Abstract
Vascular smooth muscle cell (VSMC) adhesion and migration play important roles in atherosclerosis and intimal hyperplasia. In our previous study, we found that triflavin, a nonspecific Arg-Gly-Asp (RGD)-containing peptide (also named αIIβ3 disintegrin), may have dual beneficial effects in preventing neointimal formation by acting on both platelets and VSMCs, which has created new incentives for the development of drugs with this combined action. In the present study, triflavin (10, 20, and 50 μg/mL) concentration- dependently inhibited VSMC adhesion to immobilized fibronectin (50 μg/mL). In the flow cytometric study, we found that FITC–triflavin (5 μg/mL) bound directly to VSMC membranes. In a confocal microscopic study, fibronectin (50 μg/mL) markedly stimulated protein kinase C (PKC)-α translocation from the cytosol to the membranes, which was abolished in the presence of triflavin (10 μg/mL). In conclusion, the most important findings of this study suggest that triflavin, an αIIβ3 disintegrin, inhibited immobilized fibronectin-induced cell adhesion and PKC-α translocation in VSMCs.
Introduction
Atherosclerosis and intimal hyperplasia are typical intimal diseases and are major causes of morbidity and mortality. Atherosclerosis can develop in response to endothelial injury caused by many different stimuli, including smoking, diabetes mellitus, hyperlipidemia, and hypertension (CitationLusis, 2000). Intimal hyperplasia results as a response to intimal injury after procedures such as angioplasty, peripheral arterial bypass grafting, or a carotid endarterectomy, and is a major cause of restenosis (CitationWillis et al., 2004).
Vascular smooth muscle cells (VSMCs), which normally reside in the media, are the predominant cell type in atherosclerotic plaque and intimal hyperplasia lesions. When in the intima, VSMCs proliferate and are the major producer of extracellular matrix (ECM) proteins, the predominant components of developed lesions of intimal hyperplasia and atherosclerosis (CitationWillis et al., 2004). The ECM is physiologically active and important in vascular pathophysiology. Increased deposition of the ECM is an essential component of intimal hyperplasia, and accounts for the majority of intimal substances of restenotic lesions (CitationLibby et al. 1997; CitationBatchelor et al., 1998). Although ECM deposition is recognized as prominent in chronic intimal hyperplasia, restenosis, and atherosclerosis, the acute response of the ECM to vascular injury is significantly involved in stimulating VSMC adhesion and migration. VSMC adhesion and migration are important initial steps in the development of neointimal tissues (CitationSchwartz, 1997).
The ECM is an active substrate that interacts with cells through cell matrix receptors, particularly integrins. Integrins are the principal receptors for the ECM and serve as a transmembrane link between the ECM and the actin cytoskeleton (CitationMoiseeva, 2001). Integrins are cell surface heterodimeric glycoprotein receptors that integrate the cytoskeletal activities of a cell with those of its environment by means of cell-to-cell and cell-to-ECM interactions (CitationHynes, 1992). Indeed, several integrins, including the related vitronectin receptor (αVβ3), which are present, for example, on VSMCs and platelets and recognize the Arg-Gly-Asp (RGD) sequence found in many adhesive molecules, participate in the adhesion of VSMCs to vitronectin and migration of cells toward fibronectin, laminin, and collagen types I and IV. Therefore, RGD-containing peptides can block these reactions (CitationHynes, 1992). However, platelet integrins (αIIbβ3) and several other VSMCs and vessel wall integrins (e.g. αVβ1 and αVβ5) also depend on a functional RGD sequence (CitationPytela et al., 1986; CitationSmith et al., 1990), suggesting that the nonspecific RGD blockade may target a wide variety of integrins other than just αVβ3.
Besides establishing cell–matrix interaction, integrins are cell surface receptors that can mediate the bidirectional transfer of information from outside to the inside of cells (CitationClark & Brugge, 1995). Several signaling pathways are stimulated after integrin occupation. Integrin-initiated signals stimulate the tyrosine phosphorylation of several cellular proteins, such as GTP-bound p21 ras, pp125FAK, and others (CitationKrook et al., 1993; CitationRichardson & Parsons, 1996). Ca2+-dependent signaling pathways are also activated by integrins. It was known that adhesion to fibronectin stimulates inositol lipid synthesis and inositol trisphosphate generation (CitationMcNamee et al., 1993). Activation of the Ca2+ messenger system appears to be due to stimulation of phospholipase C (PLC). These intracellular events lead to diacylglycerol (DAG) formation and the activation of protein kinase C (PKC). Moreover, the translocation of PKC from the cytosol to membranes when cells adhere to the ECM has been demonstrated, which may lead to several intracellular events, including focal adhesion formation and cell spreading (CitationHaller et al., 1998).
