
 ?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.
?Mathematical formulae have been encoded as MathML and are displayed in this HTML version using MathJax in order to improve their display. Uncheck the box to turn MathJax off. This feature requires Javascript. Click on a formula to zoom.Abstract
Context: Dihydromyricetin (DHM) is the most abundant and active flavonoid component isolated from Ampelopsis grossedentata (Hand-Mazz) W.T. Wang (Vitaceae) and it possesses numerous pharmacological activities. However, whether DHM affects the activity of human liver cytochrome P450 (CYP) enzymes remains unclear.
Materials and methods: The inhibitory effects of DHM on eight human liver CYP isoforms (i.e., 1A2, 3A4, 2A6, 2E1, 2D6, 2C9, 2C19 and 2C8) were investigated in vitro using human liver microsomes (HLMs).
Results: The results showed that DHM could inhibit the activity of CYP3A4, CYP2E1 and CYP2D6, with IC50 values of 14.75, 25.74 and 22.69 μM, respectively, but that other CYP isoforms were not affected. Enzyme kinetic studies showed that DHM was not only a non-competitive inhibitor of CYP3A4 but also a competitive inhibitor of CYP2E1 and CYP2D6, with Ki values of 6.06, 9.24 and 10.52 μM, respectively. In addition, DHM is a time-dependent inhibitor for CYP3A4 with KI/Kinact value of 12.17/0.057 min−1 μM−1.
Discussion and conclusion: The in vitro studies of DHM with CYP isoforms indicate that DHM has the potential to cause pharmacokinetic drug interactions with other co-administered drugs metabolized by CYP3A4, CYP2E1 and CYP2D6. Further clinical studies are needed to evaluate the significance of this interaction.
Introduction
Ampelopsis grossedentata (Hand-Mazz) W.T. Wang (Vitaceae) is a medicinal and edible plant that is widely distributed in southern China, and its tender stem and leaves have been used as a medicinal tea for the prevention and treatment of the common cold, sore throat and icteric viral hepatitis for hundreds of years (Hou et al. Citation2015; Bi et al. Citation2016; Chen et al. Citation2016). Dihydromyricetin (DHM) is the most abundant and active flavonoid component isolated from Ampelopsis grossedentata (Du et al. Citation2002; Louis et al. Citation2010; Yin et al. Citation2010; Xia et al. Citation2014; Zhong et al. Citation2014). DHM possesses numerous biological and pharmacological activities, including anti-oxidative, anti-inflammatory, hepatoprotective, lipid and blood glucose regulation, and anticancer effects (Jiang et al. Citation2014; Liu et al. Citation2014; Chen et al. Citation2015; Ji et al. Citation2015; Meng et al. Citation2015). Li (Citation2006) has found that DHM could reduce the serum levels of total cholesterol and triglycerides and increase the serum high-density lipoprotein cholesterol levels in hyperlipidemia rats after oral administration. Qin et al. (Citation2001) have also found that DHM could reduce the blood glucose levels in alloxan-induced diabetic or hyperglycaemic mice.
Cytochrome P450 (CYP) enzymes, a superfamily of haem-containing isoenzymes located primarily in hepatocytes, are important phase I enzymes in the biotransformation of xenobiotics, which include drugs, environmental pollutants, carcinogens and endogenous substrates (Wrighton and Stevens Citation1992; Yan and Caldwell Citation2001). CYP1A, CYP2C, CYP2D, CYP3A and CYP2E are major CYP enzymes in drug metabolism (Li Citation2001). Most CYP enzymes can be inhibited or induced by a variety of drugs and chemicals that can give rise to toxicity or treatment failure, and many herb–drug interactions have been generated from the concurrent use of herbal prescription and over-the-counter drugs (Yang et al. Citation2012; Lee et al. Citation2013). St. John’s wort, Ginkgo, Ginseng, and licorice have all been reported to interact with anticoagulants, antiretroviral drugs, anticancer drugs, immunosuppressants or antidepressants (Unger Citation2013; Meng & Liu Citation2014; Pirotta et al. Citation2014; Tao et al. Citation2014; Wang et al. Citation2015). As ampelopsis-grossedentata tea may be drunk by people every day, and therefore, the effects of DHM on the activity of CYP enzymes should be investigated. To the best of our knowledge, little information was available for the effects of DHM on CYP enzymes, particularly the inhibitory effects, which will increase the risk of therapeutic applications of DHM and its medical preparations (Bostikova et al. Citation2015).
