
ABSTRACT
Extracellular vesicles (EV) are secreted signaling entities that enhance various pathological processes when released in response to cellular stresses. Respiratory exposures such as cigarette smoke and air pollution exert cellular stresses and are associated with an increased risk of several chronic diseases. The aim of this review was to examine the evidence that modifications in EV contribute to respiratory exposure-associated diseases. Publications were searched using PubMed and Google Scholar with the search terms (cigarette smoke OR tobacco smoke OR air pollution OR particulate matter) AND (extracellular vesicles OR exosomes OR microvesicles OR microparticles OR ectosomes). All original research articles were included and reviewed. Fifty articles were identified, most of which investigated the effect of respiratory exposures on EV release in vitro (25) and/or on circulating EV in human plasma (24). The majority of studies based their main observations on the relatively insensitive scatter-based flow cytometry of EV (29). EV induced by respiratory exposures were found to modulate inflammation (19), thrombosis (13), endothelial dysfunction (11), tissue remodeling (6), and angiogenesis (3). By influencing these processes, EV may play a key role in the development of cardiovascular diseases and chronic obstructive pulmonary disease and possibly lung cancer and allergic asthma. The current findings warrant additional research with improved methodologies to evaluate the contribution of respiratory exposure-induced EV to disease etiology, as well as their potential as biomarkers of exposure or risk and as novel targets for preventive or therapeutic strategies.
Health hazards of respiratory exposures
The adult human lungs inhale between 10,000 and 20,000 L of air daily, which interact with the vast 70 m2 surface of the lungs (Brochu, Ducré-Robitaille, and Brodeur Citation2006; Frohlich et al. Citation2016). While this makes lungs efficient for enabling gas exchange, it also makes pulmonary tissue highly vulnerable to airborne toxicants. Airborne toxicants may originate from personal exposures such as tobacco smoking (Lee et al. Citation2017), from occupational exposures including nanoparticles, asbestos, and volatile organic chemicals (Kermanizadeh et al. Citation2016; Paciência et al. Citation2016; Umbright et al. Citation2017), and from environmental exposures such as particulate matter (PM) from outdoor air pollution (Chen and Yang Citation2018). Chronic exposure to respiratory toxicants increases markedly the risk of developing lung disease, cardiovascular disease (CVD), and cancer (Bartal Citation2001; Kelly and Fussell Citation2015).
Chronic obstructive pulmonary disease (COPD)
The link between exposure to respiratory toxicants and disease development is most evident for COPD, a disorder that is characterized by chronic and incompletely reversible airflow limitation. While there is a genetic aspect to COPD risk, the major risk factor is smoking (Lamprecht et al. Citation2011; Salvi and Barnes Citation2009). In patients without significant smoking history, indoor air pollution from burning of biomass fuels, outdoor air pollution, and occupational exposures are postulated to form environmental triggers for disease development (Lamprecht et al. Citation2011; Salvi and Barnes Citation2009). Repeated exposure over long periods of time ultimately results in the development of COPD, which is hallmarked by airway inflammation (chronic bronchitis), fibrosis, and degradation of the lung tissue (emphysema) (Demedts et al. Citation2006). Once COPD is established, accelerated lung function decline may be brought about by acute exacerbations which are sudden deteriorations of COPD symptoms that are often elicited by respiratory infections (Beasley et al. Citation2012) or exposure to high levels of air pollution (Song et al. Citation2014).
Cardiovascular disease
The relative risk of morbidity and mortality of CVD is increased by exposure to cigarette smoke (CS) or high levels of air pollution, especially in individuals with obesity and type II diabetes (Dunbar, Gotsis, and Frishman Citation2013; Franklin, Brook, and Arden Pope Citation2015; Rigotti and Clair Citation2013; Weichenthal, Hoppin, and Reeves Citation2014). In particular, the risk for vascular diseases such as atherosclerosis and venous thromboembolism is elevated in smokers compared to nonsmokers (Severinsen et al. Citation2009). The most serious complications of atherosclerosis are myocardial infarction and ischemic stroke and the most serious complication of venous thrombosis is pulmonary embolism. These form the three leading causes of cardiovascular mortality worldwide (ISTH Steering Committee for World Thrombosis Day Citation2014; Lozano et al. Citation2012). While the mechanisms underlying respiratory exposure-induced CVD are not completely understood, inflammation, endothelial dysfunction, and hypercoagulability are involved (Csordas and Bernhard Citation2013; Piazza and Goldhaber Citation2010).
Asthma and occupational asthma
Asthma is characterized by reversible airflow obstruction that is initiated by chronic and oftentimes allergic bronchial wall inflammation and bronchial wall remodeling (Pałgan and Bartuzi Citation2015). CS and air pollution were found to enhance the risk for developing asthma (Ferrante et al. Citation2014) and are thought to trigger acute exacerbations in patients with established asthma (Guarnieri and Balmes Citation2014; Wang et al. Citation2015). Moreover, occupational respiratory exposures, such as toluene diisocyanate and possibly nanoparticles, are etiological factors for the development of occupational asthma (Sharma Citation2010; Tarlo and Lemiere Citation2014).
Lung cancer
Exposure to respiratory toxicants predisposes to developing lung cancer, which has been and continues to be the most common cause of cancer-related death for several decades (Malvezzi et al. Citation2015; Rahib et al. Citation2014). A large proportion of lung cancer deaths are attributed to CS and male as well as female smokers have a 25-fold higher risk of dying from lung cancer than never-smokers (Ezzati and Lopez Citation2003; Thun et al. Citation2013). Lung cancer develops when chronic exposure to respiratory toxicants produces cell damage and consequently a dysfunction of DNA repair mechanisms and cell cycle control. Respiratory toxicant-induced inflammation further aggravates this effect. Besides uncontrolled cell proliferation, both tissue remodeling and neoangiogenesis are involved in tumor growth (Folkman Citation1971; Pickup, Mouw, and Weaver Citation2014).
Extracellular vesicles
Importantly, there is a high rate of comorbidity between different diseases associated with exposure to respiratory toxicants (Decramer and Janssens Citation2013). Further, these diseases share chronic inflammation and oxidative stress as common hallmarks (Barua, Sharma, and Dileepan Citation2015; Crotty Alexander, Shin, and Hwang Citation2015; Kelly and Fussell Citation2015; Siasos et al. Citation2014; Valavanidis et al. Citation2013), suggesting some overlap in the biological processes underlying disease development. Extracellular vesicles (EV) are secreted signaling entities whose production is modulated by pro-inflammatory and oxidative stimuli in vitro. It is well known that EV are involved in the pathogeneses of CVD and cancer. They were also proposed to contribute to the development of chronic lung disease (Fujita et al. Citation2015b; Loyer et al. Citation2014; Minciacchi, Freeman, and Di Vizio Citation2015). The purpose of this review was to examine the relationship of EV secretion, composition, and function with airborne environmental risk factors for developing lung disease, CVD, and cancer.
EV are small membrane-enclosed vesicles that are postulated to be released by all cell types and are detected in a variety of human body fluids, including nasal lavage fluid (Lasser et al. Citation2011), bronchoalveolar lavage fluid (BALF) (Admyre et al. Citation2003), and blood (Wolf Citation1967). These vesicles carry a complex cargo of proteins, RNA, and lipids that makes them potent entities of intercellular communication (Van Niel, D’Angelo, and Raposo Citation2018). EV-borne proteins bind to cell surface receptors and trigger intracellular signal transduction (Camussi et al. Citation2011). In addition, EV fuse with the membrane of their target cells and thereby deliver functional RNA and proteins that modulate target cellular activity (Camussi et al. Citation2011). Finally, certain EV-bound proteins exert functions in the extracellular space, such as remodeling of the extracellular matrix (Lacroix and Dignat-George Citation2013).
Heterogeneity of Extracellular vesicles
Importantly, EV are not a homogenous population (Kowal et al. Citation2016). While EV nomenclature is still controversial (Gould and Raposo Citation2013), it is generally recognized that there are three major EV subtypes. The first subtype, exosomes, is formed by inward budding of the endosomal membrane to form intraluminal vesicles, and subsequently released to the extracellular space by fusion of multivesicular endosomes (MVE) with the plasma membrane (Abels and Breakefield Citation2016). The second subtype, commonly referred to as microvesicles (or microparticles or ectosomes), arises by outward budding of the plasma membrane (Abels and Breakefield Citation2016). The third EV type is apoptotic bodies, large membrane vesicles that are formed by apoptotic membrane blebbing and that contain DNA fragments and cellular organelles (Thery, Ostrowski, and Segura Citation2009). This review focuses on the first two EV subtypes, exosomes and microvesicles, whose key properties are summarized in . Apoptotic bodies are not considered because these are formed only during apoptotic cell fragmentation.
Table 1. Key properties of exosomes and microvesicles.
Exosomes
Due to their endosomal origin, exosomes (50–150 nm) are smaller than microvesicles (100–1000 nm). Further, exosomes are enriched in members of the endosomal sorting complexes required for transport (ESCRT, e.g. Alix, TSG101) and in tetraspanins such as CD9, CD63, and CD81. As exosomes are generally smaller than the resolution limit of conventional optical techniques (light microscopy, flow cytometry scatter) (Van Der Pol et al. Citation2010), highly dedicated technology is required for their detection, including electron microscopy, nanoparticle tracking analysis (NTA) (Sokolova et al. Citation2011), tunable resistive pulse sensing (TRPS) (Maas, De Vrij, and Broekman Citation2014), and fluorescence-triggered high-resolution flow cytometry (Nolte-’T Hoen et al. Citation2012). Alternatively, exosomes may be characterized by bulk analysis techniques, such as Western blotting or bead-based flow cytometry, a technique in which EV are linked to large beads and subsequently stained for exosome marker proteins (Suarez et al. Citation2017; Volgers et al. Citation2017). The most common method for isolation of exosomes is ultracentrifugation at 100,000 × g (Gardiner et al. Citation2016).