Several disintegrin peptides have been reported (CitationGould et al., 1990). These peptides all contain RGD and bind with high affinity to integrins on the cell surface. Triflavin, an αIIbβ3 disintegrin, was purified from Trimeresurus flavoviridis snake venom (CitationSheu et al., 1992, Citation1997b, Citation1999). Its primary structure consists of 70 amino acid residues with an RGD sequence at position 49–51 (CitationHuang et al., 1991a). We previously reported that triflavin inhibits platelet aggregation by interfering with the interaction of fibrinogen and αIIbβ3 integrin, and has a powerful influence on the antithrombotic effect and antiangiogenic activity in vivo and in vitro (CitationSheu et al., 1994, Citation1997a). Furthermore, triflavin can inhibit neointimal hyperplasia in a rat model of balloon angioplasty, indicating that triflavin may act on both platelets and VSMCs (CitationSheu et al., 2001). Therefore, the present study was further designed to directly evaluate the inhibitory effects of triflavin on cell adhesion and PKC-α translocation in VSMCs.
Materials and methods
Materials
Trimeresurus flavoviridis venom was purchased from Latoxan (Rosans, France) and stored at −20°C. Triflavin was purified from the venom of T. flavoviridis as previously described (CitationHuang et al., 1991b). Wistar rats were purchased from the Experimental Animal Center of National Taiwan University, College of Medicine (Taipei, Taiwan). Dulbecco’s modified Eagle’s medium (DMEM), trypsin (0.25%), l-glutamine, and penicillin/streptomycin were all purchased from Gibco (Gaithersburg, MD). Fetal bovine serum (FBS) was purchased from PAA Laboratories (Linz, Austria). Fibronectin, poly-l-lysine, and toluidine blue were obtained from Sigma (St. Louis, MO). The anti-PKC-α monoclonal antibody (mAb) was purchased from Cell Signaling (Beverly, MA). Horseradish peroxidase-conjugated secondary antibodies were obtained from Amersham (Buckinghamshire, UK).
VSMC isolation and culture
VSMCs were enzymatically dispersed from male Wistar rats (250–300 g). Thoracic aortas from Wistar rats were removed and stripped of endothelium and adventitia. VSMCs were obtained by a modification of the combined collagenase and elastase digestion method (CitationPauly et al., 1997). These cells were grown in DMEM supplemented with 20 mM HEPES [4-(2-hydroxyethyl)-1- piperazineethanesulfonic acid], 10% FBS, 1% penicillin/streptomycin, and 2 mM glutamine at 37°C in a humidified atmosphere of 5% CO2. The growth medium was changed every 2–3 days until cells had reached confluency. The growth medium was removed, and the monolayer was rinsed with phosphate-buffered saline (PBS). A trypsin–EDTA (ethylenediaminetetraacetic acid) solution was added, and the monolayer was incubated at 37°C for 2 min. The culture dishes were observed under a phase-contrast microscope until the cells had detached. Cells were removed with 10 mL of DMEM and centrifuged at 900 g for 7 min. The pellet was re-suspended in DMEM in a culture dish, and cells () from passages 4–8 were used in all experiments.
Figure 1. Morphological photograph of primary cultured rat aortic vascular smooth muscle cells. The black bar represents 100 μm.
Adhesion assay
The efficiency of cell attachment was determined by measuring the number of cells that adhered to the substrate (immobilized fibronectin), as described previously with a few modifications (CitationKintscher et al., 2000). The adhesive substrate, fibronectin, was diluted in sterile water. Aliquots of 50 μL of fibronectin (50 μg/mL) were added to 96-well plates, which were placed overnight at room temperature. Wells were blocked with 0.5% BSA for 1 h. Cells were starved for 24 h then detached by trypsinization, washed once in DMEM without FBS, and centrifuged for 7 min at 900 g. Cells were re-suspended in medium without FBS and phenol red, and were then incubated with triflavin for 20 min followed by gentle mixing. In total, 105 VSMCs were placed in each well and allowed to adhere at 37°C for 60 min. Non-adherent cells were rinsed off with PBS, and the remaining cells were fixed with 4% paraformaldehyde for 5 min, then stained with 0.5% toluidine blue in 4% paraformaldehyde for 5 min, and rinsed with PBS. Cells were solubilized by the addition of 100 μL of 2% sodium dodecylsulfate (SDS), and quantified in a microtiter plate reader at 550 nm. Experiments described were performed in triplicate and repeated three times.
