Abstract
A series of bone-targeting prodrugs, dendritic L-Asp and L-Glu peptides Naproxen conjugates have been synthesized in a convergent approach and well characterized by NMR and MS techniques. Their solubility in water and hydroxyapatite affinity were evaluated in in vitro conditions. All the prodrugs were water soluble and exhibited high affinity to HAP. Compound NAP-G2-Asp was found more potent in HAP binding. The efficient release of the active drug moiety (naproxen) occurred by the cleavage of an amide bond in physiological conditions. These results indicated that the dendritic peptides might become a delivery system for bone tissues and provided an effective entry to the development of new bone-targeting molecules.
Introduction
Site-specific drug delivery via a prodrug has generated considerable interest for enhancing the potency or diminishing the side-effects of a drug. However, it is generally difficult to deliver and retain drugs in the bone tissues because they lack circulating systems and the blood flow is very low (CitationHirabayashi et al., 2001). Bone is composed of some unique materials, the main component of which is the inorganic compound hydroxyapatite (HAP), so the HAP may be a promising target for selective drug delivery to bone (CitationWang et al., 2005). Recently, linear acidic oligopeptides which posses Asp or Glu repetitive sequences have been identified as the high-affinity binding site for HAP (CitationMurphy et al., 2007; CitationIshizaki et al., 2009). The exact mechanisms of the preferential binding to bone is that the structures with multi-carboxy groups provide ionic interaction between negative charges and calcium ions in the mineral component of bone (HAP) and the affinities of these peptides only depend on the number of amino acid residues, but not on their optical characters (L or D) or their species (Asp or Glu) (CitationSarig, 2004).
On the basis of this mechanism, since dendritic peptides are highly branched, spherical macromolecules with particular characteristics to dendrimers and peptides, the dendritic peptides are especially attractive. Compared with linear peptides, the dendrtic peptides have the following peculiar features including: more negatively charged groups (carboxy groups) in periphery (CitationChen et al., 2004), water solubility (CitationCheng & Xu, 2008), architectonic flexibility, biocompatibility, and low cytotoxicity (CitationCrespo et al., 2005; CitationHe & Guo, 2008). Dendrimers also have several pharmaceutical applications including the enhancement of drug solubility (CitationVillalonga-Barber et al., 2008) and permeability (CitationNajlah et al., 2006). Therefore, we proposed dendritic Asp or Glu peptides as potential bone-targeting moieties and drug carriers. We aimed to investigate the bone-targeting potency of the dendritic peptides and prepare a sphere-type monodispersed dendritic peptide with a larger surface (CitationSingh, 2008) as a drug carrying candidate for bone-targeting.
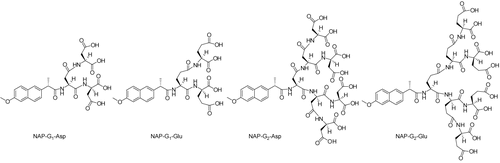
In present paper, we prepared new dendritic molecules composed of L-Asp and L-Glu amino acid residues linked through an amide linkage, L-amido acids were selected because the L-amido acid peptides were more biologically labile and enzymatically degraded (CitationOtvos, 2008). The advantageous properties are very beneficial to drug release in osseous tissues. Naproxen was selected as a model drug having low aqueous solubility and its carboxylic acid group can facilitate conjugation with dendritic peptides directly by an amide bond as biolabile linkers. The structure of the target molecule was shown in . A series of in vitro evaluations of these conjugates including water solubility, the HAP binding, and the drug release are also reported. The results of the study may be able to furnish some valuable information on new bone-targeting drug models.
Figure 1. The structure of the target molecules.
Experimental
The reactions requiring anhydrous conditions were performed in an Ar or N2 atmosphere. The chemicals and solvents were A.R. grade and purified by standard techniques. Thin layer chromatography (TLC): silica gel plates GF254; Compounds were visualized by irradiation with UV light and/or by treatment with a solution of phosphomolybdic acid (20% wt. in ethanol) followed by heating. 1H NMR analysis was performed by the INOVA VARIAN 400 MHz spectrometer using CDCl3 or D2O as a solvent at room temperature. ESI mass spectrum was recorded with Agilent 1946B ESI-MS instrument and the UV analysis was carried out by the Cary 100 UV-Vis spectrophotometer. The Hydroxyapatite was purchased from Shanghai Institute of Biochemistry with Surface Area 10 m2/g and Average particle size 30 μm.
