
Abstract
Context: Losartan and berberine (BBR) are often simultaneously used for the treatment of senile diabetic nephropathy in clinics. However, the potential herb–drug interaction between losartan and BBR is unknown.
Objective: This study investigates the influence of BBR on the pharmacokinetics of losartan and EXP3174, and investigates the effects of BBR on the metabolic stability of losartan.
Materials and methods: The pharmacokinetic profiles losartan and EXP3174 of orally administered losartan (10 mg/kg) with and without pretreatment with BBR (20 mg/kg) within 24 h were determined in Sprague-Dawley rats. The inhibitory effects of BBR on the metabolic stability of losartan were investigated using rat liver microsomes.
Results: The Cmax (1.26 ± 0.37 versus 1.96 ± 0.45 mg/L) and the AUC(0–t) (8.25 ± 0.89 versus 12.70 ± 1.42 mg h/L) of losartan were significantly (p < 0.05) increased by BBR compared to the control, while the Cmax (0.97 ± 0.15 versus 0.77 ± 0.06 mg/L) of EXP3174 was significantly decreased compared to the control (p < 0.05). The Tmax of losartan was prolonged from 0.41 ± 0.12 to 0.52 ± 0.18 h, but the difference was not significant. However, the Tmax of EXP3174 was decreased significantly (p < 0.05) from 8.14 ± 0.36 to 3.33 ± 0.28 h. The metabolic stability of losartan was increased from 37.4 to 59.6 min.
Discussion and conclusion: We infer that BBR might increase the plasma concentration of losartan and decrease the concentration of EXP3174 through inhibiting the activity of CYP3A4 or CYP2C9.
Keywords:
Introduction
Losartan is the first nonpeptide angiotensin II receptor blocker used for the treatment of hypertension (Klishadi et al. Citation2015). After oral administration, losartan can be absorbed quickly and transformed in to its active metabolite EXP3174 mediated by cytochrome P450 enzymes CYP3A4 and CYP2C9, which is about 10-fold more potent than its parent drug (Rincon et al. Citation2015). Thus, the clinical hypotensive activity is predominantly mediated by the active metabolite EXP3174, although losartan itself exhibits good efficacy (Varshney et al. Citation2013). Because of its good anti-hypertension effect, losartan has been one of the anti-hypertension drugs most frequently used for the prevention and control of hypertension in the clinic (Yasar et al. Citation2008; Yang et al. Citation2011). As we know, losartan is also a substrate of P-glycoprotein (P-gp) (Yang et al. Citation2011). Considering that losartan is a substrate of both CYP enzymes and P-gp, modulation of CYP and P-gp activities may cause significant changes in the pharmacokinetic profiles of losartan and its active metabolite EXP3174.
Berberine (BBR) is an important and typical constituent in Rhizoma coptidis (Coptis chinensis Franch. [Ranunculaceae]), which possesses a variety of activities including anti-tumour, anti-inflammation, anti-atherosclerosis, and treating infectious diarrhea (Orvos et al. Citation2015; Park et al. Citation2015). Recent studies show that BBR could be used for the controlling arrhythmia, lowering blood lipid, lowering blood pressure and reducing blood sugar in clinical. It could also effectively promote regeneration in islet cells and contributes to recovery of islet function (Cui et al. Citation2009; Shen et al. Citation2012). Therefore, BBR was widely used for the treatment of diabetes, hyperlipidemia and hypertension (Pang et al. Citation2015). Losartan and BBR are often simultaneously used for the treatment of senile diabetic nephropathy in the clinics of China. However, many herb–drug interactions resulting from concurrent use of herbal drugs with over-the-counter drugs may cause adverse reactions such as toxicity and treatment failure.
Therefore, it is an urgent need to investigate the potential herb–drug interaction between losartan and BBR to avoid the occurrence of adverse reactions. To the best of our knowledge, there are little data available for the pharmacokinetic interaction between BBR and losartan. The aim of this study was to develop a simple and reliable LC–MS method for the determination of losartan and EXP3174 in rat plasma, compare the pharmacokinetics of losartan and EXP3174 after oral administration of single losartan and both losartan and BBR, investigate the inhibitory effects of BBR on the metabolic stability of losartan, and guide clinical medication of losartan and BBR to avoid the occurrence of adverse reactions.
