Abstract
Membrane grafting of methoxypoly(ethylene glycol) [mPEG] to allogeneic leukocytes attenuates allorecognition and significantly reduces the risk of graft-versus-host disease in mice. To optimize the immunological efficacy of polymer grafting, murine splenocytes were modified using three differing linker chemistries: CmPEG (5 kDa), BTCmPEG (5 and 20 kDa) and TmPEG (5 kDa). In vitro immunocamouflage efficacy was examined by flow cytometic analysis of leukocyte markers and mixed lymphocyte reactions (MLR). In contrast to CmPEG and BTCmPEG, TmPEG exerted significant cellular toxicity. Flow cytometric analysis demonstrated that both CmPEG and BTCmPEG were highly effective at camouflaging cell surface markers while TmPEG was ineffective. Furthermore, CmPEG and BTCmPEG dramatically blocked MLR allorecognition and cellular proliferation. Polymer length was the most critical factor in the immunocamouflage of cells with the BTCmPEG 20 kDa being the most effective. In contrast to other immunomodulatory approaches, immunocamouflage of leukocytes yields a multivalent effect globally interfering with attachment, allorecognition, presentation and costimulation pathways.
INTRODUCTION
Methoxypoly(ethylene glycol) [mPEG] has been widely used to covalently modify purified proteins since the mid-1970s [Citation[1], Citation[2]]. Consequent to protein derivatization, pegylated proteins and liposomes were observed to have reduced or absent immunogenicity while exhibiting normal enzymatic function and improved vascular retention times [Citation[1-4]]. As a result of these attributes, mPEG-modified proteins and liposomes are currently utilized in multiple therapeutic situations. For example, mPEG has been grafted to bovine adenosine deaminase for use in enzyme replacement therapy of adenosine deaminase deficient severe combined immunodeficiency patients [Citation[5]]. This life-long therapeutic intervention has been crucial for those patients who are not suitable candidates for bone marrow transplantation. Similarly, mPEG-modified asparaginase has demonstrated great promise in the treatment of acute leukemias [Citation[6], Citation[7]]. Other clinical usages of pegylated proteins include mPEG-bilirubin oxidase for jaundice as well as PEG-hemoglobin as a blood substitute [Citation[8], Citation[9]].
More recently, the covalent grafting of mPEG to membrane proteins of intact cells has been used to attenuate antigenic and immunologic recognition of allogeneic red blood cells (RBC), white blood cells (WBC) and pancreatic islets [Citation[10-26]]. Importantly, these studies demonstrated that pegylated RBC had normal in vivo survival in murine transfusion models at immunoprotective levels of derivatization [Citation[10-21]]. Also of interest was the finding that the membrane grafting of mPEG to human and murine immune cells resulted in the attenuation or abrogation of allogeneic recognition [Citation[10-21]]. The immunomodulatory effect of mPEG grafting to immune cells arises from the immunocamouflage of numerous adhesion, allorecognition and costimulatory molecules on the T lymphocyte and antigen presenting cell [Citation[10-21]]. The potency of this immunocamouflage was further demonstrated by the experimental finding that modification of donor lymphocytes prevents transfusion-mediated graft versus host disease in a murine model [Citation[10-21]].
The efficacy of immunocamouflage is dependent upon both the density and depth of the mPEG brush border [Citation[10-21]]. The density of the brush border is a direct function of the linker chemistry used to graft the mPEG to the membrane proteins as well as the concentration of activated mPEG used for derivatization. The depth of the brush border is a direct function of the polymer length. While decades of study have demonstrated that mPEG itself exhibits no significant acute or chronic toxicity [Citation[27-31]], the linker chemistry has the potential to exert toxic effects. Previously, minimal attention has been paid to the linker chemistry toxicity when working on the derivatization of purified proteins unless the chemistry affected protein function. However, when working with intact, viable cells, the potential toxicity of the linker chemistry is of crucial importance.
As a result of the above concerns, comparative studies of differing linker chemistries and polymer lengths on cell viability as well as the efficacy of mPEG-mediated immunocamouflage were conducted using murine splenocytes. The differential effects of the various linker chemistries on cell viability and immunocamouflage were assessed using mixed lymphocyte reactions (MLR) and flow cytometric analysis of cell viability and the immunocamouflage of membrane proteins involved in lymphocyte adhesion and activation pathways. As will be shown, polymer length is the primary factor in imparting immunocamouflage, but this in turn is dependent upon the efficacy of membrane grafting as governed by the linker chemistry employed. Furthermore, some linker chemistries carry significant cellular toxicity concerns.