Chemistry
General procedure for the synthesis of the compound 3a
Boc-L-Asp (1.84 g, 8 mmol), NH2-Asp-Obzl2·TsOH (7.7 g, 16 mmol) and N-methylmorpholine (1.6 ml, 16 mmol) dissolved in CH2Cl2. Dicyclohexylcarbodiimide, DCC (3.2 g, 16 mmol) and N-Hydroxysuccinimide, HoSu (1.8 g, 16 mmol) were added into the solution. The reaction mixture was stirred under Ar at room temperature for 24 h. The dicyclohexylurea (DCU) was removed by filtration and the crude product was purified on recrystallization in methanol and ethyl ether. Yield: 67%. M.P. 78–80°C.
Data
1H NMR (400 MHz, CDCl3): 1.41 (s, 9H, Boc-H), 2.73–2.98 (m, 6H, Asp-βCH2), 4.72–4.90 (m, 3H, Aspα-CH), 5.2 (s, 8H, ph-CH2), 7.12 (brs, 20H, ph-H). ESI MS (m/z): calcd. for 823.3. obsd. 846.6 ([M+Na]+).
General procedure for synthesis of the compound 5a
To the solution of trifluoroacetic acid TFA/anhydrous CH2Cl2 (1:1, v/v), 3a (0.82 g,1 mmol) was added at room temperature under N2. When the protecting groups were completely removed, the solvent was rotary evaporated (yield: 99%). The residue was dissolved in CH2Cl2 then Naproxen (0.23 g, 1 mmol), DCC (0.21 g, 1 mmol) and 4-Dimethylaminopyridine DMAP (0.01 g, 0.1 mmol) was added. The mixture was stirred at room temperature overnight. The dicyclohexylurea (DCU) was removed by filtration and the crude product was purified by silica gel column chromatography using cyclohexane and ethyl acetate (7:3, v/v) as an eluent. Yield: 74%. M.P. 150–152°C.
Data
1H NMR (400 MHz, CDCl3): 1.44 (d, 3H, J = 7.2 Hz, NapCH3), 2.66–2.94 (m, 6H, Asp-βCH2), 3.82–3.96 (brs, 4H, NapOCH3+CH), 4.66–4.88 (m, 3H, Asp-αCH), 5.2 (s, 8H, ph-CH2), 7.15–7.71 (m, 6H, Nap-phH). 7.12 (brs, 20H, ph-H).
General procedure for synthesis of the compound NAP-G1-Asp
A mixture of 5a (0.18 g, 0.2 mmol) and 10% Pd/C ( 100 mg) in CH3OH was stirred at room temperature under a H2 atmosphere. After 24 h the mixture passed through a membrane filter to remove the catalyst, then evaporated under reduced pressure. After concentration, the residue was triturated with pure ethyl ether to afford NAP-G1-Asp as white powders. Yield: 90%. M.P. > 200°C.
Data
1H NMR (400 MHz, D2O): 1.47 (d, 3H, J = 7.2Hz, NapCH3), 2.62–2.90 (m, 6H, Asp-βCH2), 3.8–3.96 (brs, 4H, NapOCH3+CH), 4.62–4.81 (m, 3H, Asp-αCH), 7.15–7.80 (m, 6H, Nap-phH). ESI-MS (m/z): calcd. for 575.2. obsd. 598.0 ([M+Na]+). Anal. calcd. for C26H29N3O12: C, 54.26, H, 5.08, N,7.30. found: C, 54.07, H, 4.93, N, 7.47.