Flow cytometric analysis
VSMCs were prepared as described above. Aliquots of a VSMC suspension (106 cells/mL) were preincubated with triflavin (10 μg/mL) or EDTA (10 mM) for 5 min, followed by the addition of FITC (fluorescein isothiocyanate)–triflavin (5 μg/mL). The suspensions were then incubated for another 5 min, and the volume was adjusted to 1 mL/tube with a PBS solution. The suspensions were then assayed for fluorescein-labeled VSMCs using a flow cytometer (Beckman Coulter® EPICS® XL™). Data were collected from 10,000 VSMCs per experimental group. All experiments were repeated at least three times to ensure reproducibility.
Confocal microscopic analysis
Aliquots of 100 μL of poly-l-lysine (0.01%) and fibronectin (50 μg/mL) were added to a chamber slide, which was placed overnight at room temperature. VSMCs were prepared as described above. Aliquots of VSMCs were preincubated with triflavin (10 μg/mL) followed by gentle mixing for 20 min. In total, 105 VSMCs were placed on each chamber slide and allowed to adhere at 37°C for 10 min. Non-adherent cells were rinsed off with PBS, and the remaining cells were fixed with 4% paraformaldehyde for 30 min and permeabilized with 80% methanol for 15 min. After incubation with 3% skimmed milk in PBS for 60 min, the preparation was incubated for 1 h with an anti-PKC-α mAb (1:80). Cells were then washed three times with PBS and exposed to the secondary antibody (FITC-conjugated anti-mouse immunoglobulin G) (IgG) at 1:100, 1% bovine serum albumin (BSA)/PBS) for 60 min. The preparation was mounted with mounting buffer (Vector Laboratories, Burlingame, CA) under a glass coverslip on a Leica TCS SP5 confocal spectral microscope imaging system with use of an argon/ krypton laser.
Statistical analysis
Experimental results are expressed as the mean ± SEM and are accompanied by the number of observations. Data were assessed by the method of analysis of variance (ANOVA). If this analysis indicated significant differences among the group means, then each group was compared using the Newman–Keuls method. A p-value of less than 0.05 was considered statistically significant.
Results
Effect of triflavin on VSMC adhesion to immobilized fibronectin
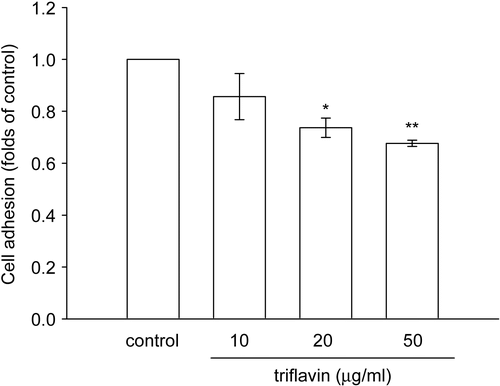
To examine whether triflavin affects VSMC adhesion to the ECM, a cell adhesion assay was performed using VSMCs. As shown in and , triflavin (10, 20, and 50 μg/mL) concentration-dependently inhibited VSMC adhesion to immobilized fibronectin (50 μg/mL) by approximately 14.3, 26.3, and 32.4% compared to the control group (treated with PBS), respectively.
Figure 2. Microscopic analysis of the inhibitory effect of triflavin in vascular smooth muscle cell (VSMC) adhesion to immobilized fibronectin. Cells (105 cell/well) were pretreated with (A) PBS or triflavin (B, 10; C, 20; D, 50 μg/mL) and incubated for 1 h to allow adhesion to fibronectin (50 μg/mL)-precoated wells. Non-adherent cells were rinsed off by PBS, and the remaining cells were fixed and photographed under a phase-contrast microscope. The black bar represents 50 μm.
