Abstract
Objective of this study was to prepare a drug delivery system for therapeutic peptides that are degraded in the upper part of the gastrointestinal tract due to degradation activity of the enzymes. Delivering peptide to the colon in which enzymatic activity is low is next hope for absorption of these agents. Pectin, a naturally occurring water soluble polysaccharide, as a matrix for peptide delivery was studied. Degradation of pectin by the colonic enzymes makes it suitable for colon-specific delivery of drugs. Bovine serum albumin (BSA) was used as a model peptide. Calcium pectinate beads were prepared by extruding BSA-loaded pectin solution to an agitating calcium chloride solution, and gelled spheres were formed instantaneously by an ionotropic gelation reaction. The effect of several factors such as concentration of pectin, concentration of calcium chloride, and total drug loading on the pattern of drug release in the dissolution medium was studied. Prepared beads showed good resistance in the release medium. The entrapment efficiency of the beads was high (between 63% and 99%). Entrapment efficiency of BSA was reversely dependent to the amount of the drug loaded in the beads. The amount of BSA loaded on the beads affects pattern of drug release. The concentration of the pectin showed the highest impact on the rate of drug release. Presence of the pectiolytic enzymes facilitated the drug release from the beads.
Targeting pharmaceuticals to the colon is valuable for local or systemic drug delivery. To deliver a compound in a non degradable form to the last part of the gastrointestinal tract (GIT), it must first pass through the stomach, and the upper part of the intestine and must use the characteristics of the colon to specifically release the drug in this part of the digestive tract (Watts and Illum Citation1997).
Natural polysaccharides are now extensively used for the development of solid dosage forms for delivery of drugs to the colon (Chourasia and Jain Citation2004). Presence of a large amount of polysaccharidases in the human colon is the main rational for use of these natural compounds. This may be regarded as the main advantage of polysaccharides for colon specific delivery. Their low prices, and wide availability in a varity of structures with different properties are other advantages of these agents. They easily can be modified chemically and biochemically and are highly stable, safe, nontoxic, hydrophilic, gel forming, and biodegradable, which suggests their use in targeted drug delivery systems.
Apart from specific treatments recommended for pathological conditions in colon, other therapeutic agents such as proteins, peptides, and diagnostic agents are intended for delivery in the colon (Krishnaiah et al. Citation2003). Attempts to delivering these therapeutic agents in the colon are due to the capacity of this part of the gastrointestinal tract to absorb these drugs. Peptide drugs cannot be directly administered orally because of their susceptibility to the enzymes present in the small intestine. It has been reported that such peptide drugs can be administered orally if they are coated first with a polymer (Saffron et al. Citation1990). Saffron and coworkers have reported that when the azopolymer-coated drugs reach the colon, the indigenous microflora reduces the azo bonds and breaks the cross-link that leads to the degradation of the polymer film, thereby releasing the drug into the lumen of the colon. However, there are some problems related to this system, the most important of which is the safety of the azopolymer that is a synthetic polymer and has not been fully evaluated.
There have been many approaches to achieve site specificity in the colon in which the specific characteristics of the colon such as pH (Gupta, Beckert, and Price Citation2001), microflora (Sinha and Kumria Citation2003), enzymes reducing (Uribe Citation1998), and transit time (Atyabi et al. Citation1996a, Citation1996b) have been addressed. However, the method by which the drug release is triggered by the colonic microflora appears to be more interesting with regard to selectivity. Some polysaccharides have been investigated in this regard (Watts and Illum Citation1997). Pectin, a natural polysaccharide, consists of partially methoxylated poly α -(1-4)-D-galacturonic acid and is completely digested by colonic bacteria (Cummings et al. Citation1979). Many studies have shown that pectin has potential value for delivering therapeutics to the colon (Liu et al. Citation2003; Ashford et al. Citation1993). Pectinate gelisphere system also has been studied for linear drug release (Pillay, Dankwerts, and Fassihi Citation2002). The main problem associated with polysaccharides is their solubility and swelling in an aqueous media. This causes early release of the active agents targeted to the colon. The swelling and solubility of pectin must be modified and reduced without compromising its biodegradability (Gliko-Kabi et al. Citation2000).
