Abstract
In our pursuit to find potent anticancer drugs, we have covalently bonded free radical tempol to chlorambucil giving a chlorambucil-tempol (CT) adduct in which both of the anticancer active sites in tempol and chlorambucil were left intact. Analysis using NMR, Maldi-TOF, and EPR verified the designed chemical structure. Because the CT adduct is more hydrophobic than chlorambucil, its delivery also was investigated using fluoroalkyl double-ended poly (ethylene glycol) (Rf-PEG) micelles. Results from EPR spectra and19 F and1 H NMR spin lattice relaxation times show that the Rf-PEG micelles are able to encapsulate CT into the Rf cores of the micelles.
Keywords :
Chlorambucil (4-[bis(2-chlorethyl)amino] benzenebutanoic acid) is a potent therapeutic belonging to the nitrogen mustard group of alkylating agents (Farmer Citation1987). It is commercially known as Leukeran and is available in tablet form for oral administration. Chlorambucil is able to kill or control neoplastic cells; hence, it is used in chemotherapy to treat chronic lymphocytic leukemia, giant follicular lymphoma, and Hodgkin's disease (Mclean et al. Citation1980; Proctor, White, and Jones Citation2005). On a pharmacokinetic basis, chlorambucil has great in vitro stability is rapid, and is predictable in systemic availability after oral dosing. Its extremely low urinary excretion makes it a more predictable alkylating agent for clinical use than melaphan (Alberts et al. Citation1980), for example. It was found to be the slowest acting and generally least toxic of the alkylating agents.
Alkylation of DNA results in breaks in DNA chemical bonds and cross-linking of the twin strands as well, thus interfering with DNA replication and transcription of RNA (Pietschmann Citation2001). Also the N-alkyl group is responsible for the cytotoxicity of DNA (Panasci et al. Citation2001).
Many derivatives of chlorambucil have been synthesized to improve its therapeutic effect (Bielawska, Bielawski, and Muszynska Citation2004; Coggiola et al. Citation2005; Parker et al. Citation2005). In hopes of increasing chlorambucil's potency to cancer cells, we have covalently bonded tempol to chlorambucil by esterification of the carboxylic acid group of chlorambucil with the hydroxyl group of tempol to make the chlorambucil tempol (2,2,6,6-tetramethylpiperidin-N-oxyl-4-yl-4-[bis(2-chloroethyl)amino]phenylbutanoate) (CT) adduct (Prabhutendolkar et al. Citation2005). The structure of this product was fully verified by analytical techniques of nuclear magnetic resonance (NMR), Matrix-Assisted Laser Desorption Ionization-Time of Flight (Maldi-TOF) and electron paramagnetic resonance (EPR). Tempol (4-hydroxy-2,2,6,6-tetramethylpiperidine-N-oxyl), a hydroxy derivative of tempo, is widely used as a spin labeling agent for EPR studies in biological systems (Mehlhorn and Packer Citation1983). The unpaired electron is primarily located in the pπ -orbital on the nitrogen atom (McConnel Citation1976). Interestingly, tempol also has an anticancer property.
There are evidences that it had a substantial, reproducible neuroprotective effect when given during reperfusion in a rat model of transient focal ischemia (Kwon et al. Citation2004). Tempol decreased the toxicity of 6-mercaptopurine and potentiated its antitumor effect in mice with transplantable adenocarcinoma 755 (Konovalova et al. Citation1996). Tempol impairs mitochondrial function and induces apoptosis in a number of tumor cell lines through free radical–dependent mechanisms (Monti et al. Citation2001). Tempol has found to synergize with Temozolomide, another alkylating anticancer drug, in two human glioblastoma cell lines, U87MG and U373MG (Ravizza et al. Citation2004). Therefore, we anticipate higher potency of CT to cancer cells since both the active sites in the free radical and chlorambucil were left intact in the compound so made. Cytotoxicity assay of CT to cancer cells is in process, and the preliminary results to human KB and MCF-7 cancer cells are promising.