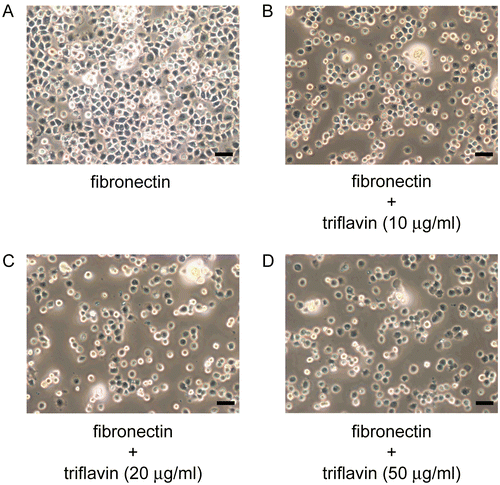
Figure 3. Effects of triflavin on vascular smooth muscle cell (VSMC) adhesion to immobilized fibronectin. Cells (105 cell/well) were pretreated with triflavin (10, 20, and 50 μg/mL) and incubated for 1 h to allow adhesion to fibronectin-precoated wells. The number of adhering cells was evaluated as described in “Materials and methods”. Data are presented as the mean ± SEM (n = 3). *p < 0.01 and **p < 0.001, compared to the control group (PBS treatment).
Effect of FITC–triflavin binding to VSMCs
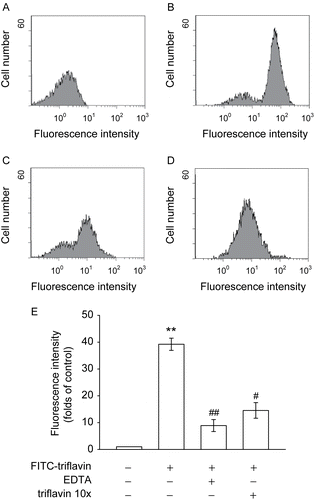
The RGD sequence is the cell-recognizing domain in a variety of adhesive proteins including fibronectin, fib rinogen, vitronectin, and collagen type I (CitationSheu & Huang, 1996). shows that FITC–triflavin (5 μg/mL) bound markedly to VSMCs compared with the control group (FITC only) (). EDTA (10 mM) () or cold triflavin (50 μg/mL) () obviously abolished FITC–triflavin binding to VSMCs by approximately 62.9 and 77.3%, respectively, indicating that this binding is divalent cation-dependent and triflavin-specific.
Figure 4. Flow cytometric analysis of FITC–triflavin binding to vascular smooth muscle cells (VSMCs). Cells were treated with (A) FITC only or (B) FITC–triflavin (5 μg/mL); or cells were pretreated with (C) EDTA (10 mM) or (D) cold triflavin (50 μg/mL), followed by the addition of FITC–triflavin (5 μg/mL) as described in “Materials and methods”. Profiles are representative examples of three similar experiments. Data (E) are presented as folds of the control group (FITC only) and expressed as the mean ± SEM (n = 3). **p < 0.001, compared to the control group (FITC only); #p < 0.01 and ##p < 0.001, compared to the FITC–triflavin group.
Effect of triflavin on PKC-α translocation in VSMCs stimulated by immobilized fibronectin
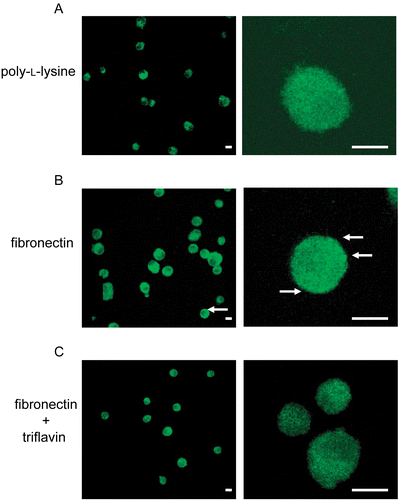
We further evaluated the inhibitory effects of triflavin on fibronectin-induced PKC-α translocation in VSMCs by a confocal microscopic study. shows that focal spots were only rarely present on immobilized poly-l-lysine (0.01%). On the other hand, immobilized fibronectin (50 μg/mL) markedly stimulated the basolateral distribution of PKC-α, which was observed by an increase in PKC-α immunoreactivity in distinct focal spots at the cell membrane (). Triflavin (10 μg/mL) significantly abolished these focal spots in VSMC membranes (). These findings strongly indicate that fibronectin can stimulate PKC-α translocation on VSMCs, and this translocation can be inhibited by triflavin.
