
Abstract
Objective
To evaluate serum neurofilament light chain (sNfL) as biomarker of disease onset, progression and treatment effect in hereditary transthyretin (ATTRv) amyloidosis patients and TTR variant (TTRv) carriers.
Methods
sNfL levels were assessed longitudinally in persistently asymptomatic TTRv carriers (N = 12), persistently asymptomatic ATTRv amyloidosis patients (defined as asymptomatic patients but with amyloid detectable in subcutaneous abdominal fat tissue) (N = 8), in TTRv carriers who developed polyneuropathy (N = 7) and in ATTRv amyloidosis patients with polyneuropathy on treatment (TTR-stabiliser (N = 20) or TTR-silencer (N = 18)). Polyneuropathy was confirmed by nerve conduction studies or quantitative sensory testing. sNfL was analysed using a single-molecule array assay.
Results
sNfL increased over 2 years in persistently asymptomatic ATTRv amyloidosis patients, but did not change in persistently asymptomatic TTRv carriers. In all TTRv carriers who developed polyneuropathy, sNfL increased from 8.4 to 49.8 pg/mL before the onset of symptoms and before polyneuropathy could be confirmed neurophysiologically. In symptomatic ATTRv amyloidosis patients on a TTR-stabiliser, sNfL remained stable over 2 years. In patients on a TTR-silencer, sNfL decreased after 1 year of treatment.
Conclusion
sNfL is a biomarker of early neuronal damage in ATTRv amyloidosis already before the onset of polyneuropathy. Current data support the use of sNfL in screening asymptomatic TTRv carriers and in monitoring of disease progression and treatment effect.
Introduction
Hereditary transthyretin (ATTRv) amyloidosis is an autosomal dominant disease with a progressive and fatal course when left untreated. ATTRv amyloidosis is caused by mutations in the gene encoding transthyretin (TTR) causing the tetrameric TTR protein to become unstable, to dissociate into amyloidogenic monomers and to aggregate into insoluble fibrils that deposit in the extracellular space of tissues and organs. The age of symptom onset varies between the second and ninth decade of life and peripheral and autonomic neuropathy are usually among the first disease manifestations [Citation1, Citation2].
Liver transplantation was the only treatment for ATTRv amyloidosis patients for a long time [Citation3]. However, in the last decade, several drugs became available for the treatment of ATTRv amyloidosis. These drugs can be categorised into two groups: the TTR-stabilisers, preventing the TTR protein from dissociation into amyloidogenic monomers, and the TTR gene silencers, decreasing the production of TTR by the liver [Citation4–7]. These treatments are most beneficial when initiated early in the disease course. Therefore, systematic and regular monitoring of asymptomatic variant (TTRv) carriers and ATTRv amyloidosis patients to identify early stages of disease is recommended to improve the prognosis [Citation2, Citation8–10]. In addition, because more treatment options are available to date, it has become prudent and rational to monitor the effect of treatment [Citation10].
At present, several neurophysiological tests are used to monitor asymptomatic TTRv carriers and ATTRv amyloidosis patients. Nerve conduction studies (NCS) are the most commonly used objective measure for the evaluation of polyneuropathy. NCS are a sensitive and specific method of assessing disease in the large myelinated nerve fibres and can give useful diagnostic information regarding the underlying pathophysiology of the polyneuropathy [Citation11]. However, NCS have limited sensitivity for axonal damage in early disease stages, are only able to measure large nerve fibres and are not able to detect small fibre neuropathy, are time-consuming, and to some degree burdensome for patients [Citation8, Citation11]. Quantitative sensory testing (QST) is a non-invasive method to evaluate the function of small sensory fibres and can, therefore, provide evidence for small fibre neuropathy, however, this test is limited by its subjective nature [Citation8, Citation12]. The sudoscan is an objective test used in the evaluation of small fibre neuropathy but is limited to sympathetic C nerve fibres of the autonomic nervous system [Citation13]. Autonomic neurofunction testing is also used in the evaluation of ATTRv patients to detect autonomic neuropathy, but does not provide information about the peripheral nerves [Citation10, Citation14]. Another modality to assess nerve involvement is a sural nerve biopsy. However, this is an invasive procedure with as consequence permanent cutaneous anaesthesia in the area previously innervated by the biopsied nerve. The diagnostic yield depends on the expertise of the performing centre and the TTR gene variant [Citation15, Citation16]. Skin biopsy is a simple and effective but also invasive tool to detect amyloid and small nerve fibre loss [Citation17–20].
Neurofilament light chain (NfL) is a neuron-specific cytoskeletal protein released in the blood or cerebrospinal fluid during axonal damage. Recent studies showed that serum neurofilament light chain (sNfL) is a biomarker for polyneuropathy and also correlates with the severity of polyneuropathy in ATTRv amyloidosis [Citation21–23]. sNfL may be a sensitive biomarker for early neuronal damage in ATTRv amyloidosis as sNfL was already increased in early symptomatic ATTRv and immunoglobulin light chain (AL) amyloidosis patients without clinically overt polyneuropathy compared to healthy controls [Citation21–25] and sNfL could be helpful to monitor treatment effect [Citation26, Citation27]. Currently, we do not know the course of sNfL levels in asymptomatic TTRv carriers who develop polyneuropathy and studies on the effect of treatment on sNfL in symptomatic ATTRv patients are scarce and restricted to the effect of TTR-gene-silencers.