MATERIALS AND METHODS
Isolation of Mouse Splenocytes
All animal studies were done in accordance with NIH, CIHR and Institutional guidelines for the care and use of laboratory animals. Spleens were recovered from two H-2 disparate mouse strains [Balb/c (H-2Kd) and C57BL/6 (H-2Kb)] and single cell suspensions were prepared [Citation[19]]. The recovered splenocytes were washed once with Hepes (Sigma Chemical Co., St. Louis, MO)/RPMI 1640 (Life Technologies, Rockville, Maryland)/and 1% Human Albumin (Sigma Chemical Co., St. Louis, MO). Red blood cells were removed using an ammonium chloride lysing reagent (Pharmingen, San Diego, CA) followed by two washes with Hepes/RPMI 1640 with 1% human albumin. Cells were counted on a hemacytometer and resuspended in Hepes/RPMI 1640 with 1% human albumin.
Cell Derivatization
As shown in , splenocytes were derivatized using three different linker chemistries: cyanuric chloride activated methoxypoly(ethylene glycol) (CmPEG; 5 kDa); methoxypoly(ethylene glycol)-1 Benzotriazolyl-Carbonate (BTCmPEG; 5 and 20 kDa) and O-Tresyl-O'-methylpolyethylene glycol (TmPEG; 5 kDa). In all cases, murine splenocytes were derivatized with the 0–5 mM mPEG at a cell concentration of 5.5 × 107 cells per ml as previously described [Citation[10-21]]. Cells were incubated with activated mPEG for 60 minutes at room temperature in isotonic alkaline phosphate buffer (PBS; 50 mM K2HPO4 and 105 mM NaCl; pH 8.0) and washed twice with Hepes/RPMI 1640 with 1% human albumin. An additional wash in AIM V media was done if preparing cells for MLR or, alternatively, two washes in PBS containing 1.5 mM sodium azide for use in flow cytometric analysis.
Table 1. Linker and Polymer Chemistry. Variable linker chemistries were examined though all chemistries used targeted lysine (more correctly, epsilon amino acid) residues. The efficiency of binding and the efficacy of the induced immunocamouflage were examined via flow cytometry and mixed lymphocyte reactions. The BTCmPEG 5000 and BTCmPEG 20000 were further analyzed to determine the effect of polymer size on the induced immunocamouflage
Mixed Lymphocyte Reactions (MLR)
One-way MLR were conducted using splenocytes from two H-2 disparate mouse strains, Balb/c (H-2Kd) and C57BL/6 (H-2Kb) [Citation[16], Citation[19], Citation[20]]. In a one-way MLR, one population of splenocytes was gamma irradiated (2 Gy) and acted as allogeneic stimulator cells. Stimulator and responder splenocytes (5.12 × 106 cells per well) were co-incubated in triplicate in 96-well plates at 37°C, 5% CO2 for 3 days. On day 3, all wells were pulsed with 3H-thymidine and incubated for 24 hours at 37°C, 5% CO2. Cellular DNA was collected on filter mats using a Skatron cell harvester (Suffolk, UK) and cellular proliferation was measured by 3H-thymidine incorporation.
Cell Viability and DNA Laddering
The viability of unmodified and modified cells was determined by propidium iodide exclusion assay on the flow cytometer [Citation[32]]. Cell viability was determined at 0, 24, 48, and 72 hours post-derivatization. In addition, cell viability was assessed both in the absence and presence of allogeneic stimulator cells to determine if allostimulation would affect viability. Similarly, viability of the control and mPEG-grafted populations was determined pre– and post–mitogen stimulation.
To assess the induction of apoptosis, an apoptotic DNA-Laddering kit (Boehringer Mannheim, Germany) was used. Approximately 2 × 106 cells per sample were collected for DNA purification as per the manufacturer's directions. DNA preparations from the samples were analyzed via agarose gel electrophoresis using a 1% agarose gel in TBE-buffer (0.10 M Sodium Acetate, 0.20 M EDTA, 0.80 M Tris Base, 36.5 mL glacial acetic acid, bring up to 1L; pH 8.0; dilute to 1X) at 75 V for approximately 2–3 hours. DNA molecular weight markers (Boehringer Mannheim, Germany) and a positive control of apoptotic U937 cells were similarly run. Ethidium bromide was used to stain the DNA for apoptotic laddering. DNA laddering gels were analyzed using Image J analysis software (NIH, Bethesda, MD).
Mitogen Stimulation
Control and mPEG-modified splenocytes were incubated in triplicate with phytohemagglutinin (PHA) (Sigma; 0.5 µg/mL) or a combination of phorbol 12-myristate 13-acetate (PMA; 50 µg/mL;) and Ionomycin (2 µg/mL ) at 37°C, 5% CO2 for 3 days [Citation[33], Citation[34]]. As in the MLR studies, all wells were pulsed with 3H-Thymidine and incubated for 24 hours at 37°C, 5% CO2 before cellular proliferation was measured by scintillation counting.