General procedure for synthesis of the compound 6a
The compound 4a (1.64 g, 2 mmol), Boc-L-Asp (0.23 g, 1 mmol) o-Benzotriazol-1-yl- tetramethyluronium hexafluorophosphate, HBTU (0.75 g, 2 mmol), Hydroxy benzotriazole, Hobt (0.27 g, 2 mmol) were dissolved in dry DMF under N2. N,N-Diisopropylethylamine, DIPEA (0.3 ml, 2 mmol) was then added and the reaction was stirred for 24 h at room temperature. The solvent was evaporated and the slurry was partitioned between ethyl acetate and water. The aqueous layer was further extracted with ethyl acetate and the combined organic layers were dried and evaporated to give a yellow oil. The crude product was purified by silica gel chromatography column using CH2Cl2 and CH3OH (50:1, v/v) as an eluent. Yield: 57%. Colorless oil.
Data
1H NMR (400 MHz, CDCl3): 1.40 (s, 9H, Boc-H), 2.69–2.94 (m, 14H, Asp-βCH2), 4.72–4.94 (m, 7H, Asp-α-CH), 5.14 (s, 16H, ph-CH2), 7.25 (s, 40H, ph-H). ESI-MS (m/z): calcd. for 1643.6. obsd. 1644.7 ([M+H]+).
General procedure for synthesis of the compound 8a
To the solution of TFA/anhydrous CH2Cl2 (1:1, v/v), 6a (0.82 g, 0.5 mmol) was added at room temperature under N2. When the protecting groups were completely removed, the solvent was rotary evaporated (yield: 99%). The residue was dissolved in CH2Cl2 then Naproxen (0.11 g, 0.5 mmol), DCC (0.10 g, 0.5 mmol) and DMAP (0.01 g, 0.1 mmol) was added. The mixture was stirred at room temperature overnight. The dicyclohexylurea (DCU) was removed by filtration and the crude product was purified by silica gel column chromatography using CH2Cl2 and CH3OH (50:1, v/v) as an eluent. Yield: 62%. Colorless oil.
Data
1H NMR (400 MHz, CDCl3): 1.46 (d, 3H, J = 6.0 Hz, NapCH3), 2.68–2.80 (m, 14H, Asp-βCH2), 3.87–3.98 (brs, 4H, NapOCH3+CH), 4.60–4.92 (m, 7H, Asp-αCH), 5.20 (s, 16H, ph-CH2 ), 7.22–7.85 (m, 6H, Nap-phH), 7.30 (br, 40H, ph-H).
General procedure for synthesis of the compound NAP-G2-Asp
A mixture of 8a (0.34 g, 0.2 mmol) and 10% Pd/C ( 100 mg) in CH3OH was stirred at room temperature under a H2 atmosphere. After 24 h the mixture passed through a membrane filter to remove the catalyst, then evaporated under reduced pressure. After concentration, the residue was triturated with pure ethyl ether to afford NAP-G2-Asp as white powders. Yield: 89%. M.P. > 200°C.
Data
1H NMR (400 MHz, D2O): 1.41 (d, 3H, J = 6.0 Hz, NapCH3), 2.42–2.81 (m, 14H, Asp-βCH2), 3.85–3.95 (brs, 4H, NapOCH3+CH), 4.54–4.82 (m, 7H, Asp-αCH), 7.32–7.85 (m, 6H, Nap-phH). ESI-MS (m/z): calcd. for 1035.3. obsd. 1058.1 ([M+Na]+). Anal. calcd. for C42H49N7O24: C, 48.70, H, 4.77, N,9.47. found: C, 48.57, H, 4.90, N, 9.51.
General procedure for synthesis of the compound 3b
Same procedure as described above for preparation of the dendron 3a. Yield: 55%.
Data
1H NMR (400 MHz, CDCl3): 1.40 (s, 9H, Boc-H), 2.49–2.65 (m, 12H, Glu-βCH2+Glu-γCH2), 4.42–4.58 (m, 3H, Glu-αCH), 5.13 (s, 8H, ph-CH2), 7.26 (brs, 20H, ph-H). ESI MS (m/z): calcd. for 865.4. obsd. 866.6 ([M+H]+).
General procedure for synthesis of the compound 5b
Same procedure as described above for preparation of the dendron 5a. Yield: 78%.
