
ABSTRACT
Introduction
Fcγ-receptors (FcγR) are membrane receptors expressed on a variety of immune cells, specialized in recognition of the Fc part of immunoglobulin G (IgG) antibodies. FcγRIIA-dependent platelet activation in platelet factor 4 (PF4) antibody-related disorders have gained major attention, when these antibodies were identified as the cause of the adverse vaccination event termed vaccine-induced immune thrombocytopenia and thrombosis (VITT) during the COVID-19 vaccination campaign. With the recognition of anti-PF4 antibodies as cause for severe spontaneous and sometimes recurrent thromboses independent of vaccination, their clinical relevance extended far beyond heparin-induced thrombocytopenia (HIT) and VITT.
Areas covered
Patients developing these disorders show life-threatening thromboses, and the outcome is highly dependent on effective treatment. This narrative literature review summarizes treatment options for HIT and VITT that are currently available for clinical application and provides the perspective toward new developments.
Expert opinion
Nearly all these novel approaches are based on in vitro, preclinical observations, or case reports with only limited implementation in clinical practice. The therapeutic potential of these approaches still needs to be proven in larger cohort studies to ensure treatment efficacy and long-term patient safety.
1. Introduction
Fcγ-receptors (FcγR) are membrane receptors expressed on a variety of immune cells, specialized in recognition of the Fc part of immunoglobulin G (IgG) antibodies. These receptors exert pivotal functions of the immune system by recognition of immune complexes, e.g. bacteria or other particles opsonized by IgG [Citation1] and subsequent mediation of clearance of these immune complexes i.e. by phagocytosis [Citation2]. FcγR-dependent cell activation requires downstream signaling after FcγR crosslinking ().
Figure 1. Signaling cascade events during FcγRIIA-mediated platelet activation.
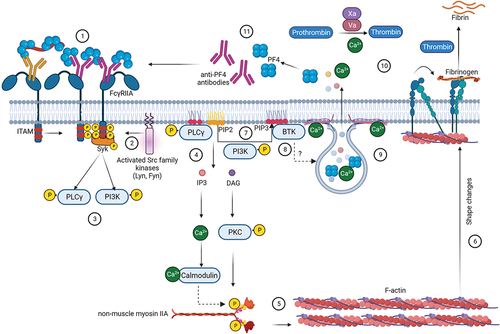
First step in the signaling cascade is clustering of FcγR by immune complexes followed by activation of intracellular domains containing immunoreceptor tyrosine-based activation motifs (ITAMs) [Citation3]. Src family kinases (SFKs), such as Lyn and Fyn phosphorylate ITAMs [Citation4], followed by recruitment and activation of the spleen tyrosine kinase (Syk) [Citation5]. Subsequently several proteins, such as phosphoinositide-3 (PI3) kinase, are activated. Generation of phosphatidylinositol-(3,4,5)-triphosphate (PIP3) by PI3 kinase recruits pleckstrin homology containing proteins such as phosphoinositide phospholipase Cγ (PLCγ) and Bruton tyrosine kinase (BTK). PLCγ acts on phosphatidylinositol-(4,5)-bisphosphate (PIP2) to generate inositol triphosphate (IP3) and diacetyl glycerol (DAG). IP3 induces calcium mobilization while DAG activates downstream proteins such as protein kinase C (PKC), which together induce activation and aggregation of platelets [Citation5,Citation6]. In particular, non-muscular myosin IIa light chain is phosphorylated by PKC and calcium-calmodulin-dependent activation of myosin light-chain kinase [Citation6], triggers cytoskeletal reorganization and activation of integrins, particularly αIIbβ3 (also known as glycoprotein IIb/IIIa [GPIIb/IIIa] receptor). This facilitates binding of fibrinogen [Citation7] and propagation of platelet activation. Furthermore, released calcium binds to SNARE proteins and initiate alpha and dense granule exocytosis [Citation8]. The vesicles release their content including calcium, which propagates the coagulation cascade. Secretion of the alpha granule protein platelet factor 4 (PF4) is especially relevant for anti-PF4 antibody-mediated disorders, as this allows formation of additional immunocomplexes enhancing FcγR-mediated activation of additional platelets.
FcyRs can be divided into three subclasses: FcyRI, FcyRII, and FcyRIII based on their structure and IgG-affinity, respectively with different tissue distributions [Citation9]. It is important to note, that only FcγRI activation is able to be triggered by monomeric IgG (high affinity) while activation of the other two receptors is highly dependent on the binding of multiple IgG molecules within an immune complex (low affinity) [Citation10]. FcγRII are distinguished further into FcγRIIA, which is an activating receptor (also known as CD32a) or FcγRIIB (CD32b), which is an inhibiting receptor.
Binding of immune complexes to FcγRIIA plays a pivotal role in autoimmune disorders, e.g. systemic lupus erythematosus or autoimmune arthritis [Citation11,Citation12]. With its high expression on platelets, FcγRIIA has an important role in antibody-mediated prothrombotic disorders and thromboinflammation. Thromboinflammation is described as derailment of the tightly orchestrated interplay between humoral and cellular signaling pathways of immune defense and coagulation, associated with increased risks for thromboembolic events as most recently clinically illustrated in the context of COVID-19 disease [Citation13].
FcγRIIA is also highly relevant for complications observed during the COVID-19 vaccination campaign. A small fraction of recipients [Citation14] of adenovirus vector-based vaccines developed thrombocytopenia and thrombosis at unusual sites, associated with platelet activating anti-PF4 antibodies, now termed vaccine-induced immune thrombocytopenia and thrombosis (VITT) [Citation15]. Interestingly, VITT has intriguing similarities with the well-known adverse drug effect heparin-induced thrombocytopenia (HIT). Both disorders are associated with platelet-activating (auto-) antibodies against PF4. In HIT, FcγRIIA-dependent platelet activation is induced by immunocomplexes of anti-PF4 antibodies bound to PF4/heparin-complexes [Citation16,Citation17]. In VITT anti-PF4 antibodies directly complex with PF4 and induce FcγRIIA-dependent, but heparin-independent platelet activation [Citation18].
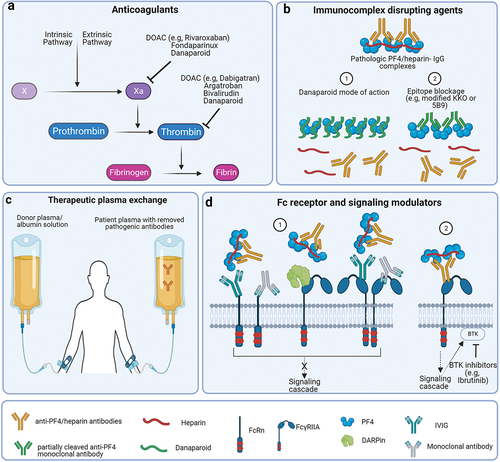
Patients developing these disorders show life-threatening symptoms. Patient outcome is highly dependent on rapid recognition and effective treatment. Despite being a prothrombotic disorder, anticoagulation is not the only treatment approach. This review summarizes current therapeutic approaches of HIT and VITT beyond anticoagulation, with special focus on the modulation of the FcγRIIA involvement in their pathogenesis. A general overview of these treatments is shown in .
Figure 2. Different therapeutic interventions in HIT.
2. Anticoagulation
For both VITT and HIT, a mainstay of treatment is anticoagulation with different possible options, as summarized in several treatment guidelines [Citation19–21]. Central aspects of anticoagulation are described in more detail in the following paragraphs and listed in .
Table 1. Treatment options for anticoagulation in HIT and VITT.
2.1. Alternative anticoagulants
HIT is a prothrombotic disorder that is induced by administration of heparin with an incidence of 0.1% (low molecular weight heparin, LMWH) up to 7% (unfractionated heparin, UFH), dependent on the type of heparin administered and other factors, including the underlying disease and duration of heparin exposure [Citation19]. In HIT, discontinuation of heparin is most important to interrupt generation of PF4/heparin complexes. In HIT patients, regardless of whether thrombosis is present or not, therapeutic anticoagulation with an alternative anticoagulant is recommended as the prothrombotic effect of HIT is so strong that prophylactic dose anticoagulation is not sufficient to prevent new thrombosis. This is also reflected by the high risk for progression to thrombosis in patients with acute HIT (>50%) in the absence of therapeutic dose anticoagulation [Citation27–30].
In principle, the choice of the alternative anticoagulant () is driven by many modalities like severity of HIT or VITT, risk of bleeding or need for surgery, mode of elimination (some drugs are contraindicated in patients with liver or renal failure), available monitoring tools, as well as availability and costs.
Direct oral anticoagulants (DOACs) such as rivaroxaban, apixaban, and dabigatran do not interfere with PF4. They are easy to use, do not require regular monitoring, and do not show cross-reactivity with HIT antibodies, owing to DOACs completely different structure compared to heparin [Citation31]. DOACs are primarily recommended for clinically stable patients. In patients on intensive care unit (ICU) or with potential urgent need for surgery, therapeutic options with a shorter half-life such as argatroban or bivalirudin may be preferred [Citation19]. Although there are no large comparative studies of DOACs with other anticoagulants in HIT and VITT, an increasing number of cases have been reported of successful use of DOACs in HIT. A clinical study of 46 probable HIT patients who were treated with rivaroxaban (25 patient received it as primary, 21 patients as secondary therapy) indicates a high efficacy as recurrent thrombosis only appeared in 2.2% of the patients [Citation32]. In addition, none of the patients developed major bleeding [Citation32]. Also in VITT, DOACs have been shown to prevent new thrombotic events in a study including eleven patients with pre-VITT (median age 48 years, 81.8% females) [Citation33] and are also recommended for acute therapy in VITT [Citation34]. However, there are theoretical concerns on the variable drug levels, which may result in under-anticoagulation at trough-levels [Citation35]. In both HIT and VITT, DOACs seem to be excellent anticoagulants once massive thrombin generation has been controlled.
