
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Combination chemotherapy is widely exploited for suppressing drug resistance and achieving synergistic anticancer efficacy in the clinic. In this paper, the nanostructured targeting methotrexate (MTX) plus pemetrexed (PMX) chitosan nanoparticles (CNPs) were developed by modifying methoxy polye (thylene glycol) (mPEG), in which PEGylation CNPs was used as stealth nanocarriers (PCNPs) and MTX was employed as a targeting ligand and chemotherapeutic agent as well. Studies were undertaken on human lung adenocarcinoma epithelial (A549) and Lewis lung carcinoma (LLC) cell lines, revealing the anti-tumor efficacy of nanoparticle drug delivery system. The co-delivery nanoparticles (MTX-PMX-PCNPs) had well-dispersed with sustained release behavior. Cell counting kit-8 (CCK8) has been used to measure A549 cell viability and the research showed that MTX-PMX-PCNPs were much more effective than free drugs when it came to the inhibition of growth and proliferation. Cell cycle assay by flow cytometry manifested that the MTX-PMX-PCNPs exhibited stronger intracellular taken up ability than free drugs at the same concentration. In vivo anticancer effect results indicated that MTX-PMX-PCNPs exhibited a significantly prolong blood circulation, more tumoral location accumulation, and resulted in a robust synergistic anticancer efficacy in lung cancer in mice. The results clearly demonstrated that such unique synergistic anticancer efficacy of co-delivery of MTX and PMX via stealth nanocarriers, providing a prospective strategy for lung cancer treatment.
Introduction
The major public health issue in the world is cancer, and lung cancer has the highest mortality among cancers (Siegel et al., Citation2017). Most carcinoma of lungs (57%) could not be diagnosed at early stage, attributed to the typically asymptomatic during this period. The 5-year survival rate of small cell lung cancer (SCLC) was 7% and that of nonsmall cell lung cancer (NSCLC) was 21% (Miller et al., Citation2016). Most patients with SCLC and stage IIIB/IV of NSCLC receive chemotherapy. However, chemotherapy is not curative for metastatic lung cancer, but just may alleviate symptoms or prolong life for weeks (Weeks et al., Citation2012). Chemotherapy is still undesirable, due to the shortages including insufficient intracellular uptake at tumor tissues, nonspecific target site concentrations and serious systemic toxicity of chemotherapeutic agents, or even emerging drug resistant cell lines (Brannon-Peppas & Blanchette, Citation2012). Targeted therapies are standard care for adenocarcinomas with defined alterations, for instance, epidermal growth factor receptor (EGFR) mutations and anaplastic lymphoma kinase (ALK) translocations, but these ineffective for SCLC. In addition, immunotherapy drugs, in response to immune checkpoint blockade, have been approved to develop personalized immunotherapeutics for NSCLC (Tan et al., Citation2016). Nevertheless, EGFR inhibitors could cause side effect, including severe acneiform rash and hematologic toxicity (Srinivas et al., Citation2016). Immunotherapy drugs for lung cancer treatment could bring about several immune mediated toxicities, such as colitis, nephritis, pneumonitis and endocrinopathy (Miller et al., Citation2016). Especially, lung cancer treatment using a single anti-cancer agent may be largely unsuccessful owing to the complex nature of cancer, such as drug resistance, which is a leading cause of chemotherapy failure (Ramirez et al., Citation2016; Wang et al., Citation2016b). It is urgent to exploit novel drug delivery systems for lung cancer.
Pemetrexed (PMX) is a structurally novel multi-targeted antifolate for lung cancer and malignant pleural mesothelioma treatment (Wibowo et al., Citation2013; Kemp et al., Citation2016), which exerts strong inhibitory effect on thymidylate synthase (TS). In addition, it also observably restrain other folate requiring enzymes, such as glycinamide ribonucleotide formyltransferase (GARFT), dihydrofolate reductase (DHFR) and aminoimidazole carboxamide ribonucleotide formyl transferase (AICARFT) (Chattopadhyay et al., Citation2007). It is a hydrophilic molecule with very limited passive diffusion because it carries two negative charges which generate from the glutamate moiety of the molecule at physiological pH. Furthermore, due to its low molecular, PMX would be quickly eliminated from neoplastic tissue to blood circulation after administration (Ando et al., Citation2015). Recently, considerable efforts have been directed toward novel PMX delivery systems to achieve the desired therapeutic effect, such as nanoparticles (Lu et al., Citation2016), prodrug (Chan et al., Citation2014) and liposomes (Ando et al., Citation2015). However, co-delivery nanocarriers of multiple target and self-targeting synergistic antitumor effects have not been reported before.
Methotrexate (MTX) is traditional chemotherapeutic agent for oncotherapy. The dominate mechanisms of it are DHFR inhibiting and folate receptor (FR) binding. FR is a surface biomarker overexpression on tumor cell membrane (Khan et al., Citation2012). Many targeted approaches for MTX have been exploited to reduce adverse effects, improve bioavailability and maximize treatment effect (Jain et al., Citation2015; Farjadian et al., Citation2016; Guo et al., Citation2018; Liu et al., Citation2017). Earlier paper demonstrated that MTX-mediated nanoparticle drug delivery system would be a predominant targeted anticancer chemotherapeutic formulation (Chen et al., Citation2014; Li et al., Citation2017a, Citation2017b; Singh & Subudhi, Citation2016).
In animal models, nanoparticles offer an opportunity to alter the pharmacokinetics profile of chemotherapeutic drugs, improve the therapeutic index, reduce off-target toxicity and obtain optimal synergistic anti-tumor efficacy. Despite desirable preclinical research, monotherapy nanomedicines remain largely unsuccessful for producing enhanced response rates on traditional chemotherapy in clinical trials (Cheng et al., Citation2013; He et al., Citation2015). Recent efforts (Yan et al., Citation2016; Kohay et al., Citation2017) are underway to exploit nanocarriers for efficiently delivering multiple chemotherapeutic agents to boost target section convergences and control release traits (Hu et al., Citation2016). Chitosan (CS) has nontoxicity, biodegradability and good biocompatibility. Also, it can be easily processed into the form of nanoparticles for drug delivery (Anitha et al., Citation2014). Stealth nanoparticles modified with PEGylated had been characterized for prolonged circulation and improved biodistribution of anti-cancer agents (Li et al., Citation2013), making superior accumulation in tumor via a process of enhancing permeability and retention (EPR) effect.
