
Abstract
Context: Schizophrenia is a heterogenous neurological disorder, which has been hypothetically linked to oxidative imbalance and associated behavioral perturbations. Preliminary evidence from animal models predictive of human psychosis suggests that Terminalia ivorensis A. Chev. (Combretaceae) has antipsychotic-like activity in mice.
Objective: This study investigates the neuroprotective property of the ethanol stem bark extracts of T. ivorensis (EETI) in reversal treatment of ketamine-induced schizophrenia-like behaviors and oxidative alteration in adult male Swiss albino mice.
Materials and methods: Animals were divided into six treatment groups (n = 5). Animals received distilled water or ketamine (20 mg/kg) once daily intraperitoneally (i.p.) for 14 days, and from the 8th to the 14th day, they were treated with EETI (125, 250 or 500 mg/kg), risperidone (RIS) or vehicle orally once daily. Behaviors related to positive (locomotor activity) and cognitive (Y maze) symptoms of schizophrenia were assessed. Glutathione (GSH) levels, superoxide dismutase (SOD) and catalase (CAT) activities, including malondialdehyde (MDA) concentration were measured in mice whole brains.
Result: The LD50 of EETI was 2236.06 mg/kg, p.o. body weight. EETI (125, 250 or 500 mg/kg, p.o.) demonstrated significant (p < 0.05) inhibition of ketamine-induced hyperlocomotion and cognitive dysfunction. The extract decreased MDA concentration (39.0, 62.6 and 67.5%) in a dose-dependent manner. Moreover, EETI significantly (p < 0.05) reversed the depletion of GSH, and increased activities of SOD and CAT in brain tissues.
Discussion and conclusion: These findings suggest that EETI probably exert its antipsychotic-like activity, via a neuroprotective compensatory mechanism of action, and as such, could be relevant in the management of schizophrenia.
Introduction
Schizophrenia is a heterogenous chronic neurological disease that affects approximately 1% of the world’s population (Picchioni & Murray Citation2007) making it the seventh most costly medical illness (Freedman Citation2003). It is characterized by severe behavioral perturbations including positive (e.g., hallucinations, delusions, disorganized speech and thought), negative (e.g., blunted affect and social isolation), and cognitive symptoms (e.g., executive and memory dysfunction) (Andreasen Citation1990; Larson et al. Citation2010). The severity of symptoms of schizophrenia depends on the antioxidant levels (Pavlovic et al. Citation2002). For instance, the severity of symptoms is usually associated with the decreased antioxidant level (Pazvantoglu et al. Citation2009). Various antioxidants are related to the positive, negative and cognitive symptoms, poor premorbid functions and computed tomography abnormalities, since oxidative stress is considered a potent intracellular signaling mechanism that induces changes in the dopamine D2 receptor (Pazvantoglu et al. Citation2009).
The brain is known to be the most metabolically active organ of the human body, which generates a high load of reactive oxygen moieties (Wood et al. Citation2009). It weighs only 2% of the body mass, but consumes 20% of body oxygen and 25% of body glucose at rest. Reactive oxygen species (ROS) produced in any tissue is directly proportional to its oxygen consumption (Ikonomidou & Kaindl Citation2011). Since the brain is continuously under oxidation/antioxidant process, it is prone to oxidative stress (Wood et al. Citation2009). This burden is increased by a number of factors like volume of neurotransmission and reduction–oxidation (redox) property of dopamine, and redox regulated activity at glutamate receptors (Bókkon & Antal Citation2011).
A clear mechanism underlying the pathogenesis of schizophrenia remains unknown. However, oxidative stress which is a condition arising from an imbalance between toxic ROS and antioxidants (Halliwell Citation2006; Wood et al. Citation2009), and as a consequence of aberrant reduction–oxidation (redox) control has become an attractive hypothesis for explaining, at least in part, the pathophysiology of schizophrenia (Wood et al. Citation2009; Zhang et al. Citation2010). This hypothesis has theoretical appeal, as the brain is considered particularly vulnerable to oxidative damage for several reasons such as its comparatively high oxygen utilization (hence generation of free radical by-products), low levels of protective antioxidant enzymes, high proportion of readily oxidizable membrane polyunsaturated fatty acids (PUFAs), and the presence of redox-catalytic metals such as iron and copper (Halliwell Citation2006). The brain is also susceptible to secondary and self-perpetuating damage from oxidative cellular injury or necrosis, via the neurotoxic effects of released excitatory amines (mainly glutamate) and iron, and the activated inflammatory response (Valko et al. Citation2007).
Terminalia ivorensis A. Chev. (Combretaceae) is an indigenous plant with a potential to treat schizophrenia due to its wide ethnomedicinal uses and rich phytochemical constituents (Foli Citation2009). It is commonly called idigbo in Nigeria (Coulibaly et al. Citation2010). Other common names of T. ivorensis include black afara, black bark, yellow terminalia, shingle wood, brimstone and Satin wood. It is known in French as Framire; Amire in Ghana (Coulibaly et al. Citation2010).
This plant species treats burns, bruises, sores, cough, diarrhea, hypertension, diabetes, and tooth decay (Coulibaly et al. Citation2010). In Nigeria, the stem bark is used as antimalarial and tranquilizer (Lawal et al. Citation2010). Many ethnopharmacological studies carried out in Africa revealed that T. ivorensis showed great promise as it possesses trypanocidal (Agbedahunsi et al. Citation2006), antioxidant activities (Ponou et al. Citation2010; Adiko et al. Citation2013). Other biological activities associated with the plant include antibacterial activity (Malcom & Sofowora Citation1969), anti-inflammatory and anti-arthritis (Iwu & Anyawu Citation1982).
T. ivorensisis rich in several phytochemicals such as terminolic, betulinic, ellergicglycirrhetic acids and quarcetin, sericic acid and lonchoterpene (Iwu & Anyawu Citation1982), polyphenols, flavonoids, saponins, steroids (Ponou et al. Citation2010), punicalagin and punicalin (Adiko et al. Citation2013), many of which are sources of a wide range of biological activities such as antioxidant, anti-neuroinflammatory and neuroprotection (Ponou et al. Citation2010; Wang et al. Citation2013).
Recently, it has been demonstrated from preliminary studies that administration of the ethanol extract of T. ivorensis stem bark in animal models predictive of human psychosis in mice suggest that this plant species possesses antipsychotic-like activity. In this context and based on the beneficial effects of the phytochemicals of the plant as antioxidants, we hypothesized that T. ivorensis could protect against oxidative stress in schizophrenia. Therefore, this present study was designed to evaluate the neuroprotective effects of the ethanol extract of T. ivorensis stem bark (EETI), as its probable mechanism of action in schizophrenia, in the reversal treatment of ketamine-induced schizophrenia-like behaviors and oxidative damage in mice as an animal model predictive of human psychosis and oxidative stress.