Fondaparinux is also widely used for anticoagulation in HIT, with little experience in VITT. Although fondaparinux is the pentasaccharide of heparin which binds to antithrombin (and therefore characterized as indirect FXa inhibitor) it is not able to generate the HIT antigen by crosslinking two PF4 molecules [Citation36]. Fondaparinux is too short to bridge two PF4 molecules and neutralize the charge cloud around them, an essential step in the generation of HIT antigens [Citation37]. Furthermore, in vitro experiments demonstrated that ~ 97% of HIT sera do not activate platelets or induce platelet aggregation in the presence of fondaparinux [Citation38]. A systematic review, pertaining to the treatment of HIT with fondaparinux found an incidence of recurrent thrombosis, major bleeding and mortality of 6.5%, 16.9% and 3.2% respectively among 154 HIT patients [Citation39]. Cross-reactivity of HIT antibodies with PF4/fondaparinux complexes [Citation25,Citation38] has been reported but this is extremely unlikely. Rather, PF4/fondaparinux complexes can rarely trigger autoimmune HIT antibodies, which then bind to PF4 even in the absence of any polyanion [Citation23]. Fondaparinux has also been used to anticoagulate VITT patients and reported to reduce the formation of thrombi and procoagulant platelets but with no effect on immunocomplexes formed between PF4 and anti-PF4 antibodies [Citation40,Citation41]. Due to its long half-life, fondaparinux may not be the first choice anticoagulant in patients with a high risk of bleeding or urgent need for surgery. Due to the same reason, low trough-levels are less a concern as with DOACs.
Argatroban, a synthetic direct thrombin-inhibitor (DTI) that binds to the active site of thrombin is approved for anticoagulation in patients with HIT. It is given by intravenous infusion to maintain constant drug levels. It showed efficacy in a cohort study that compared HIT patients: 160 with isolated thrombocytopenia (mean age 61.3 years, 57% female) and 144 with thrombocytopenia and thrombosis (mean age 61.5 years, 50% female) after the administration of argatroban with historical control subjects: 147 with isolated thrombocytopenia (mean age 66.1 years, 44% female) and 46 with thrombocytopenia and thrombosis (mean age 65.7 years, 39% female) [Citation42]. Main readout parameters of this study were activated partial thromboplastin time (aPTT) and a rise in platelet count [Citation42]. Additionally, it was safe and effective in a case series of 14 patients with HIT antibodies after cardiovascular surgery (mean patient age 61.2 years, 64.3% females), with only one patient with postoperative hemorrhage which could also be managed [Citation43]. Recently, argatroban has also been used successfully in a case study reporting the treatment of a 29-year-old male VITT patient [Citation44], in conjunction with other therapeutic agents such as intravenous immunoglobulins (IVIGs). Due to its short half-life it is often the preferred choice in patients with high bleeding risk or potential need of surgery. In contrast to most of the other alternative anticoagulants, argatroban does not accumulate in patients with renal failure. As argatroban has to be given intravenously and needs to be monitored by aPTT levels, it is usually reserved for acute patients in the ICU.
Unlike argatroban, which acts only at the active site of thrombin, bivalirudin is a short polypeptide bivalent DTI, which inhibits thrombin by interacting at two distinct sites, i.e. at exosite 1 and the active site [Citation45]. Bivalirudin is enzymatically inactivated in the plasma by cleaving. Considering its short half-life (25 min) and its predominant non-organ clearance metabolism, bivalirudin is a choice for critically ill patients who might experience organ failure [Citation46]. The therapeutic indications of bivalirudin include cardiac surgery in patients with HIT, where it showed to reduce major bleeding. Anecdotal reports indicate that bivalirudin is also an effective anticoagulant in VITT [Citation47].
All DTIs bear the risk of a false long aPTT in patients with prothrombin deficiency (vitamin K deficiency; sepsis; disseminated coagulopathy; liver disease). Monitoring based solely on the aPTT may lead to under-anticoagulation [Citation48]. Using a direct anti-thrombin assay is a potential solution but requires extensive validation in the laboratory.
2.2. Vitamin K antagonists
Vitamin K antagonists (VKAs), such as warfarin, which had been the most important anticoagulant for long-term anticoagulation before the era of DOACs, must be avoided in acute HIT and VITT [Citation19,Citation47]. VKAs cause rapid depletion of protein C and protein S. The resulting pro-coagulatory imbalance increases the risk for microvascular thrombosis in situations with strong FcγRIIA-mediated platelet activation. This may result in extensive skin necrosis and venous limb gangrene [Citation49,Citation50]. Once acute HIT or acute VITT are controlled, as indicated by normalization of the platelet count at a stable plateau at two consecutive days, VKAs might be started for long-term anticoagulation [Citation20].
2.3. Heparin
Unlike in HIT, where anti-PF4/heparin antibodies bind to polar regions of PF4, anti-PF4 antibodies developed in VITT bind to the heparin binding site of PF4 [Citation51]. This suggests competitive binding of heparin and anti-PF4 antibodies to PF4. Singh et al. demonstrated that heparin interferes with the binding of PF4 and anti-PF4 antibodies facilitating the inhibition of anti-PF4 antibody-mediated thrombus formation in VITT [Citation52]. The WHO recommends to use heparin as a treatment for VITT if non-heparin anticoagulants are not available [Citation26]. However, there is a small number of cases where anti-PF4 antibodies developed in VITT also show a positive response in chemiluminescent HIT assays [Citation53], suggesting that the antibodies might cross-react with PF4/heparin. Therefore, most guidelines recommend to administer non-heparin coagulants initially, at least until cross-reactivity of VITT anti-PF4 antibodies with PF4/heparin complexes has been excluded by in vitro functional tests [Citation34].
Even though anticoagulation is one of the most important pillars of therapy in HIT and VITT, both conditions might benefit from additional therapeutic approaches, which will be described in the following paragraphs.
3. Interference with the PF4 antigen
3.1. Danaparoid
Danaparoid is a low molecular weight heparinoid anticoagulant, containing a mixture of glycosaminoglycans (heparan sulfate, dermatan sulfate, and chondroitin sulfate), that acts primarily as an inhibitor of factor Xa (FXa) via activation of antithrombin [Citation54]. To a much lesser extent, it also has anti-thrombin activity via heparin-cofactor II. Danaparoid is the drug with the longest history of successful use in HIT [Citation55,Citation56]. The previously assumed cross-reactivity of danaparoid with HIT antibodies [Citation57,Citation58] is most likely reflecting the presence of anti-PF4 autoantibodies, which bind to PF4 and cause platelet activation in the absence of heparin (or danaparoid) and does not (or only very rarely) translate into clinically relevant cross-reactivity [Citation59].
Formation of PF4/polyanion complexes depends on the stoichiometric ratio between polyanions and PF4 [Citation60]. Danaparoid has a much lower activity compared to heparin (18 aFXa units/mg vs 150 aFXa units/mg). Therefore about 10 times more danaparoid molecules are present at therapeutic dose concentrations compared to heparin. This explains the important feature of danaparoid that it inhibits platelet activation by HIT antibodies independently of its anticoagulatory properties [Citation61]. Danaparoid interferes structurally with the antigen in HIT, by interacting with PF4 in a charge-dependent manner. Due to its high concentration, danaparoid is able to detach PF4/heparin complexes from the platelet surface and to induce the dissociation of preformed HIT antibody-PF4/heparin complexes, thereby inhibiting HIT antibody-induced platelet activation [Citation61] (). Furthermore, binding of PF4 or PF4/heparin complexes to cell surfaces, e.g. of monocytes or endothelial cells is inhibited by danaparoid [Citation61]. Based on these data, it was also assumed that danaparoid might also have a direct influence on VITT pathogenesis beyond its anticoagulant action. In fact, in vitro experiments show that danaparoid inhibits anti-PF4 antibody binding to PF4 and promotes dissociation of preformed complexes of PF4 and anti-PF4 antibodies [Citation52]. Danaparoid used in a cohort of six VITT patients led to significant improvement of clinical symptoms, as well as normalization of the platelet count and fibrin turnover, suggesting danaparoid as an option for anticoagulation in VITT [Citation62].
Danaparoid can be given intravenously and subcutaneously, although subcutaneous application requires rather large volumes to reach therapeutic dose levels (two 0.7 mL ampoules three times a day). Monitoring of danaparoid treatment via anti-Xa activity requires a specific calibrator, which might not be available in all laboratories [Citation63,Citation64]. Its long half-life can cause increased bleeding in case of urgent surgery. In case of severe renal impairment danaparoid achieves stable therapeutic levels with a 30% dose reduction [Citation24].
