (1) increase in GSSG formation and accumulation, followed by export and extracellular hydrolysis; (2) protein S-glutathionylation; (3) formation of GS adducts with electrophilic compounds, which are the by-products of lipid peroxidation [Citation44]. The resulting thioether reaction products are then exported and metabolised via the mercapturic acid pathway. GS adducts are also formed when electrophilic reactive metabolites are formed by xenobiotics as in the case of acetaminophen.
Hence, there are oxidative conditions or xenobiotic metabolic pathways that can induce a significant depletion of GSH as found in several studies. Asher and Guilford [Citation45] performed a review of the literature from 1980 to 2016 on the role that oxidative stress and GSH play in ear, nose, and throat (ENT) conditions. The authors concluded that many ENT conditions such as rhinitis, allergic rhinitis, chronic rhinosinusitis (CRS), CRS with polyps, otitis media with effusion, chronic otitis media (COM), COM and cholesteatoma, tympanic membrane sclerosis, tonsillitis, Meniere disease, laryngeal conditions, and chronic cough are associated with oxidative stress and decreased GSH, both locally in the affected tissues and systemically. They also suggested that the oxidative stress related to those conditions may be due to depletion of GSH, which is increased by higher levels of O2 in the upper respiratory tract [Citation45].
Decreased GSH levels are associated with the common features of aging as well as of a wide range of pathologic conditions, including neurodegenerative disorders. Notably, GSH depletion and/or alterations in its metabolism appear to be crucial in the onset of Parkinson disease [Citation46,Citation47], autism, schizophrenia, bipolar disorder, and Alzheimer’s disease [Citation48].
Significant depletion of GSH has been reported in lung fluids, such as BAL and epithelial lining (ELF) fluids, in different lung diseases. GSH is unique in that it is one of the few antioxidants in ELF that is expressed at higher levels than in plasma. Under normal conditions, GSH in ELF can range between 100 and 300 µM and increase to near millimolar levels under conditions of stress [Citation49]. A number of stimuli, including bacterial infection, disease, or smoking, can increase GSH levels. Under these circumstances, the increased GSH may be an adaptive response to these stimuli to avoid further damage to the lung. Conversely, ELF GSH levels are decreased in many progressive lung diseases, including idiopathic pulmonary fibrosis where GSH is reduced by 51% compared with normal individuals, acute respiratory distress syndrome, CF, lung transplantation, HIV infection, and late-stage COPD [Citation49].
Depletion of GSH is also involved in acetaminophen toxicity. Once formed within hepatocytes by the cytochrome P-450 pathway, NAPQI reacts as an electrophilic agent with the thiolate of GSH in a reaction catalysed by GST, resulting in a Michael adduct (GSH-NAPQI). This GSH-NAPQI conjugate is then metabolised stepwise by γ-glutamyl transpeptidase, dipeptidase, and N-acetylase, cleaving off the γ-glutamyl and glycine residues to ultimately form inert cysteine and mercapturate conjugates that are renally excreted. In the case of acetaminophen intoxication or when one of the following pathologic conditions likely to be associated with reduction in intrahepatic GSH concentrations occurs (HIV infection, chronic hepatitis C infection, CF, malnutrition, and eating disorders such as anorexia nervosa), then NAPQI escapes GSH detoxification and reacts with nucleophilic liver macromolecules, leading to a damaging effect [Citation50].
When an oxidative condition or xenobiotic exposure can affect the cellular content of GSH and consequently the natural defence, then a rational therapeutic intervention could be based on replenishing GSH. NAC boosts GSH because it is a precursor of Cys, which is the rate-limiting factor in cellular glutathione biosynthesis. The deacetylation of N-acetyl-l-amino acids is catalysed by several aminoacylases (I, II, and III) [Citation51], and NAC is hydrolysed by cytosolic acylase I. Yamauchi et al. [Citation49] measured the activity of the NAC-deacetylating enzyme (acylase) in various tissues of different species (rat, rabbit, dog, monkey, and man). Acylase activity was the highest in the kidney in all species studied. Enzyme activity in the liver was 10–22% of that in the kidney in rat, rabbit, monkey, and man. The tissue distribution of acylase I was then determined by western blotting and an immunohistochemical method and the results indicate that the kidney and liver are the main organs responsible for the biotransformation of NAC to cysteine in mammals. Consistent with this, pharmacokinetics studies have shown that NAC undergoes extensive first-pass metabolism in the liver and kidney [Citation51].