Materials and methods
Animals
Adult male Swiss albino mice weighing 20–25 g used for this study were obtained from the Central Animal House, College of Medicine, University of Ibadan. The animals were housed five per plastic cage (42 × 30 × 27 cm) in a controlled environment at room temperature (25 ± 1 °C) with a 12:12 h light/dark cycle. They were fed with standard rodent pellet food and water ad libitum throughout the experimental period. They were acclimatized for at least one week prior to commencement of the experiments. The experiments were performed after approval of the protocol by the Ethics Committee of the University of Ibadan according to the National institutes of Health Guide for Care and Use of Laboratory Animals (Publication No. 85-23, revised 1985). Also, efforts were made to minimize the suffering of the animals.
Plant materials
The stem bark samples of T. ivorensis were collected in March, 2014 at the Olupkele Forest Reserve, Ibadan, Oyo state, Nigeria. Taxonomical identification and authentication of the plant were done by Mr. O.S. Shasanya, at the herbarium section of the Forestry Research Institute of Nigeria (FRIN), Ibadan, Nigeria. A voucher specimen with identification number, FHI 109800, was deposited and compared with the reference specimen.
Extraction of plant
The stem-bark was air-dried for 6–7 weeks, and pulverized with an electric crusher. Pulverized stem-bark (200 g) was macerated in 70% ethanol (2 L) for 48 h after which it was filtered on absorbent cotton and on Whatmann 3 mm paper under sterile conditions. The filtrate was concentrated using Rotary evaporator (BUCHI® Rotavapor Model R-215, Buchi Labortechnick AG, Flawil, Switzerland) at 40 °C to give a semisolid residue. The dark brown paste obtained was dried to a constant weight (7.6 g) in a desiccator before storage in a sterilized glass vial for use.
Drug preparation and administration
Ketamine hydrochloride (Sigma-Aldrich, St. Louis, MO, USA), diluted in distilled water and administered intraperitoneally (i.p.), risperidone (RIS) (Sigma-Aldrich, St. Louis, MO, USA), dissolved in distilled water and administered per oral (p.o.). The ethanol extract of T. ivorensis stem bark (EETI) was dissolved in distilled water, were used. The EETI was administered p.o. (125, 250 or 500 mg/kg) based on the LD50 determined by Lorke (Citation1983). The doses of ketamine and RIS used in the study were chosen according to the literature (Monte et al. Citation2013). Appropriated vehicle groups were also assessed simultaneously per os with 10 mL/kg. RIS was used as a standard antipsychotic, because ketamine-induced model of schizophrenia turned out to be more responsive to atypical antipsychotic (Becker & Grecksch Citation2004).
Acute toxicity study
Again, Lorke’s (Citation1983) LD50 was also used here as the index of acute toxicity. Albino mice (20–25 g) of either sex were used. This method involved an initial dose finding procedure, in which the animals were divided into three groups of three mice. Doses of 10, 100 and 1000 mg/kg were administered p.o., in those groups respectively. The treated animals were monitored for 24 h for mortality and general behavioral change. From the results of the above procedure, four different doses of the extract (2000, 3000, 4000 and 5000 mg/kg) were chosen and administered p.o., in the four groups, respectively. The treated animals were monitored for 24 h. The LD50 was calculated as the geometric mean of the lowest dose showing death and the highest dose showing no death.
Experimental protocol
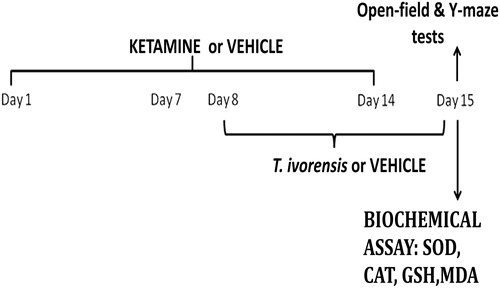
An overview of the experimental design is depicted in . This protocol was designed and modified to mimic the reversal protocol of the schizophrenia state, as previously described by Monte et al. (Citation2013). Mice were divided into six groups (n = 5/group). Group 1 was treated with vehicle (10 mL/kg, p.o.) once daily, while groups 2–6 were treated with sub-anesthetic dose of ketamine (20 mg/kg) once daily intraperitoneally (i.p.) for 14 days. From the 8th to 14th day of treatment onwards, group 2 was additionally treated with vehicle (10 mL/kg, p.o.) once daily as negative control, while group 3–5 were treated additionally with extract (125, 250, or 500 mg/kg, p.o.) once daily, and group 6 received in addition RIS (0.5 mg/kg, p.o.) once daily as a positive control, with a 30 min interval between treatments. About 24 h after the last treatment (15th day), animals were assessed for behavioral effects (motor behavior and cognitive dysfunction) on Y-maze and open-field test apparatus, respectively, before sacrificing each mice for biochemical assays ().
Figure 1. Treatment protocol.
Behavioral analysis
Open-field test (OFT)
Locomotor behavior was monitored using the open feed apparatus. The apparatus consisted of a wooden box measuring 35 × 30 × 23 cm with visible lines drawn to divide the floor into 36 (20 cm × 20 cm) squares with a frontal glass wall (Omogbiya et al. Citation2013) and placed in a sound free room. The animals were placed in the rear left square and left to explore it. The duration of ambulation(s) and number of line crossed were recorded for 5 min using a stopwatch. After each mouse session, the observation chamber was cleaned with 10% ethanol to remove residual odor.
Y-maze test (YMT)
The effect of the extract on spontaneous alternation performance was also assessed using Y-maze test (YMT), which allowed the evaluation of cognitive searching behavior, as an index for the cognitive dysfunction of schizophrenia as described by Monte et al. (Citation2013). Animals were gently placed individually in the Y-maze apparatus, which consisted of three identical arms (33 × 11 × 12 cm each) in which the arms are symmetrically separated at 120° (Dall’igna et al. Citation2007). Specifically, each mouse was placed at the end of arm A, and allowed to explore all the three arms (labeled A, B, C) freely for 5 min, taking the following parameters: the number of arm visits and sequence (alternation) of arm visits visually. An alternation was defined as entries in all three arms on consecutive occasions (Monte et al. Citation2013). The percentage of alternation was calculated as total of alternations/(total arm entries −2), as previously described (Yamada et al. Citation1996; Dall’igna et al. Citation2007). After each mice session, the observation chamber was cleaned with 10% ethanol to remove residual odor.
Biochemical assay
Superoxide dismutase assay
Superoxide dismutase (SOD) activity was measured by the method described by Misra and Fridovich (Citation1972). This method is based on the inhibition of superoxide dependent adrenaline auto-oxidation in a spectrophotometer adjusted at 480 nm. The specific activity of SOD was reported as units per mg of protein
Catalase assay
Catalase (CAT) activity was assayed by the method of Sinha (Citation1971). Hydrogen peroxide (H2O2) disappearance was continuously monitored using two cuvettes at 60 and 180 s intervals at 570 nm spectrophotometrically through its absorbance. The CAT activity was expressed as μmoles of H2O2 decomposed per minutes per mg protein.