3.2. Low-sulfated heparin
Another potentially suitable therapeutic compound with the ability to interfere with PF4/heparin complexes is 2-O,3-O-desulfated heparin (ODSH). ODSH derived from UFH was originally developed as a molecule which maintains the anti-inflammatory effects of heparin but without its anticoagulatory effects [Citation65]. ODSH still mediates charge-based interaction with PF4 by its residual sulfate groups, but does not allow close approximation of two PF4 molecules that would be needed for antigenicity [Citation31]. Because of low anticoagulatory properties, ODSH can be applied in higher concentrations without the risk of bleeding. Similar to danaparoid, these high concentrations of ODSH are able to saturate the heparin binding sites on PF4 inhibiting the formation of PF4/heparin complexes in vitro [Citation31]. ODSH displaces PF4 from the platelet surface in a similar way as danaparoid [Citation31]. Due to these characteristics, ODSH likely also interferes with the mechanism of VITT by binding PF4. However, this compound has never entered clinical application.
3.3. Partially cleaved anti-PF4 monoclonal antibodies
Monoclonal antibodies (mAb) against PF4 with a nonfunctional Fc-part have been proposed as potential treatment for HIT and VITT (). The Fc-part of IgG can be modified by different approaches like deglycosylation with specific bacterial-derived enzymes, e.g. endo-β-N-acetylglucosaminidases of Streptococcus pyogenes (EndoS) or by cleavage with specific bacterial-derived enzymes, e.g. IgG-degrading enzyme of Streptococcus pyogenes (IdeS, also known as imlifidase). EndoS deglycosylates N-glycan of the Fc-part of IgG and IdeS cleaves heavy-chain IgG at its lower hinge region, both causing loss of capability of IgG to bind to FcγR [Citation66,Citation67]. These partially cleaved antibodies still bind to their antigen, but can no longer bind to and cross-link FcγRIIA. As these mAb cannot activate FcγRs, platelet activation is prevented. Partially cleaved mAbs recognizing the PF4-binding site of HIT antibodies [Citation67,Citation68] or the PF4-binding site of VITT antibodies [Citation66] competitively block binding of the patient’s anti-PF4 antibodies to their antigen. This approach showed promising results both in vitro [Citation66–68] and in vivo in murine models [Citation67,Citation68], but is so far not applied clinically.
Furthermore, it might also be possible to directly use IdeS as a therapeutic agent to cleave HIT or VITT IgG antibodies in circulating blood [Citation69]. This approach was already successful in a murine model of immune thrombocytopenia [Citation70] and in patients with anti-human leukocyte antigen antibodies in need for organ transplantation [Citation71,Citation72]. However, precaution is needed as not only the pathogenic but nearly all IgG will be cleaved, which could increase the risk for bacterial infections in IdeS treated patients.
4. Removing anti-PF4 antibodies
Lowering the number of circulating pathogenic antibodies lowers their likelihood to interact with FcγRIIA. Therefore, decreasing the amount of pathogenic antibodies by plasmapheresis or therapeutic plasma exchange (TPE) can be used as therapeutic strategy (). TPE has been used successfully in HIT in a case report of a 55-year-old male [Citation73] and a retrospective study in 24 patients (mean age of 55.2 years, 21% female) [Citation74]. Consecutively the same approach was also applied in VITT, especially in patients non-responsive to first-line anticoagulation and high-dose IVIG therapy [Citation75–77]. The case report of Major et al. describes one 50-year-old male patient [Citation75] and Patriquin et al. observed three female patients, aged 45, 46, and 48 years respectively [Citation76], in both studies all patients survived. In the study of Pavord et al., 17 patients with severe course of VITT underwent TPE and the therapeutic approach was associated with 90% survival rate [Citation77]. Additionally, TPE may also contribute to get rid of further factors that are able to enhance thromboinflammation. i.e. immune complexes, extracellular vesicles secreted from activated platelets, and excess pro-inflammatory cytokines [Citation78]. Although antibody titers decrease rapidly during TPE, typically 2–3 TPE sessions have to be performed to achieve low enough titers of anti-PF4 antibodies. The reason is redistribution of anti-PF4 antibodies from the extravascular compartment after end of TPE [Citation79]. If TPE is performed to remove anti-PF4/heparin antibodies to allow the use of heparin during or before major cardiac surgery, it is recommended to use plasma instead of albumin. Otherwise, reduction of fibrinogen and other clotting factors increases perioperative bleeding risk [Citation80].
5. Modulation of fc receptor functions
Blocking of FcγRIIA activation or signaling is a non-anticoagulant therapeutic approach in antibody-mediated procoagulant disorders.
5.1. IVIG
IVIG is polyclonal IgG pooled from several 1000 liters of healthy donor plasma. It is used for treatment of various hematological and immunological disorders, e.g. idiopathic thrombocytopenic purpura [Citation81]. The exact underlying mechanism how therapeutic effects of IVIG may ameliorate thromboinflammation and thrombosis are still debated and may be explained by different models.
IVIG was already proposed in 1981 to block activating FcγRs making them less available for auto-antibody binding [Citation82]. A first therapeutic application of IVIG in HIT was already published decades ago in 1989 [Citation83] and different IVIGs systematically assessed for their capacity to block FcγRIIA and platelet activation by HIT antibodies in 1994 [Citation84]. Today, the efficacy of IVIG therapy is well accepted based on numerous case observations [Citation85–87], but prospective randomized studies are lacking. IVIG can therefore not be recommended for general treatment of acute HIT [Citation20], but might be suitable for a subpopulation of patients, including autoimmune and spontaneous HIT syndromes [Citation87,Citation88].
For VITT, IVIG has been successfully applied in first-line therapeutic management, as recently reviewed [Citation47]. The therapeutic success was indicated by increased platelet numbers and normalization of biomarkers for hypercoagulation (i.e. reduced circulating D-dimers and increased fibrinogen levels).
The half-life of IgG depends on binding to another receptor, the neonate Fc receptor (FcRn). IVIG competitively saturates FcRn. This prevents binding of pathogenic autoantibodies. Physiologically, binding of IgG to FcRn protects them from intracellular degradation, increasing their half-life [Citation89]. If IgG autoantibodies are not able to bind FcRn because the receptor is already occupied by IVIG, the unbound pathogenic IgG autoantibodies are prone for lysosomal degradation and their half-life is significantly reduced [Citation90].
5.2. Inhibition of Fc-receptors
Similar to the occupation with IVIG, targeting the FcRn with designed mAbs e.g. rozanolixizumab also leads to decreased half-life of pathogenic IgG antibodies due to inhibition of their recycling [Citation91].
In addition to the above-described role in antibody half-life, a substantial role of FcRn in IgG effector functions gained interest. Cellular immune responses are influenced by the cooperation between FcγRIIA and FcRn, especially when IgGs bound to their corresponding antigens form larger immune complexes. FcRn bound IgG immune complexes promote innate immunity by increased pro-inflammatory cytokine expression and the expression of tissue factor (TF) and FXa [Citation92]. However, in disorders caused by immune complexes of antibodies bound to autologous proteins, like PF4/heparin-complexes in HIT or solely PF4 in VITT, pathologic activation of FcRn and FcγRIIA occurs. To tackle this, FcRn can be blocked by an anti-FcRn mAb, disabling the Fc part of the pathogenic antibodies to bind to FcRn and preventing subsequent expression of prothrombotic TF and FXa, as well as induction of thromboembolic events by anti-PF4/heparin antibodies in vitro and in preclinical studies in murine HIT models [Citation93].
Furthermore, new designed monomeric Fc fragments displayed binding capability to both FcRn and FcγR to inhibit platelet- and complement activation in a murine model of immune thrombocytopenic purpura [Citation94] and therefore are promising to ameliorate symptoms typically observed in HIT and VITT.
5.3. Designed ankyrin repeat proteins (DARPins)
Another approach directly targeting FcγRIIA function to mitigate thromboinflammation and thrombosis are high-affinity binders specific for FcγRIIA to antagonize binding of the Fc-part of IgG in immune-complexes [Citation12]. In the last few years, DARPins as designed molecules gained interest in various biomedical research areas. DARPins are specifically engineered proteins with antibody-mimicking properties. They bind with high affinity and high specificity [Citation95] to an antigen. DARPins have no Fc-domain, preventing that they also bind to and activate Fc-receptors. Previously bispecific DARPins directed against FcγRIIB and Fcε1 bound IgG have been generated and successfully used to inhibit hypersensitive immune responses in IgE-mediated allergic reactions [Citation96]. A recently developed FcγRIIA-specific DARPin (53.2.F11) inhibits platelet activation by HIT antibodies in vitro [Citation97]. DARPins have not been applied in vivo yet for treatment of VITT or HIT.
6. Inhibition of downstream FcγRIIa signaling
6.1. Spleen tyrosine kinase (Syk) inhibition
After FcγRIIA activation, signaling includes activation of Syk [Citation98], as depicted in . To prevent subsequent downstream signaling after pathogenic FcγRIIA activation, Syk inhibitors offer therapeutic potential in HIT and VITT. The potential to treat HIT was shown by the small molecule Syk inhibitor R406 that was able to significantly reduce HIT antibody‐mediated platelet activation and monocyte procoagulant activity in vitro [Citation99]. In addition, the Syk inhibitor PRT-060318 prevented platelet aggregation induced by HIT immunocomplexes in a transgenic mouse model of HIT [Citation100]. First ex vivo approaches with VITT anti-PF4 antibodies using the Syk inhibitors entospletinib or lanraplenib showed significantly reduced procoagulant platelets and thrombus formation [Citation101]. Contrary, fostamatinib that is currently used to treat chronic immune thrombocytopenia [Citation102], only showed slight therapeutic potential in the same experimental setup [Citation101]. None of the experimentally used Syk inhibitors has been applied to treat HIT or VITT patients so far.