Several methods for confining drug release from polysaccharide based delivery systems have been proposed. One approach is employment of enteric polymer or time controlling polymers as coating material in combination with polysaccharides (Semde et al. Citation2000). The mixed coating can prevent the swelling and the solubilization of pectin during the transit from mouth to caesum (Turkoglu and Ugurlu Citation2002). Another approach for achieving site specificity to the colon is applying complexes of pectin with polycation polymers such as Eudragit RL (Semde et al. Citation2000).
The degree of esterification (DE) and degree of amidation (DA), both expressed as percentage of the carboxyl groups (esterified or amidated), are important means to classify pectins. Low methoxy pectins (with DE < 50%) form gels by the action of calcium, which cross-link the galactoronic acid chains analogously to the way calcium cross-links sodium alginate. Pectin with high degree of methoxylation (HM) is poorly soluble.
Therefore, in this study calcium pectinate was used to prepare matrix beads for colonic delivery of a polypeptide model (bovine serum albumin). The rational for the use of calcium pectinate is that it is degraded by pectinolytic enzymes in the colon. Calcium pectinate protects drug in the upper part of the GIT due to its insolubility and is not degraded by gastric or intestinal enzymes (Atyabi and Inanloo Citation2003). The effect of formulation variables on preparation and drug release characteristics of the system also was investigated.
MATERIALS AND METHODS
Nonamidated lowmethoxy pectin from citrus fruit with degree of esterification of 28% was obtained from Genu pectin (Denmark), bovine serum albumin (BSA) with average molecular weight of 66400 and pectinolytic enzyme were purchased from Merck Company (Germany). All other materials were of analytical grade.
Preparation of Calcium Pectinate Beads
Calcium pectinate beads were prepared as follows. Particular amount of pectin was dissolved in water through continuous agitation. After complete dissolving, BSA was added and left stirring for a few minutes. The mixture of polymer and drug was added to the solution of calcium chloride by a nozzle with 0.8 mm inner diameter during agitation. Prepared beads were left stirring in the medium for 30 min and then removed by filtration, washed with distilled water, and dried at room temperature. In some cases the beads were transferred to a solution of glutaraldehyde followed by washing with distilled water.
Drug Loading and Entrapment Efficiency Measurement
Drug content of the beads was determined after completely dissolving prepared calcium pectinate beads in a solution of phosphate buffer (pH 7.4). The amount of BSA was determined through a colorimetric method using Comassie brilliant blue as a reagent. The withdrawal was added to a particular concentration of Comassie blue solution mixed for 5 min and the color of the solution was analyzed at 595 nm using an ultraviolet/visible apparatus (Shimatsu, Japan). The ratio of the amount of the BSA to the initial applied amount of BSA was considered as entrapment efficiency of the beads.
Drug Release Study
Drug release profile of prepared beads was studied using USP dissolution apparatus method I (basket). A known amount of the beads was placed in a basket. Dissolution beakers were filled with the phosphate buffer solution with or without enzyme. The amount of the pectiolytic enzyme used was 300PG (activity 26000PG/ml; PG = miliequivalents of reducing groups liberated from pectin per minute per unit of enzyme). The basket containing beads was rotated at 50 rpm and temperature of the medium was fixed at 37°C ± 0.5. At appropriate time intervals, a 5-ml sample was withdrawn and replaced by the same volume of fresh medium. The sample was analyzed for the amount of the BSA. The drug release pattern was obtained by plotting the percent amount of the BSA released from the beads.
Physical Examination of Calcium Pectinate Beads
The size and shape of the beads were determined by electron microscopy as well as by the light microscope.