The purpose of this study was to investigate the effects of DHM on eight major CYP isoforms in human liver microsomes (HLMs). In vitro, phenacetin (CYP1A2), testosterone (CYP3A4), coumarin (CYP2A6), chlorzoxazone (CYP2E1), dextromethorphan (CYP2D6), diclofenac (CYP2C9), S-mephenytoin (CYP2C19) and paclitaxel (CYP2C8) were used as probe substrates to determine the effects of DHM on eight CYP enzymes. In addition, enzyme kinetic studies were conducted to determine the inhibition mode of DHM on CYP enzymes.
Materials and methods
Chemicals
DHM (≥98%) and testosterone (≥98%) were obtained from the National Institute for the Control of Pharmaceutical and Biological Products (Beijing, China). The chemical structure of DHM is shown in . d-Glucose-6-phosphate, glucose-6-phosphate dehydrogenase, corticosterone (≥98%), NADP+, phenacetin (≥98%), acetaminophen (≥98%), 4-hydroxymephenytoin (≥98%), 7-hydroxycoumarin (≥98%), 4'-hydroxydiclofenac (≥98%), sulphaphenazole (≥98%), quinidine (≥98%), tranylcypromine (≥98%), chlorzoxazone (≥98%), 6-hydroxychlorzoxazone (≥98%), paclitaxel (≥98%), 6β-hydroxytestosterone (≥98%), clomethiazole (≥98%) and furafylline (≥98%) were obtained from Sigma Chemical Co. (Chicago, IL). Montelukast (≥98%) was obtained from Beijing Aleznova Pharmaceutical (Beijing, China). Dextrorphan was purchased from Toronto Research Chemical (North York, Canada). Coumarin (≥98%), diclofenac (≥98%), dextromethorphan (≥98%) and ketoconazole (≥98%) were purchased from ICN Biomedicals (Seven Hills, Australia). 6α-Hydroxypaclitaxel and Pooled HLMs were purchased from BD Biosciences (Woburn, MA). All other reagents and solvents were of analytical reagent grade.
Assay with human liver microsomes
As shown in , to investigate the inhibitory effects of DHM on different CYP isoforms in HLM, the following probe reactions were used, according to previously described method (Zhang et al. Citation2007; Qi et al. Citation2013): phenacetin O-deethylation for CYP1A2, testosterone 6β-hydroxylation for CYP3A4, coumarin 7-hydroxylation for CYP2A6, chlorzoxazone 6-hydroxylation for CYP2E1, dextromethorphan O-demethylation for CYP2D6, diclofenac 4′-hydroxylation for CYP2C9, S-mephenytoin 4-hydroxylation for CYP2C19 and paclitaxel 6α-hydroxylation for CYP2C8. All incubations were performed in triplicate, and the mean values were utilized. The typical incubation systems contained 100 mM potassium phosphate buffer (pH 7.4), NADPH-generating system (1 mM NADP+, 10 mM glucose-6-phosphate, 1 U/mL of glucose-6-phosphate dehydrogenase, and 4 mM MgCl2), the appropriate concentration of HLM, a corresponding probe substrate and DHM (or positive inhibitor for different probe reactions) in a final volume of 200 μL.
Table 1. Isoforms tested, marker reactions, incubation conditions and Km used in the inhibition study.
The concentration of DHM was 100 μM, and the positive inhibitor concentrations were as follows: 10 μM furafylline for CYP1A2, 1 μM ketoconazole for CYP3A4, 10 μM tranylcypromine for CYP2A6, 50 μM clomethiazole for CYP2E1, 10 μM quinidine for CYP2D6, 10 μM sulphaphenazole for CYP2C9, 50 μM tranylcypromine for CYP2C19 and 5 μM montelukast for CYP2C8. Probe substrates, positive inhibitors (except for dextromethorphan and quinidine which were dissolved in water) and DHM were dissolved in methanol, with a final concentration of 1% (v/v), and 1% neat methanol was added to the incubations without inhibitor. The final microsomal protein concentration and incubation times for the different probe reactions are shown in . There was a 3-min preincubation period (at 37 °C) before the reaction was initiated by adding a NADPH-generating system. The reaction was terminated by adding a 100 μL acetonitrile (10% trichloroacetic acid for CYP2A6) internal standard mix, and the solution was placed on ice. The mixture was centrifuged at 12,000 rpm for 10 min, and an aliquot (50 μL) of supernatant was transferred for HPLC analysis. The instrument used in this study were Agilent 1260 series instrument with DAD and FLD detector (Agilent Technologies, Palo Alto, CA), and the quantitative assay for the corresponding metabolites was performed as previously reported by us (Zhang et al. Citation2016).