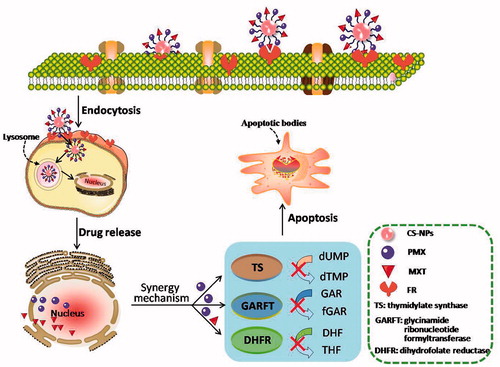
In the present research, the objectives were to develop targeting MTX plus PMX via PEGylated nanocarriers, in which MTX was a targeting ligand and chemotherapeutic agent, and to evaluate their synergistic anticancer efficacy in a carcinoma of the lungs mouse model (Chou, Citation2006, Citation2010). Human folate receptors (FRs) that transport MTX via endocytosis, proposed as targets for the specific delivery of novel classes of antifolates to tumors (). In addition, the synergistic antitumor effect of dual chemotherapeutic agents loaded nanoparticles was evaluated in vivo and in vitro.
Scheme 1. Schematic illustration for co-delivery processing of both MTX and PMX loaded PCNPS in target tumor cells by active targeting.
Materials and methods
Materials
PMX was obtained from Aladdin (China), MTX from BBI (USA). CS, 75 kDa molecular weight (MW) and 95% deacetylation degree, was provided by Zhejiang Aoxing (China). CCK8 was obtained from Dojindo Laboratories (Japan). Fetal bovine serum (FBS), trypsin, RPMI-1640 medium and Dulbecco’s modified Eagle medium (DMEM) were supplied by Gibco. Other regents were analytical or HPLC grade and were used directly without extra processing.
Cell lines and animals
The mouse Lewis lung carcinoma (LLC) and human alveolar epithelial (A549) cell lines were commercially supplied by American Type Culture Collection (USA). LLC cell lines were maintained in DMEM and A549 cell lines were kept in RPMI-1640 supplemented with 10% FBS, penicillin (100 U/ml), streptomycin (100 mg/ml), 0.1 mM non-essential amino acids, 2 mM glutamine and 1 mM sodium pyruvate at 37 °C and 5% CO2.
Four-week-old C57BL/6 mice (18–20 g) were obtained from the laboratory animal center (Shanghai, China). All procedures involving experimental animals were approved by the Committee for Animal Research of Xiamen University and complied with the Guide for the Care and Use of Laboratory Animals.
Synthesis of dual-drug-loaded nanoparticles
The synthesis method of stealth nanocarriers was in accordance with our previous reported procedure (Chen et al., Citation2014) with optimization by orthogonal experiment. In brief, 2.5 ml PMX solution (5 mg/ml) was added to 1 ml PCNPs (10 mg/ml), under the condition of catalyst, N-(3-dimethyl-aminopropyl)- N′-ethyl carbodiimide (EDC) hydrochloride. Then, the reaction mixture was magnetically stirred for 4 h with pH 4.9 at room temperature to obtain PMX-PCNPs (colorless). After that, centrifuged at 15,000 rpm at 10 °C for 20 min, dialyzed against water, and lyophilized for 24 h.
Also, the yellow MTX-PMX-PCNPs can be obtained in the same procedure above with the mass ratio of MTX and PMX is 1:1.
Nanoparticle characterization
Fourier transform infrared (FTIR) was conducted using Nicolet AVATR 360 spectrometer (Nicolet, USA) with scanning range between 4000–500 cm−1. KBr pellets were used to make the IR absorption spectra of the samples.
Morphologies of MTX-PMX-PCNPs were analyzed by transmission electron microscope (TEM) with JEM-2100 (JEOL, Japan) at 200 kV. In brief, deposited the samples onto a carbon grid and sucked dry with a filter paper. After having been dried by an infrared lamp, the sample with carbon grid was taken TEM for observing the morphology of MTX-PMX-PCNPs.
The mean hydrodynamic diameter and polgydispersity index (PDI) of drug-loaded PCNPs were determined by Marvent Nano-zs (Marvent, U.K.), based on dynamic light scattering (DLS). The samples were appropriately diluted in distilled water, and then were performed at 25 °C. Each measurement was taken three sets of 10 runs.
The zeta potential (ZP) of drug-loaded PCNPs were evaluated by determining electrophoretic mobility using Marvent Nano-zs (Marvent, U.K.). The measurements were taken in ultrapure water at 25 °C using three sets of 20 runs.
Co-loaded PCNPs entrapment efficiency and loading capacity
The amount of MTX and PMX conjugated to PEGylated nanocarriers was analyzed by a reversed phase high-performance liquid chromatography (RP-HPLC) system of Waters 1525 (Waters) having a C18 column (3.9 mm ×300 mm) operated at 25 °C with Waters 24897 UV/visible detector (Waters). The detection wavelength of MTX and PMX was 303 nm and 239 nm, respectively. The mobile phase of MTX was composed of 0.1% formic acid solution: acetonitrile (15:85 v/v) at a flow rate of 1 mL/min, and that of PMX was water with 0.1% phosphoric acid (pH 3.0)/acetonitrile (85:15, v/v) at the same flow rate. The amount of them was measured by the peak area with the standard curve. The experiment was performed in triplicates with an injection volume of 20 μL. The drug-loading capacity (LC%) and entrapment efficiency (EE%) values of MTX and PMX-loaded PCNPs were calculated by previous reported formulas (Chen et al., Citation2014).
In vitro release
In order to simulate the biological condition of body and tumor microenvironment, the drug release profile of MTX and PMX from double drug delivery nano-system was investigated at the physiological temperature of 37 °C in PBS for 144 h at pH 6.5 and 7.4. Every aliquot of MTX-PMX-PCNPs (5 mg/ml) was put in a dialysis bag (molecular cutoff =3 kDa). Then, samples were suspended in 100 ml of PBS at 37 °C with gently shaken at 100 rpm. After that, the concentration of MTX and PMX at predetermined time intervals was determined by RP-HPLC as described above and was calculated according to following formula:
(1)
(1)
Here, Mt means the amount of drug released from the nano-system at time t. Mi stands for initial loading.