RESULTS AND DISCUSSION
Many colon-specific drug delivery systems have been developed using low methoxylated amidated pectin (Kleinebudde and Sande Citation2001). The prepared beads in this study differed from those mentioned in which nonamidated lowmethoxy pectin was used as a polysaccharide for bead preparation. Low methoxylated (LM) pectin is more susceptible to degradation, and it is assumed that the presence of calcium increased the susceptibility to enzymatic attack.
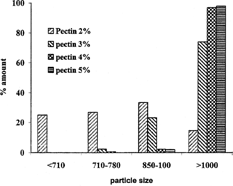
An aqueous solution of LM pectin containing BSA added drop-wise to the solution of calcium chloride and gelled spheres were formed instantaneously by ionotropic reaction between the divalent calcium ions and the negatively charged carboxyl groups of the LM pectin molecules. The shape of the beads was generally spherical. Scanning electron microscopy photograph of the beads is shown in . Some beads especially those prepared by high percent of pectin were drop shape. The encapsulation procedure used in this study resulted in bead size of 500–1000 μ. Different parameters such as concentration of calcium chloride, cross-linking time, amount of the drug, and usage of glutaraldehyde on the particle size distribution of beads were investigated. We found wide particle size distribution in samples prepared with low concentration of pectin, while different parameters had no significant effect on particle size (). However, particle size distribution is hardly dependent on the concentration of pectin solution, as more concentrated polymer solutions produced larger beads ().
FIG. 1 Scanning electron microscopic of the pectin beads.
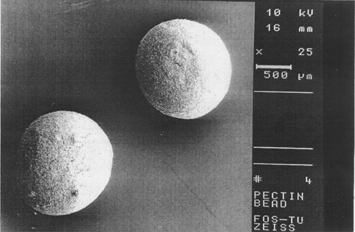
FIG. 2 Effect of the amount of the CaCl2 on the particle size distribution of the beads prepared with the solution of 2% pectin.
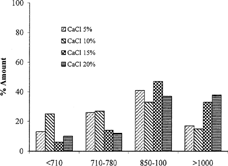
FIG. 3 Effect of the concentration of pectin on particle size distribution of beads prepared with 10% solution of CaCl2 and 50 mg BSA.
shows the entrapment efficiency of the BSA in the beads. The entrapment efficiency of the beads was so high between 63% and 99%. Entrapment efficiency of BSA is reversely dependent on the amount of the drug loaded, as attempts to load more drugs inside the beads leads to more drug loss. Such effect was observed when high calcium chloride and polymer concentration was used as well.
TABLE 1 Entrapment efficacy of different prepared formulations
Contrary to single unit drug delivery systems, multiparticular systems are spread out to the colon increasing surface area of the releasing drug and causing rapid drug release and improving absorption of drug (Pillay and Fassihi Citation1999). In this study, we attempted to overcome the drawbacks of single unit systems through use of a multiparticular system. BSA applied in this study as a model peptide is a large molecule that cannot diffuse through the pores of the matrix beads. However, BSA is released by the enzymatic degradation of the matrix. This system was found to resist the release of drug in gastric fluid but eroded and disintegrated in higher pH conditions.
The release profiles from beads tested at pH 7.4 with and without enzymes showed that calcium pectinate beads are degraded when Pectinex was present in the dissolution medium. Gelation of pectin makes it possible to reduce the penetration of water into the dosage form and hence the dissolution of the drug incorporated into it. When the degree of esterification is less than 50%, pectin forms a rigid gel by reaction with calcium salts or multivalent cations, which cross-link the galacturonic acids of the main polymer chains. Calcium pectinate is obtained by the formation of an ionic bond between the carboxylic acid groups of the pectin molecules and the calcium ions. The resulting structure has the form of an egg box, stable in solutions with a low pH. They swell in slightly basic solutions.