Materials and methods
Chemicals and reagents
Standards of losartan, BBR hydrochloride and irbesartan as an internal standard (IS) were purchased from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). Losartan carboxylic acid (EXP3174) was purchased from Toronto Research Chemicals Inc., Toronto, Canada. The chemical structures of losartan, EXP3174 and irbesartan are shown in . d-Glucose-6-phosphate, glucose-6-phosphate dehydrogenase and NADP+ were obtained from Sigma Chemical Co. (St. Louis, MO). Pooled RLM were purchased from BD Biosciences Discovery Labware (Woburn, MA). Acetonitrile and methanol were purchased from Fisher Scientific (Fair Lawn, NJ). Formic acid was purchased from Anaqua Chemicals Supply Inc. Limited (Houston, TX). Ultrapure water was prepared with a Milli-Q water purification system (Millipore, Billerica, MA). All other chemicals were of analytical grade or better.
Figure 1. The chemical structure of losartan (A), irbesartan (B) and EXP3174 (C).
Instrumentation and conditions
An Agilent 1100 liquid chromatography–mass spectrometer was used in the study. The LC system consisted of a micro-degasser, a binary pump, a high-performance well-plate autosampler and a thermostated column compartment. An Agilent G1946D mass spectrometer (Agilent) equipped with an electrospray source was connected to the LC system. The same LC separation conditions were used for LC–MS. Data acquisition and analysis were performed using Agilent ChemStation for LC/MSD version B.02.01. The sample was separated on Waters Xbridge C18 column (100 mm ×3.0 mm, i.d.; 3.5 mm) and eluted with an isocratic mobile phase: solvent A (H2O–HCOOH, 100:0.1, v/v) and solvent B (CH3OH, 55:45, v/v). The total analysis time was 8 min. The column temperature was 25 °C at a flow rate of 0.8 mL/min and injection volume of 5 μL, and the split ratio was 1:1.
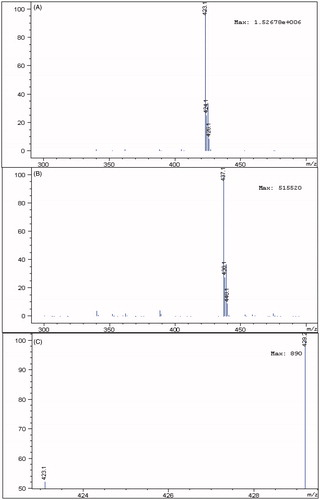
The mass conditions were optimized as follows: capillary 4000 V, nebulizer pressure 40 psig, drying gas flowing rate 10 L/min, gas temperature 350 °C and fragmentor 80 eV. Select-ion-monitoring (SIM) in the positive ion mode was used. The ions of losartan, EXP3174 and irbesartan were recorded as m/z 423.1, m/z 437.1 and m/z 429.2 for the quantification, respectively.
Sample preparation
To 100 μL aliquot of a plasma sample, 20 μL methanol and 180 μL internal standard methanol solution (0.2 μg/mL) were added and vortexed for 30 s to mix in a 1.5 mL polypropylene tube, and then centrifuged at 12,000 rpm for 10 min. The supernatant was removed into an injection vial and a 5 μL aliquot was injected into the LC–MS system for analysis.
Preparation of calibration standards, internal standard and quality control
The stock solution of losartan and EXP3174 was prepared in methanol at a concentration of 10 mg/mL. The internal standard solution was prepared in acetonitrile at a concentration of 0.2 μg/mL. Calibration standard samples for losartan and EXP3174 were prepared in blank rat plasma at concentrations of 0.01–2 μg/mL and 0.02–5 μg/mL, respectively. The quality control (QC) samples for losartan were prepared at low (20 ng/mL), medium (200 ng/mL) and high (1000 ng/mL) concentrations in the same way as the plasma samples for calibration, and QC samples for EXP3174 were prepared at low (50 ng/mL), medium (200 ng/mL) and high (2000 ng/mL) concentrations. The QC samples were stored at −40 °C until analysis.
Method validation
The method validation assays were performed according to the United States Food and Drug Administration (FDA) guidelines. Selectivity was investigated by comparing the chromatograms of six different batches of blank rat plasma with the corresponding spiked plasma to exclude interference of endogenous substances and metabolites.
The linearity of each calibration curve was determined by plotting the peak area ratio (y) of analytes to IS versus the nominal concentration (x) of analytes with weighted (1/x2) least square linear regression. The lower limit of quantification (LLOQ) was determined as the concentration of the analyte with a signal-to-noise ratio at 10 with acceptable accuracy and precision (≤20%).