The in vivo effect of NAC to improve GSH content in tissues has been tested in several animal models in both physiologic and pathologic conditions. NAC treatment increased the GSH content and GSH-to-oxidised GSH ratio in the liver of suckling piglets with hepatic damage [Citation52]. In a paraquat model of oxidative stress induced in mice, GSH depletion in liver and brain was significantly counteracted by NAC, resulting in less oxidative damage [Citation14]. Arfsten et al. [Citation53] measured radiolabelled NAC distribution and GSH tissue levels in rats. GSH concentrations were increased 20% in the skin and 50% in the liver after one dose of 1200 mg/kg NAC, whereas lung and kidney GSH were unaffected. The effect of chronic treatment with NAC was then described by Arfsten et al. [Citation54]. After 30 d with an oral dose of 600 or 1200 mg/kg/d, an increase of NAC in the kidneys and skin of 24–81% was observed [Citation54]. Based on the animal studies, it is clear that acute or chronic oral NAC treatment increases the GSH content in several tissues, including liver, kidney, skin, and brain.
Some human studies have considered the modulation of GSH levels in blood, circulating cells, and some fluids such as BAL and pulmonary ELF as a pharmacodynamic response to NAC. Some studies have found that oral treatment with NAC has the effect of increasing GSH in erythrocytes and blood lymphocytes. Pendyala and Creaven [Citation55], in a dose escalation phase 1 study dated 1995, found a significant and transient increase of GSH in peripheral blood lymphocytes after an oral dose of 800 mg/m2. Zembron-Lacny et al. [Citation15] reported that 1200 mg of NAC administered to healthy individuals for 8 d significantly increased the blood level of GSH (+33%). Kasperczyk et al. [Citation15] found that in workers exposed to lead receiving 400 and 800 mg of NAC, erythrocyte GSH concentrations were significantly increased by 5% and 6% respectively, compared with those at baseline.
Some evidence indicates the efficacy of NAC in increasing GSH content in lung fluids. When administered intravenously to eight patients with pulmonary fibrosis, NAC was found to significantly increase GSH in BAL fluid and ELF [Citation56]. It was also found that oral treatment with NAC (3 × 600 mg per day for 5 d) in non-smoking patients with idiopathic pulmonary fibrosis significantly increased GSH in BAL fluid, reaching values within the normal range. There was also a trend of increase in GSH in ELF [Citation25].
Based on these results, we can conclude that in the case of significant oxidative stress or exposure to electrophilic compounds arising from xenobiotic metabolism, the pool of GSH can be depleted, thus leaving the oxidants and electrophilic compounds free to react with biomacromolecules, leading to a damaging response. NAC given in acute or chronic regimen was found to significantly replenish the GSH pool in some areas such as liver, skin, lung, and brain, thus preventing these damaging effects.
NAC as disulphide breaking agent
NAC is an efficient reducing agent of protein disulphides through the classic thiol-disulphide interchange mechanism [Citation57]. An SN2 reaction mechanism is involved, whereby, in a single reaction step, the attacking NAC thiolate binds to the central sulphur of the disulphide and the leaving thiol (R″SH) is released via a trisulphide-like transition state structure. By considering this reaction model, the rate of the thiol-disulphide interchange reaction is strongly related to the nucleophilicity of the thiolate, and this explains the greater disulphide reducing ability of NAC compared with Cys and GSH [Citation58], which reflects the order of S nucleophilicity of the thiols: NAC > GSH > Cys. For NAC, the N-acetyl residue and carboxylated group (rather than NH3+ and − CONH- moieties) both stabilise the high electron density of the thiolate, thus increasing the nucleophilic character. Hence, while the antioxidant/radical scavenging ability of NAC, Cys, and GSH (order of activity Cys > GSH > NAC) is related to the SH acidity, which regulates the relative content of the active species (the thiolate), the reducing ability is mainly related to the SH basicity (nucleophilicity), which is the opposite of the SH acidity and hence follows the order NAC > GSH > Cys.
As demonstrated by Parker and Kharash [Citation59] the entering nucleophilic moiety (here the NAC thiolate) breaks the disulphide bonds and remains linked to the more basic sulphur atom, so displacing the more acid thiol function. By considering that the ionisation properties of a cysteine residue within a protein are roughly comparable with those of GSH, one may conclude that the SN2 reaction involving NAC and a cysteinylated protein usually liberates a cysteine molecule yielding the corresponding N-acetyl cysteinylated protein. In contrast and when the cysteinylated residues is markedly acid, the same reaction forms good amount of free protein cysteine plus NAC-Cys mixed disulphide as seen for human serum albumin Cys34 (see below). This implies a sort of protecting mechanism, since the very acid thiols which are easily oxidised in disulphide derivatives are also easily restored by circulating nucleophilic molecules.