One of the important factors influencing drug release is hydration of the matrix system. The release of drug is due to two phenomena: erosion of the coating and diffusion. When the gel layer is thick, the diffusional pathway is important and reduces the drug release. In addition the calcium pectinate coating is even more sensitive to the erosion phenomenon if the ionic strength of the gel is low. However, a gel resulting from low methoxyester pectins cross-liked with calcium ions is not eroded. High methoxy ester pectins become hydrated in the presence of calcium ions and the matrix is eroded. However, the pregelation of high methyl ester and low methyl ester pectins can lead to the matrix breaking and eroding. In these cases, the drug releases before it can reach the colon.
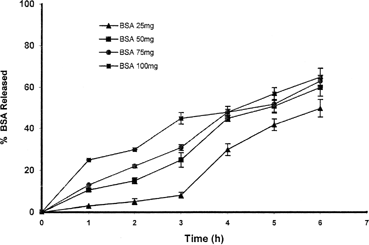
The release profiles of entrapping drug from the calcium pectinate microparticles can be changed by various factors: the amount of entrapped drug and pectin, curing time, and concentration of cross-linker. As expected, the amount of BSA loaded on the beads affects the pattern of drug release. shows the effect of the amount of drug loaded on drug release pattern. Calcium pectinate beads containing more BSA release their content more rapidly (∼50% in 3 hr) in comparison with the beads having low content of BSA (∼10% in same time). This result may be due to reduction of the ratio of the polymer to the drug in samples with high amounts of BSA. The amount of calcium chloride in the preparation medium has reverse effect on the release of the BSA from beads.
FIG. 4 Effect of the amount of the albumin on the pattern of drug release.
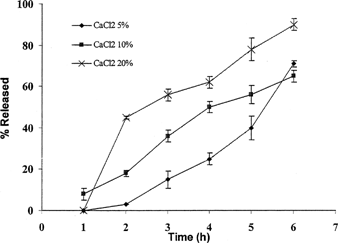
shows that drug release from beads prepared with 20% calcium chloride as cross-linker released their BSA content more rapidly than those prepared with lower amount of calcium chloride. This pattern of drug release was exactly repeated when the dissolution were studied in the dissolution medium containing Pectinex enzyme. However, the optimum concentration seems to be 10% of calcium chloride.
FIG. 5 Effect of the amount of calcium chloride on the release rate of BSA from beads prepared with 3% solution of pectin containing 50 mg BSA.
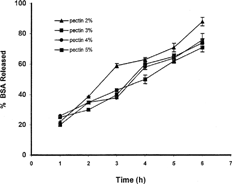
The most important factor affecting drug release from calcium pectinate beads was found to be the percent of the applied pectin, as a higher percent of pectin produced more resistant beads in the dissolution medium. As shows, beads prepared with solution of 5% pectin released only 35% of their BSA content in 6 hr treatment in dissolution medium while those prepared with the solution of 2% pectin lost about 75% of their drug content in the same time. However, the presence of the Pectinex enzyme is more effective as the dissolution pattern of particles prepared with different percents of pectin are more or less similar ().
FIG. 6 Effect of the amount of pectin on release of BSA from calcium pectinate beads in phosphate buffer medium (n = 4).
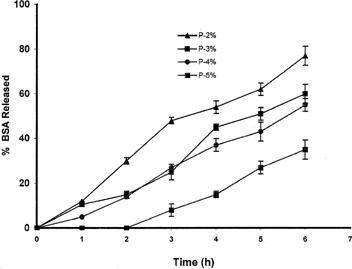
FIG. 7 Effect of the amount of pectin on release of BSA from calcium pectinate beads in medium containing enzyme (n = 4).