CD Marker Analysis
Flow cytometric studies were done on splenocytes to determine the degree of immunocamouflage of cell surface receptors, adhesion molecules, activation and costimulatory molecules [Citation[10-21]]. Experiments were done with control and mPEG modified cells resuspended at a concentration of 1.0 × 107 cells. Fluorescently labeled monoclonal anti-mouse antibodies (CD2, CD3e, CD4, CD8, CD11a, CD25, CD28, CD80, and T cell receptor; Becton Dickinson, San Jose, CA) were added as indicated. The cells were incubated for 30 minutes on ice, washed two times with 1X phosphate buffer solution (PBS) containing azide and analyzed on the flow cytometer for fluorescence intensity of specific cell surface epitopes. A minimum of 10,000 splenocytes per sample were analyzed. All samples were gated upon using mouse splenocyte parameters previously established for FACScan analysis.
RESULTS
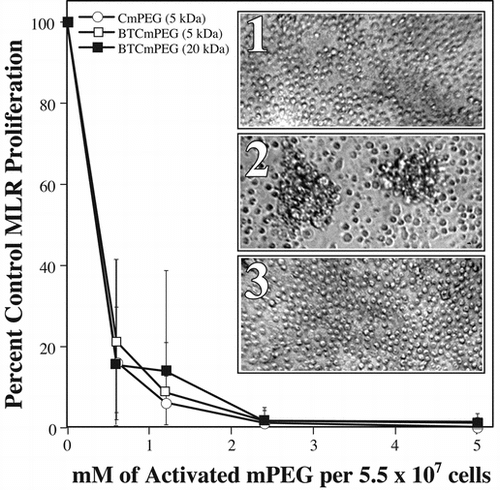
Mixed lymphocyte reactions (MLR's) were performed to determine if CmPEG (5 kDa), BTCmPEG (5 and 20 kDa), and TmPEG (5 kDa) were capable of preventing cellular proliferation in vitro. Concurrently, studies were conducted to assess whether linker chemistry and polymer length affected either the efficacy of derivatization or the degree of immunocamouflage. As shown in , 5 kDa CmPEG and both the 5 and 20 kDa BTCmPEG effectively prevented allorecognition and proliferation. Indeed, at derivatization concentrations as low as 0.6 mM per 5.5 × 107 cells both the CmPEG and BTCmPEG grafting resulted in a nearly complete abrogation of alloresponsiveness as evidenced by a > 80% decrease in 3H-thymidine incorporation. This finding was further confirmed by microscopic analysis of the MLR (). Increasing the derivatization concentration of the CmPEG and BTCmPEG to >2 mM essentially reduced the proliferation rate to that seen in resting splenocytes. Initial studies with TmPEG also suggested that it was equally (if not more) effective at blocking T cell activation and proliferation (data not shown), but for toxicological reasons, discussed below, this compound was not further pursued.
Figure 1 Covalent grafting of mPEG to splenocyte membranes significantly attenuates allorecognition of H-2 disparate cells. Shown are the proliferation responses of one-way MLR in which the responder cells have been derivatized with CmPEG 5000, BTCmPEG 5000, or BTCmPEG 20000. Insert: Photomicrographs of unstimulated splenocytes show the small rounded morphology typical of resting lymphocytes (1). In contrast, allostimulation of control (unmodified) splenocytes results in multiple foci of proliferating alloresponsive cells (2). However, as can be seen, mPEG-modified (0.6 mM) splenocytes fail to proliferate and remain morphologically unactivated (3). The data presented represented the co-culturing of 5.12 × 106 mPEG-modified Balb/c splenocytes with 5.12 × 106 irradiated C57Bl/6 splenocytes. The results shown represent the mean ± standard deviation of triplicate samples from a minimum of 4 independent experiments. Photomicrographs are representative samples.
In order to confirm that the loss of proliferation was due to mPEG-mediated immunocamouflage and not a result of cytotoxicity, viability studies were performed using propidium iodide exclusion. As shown in , neither unstimulated (i.e., no stimulator cells) CmPEG and BTCmPEG-modified splenocytes exhibited any significant cytotoxicity at immunoprotective levels (≤2.4 mM) of mPEG grafting over 72 hours in culture. In contrast, TmPEG modification yielded dead or dying cells (). The toxicity of TmPEG is likely attributable to the tri-fluoride leaving group (OSO2CH2CF3) that constitutes the linker moiety of TmPEG. Consequent to these toxicity findings of the linker chemistry used by TmPEG, this compound was not further pursued.