Therefore, in this retrospective longitudinal study, levels of sNfL in TTRv carriers and symptomatic ATTRv amyloidosis patients on treatment were measured to assess whether sNfL can detect neuronal damage before polyneuropathy can be detected by clinical assessments or NCS and to assess whether sNfL is a suitable biomarker to monitor treatment effect.
Methods
Study participants
In this retrospective study, ATTRv amyloidosis patients and asymptomatic TTRv carriers who visited the University Medical Center Groningen (UMCG) between 1 January 2000 and 1 December 2021 (N = 204) were assessed for eligibility based on information available in their electronic patient records. Patients were excluded from analysis if they had undergone (domino) liver transplantation, if they had a concomitant disorder associated with neuronal damage, if they participated in a clinical trial, if results of NCS were inconsistent, if they had ATTRv cardiomyopathy without polyneuropathy or if (follow-up) NCS or serum was not available. The selection procedure is shown in Supporting Information Figure S1.
Three different groups were formed and patients were included in one group only:
TTRv carriers (N = 12) and ATTRv amyloidosis patients (N = 8) who were all asymptomatic and remained so during the full observation period (‘merged group of persistently asymptomatic TTRv carriers and ATTRv patients’). Persistently asymptomatic TTRv carriers were defined as carriers of a pathogenic TTR gene variant without symptoms or signs of polyneuropathy and no signs of polyneuropathy on NCS and no cardiomyopathy on all occasions before and after timepoint of inclusion. Persistently asymptomatic ATTRv amyloidosis patients were defined as carriers of a pathogenic TTR variant without symptoms or signs of polyneuropathy and no signs of polyneuropathy on NCS and no cardiomyopathy on all occasions before and after timepoint of inclusion but with amyloid detectable in the subcutaneous abdominal fat tissue.
TTRv carriers who developed polyneuropathy in the observation period and, therefore, progressed to patients with symptomatic ATTRv amyloidosis (N = 7). They were defined as carriers of a pathogenic TTR variant who developed polyneuropathy. ATTRv amyloidosis was diagnosed according to current guidelines [Citation28]. Polyneuropathy was defined as symmetrical distal neuropathic symptoms or signs of sensory loss, which had to be confirmed by NCS or QST. Severity of polyneuropathy was graded according to the Polyneuropathy Disability (PND) score [Citation28].
ATTRv amyloidosis patients with polyneuropathy on treatment with a TTR-stabiliser (N = 20), TTR-silencer (N = 7) or both a TTR-stabiliser and TTR-silencer (N = 11). Patients that were treated with both a TTR-stabiliser and TTR-silencer were merged with the patients that were treated with a TTR-silencer only to become one group (N = 18).
Study design
Patients were retrospectively included if they fulfilled the criteria of one of the three groups as described above, and if at least two serum samples at different time points were available to evaluate sNfL levels at time of NCS (t = 0) with a follow-up of approximately 2 years. Patients on a TTR-silencer were included if two serum samples were available with a follow-up of approximately 1 year. N-terminal prohormone of brain natriuretic peptide (NT-proBNP), troponin T, serum creatinine and results of NCS were retrieved from the clinical records in all patients. In the persistently asymptomatic TTRv carriers and in the TTRv carriers who developed polyneuropathy, also the results of QST and subcutaneous fat tissue biopsies [Citation29] were retrieved if available. NCS and QST had been performed by one of our clinical neurophysiologists according to current guidelines.
All procedures were in compliance with the Declaration of Helsinki. The study was approved by the institutional ethical review board of the UMCG (Registration number: 201900860).
Blood sample collection, NfL measurement and reference values
In this retrospective study, blood samples from the AmyloidLines Biobank were used. Blood samples were collected between 1 January 2000 and 1 December 2021. Blood was drawn by venipuncture at the outpatient clinic of the UMCG, centrifuged at 2700 rpm for 10 min at room temperature, serum was stored within 1 h at −20 °C and subsequently stored at −80 °C within 6 months. NfL has shown to be stable in serum and plasma samples stored at −80 °C for up to 20 years [Citation30]. Measurements of NfL were performed using the NF-light Kit (Quanterix) on single-molecule array (Simoa HDX) technology (Quanterix Corp., Billerica, MA, USA) [Citation31]. The measurements were performed by certified technicians that were blinded to the clinical information at the Neurochemistry Laboratory of the Amsterdam UMC location VUmc.
Reference values including 95% confidence interval (CI) were retrieved from a large online database of individuals without a neurological disorder [Citation32] developed by the Neurochemistry lab Amsterdam. The age-dependent reference values are depicted for each individual in Supporting Information Figures S2(A–G).
Statistical analysis
Data were analysed at the UMCG. The Wilcoxon rank test was used to compare sNfL, NT-proBNP, troponin T and creatinine in the same participants at different time points. A two-sided Mann–Whitney U-test was used to compare sNfL levels between different study groups. A p-value < 0.05 was considered significant. Differences in follow-up duration between patients were adjusted by calculating the change in sNfL per month (delta sNfL), assuming a linear relationship between time and sNfL levels. Statistical analysis was performed using GraphPad Prism 8.4.2 (GraphPad Software, La Jolla, CA, USA).