CT is more hydrophobic than chlorambucil, which makes its delivery more difficult. Remarkable progress has been made during the past five years in almost all areas concerning drug delivery systems. Reduction of the toxic effects and prolongation of the activity are mainly achieved by using polymers as carriers (Chen, Huang, and Huang, Citation2000). Poly(ethylene glycol) (PEG) has been used in the delivery of chlorambucil derivatives (Caliceti et al. Citation2003; Jia, Zhang, and Huang Citation2003). PEGs are inexpensive and commercially available polymers that can be readily functionalized with different spacers and linkers (Annunziata et al. Citation2002; Caliceti et al. Citation2003). Fluoroalkyl-ended PEGs (Rf-PEGs) were designed with the properties of biocompatibility and biodegradability (Xu et al. Citation1997). Rf-PEGs also can dissociate in biocompatible organic solvents to produce a low viscosity and injectable formulation (Giyoong et al. Citation2001). A striking feature of this system is that the gel phase and the sol phase can coexist resulting in low surface erosion in an aqueous environment (Giyoong et al. Citation2002). The fluoroalkyl-ended groups on both ends of the PEG chains fold together to form hydrophobic cores of the micelles; meanwhile some of the chains cross-link between the micelles to hold together the physical network in an aqueous environment (Hennink and van Nostrum Citation2002).
These properties predict the potential of the Rf-PEG system to be delivery depot to deliver hydrophobic drugs with sustained and controlled release rates. Controlled release of a drug can only be achieved if the polymer can encapsulate the drug to a great extent and provide a controlled drug release rate into the system (Haque et al. Citation2005). Methods like microencapsulation using nanospheres (Dhoot and Whentley Citation2003) and PEG coated liposomes (Baker-Woudenberg et al. Citation2002; Dimitrakopoulos Citation2004) have been used extensively. To obtain possible therapeutic effects, the mode of delivery of CT (or any other hydrophobic drugs) is as important as its cytotoxicity. We have used sonication to encapsulate CT into the cores of the Rf-PEG micelles. We also have developed methods of EPR lineshape analysis and NMR spin lattice relaxation to monitor the encapsulation process. EPR lineshape reveals the mobility of a molecule containing the electron spin and the interactions among the electron spins with close proximity.
Spin lattice relaxation NMR is an important technique to study molecular motions in polymers (Vold et al. Citation1968). The presence of a spin relaxing agent like the free radical of tempol can have a drastic effect on T1relaxation times. Thus, 1H spin relaxation times of the backbone PEG groups and 19F relaxation times of the terminal Rf groups provide a good idea regarding the spatial loading of the electron spin–labeled drug molecules in the Rf-PEG hydrogels.
In this article, we focus on the studies of the synthesis of the CT adduct and its delivery using Rf-PEG hydrogels. The cytotoxicity assay is also reported on.
MATERIALS AND METHODS
Chlorambucil 99% was purchased from MP-Biomedicals (USA) and tempol 98% from Sigma-Aldrich (USA). DMAP (4-dimethylaminopyridine) 99% and DCC (N-N′-dicylohexyl-carbodiimide), 99%, were obtained from Acros Organics (USA). Isoasorbic acid 98% was purchased from Sigma-Aldrich. All solvents being of analytical reagent grade were procured from Fischer Scientific (USA).
Synthesis of Chlorambucil-Tempol Adduct
DCC coupling reaction was used to couple the carboxylic group of chlorambucil with the hydroxyl group of tempol with loss of water molecule as shown in . To a stirred solution of tempol (0.172 g, 1 mmol) and chlorambucil (0.304 g, 1mmol) in methylene chloride (5 ml) at 0°C under argon, DCC (0.206 g, 1 mmol) and DMAP (0.0307 g, 0.25 mmol) were added, and the reaction mixture was stirred for 12 hr at room temperature. The solid materials formed were filtered off, and the filtrate was washed with 1 M HCl (1 ml) followed by saturated NaHCO3 (2 ml) and brine (2 ml). The organic phase was dried over MgSO4 and evaporated in vacuo to give 0.4 g of CT corresponding to a yield of 87%. TLC (thin layer chromatography) of the product only showed a single spot.
FIG. 1 Route for the synthesis of chlorambucil-tempol.