Table 2. In the absence of allogeneic stimulation cell viability is unaffected by the covalent grafting of mPEG to membrane surfaces when using CmPEG 5000, BTCmPEG 5000, or BTCmPEG 20000. In contrast, TmPEG 5000 was found to exert a potent toxic effect on murine splenocytes. This toxicity was also noted on human peripheral blood mononuclear cells (data not shown). Viability was determined by flow cytometric analysis of propidium iodide exclusion. Shown are the average percentages from two experiments
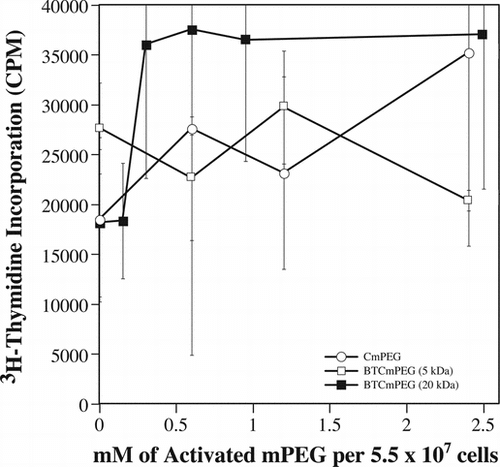
To determine whether the loss of alloresponsiveness noted in the MLRs might arise from the loss of proliferative potential due to membrane perturbations arising from the grafted CmPEG and BTCmPEG, direct mitogen stimulation was done using phorbol 12-myristate 13-acetate (PMA) in combination with ionomycin. PMA and ionomycin work in conjunction with one another to activate cellular proliferation by directly initiating intracellular signaling cascades. Importantly, these molecules, because of their very small molecular weight, readily pass through the mPEG brush border. As documented in , splenocytes modified at all concentrations of CmPEG and BTCmPEG were stimulated to levels comparable to that observed with unmodified cells. Furthermore, neither linker chemistry (CmPEG versus BTCmPEG) nor polymer size (5 versus 20 kDa) adversely affected the proliferative potential of the modified splenocytes ().
Figure 2 The proliferation potential of mPEG modified splenocytes is unaffected as demonstrated by direct mitogen stimulation. Shown are the mitogen proliferation responses of cells derivatized with CmPEG 5000 (A), BTCmPEG 5000 (B), or BTCmPEG 20000 (C). Mitogen stimulation was done using a combination of PMA (50 ng/ml) and Ionomycin (2 µg/ml). Shown are the mean ± standard deviation of triplicate samples from two independent experiments.
The mitogen proliferation studies further illuminated an interesting observation noted in the viability studies of pegylated cells that were allostimulated. As shown in , the introduction of allogeneic stimulator cells resulted in a time and mPEG-dose dependent loss of viability in both CmPEG and BTCmPEG modified cells—cells that exhibited normal viability in the absence of stimulator cells. A similar loss of viability was not observed subsequent to the mitogen stimulation of the mPEG grafted cells, arguing that proliferation itself does not result in cytotoxicity. Rather, this data suggests that the decreased viability noted following the introduction of the stimulator cells is allostimulation specific and occurs via an active process; i.e., apoptosis. Indeed, previous studies clearly demonstrate that weak or insufficient allostimulation drives lymphocytes towards an apoptotic pathway [Citation[35-41]].
Table 3. In the presence of allogeneic stimulation cell viability was significantly decreased in a time and mPEG dose dependent manner following the covalent grafting of CmPEG 5000, BTCmPEG 5000, or BTCmPEG 20000. Because of the toxicity associated with TmPEG in the absence of allogeneic stimulation, viability was not included below but approached zero in all cases.Viability was determined by flow cytometric analysis of propidium iodide exclusion. Shown are the average percentages from two experiments
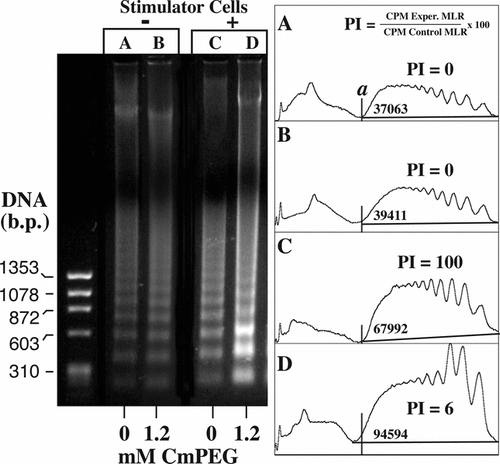
As demonstrated in , analysis of DNA laddering gels confirmed that apoptosis was preferentially induced in the mPEG-modified cells upon exposure to allogeneic stimulator cells (lanes C and D) but not in the absence of these cells (lanes A and B). Note that while apoptosis is increased in the control MLR, as denoted by the area under the demarcated curve, a significantly greater increase in DNA laddering is observed in the allostimulated MLR with the mPEG-modified responder cells. Moreover, when further assessed relative to the proliferation index (PI) of the sample, it is clear that a much greater percentage of the allostimulated non-pegylated responder cells (lane D) are undergoing apoptosis. Indeed, despite virtually no proliferation of the pegylated cells (PI = 6; see ) relative to the control MLR (PI = 100), the allostimulated mPEG splenocytes exhibit a 140% increase in the DNA laddering—especially in the very low molecular bands. While a significant increase in laddering is also seen in the control MLR upon allostimulation (lane C), much of this signal likely represent dead/dying stimulator cells.