Enzyme inhibition and kinetic studies of DHM
A 100 μM DHM was used to initially screen for its direct inhibitory effects toward different human CYP isoforms. For the CYP isoforms whose activities were strongly inhibited, secondary studies were performed to obtain the half inhibition concentration (IC50). The residual activity of the CYP isoforms were measured by adding different concentrations of DHM (0–100 μM), and then the IC50 values were calculated. Ki values were obtained by incubating various concentrations of different probe substrates (20–100 μM testosterone, 25–200 μM chlorzoxazone or 5–50 μM dextromethorphan) in the presence of 0–50 μM DHM.
Time-dependent inhibition study of DHM
To determine whether DHM could inhibit the activity of CYP3A4, CYP2E1 and CYP2D6 in a time-dependent manner, DHM (20 μM) was pre-incubated with HLMs (1 mg/mL) in the presence of an NADPH-generating system for 30 min at 37 °C. After incubation, an aliquot (20 μL) was transferred to another incubation tube (a final volume of 200 μL) containing an NADPH-generating system and probe substrates whose final concentrations were approximate to Km. Then, further incubations were performed to measure the residual activity. After being incubated for 10 and 30 min, the reactions were terminated by adding a 100 μL acetonitrile internal standard mix and then placed on ice; the corresponding metabolites was determined by HPLC.
To determine the KI and kinact values for the inactivation of CYP3A4, the incubations were conducted using higher probe substrate concentrations (approximately four-fold Km values) and various concentrations of DHM (0–50 μM) after different preincubation times (0–30 min), with a two-step incubation scheme, as described above.
Statistical analysis
The enzyme kinetic parameters for the probe reaction were estimated from the best fit line using least-squares linear regression of the inverse substrate concentration versus the inverse velocity (Lineweaver–Burk plots), and the mean values were used to calculate Vmax and Km. Inhibition data from the experiments that were conducted using multiple compound concentrations were represented by Dixon plots, and inhibition constant (Ki) values were calculated using non-linear regression according to the following equation:
where I is the concentration of the compound, Ki is the inhibition constant, S is the concentration of the substrate and Km is the substrate concentration at half the maximum velocity (Vmax) of the reaction. The mechanism of the inhibition was inspected using the Lineweaver–Burk plots and the enzyme inhibition models. The data comparison was performed using Student’s t-test and performed using IBM SPSS statistics 20 (SPSS Inc., Chicago, IL).
Results
To investigate whether the DHM affects the catalytic activity of CYP enzymes, the probe reaction assays were conducted with varied concentrations of DHM. Specific inhibitors of CYP1A2, CYP3A4, CYP2A6, CYP2E1, CYP2D6, CYP2C9, CYP2C19 and CYP2C8 were used as positive controls. As shown in , DHM did not inhibit the activities of CYP1A2, CYP2A6, CYP2C9, CYP2C19 and CYP2C8 at a concentration of 100 μM. In contrast, the activities of CYP3A4, CYP2E1 and CYP2D6 were inhibited to 87.7, 74.6 and 85.3% of their control activities, respectively.
Figure 1. The chemical structure of DHM.
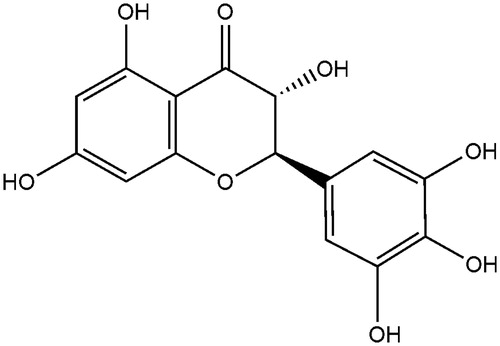
Figure 2. Inhibition of DHM on CYP450 enzymes in pooled HLMs. All data represent mean ± SD of the triplicate incubations. *p < 0.05, significantly different from the negative control. Negative control: incubation systems without DHM; DHM: incubation systems with DHM (100 μM); positive control: incubation systems with their corresponding positive inhibitors (10 μM furafylline for CYP1A2, 1 μM ketoconazole for CYP3A4, 10 μM tranylcypromine for CYP2A6, 50 μM clomethiazole for CYP2E1, 10 μM quinidine for CYP2D6, 10 μM sulphaphenazole for CYP2C9, 50 μM tranylcypromine for CYP2C19, 5 μM montelukast for CYP2C8).
