
ABSTRACT
Carbon tetrachloride (CCl4) has been extensively used and reported to produce toxicity, most notably involving the liver. Carbon tetrachloride metabolism involves CYP450-mediated bioactivation to trichloromethyl and trichloromethyl peroxy radicals, which are capable of macromolecular interaction with cell components including lipids and proteins. Radical interaction with lipids produces lipid peroxidation which can mediate cellular damage leading to cell death. Chronic exposure with CCl4 a rodent hepatic carcinogen with a mode of action (MOA) exhibits the following key events: 1) metabolic activation; 2) hepatocellular toxicity and cell death; 3) consequent regenerative increased cell proliferation; and 4) hepatocellular proliferative lesions (foci, adenomas, carcinomas). The induction of rodent hepatic tumors is dependent upon the dose (concentration and exposure duration) of CCl4, with tumors only occurring at cytotoxic exposure levels. Adrenal benign pheochromocytomas were also increased in mice at high CCl4 exposures; however, these tumors are not of relevant importance to human cancer risk. Few epidemiology studies that have been performed on CCl4, do not provide credible evidence of enhanced risk of occurrence of liver or adrenal cancers, but these studies have serious flaws limiting their usefulness for risk assessment. This manuscript summarizes the toxicity and carcinogenicity attributed to CCl4, specifically addressing MOA, dose-response, and human relevance.
Introduction
Carbon tetrachloride (CCl4) has historically been used as a feedstock for chlorofluorocarbons, as a dry-cleaning agent, fabric-spotting fluid, solvent, reagent in chemical synthesis, fire extinguisher fluid, and grain fumigant (ATSDR Agency for Toxic Substances and Disease Registry Citation2005). Currently, this chemical is employed almost exclusively as a feedstock for hydrofluorocarbons (HFCs) and hydrofluoroolefins (HFOs) (Marshall and Pottenger Citation2016). This compound is an ozone-depleting substance and thus tightly regulated under the Montreal Protocol on Substances that Deplete the Ozone Layer (the Montreal Protocol).
Carbon tetrachloride is rapidly absorbed by most routes of exposure in humans and rodents. Once absorbed, it is widely distributed among tissues, especially those with high lipid content, reaching peak concentrations within 1–6 hr, dependent upon route of administration and exposure concentration (ATSDR Agency for Toxic Substances and Disease Registry Citation2005). Inhaled CCl4 is rapidly excreted, primarily through breath (exhaled air). CCl4 is metabolized in the body, primarily by the liver, but also in kidneys, lung, and other tissues containing CYP450 enzymes (Martinez, Mourelle, and Muriel Citation1995; Slater Citation1982; Weber, Boll, and Stampfl Citation2003). The metabolism of CCl4 has been extensively studied in in vivo and in vitro mammalian systems. The initial step in the biotransformation of CCl4 is reductive dehalogenation mediated by CYP2E1 to form the trichloromethyl radical, which may be converted to the trichloromethyl peroxy radical by binding oxygen. Both radical species are responsible for the hepatotoxic effects noted with CCl4.
CCl4 was shown to induce hepatocellular tumors in rats and mice following exposure by oral, inhalation, or other parenteral routes (Andervont Citation1958; Della Porta, Terracini, and Shubik Citation1961; Edwards Citation1941; Edwards and Dalton Citation1942; Edwards, Heston, and Dalton Citation1942; JBRC Japan Bioassay Research Center Citation1998; Nagano et al. Citation1998, Citation2007a). Chronic treatment of mice with CCl4 also resulted in an increased incidence of adrenal pheochromocytomas in males and females (Nagano et al. Citation2007a). No adrenal pheochromocytomas were detected in rats following chronic exposure to CCl4. This manuscript addresses the carcinogenicity of CCl4 in rodents suggesting a mode of action (MOA) for induction of the rodent tumors by applying the International Programme on Chemical Safety (IPCS) MOA analysis evaluation of potential human relevance of rodent findings (Boobis et al. Citation2006, Citation2008).
Carcinogenicity of CCl4 in animal bioassays
Over the years, several assays were employed to address the toxicity and carcinogenicity attributed to CCl4 in rats and mice following various routes of exposure as well as one chronic study in hamsters and one study in trout (Citation1999; ATSDR Agency for Toxic Substances and Disease Registry Citation2005; IARC International Agency for Research on Cancer Solvents Citation1979). Many of these investigations do not meet the currently accepted scientific standards of protocol and interpretation. Most studies also involve oral exposure while the major route by which humans are exposed is by inhalation, particularly in an occupational setting. The inhalation exposure route has only been properly addressed in one study (JBRC Japan Bioassay Research Center Citation1998; Nagano et al. Citation1998, Citation2007a). In that study, a two-year bioassay in both rats and mice exposed to CCl4 by inhalation was performed by the Japan Bioassay Research Center (JBRC) ((Citation1998); Nagano et al. Citation1998; Nagano et al., Citation2007a). While the JBRC study is considered the most complete and thorough of the inhalation studies, the other chronic CCl4 treatment investigations, albeit using oral exposure, intraperitoneal (ip) or subcutaneous (sc) injection, will also be discussed since these experiments provide additional supportive evidence for liver carcinogenicity initiated by this compound.
2-Year inhalation bioassay in rats and mice (Nagano et al. Citation2007a)
The carcinogenicity and chronic toxicity attributed to CCl4 was evaluated in rats and mice by whole-body inhalation by the JBRC. The results of this study (JBRC Japan Bioassay Research Center Citation1998) were initially reported in a book chapter (Nagano et al. Citation1998) but were subsequently fully described in a peer-reviewed publication by the same group (Nagano et al. Citation2007a). The peer-reviewed publication by Nagano et al. (Citation2007a) will be referred to when citing this study in the remainder of this manuscript. This investigation utilized groups of 50 F344 rats and 50 BDF1 mice of both genders at exposure concentrations of 0, 5, 25, or 125 ppm for 6 hr per day, 5 days per week, for 104 weeks, with full histopathology review. Histopathologic findings of the livers in this 2-year study are presented in for rats and in for mice. There was an elevated incidence of liver tumors in rats and mice in both genders. Other liver histopathologic alterations were also seen including cirrhosis, fibrosis, and fatty changes in rats, and ceroid deposition, bile duct proliferation, and hydropic change in mice. A decrease in survival rates and corresponding reduced body weight gain were reported in both rats and mice of both genders treated at 125 ppm and in female mice treated at 25 ppm. Liver weights were significantly increased in male rats at 25 and 125 ppm, in the female rats at 25 ppm and in the only one surviving female rat at 125 ppm. Changes in liver serum chemistry alanine aminotransferase (ALT) and aspartate aminotransferase AST were found at the end of the study. However, it is difficult to separate out the hepatotoxic effects attributed to CCl4 at the end of 2-years due to masking of liver injury associated with liver tumors that were present at this time point. In male and female rats, significant elevation in number of liver adenomas, carcinomas, and combined adenomas and carcinomas were only detected at the 125 ppm exposure concentration (). There were also non-neoplastic changes in the livers of rats including fatty changes, fibrosis, and cirrhosis at the two highest exposure levels (). In male rats, there was no significant alteration in total hepatic foci at any exposure level, although basophilic foci were increased at the highest dose (). In contrast, in female rats, a significant rise in animals with foci was seen at 25 ppm, but not at 5 ppm (foci were not determined at 125 ppm).
Table 1. Incidences of selected histopathological lesions in the rats exposed to CCl4 vapor by inhalation for 2 years (Nagano et al. Citation2007a).
Table 2. Incidences of selected histopathological lesions in the mice exposed to CCl4 vapor by inhalation for 2 years (Nagano et al. Citation2007a).
In mice, there was a significant increase in incidence of liver tumors at 25 and 125 ppm CCl4 in both males and females (). In female mice exposed to 5 ppm CCl4, Nagano et al. (Citation2007a) reported a significant increase in liver adenoma levels. No marked alteration in carcinomas or combined carcinomas and adenomas was noted in female mice at 5 ppm. Unlike the rat, there was no significant change in hepatocellular foci in mice at any exposure level. The serum enzyme changes ALT, AST and lactate dehydrogenase (LDH) at the two-year sampling time were consistent with continuing hepatocellular injury and tumor burden. A 90-day inhalation study using similar treatment parameters and the same rodent strains as the chronic study from the Nagano group was also reported (Nagano et al. Citation2007b), and the results are described below.
Debate has been raised concerning the significance of the rise in hepatic adenoma levels reported in the 5 ppm CCl4-exposed female mice (Nagano et al. Citation2007a). Because there was no marked change in the incidence of hepatocellular carcinomas or combined hepatocellular tumors (adenomas plus carcinomas), this warranted a thorough evaluation of the data. Our conclusion is that the incidence of these tumors in female mice at 5 ppm should not be considered treatment related. CCl4 produced an increase in liver adenomas (8/49) over control (2/50), but there was no significant alteration in carcinomas 1/49 vs 2/50 for control or combined adenomas and carcinomas (9/49 compared to 2/50 for control) (skewed by the adenoma increase).
In reviewing the significance of the adenomas in the 5 ppm-exposed female mice, three points need to be considered. First, the historical spontaneous liver tumor incidence in BDF1 mice from 10 two-year carcinogenicity studies conducted at the JBRC showed a mean incidence of liver adenomas to be 4.4% (with a range from 2% to 8%) (Katagiri et al. Citation1998). Similarly, Yamate et al. (Citation1990) noted 6/50 liver adenomas in untreated female BDF1 mice in an additional study, but it was a lifetime study (up to 150 weeks). Finally, information provided by Dr Shoji Fukushima (personal communication), the former Director of the JBRC and the senior author on the publications by Nagano et al (Citation2007a, Citation2007b), reported the range of incidences of hepatocellular adenomas in female BDF1 mice in their lab was 2–20% with a mean of 6.4% (86 out of 1,348 mice). For hepatocellular carcinoma, the range was 0–8% with an average of 2.2% (30 out of 1,348). For combined tumors, the incidences in historical controls were in the range of 2–20% with a mean of 8.5% (114 out of 1,348). Based upon this information and reported investigations, the incidences of hepatocellular adenoma, hepatocellular carcinoma, and combined hepatocellular adenomas plus carcinomas found in female mice treated at 5 ppm in the CCl4 bioassay of Nagano et al. (Citation2007a) were within the historical range for this lab for this strain of mice.
The second point is that the significance of liver adenomas seen in the Nagano et al. (Citation2007a) bioassay needs to be reconsidered. As Haseman (Citation1983) stated, for common tumors, statistical significance for tumor incidences needs to be based upon the probability of p < .01 rather than p < .05 because of multiple comparisons and to avoid the high probability of false positives. Certainly, hepatocellular tumors in mice are a common tumor (defined by Haseman as tumors with a spontaneous incidence >1%). This statistical standard has been adopted by pharmaceutical regulatory agencies and extended by the U.S. Food and Drug Administration (U.S. FDA) to have the trend test be significant only if p < .005 rather than p < .01 (U.S. FDA United States Food and Drug Administration Citation2001). The Organization for Economic Co-operation and Development (OECD) has also accepted this standard of p < .01 for comparison of incidences of common tumors (operation and Development Citation2010). Thus, on a purely statistical basis, these tumors (even adenomas alone) were not significant and are considered not treatment related.
Third, while the Nagano et al. (Citation2007a) study noted a rise in liver adenoma rates at 5 ppm in the female BDF1 mice, the incidence of total tumors (adenomas and carcinomas combined) was not significant. Comparison of hepatocellular tumors needs to be made on the basis of total tumor incidences (adenomas plus carcinomas), not on adenomas and carcinomas separately. It is well known that the sequence of events for hepatocellular tumors in rats and mice is the formation of altered cell foci leading to adenomas leading to carcinomas (Dragan et al. Citation1993; Farber and Sarma Citation1987; Holsapple et al. Citation2006; Maronpot et al. Citation1987; Thoolen et al. Citation2010). Although these are defined entities, they are continuous, and there is often difficulty in discerning lesions that are at the border between these different diagnoses (McConnell et al. Citation1986; Quist et al. Citation2019). Adenomas have the potential to evolve into carcinomas. Given the development and progression of liver adenomas and carcinomas in rodents, the combined tumor incidence is a more accurate measurement of the tumor incidence. Based upon the biology of liver tumors, it is best to make statistical comparisons of incidences between groups based upon the incidences of animals with adenomas and/or carcinomas, rather than evaluating each one separately (McConnell et al. Citation1986; Quist et al. Citation2019). Thus, based upon statistical considerations, evaluating combined adenomas plus carcinomas and considering the historical controls, the incidences of hepatocellular adenomas, carcinomas, and combined hepatocellular tumors in the 5 ppm female mice group need to be considered not treatment related.
In addition to liver tumors, an increased incidence of pheochromocytoma of the adrenal gland at the two highest CCl4 exposure levels (25 and 125 ppm) in male mice and at the highest exposure level (125 ppm) in female mice was reported (). All of these benign adrenal tumor incidences were significant.
The rats also had high incidences of chronic progressive nephropathy (CPN), with nearly all of the animals in each group exhibiting some evidence of this common disorder in rats. There was an exposure-dependent rise in the severity score, with a significant increase at the two highest exposure levels in both males and females (). CPN was not, however, associated with an altered incidence of kidney tumors in exposed males and females. Further, CPN is not relevant to human renal toxicity (Hard, Johnson, and Cohen Citation2009).
The Nagano et al. (Citation2007a) study is the most complete and detailed investigation evaluating CCl4 carcinogenicity and is by inhalation. Other studies by other routes of administration were performed but without extensive protocols followed by Nagano et al. (Citation2007a). Nagano et al. (Citation2007a) examination in rats and mice was by inhalation and major studies in mice by oral (gavage) are summarized in . The various investigations besides Nagano et al. (Citation2007a) on CCl4 carcinogenicity are described next.
Table 3. Summary of principle chronic/carcinogenicity rodent studies of carbon tetrachloride.
Other studies in mice
An early study by Eschenbrenner and Miller (Citation1946) in which CCl4 was assessed for carcinogenicity-involved administration to male and female strain A mice by oral gavage doses of 0.1, 0.2, 0.4, 0.8, or 1.6 ml/kg (160, 320, 640, and 2,550 mg/kg) CCl4 in olive oil. Doses were administered 1–5 days apart for a total of 30 doses and the experiment was terminated after 150 days. No tumors were observed in the mice administered 1.6 ml/kg for 30 doses over a period of 30 days, but mice receiving the other doses over a period of 90 days or more all developed liver tumors. Histopathology was not performed (Eschenbrenner and Miller Citation1946). This investigation was not described in detail, and only involved observation for 150 days. Further, this investigation involved administration to strain A mice, which were determined by the National Toxicology Program (NTP) as a strain inadequate for evaluation of carcinogenicity (Maronpot et al. Citation1983). Nevertheless, the results of this study support the hepatocarcinogenicity associated with CCl4 exposure in mice.