Results
Clinical and demographic characteristics
In total, 65 patients were selected based on the inclusion and exclusion criteria: 12 persistently asymptomatic TTRv carriers and 8 persistently asymptomatic ATTRv amyloidosis patients, 7 TTRv carriers who developed polyneuropathy and 38 ATTRv patients with polyneuropathy on treatment (TTR-stabiliser (N = 20), TTR-silencer (N = 7) or both a TTR-stabiliser and TTR-silencer (N = 11)). Clinical characteristics of each group are shown in . NT-proBNP increased over a mean follow-up time of 126 months in TTRv carriers who developed polyneuropathy. NT-proBNP increased and troponin T decreased over a mean follow-up time of 25 months in 20 ATTRv patients with polyneuropathy on a TTR-stabiliser.
Table 1. Characteristics of TTRv carriers and ATTRv amyloidosis patients.
In the merged group of persistently asymptomatic TTRv carriers and ATTRv amyloidosis patients (N = 20), two patients received treatment with a TTR-stabiliser at t = 0 and eight patients at t = 1. None of the persistently asymptomatic TTRv carriers (N = 12) received treatment with a TTR-stabiliser at t = 0 whereas four carriers did at t = 1. Diflunisal was started because of a well-informed personal request of these four carriers.
In the seven TTRv carriers who developed polyneuropathy three patients already received treatment with a TTR-stabiliser at t = 0, while all seven patients received treatment at the time the PND score became I. Furthermore, all TTRv carriers who developed polyneuropathy directly started treatment with a TTR-silencer after polyneuropathy was established.
In the 38 ATTRv patients with polyneuropathy, 20 patients received treatment with a TTR-stabiliser only, 7 patients received treatment with a TTR-silencer only and 11 patients received treatment with both a TTR-stabiliser and TTR-silencer. Twelve of the 20 patients in the TTR-stabiliser only group did not yet receive this treatment at t = 0 but started on it within 3 months.
sNfL levels in the merged group of persistently asymptomatic TTRv carriers and ATTRv patients
In the merged group of persistently asymptomatic TTRv carriers and ATTRv patients the median sNfL increased from 7.6 to 8.7 pg/mL over a mean follow-up time of 26 months, which corresponds with a median increase in sNfL of 0.1 pg/mL/month.
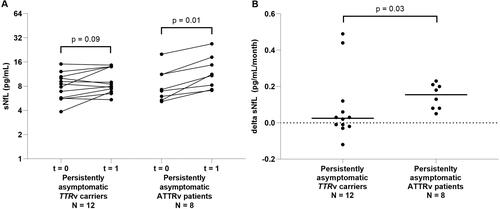
We compared the course of sNfL between persistently asymptomatic TTRv carriers and persistently asymptomatic ATTRv patients. In 12 persistently asymptomatic TTRv carriers without subcutaneous amyloid deposition, the median sNfL level did not increase. In eight persistently asymptomatic ATTRv patients, the median sNfL level increased (from 7.1 to 11.1 pg/mL) (). Accordingly, the median monthly change of sNfL (0.0 pg/mL/month) in persistently asymptomatic TTRv carriers was lower than the monthly change of sNfL (0.2 pg/mL/month) in persistently asymptomatic ATTRv amyloidosis patients ().
Figure 1. sNfL levels in the asymptomatic group. (A) sNfL levels in persistently asymptomatic TTRv carriers (amyloid negative) and in persistently asymptomatic ATTRv patients (amyloid positive) over approximately 2 years. (B) Change of sNfL per month (delta sNfL) in persistently asymptomatic TTRv carriers and in persistently asymptomatic ATTRv patients. sNfL: serum neurofilament light chain; t = 0: first timepoint at which levels of sNfL were measured; t = 1: second timepoint at which levels of sNfL were measured.
sNfL levels in TTRv carriers who developed polyneuropathy
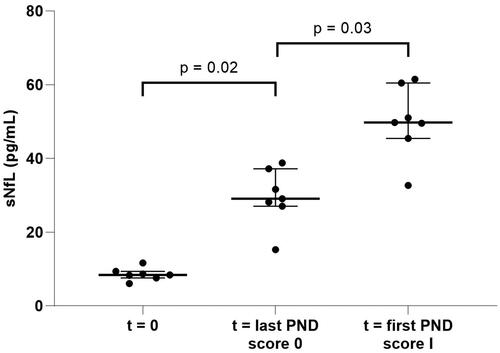
In the seven TTRv carriers who developed polyneuropathy, the median sNfL increased (p < 0.05) from 8.4 to 49.8 pg/mL after a mean follow-up of 126 months until the first PND score of I ( and ). For this group, the change of sNfL per month until the first PND score I was 0.3 pg/mL/month. The change per month between the second last PND score 0 and the last PND score 0 was 0.4 pg/mL/month (Supporting Information Figures S2(A–G), period 1; Supporting Information Table S2). The change per month between the last PND score 0 and the first PND score I was 0.9 pg/mL/month (Supporting Information Figures S2(A–G), period 2; Supporting Information Table S2). For each individual patient, age and gender-specific sNfL reference values are depicted in Supporting Information Figures S2(A–G).
Figure 2. sNfL in TTRv carriers who developed polyneuropathy. sNfL levels at t = 0 (first sample available, asymptomatic stage), t = last PND score 0 (sample at the last asymptomatic timepoint), t = first PND score I (sample at the first symptomatic timepoint where the PND score is at least I). sNfL: serum neurofilament light chain; PND: polyneuropathy disability.