Glutathione assay
Glutathione (GSH) activity was assayed by the method described by Jollow et al. (Citation1974). This method was based upon the development of a relatively stable (yellow) color when 5, 5-dithiobis-(2-nitrobenzoic acid) (DTNB) was added to sulfhydryl compounds. The chromophoric product resulting from the reaction of DTNB with the reduced GSH, 2-nitro-5-thiobenzoic acid possessed a characteristic absorbance at 412 nm and the amount of reduced GSH in the sample was proportional to the absorbance at this wavelength. The concentration of reduced GSH in the brain tissues were expressed as micromoles per gram tissue (μmol/g tissue).
Lipid peroxidation assay
The malondialdehyde (MDA) content of the lipid peroxidation was measured according to the method described by Okhawa et al. (Citation1979). This assay principle is based on the fact that lipid peroxidation generates unstable lipid peroxides that decompose to form a complex series of compounds including reactive carbonyl compounds. The polyunsaturated fatty acid peroxides produced, generate MDA upon decomposition. MDA form a 1:2 adduct with thiobarbituric acid (TBA) that gives rise to a pink color product when heated in acidic pH, with a maximum absorbance of 532 nm. The measure of lipid peroxidation was determined by thiobarbituric acid reactive substances (TBARS). Briefly, homogenates were mixed with trichloroacetic acid 30% and TBA 0.75% and heated in a boiling water bath for 45 min at 80 °C. TBARS was determined by the absorbance at 535 nm. Results were reported as nmol of TBARS per wet gram tissue.
Protein assay
According to Gornall et al. (Citation1949), the principle and method was based on the fact that proteins form a colored complex with cupric ions in an alkaline solution like Biuret reagent containing CuSO4, potassium iodide (KI) and sodium potassium tartarate. The protein and Biuret reagent form complexes with maximum absorbance at 540 nm. Brain homogenate were added to 3 mL of Biuret reagent in triplicate. The mixture was incubated at room temperature for 30 min after which the absorbance was read at 540 nm. The protein content of the samples was extrapolated from the standard curve, using bovine serum albumin as standard.
Statistical analysis
The data were expressed as mean ± standard error of mean (S.E.M). The data were analyzed using one-way analysis of variance (ANOVA) followed by post hoc test (Newman–Keul) for multiple comparisons where appropriate using GraphPadInStat® Biostatistics software (GraphPad Software, Inc., La Jolla, CA, USA version 4.0). A level of p < 0.05 was considered as statistically significant for all tests.
Results
Acute toxicity test
The LD50 of the ethanol extract of T. ivorensis stem bark was found to be 2236.06 mg/kg, p.o. body weight.
Behavioral parameters
Effect of EETI on reversal treatment of ketamine-induced hyperlocomotion
Effect of ethanol extract on reversal treatment of ketamine-induced hyperlocomotion was assessed in the open-field apparatus. Chronic ketamine treatment significantly (p < 0.05) increased locomotion (hyperlocomotion) compared to the group that received vehicle (10 mL/kg, p.o.) as indexed by the increase in the number of line crossings and reduction in ambulation time in the open-field test following 14 days treatment (). The result shows that the increased hyperlocomotion induced by ketamine (20 mg/kg, i.p.) was significantly (p < 0.05) reversed by EETI (125, 250 and 500 mg/kg, p.o.) in a dose-dependent manner following treatment from the 8th to 14th day of treatment. RIS (0.5 mg/kg, p.o.) compared to the ketamine treated group, significantly (p < 0.05) reversed the hyperlocomotion following treatment from the 8th to 14th day of treatment ().
Table 1. Effect of EETI on reversal treatment of ketamine-induced hyperlocomotion.
Effect of EETI on reversal treatment of ketamine-induced cognitive dysfunction
The effect of ethanol extract on reversal treatment of ketamine-induced cognitive dysfunction was assessed by sequence of arm entry and number of arm entries in the Y-maze apparatus. Chronic ketamine (20 mg/kg, i.p.) treatment significantly (p < 0.05) induced cognitive dysfunction (memory impairment) compared to the group that received vehicle (10 mL/kg, p.o.) only following 14 days treatment. EETI (125, 250 and 500 mg/kg, p.o.) significantly reversed in a dose-dependent manner the cognitive dysfunction following treatment from the 8th to 14th day of treatment compared to the ketamine treated group. RIS (0.5 mg/kg, p.o.) compared to the ketamine treated group, also significantly (p < 0.05) reversed the cognitive dsyfunction following treatment from the 8th to 14th day of treatment ().
Figure 2. Effect of EETI on reversal treatment of ketamine-induced cognitive dysfunction. Value represents the mean ± S.E.M of five animals/group. One way ANOVA revealed that there is significant [F (5, 24) = 13.55, p < 0.0001] difference between various treatment groups. **Denotes p < 0.05 as compared to vehicle group. *Denotes p < 0.05 as compared with ketamine group. KET: Ketamine; RIS: Risperidone; EETI: Ethanol extract of T. ivorensis stem bark.
![Figure 2. Effect of EETI on reversal treatment of ketamine-induced cognitive dysfunction. Value represents the mean ± S.E.M of five animals/group. One way ANOVA revealed that there is significant [F (5, 24) = 13.55, p < 0.0001] difference between various treatment groups. **Denotes p < 0.05 as compared to vehicle group. *Denotes p < 0.05 as compared with ketamine group. KET: Ketamine; RIS: Risperidone; EETI: Ethanol extract of T. ivorensis stem bark.](/cms/asset/abed225f-8ef6-4266-a0c8-70ebb18d85fa/iphb_a_1190382_f0002.jpg)
Biochemical parameters
Effect of EETI on reversal treatment of ketamine-induced oxidative stress
In order to consolidate the notion of a neuroprotective effect of EETI against ketamine-induced model of schizophrenia and oxidative stress, we evaluated the effects of EETI in oxidative stress parameters in the whole brain of the mice. In this context, chronic ketamine (20 mg/kg, i.p.) treatment significantly (p < 0.05) induced oxidative stress by decreasing SOD, CAT and GSH activity compared to the group treated with vehicle (10 mL/kg, p.o.) following 14 days treatment protocol. The results show that EETI (125, 250 and 500 mg/kg, p.o.) significantly reversed ketamine-induced oxidative stress by increasing SOD, CAT and GSH activity/enzyme in a dose-dependent manner following treatment from the 8th to 14th day of treatment protocol compared to ketamine (20 mg/kg, i.p.) treated group for 14 days. RIS (0.5 mg/kg, p.o.) compared to the ketamine treated group, significantly (p < 0.05) reversed ketamine-induced oxidative stress by increasing SOD, CAT and GSH activity/enzyme following treatment from the 8th to 14th day of treatment protocol ().
Figure 3. Effect of EETI on superoxide dismutase (SOD) activity in mouse brain. Value represents the mean ± S.E.M of five animals/group. One way ANOVA revealed that there is significant [F (5, 24) = 62.51, p < 0.0001] difference between various treatment groups. **Denotes p < 0.05 as compared to vehicle group. *Denotes p < 0.05 as compared with ketamine group. KET: Ketamine; RIS: Risperidone; EETI: Ethanol extract of T. ivorensis stem bark.
