
Abstract
An earlier healthy 64-year-old man with previous surgery for bilateral carpal tunnel syndrome (CTS) in his 50s, presented with dyspnoea on exertion. Cardiac amyloidosis was suspected due to “red flag” signs and symptoms. Further investigations with scintigraphy and genetic testing confirmed the diagnosis of hereditary ATTR variant (ATTRv) amyloidosis. This is the first case report of ATTRv amyloidosis in a patient of Norwegian origin and is caused by the mutation E54A (p.E74A) in the transthyretin (TTR) gene. This mutation is previously not reported in international databases. Transthyretin amyloid cardiomyopathy (ATTR-CM) is an underdiagnosed disease with a poor prognosis. Early recognition remains essential to afford the best treatment efficacy.
Main text introduction
ATTR amyloidosis is a progressive fatal disease, induced by a misfolded precursor protein. Extracellular deposition of these proteins as insoluble cross-β-sheet-rich amyloid fibrils disrupts vital organ function. ATTR-CM is an infiltrative cardiomyopathy characterised by biventricular hypertrophy and diastolic heart failure. The differential diagnosis includes cardiac amyloid light chain (AL) amyloidosis, which can be amenable to chemotherapy. Correct diagnosis is crucial with emerging novel pharmacologic treatments for ATTR amyloidosis. Increased physicians' suspicion, improved cardiovascular imaging techniques and nuclear imaging along with emerging treatment strategies have the potential to improve patient outcomes.
Case presentation with materials, methods and results
A previously healthy 64-year-old man, who had a history of an operation for bilateral carpal tunnel syndrome in his 50s, presented to the hospital with a 3 weeks history of dyspnoea on exertion, stage II New York Heart Association (NYHA), as well as 6 months with symptoms of peripheral polyneuropathy. He reported a weight loss of 10 kg without night sweats and mild peripheral oedemas. Blood pressure was 140/78 mmHg; heart rate irregular at 45–75 b.p.m, with no distention of neck veins or heart murmur. ECG showed an alternate atrioventricular block, atrial flutter and left bundle branch block (). Laboratory testing was significant for pro-b-type natriuretic peptide (NT-proBNP) at 876 ng/L (normal < 250 ng/l) troponin T was 43 ng/L (normal <14ng/L). Complete blood count and comprehensive metabolic panel were within normal limits. Transthoracic echocardiography demonstrated biventricular wall thickening, diastolic dysfunction, bi-atrial enlargement and reduced contractility with an estimated ejection fraction (EF) of 42% (). A neurologist diagnosed the patient approximately 1 month before admission with a moderate degree of demyelinating peripheral polyneuropathy. Due to positive IgG borrelia findings in serum and spinal fluid, the patient had 2 antibiotic treatments with no effect on symptoms. The patient had a family history with his mother and uncle having Charcot-Marie-Tooth (CMT) disease and died in their 60s, also presenting with heart failure symptoms. The disease was never diagnosed with genetic testing. Genetic testing of the patient for CMT was negative. Biventricular hypertrophy without an obvious cause, elevated cardiac biomarkers, symptomatic peripheral polyneuropathy and a history of bilateral carpal tunnel syndrome are typically “red flags” and raised suspicion of cardiac amyloidosis. Immunofixation electrophoresis showed no monoclonal (M) spike and serum-free kappa to lambda light chain ratio was normal. Urine immunofixation showed mildly elevated immunoglobulin kappa light chains. In the absence of an M-protein, a gammopathy was unlikely. A 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid (DPD) scintigraphy together with a SPECT/CT fusion imaging was subsequently done, showing a Perugini grade 3 tracer uptake, strongly suggestive of transthyretin amyloid (ATTR) deposition (). Abdominal fat biopsy tissue confirmed ATTR amyloidosis (). The patient presented with an alternating atrioventricular block with atrial flutter as well as some burden of non-sustained ventricular tachycardia (NSVT). In concern of pacemaker-induced left ventricular dyssynchrony leading to further reduction in stroke volume, a biventricular CRT-D was implanted. Medical treatment with loop diuretics (Bumetanide 0,5 mg q.d) was initiated. Anticoagulation was indicated due to atrial flutter and a CHADVASC score of 1, also taking into consideration the increased risk of intracardiac thrombosis in amyloidosis patients. Genetic analysis of the transthyretin (TTR) gene revealed a heterozygous mutation E54A (p.E74A) in exon 3 in TTR. This variant has not previously been reported as a cause of ATTRv amyloidosis, but several other amino acids changes in the same codon are, suggesting Glutamate (E) as an important amino acid in the 127-amino acid sequence. The patient was started on Tafamidis 61 mg q.d. During outpatient follow-up after 6 months,transthoracic echocardiography showed improved hemodynamics after biventricular CRT-D with an estimated EF of >55%. Clinical improvement was substantial and the patient resumed physical activities in a NYHA stage I. The patient received genetic counseling, and family members are planned for genetic screening.