Nanoparticle cellular uptake studies on A549 cell lines
To examine the cellular uptake capability of NPs to A549 cell lines, 1 mL (1 × 105/mL) cells were seeded into 4-well-chambered cover glasses and cultured for 12 h to attach. Then, the cells were incubated with FITC-labeled MTX + PMX, PCNPs, MTX-PCNPs, PMX-PCNPs and MTX-PMX-PCNPs for 24 h. Subsequently, were washed with PBS and followed by fixing with 4% paraformaldehyde for 20 min. After incubation with 4,6-diamidino-2-phenylindole (DAPI, 10 μg/mL) for 5 min, acted as a marker for cell nuclei, the intracellular localization of NPs was directly visualized by confocal laser scanning microscope (CLSM, Leica TCS SP5, Germany). Excitation and emission wavelengths for DAPI were 364 and 461 nm, respectively, and that for FITC were 488 and 518 nm.
CCK-8 assay
Quantitative cell viability of drug-loaded nanoparticles was evaluated with a Cell Counting Kit-8 (CCK-8) assay in A549 cells. Briefly, 4 × 103 cells were seeded in 96-well plates (three wells per group), and cultured for 24 h to attach prior to supplementation PCNPs, free MTX, free PMX, MTX + PMX, MTX-PCNPs, PMX-PCNPs and MTX-PMX-PCNPs at a concentration of 0.05, 0.5, 5, 50 and 500 μg/mL for 24, 48 and 72 h. After that, CCK-8 solution (10 µl per well) was added for further incubation 1 h at 37 °C with 5% CO2. Optical density (OD) at 450 nm was determined by a microplate reader. Cell inhibition rate and inhibitory concentration 50% (IC50) were computed pursuant to below:
(2)
(2)
The synergetic effect was further examined with combination index (CI) method and median-effect principle based on the Chou–Talalay equation (Chou, Citation2006).
Cell cycle analysis by flow cytometry
A549 cells (2 × 105/well) were seeded into 6-well plates, and treated with control, MTX + PMX, PCNPs, MTX-PCNPs, PMX-PCNPs and MTX-PMX-PCNPs for 48 h. Then, cells were centrifuged, washed twice with cold PBS and fixed with 70% precooled ethanol overnight at −20 °C. After that, the fixed cells were washed with cold PBS again and stained with PI (50 μg/mL), RNase A (100 μg/mL), 0.2% Triton-X for 30 min at 4 °C in the dark. Finally, the stained cells were detected by flow cytometer (FCM, Beckman). Cell populations categorized in sub-G1 (G0), G1, S and G2/M phases were analyzed in accordance with their DNA content with ModFit LT Mac 3.3 software (BD Biosciences).
In vivo anti-tumor activities
LLC cells were inoculated subcutaneously in C57BL/6 mice (1.08 × 107 cells per mouse). Tumor volume was measured in accordance with following formula:
(3)
(3)
(4)
(4)
Mice were divided randomly into five groups (6 mice per group), when their tumor volume was about 25 mm3 (14 d after inoculation), treated them with 200 mL of 0.9% NaCl, MTX + PMX, MTX-PCNPs, PMX-PCNPs and MTX-PMX-PCNPs 4 mg/kg (PMX-eq dose) via tail vein injection at 0, 3, 7, 14 and 21 d. Meanwhile, the tumor volume and body weight of each mouse were measured every 5 days during the whole period of treatment. Mice were sacrificed 25 days after first administration. Finally, masses were excised, weighted and photographed.
Hematoxylin and eosin staining assay
For histopathological analysis, the tumor tissues were fixed in 4% paraformaldehyde, embedded in paraffin and cut into 5 μm sections. After that, paraffin sections of tumors were stained with hematoxylin and eosin (H&E) and were visualized by an Axio Vert.A1 Zeiss microscope (Zeiss, Germany) with an Axio Cam MRC Zeiss camera.
Statistical analysis
The results were expressed as mean values ± standard deviation (SD). The statistical analysis was performed on one way analysis of variance (ANOVA) with SPSS software (Chicago). Probability (p) values < .05 indicated significance.
Results and discussion
Synthesis and characterization of nanoparticles
In order to optimize the technological parameters of nanocarriers, an orthogonal experiment was used. A series of significant factors, such as quality ratio between CS and sodium triphosphate (STPP) solution, pH value, reaction temperature and STPP drop acceleration, which could influence on the size and ZP of NPs, were taken into account. Excessive particle size may affect the EPR effect of solid tumors (Perry et al., Citation2017) and the active targeting of NPs (Wang et al., Citation2016). ZP represents a measurement of electric charge on the surface of nanoparticle, showing the physical stability of colloidal systems (Decuzzi et al., Citation2010). Then, the optimized formulation of the NPs was concluded as follows: MCS:MSTPP=5:1, pH 4.0, STPP drop acceleration 1 mL/min and reaction temperature at 10 °C. summarized the synthetic pathways of co-delivery of MTX and PMX via PEGylated copolymers.
The chemical structures of the polymers were characterized by FTIR. exhibited the FTIR spectra of MTX-PCNPs, PMX and MTX-PMX-PCNPs measured in the 500–4000 cm−1 region. Some characteristic peaks of different formulations were marked in the figure by arrows. MTX-PCNPs spectrum presents characteristic IR absorption peaks at 2888, 1659 and 1606 cm−1, whereas 3468 cm−1(amide N-H stretch) becomes wider, which is consistent with our previously reported (Chen et al., Citation2014). Peaks of the free PMX were shown at 3442 cm−1 (νN-H, νO-H), 1678 (νC=O) cm−1, 1570 cm−1 (νpyrrole) and 1623 cm−1 (δN-H), respectively. As can be observed in , compared with FTIR spectra of MTX-PCNPs, the spectra of MTX-PMX-PCNPs had a new absorption peak at 1569 cm−1 (νpyrrole of PMX). Additionally, 3470 cm−1 (amide N-H stretch), 2901 cm−1 (mPEG typical signals) and 1660 cm−1 (amide I band) became wider and left shift. Hence, these indicated that MTX and PMX were chemically combined with PCNPs.
Scheme 2. Synthetic pathways in formation of MTX-PMX-PCNPS.