Figure 5. Effects of triflavin on protein kinase C (PKC)-α translocation stimulated by fibronectin on vascular smooth muscle cells (VSMCs). Cells adhered to immobilized (A) poly-l-lysine (0.01%), (B) fibronectin (50 μg/mL), or (C) fibronectin (50 μg/mL) in the presence of triflavin (10 μg/mL). Confocal micrographs of the basolateral surface of cells are shown. Images are typical of those obtained in three separate experiments demonstrating the distribution of PKC-α (arrows) surrounding the cell membrane. Original magnifications ×100 (left) and ×630 (right); the bar indicates 10 μm.
Discussion
VSMC migration is an important and well-recognized process in the development of intimal hyperplasia and atherosclerosis, and many extracellular influences such as growth factors, the ECM, and interaction with cell surface receptors (particularly integrins) are involved. These processes result in VSMC focal adhesion, actin filament polymerization, proteolysis of the ECM, and directed migration toward increasing concentrations of chemoattractant stimuli. Furthermore, focal adhesion of VSMCs to specific ligands in the ECM environment via transmembrane integrin receptors is the initial event of this process. According to our previous study (CitationSheu et al., 2001), triflavin markedly inhibits neointimal formation in a rat model of balloon angioplasty. In the present study, we further demonstrate that triflavin directly binds to fibronectin-related receptors expressed on VSMCs, and then fibronectin-induced cell adhesion and PKC-α translocation are suppressed. These results provide new insights into the mechanisms of neointimal hyperplasia and have significant implications for disintegrin therapy for treating restenosis and atherosclerosis (CitationSheu et al., 2001).
Triflavin is known to be a nonspecific inhibitor of β3 integrins, such as αIIbβ3 and αVβ3 (CitationSheu et al., 1992). αIIbβ3 is the platelet receptor for fibrinogen, among others, and as such, is responsible for platelet aggregation and thrombus formation. αVβ3 integrin is heavily expressed by numerous cells, including endothelial cells, monocytes, and VSMCs, and is thought to play a major role in platelet-derived growth factor (PDGF)-stimulated migration of VSMCs (CitationChoi et al., 1994). Abciximab, an anti-αIIbβ3 and -αVβ3 integrin mAb, is also reported to inhibit VSMC adhesion and migration (CitationBaron et al., 2000). In the present study, we report that triflavin (10, 20, and 50 μg/mL) concentration-dependently inhibited VSMC adhesion to immobilized fibronectin, but not completely, even at the highest concentration (100 μg/mL) (data not shown), indicating that except for β3 integrin, other integrins such as β1 or several β subunits (CitationVuori & Ruoslahti, 1993; CitationWillis et al., 2004) may also be involved in fibronectin-mediated cell adhesion.
In this study, fibronectin triggered PKC-α translocation from the cytosol to cell membranes, which was significantly abolished in the presence of triflavin (). The intracellular enzyme, PKC, is a major serine–threonine kinase that has been shown to play a crucial role in growth factor- and cytokine-initiated signal transduction. PKC regulates several intracellular events, including the secretion of various proteins and cellular proliferation. It has recently become evident that PKC is not just a specific protein kinase, but a family of at least 11 kinases that are divided into three groups according to their calcium dependency and response to DAG or phorbol esters (CitationSalamanca & Khalil, 2005). The conventional PKC isotypes, including α, βI, βII, and γ, are activated by phorbol esters or DAG in a Ca2+-dependent manner, mainly localized in the cytosolic fraction of unstimulated cells, and undergo translocation to cell membranes in activated cells (CitationSalamanca & Khalil, 2005). The novel isotypes, which include δ, «, η, and θ, lack Ca2+-binding domains and are Ca2+ insensitive, but are still activated by DAG or phorbol esters. The atypical isotypes, which include λ, ι, and ζ, lack both Ca2+- and phorbol ester-binding domains. These multiple isotypes of PKC have been shown to have varying tissue distributions, biochemical regulatory abilities, and physiological functions that are cell specific. Of these isotypes, the α isotype appears to be most commonly expressed in VSMCs (CitationOkazaki et al., 2000). CitationHaller et al. (1998) reported that integrins induce intracellular signaling in VSMCs via DAG and PKC-α translocation. Integrin binding induced PKC-α translocation to focal adhesions. Their findings suggest that integrin occupation induces translocation of PKC-α, modulates the affinity of fibronectin-binding integrins, and plays a role in integrin inside-out signaling. The close association with focal adhesion kinases (FAKs) suggests that PKC-α may phosphorylate these kinases or other substrates within the focal adhesion complex. PKC-α seems to play an important role in inside-out integrin signaling and cell spreading.