The intra-day precision and accuracy of the method were assessed by determining the QC samples five times on a single day, and the inter-day precision and accuracy were estimated by determining the QC samples over three consecutive days. Relative standard deviation (RSD) and relative error (RE) were used to express the precision and accuracy, respectively.
The extraction recovery was evaluated by comparing peak areas obtained from extracted spiked samples with those of the post-extracted spiked samples. The matrix effect was evaluated by comparing the peak areas of the post-extracted spiked QC samples with those of corresponding standard solutions. These procedures were repeated for five replicates at three QC concentration levels.
Stability was examined by analysing replicates (n = 3) of the three levels of QC samples under different conditions. Short-term stability was evaluated after the exposure of QC samples to room temperature for 4 h. Post-preparative stability was conducted by reanalysing the QC samples after 24 h in the autosampler at ambient temperature. For freeze/thaw stability, the plasma samples were determined through three freeze (−40 °C)–thaw (room temperature) cycles. Long-term stability was performed by assessing the plasma samples after storage at −40 °C for 15 days.
Pharmacokinetic study
The animal facilities and protocols were approved by the Institutional Animal Care and Use Committee. Male Sprague-Dawley rats weighing 250 ± 20 g were supplied by Sino-British Sippr/BK Lab Animal Ltd. (Shanghai, China). The rats were maintained in an air-conditioned animal quarter at 22 ± 2 °C and 50 ± 10% relative humidity. Water and food (laboratory rodent chow, Shanghai, China) were allowed ad libitum. The animals were acclimatized to the facilities for 5 days, and then fasted with free access to water for 12 h prior to each experiment. Twelve rats were equally randomized to two groups (six rats in each group), including the losartan-only group (A) and the losartan and BBR group (B). The losartan and BBR fraction powders were all homogenized in 1.5% Tween 80 aqueous solution with a mortar and pestle. Animals in group A were administered according the oral dose of 10 mg/kg; and animals in group B were administered at an oral dose of BBR (20 mg/kg) and losartan (10 mg/kg). Blood samples (0.25 mL) were collected into a heparinized tube via the oculi chorioideae vein before drug administration and at 0.083, 0.25, 0.5, 1, 2, 3, 4, 6, 8, 12 and 24 h after drug administration. After centrifuge at 5000 rpm for 5 min, the supernatant was obtained and frozen at −40 °C until analysis.
Inhibitory effects of BBR on the metabolic stability of losartan
Rat liver microsomes were used to determine the phase I metabolic rate of losartan. The assay conditions and reaction mixtures were similar as reported previously (Qi et al. Citation2013, Citation2014). The reaction mixture was incubated at 37 °C for 5 min and then losartan (100 μM) was added. The effects of BBR on the metabolic rate of losartan were investigated by adding 100 μM of BBR to rat liver microsomes and preincubating for 30 min at 37 °C, and then losartan (100 μM) was added. Aliquots of 30 μL were collected from reaction volumes at 0, 1, 3, 5, 15, 30 and 60 min and 60 μL ice-cold acetonitrile containing IS was added to terminate the reaction, and then the sample preparation method was the same as the plasma sample preparation method and determined by LC–MS. The half-life (t1/2) in vitro was obtained using equation: t1/2 = 0.693/k.
Results
LC–MS optimization
Because of the complexity of plasma samples, there may be many endogenous components co-eluted with the analytes. In order to develop a sensitive and accurate LC–MS method for determination of losartan and its metabolite EXP3174 in rat plasma, the quantification analysis was performed using SIM mode owing to its high selectivity and sensitivity. Formic acid (0.1%) was added into the mobile phase to obtain better separation and increase the response of losartan and EXP3174 in the positive mode. As shown in , abundant protonated molecular ions after electrospray ionization of losartan m/z 423.1, EXP3174 m/z 437.1 and irbesartan m/z 429.2 were selected for MS detection.
Figure 2. The mass spectra of losartan (A), EXP3174 (B) and irbesartan (C).