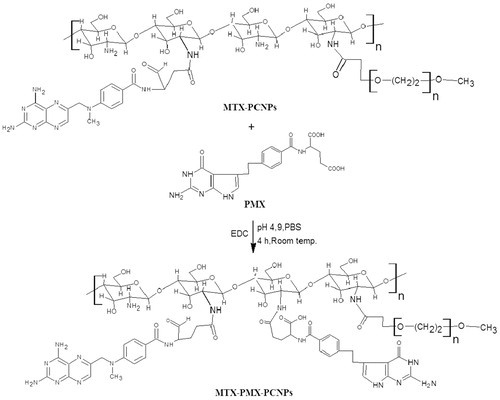
Figure 1. FTIR spectra (A); transmission electron microscopy images (B); Size distribution (C), Zeta potential (D) of MTX-PMX-PCNPs; Characteristics of NPs (E); In vitro release cumulative of MTX (F) and PMX (H) from nanocarriers, free MTX (G) and PMX (I) in PBS at pH 6.5 and 7.4 (37 °C, n = 3).
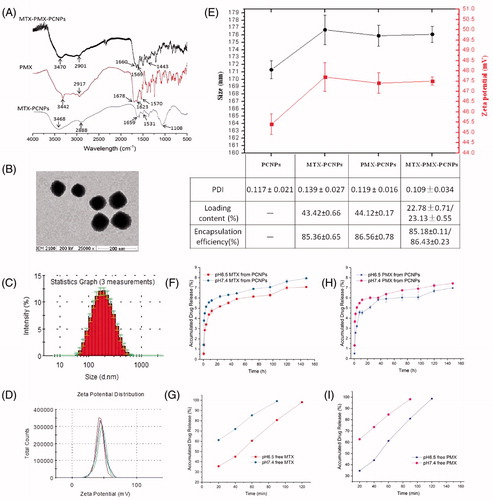
Morphology of MTX-PMX-PCNPs was observed using TEM (), which was approximately 80 nm with a subsphaeroidal shape. It is critical that drug-loading PCNPs must be well-dispersed to ensure uniform dosing before in vivo studies. Due to sticky and viscous of CS (Modaresifar et al., Citation2016), an orthogonal optimization experiment was adopted to obtain the average size with narrower PDI. Moreover, RPMI-1640 with 10% FBS was used as solvents to re-dissolve the dried PCNPs.
The mean size and surface charge of particles in demonized water, determined by DLS, were shown in . After introducing MTX and PMX into the PCNPs, final particle had an approximate diameter of 176 nm () with a narrow size distribution (PDI =0.109 ± 0.034) and a positive surface charge of about 47 mV (). Nanoparticle agents being developed vary greatly in size from 2 to 200 nm. It should be pointed out that size and zeta potential are two important characteristics of PCNPs to successfully deliver their payloads (Decuzzi et al., Citation2010). In addition, the low PDI of particle showed narrow size dispersion, and no large aggregates are observed. Notably, imaging by TEM revealed that the majority of particles were subsphaeroidal mono-dispersed about 80 nm (). The particle size measured by TEM was far less than the result of DLS (176 nm). However, this significant discrepancy is predicted. The size measured by DLS represents the hydrodynamic particle size, including the effects of hydro and dynamic. However, TEM just displays core size of the particle (Jenkins et al., Citation2013).
In the presence of EDC, the carboxyl groups of MTX/PMX were conjugated to the residual amino groups of PCNPs (). MTX-loading content (LC) of drug-loaded PCNPs was 22.78 ± 0.71%, and that of PMX is 23.13 ± 0.55%. On one hand, the high drug-loading content of the PCNPs can augment synergistic anti-tumor effect of MTX and PMX. On the other, it can strengthen the tumoral active targeting of MTX.
In vitro release study
Dialysis method was adopted to conduct the in vitro drug release behavior. The MTX and PMX release profiles from MTX-PMX-PCNPs were displayed in , performing at pH 6.5 and 7.4 PBS at 37 °C. Optimal pH levels were determined to simulate that of tumor microenvironments (pH 6.5) and physiological conditions (pH 7.4) as well. The MTX-PMX-PCNPs had a sharp sustained release mode with a characteristic of pH dependent, compared with the release process of free MTX and PMX (). MTX/PMX was almost completely released (99%) from the free MTX/PMX solution within 2 h, which confirmed that this method met the requirements of the sink condition. However, approximately 6% of MTX/PMX was released from MTX-PMX-PCNPs at the same time, probably owing to the slow hydrolysis of amide bonds. In addition, pH dependence of amide hydrolysis, the release of dual-drug from PCNPS at pH 6.5 was lower than that at pH 7.4 (). Amide bonds possess relatively high enzyme stability and are frequently used linkage (Yamashita & Hashida, Citation2013). There are differences in intracellular pH and enzymes between target and non-target tissues. Due to these physiological and biochemical differences, many efforts can be made to exploit target-specific releasing agents (Yamashita & Hashida, Citation2013). Additionally, PEG, which has high chain mobility and large excluded volume in buffered solutions, can prevent drugs release from nanocarriers (Canal et al., Citation2010; Chen et al., Citation2013; Xiong et al., Citation2016; El-Say & El-Sawy, Citation2017). More importantly, irrespective of physiological conditions or tumor microenvironments, MTX-PMX-PCNPs possess a characteristic of sustained drug release, which is a significant advantage for drug delivery systems.