Male C3H mice were treated by oral gavage with CCl4 in corn oil at doses of 200, 400, or 1,600 mg/kg body weight 3 times weekly for 10 weeks. The experiment ended after 150 days from the first treatment. The incidences of hepatomas in mice that survived the 10-week treatment period were 5/8, 4/18, and 1/6 in the 200, 400, and 1,600 mg/kg groups, respectively. No hepatomas were detected in 28 untreated controls that survived 10 weeks or in 18 vehicle controls (Kiplinger and Kensler Citation1963). There were 30 mice per group at the start of the experiment except in the 400 mg/kg group which started with 60. The lack of a dose response, short term of the study, high mortality rate, and lack of use of current histopathologic criteria are significant limitations of this study.
In a more standardized study, male and female B6C3F1 mice were administered CCl4 as a 2–5% solution in corn oil by oral gavage 5 times per week at dose levels of 1,250 or 2,500 mg/kg body weight for 78 weeks as part of the National Cancer Institute (NCI) program of carcinogenicity testing, which evolved into the National Toxicology Program (NTP) Citation(1976a) (Weisburger Citation1977). The experiment was ended after 90–92 weeks from its beginning Citation(1976a) (Weisburger Citation1977). Pooled controls (77 males and 80 females) and matched controls (18 males and 18 females) were treated with corn oil only. The reason for the two different control groups is that this experiment was designed to study the carcinogenic effects of chloroform, and CCl4 was included as a positive control. Thus, the pooled controls were part of the overall chloroform study, while the matched controls were specific only to the CCl4 part of the experiment. It should be noted that the same CCl4 exposure groups seem to have been used as a positive control for a trichloroethylene bioassay (NCI National Cancer Institute Citation1976b). All treated mice at both doses developed hepatocellular carcinomas. From the pooled control groups, 5 of 77 (6%) males and 1 of 80 females (1%) were noted to exhibit liver hepatocellular tumors (classified as carcinomas). Mice from the matched control group displayed liver carcinomas in 3 in 18 (17%) of the males and 1 in 18 (5%) of the females.
Other studies in rats
Costa et al. (Citation1963) treated rats with CCl4 by inhalation for 7 months and then were sacrificed 2–10 months after the end of the treatment. The gender, exposure concentration, and schedule of administration were not specified. Among the 30 survivors, 12 exhibited what was diagnosed as “adenocirrhosis” and 10 displayed liver nodules up to 1 cm diagnosed histopathologically as liver carcinomas. However, this study had no controls, the details were not clearly specified, and histopathologic criteria for liver lesions were not described.
In the NCI studies in which CCl4 served as a positive control for chloroform and trichloroethylene bioassays (NCI Citation1976a; Citation1976b), 50 male and 50 female Osborne-Mendel rats were treated 5 times per week by oral gavage with CCl4 in corn oil for a total of 78 weeks. Males received 47 or 94 mg CCl4/kg body weight and females 80 or 160 mg CCl4/kg body weight. The animals alive at the end of the 78-week treatment period were then maintained on control diet and terminated at 110 weeks since the beginning of the study. Hepatocellular carcinomas were present in males at 2/50 (4%) at both doses; neoplastic nodules were 2/50 (4%) and 1/50 (2%) at the low and high doses, respectively. In females, hepatocellular carcinomas were present at 4/49 (8%) and 1/49 (2%) in the low- and high-dose groups, respectively. The incidence of neoplastic nodules in females was 2/49 (4%) and 3/49 (6%) in the low- and high-dose groups, respectively. Similar to the mouse study described above, this investigation was not specifically designed as a carcinogenicity study for CCl4, but CCl4 was included as a positive control Citation(1976a) (Citation1976b; Weisburger Citation1977). It is unclear from the reports if the various studies were run together, concurrently, or separately. Compared to the corresponding study in mice, the rat was less susceptible to liver tumor induction compared to mouse.
Kawasaki (Citation1965) administered to a group of 49 male Wistar rats oral gavage doses of 2–3 ml/kg (3,200–4,800 mg/kg body weight) of CCl4 in olive oil twice weekly for 25–35 weeks and terminated 4 to 78 weeks after the end of treatment. Hepatomas were recorded in two rats, but details of the study were not adequately described and there were no controls.
Reuber and Glover (Citation1967) administered a 50% solution of CCl4 in corn oil twice weekly by sc injection to groups of 10–14 male and female Buffalo rats at ages of 4, 12, 24, and 52 weeks when treatment began, with few hepatomas observed. This was followed by another study (Reuber and Glover Citation1970) in which 1.3 ml/kg body weight (2,000 mg/kg) CCl4 as a 50% solution in corn oil was administered twice weekly by sc injection to Wistar rats, Osborn-Mendel rats, and Japanese rats. The incidences of hepatocellular carcinomas in these different strains of rats that survived 68 or more weeks were 4 of 12 (33%), 8 of 13 (60%) and 12 of 15 (80%), respectively. However, many of the details of these studies were not provided and there were few animals in each group for each strain.
Another study of CCl4 by Alpert et al. (Citation1972) sc administration of CCl4 at 1 ml/kg body weight (1,600 mg/kg body weight) involved administration twice weekly for 2 years to 30 female rats at 5–6 months of age at the start of the experiment. Eight of these 30 rats (26%) developed mammary adenocarcinomas, 3 fibroadenomas, and one developed only fibroadenoma. No mammary tumors were observed in the 15 untreated controls (Alpert et al. Citation1972). Liver tumors were not reported in this study. No information on the incidences of mammary gland tumors in historical controls was provided, which is especially critical given the high spontaneous incidences of mammary gland tumors in some strains of rats.
Frezza et al. (Citation1994) administered CCl4 by oral gavage with a treatment regimen that included administration of 0.08 ml (128 mg) per rat for the first 6 weeks, then 1.1 ml (1,753 mg) per rat for 12 weeks and concluding with 1.6 ml (2,550 mg) per rat for the remaining 12 weeks. Female Sprague-Dawley rats were utilized for this study, and livers were examined histologically. Hepatocellular carcinomas were produced in 6 of 20 (30%) rats. The treatment protocol was not standard, there were relatively small numbers of rats in the study group, and no controls were available for evaluation and comparison.
Other species
Ten male and female Syrian Golden hamsters were treated with three weekly doses of 6.25 or 12.5 μL (approximately 10 or 20 mg) CCl4 in 5% corn oil. All five hamsters of each sex that survived 10 or more weeks had liver cell carcinomas (Della Porta, Terracini, and Shubik Citation1961). However, there were no control animals in this study, the details of the study were not provided, and the number of hamsters evaluated in each treatment group was small. Tanaka, Mori, and Williams (Citation1987) administered CCl4 by oral gavage to Syrian hamsters at a dose of 0.1 mL (160 mg) per animal every 2 weeks for 30 weeks alone or after an intraperitoneal injection with 6 mg/kg body weight N-nitrosodiethylamine (NDEA). Whereas CCl4 alone produced no liver tumors in these hamsters, liver tumors occurred with incidences of 1 of 15 (7%) and 11 of 13 (85%) in the NDEA and NDEA plus CCl4 groups, respectively.
Rainbow trout were administered 3,200 or 12,800 mg CCl4/kg in the diet. Four of 44 (11%) and 3 of 34 (11%) developed hepatomas after 20 months at the two doses, respectively, and there were no liver tumors in the controls (Halver Citation1967). Details of the study were not provided, nor was a statistical comparison provided. No information regarding historical controls or actual dose was presented.
CCl4 administered combined with other agents
Hybrid mice (C57L × A F1) were treated with whole-body exposure to fast neutrons (ranging from 165 to 306 rad), followed by a single sc injection of CCl4 at a dose of 0.15 ml (240 mg) of 40% solution in sesame oil, and then followed for 2–18 months. Hepatoma levels were increased from 19% to 61% for the 2 radiation doses, with no hepatomas in the controls given only CCl4 (Cole and Nowell Citation1964). These results are similar to findings with a similar treatment schedule reported by Curtis, Tilley, and Crowley (Citation1964). However, many of the details regarding these experiments were not provided, radiation dose varied between animals, and criteria for histopathology diagnosis of tumors not stated, limiting interpretation of these studies.
In mice treated with a single dose of CCl4 followed by a single dose of N-nitrosobutylurea 8 hr later, the incidences of hepatomas and leukemias were elevated compared to mice treated with only N-nitrosobutylurea (Takizawa et al. Citation1975). Information regarding historical controls and other types of tumors in this strain of mouse have not apparently been reported. Further, the study was not designed to evaluate the carcinogenicity of CCl4 but was part of an experiment to evaluate various N-nitrosobutylurea-related chemicals. Similarly, an increased incidence of liver and kidney tumors was present in mice treated with a single dose of N-nitrosodiethylamine (NDEA) one or two days after an injection of CCl4 in mice (Pound Citation1978). However, the details of the treatment regimen were not adequately described.
A single dose of NDEA to rats and a single dose of CCl4 one day later resulted in elevated incidences of liver and kidney tumors compared to controls (Pound, Lawson, and Horn Citation1973). The details of the administration schedule and histopathology were not adequately described. Similarly, Lemonnier, Scotto, and Thuong-Trieu (Citation1974) found incidences of hepatomas were increased in rats receiving a single dose of aflatoxin followed by chronic administration of CCl4. Kanematsu (Citation1976) noted a rise in liver tumor incidences in female Donryu rats given short-term treatment with 3-methyl-4-dimethylaminoazobenzene followed by CCl4 treatment. B6C3F1 mice were treated by oral gavage with 1,600 mg/kg CCl4 dissolved in corn oil once every other week following a single dose of 15 mg/kg body weight NDEA and then terminated at 36 weeks of age. The number and volume of hepatocellular nodules were increased in the NDEA plus CCl4-treated mice compared to the NDEA alone-treated mice. However, no apparent hepatocellular nodules were detected in mice receiving only CCl4. Specific details of this study and the results were not provided, and there was no information regarding historical ranges in this lab for treatment with NDEA only (Dragani, Manenti, and Della Porta Citation1986).
Fischer 344 (F344) rats were treated with 2-acetylaminofluorene (2-AAF) in the diet for 2 weeks and then administered a single oral dose of 1,600 mg/kg CCl4 dissolved in olive oil. This was followed with phenobarbital in the diet at a concentration of 500 mg/kg for 6 weeks, followed by two-thirds partial hepatectomy. The animals were then terminated at the end of 8 weeks of treatment. Qualitative analysis of the hyperplastic nodules in the liver showed that the number and area were increased significantly in the animals that were treated with CCl4. No hyperplastic nodules were observed in animals dosed with CCl4 but not pre-treated with 2-AAF. This is a complex protocol involving relatively small numbers of animals, with wide variability in the background incidences in rats treated with 2-AAF, partial hepatectomy, or the two together, not taking into account treatment with CCl4 (Takano et al. Citation1980).
F344 rats were treated by i.p. injection 3 times per week with NDEA dissolved in 0.9% saline up to a total dose of 200 mg/kg body weight for up to 6 weeks (Zalatnai et al. Citation1991). After completion of NDEA treatment, rats were dosed by oral gavage with 0.2 ml/kg (320 mg/kg) CCl4 dissolved in corn oil twice per week for 3 months. All animals were terminated 8 months after the start of the experiment. The incidences of hepatocellular carcinomas were 17 of 17 rats in the NDEA plus CCl4 treatment group compared to 9 of 17 (52%) rats in the NDEA only treatment. No hepatocellular carcinomas were detected in a group of 15 rats (0%) that received CCl4 only (Zalatnai et al. Citation1991). There was no information on the background incidences on liver tumors in nonexposed animals, making statistical comparisons difficult. This is particularly true in the experiment by Zalatnai et al. (Citation1991), where the incidence of liver tumors was already high, with 9 of 17 (52%) in rats treated with NDEA only.
Sprague Dawley newborn rats received a single i.p. injection of 15 mg/kg body weight NDEA dissolved in 0.1 ml normal saline at one day of age (Cho and Jang Citation1993). Beginning at 3 weeks of age (weaning), female rats received i.p. injections of a 33% solution of CCl4 in 0.25 ml mineral oil twice weekly for 9 weeks. The rats were sacrificed at week 12 of age and hepatocellular nodules were determined quantitatively. The incidences of foci of cellular alteration were similar between NDEA plus CCl4 group and NDEA only (15/20 and 10/10 for the NDEA plus CCl4 and the NDEA only groups, respectively), but the incidences of neoplastic nodules were elevated with CCl4, treatment (13/20 and 0/10 for the NDEA plus CCl4 and the NDEA only groups, respectively).
Areas of GST-P-positive neoplastic nodules were significantly larger in the NDEA plus CCl4 group compared to NDEA alone (Cho and Jang Citation1993). This experiment was also poorly described, and similar to other investigations with the administration of CCl4 combined with other agents, the historical incidences were not provided. Further, histopathologic criteria for the different liver lesions were not described and diagnoses did not appear to be consistent with currently accepted standards (Thoolen et al. Citation2010). These combined assays provide some additional evidence that there is a tumorigenic effect on the liver by CCl4, however, these data are difficult to be used by themselves for risk assessment evaluation.
In a study involving C57BL/6 mice, groups of 30 male and 30 female mice were irradiated with a single dose of fast neutrons (0, 170, or 330 rads), followed by a single sc injection of 3,000 mg/kg body weight CCl4 dissolved in corn oil, and then observed over their lifetime. The incidences of liver carcinomas were increased only in female mice exposed to the high-dose (330 rads of fast neutrons) and CCl4. No liver tumors occurred in the females treated only with CCl4 without prior treatment with fast neutrons. Statistical significance was not provided, nor were the standards for evaluation of the histopathology. No liver carcinomas occurred in the females treated only with CCl4 without prior treatment with fast neutrons (Habs et al. Citation1983). In another study, ACI rats were administered 0.5 ml/kg body weight (800 mg/kg) CCl4 by oral gavage and then treated with 25 mg/kg body weight methylazoxymethanol acetate i.p. once per week for 4 weeks, and terminated 30 weeks later. There were no intestinal or liver tumors noted in rats treated only with CCl4 nor alterations in incidences of intestinal or liver tumors in rats treated with CCl4 and methylazoxymethanol acetate compared to those treated only with methylazoxymethanol acetate (Kato et al. Citation1985).
Subchronic inhalation study Nagano et al. (Citation2007b)
Since the major route of human exposure to CCl4 exposure is by inhalation, the proposed liver tumor MOA analysis relies primarily on the subchronic and chronic inhalation experiments in rats and mice performed by the JBRC (Nagano et al. Citation2007a, Citation2007b). The subchronic study involved the administration of F344 rats and BDF1 mice at concentrations of 0, 10, 30, 90, 270, or 810 ppm CCl4 vapor for 6 hr per day, 5 days per week, for 13 weeks. A rise in relative liver weights was reported for all exposed male and the ≥30 ppm exposed female rats. In mice, the relative liver weights were increased in males at ≥30 ppm and in females at 270 and 310 ppm. Indications of cell death were confirmed by both histopathology and by elevation of specific serum enzymes, including AST, ALT, and LDH. In rats, all serum liver enzymes were elevated in male rats at 270 and 810 ppm and in females at ≥30 ppm; ALT was also elevated at 90 ppm in males (). For mice, Nagano et al. (Citation2007, Citation2007) noted that not all enzymes were evaluated because of limitations on the amount of blood available for evaluation. In male mice, AST was increased at ≥270 ppm, ALT at ≥90 ppm and ALP (alkaline phosphatase) at ≥30 ppm (). In the female mice, AST and ALP were not significantly affected, but ALT was elevated at ≥90 ppm.