Microvesicles
While all the techniques that are utilized for exosome characterization may also be applied to microvesicles, this is performed less frequently because microvesicles larger than 300 nm may be more easily detected employing conventional scatter-triggered flow cytometry (Van Der Pol et al. Citation2010). Fluorescent stains commonly used for flow cytometric microvesicle detection include annexin V for the detection of the negatively charged phospholipid phosphatidylserine (PS), as well as antibodies against selectins, integrins, and cell adhesion molecules, which may provide clues regarding the cell of origin (summarized in ). Microvesicles are most commonly isolated by centrifugation at 10,000 × g.
Table 2. Overview of surface molecules that are commonly used to identify the cellular origin of microvesicles in complex biological fluids such as plasma.
Methodology
Publications published before September 2017 were searched using PubMed and Google Scholar. The search terms were (cigarette smoke OR tobacco smoke OR air pollution OR particulate matter) AND (extracellular vesicles OR exosomes OR microvesicles OR microparticles OR ectosomes). Abstracts and keywords were screened to assess whether effects of CS, CS components, PM, or other air pollutants on EV were reported. All original research articles fulfilling these criteria were included. Moreover, relevant references referred to in these publications were considered.
Systematic evaluation of the published literature
EV research is a relatively novel and rapidly developing field. As one begins to understand that the results of some early EV studies may be attributed to methodological artifacts rather than reflecting true biological properties of EV, the following criteria were examined: (1) experimental approaches and techniques used in identified studies and (2) the number of studies suggesting an involvement of respiratory exposure-induced EV in certain biological processes as an indicator for the strength of evidence. In total, 50 original research articles from 36 independent research groups were identified that assessed effects of respiratory exposures on EV. Of these, 30 studies focused on CS or CS components and 20 on PM or other air pollutants. Detailed information on each of the 50 studies is given in the supplementary material (Table S1 and Table S2).
The two oldest articles dated from 2004. Starting from the publication of the third article in 2008, there was a consistent increase in the number of articles published per year, peaking at 11 articles in 2016 (). This highlights a growing interest in the topic over the past decade. The majority of the studies were performed in vitro (26), followed by human (25) and animal studies (6) with several articles using a combination of different study types (). Congruently, the most common fluids in which EV were studied were cell culture media (22; with varying percentages of untreated or EV-depleted fetal calf serum; FCS), followed by platelet-depleted plasma (13; most commonly citrated, followed by ethylenediaminetetraacetic acid and heparinized plasma) and BALF (3; ).
Figure 1. Results of the literature search. The graphs show the number of publications (A) per year since the first publication in 2004 (B) that report on in vitro, human and animal experiments, (C) according to the biological material in which EV were studied, (D) according to the EV nomenclature used by the authors, (E) that used a specific EV characterization technique, (F) that used a specific EV isolation technique, (G) that investigated and confirmed the involvement of respiratory exposure-induced EV in the specified biological processes, and (H) according to the pathology for which the authors considered the described changes to be relevant.
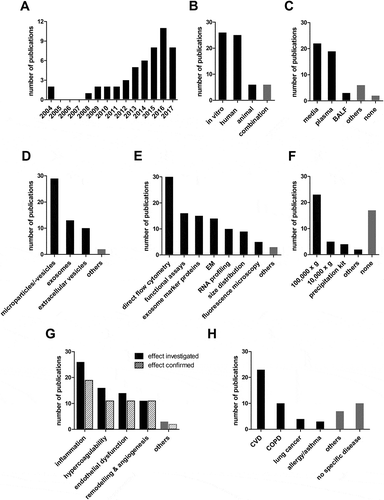
Most investigators referred to the EV in their studies as microparticles or microvesicles (29), followed by exosomes (13) and extracellular vesicles (10) (). In line with this, the most commonly applied EV characterization method was direct flow cytometry of microvesicles (30), followed by functional analyses (16), bulk detection of exosome marker proteins (15), and electron microscopy (14; ). Strikingly, the majority of the studies (27 studies, i.e., 54%) only applied a single EV characterization technique, whereas the current consensus in the field is that multiple complementary techniques need to be combined when studying EV (Lotvall et al. Citation2014). The by far most commonly employed EV isolation technique was ultracentrifugation at approximately 100,000 × g (23; ). This was often applied in experiments that referred to EV as microparticles, although it is well established that 100,000 × g centrifugation also sediments exosomes. The second most common isolation technique was centrifugation at approximately 10,000 × g (5; ), which is considered to preferentially sediment microvesicles, with a smaller proportion of exosomes than 100,000 × g centrifugation. A number of investigations applied commercial EV precipitation kits (4; ), which coprecipitate soluble, non-EV bound molecules (Taylor and Shah Citation2015). All studies employed crude centrifugation pellets rather than performing additional EV purification by density-gradient centrifugation, which is required to deplete non-vesicular high molecular weight complexes. One third of studies (17; ) did not isolate EV at all, but only measured their concentration by direct flow cytometry or observed vesicle budding at the plasma membrane by fluorescence microscopy.
Finally, the number of studies that investigated the involvement of respiratory exposure-induced EV in certain biological pathways was determined and the quantity of these studies that actually provided data in favor of such an involvement was noted. EV were proposed to be involved in biological processes such as inflammation (26/19), hypercoagulability (16/11), endothelial dysfunction (14/11), and tissue remodeling or angiogenesis (11/11; ). These biological processes were projected to be of relevance for the development of CVD (23), COPD (10), cancer (4), or allergic asthma (3; ). Taken together, a substantial amount of findings suggests a role for respiratory exposure-induced EV in the development of chronic disease.
While the available evidence is valuable, part of it was obtained using outdated and/or suboptimal methods that do not allow for attributing biological functions unambiguously to EV rather than to co-isolated contaminants. Moreover, because of non-stringent combinations of detection, isolation, and characterization methods, it is currently unclear whether certain biological effects of respiratory exposure-induced EV are exerted by microvesicles, exosomes, or both. Therefore, the generic term “EV” will be used in the following sections, in which evidence will be presented comprehensively for involvement of respiratory exposure-induced EV in biological processes that promote the development of chronic diseases.
Inflammation and immunomodulation
Inflammation is a universal attribute of respiratory exposure-associated diseases (Lee and Lawrence Citation2018). In total, 26 original research articles were identified that investigated whether and how respiratory exposure-induced EV modulated inflammatory responses.
Interference with anti-inflammatory signaling
In healthy lungs, alveolar macrophages and pulmonary epithelium act in concert to rapidly clear inhaled substances and maintain an anti-inflammatory state. EV from alveolar macrophages contribute to this anti-inflammatory state by delivering suppressor of cytokine signaling (SOCS) 1 and 3 to epithelial cells (Bourdonnay et al. Citation2015). Importantly, CS-exposed mice and human smokers exhibited decreased SOCS concentrations in BALF compared to nonsmoking controls, suggesting a loss of EV-dependent anti-inflammatory state in smokers (Bourdonnay et al. Citation2015). In addition, the anti-inflammatory effect of alveolar macrophage-derived EV depends upon their internalization by target cells, which is inhibited by the presence of CS extract (CSE) (Schneider et al. Citation2017).
Promotion of pro-inflammatory signaling
CS not only interferes with EV-dependent anti-inflammatory signaling, but also actively promotes EV-dependent pro-inflammatory signaling. Chen et al. (Citation2016) demonstrated that EV from CSE-exposed macrophages contain the pro-inflammatory alarmin high-mobility group box 1. Further, EV from CSE-exposed macrophages and epithelial cells, as well as EV isolated from BALF of smokers trigger release of interleukin-(IL-) 6, IL-8, and monocyte chemoattractant protein 1 (MCP1) by naïve pulmonary epithelial cells (Cordazzo et al. Citation2014; Héliot et al. Citation2017; Moon et al. Citation2014). EV also promote upregulation of intercellular adhesion molecule 1 (Cordazzo et al. Citation2014) and thus may contribute to inflammation and immune cell recruitment toward EV target cells.
Interference with efferocytosis
CS as well as PM exposure increases the release of phosphatidylserine (PS)+ EV from different cell types in vitro and the concentrations of circulating PS+ EV in vivo (Emmerechts et al. Citation2012b; Li et al. Citation2010; Neri et al. Citation2016; Serban et al. Citation2016). PS+ EV may contribute to inflammation, as Serban et al. (Citation2016) reported that CSE-induced PS+ EV from endothelial cells prevent efferocytosis of apoptotic cells by PS-dependent blocking of the phagocyte receptors for apoptotic cell engulfment. Importantly, deficient efferocytosis of apoptotic cells is postulated to contribute to sustained lung inflammation in COPD (McCubbrey and Curtis Citation2013) and to progression and destabilization of atherosclerotic plaques (Foks et al. Citation2016; Schrijvers et al. Citation2005).
Indirect evidence for involvement in inflammation
Exposure to CSE, PM, and other air pollutants was found to alter transcriptomic (microRNA) and, in one study, proteomic EV composition (Héliot et al. Citation2017; Lim et al. Citation2016; Pavanello et al. Citation2016; Pergoli et al. Citation2017; Rodosthenous et al. Citation2016; Sheller et al. Citation2016). Based on biological pathway analysis, these compositional changes were predicted to promote inflammation, although this was not experimentally confirmed. In one observational human study, PM exposure of young healthy individuals was associated with parallel increases in circulating endothelial cell-derived EV and various immune cells as well as pro-inflammatory molecules (Pope et al. Citation2016), yet a causal link between EV and inflammation was not established.
Allergic inflammation
Finally, respiratory exposure-induced EV may contribute to allergic inflammation and development of (occupational) asthma. In ovalbumin-sensitized mice, intratracheally instilled magnetic iron oxide nanoparticles triggered release of EV that promote dendritic cell maturation and T cell activation (Zhu et al. Citation2012). Brostrom et al. (Citation2015) found that treatment of pulmonary epithelial cells with the occupational respiratory toxicant toluene diisocyanate initiated release of autotaxin via EV, an enzyme that has been implicated in allergic airway inflammation.
