Abstract
A series of novel 2-substituted 1,2,3,4-tetrahydroeudistomin U derivatives were synthesized by the key Pictet-Spengler cyclization reaction and their in vitro. antitumor activities were evaluated using Pyricularia oryzae. Cavara and Caco-2 cancer cells. Most of the 2-alkyl-1,2,3,4-tetrahydroeudistomin U compounds (19– 23) showed potent inhibitory activities in both of the biological tests. Preliminary structure-activity relationships are also presented.
Introduction
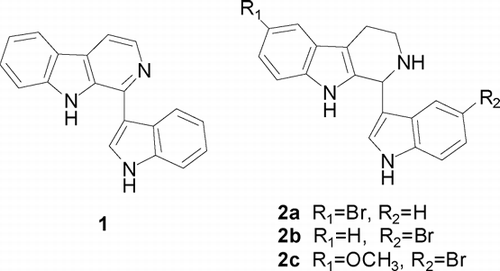
Eudistomins, originally isolated from the Caribbean tunicate Eudistoma olivaceum. Van Name (Clavelinidae), are natural marine alkaloids that show very interesting biological properties (Dong et al., Citation2003). In 1994, Francisco et al. (Badre et al., Citation1994) isolated, from Caribbean ascidian Lissoclinum fragile. Van Name (Didemnidae), a new β-carboline with a 3-indolyl substituent at C-l, eudistomin U (1) (). This alkaloid was found to be capable of binding to DNA and showed strong antibacterial activity. Our previous work had verified the potent antitumor activities of 1,2,3,4-tetrahydroeudistomin U derivatives (2a–2c) () against murine P-388 in vitro. (Dong et al., Citation2004). Recently, a series of 1,2,3,4-tetrahydro-β-carboline derivatives were reported to be CDK4-specific inhibitors (Aubry et al., Citation2006), which may cause G1 arrest of tumor cells. Several 1-aryl substituted 1,2,3,4-tetrahydro-β-carboline derivatives also showed significant cytotoxicity against murine P-388 and human tumor cell lines, KB-16, A-549, and HT-29 (Shen et al., Citation2005).
Figure 1 Structures of eudistomin U (1) and 1,2,3,4-tetrahydroeudistomin U derivatives (2a-– 2c).
In addition, a screening method detecting morphologic deformations or inhibition of mycelia germinated from conidia of Pyricularia oryzae. Cavara (Moniliaceae) has been developed and applied to evaluate the bioactivities of antimitotic and antifungal substances (Iwasaki, Citation1984; Kobayashi et al., Citation1995; Kobayashi et al., Citation1996).
In this paper, based on our previous work on eudistomin U, we synthesized a series of 2-substituted 1,2,3,4-tetrahydroeudistomin U derivatives and tested their inhibitory activities against P. oryzae. with the standard method (Kobayashi et al., Citation1996; Zhao & Zhang, Citation2005) and Caco-2 cancer cell MTT staining according to the previously reported procedures with a minor modification (Al-Allaf & Rashan, Citation1998; Zhao et al., Citation2006).
Materials and Methods
Melting points were determined without correction. All reactions were monitored by TLC (silica gel 60 F254). 1H-NMR (Bruker DPX400, Switzerland); δ values are in ppm relative to TMS as an internal standard. Mass spectra: Agilent 1100 Series LC/MSD G1946D mass spectrometer (Santa Clara, CA, USA).
General procedure for preparation of 2-substituted 1,2,3,4-tetrahydroeudistomin U derivatives
The general route for the synthesis of 2-substituted 1,2,3,4-tetrahydroeudistomin U derivatives is shown in . Schiff base 3 was prepared by refluxing tryptamine with indole-3-aldehyde and removing the water produced in the reaction. Compound 3 was treated with trifluoroacetic acid in chloroform, which underwent Pictet-Spengler cyclization to furnish 1,2,3,4-tetrahydroeudistomin U (4). Then, 4 was N.-alkylated by alkyl halides or N.-acylated by carbonyl halides in basic conditions to afford the target compounds 5–28.
Figure 2 The synthetic route of compounds 5–28. Reagents and conditions: (a) PhH, reflux (95%); (b) TFA, CHCl3, rt (58%); (c) alkyl halides, K2CO3, KI, DMF or carbonyl halides, Et3N, CH2Cl2.