![Figure 3. Effect of EETI on superoxide dismutase (SOD) activity in mouse brain. Value represents the mean ± S.E.M of five animals/group. One way ANOVA revealed that there is significant [F (5, 24) = 62.51, p < 0.0001] difference between various treatment groups. **Denotes p < 0.05 as compared to vehicle group. *Denotes p < 0.05 as compared with ketamine group. KET: Ketamine; RIS: Risperidone; EETI: Ethanol extract of T. ivorensis stem bark.](/cms/asset/afad75db-ad7c-45d5-944e-9840ebd52328/iphb_a_1190382_f0003.jpg)
Figure 4. Effect of EETI on catalase (CAT) activity in mouse brain. Value represents the mean ± S.E.M of five animals/group. One way ANOVA revealed that there is significant [F (5, 24) = 33.24, p < 0.0001] difference between various treatment groups. **Denotes p < 0.05 as compared to vehicle group. *Denotes p < 0.05 as compared with ketamine group. KET: Ketamine; RIS: Risperidone; EETI: Ethanol extract of T. ivorensis stem bark.
![Figure 4. Effect of EETI on catalase (CAT) activity in mouse brain. Value represents the mean ± S.E.M of five animals/group. One way ANOVA revealed that there is significant [F (5, 24) = 33.24, p < 0.0001] difference between various treatment groups. **Denotes p < 0.05 as compared to vehicle group. *Denotes p < 0.05 as compared with ketamine group. KET: Ketamine; RIS: Risperidone; EETI: Ethanol extract of T. ivorensis stem bark.](/cms/asset/4c7266d0-bb05-40b4-8f25-e7e23899d7cd/iphb_a_1190382_f0004.jpg)
Figure 5. Effect of EETI on glutathione (GSH) activity in mouse brain. Value represents the mean ± S.E.M of five animals/group. One way ANOVA revealed that there is significant [F (5, 24) = 26.77, p < 0.0001] difference between various treatment groups. **Denotes p < 0.05 as compared to vehicle group. *Denotes p < 0.05 as compared with ketamine group. KET: Ketamine; RIS: Risperidone; EETI: Ethanol extract of T. ivorensis stem bark.
![Figure 5. Effect of EETI on glutathione (GSH) activity in mouse brain. Value represents the mean ± S.E.M of five animals/group. One way ANOVA revealed that there is significant [F (5, 24) = 26.77, p < 0.0001] difference between various treatment groups. **Denotes p < 0.05 as compared to vehicle group. *Denotes p < 0.05 as compared with ketamine group. KET: Ketamine; RIS: Risperidone; EETI: Ethanol extract of T. ivorensis stem bark.](/cms/asset/30137fb9-7c99-409f-80be-2e4f9a5c0764/iphb_a_1190382_f0005.jpg)
The effect of EETI on reversal treatment of ketamine-induced lipid peroxidation as the consequence of oxidative stress was also evaluated. Chronic ketamine (20 mg/kg, i.p.) treatment significantly (p < 0.05) induced increased lipid peroxidation product (MDA) compared to the group treated with vehicle (10 mL/kg, p.o.) following 14 days treatment protocol. The result also shows that EETI (125, 250 and 500 mg/kg, p.o.) significantly reversed ketamine-induced oxidative stress (by decreasing lipid peroxidation product, MDA) in a dose-dependent manner following treatment from the 8th to 14th day of treatment protocol compared to ketamine (20 mg/kg, i.p.) treated group for 14 days. RIS (0.5 mg/kg, p.o.) compared to the ketamine treated group, significantly (p < 0.05) reversed ketamine-induced oxidative stress (by decreasing MDA production) following treatment from the 8th to 14th day of treatment ().
Figure 6. Effect of EETI on MDA content in mouse brain. Value represents the mean ± S.E.M of five animals/group. One way ANOVA revealed that there is significant [F (5, 24) = 24.50, p < 0.0001] difference between various treatment groups. **Denotes p < 0.05 as compared to vehicle group. *Denotes p < 0.05 as compared with ketamine group. KET: Ketamine; RIS: Risperidone; EETI: Ethanol extract of T. ivorensis stem bark.
![Figure 6. Effect of EETI on MDA content in mouse brain. Value represents the mean ± S.E.M of five animals/group. One way ANOVA revealed that there is significant [F (5, 24) = 24.50, p < 0.0001] difference between various treatment groups. **Denotes p < 0.05 as compared to vehicle group. *Denotes p < 0.05 as compared with ketamine group. KET: Ketamine; RIS: Risperidone; EETI: Ethanol extract of T. ivorensis stem bark.](/cms/asset/27b15ac1-c2ac-4540-8630-599cbc886cfa/iphb_a_1190382_f0006.jpg)
Discussion
The study shows that the administration of EETI is able to reverse changes in some oxidative stress parameters induced by ketamine in mice. Additionally, these data reinforces ketamine-induced hyperlocomotion and cognitive dysfunction, as an important tool to study the behavioral and neurochemical alterations relevant to the pathophysiology of schizophrenia (Monte et al. Citation2013). Animal models are consistently used to investigate intracellular mechanisms and pharmacological approaches involved in psychiatric disorders (Behrens & Sejnowski Citation2009; Chatterjee et al. Citation2012). However, the complexity encompassing the wide arrays of symptoms including the so-called negative and cognitive symptoms of the biological features of schizophrenia makes it difficult to mirror important aspects of the disease (Geyer & Moghaddam Citation2002; Buckley & Stahl Citation2007). Herein, we demonstrated that EETI, was capable of reversing the behavioral alterations induced by the repeated administration of ketamine in a dose-dependent manner.
Behavioral alterations and symptoms of schizophrenia
In preclinical models, hyperlocomotion is a core behavior evaluated in schizophrenia. Indeed, several studies have shown that the administration of amphetamine or apomorphine mimics the hyperlocomotion and several other behavioral and biological aspects of schizophrenia (Bourin et al. Citation1986; Omogbiya et al. Citation2013). Recently, Behrens and Sejnowski (Citation2009) and Monte et al. (Citation2013) reported that the administration of a sub-anesthetic dose of ketamine induced behavioral alterations and modified oxidative stress parameters in the mice brain. These observations point to a good face validity of the model. In addition, the behavioral and biochemical effects were normalized by treatment with RIS, pointing to a predictive validity. Moreover, several clinical and pre-clinical studies demonstrated that glutamatergic system abnormalities are involved in the pathophysiology of schizophrenia, pointing to a possible construct validity of this model. Our work corroborates and extends these data suggesting that this protocol fulfills adequate characteristics as an animal model of schizophrenia and further reinforces previous proposals that both glutamatergic system (Krystal et al. Citation1994) and oxidative stress (Wood et al. Citation2009; Zhang et al. Citation2010) might be involved in the pathophysiology of schizophrenia, which parallel those described in postmortem brains of individuals with schizophrenia (De Oliveira et al. Citation2009). Based on the similarities to the pathophysiology of schizophrenia and the widespread use of this model (Becker & Grecksch Citation2004; Chatterjee et al. Citation2012), it was therefore chosen and adapted for the present study in order to assess the reversal treatment of ketamine-induced behavioral and oxidative alteration by EETI.