6.2. Bruton Tyrosine Kinase (BTK) inhibition
After FcγRIIA activation, further downstream signaling includes activation of BTK [Citation103], as depicted in . This allows another therapeutic option focusing on thromboinflammation and the pro-thrombotic state in HIT and VITT [Citation104]. BTK inhibitors are suspected to pleiotropically impact multiple pathways of BTK signaling downstream of FcγRIIA activation, including platelet aggregation and subsequent dense granule secretion, P-selectin expression, monocyte activation, and formation of neutrophil extracellular traps [Citation104]. Several BTK inhibitors are approved for clinical use in hematologic malignancies, e.g. ibrutinib, acalabrutinib, or zanubrutinib.
Different in vitro tests showed the potential of BTK inhibition to inhibit platelet activation and aggregation. FcγRIIA-mediated platelet aggregation in whole blood was inhibited by preincubation with a variety of BTK inhibitors in a dose-dependent manner, with plasma concentrations of BTK inhibitors even lower than used for the treatment of hematologic malignancies [Citation105]. BTK inhibition also blocks platelet granule secretion and thus subsequent interaction with other blood cells like neutrophils [Citation105,Citation106]. In vitro, the BTK inhibitors ibrutinib, acalabrutinib, and fenebrutinib (a reversible BTK inhibitor) additionally showed the ability to inhibit platelet activation by HIT antibodies indicating a potential use of these drugs in the therapy of HIT [Citation105]. In a similar way, inhibition of platelet aggregation by ibrutinib, fenebrutinib, and rilzabrutinib in platelets treated with serum from VITT patients suggests low-dose BTK inhibition to be considered a therapeutic option for VITT [Citation107,Citation108].
Furthermore, inhibitory effects of BTKs on platelets may also be mediated by effects on C-type lectin-like receptor 2 (CLEC-2) signaling. CLEC-2 is abundantly expressed on platelets with a significant role in thromboinflammation [Citation109]. In vivo experiments with mice showed that low doses of ibrutinib block CLEC-2 downstream signaling of Syk and PLCγ phosphorylation [Citation110]. Both are common motives of ITAM signaling also seen in FcγRIIA-mediated platelet activation. Another receptor expressed on platelets also associated with ITAM signaling is glycoprotein VI (GPVI) that might also be targeted by BTK-inhibition to prevent platelet activation [Citation111].
The first successful treatment with ibrutinib in a 37-year-old female patient with persisting VITT-like antibodies causing arterial and venous thrombotic complications over 13 years was recently described [Citation112]. Treatment with ibrutinib 280 mg daily resulted in normalization of platelet count and D-dimer levels, and no further thrombotic complications or bleeding events occurred despite persisting platelet-activating anti-PF4 antibodies [Citation112].
Besides its role in hemostasis, BTK is a central molecule for B-cell activation and maturation and causes a reduction in circulating IgG [Citation113]. Potentially this also lowers VITT and HIT antibody titers.
In summary, first in vitro and in vivo data on inhibition of platelet activation and aggregation by BTK inhibitors in HIT and VITT showed promising results [Citation108,Citation114] and might also be well suited for other FcγRIIA-mediated disorders causing platelet aggregation. This is especially of interest as they can usually be given orally.
6.3. Selective inhibition of 12[S]-lipoxygenase (12-LOX)
12-LOX is a highly expressed oxylipin-producing enzyme in platelets demonstrated to regulate thrombosis by thrombin- and thromboxane-induced platelet aggregation [Citation115]. Another confirmed role of 12-LOX in platelet function is in FcγRIIa-mediated downstream signaling, i.e. PLCγ2 mediated mobilization of intracellular calcium [Citation116]. Therefore, 12-LOX was also identified as a suitable pharmacological target to modify platelet activation and thrombosis in vivo with 12-LOX inhibitors like ML355, also named VLX-1005 [Citation117]. First approaches in murine models of HIT showed reduction of thrombocytopenia and thrombosis induced by HIT antibodies using VLX-1005 [Citation118]. This marks pharmacological 12-LOX inhibition also as a potential treatment in other immune-mediated thrombotic disorders with thrombocytopenia like VITT.
7. GPIIb/IIIa receptor inhibition
The GPIIb/IIIa receptor (also known as integrin αIIbβ3) is the main receptor mediating platelet aggregation. GPIIb/IIIa-mediated platelet aggregation can be pharmacologically blocked by e.g. abciximab, eptifibatide and tirofiban [Citation119]. Moreover, GPIIb/IIIa receptor inhibition might be able to mitigate thromboinflammation by reduction of interleukin-1 formation and suppressed leukocyte–platelet aggregate formation [Citation119]. The GPIIb/IIIa receptor as therapeutic target in HIT has been proposed in the late 1990s [Citation120] but has not gained any clinical role. One exception is blockade of the GPIIb/IIIa receptor during cardiac surgery in patients with acute HIT to allow use of heparin [Citation121]. Similarly GPIIb/IIIa receptor inhibition was discussed for treatment of VITT [Citation122].
A general problem of this approach is that platelet aggregation but not major platelet activation with release of granules is inhibited [Citation123]. In addition, further caution is needed, as small molecules like eptifibatide and tirofiban can induce autoreactive antibodies leading to severe thrombocytopenia [Citation124].
8. Conclusion and further perspectives
First-line therapy of thrombosis and thromboinflammation in HIT and VITT is therapeutic dose anticoagulation. Escalation of treatment in severely affected patients is achieved by competitive inhibition of binding of immune complexes to FcγRIIA by IVIG as an additional established treatment option in autoimmune HIT and VITT [Citation47].
In addition to its anticoagulatory properties, danaparoid inhibits platelet activation by HIT antibodies by structurally interfering with the antigen in HIT, and in VITT. The reason for this is a much lower anticoagulatory activity compared to heparin causing more molecules to be present at therapeutic dose concentrations. This disrupts PF4/heparin complexes. A similar change in the stoichiometric balance disrupting complex formations of PF4 with polyanions is also observed in first experimental approaches with ODSH.
New approaches to inhibit the pathogenic effects of PF4 antibodies are blocking platelet Fc-receptors, i.e. FcγRIIa and FcRn by monoclonal antibodies or blocking binding of the pathogenic antibodies to PF4 and PF4/heparin complexes, respectively, by monoclonal antibodies with modified Fc-part, which cannot bind to Fc-receptors. Another potential option to block Fc-receptors are specifically designed DARPINs, which can easily be produced and effectively inhibit FcγRIIA-dependent platelet activation in vitro. Two new approaches inhibit PF4 antibody-mediated platelet activation by inhibiting downstream signaling of FcγRIIa. The first are BTK inhibitors, the other inhibition of the thromboxane platelet activation pathway by VLX-1005.
9. Expert opinion
PF4-antibody-mediated disorders are well-defined prothrombotic disorders characterized by FcγRIIA-dependent activation of platelets, monocytes, neutrophils, and indirect activaton of endothelial cells. They bridge between innate and adaptive immunity and coagulation. They are a perfect example of uncontrolled activation of the immune- and the coagulation system resulting in a life-threatening state of thromboinflammation. HIT is known since the 1960s. Understanding its pathogenesis, characterized by FcγRIIA-dependent platelet activation by immunocomplexes of anti-PF4 antibodies bound to PF4/heparin-complexes was instrumental to understand VITT. VITT is a serious adverse event triggered by adenovirus vector based COVID-19 vaccines. This has recently led to recognize anti-PF4 antibodies, which occur spontaneously, without previous heparin treatment or vaccination, as a cause of severe, sometimes recurrent thrombotic complications.
Detailed understanding of the pathogenesis of PF4-antibody-mediated disorders allowed targeted development of treatment approaches. Besides anticoagulation with a variety of excellent alternatives to heparin, i.e. direct thrombin or FXa inhibitors, understanding of PF4-antibody disorders has triggered treatments which interfere with the involved antigens. One example is danaparoid, which is on the one hand an anticoagulant, but on the other hand interferes with the antigen in PF4 disorders by binding to PF4 and disrupting PF4 complexes. This effect of danaparoid might also be relevant in patients with sepsis or post-transplantation thromboinflammation. In this regard also 2-O,3-O-desulfated heparin might be of interest, although this compound has never entered clinical application.
Blockade of FcγRIIA is well established by IVIG. Designed ankyrin repeat proteins might become a new and interesting alternative, especially as supply with IVIG is limited by human plasma donation and the worldwide increasing demand can barely be supplied already today. If the Fc-receptors cannot be blocked, inhibition of downstream Fc-receptor signaling is an interesting option. Bruton tyrosine kinase inhibitors and inhibition of the thromboxane A2 receptor signaling pathway are promising candidates.
However, nearly all novel approaches discussed above are based on in vitro and preclinical observations. The few treatment options which have already entered clinical application are based on case observations.