Reduction of Chlorambucil-Tempol-Free Radical
To characterize the structure of the CT by using NMR spectroscopy, the nitroxyl free radical was reduced using isoascorbic acid. First 3.4 mmol of the product was dissolved in 15 ml of ethanol, second 4.8 mmol of isoascorbic dissolved in 1 ml H2O was added to the solution. Reduction of the nitroxyl group to the hydroxyl was monitored by the disappearance of the pink color in the solution. The solution was then Rotovapped to remove the excess solvent and extracted with ether. The organic phase was dried over MgSO4 and evaporated in vacuo to get the reduced CT (1-hydroxy2,2,6,6-tetramethylpiperidin-4-yl-4-[bis(2-chloroethyl)amino]phenylbutanoate) with 95% yield.
Synthesis of 10KC8 (PEG MW 10,000, RfC8F17-CH2-CH2-OH) Rf-PEG
The synthesis of the Rf-PEG polymer was carried out by methods previously in literature (Xu et al. Citation1997). The product was synthesized with a yield of 80%. Purity as determined by HPLC was found to be greater than 99%.
Preparation of Drug-Loaded Sample
First, 100 mg of 10KC8 Rf-PEG and 5 mg of CT were dissolved in 1 ml methylene chloride. The homogenous mixture was allowed to air dry and after complete dissappearance of the solvent, 1 ml D2O was added. The mixture was then sonicated using a Branson sonicator to yield 0.5% of CT dissolved in 10% of the hydrogel. This gel sample was allowed to stabilize for 24 hr before NMR and EPR characterizations.
EPR Spectroscopy
The EPR spectrum of CT was done on a Bruker EMX X-band EPR spectrometer. The operating frequency of the spectrometer was 9.7 GHz. Typical experimental conditions included 20 scans at a center field of 3356.32 G and a sweep width of 100 G. The tempol and CT samples were analyzed at concentrations of 210− 6 M in methylene chloride solvent and the CT loaded hydrogel as is (0.5% in 10% 10KC8).
NMR Spectroscopy
All the NMR experiments were carried out on a Bruker Avance DRX 400 MHz instrument equipped with a 5 mm QNP probe tuned to proton and fluorine frequencies, respectively. T1 relaxation times were measured using the inversion recovery pulse sequence (Vold et al. Citation1968). Recycling delay time of 8 sec and typically 256 scans were used. The 1H chemical shifts were referenced with D2O solvent at 4.8 ppm and the 19F chemical shifts were referenced by TFA (trifluoroacetic acid) as an external standard at −78.5 ppm.
Maldi-TOF Mass Spectrometry
The product CT was analyzed on an Applied Biosystems Voyager System 4397 mass spectrometer. The matrix was 2, 5 dihydroxy benzoic acid. Reflector mode mass spectra were accumulated for 50 laser shots per sample spot with a nitrogen laser (337 nm, 4 ns pulses of 180 μ J), and a positive ion TOF detection was performed using an accelerating voltage of 20 kV. A blank spectrum to identify the matrix peaks also was carried out. The sample was prepared with a concentration of 100 pmol/μ l with the matrix of 10 mg/ml in acetonitrile solvent.
RESULTS AND DISCUSSION
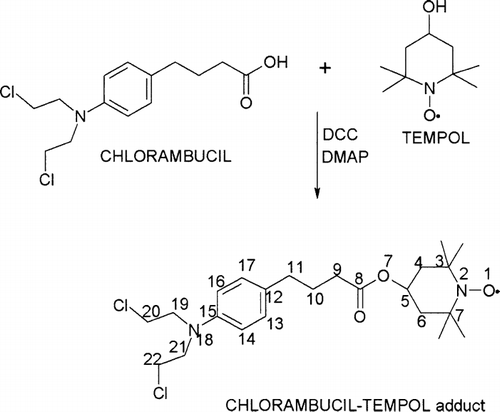
The reaction for the synthesis of the CT proceeded to completion. The free radical group was left intact as shown by the hyperfine triplet EPR spectrum (). This hyperfine triplet is characteristic for the tempol free radical due to the hyperfine coupling of 14N nucleus. For comparison, the EPR spectrum of tempol also is given in as (a) which is identical with that of CT.