The course of the sNfL levels corresponds in five patients with the disease development (Supporting Information Figures S2(C–G)). Unexplained fluctuations in sNfL levels were seen in the remaining two patients (Supporting Information Figures S2(A,B)).
sNfL levels in ATTRv patients with polyneuropathy
In the ATTRv patients with polyneuropathy who received treatment with a TTR-stabiliser (N = 20), the median sNfL did not change over a mean follow-up time of 25 months (, ). In ATTRv patients with polyneuropathy who received treatment with a TTR-silencer (N = 18), the median sNfL decreased from 61.2 to 37.7 pg/mL over a mean follow-up time of 14 months ( and ).
Figure 3. sNfL levels in ATTRv patients with polyneuropathy. (A) sNfL levels in ATTRv patients with polyneuropathy treated with a TTR-stabiliser over approximately 2 years and sNfL levels in ATTRv patients with polyneuropathy treated with a TTR-silencer over approximately 1 year. (B) Change of sNfL per month (delta sNfL) in ATTRv patients with polyneuropathy treated with a TTR-stabiliser or a TTR-silencer. Closed dots represent patients on a TTR-stabiliser or TTR-silencer only and open dots represent patients on combined treatment with a TTR-stabiliser and TTR-silencer. sNfL: serum neurofilament light chain; t = 0: first timepoint at which levels of sNfL were measured; t = 1: second timepoint at which levels of sNfL were measured.
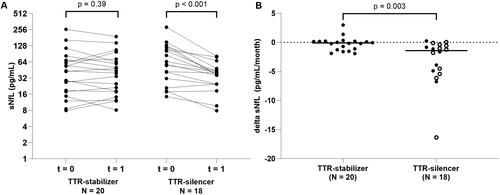
There was a difference in change of sNfL per month between ATTRv patients with polyneuropathy who received treatment with a TTR-stabiliser (median −0.1 pg/mL/month) and ATTRv patients with polyneuropathy who received treatment with a TTR-silencer (median −1.4 pg/mL/month) (p < 0.01) ().
We found no difference in change of sNfL per month between ATTRv patients with polyneuropathy who received treatment with a TTR-silencer with (median −1.2 pg/mL/month) (N = 11) or without (median −1.6 pg/mL/month) (N = 7) concomitant use of a TTR-stabiliser (p = 0.93).
Comparison of sNfL levels between study groups
At t = 0, the levels of sNfL were higher in ATTRv patients with polyneuropathy, irrespective of treatment, compared to the merged group of persistently asymptomatic TTRv carriers and ATTRv patients (all p < 0.001) as well as to the group of TTRv carriers who later developed polyneuropathy (p < 0.001 for TTR-stabiliser and p < 0.001 for TTR-silencer). The sNfL levels of ATTRv patients with polyneuropathy at both t = 0 and t = 1, irrespective of treatment, did not differ from TTRv carriers who developed polyneuropathy at t = first PND score I.
At t = 0, the sNfL levels did not differ between the merged group of persistently asymptomatic TTRv carriers and ATTRv patients and the group of TTRv carriers who later developed polyneuropathy. At t = last PND score 0 (i.e. no polyneuropathy symptoms yet), TTRv carriers who later developed polyneuropathy had higher sNfL levels (median 29.1 pg/mL) compared to the merged group of persistently asymptomatic TTRv carriers and ATTRv patients at t = 1 (median 8.7 pg/mL) ().
Figure 4. Comparison of sNfL between the asymptomatic group and TTRv carriers who developed polyneuropathy. (A) sNfL levels in the merged group of persistently asymptomatic TTRv carriers and ATTRv patients at t = 1 compared to the TTRv carriers who developed polyneuropathy at t = last PND score 0. (B) Change in sNfL per month (delta sNfL) in the merged group of persistently asymptomatic TTRv carriers and ATTRv patients between t = 0 and t = 1 compared to the delta sNfL in the TTRv carriers who developed polyneuropathy between t = last PND score 0 and t = first PND score I. sNfL: serum neurofilament light chain; PND: Polyneuropathy Disability; PNP: polyneuropathy; t = 1: second timepoint at which levels of sNfL were measured.
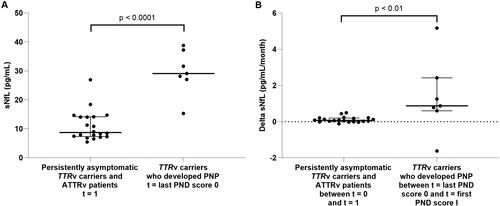
The change of sNfL per month was higher in TTRv carriers who developed polyneuropathy between t = last PND score 0 and t = first PND score I (0.9 pg/mL/month) compared to the change in the group of persistently asymptomatic TTRv carriers and ATTRv patients between t = 0 and t = 1 (0.1 pg/mL/month) (). The change of sNfL per month was not different in TTRv carriers who developed polyneuropathy between t = second last PND score 0 and t = last PND score 0 (0.4 pg/mL/month) compared to the change in the merged group of persistently asymptomatic TTRv carriers and ATTRv patients between t = 0 and t = 1 (p = 0.14).