Method validation
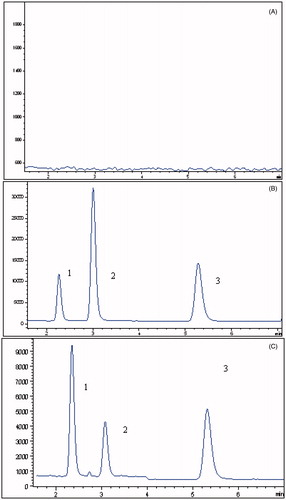
The representative chromatograms in SIM mode of losartan, EXP3174 and irbesartan are shown in . The analysis of blank rat plasma samples did not show any interference at the retention times of losartan, EXP3174 and irbesartan.
Figure 3. (A) Representative chromatograms of blank plasma; (B) blank plasma spiked with losartan (2), EXP3174 (3) and irbesartan (1); (C) rat plasma treated with BBR and losartan.
Linearity for determining losartan and EXP3174 in spiked rat plasma was prepared by eight calibration standards in five independent runs. The calibration curves were obtained with correlation coefficients (r) more than 0.999 between 0.01 and 2.00 μg/mL of losartan and between 0.02 and 5.00 μg/mL of EXP3174. The LLOQ was set at 0.01 μg/mL for losartan and 0.02 μg/mL for EXP3174 in rat plasma. The signal-to-noise ratio was >10 and the deviation was no more than 15% (n = 6).
As shown in , the precision of this method was no more than 10% RSDs for losartan and EXP3174, and the accuracy ranged from −7.10% to 9.64% for losartan and from −8.95% to 6.25% for EXP3174.
Table 1. The precision and accuracy of losartan and EXP3174 in plasma samples.
The matrix effect was examined to assess the possibility of ionization suppression or enhancement. The matrix effect ranged from 90.2% to 97.6% for losartan and EXP3174 over the three levels of QC samples. The results indicated that no obvious matrix effect was observed. The overall mean recoveries of losartan and EXP3174 in plasma at three different concentration levels were found to be 82.4–93.1% with RSD less than 10%, which indicated that the extraction procedure was consistent and reproducible.
Results of short-term stability, postpreparative stability, freeze and thaw stability and long-term stability are shown in . This result showed that the samples were stable during the routine analysis for the pharmacokinetic study of losartan and EXP3174.
Table 2. Stability of losartan and EXP3174 in plasma samples (n = 3).
Application of the method to pharmacokinetic study
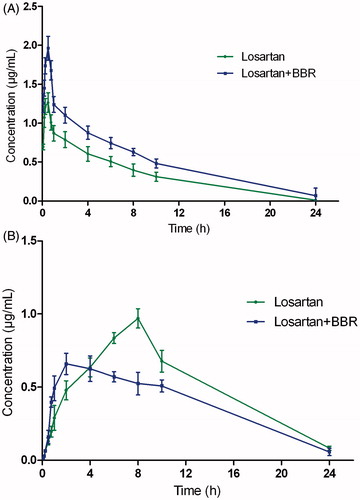
The analytical procedures described were used to quantify losartan and its metabolite EXP3174 in rat plasma samples obtained from 12 male Sprague-Dawley rats, of which six were orally administered losartan aqueous solution. The other six were orally administered losartan and BBR aqueous solution. The mean concentration–time curve of losartan and EXP3174 in the two treatments is shown in .
Figure 4. The mean concentration–time curves in rat plasma after oral administration of single losartan and both losartan and BBR, (A) losartan and (B) EXP3174.
The pharmacokinetic parameters of losartan and EXP3174 were calculated using the noncompartmental method with DAS 3.0 pharmacokinetic software (Chinese Pharmacological Association, Anhui, China). The pharmacokinetic parameters are shown in .
Table 3. Pharmacokinetic parameters of losartan and EXP3174 in male Sprague-Dawley rats following oral administration of losartan alone and both losartan and BBR.
As shown in , the parameters Cmax and AUC(0–t) for losartan in the losartan and BBR (B) group were larger than those of losartan-only group (A), and the difference was significant (p < 0.05), and the Cmax and AUC(0–t) increased approximately 55.5% and 53.9%, respectively; However, Cmax and AUC(0–t) for EXP3174 in the losartan and BBR (B) group were smaller than those of losartan-only group (A), and the difference was also significant (p < 0.05). The results indicated that the plasma concentration of losartan increased in losartan and BBR group compared with the single losartan group, and however, the plasma concentration of EXP3174 decreased.