The reducing ability of NAC toward the disulphide cross-links is clearly responsible for the mucolytic activity. Mucin polymers, the principal gel-forming proteins in mucus, are characterised by cysteine-rich domains in their N and C termini which mediate polymer extension by end-to-end disulphide linkage of mucin monomers. Mucins are also characterised by abundant cysteine-rich regions in the internal domains, which form internal cross-links upon oxidation [Citation60]. In the healthy lung, the low elastic modulus (G′) of healthy airway mucus gels indicates a low density of mucin cross-links. Lightly cross-linked mucus gels are easily transported by the mucociliary escalator. In lung disease characterised by inflammatory conditions, the oxidative burst causes oxidation of internal cysteine thiols, which, on the one hand, may contribute to antioxidant effects of mucins but, on the other hand, could modify the biophysical properties of mucins by generating disulphide cross-links between internal cysteine domains. The resulting heavily cross-linked mucus is not easily transported and accumulates to cause airflow obstruction, atelectasis, and lung infection () [Citation24]. Pathologic mucus is typically highly elastic and thought to occur as a downstream consequence of airway inflammation [Citation4]. NAC is a mucolytic agent able to reduce the heavy cross-linked mucus as already demonstrated in vitro by Sheffenr et al. [Citation61] and then in vivo by Hurst et al. in 1967 [1]. The mucolytic effect of NAC given by aerosol or by broncotracheal administration is clearly due to the direct reducing ability of NAC, which is directly transported into the mucus. NAC can also act in this area as a direct antioxidant of some oxidant species such as HOX, which are produced in abundance following the inflammatory condition and in a milieu where Cys residues and GSH molecules are greatly consumed following the oxidative burst.
Figure 2. Antioxidant and disulphide breaking activity of NAC in lung disease characterised by an inflammatory condition. The oxidative burst causes the oxidation of internal cysteine of mucins, generating disulphide cross-links between internal cysteine domains. The resulting heavily cross-linked mucus is not easily transported and accumulates to cause airflow obstruction, atelectasis, and lung infection [Citation24]. NAC is a mucolytic agent able to reduce the heavily cross-linked mucus. When Cys and GSH are depleted following the inflammatory condition, NAC can act as a direct antioxidant of some oxidant species such as HOX and NO2.
![Figure 2. Antioxidant and disulphide breaking activity of NAC in lung disease characterised by an inflammatory condition. The oxidative burst causes the oxidation of internal cysteine of mucins, generating disulphide cross-links between internal cysteine domains. The resulting heavily cross-linked mucus is not easily transported and accumulates to cause airflow obstruction, atelectasis, and lung infection [Citation24]. NAC is a mucolytic agent able to reduce the heavily cross-linked mucus. When Cys and GSH are depleted following the inflammatory condition, NAC can act as a direct antioxidant of some oxidant species such as HOX and NO2.](/cms/asset/3e04b975-9b14-4d3d-9913-bf901798d72c/ifra_a_1468564_f0002_c.jpg)
The mucolytic effect of NAC after oral treatment is more debatable because several clinical studies have shown a limited effect. This could be explained by the reduced bioavailability of NAC in the lung fluid when given orally [Citation62]. The reducing ability of NAC is given by the molecule itself, in the acetylated form, and not by the deacetylated metabolite (Cys) or by GSH. However, an increase of Cys and/or GSH induced by NAC in the lung fluid can have other effects, such as an antioxidant activity, which can prevent the formation of heavily cross-linked mucus. However, a better understanding of the distribution of NAC in lung fluids by using accurate mass spectrometry techniques is needed to better understand the direct mucolytic activity of NAC after oral administration.
The reducing ability of NAC may also be involved in the antioxidant mechanism by restoring the systemic pools of small thiols as well as of reduced protein SH groups, which regulate the redox conditions. When human plasma was incubated for 1 h with varying clinically relevant concentrations of NAC (0–1000 µg/mL), a significant increase of free Cys was observed accompanied by a decrease in Cys plasma protein binding from 85% (10 µg/mL NAC) to approximately 0% (1000 µg/mL) [Citation63]. The release of Cys bound to proteins by NAC is also involved in GSH synthesis, as demonstrated in an elegant paper by Zhou et al. [Citation64]. When stably labelled NAC was given by an intraperitoneal bolus in rats, a significant increase of GSH was found in erythrocytes. However, only less than 1% of the increased GSH was found to be labelled, and hence derived from labelled NAC, thus suggesting that GSH is formed by endogenous cysteine released by cysteinylated proteins through a thiol exchange reaction with NAC.
