
Abstract
Context: Acetaminophen overdose is regarded to a common cause of acute liver failure. The hepatotoxicity leads to mitochondrial oxidative stress and subsequent necrotic hepatocellular death.
Objective: This study examines the protective effect of metformin on acetaminophen-induced oxidative stress, inflammation and subsequent hepatotoxicity in mice.
Materials and methods: Male BALB/c mice were orally administered to acetaminophen (250 mg/kg/d) for a 7-day period. The mice received metformin (100 and 200 mg/kg/d, p.o.) for 21 days. To evaluate acetaminophen-induced oxidative stress, liver tissue level of malodialdehyde (MDA), end product of membrane lipid peroxidation, and activities of superoxide dismutase (SOD) and glutathione (GSH) were measured. Histological analysis and measurement of serum alanine aminotransferase (ALT), aspartate aminotransferase (AST) and alkaline phosphatase (ALP) were performed. Moreover, tissue concentrations of proinflammatory cytokines interleukin-6 (IL-6) and tumor necrosis factor-alpha (TNF-α), along with, C-reactive protein (CRP) were assessed.
Results: Acetaminophen caused focal hepatocyte necrosis, inflammation and fatty degeneration, as well as increased tissue levels of AST, ALT, ALP and MDA, and also decreased GSH and SOD activities. Moreover, IL-6, TNF-α and CRP levels were increased following acetaminophen hepatotoxicity. Metformin (200 mg/kg/d) significantly normalized MDA, SOD and GSH levels (p < 0.001), and exerted a hepatoprotective effect by significant decreasing ALT, AST and ALP concentrations (p < 0.001). The tissue levels of IL-6, TNF-α and CRP were markedly decreased by 21-day treatment with metformin (200 mg/kg/d) (p < 0.001).
Discussion: The results suggest metformin protects hepatocytes against acute acetaminophen toxicity. Metformin is indicated to diminish oxidative stress, proinflammatory cytokines, and hepatocyte necrosis.
Introduction
Acetaminophen (N-acetyl-p-aminophenol, APAP) is characterized as a most common used secure and beneficial painkiller and antipyretic. Despite of its beneficial effects on inflammation, it has been reported that acute overdose and chronic use of APAP lead to APAP poisoning with serious complications, including necrosis of the centrilobular cells of the liver (Bessems & Vermeulen Citation2001; James et al. Citation2003; Reid et al. Citation2005). In this regard, in 2009, the American Association of Poison Control Center offered about 56,000 emergency room visits, 26,000 hospitalizations, and over 400 deaths caused by misuse of APAP and/or drugs containing the compound (Larson et al. Citation2005; Nourjah et al. Citation2006; Bronstein et al. Citation2010; McGill et al. Citation2012).
The adverse reactions of APAP are associated with the reactive metabolite derived from hepatic cytochrome P-450 metabolism. At initial stage of metabolism, APAP is oxidized to a reactive substance, N-acetyl-p-benzoquinone imine (NAPQI), which is then conjugated and detoxified with glutathione (GSH) to produce nontoxic metabolites (Gelotte et al. Citation2007; Anon Citation2010). At noxious amount of APAP, the excessive generation of NAPQI eventually results in discharge of glutathione. In rodents, the higher doses of APAP can saturate the sulfation and glucoronidation pathways, leading to excessive formation of NAPQI (McGill et al. Citation2012). Glutathione depletion is only a cascade of intracellular events including mitochondrial oxidative stress, production of reactive oxygen and nitrogen species, activation of stress proteins and gene transcription mediators, and mobilization of the liver’s essential immune system. Ultimately, imbalance of the innumerable pathways can lead to hepatic cell loss (Kaplowitz et al. Citation2008). This condition is due to the increment of liver enzymes levels after 12 –36 h of the drug overdose (Dart et al. Citation2006). Following the alterations in cellular and biochemical indices, maximal clinical liver damages, such as jaundice, encephalopathy, coagulopathy and subsequent hepatic failure are observed (Rumack & Matthew Citation1975). To describe the mechanisms underlying these deficits, the important role of pro-inflammatory cytokines (i.e., IL-6 and TNF-α) are confirmed to be involved in APAP-induced hepatotoxicity and liver cell apoptosis (Dambach et al. Citation2006).
The previous studies have suggested that NAPQI binding to mitochondrial proteins changes the morphology and function of liver mitochondria (Das et al. Citation2010; Bajt et al. Citation2011), resulting in mitochondrial oxidative stress (McGill et al. Citation2012). The event is consistent with the opening of mitochondrial membrane permeability transition pore (MPTP) (El-Hassan et al. Citation2003; Zambon et al. Citation2006; Kon et al. Citation2004), matrix swelling, and outer membrane lysis (Das et al. Citation2010; McGill et al. Citation2012). The permeability changes is contributed to the continuous events including inner mitochondrial membrane depolarization, uncoupling of oxidative phosphorylation, release of intramitochondrial ions, and decreased ATP synthesis. The cell death occurred following the superoxide release from mitochondria (Lemasters Citation1999; Zorov et al. Citation2000; Reid et al. Citation2005). Moreover, apoptosis-inducing factor (AIF), endonuclease G (EndoG) and proapoptotic proteins, including cytochrome c and Smac/DIABLO, are released from mitochondria, followed by translocation of endonucleases to nuclei and subsequent nuclear DNA fragmentation (Lin et al. Citation2000). Finally, centrilobular hepatocyte necrosis is the endpoint of APAP toxicity, which leads to liver failure (Zhou et al. Citation2001; Kaplowitz et al. Citation2008; McGill et al. Citation2012).