Pharmacological experiments have demonstrated that sub-anesthetic doses of ketamine induce schizophrenia-like symptoms in humans as well as behavioral activation in experimental animals (Lahti et al. Citation1995). Ketamine-induced hyperlocomotion have been partially attributed to the blockade of N-methyl-d-aspartate receptors (NMDARs). Indeed, the blockade of NMDARs located on inhibitory GABAergic neurons in the limbic and subcortical brain regions leads to an increase in neuronal activity in the limbic-striatal circuits through an increase in glutamate and dopamine release; these neurochemical events relate to the positive symptoms of schizophrenia (Lorrain et al. Citation2003; Chatterjee et al. Citation2012). Hyperactivity produced by ketamine at sub-anesthetic doses is closely related to the psychotic agitation seen in patients with mental abnormality (Swerdlow et al. Citation1994). The finding that the EETI significantly reversed the hyperlocomotion induced by ketamine in a dose-dependent manner, further suggests that it has antipsychotic-like property. Increased motor activity is a validated animal paradigm employed in the evaluation of compounds suspected to have antipsychotic property (Bourin et al. Citation1986; Arnt & Skarsfeldt Citation1998). Antipsychotic agents are known to diminish spontaneous motor activity (SMA) and reverse the increase in hyperactivity in motor functions induced by ketamine (Krystal et al. Citation1994; Chatterjee et al. Citation2011). It is well known that dopaminergic mechanism plays a central role in the mediation of locomotion activity, and ketamine may influence dopamine transmission and receptor activation via multiple mechanisms (Benturquia et al. Citation2008). It is important to stress that biochemical data have shown that ketamine enhances dopamine release (Irifune et al. Citation1991) and inhibits dopamine uptake (Chatterjee et al. Citation2012) in the straitum and cortex, respectively. It has therefore been suggested that ketamine may present an indirect dopamine agonist activity and ketamine-induced behavioral stimulation may be connected with the dopamine system (Irifune et al. Citation1991). Therefore, the reversal of hyperlocomotion by EETI in ketamine-treated animals is an indication that EETI may offer some beneficial effects in the treatment of psychosis probably by affecting the glutamate-dopamine pathway. Thus, these effects corroborate with our previous findings on the behavioral effects of EETI on schizophrenia from our preliminary study.
Importantly, this study also showed that the EETI significantly reversed ketamine-induced cognitive dysfunction (learning and memory) in the YMT, as an index of cognitive function, suggesting an effect of the extract against ketamine-induced deficits in working memory. The above mentioned changes in neurotransmitter systems are associated with blockade of NMDARs in the ventral tegmental area (VTA) which promotes a decrease in dopamine release in the prefrontal cortex (PFC), which may be partially responsible for the cognitive symptoms (Seamans & Yang Citation2004). Impairment of cognitive functions and information processing following chronic NMDA receptor antagonist (e.g., phencyclidine, ketamine) has been reported in several studies in different animal species (Jentsch & Roth Citation1999; Rujescu et al. Citation2006). Learning is associated with phosphorylation by calcium/calmodulin-dependent protein kinase II (Ca2+/CaMKII). Inhibition of NMDA receptor as a ligand-gated Ca2 + channel, decreases this phosphorylation, which may explain the deficits in cognitive function induced by ketamine due to disruption of long-term potentiation (LTP) (Mouri et al. Citation2007). Also, acetylcholine plays a critical role in the initial stages of memory formation (Hasselmo Citation2006). Ketamine is also being reported to act as an antagonist to nicotinic acetylcholine receptor alpha-7nAchR (Coates & Flood Citation2001). Acetylcholine via alpha-7nAchR regulates glutamate release, which in turn, is believed to activate NMDA receptor (Chatterjee et al. Citation2012). Consistent with these observations, it was also observed in this study that ketamine impaired cognitive function in the negative control groups treated with ketamine alone following the YMT evaluation. Therefore, the finding that ketamine induces cognitive dysfunction in mice, a typical characteristic symptom of schizophrenia, further support its relevance as an animal model predictive of psychosis (Mouri et al. Citation2007; Chatterjee et al. Citation2012). In this study, EETI was able to reverse the decrease in percentage of cognition induced by ketamine, producing a cognition-enhancing effect comparable to RIS (an atypical antipsychotic) in the experimental animals thus, suggesting the ability of this extract to ameliorate the impaired working memory, set shifting and other cognitive functions that are related to schizophrenia (Aultman & Moghaddam Citation2001).
Alterations in oxidative stress and its implication in schizophrenia
Oxidative stress is an important factor involved in the pathophysiology of major neuropsychiatric disorders, including schizophrenia (Wood et al. Citation2009; Do et al. Citation2009). Increased ROS levels generate deleterious effects on signal transduction, structural plasticity and cellular resilience, mostly by inducing lipid peroxidation in membranes, damage to proteins and nucleic acids (Mahadik et al. Citation2001). In this study, the beneficial effects of EETI on the reversal treatment of ketamine-induced behavioral alterations were accompanied by alterations in oxidative balance. Hence, the administration of EETI restored GSH, SOD and CAT levels, and also ameliorated ketamine-induced lipid peroxidation. In fact, GSH is involved in the pathophysiology of schizophrenia (Wood et al. Citation2009; Zhang et al. Citation2010), and possibly together with SOD, GSH peroxidase, may work as indicators of severity of schizophrenia in acute episode (Pazvantoglu et al. Citation2009). Interestingly, resperidone displayed a great increase in GSH, SOD and CAT levels. Of note, a recent study demonstrated that resperidone treatment restored GSH levels and to a great extent reversed antioxidant defense system (Monte et al. Citation2013).
The correction of ketamine-induced oxidative imbalance by EETI administration as reported in this study may be an important feature related to the benefits of this extract in the experimental animals. This is reinforced by the fact that an imbalance in redox-state of the brain is part of the underlying pathophysiology in schizophrenia (Behrens & Sejnowski Citation2009). In this context, it was previously reported that the repetitive exposure of adult rodents to ketamine was able to increase the level of the proinflammatory cytokine interleukine-6 (IL-6) in the brain (Behrens et al. Citation2008) through depolarization increased neuronal production of cytokines specifically, IL-6 (Sallmann et al. Citation2000; Behrens et al. Citation2008), leading to activation of the superoxide producing enzyme NADPH oxidase-2 (Nox-2) (Sorce et al. Citation2010), which in turn leads to an enduring decrease of inhibitory tone (inhibitory postsynaptic potential, IPSP) in PFC (Zhang et al., Citation2008), via the loss of GABAergic phenotype of parvalbumin (PV)-interneurons in the PFC by ROS (superoxide radicals) (Behrens et al. Citation2008). The increased superoxide production caused by activation of the IL-6/Nox-2 pathway by ketamine, causes redox inactivation of the excitatory amino acid carrier-1 (EAAC1) and a decrease in cysteine transport, leading to diminishing GSH content in brain (Aoyama et al. Citation2006; Behrens et al. Citation2008). Importantly in this study, the oxidative alterations induced by ketamine were observed in the whole brain homogenates studied from biochemical assays. Indeed, cognitive symptoms of this mental disorder have been related to microglial activation and the resulting inflammatory response (Monji et al. Citation2009). Therefore, the anti-inflammatory (Iwu & Anyawu Citation1982) and antioxidant (Ponou et al. Citation2010; Adiko et al. Citation2013) properties of the stem bark of the plant may at least in part, explain its antioxidant properties observed herein.