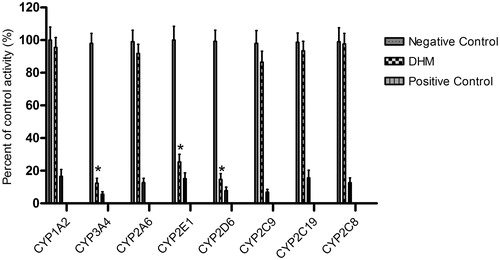
As shown in , the enzyme-inhibition study showed that inhibition of CYP3A4, CYP2E1 and CYP2D6 by DHM was concentration dependent, with IC50 values of 14.75, 25.74 and 22.69 μM, respectively.
Figure 3. Inhibitory effects of DHM on the activities of CYP3A4, CYP2E1 and CYP2D6.
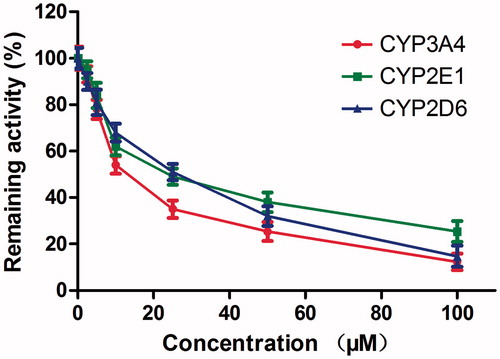
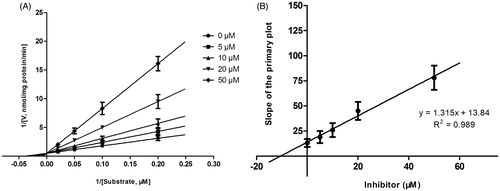
Lineweaver–Burk plots of inhibitory kinetic data suggested that the inhibition of CYP3A4 by DHM was best fit in a non-competitive manner (), whereas the inhibition of CYP2E1 () and 2D6 () by DHM was best fit in a competitive manner. The Ki values of DHM on CYP3A4 (), 2E1 () and 2D6 () were obtained from the secondary Lineweaver–Burk plot for Ki, with values of 6.06, 9.24 and 10.52 μM, respectively.
Figure 4. Lineweaver–Burk plots (A) and the secondary plot for Ki (B) of inhibition of DHM on CYP3A4 catalyzed reactions (testosterone 6β-hydroxylation) in pooled HLM. Data are obtained from a 30 min incubation with testosterone (20–100 μM) in the absence or presence of DHM (0–30 μM). The data represent the mean of the incubations (performed in triplicate).
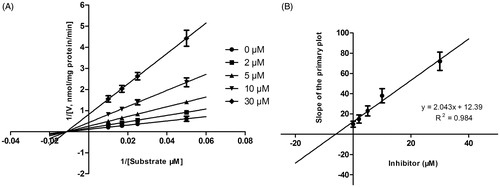
Figure 5. Lineweaver–Burk plots (A) and the secondary plot for Ki (B) of inhibition of DHM on CYP2E1 catalyzed reactions (chlorzoxazone 6-hydroxylation) in pooled HLM. Data are obtained from a 30 min incubation with chlorzoxazone (20–200 μM) in the absence or presence of DHM (0–50 μM). All data represent the mean of the incubations (performed in triplicate).
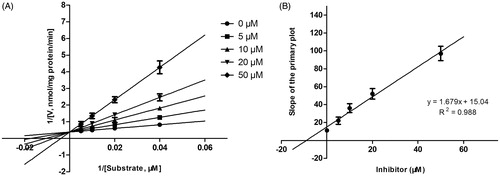
Figure 6. Lineweaver–Burk plots (A) and the secondary plot for Ki (B) of inhibition of DHM on CYP2D6 catalyzed reactions (dextromethorphan O-demethylation) in pooled HLM. Data are obtained from a 30 min incubation with dextromethorphan (5–50 μM) in the absence or presence of DHM (0–50 μM). All data represent the mean of the incubations (performed in triplicate).