Figure 1. (A, B) Electrocardiogram - Atrial flutter and left bundle branch block.
(C, D) Transthoracic echocardiography - Left ventricular hypertrophy (lVSd 12mm, PWd 10mm), along with speckled pattern in the myocardium. (E) Strain echocardiography - Speckled tracking revealed marked diminution of global longitudinal strain (-12) depicted in bulls-eye plot with apical sparing. (F, G) Tissue Doppler study revealed grade 2 left ventricular diastolic dysfunction (E/A mitral ratio 1,3, DT 210ms, E/è13,7). (H) Multiplanar single photon emission computed tomography (SPECT)- Showing myocardial tracer uptake, mostly in the left ventricle and septum with some heterogeneity in uptake intensity. (I) Pretreatment whole body 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid (DPD) scintigraphy - Showing very high 99mTc -DPD-uptake in the heart, Perugini grade 3.
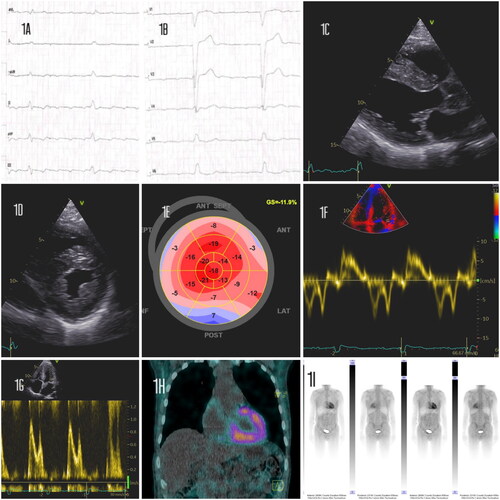
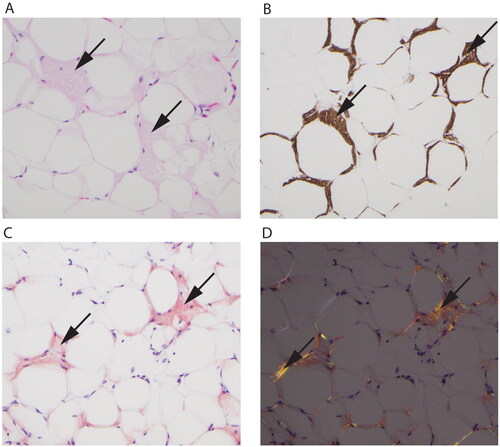
Figure 2. Fat tissue with amyloid deposition (arrows in all panels). (A) Hematoxylin eosin stain showing eosinophilic amorphous material. (B) Immunohistochemistry shows a positive reaction for transthyretin in the depositions. (C) Congo red stain is positive with orange color in the depositions and shows (D) green and orange birefringence under polarized light.