Conflicting data
While the majority of published studies suggest a role of respiratory exposure-induced EV in inflammation, several negative findings were also noted. Benedikter et al. (Citation2017) found that EV from CSE-stimulated pulmonary epithelial cells did not significantly modify IL-8 secretion by naïve cells. The same CSE-EV were less potent inducers of monocyte adhesion to endothelial cells than EV secreted by unexposed cells (Benedikter et al. Citation2017), questioning a role of CSE-induced EV in immune cell recruitment. Several human and animal studies that assessed in vivo effects of respiratory exposures were not able to link changes in circulating EV to alterations in inflammatory markers (Emmerechts et al. Citation2012a, Citation2012b; Levanen et al. Citation2013; Mobarrez et al. Citation2014). Several investigators even found that smokers displayed decreased concentrations of circulating PS+ EV compared to nonsmokers (Badrnya, Baumgartner, and Assinger Citation2014; Enjeti et al. Citation2017; Grant et al. Citation2011). In part, the failure to detect pro-inflammatory changes in EV in response to respiratory toxicants in vivo may be due to (1) use of relatively mild and acute exposures (Levanen et al. Citation2013; Mobarrez et al. Citation2014) or (2) the fact that current methods are not sensitive enough to detect subtle changes in EV and inflammatory mediators in study populations without clinical disease.
The studies that report either induction (Cordazzo et al. Citation2014; Héliot et al. Citation2017; Moon et al. Citation2014) or no induction (Benedikter et al. Citation2017) of pro-inflammatory cytokines by CS(E)-induced EV in vitro have all employed similar exposure times, EV isolation techniques, and EV target cells (bronchial epithelial cells). Yet, the divergent results might be explained by the quantity of EV used in functional assays as Héliot et al. (Citation2017) obtained their positive results with 100-fold more EV than the quantity that resulted in negative results (Benedikter et al. Citation2017). The utilization of untreated versus EV-depleted FCS in the media for target cell stimulation may also be a factor. More transparent reporting of methodological details in future studies will improve comparability and allow identifying factors responsible for divergent results.
Summary
Taken together, the majority of data suggest that exposure to CSE or PM shifts the functionality of EV secreted by mononuclear, epithelial, and endothelial cells from an anti-inflammatory toward a pro-inflammatory phenotype. Respiratory exposure-induced EV may promote monocyte and neutrophil recruitment toward EV target cells, activate pro-inflammatory signaling, and impair clearance of apoptotic cells. Such pro-inflammatory effects of respiratory exposure-induced EV are of potential relevance for atherogenesis as well as development of COPD. Some of the described pro-inflammatory effects may also have implications for allergic asthma and lung carcinogenesis. Further research is required to validate and extend current findings as well as establish whether biological processes that were identified in vitro play a role in respiratory exposure-induced disease pathogenesis in vivo.
Hypercoagulability
Hypercoagulability is an abnormal tendency to develop blood clots and is postulated to contribute significantly to cardiovascular risk in smokers. Some EV express tissue factor (TF), which is able to trigger thrombin generation via the extrinsic coagulation pathway (Van Es et al. Citation2015). The presence of PS and other negatively charged phospholipids in the membrane of EV enables assembly of coagulation factors on the EV surface and thus further promotes thrombin generation. In total, 16 studies were identified that investigated the influence of respiratory toxicants on TF+ EV, PS+ EV, or other changes in EV related to hypercoagulability.
In vitro evidence
Both short-term (15 min) and long-term (20 h) exposure of monocytes and macrophages to CSE result in elevated release of PS+ and TF+ EV, which is associated with increased procoagulant activity (Cordazzo et al. Citation2014; Li et al. Citation2010). Similarly, endothelial cells respond to CSE or PM exposure by releasing more PS+ and TF+ procoagulant EV (Neri et al. Citation2016; Serban et al. Citation2016). However, evidence for involvement of TF+ and PS+ EV in respiratory exposure-induced hypercoagulability in vivo is less clear.
In vivo evidence for TF+ Extracellular vesicles
A number of studies by one group assessed the influence of PM or ozone (O3) exposure on TF+ EV in either healthy individuals (Frampton et al. Citation2015) or type II diabetics (Frampton et al. Citation2012; Stewart et al. Citation2010). Among these, only an observational study detected a significant positive association between subacute exposure (2 and 5 days prior to sampling; Frampton et al. Citation2012) and circulating TF+ EV, whereas the other, interventional studies with a single acute exposure showed at most a trend for a positive association (Frampton et al. Citation2015; Stewart et al. Citation2010). Badrnya, Baumgartner, and Assinger (Citation2014) determined the concentration of TF+ EV in plasma of young healthy smokers and nonsmokers and found no significant difference. In another study with a broader age range, Enjeti et al. (Citation2017) reported that circulating TF+ EV were decreased in smokers compared to nonsmokers using univariate analysis, whereas there was a significant positive correlation for smoking status and TF+ EV after correcting for age. This highlights the importance of correcting for confounding factors in translational studies.
The difficulty to detect increases in TF+ EV in response to in vivo respiratory exposures may partly be explained by the small sample size and lack of correction for confounding factors in most human studies. Another problem when studying TF+ EV is the low sensitivity of antibody-based TF detection. TF concentrations required for procoagulant activity are considerably lower than the current detection limit of ELISA-based techniques (Van Es et al. Citation2015). Conventional flow cytometry does not enable detection of small EV (<300 nm) which may also express TF. Further, fluorescence intensity may not be sufficient to detect all TF+ EV >300 nm as only a limited number of TF molecules may be present within an EV due to its small size (Van Es et al. Citation2015). Thus, more sensitive methods for the detection of procoagulant EV and larger or more controlled human studies are required to establish the importance of CS- and air pollution-induced circulating TF+ EV for hypercoagulability in vivo.
In vivo evidence for PS+ Extracellular vesicles
Only one study was identified that assessed the effect of PM exposure on PS+ EV in vivo. In this human observational cohort, short-term PM exposure (hours to days) was associated with reduced, and long-term exposure (3 months to 1 year) with elevated concentrations of circulating PS+ EV (Emmerechts et al. Citation2012b). For CS exposure, while all in vitro studies reported increased levels of PS+ EV (Cordazzo et al. Citation2014; Li et al. Citation2010; Serban et al. Citation2016), all in vivo studies reported decreased concentrations in the blood circulation of smokers compared to nonsmokers (Badrnya, Baumgartner, and Assinger Citation2014; Enjeti et al. Citation2017; Grant et al. Citation2011). Whether cells indeed mount a different EV response to CS in vitro than in their physiological environment or whether the divergent findings are explained by other factors remains unclear. In vivo, PS+ EV may remain in the lung lumen rather than entering the circulation, indicating that these constituents would not be detected in plasma samples. Alternatively, PS+ EV may be rapidly phagocytosed (Serban et al. Citation2016) or absorbed by target tissues. Another potential explanation is that all in vitro studies investigated the effect of a single acute CS exposure, whereas exposure in human smokers is repeated over a long time, and adaptation may take place in healthy individuals. To elucidate whether acute human CS exposure results in increased circulating PS+ EV, PS+ EV should be analyzed in acute exposure studies such as those performed by Heiss et al. (Citation2008) and Mobarrez et al. (Citation2014). Finally, slower centrifugation speeds were used for EV isolation in the in vivo studies (10,000 × g) than in the in vitro experiments (approximately 100,000 × g), which may have resulted in analysis of different EV subpopulations. Additional research is required to establish whether PS+ EV can contribute to hypercoagulability in response to CS exposure in vivo.
Other changes in Extracellular vesicles related to hypercoagulability
Besides surface-exposed TF and PS, other EV parameters may reflect and/or promote hypercoagulability. Several investigators found that respiratory exposures are associated with increased circulating platelet-derived EV (Emmerechts et al. Citation2012a; Mobarrez et al. Citation2014; Pergoli et al. Citation2017; Xu et al. Citation2013), which are thought to reflect platelet activation and promote coagulation. Exposure to occupational and environmental PM was reported to produce changes in circulating EV microRNA cargo that were predicted to promote coagulation based upon bioinformatics analyses (Pavanello et al. Citation2016; Pergoli et al. Citation2017). Finally, respiratory exposure-induced alterations in circulating EV are associated with elevations of more classical markers for hypercoagulability such as increased coagulation factors, a greater number of platelet-leukocyte aggregates, reduced fibrinolytic factors, and enhanced thrombin generation (Emmerechts et al. Citation2012b; Pergoli et al. Citation2017; Pope et al. Citation2016).
Summary
Taken together, in vitro investigations suggest that several cell types secrete EV with PS- and TF-dependent procoagulant activity when exposed to CS or PM. In addition to PS and TF, EV also carry other cargo molecules that might promote hypercoagulability. Additional translational studies of circulating EV and EV from BALF are required to establish the importance of procoagulant EV for respiratory exposure-associated CVD and lung disease.
Endothelial dysfunction
While the healthy endothelium maintains vascular tone and blood fluidity, endothelial dysfunction is highlighted by impaired endothelium-dependent vasodilation and a shift of the endothelial secretome from anti-inflammatory and anticoagulant toward more pro-inflammatory and procoagulant factors. CS and PM exposure are both well known to produce endothelial dysfunction (Bourdrel et al. Citation2017; Messner and Bernhard Citation2014). Endothelial dysfunction is one of the earliest vascular changes in atherosclerosis and proposed to actively promote atherogenesis (Gimbrone and García-Cardeña Citation2016). Emphysema formation in COPD patients is associated with endothelial cell stress and cell death in the pulmonary capillaries, leading to impaired vessel function (McCubbrey and Curtis Citation2013; Takahashi et al. Citation2012). Endothelial damage may actively contribute to the pathogenesis of COPD (Barr Citation2011). Hence, it may mechanistically explain the high rate of comorbidity between COPD and CVD (Clarenbach et al. Citation2013). Increased concentrations of circulating endothelial-derived EV are thought to both result from and promote endothelial dysfunction (Sabatier et al. Citation2009). As such, EV are considered to act as surrogate markers for endothelial stress and may also reflect endothelial inflammation, coagulability, and vascular tone (Burger and Touyz Citation2012). A total of 14 studies were identified that assessed the influence of respiratory toxicants on circulating endothelial-derived EV in vivo or on EV secretion by endothelial cells in vitro.