Table 4. Hematological, blood biochemical, and urinary parameters of rats exposed to CCl4 vapor by inhalation for 13 weeks (Nagano et al. Citation2007b).
Table 5. Hematological and blood biochemical parameters of mice exposed to CCl4 vapor by inhalation for 13 weeks (Nagano et al. Citation2007b).
The reported histopathologic findings are most critical in defining the MOA and relative exposures for producing adverse effects. As presented in , fatty change, the most characteristic finding related to CCl4 toxicity, was present in all male rats at ≥30 ppm and in 2/10 at 10 ppm. The female rats all exhibited fatty change at doses ≥30 ppm and also in 2/10 at 10 ppm. Fibrosis was also present in the male at 270 and 810 ppm and in the female rats at ≥90 ppm. Cirrhosis was present in the male at 810 ppm, in 2/10 male rats at 270 ppm, and in the female rats at 270 and 810 ppm. At this early time point, there was already evidence of altered hepatocellular foci in 3 of 10 male rats at 270 ppm and 10 of 10 male rats at 810 ppm. In the female rats, altered hepatocellular foci were present in 3 of 10 at 90 ppm, and 10 of 10 at 270 and 810 ppm.
Table 6. Incidences and severities of selected histopathological lesions in rats exposed to CCl4 vapor by inhalation for 13 weeks (Nagano et al. Citation2007b).
In mice, there was also evidence of fatty changes in liver of males at ≥10 ppm and females at ≥30 ppm (). Liver cell architectural collapse was found in nearly all male and female mice at ≥30 ppm CCl4. Altered cell foci were present in 5 of 10 and 7 of 10 male mice at 270 and 810 ppm, respectively; and in 2 of 10 female mice at 270 ppm. Data demonstrate that there was a rise in cytotoxicity at doses of ≥30 ppm in male and female rats and mice, with some evidence of liver toxicity even at 10 ppm. It should be noted that the lowest exposure level in the 90-day study was 10 ppm, whereas 5 ppm was the lowest exposure level in the two-year bioassay (Nagano et al. Citation2007). Thus, there appears to be a significant correspondence between exposures that induced cell death and fatty changes at 90 days and development of liver tumors following exposure for 2 years.
Table 7. Incidences and severities of selected histopathological lesions in mice exposed to CCl4 vapor by inhalation for 13 weeks (Nagano et al. Citation2007b).
MOA for liver tumors
The rodent liver is the most common target for neoplasm formation in rodent chronic bioassays of chemical carcinogens (Holsapple et al. Citation2006). As such, considerable study has been devoted to understanding the MOA underlying liver carcinogenesis in rodents (Cohen Citation2010; Holsapple et al. Citation2006; Klaunig and Wang Citation2018). It is generally accepted that for the rodent liver tumor induction by chemicals, there are two general categories of MOA: DNA Reactivity/Genotoxicity and Non-Genotoxicity (). Genotoxic agents exhibit DNA reactivity and mutation, while the hallmark of non-genotoxic agents is the increase in growth (enhanced cell proliferation/decreased apoptosis) through receptor activated mitogenicity or cytotoxicity with consequent regenerative proliferation (Cohen Citation2010). The nongenotoxic liver tumor MOA may be further divided into those carcinogens that initiate nuclear receptor activation and those that are non-receptor mediated processes (). Cytotoxicity with regenerative proliferation as a MOA may or may not be induced by a specific receptor-mediated process.
Table 8. Modes of action for hepatocellular carcinogenesis (modified from Cohen Citation2010).
MOA for CCl4-induced rodent liver tumors
Established rodent liver MOAs are provided in , and the proposed MOA for CCl4-induced rodent liver tumors based upon a review of the extensive database of studies involving CCl4 and rodent liver effects is presented in , utilizing the EPA/Health Canada (Meek et al. Citation2003; Seed et al. Citation2005) and IPCS (Boobis et al. Citation2006, Citation2008; Sonich-Mullin et al. Citation2001) MOA/human relevance framework.
Table 9. Proposed mode of action for CCl4-induced rodent tumors.
The observed rodent liver tumors induced by CCl4 have consistently been shown to involve hepatocellular cytotoxicity (cell death) with a resulting compensatory proliferation (hyperplasia) (Nagano et al. Citation2007a, Citation2007b; Manibusan et al. Citation2007). The hepatotoxic effect of CCl4 was demonstrated in rodents through various routes of administration including oral (gavage), ip and sc injection, and inhalation. The postulated key events for CCl4 -mediated liver carcinogenicity involve ():
Figure 1. Mode of action for carbon tetrachloride carcinogenesis.
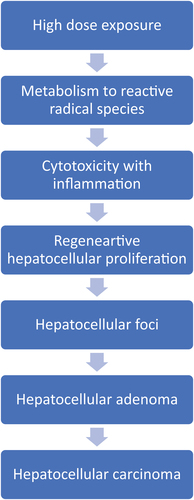
Metabolism of CCl4 to trichloromethyl and trichloromethyl peroxy radicals, resulting in
Hepatocellular cytotoxicity (cell death), followed by
Induction of hepatocyte proliferation via compensatory hyperplasia, leading to
Induction of preneoplastic cells with the production of hepatocellular foci, leading to
Formation of hepatocellular neoplasms (adenomas and carcinomas).
The sequence from foci to adenomas to carcinomas is the well-documented progression of events for hepatocellular malignancies in rats and mice that was noted with a variety of different rodent liver chemical carcinogens administered by various routes and involving various MOAs (Dragan et al. Citation1993; Farber and Sarma Citation1987; Maronpot et al. Citation1987; Thoolen et al. Citation2010). Therefore, the main focus of our discussion on the CCl4 MOA involves the relationship with cytotoxicity and resulting necrosis and regenerative proliferation. Each key event in this MOA is dose-dependent, both with the concentration of CCl4 and duration of exposure, and occurs at relatively high CCl4 exposures, resulting in elevated in liver tumor incidence in this high exposure range. The experimental literature support for each key event is provided below.
Key event 1: metabolism of CCl4 to reactive radical species
Central to the proposed CCl4 MOA is the metabolism of CCl4 to reactive radicals. The metabolism of CCl4 was determined in both in vivo and in vitro mammalian systems. CCl4 is metabolized primarily in the liver. The initial step in the biotransformation of CCl4 is reductive dehalogenation mediated by CYP2E1 to form the trichloromethyl radical, which may be converted to the trichloromethyl peroxy radical by binding oxygen (Packer, Slater, and Willson Citation1978; Lai et al. Citation1979; Reinke and Janzen Citation1991; Slater Citation1981, Citation1982). Both of these radicals participate in the observed hepatocyte toxicity attributed to CCl4 by binding directly to microsomal lipids and proteins (Ansari et al. Citation1982; Fanelli and Castro Citation1995), as well as the heme portion of CYP450 (Noguchi et al. Citation1982a, Citation1982b).
While CYP2E1 is the primary enzyme associated with CCl4 bioactivation, CYP3A was reported to be involved in its metabolism under high-dose conditions (Raucy, Kraner, and Lasker Citation1993). The linkage between metabolism of CCl4 by CYP2E1 and resulting liver toxicity was examined using inducers and inhibitors of CYP450. Sipes, Krishna, and Gillette (Citation1977) found that CCl4 metabolism was inducible by phenobarbital or ethanol. Studies with general CYP450 inhibitors such as SKF-525A inhibited the metabolism of CCl4 and also prevented liver damage (Letteron et al. Citation1990). Similarly, Wong, Chan, and Lee (Citation1998) using CYP2E1 null mice examined the role of CYP2E1 in CCl4-induced liver toxicity. Using wild-type mice, Wong, Chan, and Lee (Citation1998) demonstrated that CCl4 induced a significant rise in serum ALT and AST along with centrilobular necrosis in the liver 24 hr following an i.p. dose of 1 ml/kg CCl4. In contrast, the same CCl4 treatment to CYP2E1 null mice exerted no marked effect on liver enzymes or histopathology. Several investigators reported that liver cell lines that over-express CYP450 enzymes contain elevated levels of CCl4-induced cytotoxicity (Dai and Cederbaum Citation1995; Takahashi et al. Citation2002).
Key event 2: hepatocellular cytotoxicity (cell death) and necrosis
Liver cell toxicity attributed to CCl4 is related to its metabolism and production of trichloromethyl and trichloromethyl peroxy radicals. These radicals are highly reactive, producing cell injury by lipid peroxidation and covalent binding to cellular macromolecules (Castro, Díaz Gómez, and Castro Citation1989; Diaz Gomez and Castro Citation1980; Reinke and Janzen Citation1991). Studies using radiolabeled CCl4 noted binding to cellular macromolecules following bioactivation in various in vitro and in some in vivo systems, with membrane phospholipids displaying the highest rate of binding (Castro and Diaz Gomez Citation1972; Castro, Díaz Gómez, and Castro Citation1989). Covalent DNA binding was also detected both in vivo and in vitro investigations using C14 labeled CCl4 (Castro, Díaz Gómez, and Castro Citation1989; Rocchi, Prodi, and Grilli Citation1973). The significance of these studies is very limited due to (1) the DNA isolation methodology used, (2) concerns regarding the isolated DNA purity, and (3) possible contamination with other cellular components. In addition, the reported DNA binding of CCl4 in these studies did not correlate with its lack of mutagenicity. Radiolabelled pulse experiments demonstrated that the trichloromethyl peroxy radical is more reactive than the trichloromethyl radical to cellular macromolecules (Packer, Slater, and Willson Citation1978). Of the cellular components targeted by CCl4-produced radical species induction of lipid peroxidation is the most consistent finding detected. The resulting lipid peroxidation produces conjugated dienes, such as 4-hydroxynonenal (4-HNE), as well as reactive aldehydes such as malonaldehyde (MDA) (Lai et al. Citation1979; Recknagel, and Glende Citation1973; Slater Citation1981). Cellular calcium homeostasis is also affected by CCl4 exposure; likely due to inhibition of active calcium transport within cells (i.e., endoplasmic reticulum and mitochondria membranes) and across the plasma membrane, resulting in elevated intracellular calcium levels (Kroner Citation1982; Long and Moore Citation1986; Racay et al. Citation1997). The influence of CCl4 on membrane integrity and on active transport appears to be secondary to lipid peroxidation (Racay et al. Citation1997). With the disruption of normal calcium transport, increased intracellular calcium levels activate a number of cytosolic and lysosomal enzymes including proteases and phospholipases which further breakdown the function and structure of the membrane. Using pretreatment with deferoxamine, an iron chelator and inhibitor of iron-catalyzed lipid peroxidation attenuated CCl4 hepatotoxicity in mice was found (Younes and Siegers Citation1985). This effect was also shown not to be attributed to inhibition of metabolic activation of CCl4 (Younes and Siegers Citation1985). A similar effect was noted for acetaminophen-induced hepatotoxicity. Numerous investigators demonstrated the occurrence of lipid peroxidation following CCl4 exposure, either by detection of conjugated dienes (a characteristic marker of lipid peroxidation) or liver lipids (Lee, McCay, and Hornbrook Citation1982; Letteron et al. Citation1990; Racay et al. Citation1997; Slater Citation1982). Hartley et al. (Citation1999) reported a 5-fold rise in serum ALT at 6 hr post treatment in rats dosed by gavage (1 ml/kg) CCl4, which peaked at 36 hr with a 32-fold increase in ALT over control. Between 18 and 36 hr post-treatment, TBARS (lipid peroxide-derived aldehydes) values in liver homogenates of treated rats were elevated to a maximal level of 2.5-fold over controls. Malondialdehyde-amine and 4-hydroxynonenal sulfhydryl protein adducts were also detected, indicating lipid, protein and nucleic acids as targets for the trichloromethyl radicals following CCl4 metabolism (Hartley et al. Citation1999).
Key event 3: induction of hepatocyte proliferation via compensatory hyperplasia
As a result of cytotoxicity in the liver of CCl4-treated animals, significant compensatory regenerative hepatocellular proliferation occurs to compensate for the necrotic or damaged tissue. There is a reliable correlation (particularly at higher exposures, i.e. >5 ppm by inhalation) between occurrence of hepatotoxicity and/or regenerative/proliferative lesions and development of tumors (Nagano et al. Citation2007, Citation2007). Investigators have linked CCl4-induced cytotoxicity with chronically stimulated cellular proliferation (Doolittle, Muller, and Scribner Citation1987; Nakata et al. Citation1985). Findings from the JBRC studies are consistent with the hypothesis that liver tumors occur only at exposure levels that produced hepatotoxicity in both rats and mice. The dose-response for liver tumors in rats and mice in the 2-year inhalation study (Nagano et al. Citation2007a) which is comparable to the dose-response for the features of cytotoxicity in the 90-day inhalation studies (Nagano et al. Citation2007b). Several subchronic inhalation and oral (gavage) investigations demonstrated that CCl4-mediated hepatic toxicity and regeneration (Benson and Springer Citation1999; Lee, Cameron, and Archer Citation1998; Nakata et al. Citation1985). In rodents exposed to CCl4 vapor for up to 12 weeks, the Lowest-Observed-Adverse-Effect-Concentrations (LOAECs) for tissue damage were found at concentrations ranging from 4 to 42 ppm (adjusted to continuous exposure) and for hyperplasia/regeneration at concentrations ranging from 4 to 20 ppm (adjusted) depending upon species, with the mouse more sensitive to the toxicity of CCl4 than the hamster, and the rat least sensitive (Benson and Springer Citation1999). Thus, the results of subchronic exposure studies are consistent with data from the JBRC study in rats, showing cytotoxicity at ≥10 ppm and hyperplasia/proliferation at ≥30 ppm after 13 weeks of exposure (Nagano et al. Citation2007b), and cytotoxicity and hyperplasia/regeneration at ≥25 ppm after 104 weeks of exposure (Nagano et al. Citation2007a). In rats and mice exposed orally to CCl4 for up to 12 weeks, the LOAELs for tissue necrosis and hyperplasia/regeneration ranged from 8.6 to 80 mg/kg-day and 12 to 71 mg/kg-day (adjusted for continuous), respectively, depending on species (Benson and Springer Citation1999). Based upon the findings in these subchronic and longer studies, the NOAEL for the liver cytotoxic, proliferative and tumorigenic effects following inhalation of CCl4 in mice and rats appears to be 5 ppm, based upon 5 days per week administration.