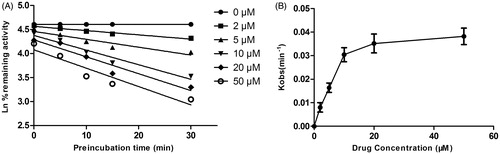
As shown in , after pre-incubation of DHM with HLM for 30 min, the activity of CYP3A4 decreased with the incubation time; however, the activity of CYP2E1 and CYP2D6 was not affected. To characterize the time-dependent inhibition of CYP3A4 by DHM, inactivation parameters of KI and Kinact values were calculated using non-linear regression analysis in HLM. As calculated from the inactivation plot of , the KI/Kinact value for CYP3A4 was 12.17/0.057 min−1 μM−1. The Kinact values imply that approximately 5.7% of CYP3A4 is inactivated each minute when a saturating concentration of DHM is incubated with HLM.
Figure 7. Time-dependent inhibition investigations of CYP3A4, 2E1 and 2D6 catalyzed reactions by DHM (20 μM). All data represent the mean of the incubations (performed in triplicate).
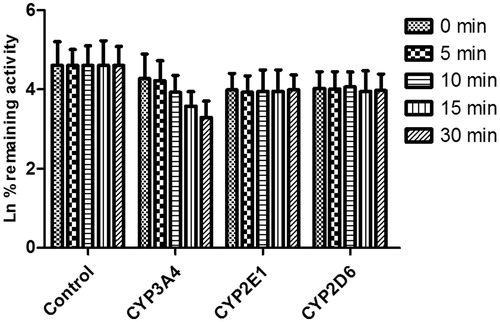
Figure 8. Time (0–30 min) and concentration (0–50 μM)-inactivation of microsomal CYP3A4 activity by DHM in the presence of NADPH. The initial rate constant of inactivation of CYP3A4 by each concentration (Kobs) was determined through linear regression analysis of the natural logarithm of the percentage of remaining activity versus pre-incubation time (A). The KI and Kinact values were determined through non-linear analysis of the Kobs versus the DHM concentration (B).
Discussion
In clinical practice, many patients undergo multiple-drug therapy. Multiple-drug therapy possesses several advantages, such as simultaneously treatment of diseases, or multi-drug therapy for the treatment complex chronical disorders, resulting in a better treatment outcome compared to monotherapy. However, many herb–drug interactions resulting from concurrent use of herbal drugs with prescription and over-the-counter drugs may cause adverse reactions such as toxicity and treatment failure (Zhou et al. Citation2004). The most common causes of herb–drug interactions are modification of the enzyme activity of cytochrome P450 enzymes, specifically through inhibitory effects. Inhibition of CYP enzymes in vivo may result in unexpected elevations in the plasma concentrations of concomitant drugs, leading to adverse effects (Hu et al. Citation2015; Wei et al. Citation2015). Therefore, regulatory authorities require preclinical (in vitro) and clinical (in vivo) interaction studies in drug development.
As ampelopsis-grossedentata tea is one of the most widely consumed drinks with numerous pharmacological activities, and therefore, it is essential to investigate the inhibitory effects of DHM on the major CYP enzymes. To the best of our knowledge, this study is the first to investigate the effects of DHM on the metabolism of probe substrates of several CYP isoforms, including CYP1A2, CYP3A4, CYP2A6, CYP2E1, CYP2D6, CYP2C9, CYP2C19 and CYP2C8. The CYP3A subfamily is one of the dominant CYP enzymes in the liver and extra-hepatic tissues, such as the intestines, and it plays an important role in the oxidation of xenobiotics and contributes to the biotransformation of approximately 60% of currently used therapeutic drugs (Pandit et al. Citation2011). Human CYP3A4 is one of the most abundant drug-metabolizing CYP isoforms in human liver microsomes, accounting for approximately 40% of the total CYP enzymes (Zhou Citation2008). In fact, characterization of the CYP3A4 isoform responsible for the metabolism of drugs and herbal constituents is important for identifying potential drug–drug or herb–drug interactions in humans. The present study showed that DHM had inhibitory effects in vitro on CYP3A4 isoform, with Ki and IC50 values of 6.06 μM and 14.75 μM, respectively. The results suggested that DHM was a weak CYP3A4 inhibitor, and the potential of herb–drug interaction with CYP3A4 would also be low. However, the results also indicated that DHM is a time-dependent inhibitor for CYP3A4 with KI/Kinact value of 12.17/0.057 μM−1 min−1, which revealed that DHM would inhibit the activity of CYP3A4 with increase of preincubation time of concentration. Therefore, in order to avoid adverse drug interactions, it is recommended that ampelopsis-grossedentata tea should not be used with other drugs metabolized by CYP3A4.