Discussion
This study shows that (1) sNfL levels can be used to detect neuronal damage before polyneuropathy symptom onset and before polyneuropathy can be confirmed by NCS and (2) sNfL levels can be used to monitor the effect of treatment in ATTRv amyloidosis patients. In addition, this study confirms previous studies showing that sNfL levels are increased in ATTRv patients with polyneuropathy.
Firstly, we longitudinally evaluated levels of sNfL in persistently asymptomatic TTRv carriers and persistently asymptomatic ATTRv amyloidosis patients (based on amyloid detected in their subcutaneous abdominal fat tissue). At baseline, levels of sNfL were comparable to what we previously found in a cross-sectional study in TTRv carriers and healthy age- and sex-matched controls [Citation23] and levels corresponded with previously published reference values [Citation32, Citation33]. In this study, we show that levels of sNfL increase over time in persistently asymptomatic ATTRv patients but not in the persistently asymptomatic TTRv carriers over a timespan of approximately 2 years. The increase in levels of sNfL (25% per year) in persistently asymptomatic ATTRv patients is much higher than can be explained by ageing alone as a large study on individuals without a neurological disorder showed an increase of 2.2% per year [Citation33, Citation34] and the Swiss sNfL reference database showed an increase of 2.1% per year in individuals 30–50 years up to 3.3% in individuals > 60 years [Citation24]. Creatinine levels slightly increased over time in this merged group of persistently asymptomatic TTRv carriers and persistently asymptomatic ATTRv amyloidosis patients. As renal function affects sNfL levels, this may have influenced results [Citation35]. At the Groningen Amyloidosis Center of Expertise (GrACE), diflunisal is prescribed to asymptomatic ATTRv patients in whom amyloid has been shown in a biopsy and sometimes diflunisal is prescribed on well-informed personal request of a TTRv carrier. This explains why all asymptomatic ATTRv amyloidosis patients and four out of twelve asymptomatic TTRv carriers used diflunisal. Diflunisal retards disease progression [Citation5], and therefore, it is likely that diflunisal influenced results. The increase in levels of sNfL would probably have been more pronounced if the asymptomatic ATTRv patients were not using diflunisal. A recent large cross-sectional study on sNfL levels in ATTRv amyloidosis, showed no significant differences in sNfL when stratifying ATTRv amyloidosis patients based on genotype or phenotype. In a multivariate regression analysis, neuropathy severity appeared to be the only significant predictor of sNfL levels in contrast to age, sex and disease duration [Citation36]. Based on this study, it seems unlikely that factors other than renal function and the use of diflunisal, confounded our results. Our findings show that sNfL can be used to detect neuronal damage before the onset of polyneuropathy symptoms and support the generally accepted concept that amyloid deposition is first in the order of events leading to neuronal damage in ATTRv amyloidosis [Citation16].
Secondly, we longitudinally studied sNfL levels over the disease course from initially asymptomatic TTRv carriers or asymptomatic ATTRv patients, to symptomatic ATTRv patients with polyneuropathy. In these initially asymptomatic individuals, levels of sNfL began to rise years before the onset of symptoms and before polyneuropathy could be confirmed by NCS. Levels of sNfL were already above the 95% percentile of normal [Citation32] 5.5 years (range 3.0–7.6 years) before the onset of symptoms (PND score I). The course of sNfL in persistently asymptomatic TTRv carriers roughly corresponds to what could be expected in healthy controls. But once amyloid became detectable in the subcutaneous abdominal fat tissue, thus transforming carriers to asymptomatic ATTRv amyloidosis patients, sNfL levels started to rise faster than could be expected based on ageing alone and increased more or less exponentially before the first symptoms of polyneuropathy appeared. One patient in the group of TTRv carriers who developed polyneuropathy manifested with cardiomyopathy during follow-up. It is unlikely that this has influenced the results as another study of our group showed that cardiac involvement is not an independent predictor of sNfL levels (unpublished) [Citation37]. The findings from our analysis of persistently asymptomatic TTRv carriers, asymptomatic ATTRv amyloidosis patients and asymptomatic TTRv carriers that became ATTRv patients with polyneuropathy collectively support the use of sNfL and subcutaneous abdominal fat tissue biopsies in monitoring for disease onset in TTRv carriers. However, the sensitivity of subcutaneous abdominal fat tissue biopsies and, therefore, the value of biopsies in monitoring highly depends on the skills and experience of the laboratory that processes and assesses the biopsies [Citation38]. In contrast, sNfL levels can be assessed by using accessible immunoassays, e.g. the Simoa assay, but also by using routine laboratory technologies, such as Lumipulse [Citation39], which allows straightforward reliable and sensitive longitudinal quantification of serum and plasma NfL.
Thirdly, we longitudinally evaluated sNfL levels in ATTRv amyloidosis patients with polyneuropathy that received treatment with a TTR-stabiliser, a TTR-silencer or a TTR-stabiliser and TTR-silencer. Higher baseline levels of sNfL in patients that received treatment with a TTR-silencer indicated more severe polyneuropathy in these patients as compared to the patients that received treatment with a TTR-stabiliser. Indeed, this was reflected by a higher PND score in these patients. sNfL levels reported in our study correspond to what has been found in previous studies in ATTRv amyloidosis [Citation21–23, Citation26, Citation27]. The levels of sNfL remained stable in the group that received treatment with a TTR-stabiliser and decreased in patients who received treatment with a TTR-silencer. The effect of TTR-stabilisers on sNfL has not been studied before. Our findings on the effect of a TTR-silencer on sNfL are in line with previous studies of sNfL in patients that participated in the Phase 3 study of patisiran and the patisiran Global Open Label Extension study [Citation26, Citation27, Citation40]. In these studies, the change in plasma NfL correlated with a change in the modified Neuropathy Impairment +7 score indicating that decreasing levels of NfL are associated with improvement in polyneuropathy.