Induction of endothelial Extracellular vesicles by respiratory exposures
A relatively large body of evidence supports that respiratory exposures induce endothelial EV release. CS exposure of mice and rats enhanced circulating endothelial-derived EV levels (Liu et al. Citation2014; Serban et al. Citation2016). Transient exposure of healthy humans to second-hand or mainstream CS, and chronic smoking are associated with elevated numbers of circulating endothelial-derived EV (Gordon et al. Citation2011; Heiss et al. Citation2008; Mobarrez et al. Citation2014). Smoking cessation reverts the rise in endothelial-derived EV, but only in individuals in whom lung damage has not yet been established (Strulovici-Barel et al. Citation2016). PM exposure was found to increase EV release by cultured endothelial cells (Neri et al. Citation2016). Observational studies in healthy volunteers provided evidence that high levels of outdoor air pollution or PM occurred concomitantly with elevated concentrations of endothelial-derived EV, especially in subjects that were overweight (Bonzini et al. Citation2017; Pope et al. Citation2016).
Association of endothelial Extracellular vesicles with biomarkers of damage
Increases in circulating endothelial-derived EV in response to human CS exposure were correlated with other markers of endothelial dysfunction, such as impaired flow mediated dilation and elevated circulating endothelial progenitor cells (Heiss et al. Citation2008; Mobarrez et al. Citation2014). Some investigators proposed a link between elevated circulating endothelial-derived EV and endothelial apoptosis, especially for CD62E+ endothelial EV (Gordon et al. Citation2011; Liu et al. Citation2014; Strulovici-Barel et al. Citation2016). It was also shown that endothelial-derived EV are associated with emphysematous lung damage and diminished lung function in smokers (Gordon et al. Citation2011; Strulovici-Barel et al. Citation2016).
Non-endothelial Extracellular vesicles in endothelial dysfunction
Finally, there is evidence that EV of non-endothelial origin contribute to endothelial dysfunction in response to respiratory exposures. Xu et al. (Citation2013) showed that mice exposed to carbon monoxide, an important component of CS and air pollution, exhibited increased endothelial permeability in the brain and skeletal muscle, which was mediated by neutrophil-derived EV.
Summary
Taken together, data suggest that exposure to respiratory toxicants is associated with elevated concentrations of circulating endothelial-derived EV in animals and humans, which was determined by direct flow cytometry for various endothelial marker proteins. While endothelial-derived EV were proposed as mediators of and surrogate markers for endothelial dysfunction, no functional characterization has yet been performed for endothelial-derived EV secreted in response to respiratory exposures.
Tissue remodeling and angiogenesis
Respiratory exposure-associated diseases are hallmarked by modifications of tissue structure. Epithelial-to-mesenchymal transition and remodeling of the extracellular matrix are important processes in COPD and lung cancer development (Houghton Citation2013). Thickening of the bronchial subepithelial layer and fibroblast-to-myofibroblast differentiation are also involved in COPD and asthma pathogenesis (Al-Muhsen, Johnson, and Hamid Citation2011; Pałgan and Bartuzi Citation2015; Salazar and Herrera Citation2011). Finally, extracellular matrix degradation may contribute to (1) emphysema formation (Salazar and Herrera Citation2011) and (2) destabilization of atherosclerotic plaques, enhancing risk for cardiovascular complications (Messner and Bernhard Citation2014). Another biological process that is related to and dependent upon tissue remodeling is neovascularization. In lung cancer, tumor-associated angiogenesis enables oxygen and nutrient supply to sustain tumor growth (Folkman Citation1971). In atherosclerosis, the formation of leaky intraplaque neovessels contributes to plaque destabilization (Michel et al. Citation2014). Finally, in asthma, subepithelial angiogenesis promotes thickening of the subepithelial layer and facilitates infiltration of immune cells into the bronchial wall (Pałgan and Bartuzi Citation2015). Eleven studies were identified that investigated whether EV secreted in response to respiratory exposures promote tissue degradation, remodeling, or angiogenesis. The evidence is largely derived from in vitro studies using CSE exposure.
Extracellular vesicles-associated proteolytic enzymes
Several investigators demonstrated that cell exposure to CSE produced an increased release of proteolytic enzymes in an EV-associated form. Li et al. (2013) noted that CSE exposure of monocytes and monocyte-derived macrophages induced a threefold rise in EV-associated matrix metalloproteinase (MMP) 14-dependent gelatinolytic and collagenolytic activity. Folkesson et al. (Citation2015) found that, when exposed to CSE, neutrophil-like HL60 cells secreted EV-associated and proteolytically active a disintegrin and metalloproteinase (ADAM)10 and ADAM17. Finally, EV released by lung epithelial cells in response to CSE promoted production of MMP1 by EV target cells (Moon et al. Citation2014). Xu and Deng (Citation2004) demonstrated that CS-induced secretion of EV-bound proteolytic calpains was associated with increased migration and invasiveness of the EV-secreting lung carcinoma cells.
Fibroblast-to-myofibroblast differentiation
A proteinase-independent contribution of CSE-induced EV to tissue remodeling was also proposed. Fujita et al. (Citation2015a) found that lung epithelial cells stimulated with CSE secrete EV which are enriched in microRNA-210. EV-dependent delivery of the microRNA to pulmonary fibroblasts resulted in inhibition of autophagy and subsequent differentiation of the fibroblasts into myofibroblasts.
Respiratory exposure-induced Extracellular vesicles and angiogenesis
Concerning angiogenesis, enhanced release of proteolytic enzymes in an EV-associated form may promote endothelial cell migration by degrading the extracellular matrix. Further, EV from CSE-treated lung epithelial cells promote secretion of the proangiogenic vascular endothelial growth factor (VEGF) by EV target cells (Liu et al. Citation2014; Moon et al. Citation2014). Liu et al. (Citation2014) demonstrated that VEGF induction was mediated by EV-associated microRNA-21 and was able to trigger endothelial tube formation.
Summary
Taken together, CSE exposure results in increased release of proteolytic enzymes by various cell types in either an EV-associated or EV-dependent manner. These EV-bound or EV-induced proteolytic enzymes may promote tissue remodeling by degrading extracellular matrix constituents. CSE-induced EV may further promote tissue remodeling by initiating fibroblast to myofibroblast differentiation and endothelial tube formation. Yet, no apparent studies were identified that investigated the effect of air pollution on EV-dependent tissue remodeling. Further, there is currently a lack of translational studies on the implications of respiratory exposure-induced proteolytic EV for tissue remodeling in vivo.
Synopsis and identification of knowledge gaps
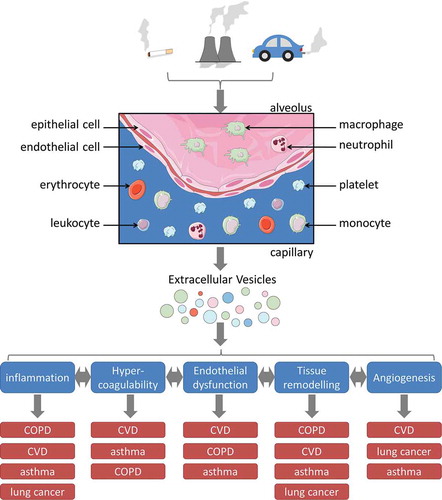
Respiratory exposures such as CS and PM promote the release of EV from various cell types including lung epithelial cells, monocytes, and endothelial cells. Further, respiratory exposures produce alterations in EV composition, which promote inflammation, hypercoagulability, endothelial dysfunction, tissue remodeling, and neovascularization. While most of the cited studies focused on relevance of respiratory toxicant-induced EV for one specific respiratory exposure-associated disorder, this review provides an integrative view of the findings from different fields. Importantly, respiratory exposure-induced EV may be involved in the pathogeneses of COPD, CVD, asthma, and lung cancer by modulating each of the above-mentioned biological processes (illustrated in ).
Figure 2. Schematic representation of how respiratory exposure-induced EV may contribute to the pathogenesis of chronic diseases. Respiratory toxicants arise from CS, as well as occupational or environmental sources. Upon inhalation, they come in contact with several cell types of the lungs, including epithelial cells, endothelial cells, alveolar macrophages, monocytes, and circulating blood cells. Exposure to respiratory toxicants causes increased release and altered composition of EV from different cellular sources. The respiratory exposure-induced EV may either remain in the lung lumen or are disseminated via the blood circulation. Locally at the site of exposure as well as systemically, they may promote inflammation, hypercoagulability, endothelial dysfunction, tissue remodeling, and angiogenesis, all of which are interrelated and can further enhance each other. By promoting these biological processes, EV may contribute to the mechanistic link between respiratory exposures and the pathogenesis of respiratory exposure-associated diseases such as COPD, CVD, asthma, and lung cancer. For each biological process, the diseases are sorted according to the strength of evidence for involvement of the respective process in their pathogenesis. This figure contains elements from Servier Medical Art.
Technical requirements for future studies
Although the presented findings are highly intriguing, it needs to be kept in mind that part of the studies were performed with outdated and/or suboptimal methodology for EV characterization and need to be validated using more up-to-date methodology according to recent recommendations (Coumans et al. Citation2017; Lotvall et al. Citation2014). Similar effects to the ones reviewed here for EV are also mediated directly by respiratory exposures or by non-EV associated molecules such as pro-inflammatory cytokines, coagulation factors, and proteolytic enzymes. Therefore, an important challenge for future studies will be establishing the relative importance of EV and free secreted molecules, which will require extensive side-by-side comparisons of isolated EV and EV-depleted biological fluids.
Requirement for translational studies
Another aspect that should be in the focus of future studies is strengthening of the translational link between functional in vitro experiments and observational human studies. It is striking that the literature largely consists of mechanistic in vitro studies that have not been verified in vivo on the one hand and observational human studies without strong mechanistic foundation on the other hand. The integrative view that this review provides of studies from different fields may help to determine relevant biological readouts for future translational studies, which need to focus on the functional characterization of circulating EV as well as EV from the lung lumen.
Circulating Extracellular vesicles
There is ample evidence showing that toxic respiratory exposures enhance the concentrations of circulating EV. This has been most thoroughly investigated for EV of endothelial origin, but also leukocyte-, erythrocyte-, and platelet-derived EV appear to be affected. However, functional analyses of circulating EV lack almost completely. Moreover, no clear link has yet been established between concentration and composition of circulating EV in healthy exposed individuals and the risk of developing respiratory exposure-associated diseases. It should also be explored whether circulating EV causally contribute to the high rate of comorbidity between COPD, lung cancer, and CVD. With improved techniques for detection, isolation, and characterization, circulating EV may develop into valuable biomarkers for cardiovascular risk in healthy individuals and patients with other chronic inflammatory disorders.