Cellular damage (necrosis) described above leads to reparative cellular proliferation. Benson and Springer (Citation1999) examined F344 rats, B6C3F1 mice, and Syrian hamsters (10 males/species) exposed by inhalation to CCl4 vapor at 0, 5, 20, or 100 ppm, 6 hr/day, 5 days/week for 12 weeks. Serum levels of ALT and SDH (markers of membrane damage) were significantly increased in mice at concentrations ≥20 ppm, and in rats and hamsters at 100 ppm. Bromodeoxyuridine (BrdU) labeling, a measure of cell proliferation, were also significantly elevated in mice at ≥20 ppm and hamsters at 100 ppm. In mice, the % BrdU positive hepatocytes at 12 weeks were approximately 20% at 20 ppm exposure concentration and 60% at 100 ppm. In hamsters at 100 ppm, the % BrdU positive hepatocytes at 12 weeks were approximately 40%. Evidence indicates that the occurrence of hepatocellular proliferation only at exposures which also produced necrotic damage and liver serum enzyme release. Lee, Cameron, and Archer (Citation1998) examined the time course and distribution of toxicity and repair in livers of male Sprague-Dawley rats at time points of 24, 36, and 48 hr following a single oral gavage dose of 40 or 400 mg/kg CCl4 in corn oil. The high dose (400 mg/kg) produced extensive hepatocyte damage in the perivenous-to-mid-lobular zones of the liver. Administration of 40 mg/kg induced regenerative hepatocyte proliferation, as indicated by a significant elevation in BrdU-positive cells in the periportal zone (the site of necrosis) at 24 hr, increasing at 36 hr, and plateauing at 48 hr. BrdU-positive cells were near the portal tract at the 24 hr sampling time. At later sampling times labeling was seen increasingly in the outer periportal and mid-lobular zones. Evidence of regeneration of livers in animals treated with CCl4 appears within 48 hr of dosing. Data are in agreement with other reports (Benson and Springer Citation1999; Doolittle, Muller, and Scribner Citation1987) that when hepatocyte death occurs following CCl4 treatment, the remainder of the liver undergoes compensatory hyperplasia replacing necrotic cells.
For calculating the oral RfD for CCl4, the liver was determined to be the most sensitive target organ for toxicity across routes of exposure and animal species (U.S. EPA United States Environmental Protection Agency Citation2010, Citation2020). The previous oral RfD of 0.0007 mg/kg-day for CCl4 (see IRIS, U.S. EPA Citation2010) was based upon the 12-week study by Bruckner et al. (Citation1986). In the 2010 IRIS (U.S. EPA United States Environmental Protection Agency Citation2010) document on CCl4, this same study (Bruckner et al. Citation1986) was used to derive the current RfD. In this study, male Sprague-Dawley rats were treated via gavage with 0, 1, 10, or 33 mg/kg of CCl4 for 5 days/week for 12 weeks. Blood was taken throughout the treatment period for serum liver and kidney markers. Both kidney and liver were examined by histopathology at the study termination. An increase in serum sorbitol dehydrogenase (SDH) was reported at doses of 10 mg/kg, but not 1 mg/kg. No marked renal effects were observed at any dose. The Bruckner et al. (Citation1986) study revealed a no-observed-adverse-effect level (NOAEL) of 1 mg/kg and a LOAEL of 10 mg/kg in rats administered CCl4 by gavage for 5 days/week at doses of 0.71 and 7.1 mg/kg-day (adjusted for daily treatment), respectively. In mice, Condie et al. (Citation1986) noted a NOAEL of 1.2 mg/kg and a LOAEL of 12 mg/kg in gavage treated mice (0.86 and 8.6 mg/kg-day, respectively (adjusted for daily exposure). In the rat and the mouse investigations, a LOAEL of 10–12 mg/kg (daily dose of 7–9 mg/kg-day) resulted in a rise in serum liver enzyme activity.
In strain A mice dosed with 2550 mg/kg of CCl4 in olive oil, necrosis was detectable in half the hepatocytes at 24 hr and mitotic activity appeared 48 hr after dosing (Eschenbrenner and Miller Citation1946). Wistar rats treated with 5 ml/kg (7,970 mg/kg) exhibited peak ALT levels at 24 hr, peak AST levels at 48 hr, and significantly elevated levels for activities of DNA-synthesizing enzymes thymidine kinase and thymidylate synthetase at 48 and 72 hr (Nakata et al. Citation1985). Activity of these DNA-synthesizing enzymes were reduced 96 hr after treatment. Doolittle, Muller, and Scribner (Citation1987) examined the relationship between hepatic toxicity induced by CCl4 and DNA synthesis in the liver of male CD-1 mice. Mice were dosed by gavage with CCl4 at a dose of 20 or 25 mg/kg for either one day (25 mg/kg) or daily for 7 days (20 mg/kg). A single dose of CCl4 at 25 mg/kg did not markedly alter DNA replication, or activities of AST and ALT. However, treatment with 20 mg/kg CCl4 daily for 7 days produced a 10-fold increase in hepatocyte replication as well as the levels of liver enzymes (AST, ALT). Data indicate that induction of replicative DNA synthesis in liver after CCl4 treatment is a consequence of hepatotoxicity.
The influence of CCl4 on liver gene expression was examined in vivo in rats and mice and in vitro in rodent and human liver cell cultures. Three of these studies are highlighted below (Columbano et al. Citation1997; Fountoulakis et al. Citation2002; Holden et al. Citation2000). There is a general consistency of the results from these studies with many of the upregulated genes following CCl4 treatment being related to stress, DNA damage and repair, cell proliferation, and signal transduction. However, a limitation of these experiments is that only a single time point was usually examined. Fountoulakis et al. (Citation2002) treated Wistar rats with a single low dose (0.25 ml/kg; 400 mg/kg CCl4) or single high dose (2 ml/kg; 3,190 mg/kg CCl4) by gavage in corn oil. Controls received corn oil only. Rats were sampled after 6 and/or 24 hr post treatment. At the low dose, AST and ALT serum levels were unchanged from control after 6 and 24 hr. In contrast, in the high-dose group, AST and ALT levels were significantly increased (approximate 3-fold) after 6 hr and almost 10-fold from control. Histopathology correlated with the serum liver enzyme levels. Protein analysis examined 192 proteins from an enriched mitochondrial fraction of the liver at both doses. Of these, 18 proteins demonstrated a fall in expression levels and three were upregulated. Stress-related proteins, including catalase and uricase, were upregulated. Downregulated proteins included enzymes related to the metabolism of lipids and amino acids, including 2-macroglobulin and senescence marker protein (SMP-30), both of which have been linked to aging and hepatocyte toxicity. Upregulated genes were related to a stress response, DNA and protein damage (such as proteasome components, heat-shock proteins, heme oxygenase, DNA-damage-inducible genes, and apoptosis-related genes). In addition, CCl4 treatment showed downregulation of genes for GST-ya and GST-yb expression and upregulation of apoptosis-related genes. Genes involved in metabolic pathways, including some P450, were downregulated in the CCl4-treated animals. Bulera et al. (Citation2001), using an Affymetrix GeneChip array with 1600 genes, also found upregulation of GST-yb expression but reported no significant changes in apoptotic gene expression 24 hr after a single dose of CCl4 to male Wistar rats at a dose of 0.5 ml/kg (800 mg/kg).
Columbano et al. (Citation1997) compared differences in gene expression in livers of female CD-1 mice with compensatory hyperplasia induced by partial hepatectomy (PH) or CCl4 with hyperplasia mediated by mitogenic non-genotoxic carcinogen TCPOBOP. Using female CD-1 mice, compensatory hyperplasia was initiated by 2/3 partial hepatectomy or by treatment with a single gavage dose of CCl4 (2 mg/kg). TCPOBOP treatment (3 mg/kg) was also via a single gavage dose. With CCl4 treatment, the expression of several cell proliferation-related genes (c-fos, c-jun and c-myc) as well as LRF-1, IGFBP-1 and PRL-1 were upregulated. The timing and the amount of enhanced expression of these genes were similar to that following PH. In contrast, the Columbano et al. (Citation1997) noted that genes involved after PH and CCl4 treatment were different that those observed following TCPOBOP treatment. These results, together with other studies (Coni et al.,Citation1989; Menegazzi et al., Citation1997), and Columbano et al. (Citation1997) showed that activation of transcription factors such as AP-1, NF-kB or immediate early genes (c-fos, c-jun, LRF-1, egr-1 and c-myc) indicated differences in gene expression patterns and timing between mitogenic nongenotoxic carcinogens and CCl4 or PH treatment.
Holden et al. (Citation2000) examined gene expression in CCl4-treated cultured hepatoma cells using DNA microarrays. Holden et al. (Citation2000) reported that 47 different genes were either upregulated or downregulated by more than 2-fold after CCl4 treatment. Of note was the upregulation of proinflammatory cytokine interleukin-8 (IL-8), which was elevated over 7-fold compared with control. IL-8 protein levels were also measured and treatment with CCl4 produced a time-dependent rise in IL-8 protein expression which followed the increase in IL-8 gene expression. The IL-8 gene expression correlated with decrease in cell viability as evidenced by LDH release.
In recent years, more sophisticated and extensive gene expression platforms have been developed. Several large databases are now available that enable sharing of RNA sequences (RNA-Seq), pathway analysis, and general gene expression results from experimental studies. These include DrugMatrix which contains results from thousands of experiments where rats or primary rat hepatocytes were treated with over 600 therapeutic, industrial, or environmental chemicals (TG-Gates, a public rat DNA microarray database of studies). In addition, with new advances major efforts are underway to utilize next-generation RNA-Seq as well as conventional DNA microarrays to identify differentially expressed protein-coding genes (DEGs) in treated versus control liver. These approaches have been involved in predicting potential rodent and human carcinogens using short term in vivo and in vitro results as well as examining ways to distinguish genotoxic versus nongenotoxic MOA of a chemical (Furihata and Suzuki Citation2023). A review of the databases where CCl4 was employed has not revealed major new differences in gene expression findings from those described above. These all support the overall MOA of cytotoxicity with consequent regenerative proliferation for CCl4-induced hepatocellular carcinogenesis. More detailed analyses of the extensive databases that are available are beyond the scope of this review.
Key event 4: Induction of preneoplastic cells with the production of hepatocellular foci
The subchronic inhalation study by Nagano et al. (Citation2007b) illustrates the relationship between cell damage (death) (assessed by both serum enzymes and histopathology) and resulting increased cell proliferation and focal lesion formation. In rats exposed to CCl4 for 13 weeks, histopathological alterations indicative of cellular damage, inflammation proliferative and regenerative liver changes were observed at exposure levels at 270 ppm, but significantly elevated fatty change was detected at >10 ppm. At ≥270 ppm, preneoplastic eosinophilic and basophilic foci were also observed. Similarly in mice exposed to CCl4 via inhalation for 13 weeks, dose-dependent histopathological findings including cytotoxicity, hepatocyte proliferation, and preneoplastic lesions (foci) were found. At ≥30 ppm, the mice exhibited significantly increased necrotic loss of hepatocytes, accompanied by hepatocyte proliferation as well as proliferation of bile ducts and oval cells; at ≥270 ppm, the incidences of atypia and altered cell foci were significantly elevated. The appearance of preneoplastic foci at this early exposure (90 days) time frame demonstrates the start of the progression of hepatocytes from normal to preneoplastic to neoplastic. At this early time point, there was already evidence of altered cell foci in 3 of 10 male rats at 270 ppm and 10 of 10 male rats at 810 ppm. In the females, altered cell foci were present in 3 of the 10 female rats at 90 ppm and in all 10 of the female rats at 270 and 810 ppm. Altered cell foci were present in 5 of 10 and 7 of 10 male mice at 270 and 810 ppm, respectively, and in 2 of 10 female mice at 270 ppm. Data demonstrate that there was a rise in cytotoxicity at doses of 30 ppm and above. Thus, there was strong concordance between the finding of cell death and fatty change at 90 days corresponding to the development of liver tumors at later time points. Although not all eventual liver neoplastic doses of CCl4 in this short exposure study reported foci to be present, these findings coupled with the presence of foci in the 2-year chronic assay confirm this intermediate stage in the progression to neoplasia. Hepatocellular foci are known as a precursor of hepatocellular adenomas in rodents (Farber and Sarma Citation1987; Maronpot et al. Citation1987; Thoolen et al. Citation2010). In many long-term bioassays, foci were replaced by adenomas and/or carcinomas, with few foci detected at 18–24 month time points (Maronpot et al. Citation1987).
Key event 5: Formation of hepatocellular neoplasms (adenomas and carcinomas)
This key event is self-explanatory in that chronic treatment of rodents with CCl4 at exposures that induce hepatocyte necrosis and resulting compensatory hepatocyte cell proliferation produces hepatic adenomas and carcinomas which represent progression of hepatocellular foci to neoplasia (Nagano et al. Citation2007a, Citation2007b). This progression from preneoplastic foci to neoplasia is a well-established sequence for hepatocellular carcinogenesis in rodents (Farber and Sarma Citation1987; Holsapple et al. Citation2006; Maronpot et al. Citation1987).
Temporality
Temporality clearly was found by Nagano et al (Citation2007a, Citation2007b), as well as other investigators (Benson and Springer Citation1999; Doolittle, Muller, and Scribner Citation1987) for CCl4-induced liver tumor MOA Key Events. Cell toxicity, fatty change, cell necrosis, and elevated liver serum enzymes occur early after CCl4 treatment, which are followed by compensatory/regenerative proliferation as evidenced by enhanced cell replication labeling indices. There is subsequent development of hepatic altered cell foci, adenomas and ultimately carcinomas. As indicated above, this sequence of events is typical for the development of hepatocellular tumors in rats and mice (Farber and Sarma Citation1987; Maronpot et al. Citation1987; Thoolen et al. Citation2010).
Strength, consistency, and specificity
The strength, consistency, and specificity of the findings are also reliable. The observations were consistent in Nagano et al (Citation2007a, Citation2007b), as well as in numerous other studies described above examining the findings of cell toxicity and regenerative proliferation. This was demonstrated following various routes of administration as well as at various doses or exposure concentrations. Critically, the exposure–response relationship appears to be similar for cell toxicity and for tumor development, with the NOAEC being 5 ppm from inhalation exposure (Nagano et al. Citation2007a, Citation2007b).
Biological plausability
The biological response of the proposed MOA for CCl4-induced rodent liver tumors is also highly plausible since cell toxicity (cell death) with consequent regenerative proliferation leading to tumor development is a frequent MOA not only for hepatocarcinogens but also for induction of tumors in other organs. The chemical most closely related to the CCl4 findings with tumor development in rats and mice is chloroform (Andersen et al. Citation2000). Cell toxicity has been well-described for chloroform leading ultimately to compensatory/regenerative proliferation, altered cell foci, adenomas, and carcinomas. The findings of tumors in chloroform-treated animals only occur at doses or exposures that also produced cytotoxicity. Doses or exposures which did not produce cytotoxicity exerted no marked tumorigenic response. This represents a threshold MOA (Andersen et al. Citation2000; Meek et al. Citation2003). The findings for CCl4 in the liver are clearly coherent, since liver tumors are consistently found not only following inhalation exposure but also following other routes of exposure as described above. In contrast to chloroform, CCl4 did not initiate tumors in the kidney.