Modulation of thromboinflammation might be important beyond PF4-disorders. Potential applications might be modulation of post-ischemia reperfusion after acute myocardial infarction [Citation125] or stroke [Citation126], which currently cannot be treated effectively by standard approaches using dual anti-platelet therapies. However, as shown in this review article, the approaches to modulate PF4 antibody-mediated thromboinflammation are highly specific and adapted to the pathogenesis of PF4-antibody mediated disorders. Thromboinflammation after myocardial infarction or stroke most likely requires therapeutic interventions adapted to their pathogenesis. As exemplified for PF4-antibody-mediated disorders, detailed understanding of the underlying molecular mechanism of thromboinflammation in different clinical scenarios is instrumental in designing new therapeutic interventions.
Article highlights
Anticoagulation is a mainstay of treatment in HIT and VITT, but some patients require additional treatment.
Besides anticoagulation, danaparoid inhibits PF4/heparin-complex-formation and PF4-binding to anti-PF4 antibodies.
Additional therapeutic strategies include: therapeutic plasma exchange to lower anti-PF4 antibody plasma concentration; modulation of Fc-receptor function, e.g. by intravenous immunoglobulins (IVIGs), and downstream signaling, e.g. by Bruton Tyrosin Kinase inhibition.
Potential novel approaches in preclinical development are partially cleaved anti-PF4 monoclonal antibodies against PF4/heparin complexes, PF4, or FcγRIIa and FcRn; and designed ankyrin repeat proteins (DARPins) blocking FcγRIIa.
List of abbreviations
| 12-LOX | = | 12[S]-lipoxygenase |
| aPTT | = | activated partial thromboplastin time |
| BTK | = | Bruton tyrosine kinase |
| CLEC-2 | = | C-type lectin-like receptor 2 |
| DAG | = | diacetyl glycerol |
| DARPins | = | Designed ankyrin repeat proteins |
| DOACs | = | direct oral anticoagulants |
| DTI | = | direct thrombin-inhibitor |
| EndoS | = | endo-β-N-acetylglucosaminidases of Streptococcus pyogenes |
| FcRn | = | neonate Fc receptor |
| FcγR | = | Fcγ-receptors |
| FXa | = | factor Xa |
| GPIIb/IIIa | = | glycoprotein IIb/IIIa |
| GPVI | = | glycoprotein VI |
| HIT | = | heparin-induced thrombocytopenia |
| ICU | = | intensive care unit |
| IdeS | = | IgG-degrading enzyme of Streptococcus pyogenes |
| Ig | = | immunoglobulin |
| IP3 | = | inositol triphosphate |
| ITAMs | = | immunoreceptor tyrosine-based activation motifs |
| IVIGs | = | intravenous immunoglobulins |
| LMWH | = | low molecular weight heparin |
| mAb | = | monoclonal antibodies |
| ODSH | = | 2-O,3-O-desulfated heparin |
| PF4 | = | platelet factor 4 |
| PI3 | = | phosphoinositid-3 |
| PIP2 | = | phosphatidylinositol-(4,5)-bisphosphate |
| PIP3 | = | phosphatidylinositol-(3,4,5)-triphosphate |
| PKC | = | protein kinase C |
| PLCγ | = | phosphoinositide phospholipase Cγ |
| SFKs | = | Src family kinases |
| Syk | = | spleen tyrosine kinase |
| TF | = | tissue factor |
| TPE | = | therapeutic plasma exchange |
| UFH | = | unfractionated heparin |
| VITT | = | vaccine-induced immune thrombocytopenia and thrombosis |
| VKAs | = | vitamin K antagonists |
Declaration of interests
A Greinacher reports grants and nonfinancial support from Aspen, Boehringer Ingelheim, MSD, Bristol Myers Squibb (BMS), Paringenix, Bayer Healthcare, Gore, Inc., Rovi, Sagent, Biomarin/Prosensa, personal fees from Aspen, Boehringer Ingelheim, MSD, Macopharma, BMS, Chromatec, Werfen (Instrumentation Laboratory), and nonfinancial support from Boehringer Ingelheim, Portola, Ergomed, GTH e.V.
All other authors have no relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript. This includes employment, consultancies, honoraria, stock ownership or options, expert testimony, grants or patents received or pending, or royalties.
Author contributions
Conceptualization, L Müller and A Greinacher; resources, L Schönborn and A Greinacher; writing – original draft preparation, L Müller and V A S Dabbiru; writing – review and editing, L Schönborn and A Greinacher; visualization, V A S Dabbiru; supervision, A Greinacher; project administration, A Greinacher; funding acquisition, A Greinacher and L Schönborn.
All authors have read and agreed to the published version of the manuscript.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Therapeutic_strategies_in_FcγIIa_Receptor_alphabeticalListofAbbreviations (1).docx
Download MS Word (480 B)Acknowledgments
L Schönborn was supported within the Gerhard-Domagk-Research-Program by the University Medicine Greifswald and by the American Society of Hematology with a Global Research Award. Figures in this article were created with BioRender.com.
Supplementary Material
Supplemental data for this article can be accessed online at https://doi.org/10.1080/14656566.2024.2328241
Additional information
Funding
References
- Sobota A, Strzelecka-Kiliszek A, Gładkowska E, et al. Binding of IgG-opsonized particles to Fc gamma R is an active stage of phagocytosis that involves receptor clustering and phosphorylation. J Immunol 2005;175(7):4450–4457. doi: 10.4049/jimmunol.175.7.4450
- Daëron M Fc receptor biology. Annu Rev Immunol 1997;15:203–234 1. doi: 10.1146/annurev.immunol.15.1.203
- Tridandapani S, Siefker K, Teillaud J-L, et al. Regulated expression and inhibitory function of fcgamma RIIb in human monocytic cells. J Biol Chem 2002;277(7):5082–5089. doi: 10.1074/jbc.M110277200
- Mkaddem SB, Murua A, Flament H, et al. Lyn and fyn function as molecular switches that control immunoreceptors to direct homeostasis or inflammation. Nat Commun. 2017;8(1):246. doi: 10.1038/s41467-017-00294-0
- Mkaddem SB, Benhamou M, Monteiro RC. Understanding fc receptor involvement in inflammatory diseases: from mechanisms to new therapeutic tools. Front Immunol. 2019;10:811. doi: 10.3389/fimmu.2019.00811
- Walker TR, Watson SP Synergy between Ca2+ and protein kinase C is the major factor in determining the level of secretion from human platelets. Biochem J 1993;289 (Pt 1):277–282. doi: 10.1042/bj2890277
- Qiao J, Al-Tamimi M, Baker RI, et al. The platelet Fc receptor, FcγRIIa. Immunol Rev. 2015;268(1):241–252. doi: 10.1111/imr.12370
- Golebiewska EM, Poole AW. Platelet secretion: from haemostasis to wound healing and beyond. Blood Rev. 2015;29(3):153–162. doi: 10.1016/j.blre.2014.10.003
- van de Winkel JG, Capel PJ Human IgG Fc receptor heterogeneity: molecular aspects and clinical implications. Immunol Today 1993;14(5):215–221. doi: 10.1016/0167-5699(93)90166-I
- Harrison PT, Davis W, Norman JC, et al. Binding of monomeric immunoglobulin G triggers Fc gamma RI-mediated endocytosis. J Biol Chem. 1994;269(39):24396–24402. doi: 10.1016/S0021-9258(19)51097-3
- Hogarth PM. Fc receptors are major mediators of antibody based inflammation in autoimmunity. Curr Opin Immunol. 2002;14(6):798–802. doi: 10.1016/S0952-7915(02)00409-0
- Pietersz GA, Mottram PL, van de Velde NC, et al. Inhibition of destructive autoimmune arthritis in FcgammaRIIa transgenic mice by small chemical entities. Immunol Cell Biol 2009;87(1):3–12. doi: 10.1038/icb.2008.82
- Stefanski A-L, Nitschke E, Dörner T. Thromboinflammation: Dynamik physiologischer und pathologischer Wechselwirkungen von Entzündung und Koagulation. Aktuelle Rheumatologie. 2022;47(6):478–482. doi: 10.1055/a-1947-5200
- Pai M. Epidemiology of VITT. Semin Hematol 2022;59(2):72–75. doi: 10.1053/j.seminhematol.2022.02.002
- Warkentin TE. Vaccine-induced immune thrombotic thrombocytopenia (VITT). Semin Hematol. 2022;59(2):57–58. doi: 10.1053/j.seminhematol.2022.03.004
- Arepally GM, Ortel TL. Clinical practice. Heparin-induced thrombocytopenia. N Engl J Med. 2006;355(8):809–817. doi: 10.1056/NEJMcp052967