Extracellular vesicles from the lung lumen
Translational studies investigating the contribution of respiratory exposure-induced EV to the pathogenesis of pulmonary diseases also need to focus on EV in the lung lumen, for instance by isolating them from BALF or sputum. In this context, TF+ EV are of particular interest as they may promote fibrin deposition in alveoli and activation of pro-inflammatory signaling via protease activated receptors (Bastarache et al. Citation2009; Witkowski, Landmesser, and Rauch Citation2016). In the lung, EV may be derived from other cellular sources than in plasma, with a larger proportion of EV from epithelial cells and alveolar macrophages. Therefore, these EV may have different functions than circulating EV.
Conclusions
Two lines of evidence point toward an involvement of EV in respiratory exposure-associated diseases, such as CVD, COPD, and lung cancer. First, in vitro studies demonstrated that exposures to respiratory toxicants such as CS and PM enhance EV release and modify EV composition and functions, promoting inflammation, tissue remodeling, and hypercoagulability. Second, observational human studies reported that circulating EV, particularly of endothelial origin, were elevated in individuals exposed to CS or PM. Yet, functional implications of these circulating EV have not been evaluated. Translational studies are required to bridge these findings and establish whether EV constitute important etiological factors in respiratory exposure-induced disease.
Declaration of interest
The authors declare that they have no actual or potential competing financial or other interests.
supplementary_material.pdf
Download PDF (892.8 KB)Supplementary material
Supplemental data for this article can be accessed here.
Additional information
Funding
References
- Abels, E. R., and X. O. Breakefield. 2016. Introduction to extracellular vesicles: Biogenesis, RNA cargo selection, content, release, and uptake. Cellular and Molecular Neurobiology 36:301–12. doi:10.1007/s10571-016-0366-z.
- Admyre, C., J. Grunewald, J. Thyberg, S. Gripenback, G. Tornling, A. Eklund, A. Scheynius, and S. Gabrielsson. 2003. Exosomes with major histocompatibility complex class II and co-stimulatory molecules are present in human BAL fluid. The European Respiratory Journal 22:578–83. doi:10.1183/09031936.03.00041703.
- Al-Muhsen, S., J. R. Johnson, and Q. Hamid. 2011. Remodeling in asthma. Journal of Allergy and Clinical Immunology 128:451–462; quiz 463–454. doi:10.1016/j.jaci.2011.04.047.
- Badrnya, S., R. Baumgartner, and A. Assinger. 2014. Smoking alters circulating plasma microvesicle pattern and microrna signatures. Thromb Haemost 112:128–36. doi:10.1160/TH13-11-0977.
- Barr, R. G. 2011. The epidemiology of vascular dysfunction relating to chronic obstructive pulmonary disease and emphysema. Proceedings of the American Thoracic Society 8:522–27. doi:10.1513/pats.201101-008MW.
- Bartal, M. 2001. Health effects of tobacco use and exposure. Monaldi Archives for Chest Disease 56:545–54.
- Barua, R. S., M. Sharma, and K. N. Dileepan. 2015. Cigarette smoke amplifies inflammatory response and atherosclerosis progression through activation of the H1R-TLR2/4-COX2 axis. Frontiers in Immunology 6:572. doi:10.3389/fimmu.2015.00572.
- Bastarache, J. A., R. D. Fremont, J. A. Kropski, F. R. Bossert, and L. B. Ware. 2009. Procoagulant alveolar microparticles in the lungs of patients with acute respiratory distress syndrome. American Journal of Physiology-Lung Cellular and Molecular Physiology 297:L1035–1041. doi:10.1152/ajplung.00214.2009.
- Beasley, V., P. V. Joshi, A. Singanayagam, P. L. Molyneaux, S. L. Johnston, and P. Mallia. 2012. Lung microbiology and exacerbations in COPD. International Journal of Chronic Obstructive Pulmonary Disease 7:555–69. doi:10.2147/COPD.S28286.
- Benedikter, B. J., F. G. Bouwman, T. Vajen, A. C. A. Heinzmann, G. Grauls, E. C. Mariman, E. F. M. Wouters, P. H. Savelkoul, C. Lopez-Iglesias, and R. R. Koenen. 2017. Ultrafiltration combined with size exclusion chromatography efficiently isolates extracellular vesicles from cell culture media for compositional and functional studies. Scientific Reports 7:15297. doi:10.1038/s41598-017-15717-7.
- Bonzini, M., L. Pergoli, L. Cantone, M. Hoxha, A. Spinazze, L. Del Buono, C. Favero, M. Carugno, L. Angelici, L. Broggi, A. Cataneo, A. C. Pesatori, and V. Bollati. 2017. Short-term particulate matter exposure induces extracellular vesicle release in overweight subjects. Environmental Research 155:228–34. doi:10.1016/j.envres.2017.02.014.
- Bourdonnay, E., Z. Zaslona, L. R. Penke, J. M. Speth, D. J. Schneider, S. Przybranowski, J. A. Swanson, P. Mancuso, C. M. Freeman, J. L. Curtis, and M. Peters-Golden. 2015. Transcellular delivery of vesicular SOCS proteins from macrophages to epithelial cells blunts inflammatory signaling. The Journal of Experimental Medicine 212:729–42. doi:10.1084/jem.20141675.
- Bourdrel, T., M.-A. Bind, Y. Béjot, O. Morel, and J.-F. Argacha. 2017. Cardiovascular effects of air pollution. Archives of Cardiovascular Diseases 110:634–42. doi:10.1016/j.acvd.2017.05.003.
- Brochu, P., J.-F. Ducré-Robitaille, and J. Brodeur. 2006. Physiological daily inhalation rates for free-living individuals aged 1 month to 96 years, using data from doubly labeled water measurements: A proposal for air quality criteria, standard calculations and health risk assessment. Human and Ecological Risk Assessment: an International Journal 12:675–701. doi:10.1080/10807030600801550.
- Brostrom, J. M., Z. W. Ye, A. Axmon, M. Littorin, H. Tinnerberg, C. H. Lindh, H. Zheng, A. Ghalali, U. Stenius, B. A. Jonsson, and J. Hogberg. 2015. Toluene diisocyanate: Induction of the autotaxin-lysophosphatidic acid axis and its association with airways symptoms. Toxicology and Applied Pharmacology 287:222–31. doi:10.1016/j.taap.2015.06.006.
- Burger, D., and R. M. Touyz. 2012. Cellular biomarkers of endothelial health: Microparticles, endothelial progenitor cells, and circulating endothelial cells. Journal of the American Society of Hypertension 6:85–99. doi:10.1016/j.jash.2011.11.003.
- Camussi, G., M. C. Deregibus, S. Bruno, C. Grange, V. Fonsato, and C. Tetta. 2011. Exosome/microvesicle-mediated epigenetic reprogramming of cells. American Journal of Cancer Research 1:98–110.
- Chen, -C.-C., and C.-Y. Yang. 2018. Association between gaseous air pollution and hospital admissions for hypertension in Taipei, Taiwan. Journal of Toxicology and Environmental Health, Part A 81:53–59. doi:10.1080/15287394.2017.1395573.
- Chen, Y., G. Li, Y. Liu, V. P. Werth, K. J. Williams, and M.-L. Liu. 2016. Translocation of endogenous danger signal HMGB1 from nucleus to membrane microvesicles in macrophages. Journal of Cellular Physiology 231:2319–26. doi:10.1002/jcp.25352.
- Clarenbach, C. F., O. Senn, N. A. Sievi, G. Camen, A. J. Van Gestel, V. A. Rossi, M. A. Puhan, R. Thurnheer, E. W. Russi, and M. Kohler. 2013. Determinants of endothelial function in patients with COPD. The European Respiratory Journal 42:1194–204. doi:10.1183/09031936.00144612.
- Cordazzo, C., S. Petrini, T. Neri, S. Lombardi, Y. Carmazzi, R. Pedrinelli, P. Paggiaro, and A. Celi. 2014. Rapid shedding of proinflammatory microparticles by human mononuclear cells exposed to cigarette smoke is dependent on ca2+ mobilization. Inflammation Research 63:539–47. doi:10.1007/s00011-014-0723-7.
- Coumans, F. A. W., A. R. Brisson, E. I. Buzas, F. Dignat-George, E. E. E. Drees, S. El-Andaloussi, C. Emanueli, A. Gasecka, A. Hendrix, A. F. Hill, R. Lacroix, Y. Lee, T. G. Van Leeuwen, N. Mackman, I. Mager, J. P. Nolan, E. Van Der Pol, M. Pegtel, S. Sahoo, P. R. M. Siljander, G. Sturk, O. De Wever, and R. Nieuwland. 2017. Methodological guidelines to study extracellular vesicles. Circulation Research 120:1632–48. doi:10.1161/CIRCRESAHA.117.309417.
- Crotty Alexander, L. E., S. Shin, and J. H. Hwang. 2015. Inflammatory diseases of the lung induced by conventional cigarette smoke: A review. Chest 148:1307–22. doi:10.1378/chest.15-0409.
- Csordas, A., and D. Bernhard. 2013. The biology behind the atherothrombotic effects of cigarette smoke. Nature Reviews. Cardiology 10:219–30. doi:10.1038/nrcardio.2013.8.
- Decramer, M., and W. Janssens. 2013. Chronic obstructive pulmonary disease and comorbidities. The Lancet Respiratory Medicine 1:73–83. doi:10.1016/S2213-2600(12)70060-7.
- Demedts, I. K., T. Demoor, K. R. Bracke, G. F. Joos, and G. G. Brusselle. 2006. Role of apoptosis in the pathogenesis of COPD and pulmonary emphysema. Respiratory Research 7:53. doi:10.1186/1465-9921-7-53.
- Dunbar, A., W. Gotsis, and W. Frishman. 2013. Second-hand tobacco smoke and cardiovascular disease risk: An epidemiological review. Cardiology in Review 21:94–100. doi:10.1097/CRD.0b013e31827362e4.