FIG. 2 EPR spectra of tempol (a) and CT (b) in methylene chloride solvents and (c) 0.5% CT in 10% 10KC8 hydrogel at 293.2 K.
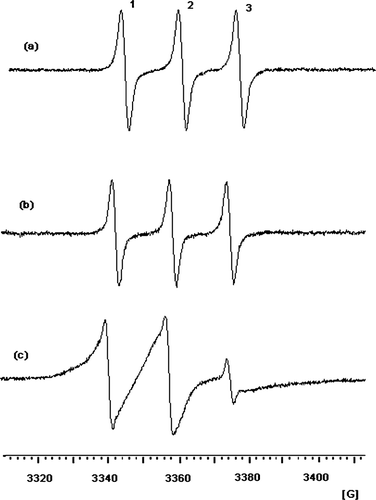
This being a novel product, structural characterization by NMR was needed. However, the presence of the free radical does not permit NMR analysis. Thus, we have reduced the nitroxyl group to its corresponding hydroxy. The 1H NMR spectrum of the reduced product is shown in and the chemical shifts are listed in . All the peaks for the product are visible although with a slight trace of solvent impurity. The peaks belonging to the chlorambucil moiety are well characterized with the parasubstituted aromatic protons appearing in the region between 6.5 to 7.03 ppm, the N, N-diethyl chloride peaks in the region between 3.5 and 3.7 ppm, and the peaks belonging to the butanoic acid chain in the region of 1.75–1.93 ppm and 2.2–2.5 ppm. The dimethyl groups of tempol appear together as a singlet at 1.3 ppm and the methylene groups at 1.75–2.07 ppm. The proton signal (broad peak between 8 to 10 ppm) of the reduced nitroxyl radical could not be clearly observed due to the proton exchange with the solvent deuterium.
FIG. 3 1H NMR spectrum of the nitroxyl-reduced CT in CDCl3 solvent referenced with TMS to 0 ppm at 299.3 K.
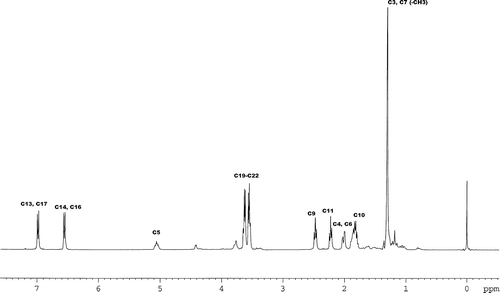
TABLE 1 Chemical shifts in ppm of the nitroxyl-reduced CT with multiplicities in parenthesis
Maldi-TOF mass spectrometry was used to fully analyze the molecule. Because the Maldi-TOF is a soft ionization technique, it shows the mass peak of the compound as an m/z ratio. In addition to the mass peaks of the sample, peaks of the matrix and daughter ions also are present. The m/z of 458.387 in the Maldi-TOF spectrum () shows the mass of the CT. The EPR, NMR, and Mass data prove that we have succeeded in synthesizing the spin-labeled drug.
FIG. 4 Maldi-TOF of the CT in 2,5-dihydroxybenzoic acid matrix.
Because the CT product is hydrophobic, it has poor water solubility, so we have used an Rf-PEG as a candidate for its delivery. Use of micelles in drug delivery is a well-established technique. Polymeric micelles can well incorporate poorly water soluble pharmaceuticals and efficiently deliver them into pathological areas via the enhanced permeability and retention effect. The drugs can be incorporated into the inner cores of micelles by covalent bonding or noncovalent bonding. Furthermore, the two-phase property of Rf-PEGs is useful to carry hydrophobic drugs with greater stability and sustained drug-release rates.
In this study, we also used the electron spin label as a probe to obtain an insight into the delivery mechanism. The EPR spectrum of CT loaded in the 10KC10 Rf-PEG hydrogel is shown in . Compared with spectrum (b) from , it shows much broader peaks indicating much slower molecular tumbling as a result of slow micelle tumbling in the gel. The EPR signal from CT dissolved in water is a few orders lower due to the low solubility, and the EPR spectrum of CT solid powder shows a single broad lineshape due to the electron-electron spin interaction. Thus, we believe this signal is from the CT encapsulated in the cores of the Rf-PEG micelles.