As anticipated above, circulating NAC can affect the mercaptoalbumin content of human serum albumin (HSA) by reducing the corresponding cysteinylated form. Albumin, the most abundant protein in plasma, is characterised by only one free cysteine residue, Cys34, which constitutes the largest pool of thiols in the circulation (80%) [Citation65]. In healthy adults, about 70–80% of the Cys34 in albumin contains a free sulphydryl group (mercaptoalbumin, HSA-SH), the rest forms a disulphide with several compound thiols, such as cysteine, homocysteine, or glutathione; the predominant modification is S-cysteinylated albumin (HSA-Cys) [Citation66]. HSA represents the main antioxidant of plasma and extracellular fluids, and this effect is mainly due to the Cys34 residue, which is able to scavenge several oxidants, such as hydroxyl and peroxyl radicals, hydrogen peroxide, peroxynitrite [Citation67], and to form covalent adducts with lipid-oxidation electrophilic by-products, such as 4-hydroxy-trans-2-nonenal [Citation68]. The high radical scavenging and carbonyl quenching efficacy of Cys34 is explained by considering: (1) the high acidity, due to the ability of the surrounding amino acids to stabilise the thiolate anion, and (2) the significant solvent accessibility of the thiolate anion. The antioxidant activity of Cys34 is demonstrated by different ex vivo studies to show that oxidative stress conditions are associated with a reduction of mercaptoalbumin and by a concomitant increase of the cysteinylated form as well as of other mixed disulphides and or higher oxidation states such as sulphinic/sulphonic acid derivatives [Citation69–71].
The molecular interaction between HSA and NAC was investigated by Harada et al. [Citation72] who reported that when NAC was added to isolated HSA, HSA-Cys and HSA-SH rapidly decreased and increased, respectively, while the HSA-NAC conjugate formed much more slowly. The results lead the authors to suggest that NAC binds HSA in a two-step process. In the first step, NAC rapidly reacts with the disulphide bond of HSA-Cys, resulting in the dissociation process and HSA-SH formation. As said above and due to the high acidity of Cys34, NAC binds preferentially Cys, forming the Cys-NAC disulphide. In a second step, low-molecular-weight disulphides such as cysteine, NAC-NAC, or Cys-NAC bind to free SH of HSA-SH, forming HSA-NAC or eventually regenerating HSA-Cys. The rate constant of the dissociation and binding are quite different; the former is 1.3 h−1 and the latter is 0.003 h−1 for NAC or less for Cys (0.00107 h−1). Hence, NAC rapidly reduces the disulphide bond of HSA-Cys, forming HSA-SH and then binding much more slowly with HSA-SH, forming the corresponding disulphide. Supporting the ability of NAC to restore protein thiols, Fu et al. [Citation73] reported that NAC treatment for 3 d at a dose of 300 mg/kg/d significantly reduced the level of cysteinylated plasma proteins.
The ability of NAC to restore thiol proteins and in particular mercaptoalbumin in plasma is an interesting mechanism and needs to be further studied considering the pivotal role of mercaptoalbumin, which undergoes significant cysteinylation under different physio-pathologic conditions.
Conclusion
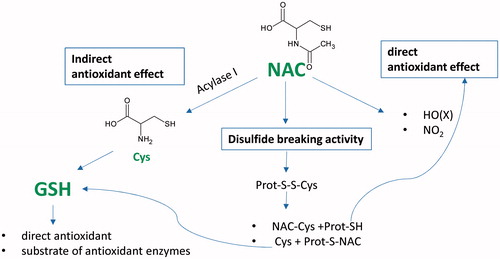
NAC is a well-established antioxidant and disulphide breaking agent as demonstrated by several in vitro and in vivo studies conducted in animals and humans. In the present paper, the main molecular mechanisms regarding the antioxidant and reducing activity of NAC have been summarised and critically reviewed (). In biological matrices, a direct antioxidant effect toward some radical and non-radical oxidants must be excluded as a main mechanism, taking into account the concentrations and reaction rates of NAC and of the competing endogenous enzymatic and non-enzymatic antioxidants. The in vivo antioxidant activity demonstrates that NAC acts as a GSH precursor, which in turn is a well-known direct antioxidant and a substrate of several antioxidant enzymes. This mechanism is supported by the ability of NAC to replenish depleted GSH pools as demonstrated in several in vivo studies. Moreover, in some conditions characterised by depletion of endogenous Cys and GSH, NAC can act also as a direct antioxidant agent for some oxidant species such as NO2 and HOX. An additional mechanism for the antioxidant activity of NAC has been considered more recently: the activity of NAC in breaking thiolated proteins. This releases free thiols, which have a better antioxidant activity than NAC and boost the synthesis of GSH, as well as reduced proteins, which in some cases, such as for mercaptoalbumin, have an important direct antioxidant activity. As well as being involved in the antioxidant mechanism, the reducing action of NAC also explains the mucolytic activity due to the effect of NAC in reducing heavily cross-linked mucus glycoproteins.