Cellular uptake study
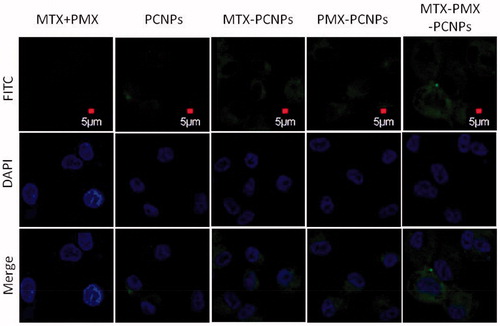
Intracellular uptake behaviors of MTX + PMX, PCNPs, MTX-PCNPs, PMX-PCNPs and MTX-PMX-PCNPs were conducted in A549 cells. Cellular localization of FITC-labeled different formulations was evaluated by CLSM. In terms of clinical applications, a rational design of drug delivery system should possess not only sustained drug release in vitro, but also effective intracellular uptake upon exposure to tumor tissues. As can be seen from , there was hardly any fluorescence signal arisen in MTX + PMX group, which demonstrated that free drugs are rapidly excreted by tumor cells in the absence of nano carriers (PCNPs). After loading on nano carriers, the distribution of drugs depends on the physical and chemical characteristics of their carriers. In addition, intracellular uptake of NPs depends on the size and the hydrophobicity of the carrier. In general, nanoparticles with suitable size of 100–200 nm have ability to enter cells by receptor-mediate endocytosis, while over 200 nm by means of phagocytosis (Win & Feng, Citation2005). Thus, it demonstrated that drug-loaded PCNPs of 176 nm possessed excellent properties in term of cellular uptake. Additionally, there was comparable fluorescence intensity between MTX-PCNPs and PMX-PCNPs, slightly stronger than that of PCNPs. It might imply that the PMX modification also boosts the cellular uptake efficacy. PMX, as the structural analog of folic acid, can utilize folate influx transporters into tumor cell (Chattopadhyay et al., Citation2007). It is noteworthy that the stronger green fluorescence signals, assembling around the nucleus (in blue) of A549 cells, were observed in MTX-PMX-PCNPs than the others. In the presence of PCNPs, PMX displays a high affinity with the FR (1 nM) by a receptor mediated endocytosis (Wibowo et al., Citation2013). MTX can be induced to internalize by means of receptor/ligand complex-mediated endocytosis. Also, compared with a single drug, drug combination therapy may overcome the drug resistance of tumor cells. It is conceivable that MTX and PMX loaded PCNPs had synergistic character in cellular uptake.
Figure 2. CLSM micrograph of cellular uptake performed on A549 cells following 24 h incubated with fluorescently labeled MTX + PMX, PCNPs, MTX-PCNPs, PMX-PCNPs and MTX-PMX-PCNPs. Bright field image shows the morphologic correspondence of A549 cells. Nuclei stained with DAPI are blue and different formulations in green.
Cell viability assays
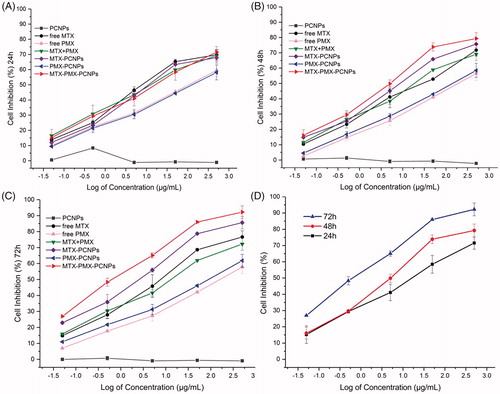
In this section, exposure of A549 cells to PCNPs, free MTX, free PMX, MTX + PMX, MTX-PCNPs, PMX-PCNPs and MTX-PMX-PCNPs at different concentration for 24,48 and 72 h by CCK-8 assay in . It must be mentioned that A549 viability was hardly affected by PCNP within investigated range of concentrations during the study period, indicating that the drug carrier itself is nontoxic to A549 cells and had good biocompatibility. In contrast to PCNPs group, MTX-PMX-PCNPs reduced A549 proliferation in a dose-dependent manner. As showed, after culture for 24 h, fewer cells were detected in free drug groups, compared with the same dose of drug-loaded nanoparticles groups. Then, after culture for 48 h (), the inhibitory effect on cell proliferation of free drug group was slightly weaker than drug-loaded nanoparticles group. However, there was a reversal after culture for 72 h (), that is, the inhibitory effect on cell proliferation of free drug group was significantly weaker than drug-loaded nanoparticles group, which may attributed to loaded drugs release from the carrier increase gradually and free drugs decreased by the metabolism (Yamashita & Hashida, Citation2013). Higher inhibition efficiency depends on much more drug transferring into the rumor cells. At intracellular site, free drugs are available by the mechanism of passive diffusion. However, they are pumped out of the cytoplasm by the P-glycoprotein before become effective (Ramirez et al., Citation2016). On the contrary, drug-loaded nanoparticles had obvious cytotoxicity due to nano-scale effect with a mechanism of cellular internalization by endocytosis (Cheng et al., Citation2013). Drug-loaded nanoparticles can inhibit proliferation and induce apoptosis of cancer cell. Intracellular mechanisms of many anticancer drugs are mediated by endocytosis. In addition, PCNP or no PCNP, MTX combined with PMX showed a stronger effect on decreasing cell proliferation than MTX or PMX alone, indicating that MTX and PMX had synergistic antitumor effects.
The assessment of different formulations following 72 h exposures using A549 cell lines was presented in . IC50 values of free MTX, free PMX, MTX + PMX, MTX-PCNPs, PMX-PCNPs and MTX-PMX-PCNPs were 4.78 ± 0.17, 25.64 ± 0.22, 8.81 ± 0.09, 1.56 ± 0.06, 11.65 ± 0.19 and 0.76 ± 0.04 μg/mL, respectively. A549 cells were most sensitive to MTX-PMX-PCNPs than the other formulations. The IC50 values of drug-loaded nanoparticles were significantly lower than free drug (p < .01). Besides, the IC50 values of free PMX was higher than that of free MTX, which might be the consequence of poor cellular uptake of the free PMX caused by its own hydrophilicity. PCNPs or not, MTX combined with PMX possessed much lower inhibitory concentration compares with MTX or PMX alone. The ability of drugs inducing apoptosis could be evaluated by IC50. That is, the lower IC50 values, the stronger ability to induce cell apoptosis. Furthermore, the combination was further examined using CI method (Chou, Citation2006, Citation2010) in . The CI value of MTX-PMX-PCNPs was 0.10 ± 0.03 (vs. free single drug) and 0.28 ± 0.06 (vs. single drug loading PCNPs), representing strong synergism (Chou, Citation2006). While, the CI value of MTX + PMX was 1.12 ± 0.11, indicating slight antagonism (Chou, Citation2006). MTX has found widespread clinical use as anti-proliferative agents by inhibiting the folate-dependent enzyme and subsequently hindering nucleic synthesis and cell division (Khan et al., Citation2012). However, the acquired or intrinsic resistance to MTX has been often observed from clinical practice. These results suggested that the drug combination was required for anticancer therapies. In this paper, MTX also served as a lung tumor targeting ligand, acting by FR-mediated endocytosis. PMX exerted its effect by means of inhibiting multiple enzymes in the folate cascade (Chattopadhyay et al., Citation2007). Combined MTX with PMX can obtain synergistic antitumor effect in multiple ways, such as inhibiting folate metabolism, increasing cell sensitivity to antitumor agents and disturbing nucleic synthesis.