No difference in sNfL levels was found between patients treated with a TTR-silencer and patients treated with a combination of a TTR-stabiliser and TTR-silencer (p = 0.9), despite the potentially synergistic action of these treatments [Citation41, Citation42]. Studies designed to compare combined treatment with monotherapy are desirable.
Limitations
This study was designed to establish the course of sNfL in persistently asymptomatic TTRv carriers, in ATTRv amyloidosis patients and in TTRv carriers who progressed to ATTRv amyloidosis patients with polyneuropathy. For this purpose, patients were retrospectively selected from the Groningen Amyloidosis Cohort. Apart from the retrospective nature of this study, other limitations to this study should be mentioned.
From our cohort, seven TTRv carriers could be selected that developed polyneuropathy during the course of follow-up at our centre. This is a unique but small cohort and although all patients showed an increase in sNfL before symptoms of polyneuropathy appeared, two of these patients showed fluctuations in sNfL that we cannot explain based on the medical records. All seven patients used a TTR-stabiliser from a certain point of time during follow-up, and this may have affected the results. As this group of patients is small, we were not able to investigate sNfL in relation to genotype. Prospective longitudinal investigation of a larger number of TTRv carriers with a variety of genotypes is warranted to specify the dynamics of both sNfL and cardiac biomarkers in TTRv carriers that progress to symptomatic ATTRv amyloidosis patients over time.
Follow-up time was not identical in the different groups. To correct for differences in follow-up time, the change of sNfL per month was calculated. Although this allows for comparison of the change across groups, this method assumes a linear relationship between sNfL and time which could be nonlinear in reality. The change over time was calculated over short intervals to approximate reality as closely as possible.
This study aimed to study sNfL in relation to large fibre neuropathy. Therefore, patients with large fibre neuropathy established by NCS were included in this study. Considering the results of this study it would also have been valuable to study sNfL in relation to measures of small fibre and autonomic neuropathy. Unfortunately, results of neurophysiological assessments of small fibre and autonomic neuropathy were not available for part of the patients leaving us unable to perform a meaningful analysis. Further research on sNfL in relation to small fibre and autonomic neuropathy in ATTRv amyloidosis is warranted.
Conclusion
We showed that sNfL is a biomarker for the detection of neuronal damage even in a pre-symptomatic stage in TTRv carriers and ATTRv amyloidosis patients, and it is a useful biomarker to monitor treatment effect in ATTRv amyloidosis patients. sNfL levels are stable and comparable to healthy controls in persistently asymptomatic TTRv carriers. sNfL levels start to rise much faster than can be expected based on ageing alone, when amyloid disease becomes histologically detectable and sNfL levels are already above the 95th percentile of the normal range some years before the onset of polyneuropathy symptoms. In ATTRv amyloidosis patients with polyneuropathy who receive treatment with a TTR-stabiliser, sNfL levels remain stable over the course of 2 years whereas in patients who receive treatment with a TTR-silencer, the sNfL levels decrease.
Our data support the use of sNfL in screening TTRv carriers and in monitoring disease progression and effect of treatment in ATTRv amyloidosis patients. Larger studies are warranted to confirm and specify the results presented here and to investigate sNfL in relation to small fibre neuropathy, autonomic neuropathy and genotype.
| Abbreviations | ||
| AL | = | immunoglobulin light chain amyloid |
| ATTRv | = | hereditary transthyretin amyloid |
| CI | = | confidence interval |
| eGFR | = | estimated glomerular filtration rate |
| GrACE | = | Groningen Amyloidosis Center of Expertise |
| NCS | = | nerve conduction studies |
| NfL | = | neurofilament light chain |
| NT-proBNP | = | N-terminal pro-brain-type natriuretic peptide |
| PND | = | polyneuropathy disability |
| PNP | = | polyneuropathy |
| QST | = | quantitative sensory testing |
| Simoa | = | single-molecule array |
| sNfL | = | serum neurofilament light chain |
| TTR | = | transthyretin |
| TTRv | = | transthyretin variant carrier |
| UMCG | = | University Medical Center Groningen |
Supplemental Material
Download MS Word (18.3 KB)Supplemental Material
Download MS Word (2.3 MB)Acknowledgement
The authors have no acknowledgements to share.
Disclosure statement
The UMCG, which employs H.L.A. Nienhuis, received consultancy fees from Pfizer and Alnylam. The other authors report no competing interests.
Data availability statement
The datasets generated during and/or analysed during this study are available from the corresponding author on reasonable request.
Additional information
Funding
References
- Samuelsson K, Jovanovic A, Egervall K, et al. Hereditary transthyretin amyloidosis in Sweden: comparisons between a non-endemic and an endemic region. Amyloid. 2022;29(4):220–227. doi: 10.1080/13506129.2022.2065191.
- Conceição I, González-Duarte A, Obici L, et al. Red-flag symptom clusters in transthyretin familial amyloid polyneuropathy. J Peripher Nerv Syst. 2016;21(1):5–9. doi: 10.1111/jns.12153.