Discussion
Cardiac amyloidosis arises from misfolded transthyretin (ATTR) or immunological light-chain (AL) aggregation [Citation1]. In untreated AL cardiac amyloidosis, median survival is about 6 months from the onset of heart failure with early diagnosis and treatment as the most important factor for survival. In ATTR-CM the median survival from the time of diagnosis is reported to be about 4 years depending on the disease stage defined by NT-proBNP and eGFR levels [Citation2]. TTR, the transporter of thyroxine- and retinol-binding protein, circulates as a stable tetramer, secreted from the liver. In ATTR amyloidosis, the tetramer becomes unstable and disassociates into monomers, which polymerize into insoluble fibrils. This instability can be the result of a point variation in the TTR-gene (ATTRv) or age-related instability of unmutated wild-type TTR (ATTRwt) [Citation3]. In ATTRv amyloidosis, there are single amino acid mutations in the 127 amino-acid sequences that destabilize the hetero-tetramer. The phenotype is dependent on the nature of the underlying mutation with over 100 different mutations known to involve the TTR-gene. The p.Val30Met mutation is the most common worldwide [Citation4]. In ATTR amyloidosis the heart is commonly involved with up to 70% having peripheral neuropathy. The infiltration of amyloid fibrils in the cardiac interstitial space produces significant thickening of both ventricles with associated stiffness resulting in impaired diastolic relaxation and restrictive physiology [Citation5]. The atria, valves and conducting system are also typically involved resulting in sequelae such as atrial fibrillation, valvular regurgitation and various degrees of heart block. The clinical symptoms of cardiac amyloidosis are vague and nonspecific with symptoms of congestive heart failure, exertional dyspnea, fatigue, weight loss and peripheral oedema [Citation6]. Initial extra-cardiac manifestations may include carpal tunnel syndrome and spinal stenosis and these often precede the cardiac complications by several years [Citation7]. Diagnosing ATTR-CM requires a high index of suspicion due to the heterogeneity of manifestations. A combination of cardiac imaging, ECG, histopathology and laboratory tests is required. Persistent elevations in cardiac biomarkers are commonly observed and may reflect small vessel ischemia or direct myocyte toxicity. Increased left ventricular wall thickness (>12mm) in the presence of a low-QRS voltage and “pseudoinfarction” pattern on ECG should prompt consideration of cardiac amyloidosis and can differentiate ATTR-CM from hypertensive or hypertrophic cardiomyopathy. However, only 25–40% of patients with ATTR-CM meet low voltage criteria [Citation8]. The distinctive pattern of reduced longitudinal strain in the basal and mid-wall with” apical sparing”, has a good diagnostic accuracy for differentiating amyloid heart disease from other aetiologies [Citation9]. Endomyocardial biopsy remains the gold standard for ATTR-CM diagnosis and is nearly 100% sensitive and specific if biopsy specimens are collected from multiple sites and tested for amyloid deposits by Congo red staining [Citation10]. An imaging modality that can accurately diagnose ATTR-CM and differentiate from AL without the need for invasive cardiac biopsy is nuclear scintigraphy using bone-avid radiotracers [Citation11]. The mechanism underlying the myocardial retention of these tracers has been attributed to the presence of clouds containing myriads of microcalcifications which are more common in ATTR than AL cardiac tissue [Citation12]. The Perugini scale is a semi-quantitative method of scoring cardiac uptake with grade 2 or 3 having 86% specificity and 99% sensitivity for ATTR-CM in the absence of a monoclonal protein by serum and urine testing [Citation13]. The treatment of ATTR-CM is two-fold; first, treatment of symptoms from cardiac dysfunction, most commonly diastolic in nature, and second, to slow or stop progressive amyloid deposits. Previously treatable only by organ transplantation, effective pharmacologic strategies are emerging for ATTR-CM. One approach is the use of TTR tetramer stabilizers (Tafamidis), which bind the thyroxine-binding sites of TTR and stabilize its tetrameric structure preventing amyloid fibrillogenesis and de novo deposition. Tafamidis has been shown to slow the progressive peripheral neurological impairment in ATTR polyneuropathy [Citation14]. Phase 3 trial (ATTR-ACT trial) demonstrated the benefits of ATTR–CM with a reduction in all-cause mortality and cardiovascular hospitalizations for subjects in NYHA I-II, highlighting the importance of early diagnosis to optimize the benefit from medical treatment [Citation15]. In ATTRv amyloidosis genetic testing of relatives allows preventive follow-up already before symptoms.