In conclusion, triflavin, an αIIβ3 disintegrin, exhibited more-potent activity at inhibiting immobilized fibronectin-induced cell adhesion and PKC-α translocation in VSMCs. Nowadays, the RGD-containing peptides such as eptifibitide and tirofiban have been employed in clinic for the treatment of patients undergoing percutaneous coronary intervention and patients with acute coronary syndrome (CitationTricoci et al., 2007). This present study provides new insights into potential disintegrin therapy for the treatment of restenosis and atherosclerosis.
Acknowledgements
This work was supported by a grant from the National Science Council of Taiwan (NSC 94-2321-B-038-001) and one from the Taipei Medical University–Mackay Memorial Hospital (94 MMH-TMU-05). The first two authors (C.-J.W. and C.-Y.H) contributed equally to this work. The authors alone are responsible for the content and writing of the paper.
References
- Baron JH, Moiseeva EP, de Bono DP, Abrams KR, Gershlick AH (2000): Inhibition of vascular smooth muscle cell adhesion and migration by c7E3 Fab (abciximab): A possible mechanism for influencing restenosis. Cardiovasc Res 48: 464–472.
- Batchelor WB, Robinson R, Strauss BH (1998): The extracellular matrix in balloon arterial injury: A novel target for restenosis prevention. Prog Cardiovasc Dis 41: 35–49.
- Choi ET, Engel L, Callow AD, Sun S, Trachtenberg J, Santoro S, Ryan US (1994): Inhibition of neointimal hyperplasia by blocking alpha V beta 3 integrin with a small peptide antagonist GpenGRGDSPCA. J Vasc Surg 19: 125–134.
- Clark EA, Brugge JS (1995): Integrins and signal transduction pathways: the road taken. Science 268: 233–239.
- Gould RJ, Polokoff MA, Friedman PA, Huang TF, Holt JC, Cood JJ (1990): Disintegrins: a family of integrin inhibitory proteins from viper venoms. Proc Soc Exp Biol Med 195: 168–171.
- Haller H, Lindschau C, Maasch C, Olthoff H, Kurscheid D, Luft FC (1998): Integrin-induced protein kinase C alpha and C epsilon translocation to focal adhesions mediates vascular smooth muscle cell spreading. Circ Res 82: 157–165.
- Huang TF, Sheu JR, Teng CM (1991a): A potent antiplatelet peptide, triflavin, from Trimeresurus flavoviridis snake venom. Biochem J 277: 351–357.
- Huang TF, Sheu JR, Teng CM, Chen SW, Liu CS (1991b): Triflavin, an antiplatelet Arg-Gly-Asp-containing peptide, is a specific antagonist of platelet membrane glycoprotein IIb/IIIa complex. J Biochem 109: 328–334.
- Hynes RO (1992): Integrins: versatility, modulation, and signaling in cell adhesion. Cell 69: 11–25.
- Kintscher U, Kappert K, Schmidt G, Doerr G, Grill M, Wollert-Wulf B, Graefe M, Fleck E, Graf K (2000): Effects of abciximab and tirofiban on vitronectin receptors in human endothelial and smooth muscle cells. Eur J Pharmacol 390: 75–87.
- Krook A, Rapoport MJ, Anderson S, Pross H, Zhou YC, Denhardt DT, Delovitch TL, Haliotis T (1993): p21ras and protein kinase C function in distinct and interdependent signaling pathways in C3H 10T1/2 fibroblasts. Mol Cell Biol 13: 1471–1479.
- Libby P, Sukhova G, Lee RT, Liao JK (1997): Molecular biology of atherosclerosis. Int J Cardiol. 62(Suppl 2): S23–S29.
- Lusis AJ (2000): Atherosclerosis. Nature 407: 233–241.
- McNamee HP, Ingber DE, Schwartz MA (1993): Adhesion to fibronectin stimulates inositol lipid synthesis and enhances PDGF-induced inositol lipid breakdown. J Cell Biol 121: 673–678.