- Hazenberg BPC. Amyloidosis: a clinical overview. Rheum Dis Clin North Am. 2013;39(2):323–345. doi: 10.1016/j.rdc.2013.02.012.
- Adams D, Gonzalez-Duarte A, O’Riordan WD, et al. Patisiran, an RNAi therapeutic, for hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):11–21. doi: 10.1056/NEJMoa1716153.
- Berk JL, Suhr OB, Obici L, et al. Repurposing diflunisal for familial amyloid polyneuropathy: a randomized clinical trial. JAMA. 2013;310(24):2658–2667. doi: 10.1001/jama.2013.283815.
- Benson MD, Waddington-Cruz M, Berk JL, et al. Inotersen treatment for patients with hereditary transthyretin amyloidosis. N Engl J Med. 2018;379(1):22–31. doi: 10.1056/NEJMoa1716793.
- Coelho T, Maia LF, Da Silva AM, et al. Long-term effects of tafamidis for the treatment of transthyretin familial amyloid polyneuropathy. J Neurol. 2013;260(11):2802–2814. doi: 10.1007/s00415-013-7051-7.
- Adams D, Suhr OB, Hund E, et al. First european consensus for diagnosis, management, and treatment of transthyretin familial amyloid polyneuropathy. Curr Opin Neurol. 2016;29 Suppl 1(Suppl 1):S14–S26. doi: 10.1097/WCO.0000000000000289.
- Obici L, Kuks JB, Buades J, et al. Recommendations for presymptomatic genetic testing and management of individuals at risk for hereditary transthyretin amyloidosis. Curr Opin Neurol. 2016;29 Suppl 1(Suppl 1):S27–S35. doi: 10.1097/WCO.0000000000000290.
- Adams D, Ando Y, Beirão JM, et al. Expert consensus recommendations to improve diagnosis of ATTR amyloidosis with polyneuropathy. J Neurol. 2021;268(6):2109–2122. doi: 10.1007/s00415-019-09688-0.
- Dyck PJ, Davies JL, Litchy WJ, et al. Longitudinal assessment of diabetic polyneuropathy using a composite score in the rochester diabetic neuropathy study cohort. Neurology. 1997;49(1):229–239. doi: 10.1212/wnl.49.1.229.
- Dyck PJ, Kennedy WR, Kesserwani H, et al. Limitations of quantitative sensory testing when patients are biased toward a bad outcome. Neurology. 1998;50(5):1213–1213. doi: 10.1212/wnl.50.5.1213.
- Casellini CM, Parson HK, Richardson MS, et al. Sudoscan, a noninvasive tool for detecting diabetic small fiber neuropathy and autonomic dysfunction. Diabetes Technol Ther. 2013;15(11):948–953. doi: 10.1089/dia.2013.0129.
- Low PA, Tomalia VA, Park KJ. Autonomic function tests: some clinical applications. J Clin Neurol. 2013;9(1):1–8. doi: 10.3988/jcn.2013.9.1.1.
- Luigetti M, Romozzi M, Bisogni G, et al. hATTR pathology: nerve biopsy results from italian referral centers. Brain Sci. 2020;10(11):780. doi: 10.3390/brainsci10110780.
- Fernandes A, Coelho T, Rodrigues A, et al. Clinicopathological correlations of sural nerve biopsies in TTR Val30Met familial amyloid polyneuropathy. Brain Commun. 2019;1(1):fcz032.
- Masuda T, Ueda M, Misumi Y, et al. Reduced intraepidermal nerve fibre density in patients with hereditary transthyretin amyloidosis. Amyloid. 2019;26(sup1):79–80. doi: 10.1080/13506129.2019.1583198.
- Ebenezer GJ, Liu Y, Judge DP, et al. Cutaneous nerve biomarkers in transthyretin familial amyloid polyneuropathy. Ann Neurol. 2017;82(1):44–56. doi: 10.1002/ana.24972.
- Leonardi L, Adam C, Beaudonnet G, et al. Skin amyloid deposits and nerve fiber loss as markers of neuropathy onset and progression in hereditary transthyretin amyloidosis. Eur J Neurol. 2022;29(5):1477–1487. doi: 10.1111/ene.15268.
- Leonardi L, Galosi E, Vanoli F, et al. Skin biopsy and quantitative sensory assessment in an italian cohort of ATTRv patients with polyneuropathy and asymptomatic carriers: possible evidence of early non-length dependent denervation. Neurol Sci. 2022;43(2):1359–1364. doi: 10.1007/s10072-021-05434-5.
- Kapoor M, Foiani M, Heslegrave A, et al. Plasma neurofilament light chain concentration is increased and correlates with the severity of neuropathy in hereditary transthyretin amyloidosis. J Peripher Nerv Syst. 2019;24(4):314–319. doi: 10.1111/jns.12350.
- Maia LF, Maceski A, Conceição I, et al. Plasma neurofilament light chain: an early biomarker for hereditary ATTR amyloid polyneuropathy. Amyloid. 2020;27(2):97–102. doi: 10.1080/13506129.2019.1708716.
- Louwsma J, Brunger AF, Bijzet J, et al. Neurofilament light chain, a biomarker for polyneuropathy in systemic amyloidosis. Amyloid. 2021;28(1):50–55. doi: 10.1080/13506129.2020.1815696.