Data
1H NMR (400 MHz, CDCl3): 1.42 (d, 3H, J = 7.2 Hz, NapCH3), 2.31–2.45 (m, 12H, Glu-βCH2+Glu-γCH2), 3.85–3.90 (brs, 4H, NapOCH3+CH), 4.42–4.55 (m, 3H, Glu-αCH), 5.21 (s, 8H, ph-CH2), 7.19–7.87 (m, 6H, Nap-phH), 7.31(brs, 20H, ph-H).
General procedure for synthesis of the compound NAP-G1-Glu
Same procedure as described above for preparation of the dendron Nap-G1-Asp. Yield: 90%.
Data
1H NMR (400 MHz, D2O): 1.34 (d, 3H, J = 7.2 Hz, NapCH3), 2.40–2.55 (m, 12H, Glu-βCH2+Glu-γCH2), 3.85–3.90 (brs, 4H, NapOCH3+CH), 4.44–4.50 (m, 3H, Glu-αCH), 7.09–7.87 (m, 6H, Nap-phH). ESI-MS (m/z): calcd. for 617.2. obsd. 640.0 ([M+Na]+). Anal. calcd. for C29H35N3O12: C, 56.40, H, 5.71, N,6.80. found: C, 56.68, H, 5.67, N, 6.71.
General procedure for synthesis of the compound 6b
Same procedure as described above for preparation of the dendron 6a. Yield: 49%.
Data
1H NMR (400 MHz, CDCl3): 1.42 (s, 9H, Boc-H), 2.33–2.60 (br, 28H, Glu-βCH2+Glu-γCH2), 4.46–4.71 (m, 7H, Glu-αCH), 5.2 (s, 16H, ph-CH2), 7.18 (s, 40H, ph-H). ESI-MS (m/z): calcd. for 1741.7 obsd. 1742.9 ([M+H]+).
General procedure for synthesis of the compound 8b
Same procedure as described above for preparation of the dendron 8a. Yield: 69%.
Data
1H NMR (400 MHz, CDCl3): 1.44 (d, 3H, J = 6.0 Hz, NapCH3), 2.44–2.64 (m, 28H, Glu-βCH2+ Glu-γCH2), 3.90–3.96 (brs, 4H, NapOCH3+CH), 4.45–4.66 (m, 7H, Glu-αCH), 5.15 (s, 16H, ph-CH2), 7.29–7.78 (m, 6H, Nap-phH), 7.30 (br, 40H, ph-H).
General procedure for synthesis of the compound NAP-G2-Glu
Same procedure as described above for preparation of the dendron NAP-G2-Glu. Yield: 89%.
Data
1H NMR (400 MHz, D2O): 1.45 (d, 3H, J = 6.0 Hz, NapCH3), 2.51–2.62 (m, 28H, Glu-βCH2+ Glu-γCH2), 3.89–3.92 (brs, 4H, NapOCH3+CH), 4.52–4.59 (m, 7H, Glu-αCH), 7.29–7.78 (m, 6H, Nap-phH). ESI-MS (m/z): calcd. for 1133.4. obsd. 1156.0 ([M+Na]+). Anal. calcd. for C49H63N7O24: C, 51.90, H, 5.60, N, 8.65. found: C, 51.68, H, 5.67, N, 8.71.
Solubility assessment
The solubility of dendritic peptide conjugates was determined at pH 3, 5, and 7 in phosphate buffers (0.05 M NaH2PO4). The method used for sample preparation was similar to each system (CitationTang et al., 2008), i.e. excess conjugates were added to 5 ml of each test solution to ensure the drug solution reaching saturation and the solution was mechanically shaken for 10 min at 37°C. Then the solutions were centrifuged at 5000 rpm for 1 min and the absorbance of the naproxen conjugate was tested at the characteristic wavelength 273 nm. The data was presented in .
Table 1. Water-solubility of these conjugates.