Figure 3 Apoptosis is preferentially induced in mPEG-modified cells upon challenge with allogeneic stimulator cells. In contrast, in the absence of allostimulation, control (lane A) and mPEG-modified (lane B) murine splenocytes demonstrate comparable viability (Table ) and DNA laddering profiles as shown by ethidium bromide staining and densitometry. Shown is a representative ethidium bromide stained agarose gel showing the DNA laddering patterns after 48 incubation in the absence (−) and presence (+) of allogeneic stimulators. The mPEG-modified cells were derivatized in the presence of 1.2 mM CmPEG 5000. Also shown are densitometric analysis of the agarose gel. Area analysis (a) of the lower banding areas are given as is the average proliferation index (PI) of the MLR as determined by Figure . PI = (CPM Exp. MLR/CPM Control MLR) × 100.
The efficacy of immunocamouflage was further examined by flow cytometric analysis of the CmPEG and BTCmPEG grafted cells. CmPEG-modification of splenocytes resulted in the dramatic camouflaging of cell surface molecules of both the TCR and CD4 ( and , respectively). In unmodified murine splenocytes, 60% of the cells were TCR positive; however, following derivatization with 2.4 mM CmPEG, only 5% of the same population were TCR positive. Similar results were observed with the CD4 receptor (). In the unmodified splenocyte population 41% of cells were CD4 positive while only 4% were positive subsequent to derivatization with 2.4 mM of CmPEG. Importantly, as shown in , mPEG grafting results in a global camouflaging effect on multiple adhesion, activation, and co-stimulatory proteins. The effect of linker chemistry and polymer size was also demonstrated by these flow cytometric analyses of the CmPEG 5000, BTCmPEG 5000 and BTCmPEG 20000 modified cells. As shown in and , the BTC linker chemistry was generally equal to or more efficient in modifying the murine splenocytes than the cyanuric chloride chemistry. However, some significant exceptions were noted. For example, at 2.4 mM of derivatization, CmPEG 5000 was able to diminish the antibody binding of the class II molecule by 71% while the BTCmPEG 5000 demonstrated only a 41% decrease in class II binding. However, in all cases the BTCmPEG 20000 was the most efficient at inducing immunocamouflage when expressed on a molar ratio. This likely reflects the much larger radius of gyration (Rg) of the 20 kDa polymer, which is crucial in the development of membrane immunocamouflage ().
Figure 4 mPEG grafting effectively immunocamouflages membrane proteins as demonstrated by the loss of antibody recognition of the murine T Cell Receptor (TCR) and CD4 molecule. Shown are representative flow cytometric dot plots of control and CmPEG 5000 (2.4 mM) derivatized murine splenocytes.
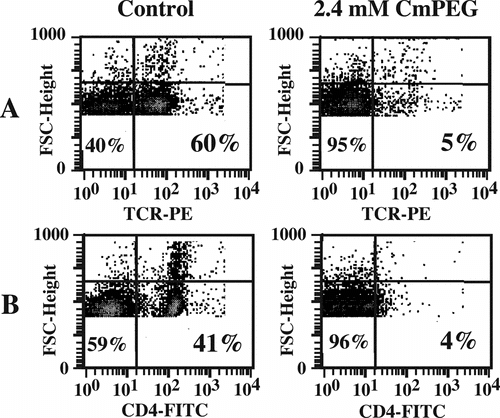
Figure 5 Differential effects of linker chemistry and polymer length on the immunocamouflage of membrane proteins. Shown are the percent positive cells for CD11a, MHC Class II, CD4 and the T cell receptor (TCR) following derivatization with CmPEG 5000, BTCmPEG 5000, and BTCmPEG 20000. The values shown are normalized to the unmodified control population.
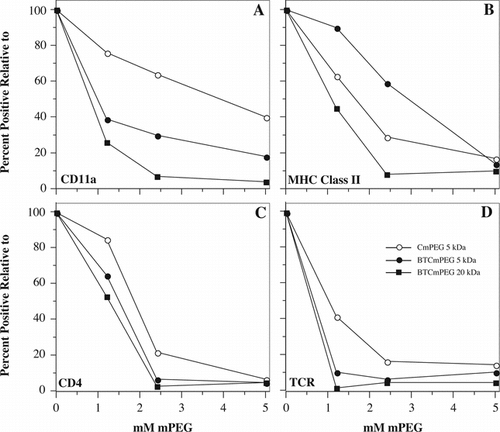
Figure 6 Mechanism of immunocamouflage. As shown, rigid linear molecules have a small radius of gyration (Rg) and fail to effectively camouflage (shaded area, a and b) cell surface proteins and carbohydrates (“antigens”). In contrast, the linear, but highly flexible, mPEG polymer yields a large zone of steric occlusion (shaded areas c and d). As noted, the immunocamouflaged area effectively covers membrane antigens. The size of the area of protection is dependent up on the length (L) of the polymer and its resultant Rg.