Dose-response concordance
The exposure–response relationship between hepatic cytotoxicity and tumor formation for CCl4 is best demonstrated by the 13-week and two-year inhalation JBRC rodent studies, with liver histopathological changes examined at 13 weeks and liver histopathologic effects examined at the end of the two-year study (Nagano et al. Citation2007a, Citation2007b). The CCl4 concentrations evaluated were 0, 10, 30, 90, 270, or 810 ppm in the 13-week study and 0, 5, 25, or 125 ppm in the two-year study. In rats exposed for 13 weeks, histopathological changes indicative of cellular damage (“fatty change”) and inflammation were detected in all CCl4 treatment groups. At ≥30 ppm CCl4, proliferative (increased mitoses) and regenerative (fibrosis, proliferative ducts, cirrhosis) responses occurred. At ≥270 ppm, eosinophilic and basophilic foci, which are associated with hyperplastic or preneoplastic changes, were noted. Liver tumors in rats were observed at an exposure level associated with hepatotoxicity following subchronic and chronic exposure; tumors were not found at an exposure level below the level that induced cytotoxicity (<10 ppm for 13-week exposure and 5 ppm for 104-week exposure). A similar but less consistent exposure–response relationship for cytotoxicity and tumor formation was noted for mice (Nagano et al. Citation2007a, Citation2007b). In mice exposed for 13 weeks, exposure-dependent histopathological findings indicative of cytotoxicity, damage, proliferation, and preneoplastic changes were found. Histopathological findings indicative of fatty change were observed in male mice exposed to ≥10 ppm and in female mice exposed to ≥30 ppm CCl4. In male and female mice exposed to ≥30 ppm, a significantly elevated incidence of liver collapse was detected. Liver collapse was characterized by shrunken parenchymal tissue in the centrilobular zone, presumably resulting from necrotic loss of hepatocytes, and accompanied by proliferation of bile ducts and oval cells. In male and female mice exposed to ≥270 ppm, the incidences of nuclear enlargement of hepatocytes with atypia and altered cell foci were significantly increased. The incidence of liver adenomas and carcinomas in male and female (see discussion above) mice in the 104-week study was elevated compared to concurrent controls at ≥25 ppm, an exposure level that also produced cytotoxicity; it is also similar to an exposure level (30 ppm) that produced a proliferative response in the 13-week study.
Alternative MOAs
A critical component of the MOA analysis is an evaluation of possible alternative MOA. The MOA for liver carcinogenesis () in rodents and in humans was delineated in various reviews (Cohen Citation2010; Holsapple et al. Citation2006; Klaunig and Wang Citation2018). These include DNA reactive and non-DNA reactive MOA. The non-DNA reactive MOA is either receptor-mediated or non-receptor-mediated. Receptor-mediated MOA includes estrogen stimulation and cytotoxicity secondary to specific reactions, such as those related to HMG-CoA reductase inhibition of various enzyme in the porphyrin-heme synthesis pathway. Other MOA that are receptor-mediated appear to be rodent-specific and include PPARα activation (peroxisome proliferation) and cytochrome enzyme induction (constitutive androstane receptor (CAR), pregnane X receptor (PXR), aryl hydrocarbon receptor (AHR)). Non-receptor mediated MOA includes cytotoxicity, infections, iron or copper overload and increased apoptosis (for example, fumonisin B1), as well as several inherited disorders in humans (Cohen Citation2010).
Genotoxic mode of action
There is an extensive body of literature on the genotoxicity evaluation of CCl4 employing various in vitro and in vivo assays (ATSDR Agency for Toxic Substances and Disease Registry Citation2005; Eastmond Citation2008; IPCS International Programme on Chemical Safety Citation1999; McGregor and Lang Citation1996; U.S. EPA United States Environmental Protection Agency Citation2010, Citation2020). In general, most reported investigations were negative or produced equivocal results regarding DNA reactivity, mutagenicity, and clastogenicity. Those reporting positive genotoxic responses tended to show a close association between these effects and high levels of cytotoxicity. The available data indicate that CCl4 is not DNA-reactive. CCl4 potential genotoxic effects are related to the fact that under highly cytotoxic conditions, metabolism of this chemical produces reactive trichloromethyl and trichloromethyl peroxy radicals which interact with cellular lipids. The resulting oxidative and lipid peroxidative damage including the lipid peroxidation by-products malonaldehyde (MDA) and 4-hydroxyalkynonenal (4-HNE) display the potential to interact with cellular macromolecules at the site of their generation (i.e., cytoplasm). In contrast to the mostly negative results obtained with Salmonella tester strains, in the WP2 strain of E. coli, which is highly sensitive to oxidative mutagens, CCl4 produced a 3-fold increase in mutations both with and without S9 (Araki et al. Citation2004). Lipid peroxidation by-products may also modify DNA repair indirectly to elevate mutation frequency (Hartley et al. Citation1999; Krokan et al. Citation1985). However, most genotoxicity studies were conducted at relatively high doses of CCl4 to optimize the detection of weak responses. As such, little data are available to assess genotoxicity at lower, non-cytotoxic CCl4 doses. The consensus (ATSDR Agency for Toxic Substances and Disease Registry Citation2005; Eastmond Citation2008) has been that the genotoxic results are secondary to the liver toxicity and cell death. Mutagenic as well as other genotoxic effects might reflect indirect effects resulting from oxidative and lipid peroxidative damage occurring during necrosis or apoptosis and exhibit clear thresholds. Finally, there is no quantitative linkage between the extent of lipid byproduct binding to liver DNA and liver carcinogenic effects. USEPA OPPT (U.S. EPA United States Environmental Protection Agency Citation2020) has concluded that on balance the available in vivo database did not demonstrate genotoxic potential for CCl4 although acknowledging that available studies were not optimal for evaluating such effects.
Receptor mediated MOA for CCl4
There are limited available data available showing a required role for nuclear receptor activation in the CCl4 MOA.Yamazaki et al. (Citation2005) examined a possible role of the nuclear constitutive androstane receptor (CAR) in CCl4-induced hepatotoxicity using CAR null knock out mice. In CCl4 treated CAR-/- (null) mice versus CAR+/+ (wild type), the loss of CAR mediated function resulted in a negligible effect on liver toxicity. Yamazaki et al. (Citation2005) concluded that CCl4 liver toxicity occurs despite an absence of CAR, and CAR-controlled CYP2B or CYP3A expression may only play a supplementary role in CCl4 metabolism. Therefore, the CAR activation liver tumor MOA does not appear to contribute to CCl4 tumor induction. A survey of the literature failed to demonstrate any other nuclear receptor involvement in carcinogenicity of CCl4.
Immunosuppression
There is no evidence for immunosuppression in CCl4-treated rodents. Further, liver tumors are not increased in immunosuppressed rodents or humans (Krynitz et al. Citation2013; Lebrec et al. Citation2016; Penn Citation1988).
In contrast, hepatic inflammatory effects (such as fibrosis and cirrhosis) of CCl4 were reported in rats and mice by Nagano et al. (Citation2007). These appear to be secondary to hepatotoxicity and liver regeneration. The liver compensatory hyperplasia/regenerative process involves complex interactions among several cell types and cell mediators, including the DNA synthesis and hepatocyte proliferation and release of serum-borne growth factors (hepatotropic factors) that act directly on liver cells to induce mitosis (Luster et al. Citation2000). Results of studies on the effects of hepatotropic factors indicate that inflammatory effects of CCl4, and other hepatotoxic chemicals, may be mediated by tumor growth factor (TGF)-β1 released from the liver during the regenerative process (Delaney and Kaminski Citation1993; Jeon et al. Citation1997). Therefore, inflammation-generated cytokines may contribute to the subsequent regenerative sequelae following the CCl4 exposure, a usual recurrence associated with epithelial damage and associated inflammation (Cohen Citation1999). Kupffer cells are also involved after the initial cytotoxicity whereby their activation releases active mediators including prostaglandins, reactive oxygen species, and cytokines (Luckey and Petersen Citation2001). These mediators are involved in the inflammatory response and fibrotic response following CCl4 hepatic injury. TGF-β1 release by the Kupffer cells is a possible link between the other hepatic inflammation events and Kupffer cell activation. In conclusion, as it relates to the CCl4 liver tumor MOA, the reported inflammatory changes following CCl4 exposure mostly occur post-cell injury and cell death and do not show a direct cause and effect relationship for liver tumors formed. Without the hepatocyte injury, death and resulting necrosis, the inflammatory changes would most likely not occur.
Epidemiology/Human effects (liver)
Most of the human case reports of acute high inhalation exposure to CCl4 describe a consistent pattern of CCl4-initiated toxicity that includes initial dizziness and nausea, followed by abdominal discomfort, liver, and kidney effects, with subsequent renal failure and death (Manno et al. Citation1996; New et al. Citation1962; Ruprah, Mant, and Flanagan Citation1985). Despite consistent findings across human acute studies, most case reports lack reliable quantitative data.
Epidemiological data on non-cancer effects from repeated inhalation exposures are also limited. A cross-sectional study was conducted at three chemical plants in northwest England to assess liver function in CCl4-exposed workers (Tomenson et al. Citation1995). This investigation included 135 CCl4-exposed workers and 276 unexposed workers from two manufacturing sites, one where CCl4 was used and one where CCl4was not utilized. The study participants were given a questionnaire that collected medical history, alcohol consumption, and length of service in a job exposed to CCl4. Blood samples were obtained and analyzed for serum enzymes indicative of liver function or injury. The exposure assessment was based upon historical personal monitoring data for various jobs at the three plants. Subjects were placed into one of three exposure categories (low, medium, or high), according to their jobs at the time of the study. If there was a lack of monitoring data, an industrial hygienist classified the exposure qualitatively based upon comparison with similar exposure groups. The findings showed a significant difference between CCl4 exposed and unexposed workers based upon multivariate analysis of several liver serum enzymes. However, no significant evidence of an exposure-response effect of serum enzymes was apparent. When each serum enzyme was individually assessed, there were no apparent differences between CCl4 exposed and non-exposed workers for the serum chemistry parameters. The serum liver enzyme markers ALP and gamma-glutamyl transpeptidase (GGT) exhibited elevation in the exposed groups but with no exposure–response relationship. The exposed workers did not present with any clinically evident disease that could be associated with CCl4 exposure. A follow-up study at one of the sites with the highest exposure level of CCl4 confirmed no evidence of any further changes in liver function variables or clinical disease in the exposed workers.
Epidemiology studies attempted to evaluate the relationship of CCl4 exposure with liver cancer, including some case reports. However, these case reports on liver tumors illustrate the difficulty of such evaluations, since individuals reported in these investigations possessed various confounding factors, such as alcohol abuse, liver diseases such as biliary tract disorders and possibly biliary cirrhosis (Johnstone Citation1948; Tracey and Sherlock Citation1968). Many of the older studies did not take into account adequate diagnosis of underlying liver diseases or the important confounding factors such as alcohol, nonalcoholic steatohepatitis (NASH), hepatitis virus infection, inherited disorders, or exposure to multiple chemicals in addition to CCl4 (Blair et al. Citation1990, Citation1998; Cantor et al. Citation1995; Blair, Decoufle, and Grauman Citation1979; Checkoway et al. Citation1984, Citation1984; Kauppinen et al. Citation2003; Kubale et al. Citation2005). Overall, there is essentially no reliable evidence for a carcinogenic effect of CCl4 for the liver, or for cancer in total. However, there have not been adequate studies addressing these issues that control for multiple factors needed for a proper evaluation.
Pheochromocytoma in mice
Benign pheochromocytomas were reported in mice exposed to CCl4 in the two-year inhalation study (Nagano et al. Citation2007a). These tumors were also found to be increased in mice when dosed by oral gavage in the NCI study in which CCl4 was used as a positive control for liver tumors (Citation(1976a) (Weisburger Citation1977). Benign pheochromocytomas are tumors that originate in chromaffin cells of the adrenal gland medulla and secrete excessive amounts of catecholamines, usually epinephrine or norepinephrine (DeLellis et al. Citation2004). In the Nagano et al. (Citation2007a) experiment, an elevated incidence of pheochromocytomas of the adrenal gland occurred at the two highest exposure levels (25 and 125 ppm) in male mice and at the highest exposure level (125 ppm) in female mice (); no pheochromocytomas occurred in mice at the lower doses or controls (). These tumors were seen only in the exposed mice but not rats ().
Studies on the mechanism by which CCl4 induces effects in the adrenal gland are limited to short-term investigations of CCl4-induced enzyme activation in adrenal tissue. In experimental animals, acute exposure to CCl4 adrenal necrosis was produced with effects localized to the zona reticularis, the innermost region of the cortex adjacent to the medulla, but not showing toxicity in the medulla (Brogan and Colby Citation1983). This localization of toxicity appears to be the result of greater activation of CCl4 by microsomal enzymes in the zona reticularis (Colby et al. Citation1994). In vitro studies noted that preincubation of adrenal microsomes with 1-aminobenzotriazole (ABT), a CYP450 suicide inhibitor, prevented the effects of CCl4 on lipid peroxidation and covalent binding (Colby et al. Citation1994). Based upon these findings, it would appear that CCl4 metabolism might play a role in induction of effects in the adrenal gland similar to liver, but evidence is limited. More likely for the medulla, itis an indirect effect secondary to stress (Greim et al. Citation2009).
In general, few chemicals were reported to induce pheochromocytomas in mice (Greim et al. Citation2009; Rosol et al. Citation2001; Tischler, Powers, and Alroy Citation2004). Of over 500 technical reports issued by the NTP, only 7 chemicals were associated with pheochromocytomas in B6C3F1 mice (Greim et al. Citation2009; Tischler, Powers, and Alroy Citation2004). Greim et al. (Citation2009) specifically identified 9 chemicals that exhibited an increased incidence of mouse pheochromocytomas. Similarly, pheochromocytomas in CD-1 mice are very uncommon, with incidences of less than 1% in various experiments involving 2163 mice, with the incidence higher in females than males and mostly benign (Petterino et al. Citation2015). The incidences of pheochromocytomas at the JBRC (where the long-term inhalation bioassay on CCl4 was performed) in BDF1 mice were 0.3% (8 of 2645, maximum of 1 per study) and 0.3% (7 of 2646, maximum of 1 per study) in females (Dr Shoji Fukushima, personal communication). Tischler, Powers, and Alroy (Citation2004) noted no apparent common denominator among the chemicals that induced mouse pheochromocytomas. Greim et al. (Citation2009) hypothesized several potential MOA for induction of mouse pheochromocytomas that included endocrine disturbance, impairment of mitochondrial function, uncoupling of oxidative phosphorylation, hepatotoxicity, and nephrotoxicity leading to impaired calcium homeostasis. By examining these diverse cellular targets and effects, it appears that these share a general toxic, stress-related mechanism of action (Everds et al. Citation2013; Greim et al. Citation2009). In most long-term studies, pheochromocytomas occur together with other tumors or toxic effects in other organs. In mice, pheochromocytomas occurred predominantly in combination with a marked hepatotoxic effect and liver carcinomas, as occurred with CCl4.