Metformin, a widely used dimethylbiguanide anti-hyperglycemic agent, is shown to exert antioxidant effects (Davidson & Peters Citation1997; Poon et al. Citation2003; Tavafi Citation2013a) along with an increase in the reduced liver and blood levels of glutathione (GSH) in diabetes (Ewis & Abdel-Rahman Citation1997; Poon et al. Citation2003; Tavafi Citation2013b). Moreover, metformin reduces the activities of plasma xanthine oxidase and erythrocyte glutathione peroxidase, suggesting its protective effect against oxidative stress (Cosic et al. Citation2001; Poon et al. Citation2003). There are evidences that metformin protects the mitochondrial respiratory chain in oxidative stress conditions and cell death (Gobe et al. Citation2013; Rafieian-Kopaei et al. Citation2013; Spasovski Citation2013). According to the literature, it is hypothesized that metformin can exert protective effects against oxidative stress-induced injury through various mechanisms. For instance, treatment with metformin maintains the hepatic mitochondrial glutathione redox status, which is essential for cell survival (Meister & Anderson Citation1983; Poon et al. Citation2003).
According to high incidence of APAP poisoning and the resulted severe liver damages, finding hepatoprotective agents seems to be necessary. Therefore, the current study aimed to examine the hepatoprotective effect of metformin in APAP-induced toxicity in mice. The study attempts were to investigate the role of metformin in normalization of liver injury-related enzymes, oxidant/antioxidant enzymes balance and proinflammatory cytokines for protecting against the liver damage induced by acute APAP exposure.
Materials and methods
Drugs
The following compounds were administered in the present study: APAP (Acetaminophen; Tocris Bioscience, Ellisville, MO), metformin (Tocris Bioscience, Ellisville, MO), N-acetylcysteine (Sigma Aldrich, St. Louis, MO).
The drugs were freshly dissolved in normal saline 0.9% and were daily administered through oral gavage in a constant volume of 10 ml/kg body weight during a 7-day (for APAP) and 21-day (for metformin) period. Also, NAC (dissolved in normal saline 0.9%) was intraperitoenally injected 1 day after a 7-day period of exposure to APAP (constant volume of 10 ml/kg body weight).
Animals
Ninety male BALB/C mice (4–5 weeks old) weighing 22–25 g were obtained from the Animal house of the hepatitis research center, Lorestan University of Medical Sciences, and were placed in cages at room temperature (20 ± 2 °C), relative humidity of approximately 50% and a 12 h dark/light cycle with access to water and standard food ad libitum. All experiments were performed in accordance with the institutional animal care and use committee (Lorestan University of Medical Sciences) guidelines. All protocols in the investigation were approved by the Ethics Committee of Lorestan University of Medical Sciences.
Experimental design
The animals were randomly divided into six main groups of 15 mice each. All groups (except for control group) were treated with APAP at a dose of 250 mg/kg body weight to induce liver damage through oral gavage for a 7-day period (Henderson et al. Citation2000). Then, the mice were subjected to oral treatment of different doses of metformin once daily for 21 days, and single oral administration of N-acetylcysteine (NAC) as follows:
Group 1: Control animals, receiving normal saline solution 0.9%;
Group 2: APAP-exposed animals, receiving normal saline solution 0.9%;
Group 3: APAP-exposed animals, receiving metformin at dose of 50 mg/kg/d, p.o. (Choi et al. Citation2006);
Group 4: APAP-exposed animals, receiving metformin at dose of 100 mg/kg/d, p.o. (Choi et al. Citation2006);
Group 5: APAP-exposed animals, receiving metformin at dose of 200 mg/kg/d, p.o. (Choi et al. Citation2006);
Groups 6–8: Animals receiving metformin at doses of 50, 100 and 200 mg/kg/d, p.o., separately.
Group 9: APAP-exposed animals, receiving a single dose of N-acetylcysteine (100 mg/kg, p.o.) (Choi et al. Citation2013);
All doses and the time interval between drugs administration and the tests were selected based on pilot studies. At the end of the experimental period, all mice were anesthetized with an intraperitoneal injection of pentobarbital (70 mg/kg) and euthanized by cervical dislocation. Then, liver and serum samples were collected and stored for further assays.
Histological analysis
After blood collection, the animals were sacrificed by decapitation and their livers were removed. The harvested livers were excised, and small sections were fixed in 10% phosphate-buffered formalin for further histological analysis. The sections were stained with hematoxylin-eosin (H&E) before investigation under blindfold conditions with standard light microscopy by a pathologist (magnification ×400).
Evaluation of liver injury markers
The blood samples were collected and centrifuged at 14,000–20,000g for 15–20 min. The obtained sera were analyzed for measurement of activities of alanine aminotransferase (ALT), aspartate aminotransferase (AST) and alkaline phosphatase (ALP), which are detected as a result of lysis or leakage from hepatocytes. The assays were performed by a colorimetric method using a biochemical analyzer (Hitachi 902 Automatic Analyzer; Hitachi, Japan), according to previously described methods (Enomoto et al. Citation2001; McGill et al. Citation2012).