Determination of HAP binding levels
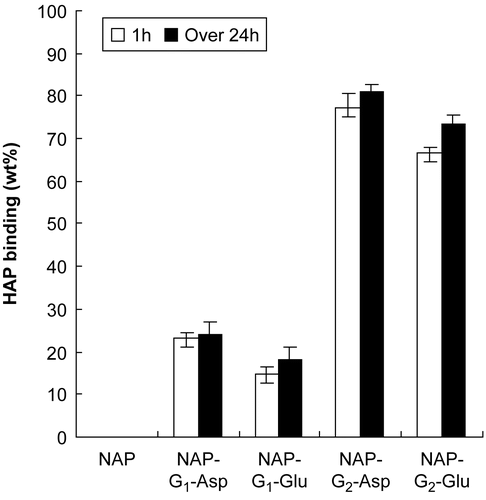
To study the binding of these conjugates to bone, an in vitro HAP binding assay was set up using in vitro HAP binding methods described by Wang et al. (2003) The conjugates were dissolved in water with various precise concentrations and the adsorption amounts were determined by a UV spectrophotometer at 273 nm to obtain the A–C linear regression equation. In tubes 25 mg/ml HAP was added to 5 ml solutions of conjugates with the precise concentrations of 500 μg/ml (C0), followed by supersonic shake for 5 min, then placed in a water bath at 37°C for 1 h and over 24 h. After the prescribed time, tubes were centrifuged for 1 min at 5000 rpm. The adsorption amounts were determined by UV at 273 nm and the ΔC was obtained by the special equation. The bound percentage was calculated by (C0 − ΔC)/C0*100% and the result was presented in .
Figure 2. Percentage of conjugates bound to HAP at 1 h or over 24 h.
Further evaluation of NAP-G2-Asp
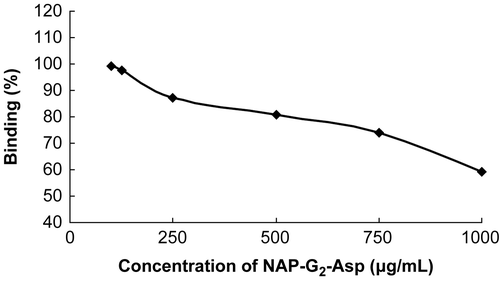
From the results in the last two experimental sections, NAP-G2-Asp seemed especially attractive for the good water-solubility and the highest level of HAP binding. To show a positive correlation and find an optimal ratio of the concentration of the NAP-G2-Asp and the HAP amount during the incubation, the NAP-G2-Asp or the HAP was kept constant, respectively, the HAP binding was determined by the same method described above. The results are presented in and .
Figure 3. Effect of HAP concentration on the degree of HAP binding (NAP-G2-Asp 500 μg/ml).
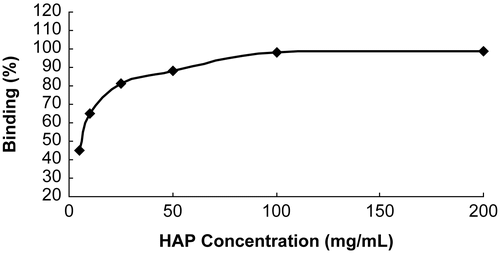
Figure 4. Effect of NAP-G2-Asp concentration on the degree of HAP binding (HAP 25 mg/ml).