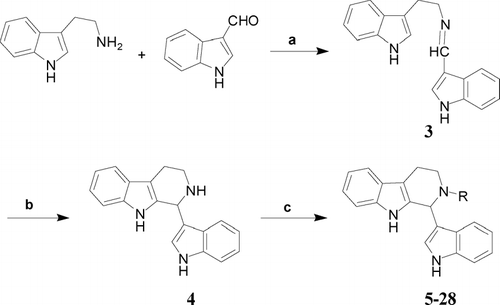
[2-(1H.-Indol-3-yl)-ethyl]-(1H.-indol-3-ylmethylene)amine (3)
A mixture of tryptamine (1.1 g, 6.8 mmol), 1H.-indole-3-carbaldehyde (0.9 g, 6.2 mmol) and benzene (60 mL) was refluxed under N2 for 2 h. Most of the solvent was removed under reduced pressure. After cooling to room temperature, the precipitation was collected by filtration, and washed. The obtained product was dried to yield 3 as a light-yellow solid (1.7 g, 94.8%), m.p. 146–148°C (Dong et al., Citation2004).
1-(1H.-Indol-3-yl)-2,3,4,9-tetrahydro-1H.-β carboline (4)
To a mixture of 3 (0.7 g, 2.4 mmol) and chloroform (5 mL) cooling in ice-bath, trifluoroacetic acid (1.96 mL) in chloroform (4 mL) was added dropwise. The reaction mixture was stirred under N2 for 22 h and diluted with ethyl acetate (15 mL). The pH value of the mixture was adjusted to 9–10 with saturated sodium carbonate aqueous solution. The mixture was extracted with ethyl acetate. The organic phases were combined, washed with saturated sodium chloride solution, and dried with magnesium sulfate anhydrous. The solution was filtered, condensed, and purified by silicon gel column chromatography to afford 4 as a light-yellow solid (0.4 g, 58%), m.p. 142–144°C (Dong et al., Citation2004).
General procedure for preparation of compounds 5–18
To a mixture of 4 (0.1 g, 0.348 mmol), triethylamine (48 μ L, 0.348 mmol), and THF (20 mL), various acyl chlorides (0.348 mmol) were added dropwise. The reaction mixture was stirred under N2 for 2 h and extracted with ethyl acetate (20 mL). The organic phase was washed with saturated sodium chloride solution and dried with magnesium sulfate anhydrous. The solution was filtered, condensed, and purified by silicon gel column chromatography to afford 5–18.
General procedure for preparation of compounds 19–28
To a mixture of 4 (0.15 g, 0.523 mmol), potassium carbonate anhydrous (72 mg, 0.523 mmol), and DMF (10 mL), various alkyl halides anhydrous (0.523 mmol) were added dropwise. The reaction mixture was stirred under N2 for 12 h and extracted with ethyl acetate (30 mL). The organic phase was washed with water and saturated sodium chloride solution and dried with magnesium sulfate anhydrous. The solution was filtered, condensed, and purified by silicon gel column chromatography to afford 19–28. The yields and identification of the compounds are listed in .
Table 1 The yield, structure identification and bioassay data of 2-substituted 1,2,3,4-tetrahydroeudistomin U derivatives.
P. oryzae. inhibitory activity assay
P. oryzae. P-2b was grown on a slant culture medium consisting of yeast extract 0.2%, soluble starch 1%, and agar 2% at 27°C. The conidia were collected 12 to 14 days after inoculation by suspending in sterilized water (10 mL). The conidia suspension was filtered to separate from mycelia. The filtrate was added to a 2% solution of yeast extract and adjusted to the concentration of 0.02%. The suspension was adjusted to 4 × 104 conidia/mL by adding sterilized water. A 96-well flat-bottomed assay plate was used for the bioassay. One column was usually used for one test material with eight different concentrations. The assay plates were incubated at 27°C for 16 h, and the shape of mycelia and inhibition of conidia germination was observed under an inverted microscope, and the minimum inhibitory concentration (MIC) values were determined by comparing with griseofulvin as a control.
MTT cytotoxicity assay
Caco-2 cells (2 × 104 cells/well) were grown in RPMI-1640 medium containing 10% (v/v) fetal calf serum, 100 μ g/mL penicillin, and 100 μ g/mL streptomycin. Cultures were propagated at 37°C in a humidified atmosphere containing 5% CO2. The synthetic compound stock solutions were prepared in DMSO. The synthetic compounds in 2% DMSO were added to the cultures at day 2 after seeding. After 48 h, 50 μ L MTT solution (2 mg/mL) per well was added to each culture medium and incubated for an additional 4 h. The optical density was measured by the microplate reader 15 min later after adding 100 μ L DMSO to each well, and the IC50 values were determined by comparing with sunitinib as a control.