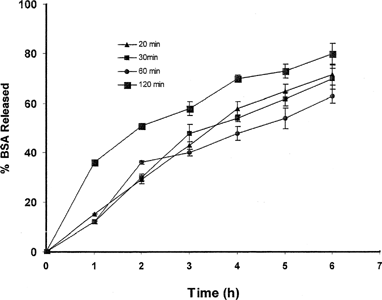
To investigate the effect of the cross-linking time on the hardness of the beads and eventually on the release properties of the beads, different cross-linking time was examined. shows that increasing the time that the beads are exposed to the calcium chloride as a cross-linker did not cause harder beads as expected. However, increasing the cross-linking time led to a faster dissolution pattern. This can be explained by the swelling of the beads is more facilitated when left in the solution for a long time.
FIG. 8 Effect of cross-linking time on release of BSA from prepared beads.
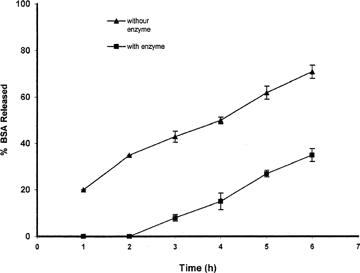
An objective for the current system was to prepare a system that was susceptible to enzymatic degradation and that release of the drug can be controlled by existing enzymes in the colon. The effect of the pectiolytic enzyme on the release of the BSA from the beads was evaluated in another set of experiments. The pectiolytic enzyme used in this study consisted of pectin methylesterase and two types of pectin depolymerase. The pectin methlesterase removes the methoxy group from the pectin while the polygalacturonase hydrolyzes the glycoside linkage next to the free carboxyl groups in the galacturonan chain by B elimination resulting in the formation of bobble bounds between C4 and C5 of the monomer. The presence of pectinolytic enzymes considerably accelerated the release of BSA from the prepared beads. This phenomenon is more apparent when a high concentration of pectin (5%) is used.
As shown in , calcium pectinate beads released their BSA content much faster when Pectinex was added to the dissolution medium. About 35% of BSA is released from beads prepared with 5% solution of pectin in phosphate buffer medium in 6 hr while this amount doubled (71%) when Pectinex was added to the medium.
FIG. 9 Effect of the presence of the pectinolytic enzymes on the pattern of BSA release from beads prepared with 5% solution of pectin and containing 50 mg BSA (n = 3).
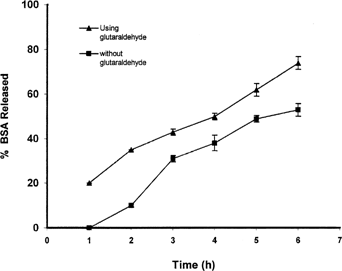
As it expected, glutaraldehyde can act as a hardening agent and as a result controls the swelling of the beads: in one experiment the prepared beads were transferred into the glutaraldehyde. shows the effect of using glutaraldehyde on drug release. Glutaraldehyde could reduce drug release from beads in the dissolution medium with enzyme, while this effect was not significant in phosphate buffer.
FIG. 10 Effect of usage of glutaraldehyde on drug release from beads prepared with 5% solution of pectin in dissolution medium containing enzyme.
CONCLUSION
In this study the capability of pectin for the preparation of a colon specific delivery was evaluated via investigation of the effect of different parameters on the preparation and in vitro drug release from beads. BSA was successfully retained and released from calcium pectinate beads whereas presence of enzyme in the dissolution medium could affect degradation of the polymer and eventually drug release from prepared beads. However, additional experiments, including biological activity of the released drug, would be required for a more reliable confirmation of this conclusion.