Another important link between NMDA hypofunction and GSH comes from the study of mismatch negativity (MMN) (Javitt et al. Citation1998). MMN is an auditory evoked potential that is elicited by deviant stimuli in an otherwise unchanging set of events (Wood et al. Citation2009), which is known to be blocked by ketamine through the blockade of event-related potentials (ERPs) generators (‘Echoic’) (Umbricht et al. Citation2000). MMN amplitude is attenuated in schizophrenia (Turetsky et al. Citation2007), leading to the possibility that increasing brain levels of GSH in schizophrenia wound improve MMN generation and other brain functions (Steullet et al. Citation2006), which serves as an index of improving cognitive functions in schizophrenia (Umbricht et al. Citation2000; Lavoie et al. Citation2008; Bitanihirwe et al. Citation2011).
Mechanistically, GSH can potentiate the activity of redox-sensitive proteins such as NMDA and dopamine receptors (Janaky et al. Citation2007) thus, modulating neurotransmitter functions of glutamatergic and dopaminergic pathways (Himi et al. Citation2003), via neuronal intra- and extra-cellular exchange of cysteine and glutamate through the cysteine-glutamate antiporter using the excitatory amino acid carrier-1 (EAAC1) (Aoyama et al. Citation2006; Behrens & Sejnowski Citation2009), since cysteine is the rate-limiting precursor for GSH synthesis (Dodd et al. Citation2008). Through this mechanism, GSH appears to modulate the binding of glutamate to NMDA receptors (Oja et al. Citation2000), or potentiate brain NMDA receptor response to glutamate in rats (Varga et al. Citation1997). Therefore, the increase in GSH levels and other antioxidants including SOD and CAT, and the resultant decrease of the consequence of oxidative stress (lipid peroxidation), as indexed by the decrease in MDA levels in the various treatment groups treated with EETI, at least in part, could explain the beneficial effects of the antipsychotic properties demonstrated by this extract in this study.
These beneficial effects of this plant is probably due to the presence of some phytochemical antioxidant constituents, contained in the plant such as flavonoids (e.g., polyphenols), saponins (triterpenoid ivorenosides A, B and C), tannins, betulinic, oleanolic acid, steroids, terminolic, ellergic, glycirrhetic acids, quarcetin, anthraquinones, sericic acid, glycosides steroids, punicalagin and punicalin (Iwu & Anyawu Citation1982; Adiko et al. Citation2013). Many of these constituents have been shown to readily cross the blood brain barrier (BBB), to exert central nervous system (CNS) activities, which includes anti-neuroinflammatory, neuroprotective, chemoprevention, antioxidation (Zijia et al. Citation2009). Flavonoids are well recognized for their antioxidant, tranquilizing and anxiolytic activities (Aguirre-Hernandez et al. Citation2010). They are known to inhibit the activity of several enzymes, including lipoxygenase, cyclooxygenase, monoxygenase, xanthine oxidase, mitochondrial succino-oxidase, nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and phospolipase A2, many which are believed to play a role in the pathophysiology of schizophrenia (Singh et al. Citation2004). Many of the biological activity of flavonoids are attributed to their antioxidant properties and free radical scavenging capability (Ponou et al. Citation2010). Therefore, the presence of these phytochemicals might be contributing to the antipsychotic activity of EETI in this study and to the tranquilizing properties of this plant used ethno-medicinally in the south-western parts of Nigeria in the management of psychosis (Lawal et al. Citation2010).
Conclusion
The study shows that ethanol stem bark extracts of T. ivorensis possess bioactive compounds that have antipsychotic potential, probably acting via a neuroprotective (antioxidant) property as its compensatory mechanism of action. The findings thus provide valuable scientific support for its traditional claims in the management of psychosis.
Acknowledgements
The authors are thankful to Mr. Olusegun Adebayo Adeoluwa of the department of Pharmacology and Therapeutics, College of Medicine and Health Sciences, Afe Babalola University, Ado Ekiti, Ekiti State, Nigeria for his commitment in the preparation of ethanol stem bark extracts of Terminalia ivorensis for the research work.
Disclosure statement
The authors report no declarations of interest.
References
- Adiko VA, Barthelemy KA, Félix ZT, Kouamé MA, Coulibali S, Léon AD. 2013. Separation and characterization of phenolic compounds from Terminalia ivoiriensis using liquid chromatography-positive electrospray ionization tandem mass spectroscopy. Afr J Biotechnol. 12:4393–4398.
- Agbedahunsi JM, IvieAnao CO, Clement OA, Simon LC. 2006. Trypanocidal properties of terminalia ivorensis. Chevalier (Combretaceae). Afr J Tradit Complement Altern Med. 3:51–56.
- Aguirre-Hernandez E, Gonzalez-Trujano ME, Martinez AL, Moreno J, Kite G, Terrazas T, Soto-Hernandez M. 2010. HPLC/MS and anxiolytic-like effect of quercetin and kaemferol flavonoids from Tilia americana var. maxicana. J Ethnopharmacol. 127:91–97.
- Andreasen NC. 1990. Methods for assessing positive and negative symptoms of schizophrenia. Mod Probl Pharmacopsychiatry. 24:73–88.
- Aoyama K, Suh SW, Hamby AM, Liu J, Chan WY, Chen Y, Swanson RA. 2006. Neuronal glutathione deficiency and age-dependent neurodegeneration in the EAAC1 deficient mouse. Nat Neurosci. 9:119–126.
- Arnt J, Skarsfeldt T. 1998. Do novel antipsychotics have similar pharmacological characteristics? A review of the evidence. Neuropsychopharmacology. 18:63–101.
- Aultman JM, Moghaddam B. 2001. Distinct contributions of glutamate and dopamine receptors to temporal aspects of rodent working memory using a clinically relevant task. Psychopharmacology (Berl). 153:353–364.
- Becker A, Grecksch G. 2004. Ketamine-induced changes in rat behaviour: a possible animal model of schizophrenia. Test of predictive validity. Prog Neuropsychopharmacol Biol Psychiatry. 28:1267–1277.
- Behrens MM, Ali SS, Dugan LL. 2008. Interleukin-6 mediates the increase in NADPH-oxidase in the ketamine model of schizophrenia. J Neurosci. 28:13957–13966.