Conclusion
This is the first case of ATTRv amyloidosis in Norway, presenting with a novel E54A(p.E74A) variant in the TTR-gene. When ATTR–CM is recognized before advanced organ dysfunction develops, patient outcomes can be improved using novel pharmacologic treatments, highlighting a new era for cardiac amyloidosis.
Consent form
Written consent was obtained from the patient for publication of this case history and images in line with COPE recommendations.
Acknowledgement
The authors thank dr. Martin P. Bogsrud for genetic counseling and dr. Melinda Raki for immunohistochemistry illustration.
Disclosure statement
No potential conflict of interest was reported by the author(s).
Additional information
Funding
References
- Dubrey SW, Hawkins PN, Falk RH. Amyloid diseases of the heart: assessment, diagnosis, and referral. Heart. 2011;97(1):75–84.
- Gillmore JD, Damy T, Fontana M, et al. A new staging system for cardiac transthyretin amyloidosis. Eur Heart J. 2018;39(30):2799–2806.
- Merlini G, Dispenzieri A, Sanchorawala V, et al. Systemic immunoglobulin light chain amyloidosis. Nat Rev Dis Primers. 2018;4(1):38.
- Maurer MS, Hanna M, Grogan M, et al. Genotype and phenotype of transthyretin cardiac amyloidosis: THAOS (transthyretin amyloid outcome survey). J Am Coll Cardiol. 2016;68(2):161–172.
- Siddiqi OK, Ruberg FL. Cardiac amyloidosis: an update on pathophysiology, diagnosis, and treatment. Trends Cardiovasc Med. 2018;28(1):10–21.
- Bhogal S, Ladia V, Sitwala P, et al. Cardiac amyloidosis: an updated review with emphasis on diagnosis and future directions. Curr Probl Cardiol. 2018;43(1):10–34.
- Aus Dem Siepen F, Hein S, Prestel S, et al. Carpal tunnel syndrome and spinal canal stenosis: harbingers of transthyretin amyloid cardiomyopathy? Clin Res Cardiol. 2019;108(12):1324–1330.
- Rapezzi C, Merlini G, Quarta CC, et al. Systemic cardiac amyloidoses: disease profiles and clinical courses of the 3 main types. Circulation. 2009;120(13):1203–1212.
- Pagourelias ED, Mirea O, Duchenne J, et al. Echo parameters for differential diagnosis in cardiac amyloidosis: a head-to-head comparison of deformation and nondeformation parameters. Circ Cardiovasc Imaging. 2017;10(3):e005588.
- Pellikka PA, Holmes DR, Jr., Edwards WD, et al. Endomyocardial biopsy in 30 patients with primary amyloidosis and suspected cardiac involvement. Arch Intern Med. 1988;148(3):662–666.
- Perugini E, Guidalotti PL, Salvi F, et al. Noninvasive etiologic diagnosis of cardiac amyloidosis using 99mTc-3,3-diphosphono-1,2-propanodicarboxylic acid scintigraphy. J Am Coll Cardiol. 2005;46(6):1076–1084.
- Thelander U, Westermark GT, Antoni G, et al. Cardiac microcalcifications in transthyretin (ATTR) amyloidosis. Int J Cardiol. 2022;352:84–91.
- Gillmore JD, Maurer MS, Falk RH, et al. Nonbiopsy diagnosis of cardiac transthyretin amyloidosis. Circulation. 2016;133(24):2404–2412.
- Coelho T, Maia LF, Martins da Silva A, et al. Tafamidis for transthyretin familial amyloid polyneuropathy: a randomized, controlled trial. Neurology. 2012;79(8):785–792.
- Maurer MS, Elliott P, Merlini G, et al. Design and rationale of the phase 3 ATTR-ACT clinical trial (tafamidis in transthyretin cardiomyopathy clinical trial). Circ Heart Fail. 2017;10(6):e003815.