Table 1. The values of IC50 and CI after the treatment with different formulations following 72 h exposure measured by CCK8 assay performed on A549 cells (**p < .01).
Cell cycle arrest
To explore whether the inhibition of cell proliferation involved cell cycle changes in A549 cells, the effects of different formulations were investigated at equivalent dose level using FCM (). The cell cycle can be assessed through five distinct phases: G0, G1, S (DNA synthesis), G2 and M (mitosis). Compared to the control group, A549 cell cycle distribution substantially retained after PCNPs culture for 48 h, indicating that the drug carrier itself had no impact on cell cycle, which are consistent with cell viability assay and previous reports (Xu et al., Citation2017). Additionally, after treated with MTX + PMX, MTX-PCNPs, PMX-PCNPs and MTX-PMX-PCNPs for 48 h, the proportion of A549 in S phase increased to 46.28, 39.64, 46.02 and 61.93%, respectively. While the percent of control group was 30.75%, lower than that of treatment groups. Interestingly, the opposite has occurred for the G2/M phase. These suggested that the formulations changed proliferating cell populations at different levels. Meanwhile, MTX-PMX-PCNPs exhibited superior S phase arrest effectiveness with significant reduction in G0/G1 and G2/M phases. PEGylated nanocarriers have imparted small-molecule chemicals to overcome disadvantages, including low solubility, cytotoxicity and nonselective biodistribution (Li et al., Citation2013). MTX and PMX have the identical mechanism of exerting pharmacological activity by competitively inhibiting the multiple target enzyme and subsequently hindering DNA and RNA synthesis, cell division inhibition and cell growth restraint (Chattopadhyay et al., Citation2007; Khan et al., Citation2012; Wibowo et al., Citation2013). Therefore, dual-drug-loaded nano-delivery system displayed the potential synergistic effect when MTX and PMX were used in combination on the cell cycle. This result was well consistent with findings of cellular uptake and cell viability assay.
Figure 3. Viability of A549 cells treated with various concentrations drugs loaded PCNPs at various concentrations for 24 h (A), 48 h (B), and 72 h (C); MTX-PMX-PCNPs at various concentrations and time (D) (n = 3).
In vivo anti-tumor efficacy
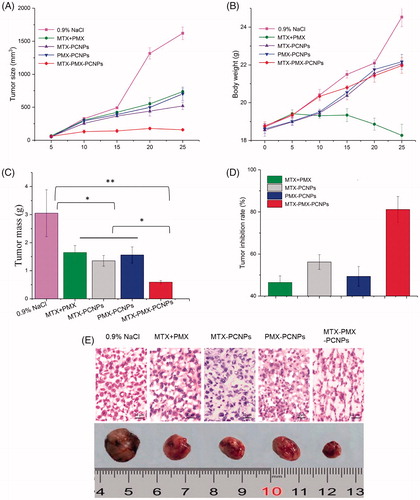
The synergistic therapeutic efficiency of dual-drug-loaded nanoparticles was assessed on C57BL/6 mice bearing a LLC advanced lung cancer with 0.9% NaCl, MTX + PMX, MTX-PCNPs, PMX-PCNPs and MTX-PMX-PCNPs. The tumor volume of mice (six mice per group) was measured for 25 days (). The tumor growth was inhibited significantly in MTX-PMX-PCNPs group. Treatment with 0.9% NaCl, MTX + PMX, MTX-PCNPs or PMX-PCNPs could not induce remarkable tumor inhibition. Meanwhile, body weight was monitored during the experiment (), which contained part of tumor growth, showed a significant fluctuation. The mice showed no obvious decreased body weight except for MTX + PMX group, which suggested that the group of free drugs might induce severe side effects. During this experimental period, all the mice were still alive.
Figure 4. Cell cycle analysis through PI staining of cells (A) treated with control, MTX + PMX, PCNPs, MTX-PCNPs, PMX-PCNPs and MTX-PMX-PCNPs. Cells cycle distribution (B) after various treatments (n = 3).
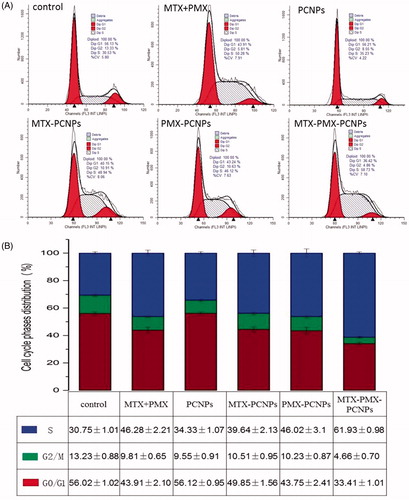
Figure 5. Tumor size (A), body weight changes (B), and weight of the solid tumors (C) of lung cancer animal model in different treatment groups within 25 days, The tumor inhibition rate for five groups upon treatment with different formulations (D), Digital photographs and H&E stained images of dissected tumors of representative mice after treated with different formulations. Sections were observed under 40× magnifications (E). Data are reported as means ± SD (n = 5). *p < 0.05, **p < 0.01.
To evaluate the potential toxicity of different formulations, an observing on animal behavior was performed as an indication of safety. Notably, compared with the experiment before administration, the activity of the mice in the positive control group (MTX + PMX) was reduced or almost immobile, the eyes were dull, the appetite decreased and the dark shiny hair become scarce and lustrous, similarly to the patients in clinic response to free MTX. Furthermore, the more frequent the dosage was, the more intense it was, indicating that the undesirable side effects of free MTX + PMX were severe. On the contrary, no undesirable side effects were presented in the MTX-PMX-PCNPs group.
To investigate in vivo antitumor effects, mice were sacrificed after 25 d. Tumors were excised and weighed (). The ultimate average tumor masses of 0.9% NaCl, MTX + PMX, MTX-PCNPs, PMX-PCNPs and MTX-PMX-PCNPs were 3.08, 1.65, 1.35, 1.56 and 0.58 g, respectively.
Afterwards, the tumor inhibition rate (TIR) for five groups was shown in . Compared to the group of 0.9% NaCl, TIR for MTX and PMX dual drug nanoparticles was 81.17%, much higher than that for MTX + PMX (46.43%), MTX-PCNPs (56.17%) or PMX-PCNPs (49.35%).