Biochemical examination
To investigate the oxidative stress injuries, the biochemical assays were performed. For this purpose, the excised liver samples were washed with ice-cold phosphate-buffered saline (PBS; 137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 2 mM KH2PO4, pH 7.4) and weighed. The liver samples were filtered using a 0.2 μm cell strainer (SPL Life Sciences, Gyeonggi-do, Korea) and syringe rubber plunger in cold PBS to obtain tissue homogenates. The homogenates were used to determine the tissue levels of malondialdehyde (MDA) and the activities of superoxide dismutase (SOD) and reduced form of glutathione (GSH).
Tissue malondialdehyde level assessment
Tissue concentrations of free malondialdehyde, an end-product and marker of lipid peroxidation, were measured as described by Ohkawa et al. (Citation1979). Each liver homogenate (200 μL) was diluted with 800 μL of PBS and mixed with 25 μL of 8.8 mg/mL butylhydroxytoluene and 500 μL of 50% trichloroacetic acid. The mixture was vortexed, incubated for 2 h on ice, and centrifuged at 2000g for 15 min (Pharmacia, Schio, Italy). The supernatant (1 mL) was transferred into a tube and mixed with 75 μL of 0.1 M EDTA and 250 μL of 0.05 M 2-thiobarbituric acid. The latter was boiled for 15 min and allowed to cool at room temperature. Finally, the samples were spectrophotometrically analyzed and the absorbance was measured twice at 532 and 600 nm (Yazdani et al. Citation2016).
Superoxide dismutase activity assessment
The activity of tissue superoxide dismutase, a most important antioxidative enzyme, was measured based on the Paoletti and Mocali (Citation1990) method. A master solution was prepared by adding 0.1 M PBS, 0.15 mg/mL sodium cyanide in 0.1 M ethylenediamine tetraacetic acid (EDTA), 1.5 mM nitroblue tetrazolium and 0.12 mM riboflavin. In brief, 100 μL of a serial dilution of liver homogenates were pipetted into a 96-well plate and mixed with 200 μL of the master solution. Ultimately, the absorbance was measured at 560 nm using spectrophotometer (ELISA reader) and the activity was expressed as SOD unit/mg protein (Yazdani et al. Citation2016).
Glutathione level assessment
The glutathione level was determined using a Glutathione Assay Kit (Sigma-Aldrich, MI, USA). Briefly, 10 mL of liver homogenate and 150 μL of working solution (1.5 mg/mL DTNB, 6 U/mL glutathione reductase, and 1 × assay buffer) were added to a 96 well-plate and mixed thoroughly. The plate was incubated for 5 min before the 50 mL of NADPH solution (0.16 mg/mL) was added to each well. The absorbance was spectrophotometrically measured at 412 nm (Paglia & Valentine Citation1967).
Assessment of proinflammatory cytokines and factors
After collection of blood samples, they were centrifuged at 3500g for 15 min to achieve the sera. Then, the concentrations of C-reactive protein (CRP), interleukin 6 (IL-6) and tumor necrosis factor-α (TNF-α) were measured using CRP, IL-6 and TNF-α ELISA kits (Abcam, Cambridge, MA) using the Liu et al. (Citation2001) method (Saeedi Saravi et al. Citation2016).
Statistical analysis
All data were expressed as mean ± SD. Statistical analysis was performed using SPSS 16.0 statistical software (Chicago, IL). The differences between 3 or more groups, the experimental groups, were analyzed by one-way analysis of variance (ANOVA), followed by Tukey's post test. Evaluation of the effects of two variables (APAP-induced hepatotoxicity vs. control and type of treatment) was performed using two-way ANOVA followed by the Bonferroni post test. p-Value <0.05 was considered statistically significant.
Results
Histological findings
Presence of APAP-induced hepatotoxicity was confirmed by H&E histological analysis. The H&E staining of liver tissues sampled from APAP-induced hepatotoxic mice have demonstrated focal hepatocyte necrosis as well as increased infiltration of the inflammatory cells into the portal tract. Also, fatty degeneration and central vein dilation were observed in the histological examination (, ). Otherwise, metformin at dose of 200 mg/kg as well as NAC at dose of 100 mg/kgmarkedly reversed the hepatocellular damages induced by APAP ().
Figure 1. A and B: H&E staining of liver tissues isolated from CCl4-exposed rats demonstrates focal hepatocyte necrosis, as well as, increased infiltration of the inflammatory cells into the portal tract in the APAP-induced hepatotoxic mice; Fatty degeneration and central vein dilation were obvious (magnification ×400). C and D: No hepatocellular necrosis and APAP-induced damage was observed in metformin (200 mg/kg)- and NAC (100 mg/kg)-treated mice (magnification ×200).
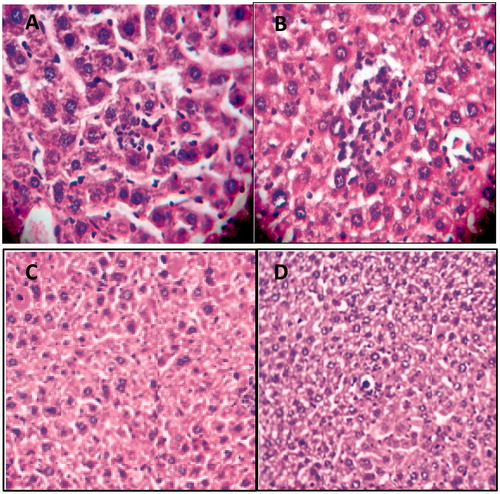
Table 1. Histopathological evaluation of liver tissue obtained from control, APAP-induced hepatotoxic treated with either normal saline or metformin 200 mg/kg.