Inhibitory effects of BBR on the metabolic stability of losartan
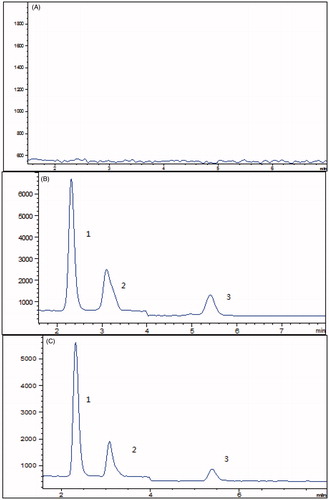
As we know, the metabolism of losartan was mainly mediated by CYP450, and therefore, in this research, the effects of BBR on the metabolic rate of losartan were investigated. The representative chromatograms of determination are shown in . The results showed that the metabolic stability of losartan was 37.4 min, while the metabolic rate was prolonged (59.6 min) in the presence of BBR. The results indicated that BBR could inhibit the metabolic stability of losartan.
Figure 5. (A) Representative chromatograms of blank microsome incubation solutions; (B) blank microsome incubation solution spiked with losartan (2), EXP3174 (3) and irbesartan (1); (C) blank microsome incubation solution treated with BBR and losartan.
Discussion
A multitude of drugs has been taken off the market after successful clinical trials (Lehmann Citation2013). One reason for this is the incidence of adverse drug–drug interactions (Li Citation2001). It is now well known that a drug can inhibit the metabolic stability of another drug, leading to a higher than intended plasma level (Wrighton & Stevens Citation1992; Zhang et al. Citation2007; Ye et al. Citation2011). The major clinical consequence of inhibitory drug–drug interactions is undesired drug toxicity (Gouws et al. Citation2012; Qi et al. Citation2013). For example, the antifungal ketoconazole, a potent inhibitor of CYP3A4, causes drug–drug interactions with drugs that are substrates of CYP3A4 (Xiaoyang et al. Citation2015). However, undesired drug–drug interactions can also be caused by transporters (Kim et al. Citation1998; Li et al. Citation2014; Joyce et al. Citation2015; Wen et al. Citation2016). Some drug interactions, previously believed to be P450-mediated, are now considered at least in part due to the inhibition of transport proteins. For example for talinolol, which undergoes little metabolism, the increased bioavailability in the presence of verapamil is most likely due to inhibition of P-gp by verapamil (Nguyen et al. Citation2014).
Losartan was administrated in a tablet form, and therefore its absorption and metabolism may be affected by the activity of the excretory transporters and drug metabolizing enzymes. Several researches have indicated that the absorption of losartan in small intestinal mucosa was mainly mediated by P-gp, and losartan was mainly metabolized by CYP3A4 and CYP2C19 (Yun et al. Citation1995; Soldner et al. Citation2000). Therefore, the potential drug–drug interaction of losartan mediated by CYP450 and P-gp might happen. As BBR and losartan was usually co-administered to the patients for the treatment of senile diabetic nephropathy in the clinic in china. Therefore, it is an urgent need to investigate the potential herb–drug interaction between BBR and losartan.
Previous studies have reported that the pharmacokinetic profiles of losartan and EXP3174 in rats were similar to humans, and therefore, using rat to speculate its potential herb–drug interaction was rationale (Yoshitani et al. Citation2002). In this research, a sensitive and reliable LC–MS method was developed for the determination of losartan and EXP3174 in rat plasma, method validation was also fully validated. The plasma concentration of losartan and EXP3174 was determined using the LC–MS method after oral administration of losartan (10 mg/kg) or both losartan (10 mg/kg) and BBR (20 mg/kg), and the pharmacokinetics profiles of losartan and EXP3174 were calculated using Das 3.0. The results indicated that the pharmacokinetics profiles of losartan and EXP3174 changed significantly compared to the control (p < 0.05), and potential herb–drug interaction might happen when they are co-administered. As losartan was a substrate of P-gp and metabolized by CYP3A4 and CYP2C9, therefore we think that BBR could change the pharmacokinetic profiles through changing the activity of P-gp, CYP3A4 or CYP2C9. Choi et al. have also reported that when losartan and licochalcon A was administered, the pharmacokinetic profiles of losartan were changed significantly (p < 0.05), and the experiment also showed that licochalcon A might change the bioavailability of losartan through decreasing P-gp mediated efflux and metabolism mediated by CYP3A4 or CYP2C9 (Choi & Choi Citation2013). Several researches have indicated that BBR has little influence on the activity of P-gp (Zhang et al. Citation2011), and therefore, it was impossible for BBR to change the pharmacokinetics profiles of losartan and EXP3174 through affecting the activity of P-gp. We speculated that BBR affects its pharmacokinetics might through inhibiting the activity of CYP3A4 or CYP2C9 as researches have reported that BBR was an inhibitor of CYP3A4. Therefore, in this research, BBR on the metabolic stability of losartan was investigated using rat liver microsomes, and the results indicated that the metabolic stability was prolonged with the pretreatment of BBR. After BBR and losartan was co-administered to rats, the plasma concentration of losartan was increased and EXP3174 was decreased, the results were also consistent with the metabolism of losartan which was inhibited, and so the concentration of EXP3174 was decreased. Therefore, the changed pharmacokinetics profiles of losartan and EXP3174 may be due to the inhibition of CYP3A4 or CYP2C9. The present study reminds us that the herb–drug interaction between BBR and losartan might happen, and further evaluation in clinical studies is necessary.