- Moiseeva EP (2001): Adhesion receptors of vascular smooth muscle cells and their functions. Cardiovasc Res 52: 372–386.
- Okazaki J, Mawatari K, Liu B, Kent KC (2000): The effect of protein kinase C and its alpha subtype on human vascular smooth muscle cell proliferation, migration and fibronectin production. Surgery 128: 192–197.
- Pauly RR, Bilato C, Cheng L, Monticone R, Crow MT (1997): Vascular smooth muscle cell cultures. Meth Cell Biol 52: 133–154.
- Pytela R, Pierschbacher MD, Ginsberg MH, Plow EF, Ruoslahti E (1986): Platelet membrane glycoprotein IIb/IIIa: member of a family of Arg-Gly-Asp specific adhesion receptors. Science 231: 1559–1562.
- Richardson A, Parsons T (1996): A mechanism for regulation of the adhesion associated protein tyrosine kinase pp125FAK. Nature 380: 538–540.
- Salamanca DA, Khalil RA (2005): Protein kinase C isoforms as specific targets for modulation of vascular smooth muscle function in hypertension. Biochem Pharmacol 70: 1537–1547.
- Schwartz SM (1997): Perspectives series: Cell adhesion in vascular biology. Smooth muscle migration in atherosclerosis and restenosis. J Clin Invest 99: 2814–2816.
- Sheu JR, Chao SH, Yen MH, Huang TF (1994): In vivo antithrombotic effect of triflavin, an Arg-Gly-Asp-containing peptide on platelet plug formation in mesenteric microvessels of mice. Thromb Haemost 72: 617–621.
- Sheu JR, Huang TF (1996): Triflavin, an Arg-Gly-Asp-containing peptide, inhibits B16-F10 mouse melanoma cell adhesion to matrix proteins via direct binding to tumor cells. J Biomed Sci 3: 359–364.
- Sheu JR, Hung WC, Wu CH, Ma MC, Kan YC, Lin CH, Lin MS, Luk HN, Yen MH (1999): Reduction in lipopolysaccharide-induced thrombocytopenia by triflavin in a rat model of septicemia. Circulation 99: 3056–3062.
- Sheu JR, Teng CM, Huang TF (1992): Triflavin, an RGD-containing antiplatelet peptide, binds to GpIIIa of ADP-stimulated platelets. Biochem Biophys Res Commun 189: 1236–1242.
- Sheu JR, Wu CH, Chen YC, Hsiao G, Lin CH (2001): Mechanisms in the inhibition of neointimal hyperplasia with triflavin in a rat model of balloon angioplasty. J Lab Clin Med 137: 270–278.
- Sheu JR, Yen MH, Kan YC, Hung WC, Chang PT, Luk HN (1997a): Inhibition of angiogenesis in vitro and in vivo: comparison of the relative activities of triflavin, an Arg-Gly-Asp-containing peptide and anti-αVβ3 integrin monoclonal antibody. Biochim Biophys Acta 1336: 445–454.
- Sheu JR, Yen MH, Hung WC, Lee YM, Su CH, Huang TF (1997b): Triflavin inhibits platelet-induced vasocontraction in deendothelialized aorta. Arterioscle Thromb Vacs Biol 17: 3461–3468.
- Smith JW, Ruggeri ZM, Kunicki TJ, Cheresh DA (1990): Interactions of integrins αVβ3 and glycoprotein IIb/IIIa with fibrinogen. Differential peptide recognition accounts for distinct binding sites. J Biol Chem 265: 12267–12271.
- Tricoci P, Peterson ED, Chen AY, Newby LK, Harrington RA, Greenbaum AB, Cannon CP, Gibson CM, Hoekstra JW, Pollack CV Jr, Ohman EM, Gibler WB, Roe MT (2007): Timing of glycoprotein IIb/IIIa inhibitor use and outcomes among patients with non-ST-segment elevation myocardial infarction undergoing percutaneous coronary intervention (results from CRUSADE). Am J Cardiol 99: 1389–1393.
- Vuori K, Ruoslahti E (1993): Activation of protein kinase C precedes alpha 5 beta 1 integrin-mediated cell spreading on fibronectin. J Biol Chem 268: 21459–21462.
- Willis AI, Pierre-Paul D, Sumpio BE, Gahtan V (2004): Vascular smooth muscle cell migration: current research and clinical implications. Vasc Endovasc Surg 38: 11–23.