CYP2E1 and CYP2D6 play an important role in the metabolism of many drugs (Chiangsom et al. Citation2016; Ryu et al. Citation2016). Our study showed that DHM competitively inhibited human liver microsomal CYP2E1 and CYP2D6 activity. Therefore, DHM should also be used carefully with drugs metabolized by CYP2E1 and CYP2D6 in order to avoid possible drug interactions even though DHM was an inhibitor for these two CYP isoforms.
As we know, in vitro data are essential for understanding a potential enzyme inhibition and DDI in vivo. However, an observed in vitro inhibition of a CYP enzyme does not mean that the drug will cause clinically relevant interactions. Many other factors might influence drug interactions mediated by CYP inhibition, including the contribution of the hepatic clearance to the total clearance of the affected drug, the fraction of the hepatic clearance which is subject to metabolic inhibition, and the ratio of the inhibition constant (Ki) over the in vivo concentration of the inhibitor (Ito et al. Citation1998; Ericsson et al. Citation2014). Therefore, further in vivo studies are needed to identify the interactions of DHM with CYP isoform in humans.
The results of this study indicate that DHM may influence the in vitro metabolism of drugs that are substrates of CYP3A4, CYP2E1 and CYP2D6. Some research articles (Zhang et al. Citation2007; Tong et al. Citation2015) have investigated the pharmacokinetic profiles of DHM in rats, and the results indicated that the Cmax values of DHM in rats reached 1.7 μg/mL after oral administration of a dose of 1564.8 mg/kg of Rattan Tea decoction. Therefore, the rat plasma concentrations of DHM was similar to the Ki and IC50 values of DHM determined in this study, and herb–drug interaction might occur if DHM were co-administered with the substrates of the CYP3A4, CYP2E1 and CYP2D6. However, due to the pharmacokinetic differences of human and rats, further in vivo studies are needed to identify the interactions of DHM with CYP isoform in humans.
UDP-glucuronosyltransferases also plays an important role in the biotransformation of drugs, while the inhibitory effects of DHM on the activity of UDP-glucuronosyltransferases were not investigated. In future research, the effects of DHM on the activity of UDP-glucuronosyltransferases should also be investigated in vitro and in vivo.
In conclusion, the effects of DHM on the activity of CYP enzymes were systematically investigated. The results showed that DHM could inhibit the activity of CYP3A4, CYP2E1 and CYP2D6, while the activity of other CYP enzymes was not affected. Therefore, we suggested that DHM has the potential to cause pharmacokinetic drug interactions with other co-administered drugs metabolized by CYP3A4, CYP2E1 and CYP2D6, and it should be cautious when ampelopsis-grossedentata tea used with these drugs.
Disclosure statement
The authors have declared no conflict of interest.
Additional information
Funding
References
- Bi W, He C, Ma Y, Shen J, Zhang LH, Peng Y, Xiao P. 2016. Investigation of free amino acid, total phenolics, antioxidant activity and purine alkaloids to assess the health properties of non-Camellia tea. Acta Pharm Sin B. 6:170–181.
- Bostikova Z, Moserova M, Pavek P, Stiborova M, Hodek P. 2015. Role of dihydromyricetin in cytochrome P450-mediated metabolism and carcinogen activation. Neuro Endocrinol Lett. 36 Suppl 1:46–52.
- Chen J, Wu Y, Zou J, Gao K. 2016. α-Glucosidase inhibition and antihyperglycemic activity of flavonoids from Ampelopsis grossedentata and the flavonoid derivatives. Bioorg Med Chem. 24:1488–1494.
- Chen S, Zhao X, Wan J, Ran L, Qin Y, Wang X, Gao Y, Shu F, Zhang Y, Liu P, et al. 2015. Dihydromyricetin improves glucose and lipid metabolism and exerts anti-inflammatory effects in nonalcoholic fatty liver disease: a randomized controlled trial. Pharmacol Res. 99:74–81.
- Chiangsom A, Lawanprasert S, Oda S, Kulthong K, Luechapudiporn R, Yokoi T, Maniratanachote R. 2016. Inhibitory and inductive effects of Phikud Navakot extract on human cytochrome P450. Drug Metab Pharmacokinet. 31:210–217.