Funding information
This study was supported by National Nature Science Foundation of China (30700387).
Disclosure statement
The authors have declared no conflict of interest.
References
- Choi JS, Choi DH. 2013. Effects of licochalcon A on the pharmacokinetics of losartan and its active metabolite, EXP-3174, in rats. Die Pharmazie. 68:882–888.
- Cui G, Qin X, Zhang Y, Gong Z, Ge B, Zang YQ. 2009. Berberine differentially modulates the activities of ERK, p38 MAPK, and JNK to suppress Th17 and Th1 T cell differentiation in type 1 diabetic mice. J Biol Chem. 284:28420–28429.
- Gouws C, Steyn D, Du Plessis L, Steenekamp J, Hamman JH. 2012. Combination therapy of Western drugs and herbal medicines: recent advances in understanding interactions involving metabolism and efflux. Expert Opin Drug Metab Toxicol. 8:973–984.
- Joyce H, McCann A, Clynes M, Larkin A. 2015. Influence of multidrug resistance and drug transport proteins on chemotherapy drug metabolism. Expert Opin Drug Metab Toxicol. 11:795–809.
- Kim RB, Fromm MF, Wandel C, Leake B, Wood AJ, Roden DM, Wilkinson GR. 1998. The drug transporter P-glycoprotein limits oral absorption and brain entry of HIV-1 protease inhibitors. J Clin Invest. 101:289–294.
- Klishadi MS, Zarei F, Hejazian SH, Moradi A, Hemati M, Safari F. 2015. Losartan protects the heart against ischemia reperfusion injury: sirtuin3 involvement. J Pharm Pharm Sci: a publication of the Canadian Society for Pharmaceutical Sciences, Societe canadienne des sciences pharmaceutiques. 18:112–123.
- Lehmann K. 2013. Drug interactions—principles, examples and clinical consequences. Additional important drug interactions. Dtsch Arztebl Int. 110:133.
- Li AP. 2001. Screening for human ADME/Tox drug properties in drug discovery. Drug Discov Today. 6:357–366.
- Li Z, Wang K, Zheng J, Cheung FS, Chan T, Zhu L, Zhou F. 2014. Interactions of the active components of Punica granatum (pomegranate) with the essential renal and hepatic human solute carrier transporters. Pharm Biol. 52:1510–1517.
- Nguyen MA, Staubach P, Wolffram S, Langguth P. 2014. Effect of single-dose and short-term administration of quercetin on the pharmacokinetics of talinolol in humans – implications for the evaluation of transporter-mediated flavonoid–drug interactions. Eur J Pharm Sci. 61:54–60.
- Orvos P, Virag L, Talosi L, Hajdu Z, Csupor D, Jedlinszki N, Szel T, Varro A, Hohmann J. 2015. Effects of Chelidonium majus extracts and major alkaloids on hERG potassium channels and on dog cardiac action potential – a safety approach. Fitoterapia. 100:156–165.
- Pang B, Zhao LH, Zhou Q, Zhao TY, Wang H, Gu CJ, Tong XL. 2015. Application of berberine on treating type 2 diabetes mellitus. Int J Endocrinol. 2015:905749.
- Park SH, Sung JH, Kim EJ, Chung N. 2015. Berberine induces apoptosis via ROS generation in PANC-1 and MIA-PaCa2 pancreatic cell lines. Braz J Med Biol Res. 48:111–119.