- Emmerechts, J., V. De Vooght, S. Haenen, S. Loyen, S. Van Kerckhoven, B. Hemmeryckx, J. A. Vanoirbeek, P. H. Hoet, B. Nemery, and M. F. Hoylaerts. 2012a. Thrombogenic changes in young and old mice upon subchronic exposure to air pollution in an urban roadside tunnel. Thromb Haemost 108:756–68. doi:10.1160/TH12-03-0161.
- Emmerechts, J., L. Jacobs, S. Van Kerckhoven, S. Loyen, C. Mathieu, F. Fierens, B. Nemery, T. S. Nawrot, and M. F. Hoylaerts. 2012b. Air pollution-associated procoagulant changes: The role of circulating microvesicles. Journal of Thrombosis and Haemostasis 10:96–106. doi:10.1111/j.1538-7836.2011.04557.x.
- Enjeti, A. K., A. Ariyarajah, A. D’Crus, M. Seldon, and L. F. Lincz. 2017. Circulating microvesicle number, function and small RNA content vary with age, gender, smoking status, lipid and hormone profiles. Thrombosis Research 156:65–72. doi:10.1016/j.thromres.2017.04.019.
- Ezzati, M., and A. D. Lopez. 2003. Estimates of global mortality attributable to smoking in 2000. The Lancet 362:847–52. doi:10.1016/S0140-6736(03)14338-3.
- Ferrante, G., R. Antona, V. Malizia, L. Montalbano, G. Corsello, and S. La Grutta. 2014. Smoke exposure as a risk factor for asthma in childhood: A review of current evidence. Allergy and Asthma Proceedings 35:454–61. doi:10.2500/aap.2014.35.3789.
- Foks, A. C., D. Engelbertsen, F. Kuperwaser, N. Alberts-Grill, A. Gonen, J. L. Witztum, J. Lederer, P. Jarolim, R. H. DeKruyff, G. J. Freeman, and A. H. Lichtman. 2016. Blockade of Tim-1 and Tim-4 enhances atherosclerosis in low-density lipoprotein receptor–deficient mice. Arteriosclerosis, Thrombosis, and Vascular Biology 36:456–65. doi:10.1161/ATVBAHA.115.306860.
- Folkesson, M., C. Li, S. Frebelius, J. Swedenborg, D. Wagsater, K. J. Williams, P. Eriksson, J. Roy, and M. L. Liu. 2015. Proteolytically active ADAM0 and ADAM7 carried on membrane microvesicles in human abdominal aortic aneurysms. Thromb Haemost 114:1165–74. doi:10.1160/TH14-10-0899.
- Folkman, J. 1971. Tumor angiogenesis: Therapeutic implications. The New England Journal of Medicine 285:1182–86. doi:10.1056/NEJM197111182852108.
- Frampton, M. W., J. Bausch, D. Chalupa, P. K. Hopke, E. L. Little, D. Oakes, J. C. Stewart, and M. J. Utell. 2012. Effects of outdoor air pollutants on platelet activation in people with type 2 diabetes. Inhalation Toxicology 24:831–38. doi:10.3109/08958378.2012.724117.
- Frampton, M. W., A. Pietropaoli, M. Dentler, D. Chalupa, E. L. Little, J. Stewart, L. Frasier, D. Oakes, J. Wiltshire, R. Vora, and M. J. Utell. 2015. Cardiovascular effects of ozone in healthy subjects with and without deletion of glutathione-s-transferase M1. Inhalation Toxicology 27:113–19. doi:10.3109/08958378.2014.996272.
- Franklin, B. A., R. Brook, and C. Arden Pope 3rd. 2015. Air pollution and cardiovascular disease. Current Problems in Cardiology 40:207–38. doi:10.1016/j.cpcardiol.2015.01.003.
- Frohlich, E., A. Mercuri, S. Wu, and S. Salar-Behzadi. 2016. Measurements of deposition, lung surface area and lung fluid for simulation of inhaled compounds. Frontiers in Pharmacology 7:181. doi:10.3389/fphar.2016.00181.
- Fujita, Y., J. Araya, S. Ito, K. Kobayashi, N. Kosaka, Y. Yoshioka, T. Kadota, H. Hara, K. Kuwano, and T. Ochiya. 2015a. Suppression of autophagy by extracellular vesicles promotes myofibroblast differentiation in COPD pathogenesis. Journal of Extracellular Vesicles 4:28388. doi:10.3402/jev.v4.28388.
- Fujita, Y., N. Kosaka, J. Araya, K. Kuwano, and T. Ochiya. 2015b. Extracellular vesicles in lung microenvironment and pathogenesis. Trends in Molecular Medicine 21:533–42. doi:10.1016/j.molmed.2015.07.004.
- Gardiner, C., D. Di Vizio, S. Sahoo, C. Théry, K. W. Witwer, M. Wauben, and A. F. Hill. 2016. Techniques used for the isolation and characterization of extracellular vesicles: Results of a worldwide survey. Journal of Extracellular Vesicles 5:32945. doi:10.3402/jev.v5.32945.
- Gimbrone, M. A.Jr., and G. García-Cardeña. 2016. Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circulation Research 118:620–36. doi:10.1161/CIRCRESAHA.115.306301.
- Gordon, C., K. Gudi, A. Krause, R. Sackrowitz, B.-G. Harvey, Y. Strulovici-Barel, J. G. Mezey, and R. G. Crystal. 2011. Circulating endothelial microparticles as a measure of early lung destruction in cigarette smokers. American Journal of Respiratory and Critical Care Medicine 184:224–32. doi:10.1164/rccm.201012-2061OC.
- Gould, S. J., and G. Raposo. 2013. As we wait: Coping with an imperfect nomenclature for extracellular vesicles. Journal of Extracellular Vesicles 2:20389. doi:10.3402/jev.v2i0.20389.
- Grant, R., E. Ansa-Addo, D. Stratton, S. Antwi-Baffour, S. Jorfi, S. Kholia, L. Krige, S. Lange, and J. Inal. 2011. A filtration-based protocol to isolate human plasma membrane-derived vesicles and exosomes from blood plasma. Journal of Immunological Methods 371:143–51. doi:10.1016/j.jim.2011.06.024.
- Guarnieri, M., and J. R. Balmes. 2014. Outdoor air pollution and asthma. The Lancet 383:1581–92. doi:10.1016/S0140-6736(14)60617-6.
- Heiss, C., N. Amabile, A. C. Lee, W. M. Real, S. F. Schick, D. Lao, M. L. Wong, S. Jahn, F. S. Angeli, P. Minasi, M. L. Springer, S. K. Hammond, S. A. Glantz, W. Grossman, J. R. Balmes, and Y. Yeghiazarians. 2008. Brief secondhand smoke exposure depresses endothelial progenitor cells activity and endothelial function: Sustained vascular injury and blunted nitric oxide production. Journal of the American College of Cardiology 51:1760–71. doi:10.1016/j.jacc.2008.01.040.
- Héliot, A., Y. Landkocz, F. Roy Saint-Georges, P. Gosset, S. Billet, P. Shirali, D. Courcot, and P. J. Martin. 2017. Smoker extracellular vesicles influence status of human bronchial epithelial cells. International Journal of Hygiene and Environmental Health 220:445–54. doi:10.1016/j.ijheh.2016.12.010.
- Houghton, A. M. 2013. Mechanistic links between COPD and lung cancer. Nature reviews. Cancer 13:233–45. doi:10.1038/nrc3477.
- ISTH Steering Committee for World Thrombosis Day. 2014. Thrombosis: A major contributor to the global disease burden. Journal of Thrombosis and Haemostasis : JTH 12:1580–90. doi:10.1111/jth.12698.
- Kelly, F. J., and J. C. Fussell. 2015. Air pollution and public health: Emerging hazards and improved understanding of risk. Environ Geochem Health 37:631–49. doi:10.1007/s10653-015-9720-1.
- Kermanizadeh, A., I. Gosens, L. MacCalman, H. Johnston, P. H. Danielsen, N. R. Jacobsen, A. G. Lenz, T. Fernandes, R. P. Schins, F. R. Cassee, H. Wallin, W. Kreyling, T. Stoeger, S. Loft, P. Moeller, L. Tran, and V. Stone. 2016. A multilaboratory toxicological assessment of a panel of 10 engineered nanomaterials to human health–ENPRA project–The highlights, limitations, and current and future challenges. Journal of Toxicology and Environmental Health, Part B 19:1–28. doi:10.1080/10937404.2015.1126210.
- Kowal, J., G. Arras, M. Colombo, M. Jouve, J. P. Morath, B. Primdal-Bengtson, F. Dingli, D. Loew, M. Tkach, and C. Théry. 2016. Proteomic comparison defines novel markers to characterize heterogeneous populations of extracellular vesicle subtypes. Proceedings of the National Academy of Sciences of the United States of America 113:E968–E977. doi:10.1073/pnas.1521230113.
- Lacroix, R., and F. Dignat-George. 2013. Microparticles: New protagonists in pericellular and intravascular proteolysis. Seminars in Thrombosis and Hemostasis 39:33–39. doi:10.1055/s-0032-1333310.
- Lamprecht, B., M. A. McBurnie, W. M. Vollmer, G. Gudmundsson, T. Welte, E. Nizankowska-Mogilnicka, M. Studnicka, E. Bateman, J. M. Anto, P. Burney, D. M. Mannino, and S. A. Buist. 2011. COPD in never smokers: Results from the population-based burden of obstructive lung disease study. Chest 139:752–63. doi:10.1378/chest.10-1253.
- Lasser, C., S. E. O’Neil, L. Ekerljung, K. Ekstrom, M. Sjostrand, and J. Lotvall. 2011. RNA-containing exosomes in human nasal secretions. American Journal of Rhinology & Allergy 25:89–93. doi:10.2500/ajra.2011.25.3573.
- Lee, F., and D. A. Lawrence. 2018. From infections to anthropogenic inflicted pathologies: Involvement of immune balance. Journal of Toxicology and Environmental Health, Part B 21:24–46. doi:10.1080/10937404.2017.1412212.