- Greinacher A. CLINICAL PRACTICE. Heparin-induced thrombocytopenia. N Engl J Med. 2015;373(3):252–261. doi: 10.1056/NEJMcp1411910
- Greinacher A, Selleng K, Palankar R, et al. Insights in ChAdOx1 nCoV-19 vaccine-induced immune thrombotic thrombocytopenia. Blood. 2021;138(22):2256–2268. doi: 10.1182/blood.2021013231
- Cuker A, Arepally GM, Chong BH, et al. American Society of Hematology 2018 guidelines for management of venous thromboembolism: heparin-induced thrombocytopenia. Blood Adv. 2018;2(22):3360–3392. doi: 10.1182/bloodadvances.2018024489
- Gruel Y, de ME, Pouplard C, et al. Diagnosis and management of heparin-induced thrombocytopenia. Anaesth Crit Care Pain Med. 2020;39(2):291–310. doi: 10.1016/j.accpm.2020.03.012
- Pavord S, Hunt BJ, Horner D, et al. Vaccine induced immune thrombocytopenia and thrombosis: summary of NICE guidance. BMJ. 2021;375:n2195. doi: 10.1136/bmj.n2195
- Carré J, Jourdi G, Gendron N, et al. Recent advances in anticoagulant treatment of immune thrombosis: a focus on direct oral anticoagulants in heparin-induced thrombocytopenia and anti-phospholipid syndrome. Int J Mol Sci. 2021;23(1):93. doi: 10.3390/ijms23010093
- Warkentin TE, Lim W. Can heparin‐induced thrombocytopenia be associated with fondaparinux use? Reply to a rebuttal. J Thromb Haemost. 2008;6(7):1243–1246. doi: 10.1111/j.1538-7836.2008.02972.x
- Greinacher A, Selleng K, Warkentin TE. Autoimmune heparin-induced thrombocytopenia. J Thromb Haemost. 2017;15(11):2099–2114. doi: 10.1111/jth.13813
- Manji F, Warkentin TE, Sheppard J-A, et al. Fondaparinux cross-reactivity in heparin-induced thrombocytopenia successfully treated with high-dose intravenous immunoglobulin and rivaroxaban. Platelets 2019;31(1):124–127. doi: 10.1080/09537104.2019.1652263
- WHO. Guidance for clinical case management of thrombosis with thrombocytopenia syndrome (TTS) following vaccination to prevent coronavirus disease (COVID-19). Geneva: World Health Organization; 2023. https://www.who.int/publications/i/item/9789240061989
- Farner B, Eichler P, Kroll H, et al. A comparison of danaparoid and lepirudin in heparin-induced thrombocytopenia. Thromb Haemost 2001;85(6):950–957. doi: 10.1055/s-0037-1615946
- Fathi M. Heparin-induced thrombocytopenia (HIT): identification and treatment pathways. Gcsp 2018;2018(2):15. doi: 10.21542/gcsp.2018.15
- Nilius H, Kaufmann J, Cuker A, et al. Comparative effectiveness and safety of anticoagulants for the treatment of heparin-induced thrombocytopenia. American J Hematol 2021;96(7):805–815. doi: 10.1002/ajh.26194
- Warkentin TE, Kelton JG. A 14-year study of heparin-induced thrombocytopenia. Am J Med. 1996;101(5):502–507. doi: 10.1016/S0002-9343(96)00258-6
- Krauel K, Hackbarth C, Fürll B, et al. Heparin-induced thrombocytopenia: in vitro studies on the interaction of dabigatran, rivaroxaban, and low-sulfated heparin, with platelet factor 4 and anti-PF4/heparin antibodies. Blood. 2012;119(5):1248–1255. doi: 10.1182/blood-2011-05-353391
- Warkentin TE, Pai M, Linkins L-A. Direct oral anticoagulants for treatment of HIT: update of Hamilton experience and literature review. Blood. 2017;130(9):1104–1113. doi: 10.1182/blood-2017-04-778993
- Salih F, Schönborn L, Kohler S, et al. Vaccine-induced thrombocytopenia with severe headache. N Engl J Med. 2021;385(22):2103–2105. doi: 10.1056/NEJMc2112974
- Oldenburg J, Klamroth R, Langer F, et al. Updated GTH statement on vaccination with the AstraZeneca COVID-19, 22.03.2021. [cited 2024 Mar 5]. Available from: https://gth-online.org/wp-content/uploads/2021/03/GTH_Stellungnahme_AstraZeneca_engl._3_22_2021.pdf
- Toorop MMA, van Rein N, Nierman MC, et al. Inter- and intra-individual concentrations of direct oral anticoagulants: the KIDOAC study. J Thromb Haemost. 2022;20(1):92–103. doi: 10.1111/jth.15563
- Kreimann M, Brandt S, Krauel K, et al. Binding of anti-platelet factor 4/heparin antibodies depends on the thermodynamics of conformational changes in platelet factor 4. Blood 2014;124(15):2442–2449. doi: 10.1182/blood-2014-03-559518
- Nguyen T-H, Greinacher A, Delcea M. Quantitative description of thermodynamic and kinetic properties of the platelet factor 4/heparin bonds. Nanoscale. 2015;7(22):10130–10139. doi: 10.1039/C5NR02132D
- Savi P, Chong BH, Greinacher A, et al. Effect of fondaparinux on platelet activation in the presence of heparin-dependent antibodies: a blinded comparative multicenter study with unfractionated heparin. Blood. 2005;105(1):139–144. doi: 10.1182/blood-2004-05-2010
- Linkins L-A, Hu G, Warkentin TE. Systematic review of fondaparinux for heparin-induced thrombocytopenia: when there are no randomized controlled trials. Res Pract Thromb Haemost. 2018;2(4):678–683. doi: 10.1002/rth2.12145
- Arnold DM. Heparin or nonheparin anticoagulants for VITT. Blood. 2022;139(23):3358–3359. doi: 10.1182/blood.2022016423
- Bauersachs RM. Fondaparinux sodium: recent advances in the management of thrombosis. J Cardiovasc Pharmacol Ther. 2023;28:10742484221145010. doi: 10.1177/10742484221145010
- Lewis BE, Wallis DE, Berkowitz SD, et al. Argatroban anticoagulant therapy in patients with heparin-induced thrombocytopenia. Circulation. 2001;103(14):1838–1843. doi: 10.1161/01.CIR.103.14.1838
- Koster A, Buz S, Hetzer R, et al. Anticoagulation with argatroban in patients with heparin-induced thrombocytopenia antibodies after cardiovascular surgery with cardiopulmonary bypass: first results from the ARG-E03 trial. J Thorac Cardiovasc Surg. 2006;132(3):699–700. doi: 10.1016/j.jtcvs.2006.04.034
- Graf T, Thiele T, Klingebiel R, et al. Immediate high-dose intravenous immunoglobulins followed by direct thrombin-inhibitor treatment is crucial for survival in sars-covid-19-adenoviral vector vaccine-induced immune thrombotic thrombocytopenia VITT with cerebral sinus venous and portal vein thrombosis. J Neurol. 2021;268(12):4483–4485. doi: 10.1007/s00415-021-10599-2
- Di Nisio M, Middeldorp S, Büller HR. Direct thrombin inhibitors. N Engl J Med. 2005;353(10):1028–1040. doi: 10.1056/NEJMra044440
- Warkentin TE, Greinacher A, Koster A. Bivalirudin. Thromb Haemost 2008;99(5):830–839. doi: 10.1160/TH07-10-0644
- Gabarin N, Arnold DM, Nazy I, et al. Treatment of vaccine-induced immune thrombotic thrombocytopenia (VITT). Semin Hematol. 2022;59(2):89–96. doi: 10.1053/j.seminhematol.2022.03.002
- Warkentin TE. Anticoagulant failure in coagulopathic patients: PTT confounding and other pitfalls. Expert Opin Drug Saf. 2014;13(1):25–43. doi: 10.1517/14740338.2013.823946
- Srinivasan AF, Rice L, Bartholomew JR, et al. Warfarin-induced skin necrosis and venous limb gangrene in the setting of heparin-induced thrombocytopenia. Arch Internal Med. 2004;164(1):66–70. doi: 10.1001/archinte.164.1.66
- Warkentin TE, Elavathil LJ, Hayward CP, et al. The pathogenesis of venous limb gangrene associated with heparin-induced thrombocytopenia. Ann internal med 1997;127(9):804–812. doi: 10.7326/0003-4819-127-9-199711010-00005
- Huynh A, Kelton JG, Arnold DM, et al. Antibody epitopes in vaccine-induced immune thrombotic thrombocytopaenia. Nature. 2021;596(7873):565–569. doi: 10.1038/s41586-021-03744-4
- Singh A, Toma F, Uzun G, et al. The interaction between anti-PF4 antibodies and anticoagulants in vaccine-induced thrombotic thrombocytopenia. Blood. 2022;139(23):3430–3438. doi: 10.1182/blood.2021013839
- Schönborn L, Esteban O, Wesche J, et al. Heparin- and vaccine-independent anti-platelet factor 4 Immunothrombosis, 2023. Available at SSRN: https://ssrn.com/abstract=4519530
- Garcia DA, Baglin TP, Weitz JI, et al. Parenteral anticoagulants: antithrombotic therapy and prevention of thrombosis, 9th ed: American college of chest physicians evidence-based clinical practice guidelines. Chest 2012;141(2 Suppl):e24–e43. doi: 10.1378/chest.11-2291