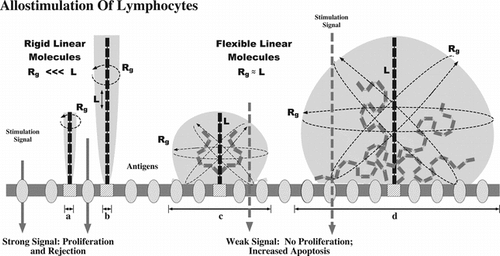
Table 4. The covalent grafting of mPEG to membrane proteins results in the global camouflage of multiple proteins crucial for effective allorecognition of foreign tissue. Values shown are “Percent Positive Cells” relative to the unmodified control (= 100). The results given are the mean of a minimum of 3 separate experiments
DISCUSSION
Our studies of four different mPEG compounds clearly demonstrate that the selection of appropriate, non-toxic linker chemistries as well as polymer chain length are crucial factors in the use of stealth technology on intact, viable cells. While all the activated mPEG compounds studied [CmPEG 5000, BTCmPEG 5000, BTCmPEG 20000, and TmPEG 5000] targeted lysine residues on the exterior face of membrane proteins, significant differences were noted in both cellular toxicity and the efficacy of immunocamouflage.
Unlike the CmPEG or BTCmPEG, the TmPEG was of particular interest in that its linker chemistry results in a direct, covalent linkage between the mPEG polymer chain and the protein, thereby removing any biological consequence that the linker group might have on the membrane surface () [Citation[42]]. Indeed, initial screening of the TmPEG 5000 in the MLR assay appeared promising as no proliferation was noted upon allogeneic stimulation. However, subsequent studies demonstrated that the OSO2CH2CF3 leaving group was highly toxic, thereby accounting for the loss of proliferation as dead cells don't divide. Furthermore, flow cytometric analysis of the TmPEG-modified cells also demonstrated that this linker chemistry was very poor at camouflaging cell surface epitopes due to its poor derivatization of membrane proteins. For example, while both the CmPEG 5000 and BTCmPEG 5000 resulted in an almost 65% decrease in detectable CD3ε, the TmPEG yielded only a 25% decrease and killed the cells as well. Consequently, further use of this compound was avoided.
The derivatization efficiency of the CmPEG 5000 and BTCmPEG 5000 were fairly similar, though the BTCmPEG was, on average, slightly better (; ). What was clearly observed was that polymer length ultimately determined the efficacy of the mPEG-mediated immunocamouflage. As shown in , the rigid linear molecules lack any significant radius of gyration (Rg) resulting in a poor or absent camouflaging (a, b) of the membrane antigens. In contrast, while methoxypoly(ethylene glycol) is a linear molecule, it has a high degree of intra-chain flexibility due to the repeating, highly mobile, ethoxy units. Based on its expansive Rg, as well as the heavy hydration of the polymer chain itself, the grafted mPEG is able to sterically occlude a large three dimensional (3D) volume (). Consequent to the space filling capacity of mPEG, the immunocamouflage of surface membrane proteins and carbohydrates (antigens in ) occurs. However, as denoted, the longer the mPEG polymer chain (c, d) the larger the membrane surface area covered by this steric shield (c, d and stippled area). Importantly, the larger polymers allow for a more efficient immunocamouflage of the cell membrane at equimolar concentrations. This is clearly observed with all the membrane markers examined (). Furthermore, our previous studies on red blood cells demonstrate that the larger polymers (e.g., BTCmPEG 20000) also improve the viability and circulation time of mPEG-modified cells relative to the shorter polymer chains. Indeed, at derivatization concentrations in excess of 2 mM BTCmPEG 20000, normal RBC in vivo survival times were noted in murine transfusion models [Citation[10-21]].
Interestingly, despite the incomplete camouflaging of immunologically important adhesion, recognition, activation and stimulation pathways on T lymphocytes and antigen presenting cells, the CmPEG 5000, BTCmPEG 5000, and the BTCmPEG 20000 were all highly effective in actually inhibiting allorecognition as demonstrated by the MLR studies ( and ). Even slight derivatization (≥0.20 mM mPEG species per 5.5 × 107 cells) of the responder cells resulted in significantly (p ≤ 0.0001) decreased or even absent proliferation as evidenced by the loss of 3H-thymidine incorporation. At slightly higher derivatization dosages (1.2 mM), a virtually complete blockade of T cell activation was observed for the CmPEG and BTCmPEGs. Clearly these data suggest that it is not necessary to completely “encase” allogeneic cells in a polymer envelope. Rather the data suggest that even light levels of cell or tissue pegylation may be sufficient to weaken the multiple interaction between H-2/MHC disparate cell populations (e.g., T cell and antigen presenting cell), thereby conferring an immunologically stealthy phenotype. Indeed, a large body of research clearly indicates that a biologically prolonged interaction between the T lymphocyte and the antigen presenting cell is necessary to efficiently transmit the allorecognition and stimulation signals for T lymphocyte activation () [Citation[43-48]]. However, pegylation of the membrane surface interferes with both the stability and longevity of these receptor-ligand interactions, there-by preventing allorecognition and favoring immunological anergy due to the apoptosis of the alloresponsive cell population.