- Lee, S.-B., J.-H. Kim, M.-H. Cho, E.-S. Choe, K.-S. Kim, and S.-M. Shim. 2017. Impact of commercial cigarette smoke condensate on brain tissue co-cultured with astrocytes and blood-brain barrier endothelial cells. Journal of Toxicology and Environmental Health, Part A 80:533–41. doi:10.1080/15287394.2017.1355863.
- Levanen, B., N. R. Bhakta, P. Torregrosa Paredes, R. Barbeau, S. Hiltbrunner, J. L. Pollack, C. M. Skold, M. Svartengren, J. Grunewald, S. Gabrielsson, A. Eklund, B.-M. Larsson, P. G. Woodruff, D. J. Erie, and A. M. Wheelock. 2013. Altered microrna profiles in bronchoalveolar lavage fluid exosomes in asthmatic patients. The Journal of Allergy and Clinical Immunology 131:894–903. doi:10.1016/j.jaci.2012.11.039.
- Li, C. J., Y. Liu, Y. Chen, D. Yu, K. J. Williams, and M. L. Liu.. 2013. “Novel proteolytic microvesicles released from human macrophages after exposure to tobacco smoke.” Am J Pathol 182:1552–1562. doi:10.1016/j.ajpath.2013.01.035.
- Li, M., D. Yu, K. J. Williams, and M.-L. Liu. 2010. Tobacco smoke induces the generation of procoagulant microvesicles from human monocytes/macrophages. Arteriosclerosis, Thrombosis, and Vascular Biology 30:1818–24. doi:10.1161/ATVBAHA.110.209577.
- Lim, J.-H., M.-K. Song, Y. Cho, W. Kim, S. O. Han, and J.-C. Ryu. 2016. Expression of exosomal and cellular micrornas: As biomarkers for toluene, ethylbenzene, xylene (TEX) exposure. Molecular Cellular Toxicogical 12:359–69. doi:10.1007/s13273-016-0040-z.
- Liu, H., L. Ding, Y. Zhang, and S. Ni. 2014. Circulating endothelial microparticles involved in lung function decline in a rat exposed in cigarette smoke maybe from apoptotic pulmonary capillary endothelial cells. Journal of Thoracic Disease 6:649–55. doi:10.3978/j.issn.2072-1439.2014.06.26.
- Lotvall, J., A. F. Hill, F. Hochberg, E. I. Buzas, D. Di Vizio, C. Gardiner, Y. S. Gho, I. V. Kurochkin, S. Mathivanan, P. Quesenberry, S. Sahoo, H. Tahara, M. H. Wauben, K. W. Wetmer, and C. Thery. 2014. Minimal experimental requirements for definition of extracellular vesicles and their functions: A position statement from the international society for extracellular vesicles. Journal of Extracellular Vesicles 3:26913. doi:10.3402/jev.v3.26913.
- Loyer, X., A.-C. Vion, A. Tedgui, and C. M. Boulanger. 2014. Microvesicles as cell-cell messengers in cardiovascular diseases. Circulation Research 114:345–53. doi:10.1161/CIRCRESAHA.113.300858.
- Lozano, R., M. Naghavi, K. Foreman, S. Lim, K. Shibuya, V. Aboyans, J. Abraham, T. Adair, R. Aggarwal, S. Y. Ahn, et al. 2012. Global and regional mortality from 235 causes of death for 20 age groups in 1990 and 2010: A systematic analysis for the global burden of disease study 2010. Lancet 380:2095–128. doi:10.1016/S0140-6736(12)61728-0.
- Maas, S. L., J. De Vrij, and M. L. Broekman. 2014. Quantification and size-profiling of extracellular vesicles using tunable resistive pulse sensing. Journal of Visualized Experiments : JoVE e51623. doi:10.3791/51623.
- Malvezzi, M., P. Bertuccio, T. Rosso, M. Rota, F. Levi, C. La Vecchia, and E. Negri. 2015. European cancer mortality predictions for the year 2015: Does lung cancer have the highest death rate in EU women? Annals of Oncology 26:779–86. doi:10.1093/annonc/mdv001.
- McCubbrey, A. L., and J. L. Curtis. 2013. Efferocytosis and lung disease. Chest 143:1750–57. doi:10.1378/chest.12-2413.
- Messner, B., and D. Bernhard. 2014. Smoking and cardiovascular disease: Mechanisms of endothelial dysfunction and early atherogenesis. Arteriosclerosis, Thrombosis, and Vascular Biology 34:509–15. doi:10.1161/ATVBAHA.113.300156.
- Michel, J. B., J. L. Martin-Ventura, A. Nicoletti, and B. Ho-Tin-Noe. 2014. Pathology of human plaque vulnerability: Mechanisms and consequences of intraplaque haemorrhages. Atherosclerosis 234:311–19. doi:10.1016/j.atherosclerosis.2014.03.020.
- Minciacchi, V. R., M. R. Freeman, and D. Di Vizio. 2015. Extracellular vesicles in cancer: Exosomes, microvesicles and the emerging role of large oncosomes. Seminars in Cell & Developmental Biology 40:41–51. doi:10.1016/j.semcdb.2015.02.010.
- Mobarrez, F., L. Antoniewicz, J. A. Bosson, J. Kuhl, D. S. Pisetsky, and M. Lundback. 2014. The effects of smoking on levels of endothelial progenitor cells and microparticles in the blood of healthy volunteers. PLoS One 9:e90314. doi:10.1371/journal.pone.0090314.
- Moon, H.-G., S.-H. Kim, J. Gao, T. Quan, Z. Qin, J. C. Osorio, I. O. Rosas, M. Wu, Y. Tesfaigzi, and Y. Jin. 2014. CCN1 secretion and cleavage regulate the lung epithelial cell functions after cigarette smoke. American Journal of Physiology-Lung Cellular and Molecular Physiology 307:L326–L337. doi:10.1152/ajplung.00102.2014.
- Neri, T., L. Pergoli, S. Petrini, L. Gravendonk, C. Balia, V. Scalise, A. Amoruso, R. Pedrinelli, P. Paggiaro, V. Bollati, and A. Celi. 2016. Particulate matter induces prothrombotic microparticle shedding by human mononuclear and endothelial cells. Toxicology in Vitro 32:333–38. doi:10.1016/j.tiv.2016.02.001.
- Nolte-’T Hoen, E. N., E. J. Van Der Vlist, M. Aalberts, H. C. Mertens, B. J. Bosch, W. Bartelink, E. Mastrobattista, E. V. Van Gaal, W. Stoorvogel, and G. J. Arkesteijn. 2012. Quantitative and qualitative flow cytometric analysis of nanosized cell-derived membrane vesicles. Nanomedicine 8:712–20. doi:10.1016/j.nano.2011.09.006.
- Paciência, I., J. Madureira, J. Rufo, A. Moreira, and O. Fernandes Ede. 2016. A systematic review of evidence and implications of spatial and seasonal variations of volatile organic compounds (VOC) in indoor human environments. Journal of Toxicology and Environmental Health, Part B 19:47–64. doi:10.1080/10937404.2015.1134371.
- Pałgan, K., and Z. Bartuzi. 2015. Angiogenesis in bronchial asthma. International Journal of Immunopathology and Pharmacology 28:415–20. doi:10.1177/0394632015580907.
- Pavanello, S., M. Bonzini, L. Angelici, V. Motta, L. Pergoli, M. Hoxha, L. Cantone, A. C. Pesatori, P. Apostoli, A. Tripodi, A. Baccarelli, and V. Bollati. 2016. Extracellular vesicle-driven information mediates the long-term effects of particulate matter exposure on coagulation and inflammation pathways. Toxicology Letters 259:143–50. doi:10.1016/j.toxlet.2016.08.002.
- Pergoli, L., L. Cantone, C. Favero, L. Angelici, S. Iodice, E. Pinatel, M. Hoxha, L. Dioni, M. Letizia, B. Albetti, L. Tarantini, F. Rota, P. A. Bertazzi, A. S. Tirelli, V. Dolo, and A. Cattaneo. 2017. Extracellular vesicle-packaged miRNA release after short-term exposure to particulate matter is associated with increased coagulation. Particle and Fibre Toxicology 14:32. doi:10.1186/s12989-017-0214-4.
- Piazza, G., and S. Z. Goldhaber. 2010. Venous thromboembolism and atherothrombosis: An integrated approach. Circulation 121:2146–50. doi:10.1161/CIRCULATIONAHA.110.951236.
- Pickup, M. W., J. K. Mouw, and V. M. Weaver. 2014. The extracellular matrix modulates the hallmarks of cancer. EMBO Reports 15:1243–53. doi:10.15252/embr.201439246.
- Pope, C. A.3rd, A. Bhatnagar, J. P. McCracken, W. Abplanalp, D. J. Conklin, and T. O’Toole. 2016. Exposure to fine particulate air pollution is associated with endothelial injury and systemic inflammationnovelty and significance. Circulation Research 119:1204–14. doi:10.1161/CIRCRESAHA.116.309279.
- Rahib, L., B. D. Smith, R. Aizenberg, A. B. Rosenzweig, J. M. Fleshman, and L. M. Matrisian. 2014. Projecting cancer incidence and deaths to 2030: The unexpected burden of thyroid, liver, and pancreas cancers in the United States. Cancer Research 74:2913–21. doi:10.1158/0008-5472.CAN-14-0155.
- Rigotti, N. A., and C. Clair. 2013. Managing tobacco use: The neglected cardiovascular disease risk factor. European Heart Journal 34:3259–67. doi:10.1093/eurheartj/eht352.
- Rodosthenous, R. S., B. A. Coull, Q. Lu, P. S. Vokonas, J. D. Schwartz, and A. A. Baccarelli. 2016. Ambient particulate matter and micrornas in extracellular vesicles: A pilot study of older individuals. Particle and Fibre Toxicology 13:13. doi:10.1186/s12989-016-0121-0.
- Sabatier, F., L. Camoin-Jau, F. Anfosso, J. Sampol, and F. Dignat-George. 2009. Circulating endothelial cells, microparticles and progenitors: Key players towards the definition of vascular competence. Journal of Cellular and Molecular Medicine 13:454–71. doi:10.1111/j.1582-4934.2008.00639.x.