Figure 3. Overview of the antioxidant action of NAC. The antioxidant effect is due to indirect (GSH synthesis) and direct antioxidant activity, as well as disulphide breaking activity. The indirect activity refers to the ability of NAC to act as a GSH precursor, which in turn is a well-known direct antioxidant and a substrate of several antioxidant enzymes. When an oxidative stress status depletes the SH pools, NAC can act as direct scavenger of some oxidants such as NO(X) and NO2. NAC breaks thiolated proteins thus releasing free thiols, which have a better antioxidant activity than NAC and boost the synthesis of GSH and reduced proteins, which in some cases, such as for mercaptoalbumin, have an important direct antioxidant activity.
It should be pointed that some of the chemical and biochemical differences between Cys and NAC, and, in particular the antioxidant and reducing properties, are well explained by their different pKa values, which are 8.30 for Cys and 9.52 for NAC. In particular, the reduced acidity of the SH moiety of NAC on the one hand makes it more resistant to air oxidation but less potent as a direct oxidant and on the other hand it explains the greater disulphide reducing ability of NAC compared with Cys. A difference can also be found in the different cellular uptakes as demonstrated by He et al who found that NAC is superior to cysteine in replenishing intracellular cysteine in the cell [Citation74]. However, further studies on the different ability of Cys and NAC in replenishing GSH and cellular Cys are needed.
The ability of NAC to break thionylated proteins such as extracellular cysteinylated proteins can have further important biological effects by considering that such protein-thiol mixed disulphides may be involved in some pathogenetic mechanism and are now recognised as promising drug targets. Accordingly, Moreno et al. [Citation75] reported that disulphide stress may be considered as a specific type of oxidative stress in acute inflammation associated with certain mixed disulphides, particularly protein cysteinylation, and the oxidation of low-molecular-weight thiols such as cysteine, γ-glutamylcysteine, and homocysteine.
Acknowledgements
English language editing and styling assistance was provided by Edra spa, and unconditionally funded by Zambon Spa.
Disclosure statement
Francesco Sergio and Luisa Borsani are employees of Zambon SpA.
References
- Hurst GA, Shaw PB, LeMaistre CA. Laboratory and clinical evaluation of the mucolytic properties of acetylcysteine. Am Rev Respir Dis. 1967;96(5):962–970.
- Prescott LF. New approaches in managing drug overdosage and poisoning. Br Med J (Clin Res Ed). 1983;287(6387):274–276.
- Balsamo R, Lanata L, Egan CG. Mucoactive drugs. Eur Respir Rev. 2010;19(116):127–133.
- Whitehouse LW, Wong LT, Paul CJ, et al. Postabsorption antidotal effects of N-acetylcysteine on acetaminophen-induced hepatotoxicity in the mouse. Can J Physiol Pharmacol. 1985;63(5):431–437.
- Dodd S, Dean O, Copolov DL, et al. N-acetylcysteine for antioxidant therapy: pharmacology and clinical utility. Expert Opin Biol Ther. 2008;8(12):1955–1962.
- Vanderbist F, Maes P, Nève J. In vitro comparative assessment of the antioxidant activity of nacystelyn against three reactive oxygen species. Arzneimittelfor-schung. 1996;46(8):783–788.
- Kondo H, Takahashi M, Niki E. Peroxynitrite-induced hemolysis of human erythrocytes and its inhibition by antioxidants. FEBS Lett. 1997;413(2):236–238.
- Sueishi Y, Hori M, Ishikawa M, et al. Scavenging rate constants of hydrophilic antioxidants against multiple reactive oxygen species. J Clin Biochem Nutr. 2014;54(2):67–74.
- Ates B, Abraham L, Ercal N. Antioxidant and free radical scavenging properties of N-acetylcysteine amide (NACA) and comparison with N-acetylcysteine (NAC). Free Radic Res. 2008;42(4):372–377.
- Sagristá ML, García AE, Africa De Madariaga M, et al. Antioxidant and pro-oxidant effect of the thiolic compounds N-acetyl-L-cysteine and glutathione against free radical-induced lipid peroxidation. Free Radic Res. 2002;36(3):329–340.