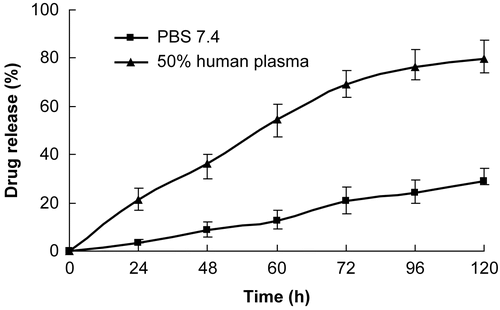
Another important factor to be considered for the conjugate is the drug releasing profile. Since the conjugate contains covalent bond between the drug and the bone-targeting peptides, it should be enzymatically and/or hydrolytically degradable to release the drug molecules in vivo (bone tissue) (CitationErez et al., 2008). An aqueous solution of NAP-G2-Asp was vigorously stirred with HAP (Concentration ratio 0.005) for 2 h. Then, the mixture was filtered and the precipitate was washed with water. The precipitate (HAP and any bound Nap-G2-Asp) was incubated in PBS pH 7.4 or 50% (v/v in PBS) human plasma (with a small amount of methanol as hydrotropy agent) at 37°C and the hydrolytic release of naproxen in the solution was monitored by RP-HPLC (Compound NAP-G2-Asp was evaluated on an Agilent 1100 HPLC system using a Waters XTerra RP18 column ( 250 mm × 4.6 mm, 5 μm). The mobile phase is a mixture of methanol and 0.05 mol/l potassium dihydrogen phosphate (adjusted to pH 3.0 with phosphoric acid) (60:40). The flow rate, the column temperature, and the detection wavelength were 1.0 ml/min, 30°C, and 240 nm. The retention time (RT) of NAP-G2-Asp is 4.041 min, and the retention time of Naproxen is 10.462 min). The percentage of drug release was deduced from the difference between the amount of bounded Nap-G2-Asp and that of regenerated drug naproxen. Error bars represented the mean and standard deviation of three independent experiments.
Results and discussion
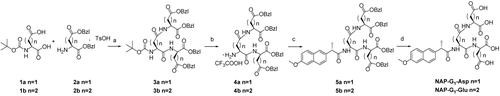
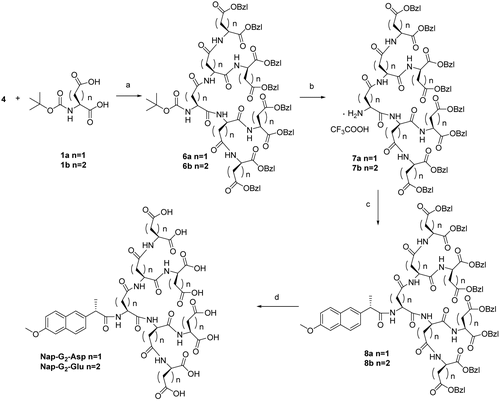
As shown in , Boc-L-Asp or Boc-L-Glu was mixed with NH2-L-Asp-OBzl2 or NH2-L-Glu-OBzl2 by treatment with DCC and HoSu at ambient temperature. Then, after deprotection of the Boc in TFA, naproxen was linked to the dendritic peptides using DCC and DMAP. The crude products were purified by silica gel column chromatography using cyclohexane and ethyl acetate (7:3, v/v) as an eluent. A conversion of compounds 5 into the corresponding Asp or Glu oligopeptides was accomplished by hydrogenolysis via 10% Pd/C, and the reactions only required filtration and evaporation to provide isolated products. In the synthesis of the second-generation dendritic molecules, for the large size of the reactant, using DCC/HoSu did not give the product in acceptable yields. Only a solid phase peptide coupling reagent HBTU/DIPEA (o-Benzotriazol-1-yl-tetramethyluronium hexafluorophosphate/N,N-Diisopropylethylamine) produced the products in good yields. The Naproxen was linked to the dendritic peptides also using DCC and DMAP. The crude products were purified by silica gel column chromatography using CH2Cl2 and CH3OH (50:1,v/v) as an eluent. Debenzylation used catalytic hydrogenolysis to get NAP-G2-Asp and NAP-G2-Glu. The synthetic route was outlined in .
Scheme 1. (a) DCC, HOSu, NMM, 3a, 67%, 3b, 55%; (b) TFA, CH2Cl2, 99%; (c) Naproxen, DCC, DMAP, 5a, 74%, 5b, 78%; (d) H2, Pd/C, 90%.
Scheme 2. (a) HBTU/Hobt, DIPEA, 6a, 57%, 6b, 49%; (b) TFA, CH2Cl2, 99% (c) Naproxen, DCC, DMAP, 8a, 62%, 8b, 69%; (d) H2, Pd/C, 89%.