To further analyze the encapsulation, 19F and 1H spin lattice relaxation NMR experiments were carried out. T1 relaxation times give information about the surrounding magnetic spins around a particular nucleus. They also give information about the mobility of the involved nuclei for the relaxation. In the micelles, the backbone PEG groups protrude to water phase and the terminal Rf groups form the cores. A paramagnetic electron spin when present in close proximity to a magnetic nucleus can alter its T1 relaxation time through varying the local magnetic field. and 5b show the proton and 19F NMR spectra of the CT loaded Rf-PEG hydrogel, respectively. All the peaks in the proton and 19F spectra appear broad because of the restricted molecular motions in the hydrogel. In spectrum (a), the broad peak appearing at 3.8 ppm corresponds to the PEG backbone and the other peak appearing at 4.8 ppm is the residual protons from the D2O solvent. In the 19F NMR spectrum, the peak at −83 ppm corresponds to the terminal -CF3 groups and the peaks around −123 ppm correspond to the CF2 groups of the Rf alcohol. Since the CF3 groups would give a better understanding about the core of the micelle, and only correspond to a single -CF3 group, the T1 relaxation times of this peak was used in our experiment. Three samples were measured. The first was the Rf-PEG hydrogel without any drug being introduced, the second with tempol introduced into the Rf-PEG hydrogel, and the third sample prepared with the CT into the Rf-PEG. The proton and 19F T1 results are tabulated in .
FIG. 5 1H NMR spectrum (a) and 19F NMR spectrum (b) of the 10KC8 hydrogel loaded with CT at 299.3 K.
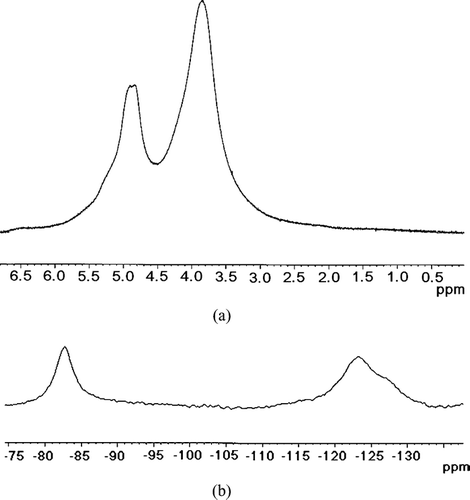
TABLE 2 Comparison of T1 relaxation times for 10KC8 hydrogel, tempol loaded 10KC8 hydrogel and CT loaded 10KC8 hydrogel
The first sample gives us an indication of the relaxation times of the original Rf-PEG. The proton relaxation times were found to be 0.68 sec and the 19F relaxation times were 0.58 sec. Tempol was expected to freely occupy all parts of the hydrogel, around the hydrophilic PEG backbone and the hydrophobic Rf groups in the core of the micelle. Thus, it is in close proximity to the PEG protons as well as the Rf fluorine's. As a result, the T1relaxation times for the 1H and 19F nuclei are decreased by 0.45 s and 0.35 s, respectively. In the CT, the free hydroxyl group of tempol and the carboxyl group in chlorambucil are replaced by the ester bond resulting in a hydrophobic molecule. Thus, this molecule was expected to be significantly attracted to the Rf cores of the micelles. Consequently, only the 19F relaxation time was affected (reduced by 0.22 sec) but that of 1H was affected insignificantly.
The T1 results show that Rf-PEGs are able to encapsulate the hydrophobic drug into the cores of the micelles, and the solubility of the drug in the water phase is insignificant. The T1 relaxation times give a strong indication about the mechanism of hydrophobic drug delivery using Rf-PEGs. The hydrophobic drug preferentially partitions into the hydrophobic cores of the micelles. These micelles thus enhance the solubility of the drug and will surely increase the duration of delivery action. This is the first time that EPR and spin lattice relaxation NMR have been used to probe the interactions of micelle-drug conjugates as compared with the traditional fluorescence probes.