- Michelucci A, De Marco A, Guarnier FA, et al. Antioxidant treatment reduces formation of structural cores and improves muscle function in RYR1Y522S/WT mice. Oxid Med Cell Longev. 2017;2017:6792694.
- Dhanda S, Kaur S, Sandhir R. Preventive effect of N-acetyl-L-cysteine on oxidative stress and cognitive impairment in hepatic encephalopathy following bile duct ligation. Free Radic Biol Med. 2013;56:204–215.
- Sandhir R, Kaur S, Dhanda S. N-acetyl-l-cysteine prevents bile duct ligation induced renal injury by modulating oxidative stress. Indian J Clin Biochem. 2017;32(4):411–419.
- Ortiz MS, Forti KM, Suárez Martinez EB, et al. Effects of antioxidant N-acetylcysteine against paraquat-induced oxidative stress in vital tissues of mice. Int J Sci Basic Appl Res. 2016;26(1):26–46.
- Zembron-Lacny A, Slowinska-Lisowska M, Szygula Z, et al. The comparison of antioxidant and hematological properties of N-acetylcysteine and alpha-lipoic acid in physically active males. Physiol Res. 2009;58(6):855–861.
- Zhang L, Xu S, Huang Q, et al. N-acetylcysteine attenuates the cuprizone-induced behavioral changes and oligodendrocyte loss in male C57BL/7 mice via its anti-inflammation actions. J Neurosci Res. 2018;96(5):803–816.
- Samuni Y, Goldstein S, Dean OM, et al. The chemistry and biological activities of N-acetylcysteine. Biochim Biophys Acta. 2013;1830(8):4117–4129.
- Soldini D, Zwahlen H, Gabutti L, et al. Pharmacokine-tics of N-acetylcysteine following repeated intravenous infusion in haemodialysed patients. Eur J Clin Pharmacol. 2005;60(12):859–864.
- Winterbourn CC. Revisiting the reactions of superoxide with glutathione and other thiols. Arch Biochem Biophys. 2016;595:68–71.
- Trujillo M, Radi R. Peroxynitrite reaction with the reduced and the oxidized forms of lipoic acid: new insights into the reaction of peroxynitrite with thiols. Arch Biochem Biophys. 2002;397(1):91–98.
- Carballal S, Bartesaghi S, Radi R. Kinetic and mechanistic considerations to assess the biological fate of peroxynitrite. Biochim Biophys Acta. 2014;1840(2):768–780.
- Hoy A, Leininger-Muller B, Kutter D, et al. Growing significance of myeloperoxidase in non-infectious diseases. Clin Chem Lab Med. 2002;40(1):2–8.
- O’Donnell C, Newbold P, White P, et al. 3-Chlorotyrosine in sputum of COPD patients: relationship with airway inflammation. COPD. 2010;7(6):411–417.
- Yuan S, Hollinger M, Lachowicz-Scroggins ME, et al. Oxidation increases mucin polymer cross-links to stiffen airway mucus gels. Sci Transl Med. 2015;7(276):276ra27.
- Meyer A, Buhl R, Magnussen H. The effect of oral N-acetylcysteine on lung glutathione levels in idiopathic pulmonary fibrosis. Eur Respir J. 1994;7(3):431–436.
- Persinger RL, Poynter ME, Ckless K, et al. Molecular mechanisms of nitrogen dioxide induced epithelial injury in the lung. Mol Cell Biochem. 2002;2002:71–80.
- Storkey C, Davies MJ, Pattison DI. Reevaluation of the rate constants for the reaction of hypochlorous acid (HOCl) with cysteine, methionine, and peptide derivatives using a new competition kinetic approach. Free Radic Biol Med. 2014;73:60–66.
- Müller B, Oske M, Hochscheid R, et al. Effect of N-acetylcysteine treatment on NO2-impaired type II pneumocyte surfactant metabolism. Eur J Clin Invest. 2001;31(2):179–188.
- Siddiqui MR, Wabaidur SM, Ola MS, et al. High-throughput UPLC-MS method for the determination of N-acetyl-l-cysteine: application in Tissue Distribution Study in Wistar rats. J Chromatogr Sci. 2016;54(7):1244–1252.
- Buettner GR, Jurkiewicz BA. Catalytic metals, ascorbate and free radicals: combinations to avoid. Radiat Res. 1996;145(5):532–541.
- Kimoto E, Tanaka H, Gyotoku J, et al. Enhancement of antitumor activity of ascorbate against Ehrlich ascites tumor cells by the copper:glycylglycylhistidine complex. Cancer Res. 1983;43(2):824–828.