- Behrens MM, Sejnowski TJ. 2009. Does schizophrenia arise from oxidative dysregulation of parvalbumininterneurons in the developing cortex? Neuropharmacol. 57:193–200.
- Benturquia N, Courtin C, Noble F, Marie-Claire C. 2008. Involvement of D1 dopamine receptor in MDMA-induced locomotor activity and striatal gene expression in mice. Brain Res.1211:1–5.
- Bitanihirwe KY, Tsung-Ung W, Woo MD. 2011. Oxidative stress in schizophrenia: An integrated approach. Neurosci Biobehav Rev. 35:878–893.
- Bókkon I, Antal I. 2011. Schizophrenia: redox regulation and volume neurotransmission. Curr Neuropharmacol. 9:289–300.
- Bourin M, Poisson L, Larousse C. 1986. Piracetam interaction with neuroleptics in psychopharmacological tests. Neuropsychobiology. 19:93–96.
- Buckley PF, Stahl SM. 2007. Pharmacological treatment of negative symptoms of schizophrenia: therapeutic opportunity or cul-de-sac? Acta Psychiatr Scand. 115:93–100.
- Chatterjee M, Ganguly S, Srivastava M, Palit G. 2011. Effect of ‘chronic’ versus ‘acute’ ketamine administration and its ‘withdrawal’ effect on behavioural alterations in mice: implications for experimental psychosis. Behav Brain Res. 216:247–254.
- Chatterjee M, Rajkumar V, Surajit G, Gautam P. 2012. Neurochemical and molecular characterization of ketamine-induced experimental psychosis model in mice. Neuropharmacology. 63:1161–1171.
- Coates KM, Flood P. 2001. Ketamine and its preservative, benzethonium chloride, both inhibit human recombinant alpha7 and alpha4beta2 neuronal nicotinic acetylcholine receptors in Xenopus oocytes. Br J Pharmacol. 134:871–879.
- Coulibaly K, Zirihi GN, N'Guessan K, Djaman AJ. 2006. Botanical study and evaluation of antifungal activity of Terminalia ivorensis and Terminalia superba (Combretaceae) on the growth in vitro of Aspergillus fumigatus. Int J Ph Sci. 2:904–912.
- Dall’igna OP, Fett P, Gomes MW. 2007. Caffeine and adenosine A(2a) receptor antagonists prevent beta-amyloid (25-35)-induced cognitive deficits in mice . Exp Neurol. 203:241–245.
- De Oliveira L, Spiazzi CM, Bortolin T. 2009. Different sub-anesthetic doses of ketamine increase oxidative stress in the brain of rats. Prog Neuropsychopharmacol Biol Psychiatry. 33:1003–1008.
- Do KQ, Cabungcal JH, Frank A, Steullet P, Cuenod M. 2009. Redox dysregulation, neurodevelopment, and schizophrenia. Curr Opin Neurobiol. 19:220–230.
- Dodd S, Dean O, Copolov DL, Malhi GS, Berk M. 2008. N-Acetylcysteine for antioxidant therapy: pharmacology and clinical utility. Expert Opin Biol Ther. 8:1955–1962.
- Foli EG. (2009). Terminalia ivorensis A. Chevalier. In: Lemmens RHMJ, Louppe D, OtengAmoako A, editors. Prota 7(2): Timbers/Bois d’œuvre 2. [CD-Rom]. PROTA, Wageningen: Netherlands.
- Freedman R. 2003. Schizophrenia. N Engl J Med. 349:1738–1749.
- Geyer MA, Moghaddam B. 2002. Animal models relevant to schizophrenia disorders. Neuropsychopharmacology. 50:690–701.
- Gornall AG, Bardawill CJ, David MM. 1949. Determination of serum proteins by means of the biuret reaction. J Biol Chem. 177:751–766.
- Halliwell B. 2006. Oxidative stress and neurodegeneration: where are we now? J Neurochem. 97:1634–1658.
- Hasselmo ME. 2006. The role of acetylcholine in learning and memory. Curr Opin Neurobiol. 16:710–715.
- Himi T, Ikeda M, Yasuhara T. 2003. Oxidative neuronal death caused by glutamate uptake inhibition in cultured hippocampal neurons. J Neurosci Res. 71:679–688.
- Ikonomidou C, Kaindl AM. 2011. Neuronal death and oxidative stress in the developing brain. Antioxid Redox Signal. 14:1535–1550.
- Irifune M, Shimizu T, Nomoto M. 1991. Ketamine-induced hyperlocomotion associated with alteration of presynaptic component of dopamine neurons in the nucleus accumbens of mice. Pharmacol Biochem Behav. 40:399–407.
- Iwu MM, Anyawu BN. 1982. Anti-inflammatory and anti-arthritic properties of Terminalia ivorensis. Fitoterapia. 52:25–34.
- Janaky R, Dohovics R, Saransaari P. 2007. Modulation of [3H]dopamine release by glutathione in mouse striatal slices. Neurochem Res. 32:1357–1364.
- Javitt DC, Grochowski S, Shelley AM, Ritter W. 1998. Impaired mismatch negativity (MMN) generation in schizophrenia as a function of stimulus deviance, probability, and interstimulus/interdeviant interval. Electroencephalogr Clin Neurophysiol. 108:143–153.
- Jentsch JD, Roth RH. 1999. The neuropsychopharmacology of phencyclidine: from NMDA receptor hypofunction to the dopamine hypothesis of schizophrenia. Neuropsychopharmacology. 20:201–225.
- Jollow DJ, Michell JR, Zampaglione N, Gillete J. 1974. Bromobenzene-induced liver necrosis. Protective role of glutathione an evidence for 3,4-bromobenzene oxide as the hepatotoxic metabolite. Pharmacology. 11:151–169.
- Krystal JH, Karper LP, Seibyl JP, Freeman GK, Delaney R, Bremner JD. 1994. Subanaesthetic effects of the non-competitive NMDA antagonist, ketamine, in humans: psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 51:199–214.
- Lahti AC, Koffel B, LaPorte D, Tamminga CA. 1995. Subanesthetic doses of ketamine stimulate psychosis in schizophrenia. Neuropsychopharmacology. 13:9–19.
- Larson MK, Walker EF, Compton MT. 2010. Early signs, diagnosis and therapeutics of the prodromal phase of schizophrenia and related psychotic disorders. Expert Rev Neurother. 10:1347–1359.
- Lavoie S, Murray MM, Deppen P. 2008. Glutathione precursor, N-acetyl-cysteine, improves mismatch negativity in schizophrenia patients. Neuropsychopharmacology. 33:2187–2199.
- Lawal IO, Uzokwe NE, Igboanugo AB, Adio AF, Awosan EA, Nwogwugwu JO. 2010. Ethno-medicinal information on collation and identification of some medicinal plants in Research Institutes of South-west Nigeria. Afr J Pharm Pharmacol. 4:001–007.
- Lorke D. 1983. A new approach to practical acute toxicity testing. Arch Toxicol. 54:275–287.