- Du Q, Cai W, Xia M, Ito Y. 2002. Purification of (+)-dihydromyricetin from leaves extract of Ampelopsis grossedentata using high-speed countercurrent chromatograph with scale-up triple columns. J Chromatogr A. 973:217–220.
- Ericsson T, Sundell J, Torkelsson A, Hoffmann KJ, Ashton M. 2014. Effects of artemisinin antimalarials on cytochrome P450 enzymes in vitro using recombinant enzymes and human liver microsomes: potential implications for combination therapies. Xenobiotica. 44:615–626.
- Hou XL, Tong Q, Wang WQ, Shi CY, Xiong W, Chen J, Liu X, Fang JG. 2015. Suppression of inflammatory responses by dihydromyricetin, a flavonoid from Ampelopsis grossedentata, via inhibiting the activation of NF-κB and MAPK signaling pathways . J Nat Prod. 78:1689–1696.
- Hu X, Huang W, Yang Y. 2015. Cytochrome P450 isoenzymes in rat and human liver microsomes associate with the metabolism of total coumarins in Fructus cnidii. Eur J Drug Metab Pharmacokinet. 40:373–377.
- Ito K, Iwatsubo T, Kanamitsu S, Nakajima Y, Sugiyama Y. 1998. Quantitative prediction of in vivo drug clearance and drug interactions from in vitro data on metabolism, together with binding and transport. Annu Rev Pharmacol Toxicol. 38:461–499.
- Ji FJ, Tian XF, Liu XW, Fu LB, Wu YY, Fang XD, Jin HY. 2015. Dihydromyricetin induces cell apoptosis via a p53-related pathway in AGS human gastric cancer cells. Genet Mol Res. 14:15564–15571.
- Jiang B, Le L, Pan H, Hu K, Xu L, Xiao P. 2014. Dihydromyricetin ameliorates the oxidative stress response induced by methylglyoxal via the AMPK/GLUT4 signaling pathway in PC12 cells. Brain Res Bull. 109:117–126.
- Lee SY, Lee JY, Kang W, Kwon KI, Park SK, Oh SJ, Ma JY, Kim SK. 2013. Cytochrome P450-mediated herb–drug interaction potential of Galgeun-tang. Food Chem Toxicol. 51:343–349.
- Li AP. 2001. Screening for human ADME/Tox drug properties in drug discovery. Drug Discov Today. 6:357–366.
- Li YS. 2006. Effects of Enshi-Ampelopsis grossede on serum lipid and blood rheology and oxygen free radical of hyperlipidemia model rats. J Hubei Inst Natl Med Ed. 23:7–9.
- Liu J, Shu Y, Zhang Q, Liu B, Xia J, Qiu M, Miao H, Li M, Zhu R. 2014. Dihydromyricetin induces apoptosis and inhibits proliferation in hepatocellular carcinoma cells. Oncol Lett. 8:1645–1651.
- Louis A, Petereit F, Lechtenberg M, Deters A, Hensel A. 2010. Phytochemical characterization of Rhododendron ferrugineum and in vitro assessment of an aqueous extract on cell toxicity. Planta Med. 76:1550–1557.
- Meng G, Yang S, Chen Y, Yao W, Zhu H, Zhang W. 2015. Attenuating effects of dihydromyricetin on angiotensin II-induced rat cardiomyocyte hypertrophy related to antioxidative activity in a NO-dependent manner. Pharm Biol. 53:904–912.
- Meng Q, Liu K. 2014. Pharmacokinetic interactions between herbal medicines and prescribed drugs: focus on drug metabolic enzymes and transporters. Curr Drug Metab. 15:791–807.
- Pandit S, Mukherjee PK, Ponnusankar S, Venkatesh M, Srikanth N. 2011. Metabolism mediated interaction of α-asarone and Acorus calamus with CYP3A4 and CYP2D6. Fitoterapia. 82:369–374.
- Pirotta M, Willis K, Carter M, Forsdike K, Newton D, Gunn J. 2014. 'Less like a drug than a drug': the use of St. John's wort among people who self-identify as having depression and/or anxiety symptoms. Complement Ther Med. 22:870–876.
- Qi XY, Liang SC, Ge GB, Liu Y, Dong PP, Zhang JW, Wang AX, Hou J, Zhu LL, Yang L, et al. 2013. Inhibitory effects of sanguinarine on human liver cytochrome P450 enzymes. Food Chem Toxicol. 56:392–397.