- Neuzil J, Thomas SR, Stocker R. Requirement for, promotion, or inhibition by alpha-tocopherol of radical-induced initiation of plasma lipoprotein lipid peroxidation. Free Radic Biol Med. 1997;22(1–2):57–71.
- Viña J, Saez GT, Wiggins D, et al. The effect of cysteine oxidation on isolated hepatocytes. Biochem J. 1983;212(1):39–44.
- Saez G, Thornalley PJ, Hill HA, et al. The production of free radicals during the autoxidation of cysteine and their effect on isolated rat hepatocytes. Biochim Biophys Acta. 1982;719(1):24–31.
- Viña J, Hems R, Krebs HA. Maintenance of glutathione content is isolated hepatocyctes. Biochem J. 1978;170(3):627–630.
- Wu MS, Lien GS, Shen SC, et al. N-acetyl-L-cysteine enhances fisetin-induced cytotoxicity via induction of ROS-independent apoptosis in human colonic cancer cells. Mol Carcinog. 2014;53(Suppl1):E119–E129.
- Zheng J, Lou JR, Zhang XX, et al. N-acetylcysteine interacts with copper to generate hydrogen peroxide and selectively induce cancer cell death. Cancer Lett. 2010;298(2):186–194.
- Zheng Z, Shetty K. Solid-state bioconversion of phenolics from cranberry pomace and role of Lentinus edodes beta-glucosidase. J Agric Food Chem. 2000;48(3):895–900.
- Meister A, Anderson ME. Glutathione. Annu Rev Biochem. 1983;52:711–760.
- Deponte M. Glutathione catalysis and the reaction mechanisms of glutathione-dependent enzymes. Biochim Biophys Acta. 2013;1830(5):3217–3266.
- Vistoli G, De Maddis D, Cipak A, et al. Advanced glycoxidation and lipoxidation end products (AGEs and ALEs): an overview of their mechanisms of formation. Free Radic Res. 2013;47(Suppl1):3–27.
- Aldini G, Carini M, Yeum KJ, et al. Novel molecular approaches for improving enzymatic and nonenzymatic detoxification of 4-hydroxynonenal: toward the discovery of a novel class of bioactive compounds. Free Radic Biol Med. 2014;69:145–156.
- Aldini G, Vistoli G, Stefek M, et al. Molecular strategies to prevent, inhibit, and degrade advanced glycoxidation and advanced lipoxidation end products. Free Radic Res. 2013;47(Suppl1):93–137.
- Giustarini D, Colombo G, Garavaglia ML, et al. Assessment of glutathione/glutathione disulphide ratio and S-glutathionylated proteins in human blood, solid tissues, and cultured cells. Free Radic Biol Med. 2017;112:360–375.
- Asher BF, Guilford FT. Oxidative stress and low glutathione in common ear, nose, and throat conditions: a systematic review. Altern Ther Health Med. 2016;22(5):44–50.
- Homma T, Fujii J. Application of glutathione as anti-oxidative and anti-aging drugs. Curr Drug Metab. 2015;16(7):560–571.
- Morris G, Anderson G, Dean O, et al. The glutathione system: a new drug target in neuroimmune disorders. Mol Neurobiol. 2014;50(3):1059–1084.
- Gu F, Chauhan V, Chauhan A. Glutathione redox imbalance in brain disorders. Curr Opin Clin Nutr Metab Care. 2015;18(1):89–95.
- Gould NS, Day BJ. Targeting maladaptive glutathione responses in lung disease. Biochem Pharmacol. 2011;81(2):187–193.
- Kalsi SS, Dargan PI, Waring WS, et al. A review of the evidence concerning hepatic glutathione depletion and susceptibility to hepatotoxicity after paracetamol overdose. Open Access Emerg Med. 2011;3:87–96.
- Anders MW, Dekant W. Aminoacylases. Adv Pharmacol. 1994;27:431–448.
- Zhang H, Su W, Ying Z, et al. N-acetylcysteine attenuates intrauterine growth retardation-induced hepatic damage in suckling piglets by improving glutathione synthesis and cellular homeostasis. Eur J Nutr. 2018;57(1):327–338.
- Arfsten DP, Johnson EW, Wilfong ER, et al. Distribution of radio-labeled N-acetyl-L-cysteine in Sprague-Dawley rats and its effect on glutathione metabolism following single and repeat dosing by oral gavage. Cutan Ocul Toxicol. 2007;26(2):113–134.
- Arfsten D, Johnson E, Thitoff A, et al. Impact of 30-day oral dosing with N-acetyl-L-cysteine on Sprague-Dawley rat physiology. Int J Toxicol. 2004;23(4):239–247.