The high degree of symmetry in these dendritic molecules enabled facile confirmation of both structure and purity by NMR techniques. For example, in the 1HNMR spectrum of Nap-G2-Asp: the naproxen protons observed the resonance signals at 1.41 (s), 3.95 (brs) ppm were clearly distinguishable from the resonances arising from the wedges (Asp oligopeptides) at 2.81 (m) and 4.82 (m) ppm. Integration of the respective areas of the core protons and β-methine protons of Asp oligopeptides confirmed a complete coupling. Also, the structures of these dendritic molecules were further verified by ESI-MS. All of the spectra displayed a very prominent peak corresponding to the dendritic molecules complexed with protons or sodium cation. Moreover, elemental analysis was also in good agreement with those signed structures.
A series of solubility experiments were carried out and the results were shown in . It was observed that the solubility of naproxen increased significantly with a dendritic peptides conjugation. This was presumabley due to the increase in the number of surface carboxy groups that were available to interact with water molecules (CitationKono, Liu, & Frechet, 1999). The results of the most effective pH conditions for solubility were also shown in and, as it can be seen, the process was pH-dependent. The solubility in water was highest at pH 7, less at pH 5, and least at pH 3. It was proposed therefore that the solubility enhancement was due to an electrostatic interaction of the surface carboxy groups and the weakly acidic conjugates were not fully ionized at low pH conditions and hence it can not freely interact electrostatically in water. The solubility of the conjugates depended on the surface area and carboxy groups of the dendritic peptides, thus a molecule of higher generation should have a higher ability to interact with water molecule than that of lower ones (CitationSvenson & Chauhan, 2008).
The data presented in clearly demonstrated the capability of naproxen-dendritic peptides binding HAP and all conjugates exhibited rapid binding in 1 h. Non-targeted naproxen hardly showed any binding, demonstrating that the dendritic peptides played an important role in the binding process and the second-generation dendritic peptides were found to be more effective in binding HAP (up to 80%).
and showed a positive correlation between the degree of binding and the HAP amount of the conjugate (CitationHengst et al., 2007): NAP-G2-Asp. A higher HAP amount resulted in higher degree of binding. When the amount of HAP present during the incubation was kept constant, an increased amount of NAP-G2-Asp resulted in a lower binding, likely due to saturation of the HAP binding sites. Altogether, these results demonstrated that maximal binding could be reached to an optimal ratio of NAP-G2-Asp to HAP. In our experimental set-up, when the concentration of NAP-G2-Asp was kept in 500 μg/ml, the max binding was reached with the HAP concentration 100 mg/ml, meanwhile, when the HAP was kept constant at 25 mg/ml, the max binding was reached with the NAP-G2-Asp concentration 125 μg/ml. This ratio was ~ 0.005 (0.5 mg/ml: 100 mg/ml).
showed drug releasing profile of HAP-bounded NAP-G2-Asp. Specifically, the drug released rapidly from the conjugate in 50% human plasma, reaching 54.4% after 60 h and almost 80% within 5 days, whereas the drug release at PBS was much slower, implying 12.6% and 29.1% in the same period, respectively. As desired, the parent drug naproxen released an in vitro bone model during the observed period. The release profile confirmed that the linkage was labile under blood circulation conditions and relatively stable in PBS (pH 7.4) to allow the transport of the prodrug to bone issues and effectively release the free active drug from the carrier.
Figure 5. Release of naproxen from HAP bound NAP-G2-Asp under physiological conditions.
Conclusion
In conclusion, novel bone-targeting and biodegradable first- and second-generation dendritic peptides naproxen conjugates were designed and prepared with amido coupling reactions. The conjugates were water-soluble and the water solubility was increased by the structures with more polar groups. The second- generation dendritic peptides were very efficient in binding HAP, generally > 70% and the bounded compound Nap-G2-Asp can slowly release parent drug in physiological condition. This suggested that this kind of dendritic peptides were worthy for further investigation to discover new bone-targeting agents. Preliminary study of biodistribution and pharmacokinetic analysis of these conjugates for bone-targeting in vivo is now under way.
Acknowledgement
This work was supported by the National Natural Science Foundation of China (No.20472055).
Declaration of interest: The authors report no conflicts of interest. The authors alone are responsible for the content and writing of the paper.