- Qin J, Zhong Z, Zhou G, Chen X. 2001. The experimental studies of dihydromyricetin on antidiabetic effect. Chin J Mod Appl Pharm. 18:351.
- Ryu CS, Oh SJ, Oh JM, Lee JY, Lee SY, Chae JW, Kwon KI, Kim SK. 2016. Inhibition of cytochrome P450 by propolis in human liver microsomes. Toxicol Res. 32:207–213.
- Tao X, Zheng L, Qi Y, Xu Y, Xu L, Yin L, Han X, Liu K, Peng J. 2014. Inhibitory effects of dioscin on cytochrome P450 enzymes. RSC Adv. 4:54026–54031.
- Tong Q, Hou X, Fang J, Wang W, Xiong W, Liu X, Xie X, Shi C. 2015. Determination of dihydromyricetin in rat plasma by LC-MS/MS and its application to a pharmacokinetic study. J Pharm Biomed Anal. 114:455–461.
- Unger M. 2013. Pharmacokinetic drug interactions involving Ginkgo biloba. Drug Metab Rev. 45:353–385.
- Wang ZY, Chen M, Zhu LL, Yu LS, Zeng S, Xiang MX, Zhou Q. 2015. Pharmacokinetic drug interactions with clopidogrel: updated review and risk management in combination therapy. Ther Clin Risk Manag. 11:449–467.
- Wei JC, Meng F, Qu K, Wang ZX, Wu QF, Zhang LQ, Pang Q, Liu C. 2015. Sorafenib inhibits proliferation and invasion of human hepatocellular carcinoma cells via up-regulation of p53 and suppressing FoxM1. Acta Pharmacol Sin. 36:241–251.
- Wrighton SA, Stevens JC. 1992. The human hepatic cytochromes P450 involved in drug metabolism. Crit Rev Toxicol. 22:1–21.
- Xia J, Guo S, Fang T, Feng D, Zhang X, Zhang Q, Liu J, Liu B, Li M, Zhu R. 2014. Dihydromyricetin induces autophagy in HepG2 cells involved in inhibition of mTOR and regulating its upstream pathways. Food Chem Toxicol. 66:7–13.
- Yan Z, Caldwell GW. 2001. Metabolism profiling, and cytochrome P450 inhibition & induction in drug discovery. Curr Top Med Chem. 1:403–425.
- Yang JM, Ip SP, Xian Y, Zhao M, Lin ZX, Yeung JH, Chan RC, Lee SS, Che CT. 2012. Impact of the herbal medicine Sophora flavescens on the oral pharmacokinetics of indinavir in rats: the involvement of CYP3A and P-glycoprotein. PLoS One. 7:e31312.
- Yin S, Sykes ML, Davis RA, Shelper T, Avery VM, Camp D, Quinn RJ. 2010. New galloylated flavanonols from the Australian plant Glochidion sumatranum. Planta Med. 76:1877–1881.
- Zhang H, Ya G, Rui H. 2016. Inhibitory effects of triptolide on human liver cytochrome P450 enzymes and P-glycoprotein. Eur J Drug Metab Pharmacokinet. 42:89–98.
- Zhang JW, Liu Y, Cheng J, Li W, Ma H, Liu HT, Sun J, Wang LM, He YQ, Wang Y, et al. 2007. Inhibition of human liver cytochrome P450 by star fruit juice. J Pharm Pharm Sci. 10:496–503.
- Zhang YS, Zhang QY, Li LY, Wang B, Zhao YY, Guo DA. 2007. Simultaneous determination and pharmacokinetic studies of dihydromyricetin and myricetin in rat plasma by HPLC-DAD after oral administration of Ampelopsis grossedentata decoction. J Chromatogr B: Analyt Technol Biomed Life Sci. 860:4–9.
- Zhong S, Kong Y, Zhou L, Zhou C, Zhang X, Wang Y. 2014. Efficient conversion of myricetin from Ampelopsis grossedentata extracts and its purification by MIP-SPE. J Chromatogr B: Analyt Technol Biomed Life Sci. 945–946:39–45.
- Zhou S, Chan E, Li SC, Huang M, Chen X, Li X, Zhang Q, Paxton JW. 2004. Predicting pharmacokinetic herb–drug interactions. Drug Metabol Drug Interact. 20:143–158.
- Zhou SF. 2008. Drugs behave as substrates, inhibitors and inducers of human cytochrome P450 3A4. Curr Drug Metab. 9:310–322.