- Pendyala L, Creaven PJ. Pharmacokinetic and pharmacodynamic studies of N-acetylcysteine, a potential chemopreventive agent during a phase I trial. Cancer Epidemiol Biomarkers Prev. 1995;4(3):245–251.
- Meyer A, Buhl R, Kampf S, et al. Intravenous N-acetylcysteine and lung glutathione of patients with pulmonary fibrosis and normals. Am J Respir Crit Care Med. 1995;152(3):1055–1060.
- Nagy P. Kinetics and mechanisms of thiol-disulfide exchange covering direct substitution and thiol oxidation-mediated pathways. Antioxid Redox Signal. 2013;18(13):1623–1641.
- Noszál B, Visky D, Kraszni M. Population, acid–base, and redox properties of N-acetylcysteine conformers. J Med Chem. 2000;43(11):2176–2182.
- Parker AJ, Kharasch N. Derivatives of sulfenic acids. XXXVI. The ionic scission of the sulfur–sulfur Bond 1 part 1. J Am Chem Soc. 1960;82:4.
- McGuckin MA, Thornton DJ, Whitsett JA, et al. Mucins and Mucus A2 – Mestecky, Jiri. Mucosal immunology. 4th ed. Boston: Academic Press; 2015. p. 231–250 (Chapter 14).
- Sheffner AL, Medler EM, Jacobs LW, et al. The in vitro reduction in viscosity of human tracheobronchial secretions by acetylcysteine. Am Rev Respir Dis. 1964;90:721–729.
- Janssen B, Hohenadel D, Brinkkoetter P, et al. Carnosine as a protective factor in diabetic nephropathy: association with a leucine repeat of the carnosinase gene CNDP1. Diabetes. 2005;54(8):2320–2327.
- Radtke KK, Coles LD, Mishra U, et al. Interaction of N-acetylcysteine and cysteine in human plasma. J Pharm Sci. 2012;101(12):4653–4659.
- Zhou J, Coles LD, Kartha RV, et al. Intravenous administration of stable-labeled N-acetylcysteine demonstrates an indirect mechanism for boosting glutathione and improving redox status. J Pharm Sci. 2015;104(8):2619–2626.
- Turell L, Radi R, Alvarez B. The thiol pool in human plasma: the central contribution of albumin to redox processes. Free Radic Biol Med. 2013;65:244–253.
- Colombo G, Clerici M, Giustarini D, et al. Redox albuminomics: oxidized albumin in human diseases. Antioxid Redox Signal. 2012;17(11):1515–1527.
- Carballal S, Alvarez B, Turell L, et al. Sulfenic acid in human serum albumin. Amino Acids. 2007;32(4):543–551.
- Aldini G, Regazzoni L, Orioli M, et al. A tandem MS precursor-ion scan approach to identify variable covalent modification of albumin Cys34: a new tool for studying vascular carbonylation. J Mass Spectrom. 2008;43(11):1470–1481.
- Nagumo K, Tanaka M, Chuang VT, et al. Cys34-cysteinylated human serum albumin is a sensitive plasma marker in oxidative stress-related chronic diseases. PLoS One. 2014;9(1):e85216.
- Regazzoni L, Del Vecchio L, Altomare A, et al. Human serum albumin cysteinylation is increased in end stage renal disease patients and reduced by hemodialysis: mass spectrometry studies. Free Radic Res. 2013;47(3):172–180.
- Carballal S, Radi R, Kirk MC, et al. Sulfenic acid formation in human serum albumin by hydrogen peroxide and peroxynitrite. Biochemistry. 2003;42(33):9906–9914.
- Harada D, Anraku M, Fukuda H, et al. Kinetic studies of covalent binding between N-acetyl-L-cysteine and human serum albumin through a mixed-disulfide using an N-methylpyridinium polymer-based column. Drug Metab Pharmacokinet. 2004;19(4):297–302.
- Fu XY, Cate SA, Chen JM, et al. Effect of N-acetyl-L-cysteine on cysteine redox status in patients with thrombotic thrombocytopenic purpura: protein disulfide bound cysteine as a biomarker of oxidative stress. Blood. 2015;126(23):1044.
- He X, Wu X, Shi W, et al. Comparison of N-acetylcysteine and cysteine in their ability to replenish intracellular cysteine by a specific fluorescent probe. Chem Commun (Camb). 2016;52(60):9410–9413.
- Moreno ML, Escobar J, Izquierdo-Álvarez A, et al. Disulfide stress: a novel type of oxidative stress in acute pancreatitis. Free Radic Biol Med. 2014;70:265–277.