Pheochromocytomas are not common in humans, with approximately 30% of tumors being of genetic origin, related to a variety of inherited disorders (Fishbein and Nathanson Citation2012; Greim et al. Citation2009; Koch, Pacak, and Chrousos Citation2002). These include such inherited disorders as multiple endocrine neoplasia (MEN) types 2A and 2B, Von Hippel-Lindau disease type 2, Von Recklinghausen neurofibromatosis type 1, paragangliomas associated with pheochromocytomas, and other rare inherited disorders. There is no indication that any of the substances that induce pheochromocytomas in animal experiments, either rats or mice, increase risk of pheochromocytomas in humans (Greim et al. Citation2009). Further, there is no evidence that chronic hypoxia (such as occurs with chronic obstructive pulmonary disease), vitamin D administration, disturbances of endocrine effects, or other MOA in rats or mice, elevate the risk of pheochromocytomas in the general population or in individuals with any of the inherited disorders associated with pheochromocytomas in humans. Essentially, non-genotoxic chemically induced pheochromocytomas in rats or mice are not quantitatively relevant to human risk. Thus, pheochromocytomas are not of relevant importance to the overall risk assessment of CCl4 as these adrenal disorders did not occur at the same doses as the increase in hepatocellular tumors.
Conclusions
Carbon tetrachloride is a hepatotoxic chemical that is known to induce hepatocellular tumors in rodents following chronic exposure but only at hepatotoxic doses which was demonstrated in several species with multiple exposure scenarios. In mice, CCl4 also induced benign adrenal pheochromocytomas. However, there is no evidence that any chemical, including CCl4, increases the risk of such tumors in humans. The pheochromocytomas in rodents are not relevant to human cancer risk.
The evidence is clear that induction of hepatic tumors in rodents is directly related to the dose of CCl4. Doses or exposures that induce cytotoxicity chronically result in chronic compensatory enhanced cell proliferation, which increases the opportunity for spontaneous errors to occur in DNA. The important component of this MOA is induction of cell injury to a level sufficient to produce cell death (necrosis) that leads to consequent hepatocyte cell proliferation. The insult needs to be chronic. Doses or exposures that do not initiate sufficient cytotoxicity to elicit compensatory hyperplasia do not start the cascade to neoplasm formation. Alternative MOAs, including genotoxicity, were excluded.
Although in theory these same processes function in humans and therefore this MOA might also apply to humans, there is no reliable epidemiological data to support liver tumor development in humans following chronic CCl4 exposure. This is related to the fact that any hepatotoxicity in humans would be for short-term since the exposure would not be continued once the liver injury was identified. While very low level chronic human exposure might occur, the levels reported are not sufficient to induce the required level of chronic hepatic toxicity needed to produce the regenerative hyperplastic response. This sequence of events is similar to that observed with chloroform which has been identified as a threshold carcinogen (Andersen et al. Citation2000; Meek et al. Citation2003). A similar assessment of risk is appropriate for CCl4. Thus, the overall MOA evidence indicates CCl4-induced rodent liver tumors do not pose a quantitative carcinogenic hazard at exposure levels that do not induce hepatotoxicity.
Statements of interest
Dr Bevan is the Science Director of the Halogenated Solvents Industry Alliance, Inc., a trade association that represents producers and users of CCl4.
Acknowledgments
We gratefully acknowledge the valuable assistance of Lisa Allen with the preparation of this manuscript. Support for the preparation of this manuscript was provided by the Halogenated Solvents Industry Alliance, Inc., but the statements and conclusions in the manuscript represent solely those of the authors.
Disclosure statement
No potential conflict of interest was reported by the authors.
Data availability statement
This is a review manuscript, and all data referred to is available in the published literature.
Additional information
Funding
References
- Albano, E., K. A. K. Lott, T. F. Slater, A. Stier, M. C. R. Symons, and A. Tomasi. 1982. Spin-trapping studies on the free-radical products formed by metabolic activation of carbon tetrachloride in rat liver microsomal fractions isolated hepatocytes and in vivo in the rat. Biochem. J. 204 (2):593–603. doi:10.1042/bj2040593.
- Alpert, A. E., A. V. Arkhangelsky, A. M. Lunts, and N. P. Panina. 1972. Experimental hepatopathies and carcinomas of the breast in rats. Bjull. Eksp. Biol. Med 74 (4):1299–301. doi:10.1007/BF00801865.
- Andersen, M. E., E. Meek, G. A. Boorman, D. J. Brusick, S. M. Cohen, Y. P. Dragan, C. B. Frederick, J. I. Goodman, G. C. Hard, E. J. O’Flaherty, et al. 2000. Lessons learned in applying the U.S. EPA’s proposed cancer guidelines to specific compounds. Toxicol. Sci. 53 (2):159–72. doi:10.1093/toxsci/53.2.159.
- Andervont, H. B. 1958. Induction of hepatomas in strain C3H mice with 4-o-tolyazoo-toluidine and carbon tetrachloride. J. Natl. Cancer Inst. 20 (2):431–38. doi:10.1093/jnci/20.2.431.
- Ansari, G. A., M. T. Moslen, and E. S. Reynolds. 1982. Evidence for in vivo covalent binding of CCl3 derived from CCl4 to cholesterol of rat liver. Biochem. Pharmacol. 31 (21):3509–10. doi:10.1016/0006-2952(82)90634-7.
- Araki, A., N. Kamigaito, T. Sasaki, and T. Matsushima. 2004. Mutagenicity of carbon tetrachloride and chloroform in Salmonella typhimurium TA98, TA100, TA1535, and TA1537, and Escherichia coli WP2uvrA/pKM101 and WP2/pKM101, using a gas exposure method. Environ. Mol. Mutagen. 43 (2):128–33. doi:10.1002/em.20005.
- ATSDR (Agency for Toxic Substances and Disease Registry). 2005. Toxicological profile for carbon tetrachloride (update), Agency for toxic substances and disease registry. Atlanta, GA: U.S. Department of Health and Human Services.
- Benson, J. M., and D. L. Springer. 1999. Improved risk estimates for carbon tetrachloride. Finalreport. New Mexico: U.S. Department of Energy. Report DE-FC04-96AL76406. Project No. 54940.
- Blair, A., P. Decoufle, and D. Grauman. 1979. Causes of death among laundry and dry cleaning workers. Am. J. Public Heath 69 (5):508–11. doi:10.2105/ajph.69.5.508.
- Blair, A., P. P. Hartge, A. Stewart, M. McAdams, and J. Lubin. 1998. Mortality and cancer incidence of aircraft maintenance workers exposed to trichloroethylene and other organic solvents and chemicals: Extended follow up. Occup. Environ. Med 55 (3):161–71. doi:10.1136/oem.55.3.161.
- Blair, A., P. A. Stewart, P. E. Tolbert, D. Grauman, F. X. Moran, J. Vaught, and J. Rayner. 1990. Cancer and other causes of death among a cohort of dry cleaners. Br. J. Ind Med 47 (3):162–68. doi:10.1136/oem.47.3.162.
- Boobis, A. R., S. M. Cohen, V. Dellarco, D. McGregor, M. E. Meek, C. Vickers, D. Willcocks, and W. Farland. 2006. IPCS framework for analyzing the relevance of a cancer mode of action for humans. Crit. Rev. Toxicol. 36 (10):781–92. doi:10.1080/10408440600977677.
- Boobis, A. R., J. E. Doe, B. Heinrich-Hirsch, M. E. Meek, S. Munn, M. Ruchirawat, J. Schlatter, J. Seed, and C. Vickers. 2008. IPCS framework for analyzing the relevance of a noncancer mode of action for humans. Crit. Rev. Toxicol. 38 (2):87–96. doi:10.1080/10408440701749421.
- Brogan, W. C., and H. D. Colby. 1983. Carbon tetrachloride (CCl4) toxicity in the guinea pig adrenal cortex. W V Med. J 79 (12):274.
- Bruckner, J. V., H. J. Kim, S. Muralidhara, and J. M. Gallo. 1990. Influence of Route and Pattern of Exposure on the Pharmacokinetics and Hepatotoxicity of Carbon Tetrachloride. ADA223461.
- Bruckner, J. V., W. F. MacKenzie, S. Muralidhara, R. Luthra, G. M. Kyle, and D. Acosta. 1986. Oral toxicity of carbon tetrachloride: Acute, subacute, and subchronic studies in rats. Fundam. Appl. Toxicol 6 (1):16–34. doi:10.1016/0272-0590(86)90260-5.
- Bulera, S. J., S. M. Eddy, E. Ferguson, T. A. Jatkoe, J. F. Reindel, M. R. Bleavins, and F. A. De La Iglesia. 2001. RNA expression in the early characterization of hepatotoxicants in wistar rats by high-density DNA microarrays. Hepatology 33 (5):1239–58. doi:10.1053/jhep.2001.23560.
- Cantor, K. P., P. A. Stewart, L. A. Brinton, and M. Dosemeci. 1995. Occupational exposures and female breast cancer mortality in the United States. J. Occup. Environ. Med. 37 (3):336–48. doi:10.1097/00043764-199503000-00011.
- Castro, J. A., and M. I. Diaz Gomez. 1972. Studies on the irreversible binding of 14C-CCl4 to microsomal lipids in rats under varying experimental conditions. Toxicol. Appl. Pharmacol 23 (4):541–52. doi:10.1016/0041-008x(72)90095-6.
- Castro, G. D., M. I. D. Díaz Gómez, and J. A. Castro. 1989. Species differences in the interaction between CCI 4 reactive metabolites and liver DNA or nuclear protein fractions. Carcinogenesis 10 (2):289–94. doi:10.1093/carcin/10.2.289.
- Checkoway, H., T. Wilcosky, P. Wolf, and H. Tyroler. 1984. An evaluation of the associations of leukemia and rubber industry solvent exposures. Am. J. Ind. Med. 5 (3):239–49. doi:10.1002/ajim.4700050307.
- Cho, K. J., and J. J. Jang. 1993. Effects of carbon tetrachloride, ethanol and acetaldehyde on diethylnitrosamine-induced hepatocarcinogenesis in rats. Cancer Lett. 70 (1–2):33–39. doi:10.1016/0304-3835(93)90071-g.
- Cohen, S. M. 1999. Infection, cell proliferation, and malignancy. In Microbes and Malignancy: Infection as a Cause of Cancer, ed. J. Parsonnet, and S. Horning, pp. 89–106. Oxford, England: Oxford University Press.
- Cohen, S. M. 2010. Evaluation of possible carcinogenic risk to humans based on liver tumors in rodent assays: The two-year bioassay is no longer necessary. Toxicol .Pathol 38 (3):487–501. doi:10.1177/0192623310363813.
- Colby, H. D., H. Purcell, S. Kominami, S. Takemori, and D. C. Kossor. 1994. Adrenal activation of carbon tetrachloride: Role of microsomal P450 isozymes. Toxicology 94 (1–3):31–40. doi:10.1016/0300-483x(94)90026-4.
- Cole, L. J., and P. C. Nowell. 1964. Accelerated induction of hepatomas in fast neutron-irradiated mice injected with carbon tetrachloride. Ann. N. Y. Acad. Sci. 114 (1):259–67. doi:10.1111/j.1749-6632.1964.tb53581.x.
- Columbano, A., G. M. Ledda-Columbano, M. Pibiri, R. Piga, H. Shinozuka, V. De Luca, F. Cerignoli, and M. Tripodi. 1997. Increased expression of c-fos, c-jun and LRF-1 is not required for in vivo priming of hepatocytes by the mitogen TCPOBOP. Oncogene 14 (7):857–63. doi:10.1038/sj.onc.1200891.
- Condie, L. W., R. D. Laurie, T. Mills, M. Robinson, and J. P. Bercz. 1986. Effect of gavage vehicle on hepatotoxicity of carbon tetrachloride in CD-1 mice: Corn oil versus tween-60 aqueous emulsion. Fundam. Appl. Toxicol 7 (2):199–206. doi:10.1016/0272/0590(86)90148-x.
- Coni P., F. Bignone, G. Pichiri, G. Ledda-Columbano, A. Columbano, P. Rao, S. Rajalakshmi and D. Sarma. 1989. “Studies on the kinetics of expression of cell cycle dependent proto-oncogenes during mitogen-induced liver cell proliferation.“ Cancer Letters 47 (1–2): 115–119. doi:10.1016/0304-3835(89)90186-9.
- Costa, A., G. Weber, F. S. O. Bartoloni, and G. Campana. 1963. Experimental cancerous cirrhosis from carbon tetrachloride in rats. Arch. DeVecchi 39:303–56.
- Curtis, H. J., J. Tilley, and C. Crowley. 1964. The elimination of chromosome aberrations in liver cells by cell division. Radiat. Res. 22 (4):730–34. doi:10.2307/3571554.
- Dai, Y., and A. I. Cederbaum. 1995. Inactivation and degradation of human cytochrome P4E1 by CCl4 in a transfected HepG2 cell line. J. Pharmacol. Exp. Ther. 275 (3):1614–22.
- Delaney, B., and N. E. Kaminski. 1993. Induction of serum-borne immunomodulatory factors in B6C3F1 mice by carbon tetrachloride. I. Carbon tetrachloride-induced suppression of helper T-lymphocyte function is mediated by a serum borne factor. Toxicology 85 (1):67–84. doi:10.1016/0300-483x(93)90083-5.
- DeLellis, R. A., R. V. Lloyd, P. U. Heitz, and C. Eng, eds. 2004. “Pathology and genetics of tumours of endocrine organs.“ In World Health Organization Classification of Tumours. Lyon, France: IARC Press.
- Della Porta, G. D., B. Terracini, and P. Shubik. 1961. Induction with carbon tetrachloride of liver-cell carcinomas in hamsters. J. Natl. Cancer Inst. 26:855–63. doi:10.1093/jnci/26.4.855.
- Diaz Gomez, M. I., and J. A. Castro. 1980. Covalent binding of carbon tetrachloride metabolites to liver nuclear DNA, proteins, and lipids. Toxicol. Appl. Pharmacol 56 (2):199–206. doi:10.1016/0041-008x(80)90290-2.
- Doolittle, D. J., G. Muller, and H. E. Scribner. 1987. Relationship between hepatotoxicity and induction of replicative DNA synthesis following single or multiple doses of carbon tetrachloride. J. Toxicol. Environ Health 22 (1):63–78. doi:10.1080/15287398709531051.
- Dragani, T. A., G. Manenti, and G. Della Porta. 1986. Enhancing effects of carbon tetrachloride in mouse hepatocarcinogenesis. Cancer Lett. 31 (2):171–79. doi:10.1016/0304-3835(86)90008-x.
- Dragan, Y. P., L. Sargent, Y. D. Xu, H. C. Pitot, and H. C. Pitot. 1993. The initiation-promotion-progression model of rat hepatocarcinogenesis. Proc. Soc. Exp. Biol. Med. 202 (1):16–24. doi:10.3181/00379727-202-43511c.