Measurement of liver injury factors
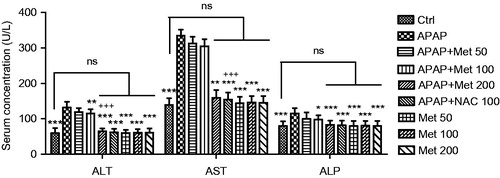
The measurement of liver injury factors such as ALT, AST and ALP enzymes levels in the studied groups were performed and the results are presented in . The examination showed a significant increase in the tissue levels of the three enzymes in control animals compared to the animals received subacute APAP with no treatment (p < 0.001). This resulted from hepatocyte lysis or leakage. The findings have demonstrated the significant hepatoprotective activity of metformin (100 and 200 mg/kg) in the animals received APAP through reduction in the levels of ALT, AST and ALP enzymes (p < 0.01 and p < 0.001, respectively), while 50 mg/kg of metformin was observed to non-significantly decrease the enhanced levels of these enzymes (p > 0.05). The results demonstrated no significant differences in the examined ALT, AST and ALP levels between APAP/metformin 200 mg/kg and APAP/NAC 100 mg/kg (p > 0.05)
Figure 2. The liver tissue levels of injury biomarkers (ALT, AST and ALP) in the control and subacute APAP-treated BALB/c mice receiving normal saline,metformin (50, 100 and 200 mg/kg) and NAC (100 mg/kg). The values are expressed as mean ± SD and analyzed using one-way ANOVA method followed by Tukey's post test. *p < 0.05, **p < 0.01, ***p < 0.001 compared to APAP/normal saline group; +++p < 0.001 compared to APAP/metformin 100 mg/kg group. ns: non-significant.
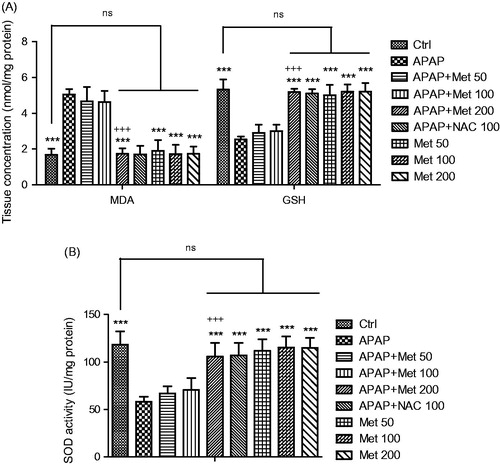
Tissue antioxidant enzymes levels
As shown in , analysis of the concentrations of liver tissue MDA and activities of SOD and GSH in all experimental groups of animals demonstrated no significant differences in the biochemical parameters between the control group and the animals treated with metformin at a dose of 200 mg/kg (p > 0.05). It is observed that there was a significant difference in the three antioxidant enzymes levels and activities between the APAP-induced hepatotoxic and control groups (p < 0.001). Moreover, metformin at a dose of 200 mg/kg, unlike the lower doses, significantly decreased the liver tissue MDA levels in the mice treated with APAP for 7 days (p < 0.001). It is demonstrated that treatment with metformin exerts its hepatoprotective effects reducing APAP-induced lipid peroxidation damages of liver tissue (p < 0.001). The hepatoprotective role of metformin is confirmed by the observation that the activity of SOD and GSH antioxidant enzymes in the mice receiving subacute APAP significantly increased following treatment with metformin (p < 0.001). On the other hand, treatment of APAP-exposed mice with NAC demonstrated no significant differences in MDA and GSH levels and SOD activity compared to APAP/metformin 200 mg/kg mice (p > 0.05)
Figure 3. The liver tissue levels of MDA and antioxidants SOD and GSH activity levels in the control and subacute APAP-treated BALB/c mice receiving normal saline, metformin (50, 100 and 200 mg/kg) and NAC (100 mg/kg). The values are expressed as mean ± SD and analyzed using one-way ANOVA method followed by Tukey's post test. ***p < 0.001 compared to APAP/normal saline group; +++p < 0.05 compared to APAP/metformin 100 mg/kg group. ns: non-significant.
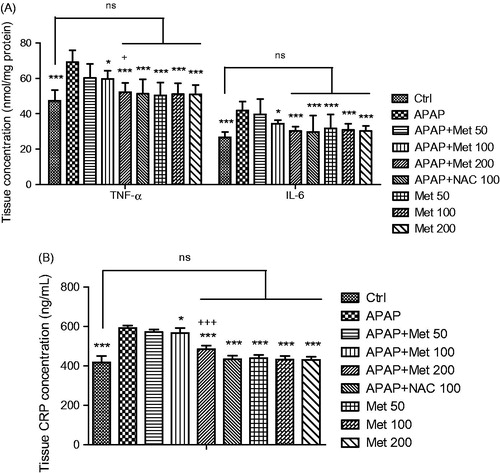
Tissue levels of pro-inflammatory factors
In addition to the antioxidant enzymes, the concentrations of pro-inflammatory factors, including interleukin-6, tumor necrosis factor-α and C-reactive protein were measured. demonstrates that the serum concentrations of CRP, IL-6 and TNF-α were enhanced following 7-days administration of APAP compared to control animals (p < 0.001). According to the results, metformin at doses of 100 and 200 mg/kg, despite 50 mg/kg, significantly decreased the factor levels in the mice that received subacute APAP (p < 0.05 and p < 0.001, respectively). Furthermore, 200 mg/kg of metformin exerted a significant hepatoprotective effect compared to the lower dose (100 mg/kg; p < 0.001 for CRP and p < 0.05 for TNF-α), while metformin at a dose of 200 mg/kg demonstrated no significant differences in the tissue levels of the pro-inflammatory cytokines IL-6 and TNF-α in comparison with the control group and NAC-treated mice which were exposed to APAP (p > 0.05).