- Qi X, Qin J, Ma N, Chou X, Wu Z. 2014. Solid self-microemulsifying dispersible tablets of celastrol: formulation development, charaterization and bioavailability evaluation. Int J Pharm. 472:40–47.
- Qi XY, Liang SC, Ge GB, Liu Y, Dong PP, Zhang JW, Wang AX, Hou J, Zhu LL, Yang L, et al. 2013. Inhibitory effects of sanguinarine on human liver cytochrome P450 enzymes. Food Chem Toxicol. 56:392–397.
- Rincon J, Correia D, Arcaya JL, Finol E, Fernandez A, Perez M, Yaguas K, Talavera E, Chavez M, Summer R, et al. 2015. Role of angiotensin II type 1 receptor on renal NAD(P)H oxidase, oxidative stress and inflammation in nitric oxide inhibition induced-hypertension. Life Sci. 124:81–90.
- Shen N, Huan Y, Shen ZF. 2012. Berberine inhibits mouse insulin gene promoter through activation of AMP activated protein kinase and may exert beneficial effect on pancreatic β-cell. Eur J Pharmacol. 694:120–126.
- Soldner A, Benet LZ, Mutschler E, Christians U. 2000. Active transport of the angiotensin-II antagonist losartan and its main metabolite EXP 3174 across MDCK-MDR1 and caco-2 cell monolayers. Br J Pharmacol. 129:1235–1243.
- Varshney E, Saha N, Tandon M, Shrivastava V, Ali S. 2013. Genotype–phenotype correlation of cytochrome P450 2C9 polymorphism in Indian National Capital Region. Eur J Drug Metab Pharmacokinet. 38:275–282.
- Wen F, Shi M, Bian J, Zhang H, Gui C. 2016. Identification of natural products as modulators of OATP2B1 using LC–MS/MS to quantify OATP-mediated uptake. Pharm Biol. 54:293–302.
- Wrighton SA, Stevens JC. 1992. The human hepatic cytochromes P450 involved in drug metabolism. Crit Rev Toxicol. 22:1–21.
- Xiaoyang L, Chenming N, Chengqing L, Tao L. 2015. Drug–drug interaction prediction between ketoconazole and anti-liver cancer drug Gomisin G. Afr Health Sci. 15:590–593.
- Yang SH, Cho YA, Choi JS. 2011. Effects of ticlopidine on pharmacokinetics of losartan and its main metabolite EXP-3174 in rats. Acta Pharmacol Sin. 32:967–972.
- Yang SH, Choi JS, Choi DH. 2011. Effects of HMG-CoA reductase inhibitors on the pharmacokinetics of losartan and its main metabolite EXP-3174 in rats: possible role of CYP3A4 and P-gp inhibition by HMG-CoA reductase inhibitors. Pharmacology. 88:1–9.
- Yasar U, Babaoglu MO, Bozkurt A. 2008. Disposition of a CYP2C9 phenotyping agent, losartan, is not influenced by the common 3435C > T variation of the drug transporter gene ABCB1 (MDR1). Basic Clin Pharmacol Toxicol. 103:176–179.
- Ye L, Wang T, Tang L, Liu W, Yang Z, Zhou J, Zheng Z, Cai Z, Hu M, Liu Z. 2011. Poor oral bioavailability of a promising anticancer agent andrographolide is due to extensive metabolism and efflux by P-glycoprotein. J Pharm Sci. 100:5007–5017.
- Yoshitani T, Yagi H, Inotsume N, Yasuhara M. 2002. Effect of experimental renal failure on the pharmacokinetics of losartan in rats. Biol Pharm Bull. 25:1077–1083.
- Yun CH, Lee HS, Lee H, Rho JK, Jeong HG, Guengerich FP. 1995. Oxidation of the angiotensin II receptor antagonist losartan (DuP 753) in human liver microsomes. Role of cytochrome P4503A(4) in formation of the active metabolite EXP3174. Drug Metab Dispos. 23:285–289.
- Zhang JW, Liu Y, Cheng J, Li W, Ma H, Liu HT, Sun J, Wang LM, He YQ, Wang Y, et al. 2007. Inhibition of human liver cytochrome P450 by star fruit juice. J Pharm Pharm Sci. 10:496–503.
- Zhang X, Qiu F, Jiang J, Gao C, Tan Y. 2011. Intestinal absorption mechanisms of berberine, palmatine, jateorhizine, and coptisine: involvement of P-glycoprotein. Xenobiotica. 41:290–296.