- Eastmond, D. A. 2008. Evaluating genotoxicity data to identify a mode of action and its application in estimating cancer risk at low doses: A case study involving carbon tetrachloride. Environ. Mol. Mutagen. 49 (2):132–41. doi:10.1002/em.20368.
- Edwards, J. E. 1941. Hepatomas in mice induced with carbon tetrachloride. J. Natl. Cancer Inst. 2:197–99. doi:10.1093/jnci/2.2.197.
- Edwards, J. E., and A. J. Dalton. 1942. Induction of cirrhosis of the liver and of hepatomas in mice with carbon tetrachloride. J. Natl. Cancer Inst. 3:19–41. doi:10.1093/jnci/3.1.19.
- Edwards, J., W. E. Heston, and A. J. Dalton. 1942. Induction of the carbon tetrachloride hepatoma in strain L mice. J. Natl. Cancer Inst. 3:297–301. doi:10.1093/jnci/3.3.297.
- Eschenbrenner, A. B., and E. Miller. 1946. Liver necrosis and the induction of carbon tetrachloride hepatomas in strain a mice. J. Natl. Cancer Inst. 6:325–41. doi:10.1093/jnci/6.6.325.
- Everds, N. E., P. W. Snyder, K. L. Bailey, B. Bolon, D. M. Creasy, G. L. Foley, T. J. Rosol, and T. Sellers. 2013. Interpreting stress responses during routine toxicity studies: A review of the biology, impact, and assessment. Toxicol. Pathol 41 (4):560–614. doi:10.1177/0192623312466452.
- Fanelli, S. L., and J. A. Castro. 1995. Covalent binding of carbon tetrachloride reactive metabolites to liver microsomal and nuclear lipid and phospholipid classes from sprague dawley and Osborne Mendel male rats. Teratogen. Carcinogen. Mutagen 15 (4):155–66. doi:10.1002/tcm.1770150402.
- Farber, E., and D. S. R. Sarma. 1987. Hepatocarcinogenesis: A dynamic cellular perspective. Lab. Invest. 56 (1):4–22.
- Fishbein, L., and K. L. Nathanson. 2012. Pheochromocytoma and paraganglioma: Understanding the complexities of the genetic background. Cancer. Genet 205 (1–2):1–11. doi:10.1016/j.cancergen.2012.01.009.
- Fountoulakis, M., M. C. de Vera, F. Crameri, F. Boess, R. Gasser, S. Albertini, and L. Suter. 2002. Modulation of gene and protein expression by carbon tetrachloride in the rat liver. Toxicol. Appl. Pharmacol 183 (1):71–80. doi:10.1006/taap.2002.9460.
- Frezza, E. E., G. E. Gerunda, F. Farinati, N. DeMaria, A. Galligioni, F. Plebani, A. Giocomin, and D. H. Van Thiel. 1994. CCl4-induced liver cirrhosis and hepatocellular carcinoma in rats: Relationship to plasma zinc, copper and estradiol levels. Hepatogastroenterology 41 (4):367–69.
- Furihata, C., and T. Suzuki. 2023. Short-term in vivo testing to discriminate genotoxic carcinogens from non-genotoxic carcinogens and non-carcinogens using next-generation RNA sequencing, DNA microarray, and qPCR. Genes. and Environ 45 (1):7. doi:10.1186/s41021-023-00262-9.
- Greim, H., A. Hartwig, U. Reuter, H. B. Richter-Reichhelm, and H. W. Thielmann. 2009. Chemically induced pheochromocytomas in rats: Mechanisms and relevance for human risk assessment. Crit. Rev. Toxicol. 39 (8):695–718. doi:10.1080/10408440903190861.
- Habs, H., K. Künstler, D. Schmähl, and L. Tomatis. 1983. Combined effects of fast-neutron irradiation and subcutaneously applied carbon tetrachloride or chloroform in C57B16 mice. Cancer Lett. 20 (1):13–20. doi:10.1016/0304-3835(83)90181-7.
- Halver, J. E. 1967. Crystalline aflatoxin and other vectors for trout hepatoma. In Trout Hepatoma Research Conference Papers, ed. J. E. Halver, and I. A. Mitchell, Washington, DC: Bureau of Sports Fisheries and Wildlife. pp. 78a
- Hard, G. C., K. J. Johnson, and S. M. Cohen. 2009. A comparison of rat chronic progressive nephropathy with human renal disease—implications for human risk assessment. Crit. Rev. Toxicol. 39 (4):332–46. doi:10.1080/10408440802368642.
- Hartley, D. P., K. L. Kolaja, J. Reichard, and D. R. Petersen. 1999. 4-Hydroxynonenal and malondialdehyde hepatic protein adducts in rats treated with carbon tetrachloride: Immunochemical detection and lobular localization. Toxicol. Appl. Pharmacol 161 (1):23–33. doi:10.1006/taap.1999.8788.
- Haseman, J. K. 1983. A reexamination of false-positive rates for carcinogenesis studies. Fundam. Appl. Toxicol 3 (4):334–39. doi:10.1016/s0272-0590(83)80148-1.
- Holden, P. R., N. H. James, A. N. Brooks, R. A. Roberts, I. Kimber, and W. D. Pennie. 2000. Identification of a possible association between carbon tetrachloride-induced hepatotoxicity and interleukin-8 expression. J. Biochem. Mol. Toxicol. 14:283–90. doi:10.1002/1099-0461(2000)14:5<283:AID-JBT7>3.0.CO;2-S.
- Holsapple, M. P., H. C. Pitot, S. H. Cohen, A. R. Boobis, J. E. Klaunig, T. Pastoor, V. L. Dellarco, and Y. P. Dragan. 2006. Mode of action in relevance of rodent liver tumors to human cancer risk. Toxicol. Sci. 89 (1):51–56. doi:10.1093/toxsci/kfj001.
- IARC (International Agency for Research on Cancer). Carbon Tetrachloride. 1999. IARC monographs on the evaluation of carcinogenic risk to humans, lyon. France 71: 401–32.
- IARC (International Agency for Research on Cancer). Solvents. 1979. IARC monographs on the evaluation of carcinogenic risk to humans, lyon. France 20: 371–402.
- IPCS (International Programme on Chemical Safety). 1999. Carbon tetrachloride. Geneva, Switzerland: International Programme for Chemical Safety.
- JBRC (Japan Bioassay Research Center). Subchronic inhalation toxicity and carcinogenicity studies of carbon tetrachloride in F344 rats and BDF1 mice (Studies Nos. 0020, 0021, 0043, and 0044). 1998. Kanagawa, Japan Industrial Safety and Health Association (Unpublished report to the Ministry of Labor). Hirasawa Hadano Kanagawa, 257 Japan.
- Jeon, Y. J., S. H. Han, K. H. Yang, and N. E. Kaminski. 1997. Induction of liver-associated transforming growth factor β1 (TGF-β1) mRNA expression by carbon tetrachloride leads to the inhibition of T helper 2 cell-associated lymphokines. Toxicol. Appl. Pharmacol 144 (1):27–35. doi:10.1006/taap.1997.8126.
- Johnstone, R. T. 1948. Occupational Medicine and Industrial Hygiene, pp. 148–58. Louis, MO: C. V. Mosby Co. St.
- Kanematsu, T. 1976. Promoting effect of carbon tetrachloride on azo-dye hepatocarcinogenesis in rats. Fukuoka. Igaku Zasshi 67:134–45.
- Katagiri, T., K. Nagano, S. Aiso, H. Senoh, Y. Sakura, T. Takeuchi, and M. Okudaira. 1998. A pathological study on spontaneous hepatic neoplasms in BDF1 mice. J. Toxicol. Pathol 11 (1):21–21. doi:10.1293/tox.11.21.
- Kato, K., T. Kawai, M. Fujii, Y. Bunai, H. Shima, and M. Takahashi. 1985. Enhancing effect of preadministration of carbon tetrachloride on methylazoxymethanol acetate-induced intestinal carcinogenesis. J. Toxicol Sci 10 (4):289–93. doi:10.2131/jts.10.289.
- Kauppinen, T., E. Pukkala, A. Saalo, and A. J. Sasco. 2003. Exposure to chemical carcinogens and risk of cancer among Finnish laboratory workers. Am. J. Ind. Med. 44 (4):343–50. doi:10.1002/ajim.10278.
- Kawasaki, H. 1965. Development of tumor in the course of spontaneous restoration of carbon tetrachloride induced cirrhosis of the liver in rats. Kurume Med. J 12 (1):37–42. doi:10.2739/kurumemedj.12.37.
- Kiplinger, G. F., and C. J. Kensler. 1963. Failure of phenoxybenzamine to prevent formation of hepatomas after chronic carbon tetrachloride administration. J. Natl. Cancer Inst. 30:837–43. doi:10.1093/jnci/30.4.837.
- Klaunig, J. E., and Z. Wang. 2018. Chemical Carcinogenesis. In Casarett & Doull’s Toxicology: The Basic Science of Poisons, ed. C. D. Klaassen, pp. 705–813. 9th ed. New York: McGraw-Hill.
- Koch, C. A., K. Pacak, and G. P. Chrousos. 2002. The molecular pathogenesis of hereditary and sporadic adrenocortical and adrenomedullary tumors. J. Clin. Endocrinol. Metab. 87 (12):5367–84. doi:10.1210/jc.2002-021069.
- Krokan, H., R. C. Grafstrom, K. Sundqvist, H. Esterbauer, and C. C. Harris. 1985. Cytotoxicity, thiol depletion and inhibition of O6-methylguanine-DNA methyltransferase by various aldehydes in cultured human bronchial fibroblasts. Carcinogenesis 6 (12):1755–59. doi:10.1093/carcin/6.12.1755.
- Kroner, H. 1982. The intracellular distribution of liver cell calcium in normal rats and one hour after administration of carbon tetrachloride. Biochem. Pharmacol. 31 (6):1069–73. doi:10.1016/0006-2952(82)90344-6.
- Krynitz, B., G. Edgren, B. Lindelof, E. Baecklund, C. Brattstrom, H. Wilczek, and K. E. Smedby. 2013. Risk of skin cancer and other malignancies in kidney, liver, heart and lung transplant recipients 1970 to 2008–a Swedish population-based study. Int. J. Cancer 132 (6):1429–38. doi:10.1002/ijc.27765.
- Kubale, T. L., R. D. Daniels, J. H. Yiin, J. Couch, M. K. Schubauer-Berigan, G. M. Kinnes, S. R. Silver, S. J. Nowlin, and P. Chen. 2005. A nested case-control study of leukemia mortality and ionizing radiation at the Portsmouth Naval Shipyard. Radiat. Res. 164 (6):810–19. doi:10.1667/rr3473.1.
- Lai E. K., P. B. McCay, N. Toshikazu and F. Kuo-Lan. 1979. “In vivo spin-trapping of trichloromethyl radicals formed from CCl4.“ Biochemical Pharmacology 28 (14): 2231–2235. doi:10.1016/0006-2952(79)90212-0.
- Lebrec, H., F. R. Brennan, H. Haggerty, D. Herzyk, C. Kamperschroer, C. C. Maier, R. Ponce, B. D. Preston, D. Weinstock, and R. D. Mellon. 2016. HESI/FDA workshop on immunomodulators and cancer risk assessment: Building blocks for a weight-of-evidence approach. Regul. Toxicol. Pharmacol. 75:72–80. doi:10.1016/j.yrtph.2015.12.018.
- Lee, V. M., R. G. Cameron, and M. C. Archer. 1998. Zonal location of compensatory hepatocyte proliferation following chemically induced hepatotoxicity in rats and humans. Toxicol. Pathol 26 (5):621–27. doi:10.1177/019262339802600505.
- Lee, P. Y., P. B. McCay, and K. R. Hornbrook. 1982. Evidence for carbon tetrachloride-induced lipid peroxidation in mouse liver. Biochem. Pharmacol. 31 (3):405–09. doi:10.1016/0006-2952(82)901089-7.
- Lemonnier, F. J., J. M. Scotto, and C. Thuong-Trieu. 1974. Disturbances in tryptophan metabolism after a single dose of Aflatoxin Bl and chronic intoxication with carbon tetrachloride. J. Natl. Cancer Inst. 53 (3):745–49. doi:10.1093/jnci/53.3.745.
- Letteron, P., G. Labbe, C. Degott, A. Berson, B. Fromenty, M. Delaforge, D. Larrey, and D. Pessayre. 1990. Mechanism for the protective effects of silymarin against carbon tetrachloride-induced lipid peroxidation and hepatotoxicity in mice. Evidence that silymarin acts both as an inhibitor of metabolic activation and as a chain-breaking antioxidant. Biochem. Pharmacol. 39 (12):2027–34. doi:10.1016/0006-2952(90)90625-u.
- Long, R. M., and L. Moore. 1986. Elevated cytosolic calcium in rat hepatocytes exposed to carbon tetrachloride. J. Pharmacol. Exp. Ther. 238 (1):186–91.
- Luckey, S. W., and D. R. Petersen. 2001. Activation of Kupffer cells during the course of carbon tetrachloride-induced liver injury and fibrosis in rats. Exp. Mol. Pathol. 71 (3):226–40. doi:10.1006/exmp.2001.2399.
- Luster, M. I., P. P. Simeonova, R. M. Gallucci, J. M. Matheson, and B. Yucesoy. 2000. Immunotoxicology: Role of inflammation in chemical-induced hepatotoxicity. Int. J. Immunopharmacol. 22 (12):1143–47. doi:10.1016/s0192-0561(00)00073-4.
- Manibusan, M. K., M. Odin, and D. A. Eastmond. 2007. Postulated carbon tetrachloride mode of action: A review. J. Environ Sci Health C Environ Carcinog Ecotoxicol Rev 25 (3):185–209. doi:10.1080/10590500701569398.
- Manno, M. M. R., C. Grossi, M. Sbrana, and C. Sbrana. 1996. Potentiation of occupational carbon tetrachloride toxicity by ethanol abuse. Human Exp. Toxicol 15 (4):294–300. doi:10.1177/096032719601500404.
- Maronpot, R. R., J. K. Haseman, G. A. Boorman, S. E. Eustis, G. N. Rao, and J. E. Huff. 1987. Liver lesions in B6C3F1 mice: The national toxicology program, experience and position. Arch. Toxicol. Suppl 10:10–26. doi:10.1007/978-3-642-71617-1_2.
- Maronpot, R. R., H. Witschi, L. H. Smith, and J. L. McCoy.1983.“Recent experience with the strain a mouse pulmonary tumor bioassay model”Short-term bioassays in the analysis of complex environmental mixtures II.ed. In M. D. Waters, S. S. Sandhu, J. Lewtas, L. Claston, N. Chernoff, and S. NesnowVol. 27.Plenum Presspp. 341–49.10.1007/978-1-4613-3611-2_24
- Marshall, K. A., and L. H., Pottenger. 2016. “Chlorocarbons and Chlorohydrocarbons.“ Kirk-Othmer Encyclopedia of Chemical Technology. 4th ed. Wiley-Interscience.
- Martinez, M., M. Mourelle, and P. Muriel. 1995. Protective effect of colchicine on acute liver damage induced by CCl4. Role of cytochrome P-450. J. Appl. Toxicol. 15 (1):49–52. doi:10.1002/jat.250150111.