Figure 4. The serum levels of pro-inflammatory factors (IL-6, TNF-α and CRP) in the control and subacute APAP-treated BALB/c mice receiving normal saline, metformin (50, 100 and 200 mg/kg) and NAC (100 mg/kg). The values are expressed as mean ± SD and analyzed using one-way ANOVA method followed by Tukey's post test. *p < 0.05, ***p < 0.001 compared to APAP/normal saline group; +p < 0.05 compared to APAP/metformin 100 mg/kg group; +++p < 0.001. ns: non-significant.
Discussion
The objective of the present study was to examine the hypothesis that metformin exerts a protective effect against APAP-induced hepatotoxicity. It has been demonstrated that metformin at a dose of 200 mg/kg significantly reverses the oxidant/antioxidant imbalance induced by APAP toxicity. In this regard, we have shown that metformin protects hepatocytes against lipid peroxidation via significant decrement of MDA levels. Therefore, metformin can diminish lipid peroxidation-induced disruption of the phospholipid bilayer membrane and cell integrity, and eventually inhibits the leakage of cytoplasmic components and the enzymes into the blood and the subsequent cell necrosis (Walker et al. Citation2000; Hadi et al. Citation2012). On the other hand, metformin increased the liver tissue levels of SOD and GSH, which were diminished in the animals exposed to APAP during a 7-day period. The reports have shown that APAP hepatotoxicity impairs the mitochondrial antioxidant defenses (SOD and GPx enzymes) (Ramachandran et al. Citation2011). It is noteworthy that, the hepatoprotective effects of metformin was consistent with NAC, as a standard drug to protect hepatocytes against cellular necrosis in APAP-induced hepatotoxicity. Therefore, the importance of finding a protective agent against mitochondrial dysfunction in APAP-overdose patients markedly returns to the close correlation between the release of biomarkers of mitochondrial damage and hepatocellular necrosis (McGill et al. Citation2012).
A body of evidence has reported a significant contribution between both intra- and extracellular ROS and liver injury (Bajt et al. Citation2011). This is considered as a main mechanism for centrilobular hepatic necrosis, leading to acute liver failure (Larson et al. Citation2005; Hanawa et al. Citation2008). As APAP, especially its reactive metabolite NAPQI, produces oxidative stress-induced hepatocyte injury, metformin is proposed to suppress the mitochondrial oxidative stress and ROS production. The effect of metformin probably exerts through inhibition of JNK activation along with stimulation of hypooxygenase (HO)-1 expression, resulting in hepatoprotection against oxidative stress-induced injuries (de La Rosa et al. Citation2005; Vishal Citation2008; Hadi et al. Citation2012; Kim et al. Citation2015). APAP-induced oxidative stress results in lipid peroxidation, oxidation of thiol-proteins, mitochondrial damage, altered calcium homeostasis, and DNA damage (Jaeschke Citation2003). Otherwise, it is believed that medical interventions can restore the scavenging capacity for ROS and peroxynitrite in mitochondria (Saito et al. Citation2010; Knight et al. Citation2002; James et al. Citation2003; Bajt et al. Citation2003; McGill et al. Citation2012). Furthermore, molecular docking analysis has declared an interaction between metformin and CAT via hydrogen bonds, leading to hepatoprotective effects against oxidative liver injury (Dai et al. Citation2014).
The results have demonstrated increased plasma levels of ALT, AST and ALP in the APAP-exposed mice, suggesting that mitochondrial damage may be related to cell necrosis. The findings are consistent with the report by McGill et al. (Citation2012). The enzymes were released from hepatocytes following the nuclear DNA damage and cell death. We observed that metformin at the same doses which normalize the oxidant/antioxidant imbalance, decreases the abnormal elevating plasma levels of ALT, AST and ALP in the APAP-induced hepatotoxic mice. This means that metformin probably decreases the nuclear DNA damage and subsequent cell death in the mouse liver after APAP overdose.
We found that a 7-day exposure to APAP causes an increased liver tissue levels of IL-6, TNF-α and CRP in the examined mice. Treatment with metformin (100 and 200 mg/kg) significantly normalized the increased levels of the proinflammatory cytokines and CRP, while no significant difference was observed compared to the control group. There is growing evidence that extensive formation of proinflammatory mediators, including proinflammatory cytokines IL-6 and TNF-α, are extensively formed and neutrophils accumulate into the liver after APAP overdose (Lawson et al. Citation2000; Das et al. Citation2010; McGill et al. Citation2012). Metformin decreases the sensitization of hepatocytes to TNF-α-induced cell death through activation of nuclear factor kappa B (NF-kB) (Schoemaker et al. Citation2002). This can protect against TNF-α-mediated inflammatory liver failure (Woudenberg-Vrenken et al. Citation2013). Furthermore, a direct anti-inflammatory effect of metformin is exerted by inhibiting NF-κB through blockade of the PI3K/Akt pathway. It is obvious that metformin demonstrates a dose-dependent inhibition of interleukin-1β-induced release of the proinflammatory cytokines IL-6 and -8 (Hadi et al. Citation2012). IL-6 and TNF-α can regulate inflammatory biomarkers such as C-reactive protein (CRP) in acute toxicity (Zambon et al. Citation2006).