- Lorrain DS, Baccei CS, Bristow LJ. 2003. Effects of ketamine and N-methyl-d-aspartate on glutamate and dopamine release in the rat prefrontal cortex: modulation by a group II selective metabotropicglutamate receptor agonist LY379268. Neuroscience. 117:697–706.
- Mahadik SP, Evans D, Lal H. 2001. Oxidative stress and role of antioxidant and omega-3 essential fatty acid supplementation in schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 25:463–493.
- Malcom SA, Sofowora EA. 1969. Antimicrobial activity of selected Niegerian folk remedies and their constituent plants. Lloydia. 32:512–517.
- Misra HP, Fridovich I. 1972. The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J Biol Chem. 247:3170–3175.
- Monji A, Kato T, Kanba S. 2009. Cytokines and schizophrenia: microglia hypothesis of schizophrenia. Psychiatry Clin Neurosci. 63:257–265.
- Monte AS, Greicy CS, Roger SM, Joanna KS, Júnia VS, Rafaela CC. 2013. Prevention and reversal of ketamine-induced schizophrenia related behavior by minocycline in mice: possible involvement of antioxidant and nitrergic pathway. J Psychopharmacol. 11:1032–1043.
- Mouri A, Noda Y, Noda A, Nakamura T, Tokura T, Yura Y, Nitta A, Furukawa H, Nabeshima T. 2007. Involvement of a dysfunctional sopamine-D1/N-methyl-d-aspartate- NR1 and Ca2+/calmodulin-dependent protein kinase II pathway in the impairment of latent learning in a model of schizophrenia induced by phencyclidine. Mol Pharmacol. 71:1598–1609.
- Oja SS, Janáky R, Varga V, Saransaari P. 2000. Modulation of glutamate receptor functions by glutathione. Neurochem Int. 37:299–306.
- Okhawa H, Ohishi N, Yagi K. 1979. Assay for lipid peroxides in animal tissues by thiobarbituric acid reaction. Anal Biochem. 95:351–358.
- Omogbiya IA, Solomon U, Aderibigbe AO, Adewale GB. 2013. Jobelyn pretreatment ameliorates symptoms of psychosis in experimental models. J Basic Clin Physiol Pharmacol. 24:331–336.
- Pavlovic D, Tamburic V, Stojanovic I. 2002. Oxidative stress as marker of positive symptoms in schizophrenia. Facta Univ. 9:157–161.
- Pazvantoglu O, Selek S, Okay IT. 2009. Oxidative mechanisms in schizophrenia and their relationship with illness subtype and symptom profile. Psychiatry Clin Neurosci. 63:693–700.
- Picchioni MM, Murray RM. 2007. Schizophrenia. BMJ. 335:91–95.
- Ponou BK, Teponno RB, Ricciutelli M, Quassinti L, Bramucci M, Lupidi G, Barboni L, Tapondjou LA. 2010. Dimeric antioxidant and cytotoxic triterpenoid saponins from Terminalia ivorensis A. Chev. Phytochemistry. 71:2108–2115.
- Rujescu D, Bender A, Hartmann AM, Ohl F, Raeder H. 2006. A pharmacological model of psychosis based on N methyl-d-aspartate receptor hypofunction: molecular, cellular, functional and behavioral abnormalities. Biol Psychiatry. 59:721–729.
- Sallmann S, Juttler E, Prinz S, Petersen N, Knopf U, Weiser T, Schwaninger M. 2000. Induction of interleukin-6 by depolarization of neurons. J Neurosci. 20:8637–8642.
- Seamans JK, Yang CR. 2004. The principal features and mechanisms of dopamine modulation in the prefrontal cortex. Prog Neurobiol. 74:1–58.
- Singh RP, Sharad S, Kapur S. 2004. Free radicals and oxidative stress in neurodegenerative diseases: relevance of dietary antioxidants. JIACM. 5:218–225.
- Sinha KA. 1971. Colorimetric assay of catalase. Anal Biochem. 47:389–394.
- Sorce S, Schiavone S, Tucci P, Colaianna M, Jaquet V, Cuomo V, Dubois-Dauphin M, Trabace L, Krause KH. 2010. The NADPH oxidase NOX2 controls glutamate release: a novel mechanism involved in psychosis-like ketamine responses. J Neurosci. 30:11317–11325.
- Steullet P, Neijt HC, Cuénod M, Do KQ. 2006. Synaptic plasticity impairment and hypofunction of NMDA receptors induced by glutathione deficit: relevance to schizophrenia. Neuroscience. 137:807–819.
- Swerdlow NR, Braff DL, Taaid N, Geyer MA. 1994. Assessing the validity of an animal model of deficient sensorimotor gating in schizophrenic patients. Arch Gen Psychiatry. 51:139–154.
- Turetsky BI, Calkins ME, Light GA, Olincy A, Radant AD, Swerdlow NR. 2007. Neurophysiological endophenotypes of schizophrenia: the viability of selected candidate measures. Schizophr Bull. 33:69–94.
- Umbricht D, Schmid L, Koller R, Vollenweider FX, Hell D, Javitt DC. 2000. Ketamine-induced deficits in auditory and visual context-dependent processing in healthy volunteers: Implications for models of cognitive deficits in schizophrenia. Arch Gen Psychiatry. 57:1139–1147.
- Valko M, Leibfritz D, Moncol J, Cronin MT, Mazur M, Telser J. 2007. Free radicals and antioxidants in normal physiological functions and human disease. Int J Biochem Cell Biol. 39:44–84.
- Varga V, Jenei Z, Janaky R. 1997. Glutathione is an endogenous ligand of rat brain N-methyl-a-aspartate (NMDA) and 2-amino-3-hydroxy-5-methyl-4-isoxazolepropionate (AMPA) receptors. Neurochem Res. 22:1165–1171.
- Wang Y, Zhang H, Yuan Q. 2013. Purification, antioxidant activity and protein-precipitating capacity of punicalin from pomegranate husk. Food Chem. 138:437–443.
- Wood SJ, Yücel M, Pantelis C, Berk M. 2009. Neurobiology of schizophrenia spectrum disorders: the role of oxidative stress. Ann Acad Med Singapore. 38:396–396.
- Yamada K, Noda Y, Hasegawa T, Komori Y, Nikai T, Sugihara H, Nabeshima T. 1996. The role of nitric oxide in dizocilpine-induced impairment of spontaneous alternation behavior in mice. J Pharm Exp Ther. 276:460–466.
- Zhang Y, Behrens MM, Lisman JE. 2008. Prolonged exposure to NMDAR antagonist suppresses inhibitory synaptic transmission in prefrontal cortex. J Neurophysiol. 100:959–965.
- Zhang M, Zhao Z, He L, Wan C. 2010. A meta-analysis of oxidative stress markers in schizophrenia. Sci China Life Sci. 53:112–124.
- Zijia Z, Liping L, Jeffrey M, Tao W, Zhengtao W. 2009. Antioxidant phenolic compounds from walnut kernels (Juglansregia L). Food Chem. 113:160–165.