REFERENCES
- Ashford M., Fell J. T., Attwood D., Sharma H., Woodhead P. An evaluation of pectin as a carrier for drug targeting to the colon. J. Control. Rel. 1993; 26: 213–220, [CSA]
- Atyabi F., Sharma H. L., Mohammad H. A. H., Fell J. T. Controlled drug release from coated floating ion exchange resin beads. J. Control. Rel. 1996a; 42: 25–28, [CSA], [CROSSREF]
- Atyabi F., Sharma H. L., Mohammad H. A. H., Fell J. T. In vivo evaluation of a novel gastric retentive formulation based on ion echange resins. J. Control. Rel. 1996b; 42: 105–113, [CSA], [CROSSREF]
- Atyabi F., Inanlo K. Calcium pectinate as a carrier for albumin delivery. Proc. 30th Int. Symp. Control. Rel. Bioac. Mater. 2003
- Chourasia M. K., Jain S. K. Polysaccharides for colon targeted drug delivery. Drug Del. 2004; 11: 129–48, [CSA], [CROSSREF]
- Cummings J. H., Southgate D. A., Branch W. J., Wigging H. S., Houston H., Jenkins D. J., Jivraj T., Hill M. J. The digestion of pectin in the human gut and its effects on calcium absorption and large bowel function. Br. Nutr. 1979; 41: 477–485, [CSA], [CROSSREF]
- Gliko-Kabir I., Yagen B., Penhasi A., Rubinstein A. Phosphated crosslinked guar for colon-specific drug delivery, I. Preparation and physicochemical characterization. J. Control. Rel. 2000; 63: 121–127, [CSA], [CROSSREF]
- Gupta V. K., Beckert T. E., Price J. C. A novel pH- and time-based multi-unit potential colonic drug delivery system. I. Development. Int. J. Pharm. 2001; 213: 83–91, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Kleinebudde T. T., Sande S. A. Extrusion/spheronization of pectin-based formulations. II. Effect of additive concentration in the granulation liquid. AAPS Pharm. Sci. Tech. 2001; 2: 27, [CSA]
- Krishnaiah Y. S. R., Indira Muzib Y., Srinivasa Rao G., Bhaskar P., Satyanarayana V. Studies on the development of colon targeted oral drug delivery systems for ornidazole in the treatment of amoebiasis. Drug Del. 2003; 10: 111–117, [CSA]
- Liu L., Fishman M. L., Kost J., Hicks K. B. Pectin-based systems for colon-specific drug delivery via oral route. Biomaterials 2003; 19: 3333–3343, [CSA], [CROSSREF]
- Pillay V., Danckwerts M. P., Fassihi R. A crosslinked calcium-alginate-pectinate-cellulose acetophthalate gelisphere system for linear drug release. Drug Del. 2002; 9: 77–86, [CSA], [CROSSREF]
- Pillay V., Fassihi R. In vitro release modulation from crosslinked pellets for site-specific drug delivery to the gastrointestinal tract: comparison of pH-responsive drug release and associated kinetics. J. Control. Rel. 1999; 59: 229–242, [CSA], [CROSSREF]
- Saffron M., Kumar G. S., Neekers D. S., Pena J., Jones R. H., Field J. B. Biodegradable azopolymer coating for oral delivery of peptide drugs. Biochem. Soc. Trans. 1990; 18: 752–754, [CSA]
- Semde R., Amighi K., Devleeschouwer M. J., Moes A. J. Studies of pectin HM/Eudragit RL/Eudragit NE film-coating formulations intended for colonic drug delivery. Int. J. Pharm. 2000; 197: 181–192, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Sinha V. R., Kumria R. Microbially triggered drug delivery to the colon. Eur. J. Pharm. Sci. 2003; 18: 3–18, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Turkoglu M., Ugurlu T. In vitro evaluation of pectin-HPMC compression coated 5-aminosalicylic acid tablets for colonic delivery. Eur. J. Pharm. Biopharm. 2002; 53: 65–73, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Uribe M., Moran S., Poo J. L., Mendez-Sanchez N., Guevara L., Garcia-Ramos G. Beneficial effect of carbohydrate maldigestion induced by a disaccharidase inhibitor (AO-128) in the treatment of chronic portal-systemic encephalopathy. A double-blind, randomized, controlled trial. Scand. J. Gastroenterol. 1998; 33: 1099–106, [PUBMED], [INFOTRIEVE], [CSA], [CROSSREF]
- Watts P. J., Illum L. Colonic drug delivery. Drug Dev. Ind. Pharm. 1997; 29: 893–913, [CSA]