In sum, an increasing body of evidence suggests that the covalent grafting of mPEG to membrane surfaces can significantly reduce the inherent antigenicity and immunogenicity of allogeneic cells [Citation[10-26]]. This immunocamouflage of cells is highly dependent on the molecular weight of the grafted mPEG and to a lesser extent on the chemical linkers used to target the protein residues. However, this does not mean that the linker chemistry is unimportant as some linkers, such as the tresylated mPEG (TmPEG), may exert significant toxicity when used to derivatize intact, viable, cells. While previous studies on red blood cells and more recently on pancreatic ß cell islets [Citation[16], Citation[26]] have demonstrated that pegylation of membranes can be accomplished without adversely affecting the structure, function or viability of the modified cell, an unexpected finding of this study was that allostimulation of pegylated splenocytes induced apoptosis. This finding suggests that it may be possible to induce an anergic state by the introduction of pegylated donor cells, thereby driving potential alloresponders into an apoptotic mode.
This work was supported by the Canadian Blood Services and the Canadian Institutes of Health Research.
REFERENCES
- Abuchowski, A., Van Es, T., Palczuk, N.C., Davis, F.F. (1977). J. Bio. Chem. 252: 3578–3581, [CSA]
- Abuchowski, A., McCoy, J.R., Palczuk, N.C., Van Es, T., Davis, F.F. (1977). J. Bio. Chem. 252: 3582–3586, [CSA]
- Jackson, C.-J., Charlton, J.L., Kuzminski, K., Lang, G.M., Sehon, A.H. (1987). Analytical Biochemistry 165: 114–27, [INFOTRIEVE], [CROSSREF], [CSA]
- Senior, J., Delgado, C., Fisher, D., Tilcock, C., Gregoriadis, G. (1991). Biochim. Biophys. Acta. 1062: 77–82, [INFOTRIEVE], [CSA]
- Chun, J.D., Lee, N., Kobayashi, R.H., Chafee, S., Hershfield, M.S., Stiehm, E.R. (1993). Ann Allergy 70: 462–466, [INFOTRIEVE], [CSA]
- Ettinger, L.J., Kurtzberg, J., Voute, P.A., Jurgens, H., Hapern, S.L. (1995). Cancer 75: 1176–1181, [INFOTRIEVE], [CSA]
- Keating, M.J., Holmes, R., Lerner, S., Ho, D.H. (1993). L-asparaginase and PEG asparaginase—past, present, and future. Leuk Lymphoma 10(Suppl.): 153–157, [INFOTRIEVE], [CSA]
- Maeda, H., Kimura, M., Sasaki, I., Hirose, Y., Konno, T. (1992). in Poly(Ethylene-Glycol) Chemistry—Biotechnical and Biomedical Applications, J.M. Harris, Ed., Plenum Press: New York, pp. 153–168.
- Nho, K., Zalipsky, S., Abuchowski, A., Davis, F.F. (1992). in Poly(Ethylene-Glycol) Chemistry—Biotechnical and Biomedical Applications, J.M. Harris, Ed., Plenum Press: New York, pp. 263–281.
- Scott, M.D., Murad, K.L., Eaton, J.W. (1997). in Advances in Blood Substitutes: Industrial Opportunities and Medical Challenges, R.M. Winslow, K.D. Vandegriff, M. Intaglietta, Eds., Birkhäuser: Boston, pp. 133–150.
- Scott, M.D., Murad, K.L., Koumpouras, F., Talbot, M., Eaton, J.W. (1997). Proc. Nat. Acad. Sci. USA 94: 7566–7571, [INFOTRIEVE], [CROSSREF], [CSA]
- Scott, M.D., Murad, K.L. (1998). Current Pharmaceutical Design 4, 423–438, [INFOTRIEVE], [CSA]
- Murad, K.L., Mahany, K.L., Kuypers, F.A., Brugnara, C., Eaton, J.W., Scott, M.D. (1999). Blood 93: 2121–2127, [INFOTRIEVE], [CSA]
- Murad, K.L., Gosselin, E.J., Eaton, J.W., Scott, M.D. (1999). Blood 94: 2135–2141, [INFOTRIEVE], [CSA]
- Scott, M.D., Bradley, A.J., Murad, K.L. (2000). Transfusion Medicine Reviews 14: 53–63, [INFOTRIEVE], [CROSSREF], [CSA]
- Chen, A.M., Scott, M.D. (2001). BioDrugs 15: 833–847, [INFOTRIEVE], [CROSSREF], [CSA]