CONCLUSION
Tempol electron spin labeled chlorambucil was successfully synthesized, as proven by the EPR, NMR and mass structural characterizations. Both chlorambucil and tempol are of therapeutic value especially in anticancer research. Because both of the active functional groups in both of the tempol and chlorambucil are preserved in CT, we anticipate its increasing the therapeutical potential as an antineoplastic agent.
Because CT is more hydrophobic than the chlorambucil, we have tested the feasibility of using an Rf-PEG to deliver the drug. The 1H and 19F T1 relaxation times show that the hydrophobic drug was encapsulated into the cores of the micelles and indicate that the Rf-PEG micelles are promising in delivering hydrophobic drugs.
This project is supported by NSF grant award #0351848. Thanks to the Department of Defense for funding the EPR spectrometer under grant d20030904hsi. Thanks to Dr. Y. Mao for his assistance in the EPR experiments.
REFERENCES
- Alberts D. S., Chang S. Y., Chen H. S., et al. 13 comparative pharmacokinetics of chlorambucil and melphalan in man; recent results in cancer research. Fortschritte der Krebsforschung 1980; 74: 124–131, [INFOTRIEVE]
- Annunziata R., Benaglia M., Cinquini M., Cozzi F. Poly(ethylene glycol)-supported aniline—a useful starting material for the soluble polymer-supported synthesis of imines and 1,2,3,4-tetrahydroquinolines. Eur. J. Org. Chem. 2002, 7: 1184–1190, [CSA], [CROSSREF]
- Baker-Woudenberg I. A., ten Kate M. T., Guo L., et al. Ciprofloxacin in polyethylene glycol-coated liposomes: efficacy in rat models of acute or chronic Pseudomonas aeruginosa infection. Antimicrob. Agents Chemother. 2002; 46(8)2575–2581, [CSA], [CROSSREF]
- Bielawska A., Bielawski K., Muszynska A. Synthesis and biological evaluation of new cyclic amidine analogs of chlorambucil. IL Farmaco. 2004; 59: 111–117, [INFOTRIEVE], [CSA], [CROSSREF]
- Caliceti P., Salmaso S., Semenzato A., et al. Synthesis and physicochemical characterization of folate-cyclodextrin bioconjugate for active drug delivery. Bioconjug. Chem. 2003; 14(5)899–908, [INFOTRIEVE], [CSA], [CROSSREF]
- Chen S., Huang Z., Huang J. Synthesis and characterization of novel kinds of polyethylene oxide drugs containing 5-fluorouracil and nitrogen mustard at one end and 4-amino-N-(2-pyrimidinyl) benzene sulfonamide at the other end. Eur. Polym. J. 2000; 36: 1703–1710, [CSA], [CROSSREF]
- Coggiola B., Pagliai F., Allegrone G., et al. Synthesis and biological activity of mustard derivatives of combretastatins. Bioorg. Med. Chem. Lett. 2005; 15: 3551–3554, [INFOTRIEVE], [CSA], [CROSSREF]
- Dhoot N., Wheatley M. Microencapsulatedliposomes incontrolled drug delivery: strategies to modulate drug release and eliminate the bhurst effect. J. Pharm. Sci. 2003; 92(3)679–689, [INFOTRIEVE], [CSA], [CROSSREF]
- Dimitrakopoulos P. Longitudinal relaxation of initially straight flexible and stiff polymers. Phys. Rev. Lett. 2004; 93(21)217801(1)–217801(4), [CSA], [CROSSREF]
- Farmer P. B. Metabolism and reactions of alkylating agents. Pharmacol. Therap. 1987; 35: 301–358, [CSA], [CROSSREF]
- Giyoong T., Kornfield J. A., Hubbell J. A., et al. Ordering transitions of fluoroalkyl-ended poly(ethylene glycol): rheology and SANS. Macromolecules 2002; 35: 4448–4457, [CSA], [CROSSREF]