This clearly demonstrated that anti-tumor effect of MTX-PMX-PCNPs was about 0.5 folders stronger than other groups. Hence, both the decrease of tumor mass and increase of tumor inhibition rate were statistically significant. It further confirmed that MTX-PMX-mPEG-CS-NPs has anti-tumor efficacy in vivo. In addition, MTX and PMX dual drug nanoparticles have a synergistic effect.
To further reveal the antitumor effect of dual drug delivery system in vivo, tumor specimens from sacrificed mice were done into paraffin sections on day 25 after drug administration, then analyzed by HE staining. As can be seen from , tumor cells in 0.9% NaCl group were closely packed with intense staining and regular array. Then, a smaller but apparently necrosis was also demonstrated in MTX + PMX, MTX-PCNPs and PMX-PCNPs groups. Whereas, in the MTX-PMX-PCNPs group, there exhibited lower nuclear-cytoplasmic ratio, more significant necrotic region with poorly defined borders and weak staining in tumor sections compared with other groups.
Nano-drug delivery system is exploited not only to maximize therapeutic efficacy, but also to possess an optimal selectivity and specificity for target sites (Yamashita & Hashida, Citation2013). For labile ingredients, polymeric NPs can enhance stability and prolong duration. Then, chemotherapeutics based on them can improve therapeutic effect and decrease cytotoxicity (El-Say & El-Sawy, Citation2017). When PEGylated PCNPs circulate in the blood, PEG affords hydrophilic chains cloud outer surface for escaping phagocytosis (Liu et al., Citation2013). Due to the EPR effect, MTX-PMX-PCNPs can reach tumor sites via leaky vessels after intravenous injection (Fang et al., Citation2011). To increase the therapeutic efficacy, macromolecular carriers are being developed on the unique pathophysiological features of the EPR effect. Macromolecules with a molecular weight over 40 kDa have a characteristic of selective leakage, accumulating in tumor tissues via blood vessels. While normal tissues do not take on this phenomenon (Fang et al., Citation2011). In addition, MTX acts as ligand mediating active tumor targeting via endocytosis mechanism. Clinically, most tumors are found in the middle or late stages and are strong dependency upon the active recruitment of new blood vessels (Hanahan & Weinberg, Citation2011). Thus, the receptor/ligand complex can work well. More importantly, PMX has the identical mechanism of exerting pharmacological activity by competitively inhibiting the multiple target enzyme (TS, GARFT and DHFR) (Chattopadhyay et al., Citation2007; Wibowo et al., Citation2013), in company with MTX inhibiting DHFR (Khan et al., Citation2012). Thus, combined MTX with PMX via PEGylated nanocarriers provides an ideal platform for a synergistic antitumor effect.
Conclusions
Here, we have described a strategy of employing PCNPs as smart nano carrier for co-delivery of MTX and PMX for lung cancer treatment. MTX-PMX-PCNPs were found to be subsphaeroidal with a controllable particle size and a property of sustained release in vitro. The dual drug loaded nano-delivery system exhibited the facilitating intracellular uptake, significant anti-proliferative activity and superior S phase arrest effectiveness in human alveolar epithelial cell lines, compared to those of free drug combination or single loading PCNPs formulations. Furthermore, the nano-scaled delivery system could improve in vivo anti-tumor of C57BL/6 mice bearing a LLC advanced lung cancer. In summary, this combination nano-delivery strategy could be considered as a prospective synergistic targeting anticancer chemotherapeutic agent, especially for carcinoma of advance lungs.
Disclosure statement
No potential conflict of interest was reported by the authors.
Additional information
Funding
References
- Ando H, Kobayashi S, Abu Lila AS, et al. (2015). Advanced therapeutic approach for the treatment of malignant pleural mesothelioma via the intrapleural administration of liposomal pemetrexed. J Control Release 220:29–36.
- Anitha A, Sowmya S, Kumar PTS, et al. (2014). Chitin and chitosan in selected biomedical applications. Prog Polym Sci 39:1644–67.
- Brannon-Peppas L, Blanchette JO. (2012). Nanoparticle and targeted systems for cancer therapy. Adv Drug Delivery Rev 64:206–12.
- Canal F, Vicent MJ, Pasut G, et al. (2010). Relevance of folic acid/polymer ratio in targeted PEG–epirubicin conjugates. J Control Release 146:388–99.
- Chan M, Gravel M, Bramoulle A, et al. (2014). Synergy between the NAMPT inhibitor GMX1777(8) and pemetrexed in non-small cell lung cancer cells is mediated by PARP activation and enhanced NAD consumption. Cancer Res 74:5948–54.
- Chattopadhyay S, Moran RG, Goldman ID. (2007). Pemetrexed: biochemical and cellular pharmacology, mechanisms, and clinical applications. Mol Cancer Ther 6:404–17.
- Chen J, Huang LQ, Lai HX, et al. (2014). Methotrexate-loaded PEGylated chitosan nanoparticles: synthesis, characterization, and in vitro and in vivo antitumoral activity. Mol Pharmaceutics 11:2213–23.
- Chen MC, Mi FL, Liao ZX, et al. (2013). Recent advances in chitosan-based nanoparticles for oral delivery of macromolecules. Adv Drug Deliv Rev 65:865–79.
- Cheng R, Meng FH, Deng C, et al. (2013). Dual and multi-stimuli responsive polymeric nanoparticles for programmed site-specific drug delivery. Biomaterials 34:3647–57.
- Chou TC. (2006). Theoretical basis, experimental design, and computerized simulation of synergism and antagonism in drug combination studies. Pharmacol Rev 58:621–81.
- Chou TC. (2010). Drug combination studies and their synergy quantification using the Chou-Talalay method. Cancer Res 70:440–6.
- Decuzzi P, Godin B, Tanaka T, et al. (2010). Size and shape effects in the biodistribution of intravascularly injected particles. J Control Release 141:320–7.
- El-Say KM, El-Sawy HS. (2017). Polymeric nanoparticles: promising platform for drug delivery. Int J Pharm 528:675–91.
- Fang J, Nakamura H, Maeda H. (2011). The EPR effect: Unique features of tumor blood vessels for drug delivery, factors involved, and limitations and augmentation of the effect. Adv Drug Deliv Rev 63:136–51.
- Farjadian F, Ghasemi S, Mohammadi-Samani S. (2016). Hydroxyl-modified magnetite nanoparticles as novel carrier for delivery of methotrexate. Int J Pharm 504:110–6.