Epidemiological studies have suggested that metformin significantly improves the viability of hepatocytes in fatty livers including nonalcoholic fatty liver disease (NAFLD) and nonalcoholic steatohepatitits (NASH) as well as hepatocarcinoma (Noto et al. Citation2012). In addition, metformin reduces the serum levels of ALT and AST, enzymes releasing from injured hepatocytes, in NASH (Angulo & Lindor Citation2002; Bugianesi et al. Citation2004; Angelico et al. Citation2007; Woudenberg-Vrenken et al. Citation2013). Although the underlying mechanisms for the protective activity of metformin in the complications is not well found, down regulation of TNF-α-mediated signaling (Lin et al. Citation2000) along with the activation of adenosine monophosphate kinase (AMPK) and AMPK-dependent signaling pathways are described as possible mechanisms for the effect (Zhou et al. Citation2001). Moreover, and the augmentation of β-oxidation of fatty acids is suggested as an essential factor, which is mediated by AMPK-dependent pathway (Zhou et al. Citation2001).
Conclusions
In conclusion, for first time, the hepatoprotective effect of metformin was demonstrated in the APAP-induced toxicity in mice. The study has shown that metformin can exert an oxidant/antioxidant balance and limit the inflammation and hepatocyte injury, and subsequently increases the cell survival in the APAP-induced hepatotoxicity. The investigation was performed to present a new treatment to attenuate the hepatotoxicity induced by APAP overdose for further interpretation to humans.
Disclosure statement
The authors declare that there is no conflict of interest.
References
- Angelico F, Burattin M, Alessandri C, Del Ben M, Lirussi F. 2007. Drugs improving insulin resistance for non-αlcoholic fatty liver disease and/or nonalcoholic steatohepatitis. Cochrane Database Syst Rev. CD005166.
- Angulo P, Lindor KD. 2002. Treatment of non-alcoholic steatohepatitis. Best Pract Res Clin Gastroenterol. 16:797–810.
- Anon. 2010. Tylenol professional product information. Fort Washington (MD): McNeil Consumer Healthcare, Division of McNEIL-PPC, Inc.
- Bajt ML, Knight TR, Farhood A, Jaeschke H. 2003. Scavenging peroxynitrite with glutathione promotes regeneration and enhances survival during acetaminophen-induced liver injury in mice. J Pharmacol Exp Ther. 307:67–73.
- Bajt ML, Ramachandran A, Yan HM, Lebofsky M, Farhood A, Lemasters JJ, Jaeschke H. 2011. Apoptosis-inducing factor modulates mitochondrial oxidant stress in acetaminophen hepatotoxicity. Toxicol Sci. 122:598–605.
- Bessems JG, Vermeulen NP. 2001. Paracetamol (acetaminophen)-induced toxicity: molecular and biochemical mechanisms, analogues and protective approaches. Crit Rev Toxicol. 31:55–138.
- Bronstein AC, Spyker DA, Cantilena LR. Jr, Green JL, Rumack BH, Giffin SL. 2010. 2009 annual report of the American association of poison control centers' national poison data system (NPDS): 27th annual report. Clin Toxicol (Phila). 48:979–1178.
- Bugianesi E, Marzocchi R, Villanova N, Marchesini G. 2004. Non-alcoholic fatty liver disease/non-alcoholic steatohepatitis (NAFLD/NASH): treatment. Best Pract Res Clin Gastroenterol. 18:1105–1116.
- Choi J, Park K-H, Kim SZ, Shin JH, Jang SI. 2013. The ameliorative effects of l-2-oxothiazolidine-4-carboxylate on acetaminophen-induced hepatotoxicity in mice. Molecules 18:3467–3478.
- Choi YH, Kim SG, Lee MG. 2006. Dose-independent pharmacokinetics of metformin in rats: hepatic and gastrointestinal first-pass effects. J Pharm Sci. 95:2543–2552.
- Cosic V, Antic S, Pesic M, Tovanovic O, Kundal S, Djordjevic VB. 2001. Monotherapy with metformin: does it improve hypoxia in type 2 diabetic patients? Clin Chem Lab Med. 39:818–821.
- Dai J, Liu M, Ai Q, Lin L, Wu K, Deng X, Jing Y, Jia M, Wan J, Zhang L. 2014. Involvement of catalase in the protective benefits of metformin in mice with oxidative liver injury. Chem Biol Interact. 216:34–42.
- Dambach DM, Durham SK, Laskin JD, Laskin DL. 2006. Distinct roles of NF-kappaB p50 in the regulation of acetaminophen-induced inflammatory mediator production and hepatotoxicity. Toxicol Appl Pharmacol. 211:157–165.
- Dart RC, Erdman AR, Olson KR, Christianson G, Manoguerra AS. 2006. Acetaminophen poisoning: an evidence based consensus guideline for out-of-hospital management. Clin Toxicol (Phila). 44:1–18.