- Bradley, A.J., Test, T.T., Murad, K.L., Mitsuyoshi, J., Scott, M.D. (2001). Transfusion 41: 1225–1233, [INFOTRIEVE], [CROSSREF], [CSA]
- Bradley, A.J., Murad, K.L., Regan, K.L., Scott, M.D. (2002). Biochimica et Biophysica Acta 1561: 147–58, [INFOTRIEVE], [CSA]
- Chen, A.M., Scott, M.D. (2003). Journal of Biomedical Materials Research 67A: 626–636, [CROSSREF], [CSA]
- Scott, M.D., Chen, A.M. (2004). Transfusion Clinique et Biologique 11: 40–46, [INFOTRIEVE], [CROSSREF], [CSA]
- Bradley, A.J., Scott, M.D. (2004). J. Chromatogr. B. 807: 163–168, [CROSSREF], [CSA]
- Armstrong, J.K., Meiselman, H.J., Fisher, T.C. (1997). Am. J. Hematol. 56: 26–28, [INFOTRIEVE], [CROSSREF], [CSA]
- Neu, B., Armstrong, J.K., Fisher, T.C., Meiselman, H.J. (2003). Biorheology 40: 477–87, [INFOTRIEVE], [CSA]
- Hortin, G.L., Lok, H.T., Huang, S.T. (1997). Artif Cells Blood Substit. Immobil. Biotechnol. 25: 487–491, [INFOTRIEVE], [CSA]
- Jeong, S.T., Byun, S.M. (1996). Art. Cells, Blood Subs., and Immob. Biotech. 24: 503–511, [CSA]
- Jang, J.Y., Lee, D.Y., Park, S.J., Byun, Y. (2004). Biomaterials 25: 3663–3669, [INFOTRIEVE], [CROSSREF], [CSA]
- Shaffer, C.B., Critchfield, F.H. (1947). Journal of the American Pharmaceutical Association 36: 152–57, [CSA]
- Smyth, H.E., Carpenter, C.P., Shaffer, C.B. (1947). Journal of the American Pharmaceutical Association 36: 157–60, [CSA]
- Molineux, G. (2003). Pharmacotherapy 23: 3S–8S, [INFOTRIEVE], [CROSSREF], [CSA]
- Greenwald, R.B., Choe, Y.H., McGuire, J., Conover, C.D. (2003). Adv. Drug. Deliv. Rev. 55: 217–50, [INFOTRIEVE], [CROSSREF], [CSA]
- Harris, J.M., Martin, N.E., Modi, M. (2001). Clin. Pharmacokinet. 40: 539–51, [INFOTRIEVE], [CROSSREF], [CSA]
- Noel, P.J., Boise, L.H., Green, J.M., Thompson, C.B. (1996). J. Immunol. 157: 636–642, [INFOTRIEVE], [CSA]
- O'Flynn, K., Krensky, A.M., Beverley, P.C., Burakoff, S.J., Linch, D.C. (1985). Nature 313: 21–27, [CSA]
- Anderson, S.J., Coleclough, C. (1993). J. Immunol. 151: 5123–5134, [INFOTRIEVE], [CSA]
- Kaufman, M., Andris, F., Leo, O. (1999). Immunol. 96: 3894–3899, [CSA]
- Noel, P.J., Boise, L.H., Green, J.M., Thompson, C.B. (1996). J. Immunol. 157: 636–642, [INFOTRIEVE], [CSA]
- Rudin, C.M., Van Dongen, J., Thompson, C.B. (1996). Curr. Opin. Hematol. 3: 35–40, [INFOTRIEVE], [CSA]
- Gudmundsdottir, H., Turka, L.A. (1999). J. Am. Soc. Nephro. 10: 1356–1365, [CSA]
- Mueller, D.L., Jenkins, M.K., Schwartz, R.H. (1989). Ann. Rev. Immunol. 7: 445, [CSA]
- Gross, J.A., Callas, E., Allison, J.P. (1992). J. of Immunol. 149: 380–388, [CSA]
- Hollander, G.A., Zuklys, S., Forster, E., Krenger, W. (1999). Transpl. Proc. 31: 25S–32S, [CROSSREF], [CSA]
- Francis, G.E., Fisher, D., Delgado, C., Malik, F., Gardiner, A., Neale, D. (1998). Int. J. Hematol. 68: 1–18, [INFOTRIEVE], [CROSSREF], [CSA]
- Davis, S., van der Merwe, A. (1996). Immunol. Today 17: 177–187, [INFOTRIEVE], [CROSSREF], [CSA]
- Collins, T.L., Kassner, P.D., Bierer, B.E., Burakoff, S.J. (1994). Curr. Opin. Immunol. 6: 385–393, [INFOTRIEVE], [CROSSREF], [CSA]
- Bierer, B.E., Hahn, W.C. (1993). Semin. Immunol. 5: 249–261, [INFOTRIEVE], [CROSSREF], [CSA]
- Blanchard, N., Hivroz, C. (2002). Biol. Cell. 94: 345–54, [INFOTRIEVE], [CROSSREF], [CSA]
- Hudrisier, D., Bongrand, P. (2002). FASEB J. 16: 477–86, [INFOTRIEVE], [CROSSREF], [CSA]
- Krogsgaard, M., Huppa, J.B., Purbhoo, M.A., Davis, M.M. (2003). Semin. Immunol. 15: 307–15, [INFOTRIEVE], [CROSSREF], [CSA]