- Guo YX, Zhang Y, Ma JY, et al. (2018). Light/magnetic hyperthermia triggered drug released from multi-functional thermo-sensitive magnetoliposomes for precise cancer synergetic theranostics. J Control Release 272:145–58.
- Hanahan D, Weinberg RA. (2011). Hallmarks of cancer: the next generation. Cell 144:646–74.
- He CB, Lu JQ, Lin WB. (2015). Hybrid nanoparticles for combination therapy of cancer. J Control Release 219:224–36.
- Hu QY, Sun WJ, Wang C, et al. (2016). Recent advances of cocktail chemotherapy by combination drug delivery systems. Adv Drug Deliv Rev 98:19–34.
- Jain A, Jain A, Garg NK, et al. (2015). Surface engineered polymeric nanocarriers mediate the delivery of transferrin-methotrexate conjugates for an improved understanding of brain cancer. Acta Biomater 24:140–51.
- Jenkins SI, Pickard MR, Furness DN, et al. (2013). Differences in magnetic particle uptake by CNS neuroglial subclasses: implications for neural tissue engineering. Nanomedicine 8:951–68.
- Kemp JA, Shim MS, Heo CY, et al. (2016). “Combo” nanomedicine: Co-delivery of multi-modal therapeutics for efficient, targeted, and safe cancer therapy. Adv Drug Delivery Rev 98:3–18.
- Khan ZA, Tripathi R, Mishra B. (2012). Methotrexate: a detailed review on drug delivery and clinical aspects. Expert Opin Drug Deliv 9:151–69.
- Kohay H, Sarisozen C, Sawant R, et al. (2017). PEG-PE/clay composite carriers for doxorubicin: Effect of composite structure on release, cell interaction and cytotoxicity. Acta Biomater 55:443–54.
- Li WJ, Zhan P, De Clercq E, et al. (2013). Current drug research on PEGylation with small molecular agents. Prog Polym Sci 38:421–44.
- Li Y, Lin JJ, Ma JY, et al. (2017a). Methotrexate-camptothecin prodrug nanoassemblies as a versatile nanoplatform for biomodal imaging-guided self-active targeted and synergistic chemotherapy. Acs Appl Mater Interfaces 9:34650–65.
- Li Y, Song L, Lin JY, et al. (2017b). Programmed nanococktail based on ph-responsive function switch for self-synergistic tumor-targeting therapy. Acs Appl Mater Interfaces 9:39127–42.
- Liu GH, Ma JY, Li Y, et al. (2017). Core-interlayer-shell Fe3O4@mSiO(2)@lipid-PEG-methotrexate nanoparticle for multimodal imaging and multistage targeted chemo-photodynamic therapy. Int J Pharm 521:19–32.
- Liu Y, Yin Y, Wang LY, et al. (2013). Surface hydrophobicity of microparticles modulates adjuvanticity. J Mater Chem B 1:3888–96.
- Lu NN, Li RT, Liu Q, et al. (2016). Antitumor and antimetastatic effects of pemetrexed-loaded targeted nanoparticles in B16 bearing mice. Drug Deliv 23:2566–74.
- Miller KD, Siegel RL, Lin CC, et al. (2016). Cancer treatment and survivorship statistics, 2016. Ca-Cancer. CA Cancer J Clin 66:271–89.
- Modaresifar K, Azizian S, Hadjizadeh A. (2016). Nano/biomimetic tissue adhesives development: from research to clinical application. Polym Rev 56:329–61.
- Perry JL, Reuter KG, Luft JC, et al. (2017). Mediating passive tumor accumulation through particle size, tumor type, and location. Nano Lett 17:2879–86.
- Ramirez M, Rajaram S, Steininger RJ, et al. (2016). Diverse drug-resistance mechanisms can emerge from drug-tolerant cancer persister cells. Nat Comms 7:10690.
- Siegel RL, Miller KD, Jemal A. (2017). Cancer Statistics, 2017. Ca-Cancer. J Clin 67:7–30.
- Singh VK, Subudhi BB. (2016). Development and characterization of lysine-methotrexate conjugate for enhanced brain delivery. Drug Deliv 23:2327–37.
- Srinivas NSK, Verma R, Kulyadi GP, et al. (2016). A quality by design approach on polymeric nanocarrier delivery of gefitinib: formulation, in vitro, and in vivo characterization. IJN 12:15–28.
- Tan WL, Jain A, Takano A, et al. (2016). Novel therapeutic targets on the horizon for lung cancer. Lancet Oncol 17:e347–E62.
- Wang X, Tang H, Wang CZ, et al. (2016). Phenylboronic acid-mediated tumor targeting of chitosan nanoparticles. Theranostics 6:1378–92.
- Wang YW, Zhang HY, Hao J, et al. (2015). Lung cancer combination therapy: co-delivery of paclitaxel and doxorubicin by nanostructured lipid carriers for synergistic effect. Drug Deliv 23, 1398–403.
- Weeks JC, Catalano PJ, Cronin A, et al. (2012). Patients' expectations about effects of chemotherapy for advanced cancer. N Engl J Med 367:1616–25.
- Wibowo AS, Singh M, Reeder KM, et al. (2013). Structures of human folate receptors reveal biological trafficking states and diversity in folate and antifolate recognition. Proc Natl Acad Sci USA 110:15180–8.
- Win KY, Feng SS. (2005). Effects of particle size and surface coating on cellular uptake of polymeric nanoparticles for oral delivery of anticancer drugs. Biomaterials 26:2713–22.
- Xiong Y, Zhao Y, Miao L, et al. (2016). Co-delivery of polymeric metformin and cisplatin by self-assembled core-membrane nanoparticles to treat non-small cell lung cancer. J Control Release 244:63–73.
- Xu YR, Asghar S, Yang L, et al. (2017). Nanoparticles based on chitosan hydrochloride/hyaluronic acid/PEG containing curcumin: In vitro evaluation and pharmacokinetics in rats. Int J Biol Macromol 102:1083–91.
- Yamashita F, Hashida M. (2013). Pharmacokinetic considerations for targeted drug delivery. Adv Drug Deliv Rev 65:139–47.
- Yan JK, Wang YZ, Zhang XF, et al. (2016). Targeted nanomedicine for prostate cancer therapy: docetaxel and curcumin co-encapsulated lipid-polymer hybrid nanoparticles for the enhanced anti-tumor activity in vitro and in vivo. Drug Deliv 23:1757–62.