- Loser V, Benkert P, Vicino A, et al. Serum neurofilament light chain as a reliable biomarker of hereditary transthyretin-related amyloidosis—a Swiss reference center experience. J Peripher Nerv Syst. 2023;28(1):86–97. doi: 10.1111/jns.12524.
- Luigetti M, Di Paolantonio A, Guglielmino V, et al. Neurofilament light chain as a disease severity biomarker in ATTRv: data from a single-centre experience. Neurol Sci. 2022;43(4):2845–2848. doi: 10.1007/s10072-021-05850-7.
- Ticau S, Sridharan GV, Tsour S, et al. Neurofilament light chain as a biomarker of hereditary transthyretin-mediated amyloidosis. Neurology. 2021;96(3):E412–E422. doi: 10.1212/WNL.0000000000011090.
- Ticau S, Aldinc E, Polydefkis M, et al. Treatment response and neurofilament light chain levels with long-term patisiran in hereditary transthyretin-mediated amyloidosis with polyneuropathy: 24-month results of an open-label extension study. Amyloid. 2023;31(1):1–11. doi: 10.1080/13506129.2023.2232520.
- Ando Y, Coelho T, Berk JL, et al. Guideline of transthyretin-related hereditary amyloidosis for clinicians. Orphanet J Rare Dis. 2013;8(1):31. doi: 10.1186/1750-1172-8-31.
- Van Gameren II, Hazenberg BPC, Bijzet J, et al. Diagnostic accuracy of subcutaneous abdominal fat tissue aspiration for destecting systemic amyloidosis and its utility in clinical practice. Arthritis Rheum. 2006;54(6):2015–2021. doi: 10.1002/art.21902.
- Schubert CR, Paulsen AJ, Pinto AA, et al. Effect of long-term storage on the reliablity of blood biomarkers for alzheimer’s disease and neurodegeneration. J Alzheimers Dis. 2022;85(3):1021–1029. doi: 10.3233/JAD-215096.
- Van Lierop ZYGJ, Verberk IMW, Van Uffelen KWJ, et al. Pre-analytical stability of serum biomarkers for neurological disease: neurofilament-light, glial fibrillary acidic protein and contactin-1. Clin Chem Lab Med. 2022;60(6):842–850. doi: 10.1515/cclm-2022-0007.
- Vermunt L, Otte M, Verberk IMW, et al. Age- and disease-specific reference values for neurofilament light presented in an online interactive support interface. Ann Clin Transl Neurol. 2022;9(11):1832–1837. doi: 10.1002/acn3.51676.
- Benkert P, Meier S, Schaedelin S, et al. Serum neurofilament light chain for individual prognostication of disease activity in people with multiple sclerosis: a retrospective modelling and validation study. Lancet Neurol. 2022;21(3):246–257. doi: 10.1016/S1474-4422(22)00009-6.
- Khalil M, Teunissen CE, Otto M, et al. Neurofilaments as biomarkers in neurological disorders. Nat Rev Neurol. 2018;14(10):577–589. doi: 10.1038/s41582-018-0058-z.
- Akamine S, Marutani N, Kanayama D, et al. Renal function is associated with blood neurofilament light chain level in older adults. Sci Rep. 2020;10(1):20350. doi: 10.1038/s41598-020-76990-7.
- Romano A, Primiano G, Antonini G, et al. Serum neurofilamant light chain: a promising biomarker for hereditary transthyretin amyloidosis? Eur J Neurol. 2024;31(1):e16070.
- Berends M, Nienhuis HLA, Brunger AF, et al. Serum neurofilament light chain (sNfL) in relation to myocardial sympathetic neuronal damage based on 123I-meta-iodobenzylguanidine (MIBG) scintigraphy in hereditary transthyretin (ATTRv) amyloidosis. Paper presented at: Cardiovascular metabolism, innervation and perfusion. The Annual European Association of Nuclear Medicine; 2022 Oct 15–19; Barcelona, Spain.
- Groothof D, Bijzet J, van den Berg MP, et al. Diagnostic value of subcutaneous abdominal fat tissue aspirates in cardiac amyloidosis. Poster presented at: Cardiac amyloidosis and other forms. The XVII International Symposium On Amyloidosis; 2020 Sep 14–18; Tarragona, Spain.
- Coppens S, Lehmann S, Hopley C, et al. Neurofilament-light, a promising biomarker: analytical, metrological and clinical challenges. Int J Mol Sci. 2023;24(14):11624. doi: 10.3390/ijms241411624.
- Polydefkis M, Aldinc E, Nienhuis H, et al. NfL levels significantly decrease in response to treatment with patisiran or vutrisiran in hATTR amyloidosis with polyneuropathy. Poster presented at: Global neurology. 147th annual meeting American Neurological Association; 2022 Oct 22–25; Chicago, IL.
- Ando Y, Adams D, Benson MD, et al. Guidelines and new directions in the therapy and monitoring of ATTRv amyloidosis. Amyloid. 2022;29(3):143–155. doi: 10.1080/13506129.2022.2052838.
- Buxbaum JN. Treatment of hereditary and acquired forms of transthyretin amyloidosis in the era of personalized medicine: the role of randomized controlled trials. Amyloid. 2019;26(2):55–65. doi: 10.1080/13506129.2019.1575201.