- Giyoong T., Kornfield J. A., Hubbell J. A., et al. Hydrogels with controlled, surface erosion characteristics self-assembly of fluoroalkyl-ended poly(ethylene glycol). Macromolecules. 2001; 34: 6409–6419, [CSA], [CROSSREF]
- Hennink W. E., van Nostrum C. F. Novel crosslinking methods to design hydrogels. Adv. Drug. Deliv. Rev. 2002; 54: 13–36, [INFOTRIEVE], [CSA], [CROSSREF]
- Haque T., Chen H., Ouyang W., et al. Superior cell delivery features of poly(ethylene glycol) incorporated alginate, chitosan, and poly-L-lysine microcapsules. Mol. Pharm. 2005; 2(1)29–36, [INFOTRIEVE], [CSA], [CROSSREF]
- Jia Z., Zhang H., Huang J. Synthesis of polyethylene glycol with sulfadiazine and chlorambucil end groups and investigation of its antitumor activity. Bioorg. Med. Chem. Lett. 2003; 13: 2531–2534, [INFOTRIEVE], [CSA], [CROSSREF]
- Konovalova N. P., D'yachkovskaya R. F., Volkova L. M., Varfolomeev V. N. The nitroxyl radical Tempol as a modulator of 6-mercaptopurine toxicity and antitumor activity. Voprosy Onkologii. 1996; 42(3)57–63, [INFOTRIEVE], [CSA]
- Kwon T., Chao D. L., Malloy K., et al. Neuroprotection by the stable nitroxide Tempol during reperfusion in a rat model of ransient focal ischemia. J. Neurotraum. 2004; 21(10)1405–1414, [CSA], [CROSSREF]
- McConnell H. M. Molecular motion in biological membranes. Spinlabeling. Theory and applications. Academic Press, New York 1976
- McLean A., Newell D., Baker G., Connors T. The metabolism of chlorambucil. Biochem. Pharmacol. 1980; 29: 2039–2047, [INFOTRIEVE], [CSA], [CROSSREF]
- Mehlhorn R. J., Packer L. Bioenergetic studies of cells with spin probes. Ann. N. Y. Acad. Sci. 1983; 414: 180–189, [INFOTRIEVE], [CSA]
- Monti E., Supino R., Colleoni M., et al. Nitroxide tempol impairs mitochondrail function and induces apoptosis in HL60 cells. J. Cell. Biochem. 2001; 82: 271–276, [INFOTRIEVE], [CSA], [CROSSREF]
- Panasci L., Paiement J. P., Christodoulopoulos G., et al. Chlorambucil drug resistence in CLL. The emerging role of DNA repair. Clin. Cancer Res. 2001; 7: 454–461, [INFOTRIEVE], [CSA]
- Parker L. L., Anderson F. M., O'Hare C. C., et al. Synthesis of novel DNA cross-linking antitumour agents based on polyazamacrocycles. Biorgan. Med. Chem. 2005; 13: 2389–2395, [CSA], [CROSSREF]
- Pietschmann C. Antibodies as selective weapons. Livingbridges 2001; 1: 54–57, [CSA]
- Prabhutendolkar A., Mathias E., Liu L., et al (2005) Study of the delivery of the hydrophobic drugs by fluoroalkyl terminated polyethylene glycol hydrogels using NMR and ESR spectroscopy. 229th ACS National Meeting, San Diego, CA, March, 13–172005
- Proctor S. J., White J., Jones G. L. An international approach to the treatment of Hodgkin's disease in the elderly: launch of Shield study progamme. Eur. J. Haematol. Suppl. 2005; 66: 63–67, [INFOTRIEVE], [CSA]
- Ravizza R., Cereda E., Monti E., Gariboldi M. The piperidine nitroxide Tempol potentiates the cytotoxic effects of temozolomide in human glioblastoma cells. Int. J. Oncol. 2004; 25: 1817–1822, [INFOTRIEVE], [CSA]
- Vold R. L., Waugh J. S., Klein M. P., Phelps D. E. Measurement of spin relaxation in complex systems. J. Chem. Phy. 1968; 48: 3831–3832, [CSA], [CROSSREF]
- Xu B., Li L., Yekta A., et al. Synthesis, characterization, and rheological behavior of polyethylene glycols end-capped with fluorocarbon hydrophobes. Langmuir. 1997; 13: 2447–2456, [CSA], [CROSSREF]