- Das J, Ghosh J, Manna P, Sil PC. 2010. Taurine protects acetaminophen-induced oxidative damage in mice kidney through APAP urinary excretion and CYP2E1 inactivation. Toxicology. 269:24–34.
- Davidson MB, Peters AL. 1997. An overview of metformin in the treatment of type 2 diabetes mellitus. Am J Med. 102:99–110.
- de la Rosa LC, Vrenken TE, Manon B, Buist-Homan M, Faber KN, Moshage H. 2005. Metformin protects primary rat hepatocytes against oxidative stress-induced apoptosis. Pharmacol Res Pers. 11:340–375.
- El-Hassan H, Anwar K, Macanas-Pirard P, Crabtree M, Chow SC, Johnson VL, Lee PC, Hinton RH, Price SC, Kass GE. 2003. Involvement of mitochondria in acetaminophen-induced apoptosis and hepatic injury: roles of cytochrome c, bax, bid, and caspases. Toxicol Appl Pharmacol. 191:118–129.
- Enomoto A, Itoh K, Nagayoshi E, Haruta J, Kimura T, O'Connor T. 2001. High sensitivity of Nrf2 knockout mice to acetaminophen hepatotoxicity associated with decreased expression of ARE-regulated drug metabolizing enzymes and antioxidant genes. Toxicol Sci. 59:169–177.
- Ewis SA, Abdel-Rahman MS. 1997. Influence of atenolol and/or metformin on glutathione and magnesium levels in diabetic rats. J Appl Toxicol. 17:409–413.
- Gelotte CK, Auiler JF, Lynch JM, Temple AR, Slattery JT, Slattery JT. 2007. Disposition of acetaminophen at 4, 6, and 8 g/day for 3 days in healthy young adults. Clin Pharmacol Ther. 81:840–848.
- Gobe GC, Morais C, Vesey DA, Johnson DW. 2013. Use of high-dose erythropoietin for repair after injury: a comparison of outcomes in heart and kidney. J Nephropathol. 2:154–165.
- Hadi NR, Swadi AA, Mohammad BI. 2012. Hepatoprotective potential of metformin against methotrexate induced hepatotoxicity. Asian J Exp Biol Sci. 3:110–117.
- Hanawa N, Shinohara M, Saberi B, Gaarde WA, Han D, Kaplowitz N. 2008. Role of JNK translocation to mitochondria leading to inhibition of mitochondria bioenergetics in acetaminophen-induced liver injury. J Biol Chem. 283:13565–13577.
- Henderson CJ, Wolf CR, Kitteringham N, Powell H, Otto D, Park BK. 2000. Increased resistance to acetaminophen hepatotoxicity in mice lacking glutathione S-transferase Pi. Proc Natl Acad Sci USA. 97:12741–12745.
- Jaeschke H. 2003. Molecular mechanisms of hepatic ischemia-reperfusion injury and preconditioning. Am J Physiol Gastrointest Liver Physiol. 284:G15–G26.
- James LP, Mayeux PR, Hinson JA. 2003. Acetaminophen-induced hepatotoxicity. Drug Metab Dispos. 31:1499–1506.
- James LP, McCullough SS, Lamps LW, Hinson JA. 2003. Effect of N-acetylcysteine on acetaminophen toxicity in mice: relationship to reactive nitrogen and cytokine formation. Toxicol Sci. 75:458–467.
- Kaplowitz N, Shinohara M, Liu ZX, Han D. 2008. How to protect against acetaminophen: don't ask for JUNK. Gastroenterology. 135:1047–1051.
- Kim YH, Hwang JH, Kim KS, Noh JR, Choi DH, Kim DK, Tadi S, Yim YH, Choi HS, Lee CH. 2015. Metformin ameliorates acetaminophen hepatotoxicity via Gadd45β-dependent regulation of JNK signaling in mice. J Hepatol. 63:75–82.
- Knight TR, Ho YS, Farhood A, Jaeschke H. 2002. Peroxynitrite is a critical mediator of acetaminophen hepatotoxicity in murine livers: protection by glutathione. J Pharmacol Exp Ther. 303:468–475.
- Kon K, Kim JS, Jaeschke H, Lemasters JJ. 2004. Mitochondrial permeability transition in acetaminophen-induced necrosis and apoptosis of cultured mouse hepatocytes. Hepatology. 40:1170–1179.
- Larson AM, Polson J, Fontana RJ, Davern TJ, Lalani E, Hynan LS, Reisch JS, Schiødt FV, Ostapowicz G, Shakil AO, et al. 2005. Acetaminophen-induced acute liver failure: results of a United States multicenter, prospective study. Hepatology. 42:1364–1372.
- Lawson JA, Farhood A, Hopper RD, Bajt ML, Jaeschke H. 2000. The hepatic inflammatory response after acetaminophen overdose: role of neutrophils. Toxicol Sci. 54:509–516.
- Lemasters JJ. 1999. V. Necrapoptosis and the mitochondrial permeability transition: shared pathways to necrosis and apoptosis. Am J Physiol. 276:G1–G6.
- Lin HZ, Yang SQ, Chuckaree C, Kuhajda F, Ronnet G, Diehl AM. 2000. Metformin reverses fatty liver disease in obese, leptin-deficient mice. Nat Med. 6:998–1003.
- Liu H, Song D, Lee SS. 2001. Role of hemeoxygenase-carbon monoxide pathway in pathogenesis of cirrhotic cardiomyopathy in the rat. Am J Physiol: Gastrointest Liver Physiol. 280:68–74.