- Salazar, L. M., and A. M. Herrera. 2011. Fibrotic response of tissue remodeling in COPD. Lung 189:101–09. doi:10.1007/s00408-011-9279-2.
- Salvi, S. S., and P. J. Barnes. 2009. Chronic obstructive pulmonary disease in non-smokers. The Lancet 374:733–43. doi:10.1016/S0140-6736(09)61303-9.
- Schneider, D. J., J. M. Speth, L. R. Penke, S. H. Wettlaufer, J. A. Swanson, and M. Peters-Golden. 2017. Mechanisms and modulation of microvesicle uptake in a model of alveolar cell communication. The Journal of Biological Chemistry 292:20897–910. doi:10.1074/jbc.M117.792416.
- Schrijvers, D. M., G. R. De Meyer, M. M. Kockx, A. G. Herman, and W. Martinet. 2005. Phagocytosis of apoptotic cells by macrophages is impaired in atherosclerosis. Arteriosclerosis, Thrombosis, and Vascular Biology 25:1256–61. doi:10.1161/01.ATV.0000166517.18801.a7.
- Serban, K. A., S. Rezania, D. N. Petrusca, C. Poirier, D. Cao, M. J. Justice, M. Patel, I. Tsvetkova, K. Kamocki, and A. Mikosz. 2016. Structural and functional characterization of endothelial microparticles released by cigarette smoke. Scientific Reports 6:31596. doi:10.1038/srep31596.
- Severinsen, M. T., S. R. Kristensen, S. P. Johnsen, C. Dethlefsen, A. Tjonneland, and K. Overvad. 2009. Smoking and venous thromboembolism: A Danish follow-up study. Journal of Thrombosis and Haemostasis 7:1297–303. doi:10.1111/j.1538-7836.2009.03490.x.
- Sharma, M. 2010. Understanding the mechanism of toxicity of carbon nanoparticles in humans in the new millennium: A systemic review. Indian Journal of Occupational and Environmental Medicine 14:3–5. doi:10.4103/0019-5278.64607.
- Sheller, S., J. Papaconstantinou, R. Urrabaz-Garza, L. Richardson, G. Saade, C. Salomon, and R. Menon. 2016. Amnion-epithelial-cell-derived exosomes demonstrate physiologic state of cell under oxidative stress. PLoS One 11:e0157614. doi:10.1371/journal.pone.0157614.
- Siasos, G., V. Tsigkou, E. Kokkou, E. Oikonomou, M. Vavuranakis, C. Vlachopoulos, A. Verveniotis, M. Limperi, V. Genimata, A. G. Papavassiliou, C. Stefanadis, and D. Tousoulis. 2014. Smoking and atherosclerosis: Mechanisms of disease and new therapeutic approaches. Current Medicinal Chemistry 21:3936–48. doi:10.2174/092986732134141015161539.
- Sokolova, V., A.-K. Ludwig, S. Hornung, O. Rotan, P. A. Horn, M. Epple, and B. Giebel. 2011. Characterisation of exosomes derived from human cells by nanoparticle tracking analysis and scanning electron microscopy. Colloids and Surfaces B: Biointerfaces 87:146–50. doi:10.1016/j.colsurfb.2011.05.013.
- Song, Q., D. C. Christiani, X. Wang, and J. Ren. 2014. The global contribution of outdoor air pollution to the incidence, prevalence, mortality and hospital admission for chronic obstructive pulmonary disease: A systematic review and meta-analysis. International Journal of Environmental Research and Public Health 11:11822–32. doi:10.3390/ijerph111111822.
- Stewart, J. C., D. C. Chalupa, R. B. Devlin, L. M. Frasier, L.-S. Huang, E. L. Little, S. M. Lee, R. P. Phipps, A. P. Pietropaoli, M. B. Taubman, M. J. Utell, and M. W. Frampton. 2010. Vascular effects of ultrafine particles in persons with type 2 diabetes. Environmental Health Perspectives 118:1692–98. doi:10.1289/ehp.1002237.
- Strulovici-Barel, Y., M. R. Staudt, A. Krause, C. Gordon, A. E. Tilley, B.-G. Harvey, R. J. Kaner, C. Hollmann, J. G. Mezey, H. Bitter, S. G. Pillai, H. Hilton, G. Wolff, C. S. Stevenson, S. Visvanathan, J. S. Fine, and R. G. Crystal. 2016. Persistence of circulating endothelial microparticles in COPD despite smoking cessation. Thorax 71:1137–44. doi:10.1136/thoraxjnl-2015-208274.
- Suarez, H., A. Gamez-Valero, R. Reyes, S. Lopez-Martin, M. J. Rodriguez, J. L. Carrascosa, C. Cabanas, F. E. Borras, and M. Yanez-Mo. 2017. A bead-assisted flow cytometry method for the semi-quantitative analysis of extracellular vesicles. Scientific Reports 7:11271. doi:10.1038/s41598-017-11249-2.
- Takahashi, T., S. Kobayashi, N. Fujino, T. Suzuki, C. Ota, M. He, M. Yamada, S. Suzuki, M. Yanai, S. Kurosawa, M. Yamaya, and H. Kubo. 2012. Increased circulating endothelial microparticles in COPD patients: A potential biomarker for COPD exacerbation susceptibility. Thorax 67:1067–74. doi:10.1136/thoraxjnl-2011-201395.
- Tarlo, S. M., and C. Lemiere. 2014. Occupational asthma. The New England Journal of Medicine 370:640–49. doi:10.1056/NEJMra1301758.
- Taylor, D. D., and S. Shah. 2015. Methods of isolating extracellular vesicles impact down-stream analyses of their cargoes. Methods 87:3–10. doi:10.1016/j.ymeth.2015.02.019.
- Thery, C., M. Ostrowski, and E. Segura. 2009. Membrane vesicles as conveyors of immune responses. Nature Reviews. Immunology 9:581–93. doi:10.1038/nri2567.
- Thun, M. J., B. D. Carter, D. Feskanich, N. D. Freedman, R. Prentice, A. D. Lopez, P. Hartge, and S. M. Gapstur. 2013. 50-year trends in smoking-related mortality in the United States. The New England Journal of Medicine 368:351–64. doi:10.1056/NEJMsa1211127.
- Umbright, C., R. Sellamuthu, J. R. Roberts, S.-H. Young, D. Richardson, D. Schwegler-Berry, W. McKinney, B. Chen, J. K. Gu, M. Kashon, and P. Joseph. 2017. Pulmonary toxicity and global gene expression changes in response to sub-chronic inhalation exposure to crystalline silica in rats. Journal of Toxicology and Environmental Health, Part A 80:1349–68. doi:10.1080/15287394.2017.1384773.
- Valavanidis, A., T. Vlachogianni, K. Fiotakis, and S. Loridas. 2013. Pulmonary oxidative stress, inflammation and cancer: Respirable particulate matter, fibrous dusts and ozone as major causes of lung carcinogenesis through reactive oxygen species mechanisms. International Journal of Environmental Research and Public Health 10:3886–907. doi:10.3390/ijerph10093886.
- Van Der Pol, E., A. G. Hoekstra, A. Sturk, C. Otto, T. G. Van Leeuwen, and R. Nieuwland. 2010. Optical and non-optical methods for detection and characterization of microparticles and exosomes. Journal of Thrombosis and Haemostasis 8:2596–607. doi:10.1111/j.1538-7836.2010.04074.x.
- Van Es, N., S. Bleker, A. Sturk, and R. Nieuwland. 2015. Clinical significance of tissue factor-exposing microparticles in arterial and venous thrombosis. Seminars in Thrombosis and Hemostasis 41:718–27. doi:10.1055/s-0035-1556047.
- Van Niel, G., G. D’Angelo, and G. Raposo. 2018. Shedding light on the cell biology of extracellular vesicles. Nature reviews. Molecular Cell Biology 19:213–28. doi:10.1038/nrm.2017.125.
- Volgers, C., B. J. Benedikter, G. E. Grauls, P. H. M. Savelkoul, and F. R. M. Stassen. 2017. Bead-based flow-cytometry for semi-quantitative analysis of complex membrane vesicle populations released by bacteria and host cells. Microbiological Research 200:25–32. doi:10.1016/j.micres.2017.04.003.
- Wang, Z., S. M. May, S. Charoenlap, R. Pyle, N. L. Ott, K. Mohammed, and A. Y. Joshi. 2015. Effects of secondhand smoke exposure on asthma morbidity and health care utilization in children: A systematic review and meta-analysis. Annals of Allergy, Asthma & Immunology 115 (5):396–401.e2. doi:10.1016/j.anai.2015.08.005.
- Weichenthal, S., J. A. Hoppin, and F. Reeves. 2014. Obesity and the cardiovascular health effects of fine particulate air pollution. Obesity (Silver Spring) 22:1580–89. doi:10.1002/oby.20748.
- Witkowski, M., U. Landmesser, and U. Rauch. 2016. Tissue factor as a link between inflammation and coagulation. Trends in Cardiovascular Medicine 26:297–303. doi:10.1016/j.tcm.2015.12.001.
- Wolf, P. 1967. The nature and significance of platelet products in human plasma. British Journal of Haematology 13:269–88. doi:10.1111/j.1365-2141.1967.tb08741.x.
- Xu, J., M. Yang, P. Kosterin, B. M. Salzberg, T. N. Milovanova, V. M. Bhopale, and S. R. Thom. 2013. Carbon monoxide inhalation increases microparticles causing vascular and CNS dysfunction. Toxicology and Applied Pharmacology 273:410–17. doi:10.1016/j.taap.2013.09.019.
- Xu, L., and X. Deng. 2004. Tobacco-specific Nitrosamine 4-(Methylnitrosamino)-1-(3-pyridyl)-1-butanone induces phosphorylation of μ- and m-Calpain in association with increased secretion, cell migration, and invasion. The Journal of Biological Chemistry 279:53683–90. doi:10.1074/jbc.M409889200.
- Zhu, M., Y. Li, J. Shi, W. Feng, G. Nie, and Y. Zhao. 2012. Exosomes as extrapulmonary signaling conveyors for nanoparticle-induced systemic immune activation. Small 8:404–12. doi:10.1002/smll.201101708.