- Harenberg J, Zimmermann R, Schwarz F, et al. Treatment of heparin-induced thrombocytopenia with thrombosis by new heparinoid. Lancet (London, England) 1983;1(8331):986–987. doi: 10.1016/S0140-6736(83)92107-4
- Wilde MI, Markham AD A review of its pharmacology and clinical use in the management of heparin-induced thrombocytopenia. Drugs 1997;54(6):903–924. doi: 10.2165/00003495-199754060-00008
- Chong BH, Magnani HN Orgaran in heparin-induced thrombocytopenia. Haemostasis 1992;22(2):85–91. doi: 10.1159/000216299
- Keng TB, Chong BH. Heparin-induced thrombocytopenia and thrombosis syndrome: in vivo cross-reactivity with danaparoid and successful treatment with r-hirudin. Br J Haematol. 2001;114(2):394–396. doi: 10.1046/j.1365-2141.2001.02943.x
- Magnani HN, Gallus A. Heparin-induced thrombocytopenia (HIT). A report of 1,478 clinical outcomes of patients treated with danaparoid (orgaran) from 1982 to mid-2004. Thromb Haemost 2006;95(6):967–981. doi: 10.1160/TH05-07-0489
- Greinacher A, Alban S, Omer-Adam MA, et al. Heparin-induced thrombocytopenia: a stoichiometry-based model to explain the differing immunogenicities of unfractionated heparin, low-molecular-weight heparin, and fondaparinux in different clinical settings. Thromb Res. 2008;122(2):211–220. doi: 10.1016/j.thromres.2007.11.007
- Krauel K, Fürll B, Warkentin TE, et al. Heparin-induced thrombocytopenia—therapeutic concentrations of danaparoid, unlike fondaparinux and direct thrombin inhibitors, inhibit formation of platelet factor 4-heparin complexes. J Thromb Haemost 2008;6(12):2160–2167. doi: 10.1111/j.1538-7836.2008.03171.x
- Myllylahti L, Pitkänen H, Magnani H, et al. Experience of danaparoid to treat vaccine-induced immune thrombocytopenia and thrombosis, VITT. Thromb J. 2022;20(1):4. doi: 10.1186/s12959-021-00362-y
- Kitchen S, Iampietro R, Woolley AM, et al. Anti xa monitoring during treatment with low molecular weight heparin or Danaparoid: inter-assay variability. Thromb Haemost 1999;82(10):1289–1293. doi: 10.1055/s-0037-1614377
- Boissier E, Senage T, Babuty A, et al. Heparin anti-xa activity, a readily available unique test to quantify apixaban, rivaroxaban, fondaparinux, and danaparoid levels. Anesthesia & Analgesia. 2021;132(3):707–716. doi: 10.1213/ANE.0000000000005114
- Rao NV, Argyle B, Xu X, et al. Low anticoagulant heparin targets multiple sites of inflammation, suppresses heparin-induced thrombocytopenia, and inhibits interaction of RAGE with its ligands. Am J Physiol Cell Physiol. 2010;299(1):C97–110. doi: 10.1152/ajpcell.00009.2010
- Vayne C, Palankar R, Billy S, et al. The deglycosylated form of 1E12, a monoclonal anti-PF4 IgG, strongly inhibits antibody-triggered cellular activation in vaccine-induced thrombotic thrombocytopenia, and is a potential new treatment for Vιττ. Blood. 2021;138(Supplement 1):582. doi: 10.1182/blood-2021-147922
- Kizlik-Masson C, Deveuve Q, Zhou Y, et al. Cleavage of anti-PF4/heparin IgG by a bacterial protease and potential benefit in heparin-induced thrombocytopenia. Blood. 2019;133(22):2427–2435. doi: 10.1182/blood.2019000437
- Sarkar A, Khandelwal S, Koma GT, et al. Treatment of thrombocytopenia and thrombosis in HIT in mice using deglycosylated KKO: a novel therapeutic? Blood Adv. 2023;7(15):4112–4123. doi: 10.1182/bloodadvances.2023009661
- Vayne C, Rollin J, Gruel Y, et al. PF4 immunoassays in vaccine-induced thrombotic thrombocytopenia. N Engl J Med. 2021;385(4):376–378. doi: 10.1056/NEJMc2106383
- Johansson BP, Shannon O, Björck L, et al. IdeS: a bacterial proteolytic enzyme with therapeutic potential. PLoS One. 2008;3(2):e1692. doi: 10.1371/journal.pone.0001692
- Jordan SC, Lorant T, Choi J, et al. IgG endopeptidase in highly sensitized patients undergoing transplantation. N Engl J Med. 2017;377(5):442–453. doi: 10.1056/NEJMoa1612567
- Lonze BE, Tatapudi VS, Weldon EP, et al. IdeS (imlifidase): a novel agent that cleaves human IgG and permits successful kidney transplantation across high-strength donor-specific antibody. Ann Surg. 2018;268(3):488–496. doi: 10.1097/SLA.0000000000002924
- Cho JH, Parilla M, Treml A, et al. Plasma exchange for heparin-induced thrombocytopenia in patients on extracorporeal circuits: a challenging case and a survey of the field. J of Clinical Apheresis 2019;34(1):64–72. doi: 10.1002/jca.21671
- Moreno-Duarte I, Cooter M, Onwuemene OA, et al. Clinical outcomes of cardiac surgery patients undergoing therapeutic plasma exchange for heparin-induced thrombocytopenia. Vox Sang. 2021;116(2):217–224. doi: 10.1111/vox.13008
- Major A, Carll T, Chan CW, et al. Refractory vaccine-induced immune thrombotic thrombocytopenia (VITT) managed with delayed therapeutic plasma exchange (TPE). J of Clinical Apheresis 2022;37(1):117–121. doi: 10.1002/jca.21945
- Patriquin CJ, Laroche V, Selby R, et al. Therapeutic plasma exchange in vaccine-induced immune thrombotic thrombocytopenia. N Engl J Med. 2021;385(9):857–859. doi: 10.1056/NEJMc2109465
- Pavord S, Scully M, Hunt BJ, et al. Clinical features of vaccine-induced immune thrombocytopenia and thrombosis. N Engl J Med. 2021;385(18):1680–1689. doi: 10.1056/NEJMoa2109908
- Rock G, Weber V, Stegmayr B. Therapeutic plasma exchange (TPE) as a plausible rescue therapy in severe vaccine-induced immune thrombotic thrombocytopenia. Transfus Apheresis Sci. 2021;60(4):103174. doi: 10.1016/j.transci.2021.103174
- Warkentin TE, Sheppard J-A, Chu FV, et al. Plasma exchange to remove HIT antibodies: dissociation between enzyme-immunoassay and platelet activation test reactivities. Blood 2015;125(1):195–198. doi: 10.1182/blood-2014-07-590844
- Jones CG, Pechauer SM, Curtis BR, et al. Normal plasma IgG inhibits HIT antibody-mediated platelet activation: implications for therapeutic plasma exchange. Blood 2018;131(6):703–706. doi: 10.1182/blood-2017-08-803031
- Nimmerjahn F, Ravetch JV Anti-inflammatory actions of intravenous immunoglobulin. Annu Rev Immunol 2008;26:513–533 1. doi: 10.1146/annurev.immunol.26.021607.090232
- Imbach P, Barandun S, d’Apuzzo V, et al. High-dose intravenous gammaglobulin for idiopathic thrombocytopenic purpura in childhood. Lancet (London, England) 1981;1(8232):1228–1231. doi: 10.1016/S0140-6736(81)92400-4
- Frame JN, Mulvey KP, Phares JC, et al. Correction of severe heparin-associated thrombocytopenia with intravenous immunoglobulin. Ann internal med 1989;111(11):946–947. doi: 10.7326/0003-4819-111-11-946
- Greinacher A, Liebenhoff U, Kiefel V, et al. Heparin-associated thrombocytopenia: the effects of various intravenous IgG preparations on antibody mediated platelet activation—a possible new indication for high dose i.V. Thromb Haemost 1994;71(5):641–645. doi: 10.1055/s-0038-1642496
- Irani M, Siegal E, Jella A, et al. Use of intravenous immunoglobulin G to treat spontaneous heparin-induced thrombocytopenia. Transfusion 2018;59(3):931–934. doi: 10.1111/trf.15105
- Padmanabhan A, Jones CG, Pechauer SM, et al. Ivig for treatment of severe refractory heparin-induced thrombocytopenia. Chest. 2017;152(3):478–485. doi: 10.1016/j.chest.2017.03.050
- Warkentin TE. High-dose intravenous immunoglobulin for the treatment and prevention of heparin-induced thrombocytopenia: a review. Exp Rev Hematol. 2019;12(8):685–698. doi: 10.1080/17474086.2019.1636645
- Mohanty E, Nazir S, Sheppard J-A, et al. High-dose intravenous immunoglobulin to treat spontaneous heparin-induced thrombocytopenia syndrome. J Thromb Haemost 2019;17(5):841–844. doi: 10.1111/jth.14411
- Roopenian DC, Christianson GJ, Sproule TJ, et al. The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis, and fate of IgG-fc-coupled drugs.J Immunol 2003;170(7):3528–3533. doi: 10.4049/jimmunol.170.7.3528
- Hansen RJ, Balthasar JP Intravenous immunoglobulin mediates an increase in anti-platelet antibody clearance via the FcRn receptor. Thromb Haemost 2002;88(6):898–899. doi: 10.1055/s-0037-1613331