- McGill Sharpe MR, Williams CD, Taha M, Curry SC, Jaeschke H. 2012. The mechanism underlying acetaminopheninduced hepatotoxicity in humans and mice involves mitochondrial damage and nuclear DNA fragmentation. J Clin Invest. 122:1574–1583.
- Meister A, Anderson ME. 1983. Glutathione. Annu Rev Biochem. 52:711–760.
- Noto H, Goto A, Tsujimoto T, et al. 2012. Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PLoS One. 7:e33411.
- Nourjah P, Ahmad SR, Karwoski C, Willy M. 2006. Estimates of acetaminophen (Paracetomal)-associated overdoses in the United States. Pharmacoepidemiol Drug Saf. 15:398–405.
- Ohkawa H, Ohishi N, Yagi K. 1979. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 95:351–358.
- Paglia DE, Valentine WN. 1967. Studies on the quantitative and qualitative characterization of erythrocyte glutathione peroxidase. J Lab Clin Med. 70:158–169.
- Paoletti F, Mocali A. 1990. Determination of superoxide dismutase activity by purely chemical system based on NAD(P)H oxidation. Meth. Enzymol. 186:209–220.
- Poon MKT, Chiu PY, Mak DHF, Ko KM. 2003. Metformin protects against carbon tetrachloride hepatotoxicity in mice. J Pharmacol Sci. 93:501–504.
- Rafieian-Kopaei M, Baradaran A, Rafieian M. 2013. Plants antioxidants: from laboratory to clinic. J Nephropathol. 2:152–153.
- Ramachandran A, Lebofsky M, Weinman SA, Jaeschke H. 2011. The impact of partial manganese superoxide dismutase (SOD2)-deficiency on mitochondrial oxidant stress, DNA fragmentation and liver injury during acetaminophen hepatotoxicity. Toxicol Appl Pharmacol. 251:226–233.
- Reid AB, Kurten RC, McCullough SS, Brock RW, Hinson JA. 2005. Mechanisms of acetaminophen-induced hepatotoxicity: role of oxidative stress and mitochondrial permeability transition in freshly isolated mouse hepatocytes. J Pharmacol Exp Ther. 312:509–516.
- Rumack BH, Matthew H. 1975. Acetaminophen poisoning and toxicity. Pediatrics. 55:871–876.
- Saeedi Saravi SS, Ghazi-Khansari M, Mehr SE, Nobakht M, Mousavi SE, Dehpour AR. 2016. Contribution of mammalian target of rapamycin in the pathophysiology of cirrhotic cardiomyopathy. World J Gastroenterol. 22:4685–4694.
- Saito C, Zwingmann C, Jaeschke H. 2010. Novel mechanisms of protection against acetaminophen hepatotoxicity in mice by glutathione and N-acetylcysteine. Hepatology. 51:246–254.
- Schoemaker MH, Ros JE, Homan M, Trautwein C, Liston P, Poelstra K, van Goor H, Jansen PL, Moshage H. 2002. Cytokine regulation of pro- and anti-apoptotic genes in rat hepatocytes: NF-kappaB-regulated inhibitor of apoptosis protein 2 (cIAP2) prevents apoptosis. J Hepatol. 36:742–750.
- Spasovski D. 2013. Renal markers for assessment of renal tubular and glomerular dysfunction. J Nephropathol. 2:23–25.
- Tavafi M. 2013a. Complexity of diabetic nephropathy pathogenesis and design of investigations. J Renal Inj Prev. 2:59–62.
- Tavafi M. 2013b. Diabetic nephropathy and antioxidants. J Nephropathol. 2:20–27.
- Vishal RT. 2008. Metformin therapy: benefits beyond glycemic control. Int J Diab Dev Ctries. 27:87–95.
- Walker TM, Rhodes PC, Westmoreland C. 2000. The differential cytotoxicity of methotrexate in rat hepatocyte monolayer and spheroid cultures. Toxicol in Vitro. 14:475–485.
- Woudenberg-Vrenken TE, de la Rosa LC, Buist-Homan M, Faber KN, Moshage H. 2013. Metformin protects rat hepatocytes against bile acid-induced apoptosis. PLoS One. 8:e71773.
- Yazdani I, Ghazi-Khansari M, Saeedi Saravi SS, Nobakht M, Majdani R, Rezayat SM, Mousavi SE, Yari A, Dehpour AR. 2016. Nortriptyline protects testes against germ cell apoptosis and oxidative stress induced by testicular ischaemia/reperfusion. Andrologia. doi:10.1111/and.12605.
- Zambon A, Gervois P, Pauletto P, Fruchart JC, Staels B. 2006. Modulation of hepatic inflammatory risk markers of cardiovascular diseases by PPAR-alpha activators: clinical and experimental evidence. Arterioscler Thromb Vasc Biol. 26:977–986.
- Zhou G, Myers R, Li Y, Chen Y, Shen X, Fenyk-Melody J, Wu M, Ventre J, Doebber T, Fujii N, et al. 2001. Role of AMP-activated protein kinase in mechanism of metformin action. J Clin Invest. 108:1167–1174.
- Zorov DB, Filburn CR, Klotz LO, Zweier JL, Sollott SJ. 2000. Reactive oxygen species (ROS)-induced ROS release: a new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J Exp Med. 192:1001–1014.