- McConnell, E. E., H. A. Sollevelt, J. A. Swenberg, and G. A. Boorman. 1986. Guidelines for combining neoplasms for evaluation of rodent carcinogenesis studies. J. Natl. Cancer Inst. 76 (2):283–89.
- McGregor, D., and M. Lang. 1996. Carbon tetrachloride: Genetic effects and other modes of action. Mutat. Res 366 (3):181–95. doi:10.1016/s0165-1110(96)90027-5.
- Meek, M. E., J. R. Bucher, S. M. Cohen, V. Dellarco, R. N. Hill, L. D. Lehman-McKeeman, D. G. Longfellow, T. Pastoor, J. Seed, and D. E. Patton. 2003. A framework for human relevance analysis of information on carcinogenic modes of action. Crit. Rev. Toxicol. 33 (6):591–653. doi:10.1080/713608373.
- Menegazzi, M., A. Carcereri-De Prati, H. Suzuki, H. Shinozuka, M. Pibiri, R. Piga, A. Columbano, and G. M. Ledda-Columbano. 1997. Liver cell proliferation induced by nafenopin and cyproterone acetate is not associated with increases in activation of transcription factors NF-kappaB and AP-1 or with expression of tumor necrosis factor alpha. Hepatology 25 (3):585–92. doi:10.1002/hep.510250316.
- Nagano, K., T. Nishizawa, S. Yamamoto, and T. Matsushima 1998. Inhalation carcinogenesis studies of six halogenated hydrocarbons in rats and mice. In: Advances in the prevention of occupational respiratory diseases: proceedings of the 9thinternational conference on occupational respiratory diseases, ed. K. Chiyotani, Y. Hosoda, and Y. Aizawa, 741–46. New York: Elsevier.
- Nagano, K., T. Sasaki, Y. Umeda, T. Nishizawa, N. Ikawa, H. Ohbayashi, H. Arito, S. Yamamoto, and S. Fukushima. 2007a. Inhalation carcinogenicity and chronic toxicity of carbon tetrachloride in rats and mice. Inhal. Toxicol 19 (13):1089–103. doi:10.1080/08958370701628770.
- Nagano, K., Y. Umeda, M. Saito, T. Nishizawa, N. Ikawa, H. Arito, S. Yamamoto, and S. Fukushima. 2007b. Thirteen-week inhalation toxicity of carbon tetrachloride in rats and mice. J. Occup Health 49 (4):249–59. doi:10.1539/joh.49.249.
- Nakata, R., I. Tsukamoto, M. Miyoshi, and S. Kojo. 1985. Liver regeneration after carbon tetrachloride intoxication in the rat. Biochem. Pharmacol. 34 (4):586–88. doi:10.1016/0006-2952(85)90195-9.
- NCI (National Cancer Institute). 1976a. Report on the carcinogenesis bioassay of chloroform. CAS No. 67-66-3. U.S. Department of Health, Education and Welfare.
- NCI (National Cancer Institute). 1976b. Report on the carcinogenesis bioassay of trichloroethylene. CAS No. 79-01-6. U.S. Department of Health, Education and Welfare.
- New, P. S., G. D., Lubash, Scherr, L., and Rubin, A. L. 1962. Acute renal failure associated with carbon tetrachloride intoxication. J. Am Med Assoc 181 903–906. doi:10.1001/jama.1962.03050360089019c
- Noguchi, T., K. L. Fong, E. K. Lai, S. S. Alexander, M. M. King, L. Olson, J. L. Poyer, and P. B. McCay. 1982b. Specificity of a phenobarbital-induced cytochrome P-450 for metabolism of carbon tetrachloride to the trichloromethyl radical. Biochem. Pharmacol. 31 (5):615–24. doi:10/1016/0006-2952(82)90440-3.
- Noguchi, T., K. L. Fong, E. K. Lai, L. Olson, and P. B. McCay. 1982a. Selective early loss of polypeptides in liver microsomes of CCl4-treated rats. Relationship to cytochrome P-450 content. Biochem. Pharmacol. 31 (5):609–14. doi:10.1016/0006-2952(82)90439-7.
- OECD (Organisation for Economic Co-operation and Development). 2010. Including Benchmark Dose And Linear Extrapolation. NOAELS And NOELS, LOAELS And LOELS.
- Packer, J. E., T. F. Slater, and R. L. Willson. 1978. Reactions of the carbon tetrachloride-related peroxy free radical (CCl3O2.) with amino acids: Pulse radiolysis evidence. Life Sci. 23 (26):2617–20. doi:10.1016/0024-3205(78)90378-8.
- Penn, I. 1988. Tumors of the immunocompromised patient. Annu. Rev. Med. 39 (1):63–73. doi:10.1146/annurev.me.39.020188.000431.
- Petterino, C., S. Naylor, S. Mukaratirwa, and A. Bradley. 2015. Adrenal gland background findings in CD-1 (Crl: CD-1(ICR)BR) mice from 104-week carcinogenicity studies. Toxicol. Pathol 43 (6):816–24. doi:10.1177/0192623315587921.
- Pohl, L. R., R. D. Schulick, R. J. Highet, and J. W. George. 1984. “Reductive-oxygenation mechanism of metabolism of carbon tetrachloride to phosgene by cytochrome P-450.“ Mol Pharmacol 25 (2): 318–21.
- Pound, A. W. 1978. Influence of carbon tetrachloride on induction of tumours of the liver and kidneys in mice by netrosamines. Br. J. Cancer 37 (1):67–75. doi:10.1038/bjc.1978.10.
- Pound, A. W., T. A. Lawson, and L. Horn. 1973. Increased carcinogenic action of dimethylnitrosamine after prior administration of carbon tetrachloride. Br. J. Cancer 27 (6):451–59. doi:10.1038/bjc.1973.57.
- Quist, E. M., G. A. Boorman, J. M. Cullen, R. R. Maronpot, A. K. Remick, J. A. Swenberg, L. Freshwater, and J. F. Hardisty. 2019. Reevaluation of hepatocellular neoplasms in CD-1 mice from a 2-year oral carcinogenicity study with permethrin. Toxicol. Pathol 47 (1):11–17. doi:10.1177/0192623318809304.
- Racay, P., P. Kaplan, V. Mezesova, and L. Lehotsky. 1997. Lipid peroxidation both inhibits Ca(2+)-ATPase and increases Ca2+permeability of endoplasmic reticulum membrane. Biochem. Mol. Biol. Int. 41 (4):647–55. doi:10.1080/15216549700201691.
- Raucy, J. L., J. C. Kraner, and J. M. Lasker. 1993. Bioactivation of halogenated hydrocarbons by cytochrome P4502E1. Crit. Rev. Toxicol. 23 (1):1–20. doi:10.3109/10408449309104072.
- Recknagel, R. O., E. A. Glende. 1973. Carbon tetrachloride hepatotoxicity: An example of lethal cleavage. Crit. Rev. Toxicol. 2 (3):263–97. doi:10.3109/10408447309082019.
- Reinke, L. A., and E. G. Janzen. 1991. Detection of spin adducts in blood after administration of carbon tetrachloride to rats. Chem. Biol. Interact. 78 (2):155–65. doi:10.1016/0009-2797(91)90011-u.
- Reuber, M. D., and E. L. Glover. 1967. Hyperplastic and early neoplastic lesions of the liver in Buffalo strain rats of various ages given subcutaneous carbon tetrachloride. J. Natl. Cancer Inst. 38 (6):891–99. doi:10.1093/jnci/38.6.891.
- Reuber, M. D., and E. L. Glover. 1970. Cirrhosis and carcinoma of the liver in male rats given subcutaneous carbon tetrachloride. J. Natl. Cancer Inst. 44 (2):419–27. doi:10.1093/jnci/44.2.419.
- Rocchi, P., G Prodi, S Grilli, and A. M. Ferreri. 1973. In vivo and in vitro binding of carbon tetrachloride with nucleic acids and proteins in rat and mouse liver. Int. J. Cancer 11 (2):419–25. doi:10.1002/ijc.2910110219.
- Rosol, T. J., J. T. Yarrington, J. Latendresse, and C. C. Capen. 2001. Adrenal gland: Structure, function, and mechanisms of toxicity. Toxicol. Pathol 29 (1):41–48. doi:10.1080/019262301301418847.
- Ruprah, M., T. G. K. Mant, and R. J. Flanagan. 1985. Acute carbon tetrachloride poisoning in 19 patients: Implications for diagnosis and treatment. Lancet 325 (8436):1027–29. doi:10.1016/s0140-6736(85)91624-1.
- Seed, J., E. W. Carney, R. A. Corley, K. Crofton, J. M. DeSesso, P. M. Foster, R. Kavlock, G. Kimmel, J. Klaunig, M. E. Meek, et al. 2005. Overview: Using mode of action and life stage information to evaluate the human relevance of animal toxicity data. Crit. Rev. Toxicol. 35 (8–9):664–72. doi:10.1080/713608373.
- Sipes, I. G., G. Krishna, and J. R. Gillette. 1977. Bioactivation of carbon tetrachloride, chloroform and bromotrichloromethane: Role of cytochrome P-450. Life Sci. 20 (9):1541–48. doi:10.1016/0024-3205(77)90446-5.
- Slater, T. F. 1981. Free radicals as reactive intermediates in tissue injury. Adv. Exp. Med. Biol. 136:575–89. doi:10.1007/978-1-4757-0674-1_42.
- Slater, T. F. 1982. Activation of carbon tetrachloride: Chemical principles and biological significance. In Free radicals, lipid peroxidation and cancer, ed. D. C. H. McBrien, and T. F, pp. 243–74. New York, NY: Academic Press.
- Sonich-Mullin, C., R. Fielder, J. Wiltse, K. Baetcke, J. Dempsey, P. Fenner-Crisp, D. Grant, M. Hartley, A. Knaap, D. Kroese, et al. 2001. IPCS conceptual framework for evaluating a node of action for chemical carcinogenesis. Regul. Toxicol. Pharmacol. 34(2):146–52. doi:10.1006/rtph.2001.1493.
- Takahashi, S., T. Takahashi, S. Mizobuchi, M. Matsumi, K. Morita, M. Miyazaki, M. Namba, R. Akagi, and M. Hirakawa. 2002. Increased cytotoxicity of carbon tetrachloride in a human hepatoma cell line overexpressing cytochrome P450 2E1. J. Int. Med. Res. 30 (4):400–05. doi:10.1177/147323000203000406.
- Takano, T., M. Tatematsu, R. Hasegawa, K. Imaida, and N. Ito. 1980. Dose-response relationship for the promoting effect of phenobarbital on the induction of liver hyperplastic nodules in rats exposed to 2-fluorenylacetamide and carbon tetrachloride. Gan 71 (4):580–81.
- Takizawa, S., H. Watanabe, Y. Naito, and S. Inoue. 1975. Preparative action of carbon tetrachloride in liver tumorigenesis by a single application of N-butylnitrosourea in male ICR/JCL strain mice. Gan 66 (6):603–14.
- Tanaka, T., H. Mori, and G. M. Williams. 1987. Enhancement of dimethylnitrosamine-initiated hepatocarcinogenesis in hamsters by subsequent administration of carbon tetrachloride but not phenobarbital or p, p ′-dichlorodiphenyltrichloroethane. Carcinogenesis 8 (9):1171–78. doi:10.1093/carcin/8.9.1171.
- Thoolen, B., R. R. Maronpot, T. Harada, A. Nyska, C. Rousseaux, T. Nolte, D. E. Malarkey, W. Kaufmann, K. Kuttler, U. Deschl, et al. 2010. Proliferative and nonproliferative lesions of the rat and mouse hepatobiliary system. Toxicol. Pathol 38 (7_suppl):5S–81S. doi:10.1177/0192623310386499.
- Tischler, A. S., J. F. Powers, and J. Alroy. 2004. Animal models of pheochromocytoma. Histol. Histopathol. 19:883–95. doi:10.14670/hh-19.883.
- Tomenson, J. A., C. E. Baron, J. J. O’Sullivan, J. C. Edwards, M. D. Stonard, R. J. Walker, and D. M. Fearnley. 1995. Hepatic function in workers occupationally exposed to carbon tetrachloride. Occup Environ. Med 52 (8):508–14. doi:10.1136/oem.52.8.508.
- Tracey, J. P., and P. Sherlock. 1968. Hepatoma following carbon tetrachloride poisoning. N Y State J .Med 68 (16):2202–04.
- U.S. EPA (U.S. Environmental Protection Agency). February 2010. “Toxicological review of carbon tetrachloride (CAS no. 56-23-5).“ In Support Of Summary Information On The Int. Risk Information System (IRIS. EPA Document #EPA/635/R-08/005F.
- U.S. EPA (U.S. Environmental Protection Agency). October 2020. “Risk evaluation for carbon tetrachloride (Methane, Tetrachloride) CASRN: 56-23-5.“ EPA. Document# EPA-740-R1-8014.
- U.S. FDA (U.S. Food and Drug Administration) May 2001. Guidance for industry. Statistical aspects of the design, analysis, and interpretation of chronic rodent carcinogenicity studies of pharmaceuticals.
- Weber, L. W. D., M. Boll, and A. Stampfl. 2003. Hepatotoxicity and mechanism of action of haloalkanes: Carbon tetrachloride as a toxicological model. Crit. Rev. Toxicol. 33 (2):105–36. doi:10.1080/713611034.
- Weisburger, E. K. 1977. Carcinogenicity studies on halogenated hydrocarbons. Environ. Health Perspect. 21:7–16. doi:10.1289/ehp.77217.
- Wong, F. W., W. Chan, and S. S. Lee. 1998. Resistance to carbon tetrachloride-induced hepatotoxicity in mice which lack CYP2E1 expression. Toxicol. Appl. Pharmacol 153 (1):109–18. doi:10.1006/taap.1998.8547.
- Yamate, J., M. Tajima, S. Kudow, and S. Sannai. 1990. Background pathology in BDF 1 mice allowed to live out their life-span. Lab. Anim (NY) 24 (4):332–40. doi:10.1258/002367790780865976.
- Yamazaki, Y., S. Kakizaki, N. Horiguchi, H. Takagi, M. Mori, and M. Negishi. 2005. Role of nuclear receptor CAR in carbon tetrachloride-induced hepatotoxicity. World J. Gastroenterol 11 (38):5966–72. doi:10.3748/wjg.v11.i38.5966.
- Younes, M., and C. P. Siegers. 1985. The role of iron in the paracetamol- and CCl4-induced lipid peroxidation and hepatotoxicity. Chem. Biol. Interact. 55:327–34. doi:10.1016/s0009-2797(85)80139-3.
- Zalatnai, A., I. Sarosi, A. Rot, and K. Lapsis. 1991. Inhibitory and promoting effects of carbon tetrachloride-induced liver cirrhosis on the diethylnitrosamine hepatocarcinogenesis in rats. Cancer Lett. 57 (1):67–73. doi:10.1016/0304-3835(91)90065-p.