Results and Discussions
The biological activity data of target compounds are summarized in . These data obviously suggested that most of the N.-alkylation derivatives exhibited more potent inhibitory activities against P. oryzae. generally compared with the N.-acylation derivatives. Compounds 19–23 with alkyl substitutes gave the best activities in this series of derivatives with the MIC value of 1.6–50 μ g/mL on P. oryzae. inhibitory activities test. This result indicated that these compounds may have potent antitumor activities. It was validated later in the MTT cytotoxicity test in vitro.. The results of the MTT assay showed that the compounds 19–23 with alkyl substitutes suppressed Caco-2 cancer cell proliferation with low IC50 value of 1.2–3.6 μ M, which may contribute to their lipophilic properties. An exceptional phenomenon was observed in that N.-acylation derivatives 11 and 12 also exhibited potent inhibitory activities against Caco-2 cancer cell, whereas they were nearly inactive against P. oryzae..
In conclusion, a series of 2-substituted 1,2,3,4-tetrahydroeudistomin U derivatives were synthesized by a facile and convenient method, and their inhibitory activities against P. oryzae. and Caco-2 cancer cells were assessed in vitro.. Among the compounds tested, compounds 19–23 with alkyl substitutes demonstrated the most potent activities.
References
- Al-Allaf T AK, Rashan L J. Synthesis and cytotoxic evaluation of the first trans.-palladium(II) complex with naturally occurring alkaloid harmine. Eur J Med Chem 1998; 33: 817–820
- Aubry C, Wilson A J, Jenkins P R, Mahale S, Chaudhuri B, Marechal J D, Sutcliffe M J. Design, synthesis and biological activity of new CDK4-specific inhibitors, based on fascaplysin. Org Biomol Chem 2006; 4: 787–801
- Badre A, Boulanger A, Aboumansour E, Banaigs B, Combaut G, Francisco C. Eudistomin-U and isoeudistomin U, new alkaloids from the Caribbean Ascidian Lissoclinum fragile.. J Nat Prod 1994; 57: 528–533
- Dong X C, Miao Y P, Lin Z G, Yu F, Wen R. Advances in studies on eudistomin marine alkaloids. Acta Pharm Sin 2003; 38: 876–880
- Dong X C, Wen R, Zheng J B. Synthesis of 1-indole substituted β-carboline alkaloid and its derivatives and evaluation of their preliminary antitumor activities. Acta Pharm Sin 2004; 39: 259–262
- Iwasaki S, Kobayashi H, Furukawa J, Namikoshi M, Okuda S, Sato Z, Matsuda I, Noda T. Studies on macrocyclic lactone antibiotics. VII. Structure of a phytotoxin “rhizoxin” produced by Rhizopus chinensis.. J Antibiot 1984; 37: 354–362
- Kobayashi H, Namikoshi M, Yoshimoto T, Yokochi T. A screening method for antimitotic and antifungal substances using conidia of Pyricularia oryzae., modification and application to tropical marine fungi. J Antibiot 1996; 49: 873–879
- Kobayashi H, Sunaga R, Furihata K, Morisaki N, Iwasaki S. Isolation and structures of an antifungal antibiotic fusarielin A and related compounds produced by a Fusarium. sp. J Antibiot 1995; 48: 42–52
- Shen Y C, Chen C Y, Hsieh P W, Duh C Y, Lin Y M, Ko C L. The preparation and evaluation of 1-substituted 1,2,3,4-tetrahydro-and 3,4-dihydro-β-carboline derivatives as potential antitumor agents. Chem Pharm Bull 2005; 53: 32–36
- Zhao L L, Zhang H P. Isolation of secondary metabolites of 9F series marine fungi and their bioactivities against Pyricularia oryzae.. Nat Prod Res Dev 2005; 17: 677–680
- Zhao M, Bi L R, Wang W, Wang C, Baudy-Floc'h M, Ju J F, Peng S Q. Synthesis and cytotoxic activities of β-carboline amino acid ester conjugates. Bioorg Med Chem 2006; 14: 6998–7010