References
- Chen, H.F., Chen, Y.Q., Orr, B.G., Holl, M.B., Majoros, I., Clarkson, B.H. (2004). Nanoscale probing of the enamel nanorod surface using polyamidoamine dendrimers. Langmuir, 20, 4168–71.
- Cheng, Y.Y., Xu, T.W. (2008). The effect of dendrimers on the pharmacodynamic and pharmacokinetic behaviors of non-covalently or covalently attached drugs. Eur J Med Chem, 43, 2291–7.
- Crespo, L., Sanclimens, G., Pons, M., Giralt, E., Royo, M., Albericio, F. (2005). Peptide and amide bond-containing dendrimers. Chem Rev, 105, 1663–81.
- Erez, R., Ebner, S., Attali, B., Shabat, D. (2008). Chemotherapeutic bone-targeted bisphosphonate prodrugs with hydrolytic mode of activation. Bioorg Med Chem Lett, 18, 816–20.
- He, G., Guo, L. (2008). Progress in dendrimers as drug carriers. Chin J Org Chem, 28, 1326–35.
- Hengst, V., Oussoren, C., Kissel, T., Storm, G. (2007). Bone targeting potential of bisphosphonate-targeted liposomes— preparation, characterization and hydroxyapatite binding in vitro. Int J Pharm, 331, 224–7.
- Hirabayashi, H., Takahashi, T., Fujisaki, J., Masunaga, T., Sato, S., Hiroi, J., Tokunaga, Y., Kimura, S., Hata, T. (2001). Bone-specific delivery and sustained release of diclofenac, a non-steroidal anti-inflammatory drug, via bisphosphonic prodrug based on the Osteotropic Drug Delivery System (ODDS). J Contr Rel, 70, 183–91.
- Ishizaki, J., Waki, Y., Takahashi-Nishioka, T., Yokogawa, K., Miyamoto, K. (2009). Selective drug delivery to bone using acidic oligopeptides. J Bone Miner Metab, 27, 1–8.
- Kono, K., Liu, M.J., Frechet, JMJ. (1999). Design of dendritic macromolecules containing folate or methotrexate residues. Bioconjugate Chem, 10, 1115–21.
- Murphy, M.B., Hartgerink, J.D., Goepferich, A., Mikos, A.G. (2007). Synthesis and in vitro hydroxyapatite binding of peptides conjugated to calcium-binding moieties. Biomacromolecules, 8, 2237–43.
- Najlah, M., Freeman, S., Attwood, D., D’Emanuele, A. (2006). Synthesis, characterization and stability of dendrimer prodrugs. Int J Pharm, 308, 175–82.
- Otvos, L. (2008). Peptide-based drug design. New York: Springer Science.
- Sarig, S. (2004). Aspartic acid nucleates the apatite crystallites of bone: a hypothesis. Bone, 35, 108–13.
- Singh, M. (2008). Simultaneous study of interfacial tension, surface tension, and viscosity of few surfactant solutions with survismeter. Surf Interface Anal, 40, 1344–9.
- Svenson, S., Chauhan, A.S. (2008). Dendrimers for enhanced drug solubilization. Nanomedicine, 3, 679–702.
- Tang, X.Y., Zhang, P., Ye, H., Zhang, C., Shen, W.B., Ping, Q.N. (2008). Water-soluble gambogic acid PEGylated prodrugs: synthesis, characterization, physicochemical properties and in vitro hydrolysis. Pharmazie, 63, 711–7.
- Villalonga-Barber, C., Micha-Screttas, M., Steele, B.R., Georgopoulos, A., Demetzos, C. (2008). Dendrimers as biopharmaceuticals: synthesis and properties. Curr Top Med Chem, 8, 1294–309.
- Wang, D., Miller, S., Sima, M., Kopeckova, P., Kopecek, J. (2003). Synthesis and evaluation of water-soluble polymeric bone- targeted drug delivery systems. Bioconjugate Chem, 14, 853–9.
- Wang, D., Miller, S.C., Kopeckova, P., Kopecek, J. (2005). Bone-targeting macromolecular therapeutics. Adv Drug Deliv Rev, 57, 1049–76.