- Smith B, Christodoulou L, Clargo A, et al. Generation of two high affinity anti-mouse FcRn antibodies: inhibition of IgG recycling in wild type mice and effect in a mouse model of immune thrombocytopenia. Int Immunopharmacol. 2019;66:362–365. doi: 10.1016/j.intimp.2018.11.040
- Pyzik M, Kozicky LK, Gandhi AK, et al. The therapeutic age of the neonatal Fc receptor. Nat Rev Immunol. 2023;23(7):415–432. doi: 10.1038/s41577-022-00821-1
- Cines DB, Zaitsev S, Rauova L, et al. FcRn augments induction of tissue factor activity by IgG-containing immune complexes. Blood. 2020;135(23):2085–2093. doi: 10.1182/blood.2019001133
- Monnet C, Jacque E, de RC, et al. The dual targeting of FcRn and FcγRs via monomeric Fc fragments results in strong inhibition of IgG-dependent autoimmune pathologies. Front Immunol. 2021;12:728322. doi: 10.3389/fimmu.2021.728322
- Plückthun A. Designed ankyrin repeat proteins (DARPins): binding proteins for research, diagnostics, and therapy. Annu Rev Pharmacol Toxicol. 2015;55(1):489–511. doi: 10.1146/annurev-pharmtox-010611-134654
- Zellweger F, Gasser P, Brigger D, et al. A novel bispecific DARPin targeting FcγRIIB and FcεRI-bound IgE inhibits allergic responses. Allergy. 2017;72(8):1174–1183. doi: 10.1111/all.13109
- Riechert V, Hein S, Visser M, et al. FcγRIIA-specific DARPins as novel tools in blood cell analysis and platelet aggregation. J Biol Chem. 2023;299(6):104743. doi: 10.1016/j.jbc.2023.104743
- Kiener PA, Rankin BM, Burkhardt AL, et al. Cross-linking of Fc gamma receptor I (Fc gamma RI) and receptor II (Fc gamma RII) on monocytic cells activates a signal transduction pathway common to both Fc receptors that involves the stimulation of p72 syk protein tyrosine kinase. J Biol Chem. 1993;268(32):24442–24448. doi: 10.1016/S0021-9258(20)80545-6
- Lhermusier T, van Rottem J, Garcia C, et al. The Syk-kinase inhibitor R406 impairs platelet activation and monocyte tissue factor expression triggered by heparin-PF4 complex directed antibodies. J Thromb Haemost. 2011;9(10):2067–2076. doi: 10.1111/j.1538-7836.2011.04470.x
- Reilly MP, Sinha U, André P, et al. PRT-060318, a novel syk inhibitor, prevents heparin-induced thrombocytopenia and thrombosis in a transgenic mouse model. Blood. 2011;117(7):2241–2246. doi: 10.1182/blood-2010-03-274969
- Zlamal J, Singh A, Uzun G, et al. PB1344 impact of spleen tyrosine kinase inhibition on vaccine-induced thrombotic thrombocytopenia antibody-induced procoagulant platelet and thrombus formation. Res Pract Thromb Haemost. 2023;7:101444. doi: 10.1016/j.rpth.2023.101444
- Connell NT, Berliner N. Fostamatinib for the treatment of chronic immune thrombocytopenia. Blood. 2019;133(19):2027–2030. doi: 10.1182/blood-2018-11-852491
- Duan R, Goldmann L, Li Y, et al. Spontaneous platelet aggregation in blood is mediated by FcγRIIA stimulation of Bruton’s tyrosine kinase. Int J Mol Sci. 2021;23(1):76. doi: 10.3390/ijms23010076
- von Hundelshausen P, Lorenz R, Siess W, et al. Vaccine-Induced Immune Thrombotic Thrombocytopenia (VITT): targeting pathomechanisms with Bruton Tyrosine Kinase Inhibitors. Thromb Haemost 2021;121(11):1395–1399. doi: 10.1055/a-1481-3039
- Goldmann L, Duan R, Kragh T, et al. Oral Bruton tyrosine kinase inhibitors block activation of the platelet Fc receptor CD32a (FcγRIIA): a new option in HIT? Blood Adv. 2019;3(23):4021–4033. doi: 10.1182/bloodadvances.2019000617
- Rigg RA, Aslan JE, Healy LD, et al. Oral administration of Bruton’s tyrosine kinase inhibitors impairs GPVI-mediated platelet function. Am J Physiol Cell Physiol. 2016;310(5):C373–80. doi: 10.1152/ajpcell.00325.2015
- Smith CW, Harbi MH, Garcia-Quintanilla L, et al. The Btk inhibitor AB-95-LH34 potently inhibits atherosclerotic plaque-induced thrombus formation and platelet procoagulant activity. J Thromb Haemost 2022;20(12):2939–2952. doi: 10.1111/jth.15899
- Weber C, von Hundelshausen P, Siess W VITT after ChAdOx1 nCoV-19 vaccination. N Engl J Med 2021;385(23):2202–2205. doi: 10.1056/NEJMc2111026
- Meng D, Luo M, Liu B. The role of CLEC-2 and its ligands in thromboinflammation. Front Immunol. 2021;12:688643. doi: 10.3389/fimmu.2021.688643
- Nicolson PLR, Nock SH, Hinds J, et al. Low-dose btk inhibitors selectively block platelet activation by CLEC-2. Haematologica 2021;106(1):208–219. doi: 10.3324/haematol.2019.218545
- Zheng TJ, Lofurno ER, Melrose AR, et al. Assessment of the effects of syk and BTK inhibitors on GPVI-mediated platelet signaling and function. Am J Physiol Cell Physiol. 2021;320(5):C902–C915. doi: 10.1152/ajpcell.00296.2020
- Lindhoff-Last E, Schönborn L, Zaninetti C, et al. Rescue therapy in chronic prothrombotic autoimmune anti-PF4 disorder. N Engl J Med. 2023;389(14):1339–1341. doi: 10.1056/NEJMc2309016
- Hemme E, Biskop D, Depuydt MAC, et al. Bruton’s tyrosine kinase inhibition by acalabrutinib does not affect early or advanced atherosclerotic plaque size and morphology in ldlr-/- mice. Vascul Pharmacol. 2023;150:107172. doi: 10.1016/j.vph.2023.107172
- Smith CW, Montague SJ, Kardeby C, et al. Antiplatelet drugs block platelet activation by VITT patient serum. Blood. 2021;138(25):2733–2740. doi: 10.1182/blood.2021012277
- Nyby MD, Sasaki M, Ideguchi Y, et al. Platelet lipoxygenase inhibitors attenuate thrombin- and thromboxane mimetic-induced intracellular calcium mobilization and platelet aggregation. J Pharmacol Exp Ther. 1996;278(2):503–509.
- Yeung J, Tourdot BE, Fernandez-Perez P, et al. Platelet 12-LOX is essential for FcγRIIa-mediated platelet activation. Blood. 2014;124(14):2271–2279. doi: 10.1182/blood-2014-05-575878
- Adili R, Tourdot BE, Mast K, et al. First selective 12-LOX inhibitor, ML355, impairs thrombus formation and vessel occlusion in vivo with minimal effects on hemostasis. Arteriosclerosis Thrombosis Vasc Biol. 2017;37(10):1828–1839. doi: 10.1161/ATVBAHA.117.309868
- Renna SA, Zhao X, Kunapuli SP, et al. Novel strategy to combat the procoagulant phenotype in heparin-induced thrombocytopenia using 12-LOX inhibition. ATVB. 2023;43(10):1808–1817. doi: 10.1161/ATVBAHA.123.319434
- Sharifi-Rad J, Sharopov F, Ezzat SM, et al. An updated review on glycoprotein IIb/IIIa inhibitors as antiplatelet agents: basic and clinical perspectives. High Blood Press Cardiovasc Prev. 2023;30(2):93–107. doi: 10.1007/s40292-023-00562-9
- Jeske WP, Walenga JM, Szatkowski E, et al. Effect of glycoprotein IIb/IIIa antagonists on the HIT serum induced activation of platelets. Thromb Res. 1997;88(3):271–281. doi: 10.1016/S0049-3848(97)00254-5
- Koster A, Kukucka M, Bach F, et al. Anticoagulation during cardiopulmonary bypass in patients with heparin-induced thrombocytopenia type II and renal impairment using heparin and the platelet glycoprotein IIb-IIIa antagonist tirofiban. Anesthesiology 2001;94(2):245–251. doi: 10.1097/00000542-200102000-00013
- Pitkänen HH, Jouppila A, Helin T, et al. COVID-19 adenovirus vaccine triggers antibodies against PF4 complexes to activate complement and platelets. Thromb Res. 2021;208:129–137. doi: 10.1016/j.thromres.2021.10.027
- Polgár J, Eichler P, Greinacher A, et al. Adenosine Diphosphate (ADP) and ADP receptor play a major role in platelet activation/aggregation induced by sera from heparin-induced thrombocytopenia patients. Blood 1998;91(2):549–554. doi: 10.1182/blood.V91.2.549
- Aster RH, Curtis BR, Bougie DW, et al. Thrombocytopenia associated with the use of GPIIb/IIIa inhibitors: position paper of the ISTH working group on thrombocytopenia and GPIIb/IIIa inhibitors. J Thromb Haemost. 2006;4(3):678–679. doi: 10.1111/j.1538-7836.2006.01829.x
- Stark K, Massberg S. Interplay between inflammation and thrombosis in cardiovascular pathology. Nat Rev Cardiol. 2021;18(9):666–682. doi: 10.1038/s41569-021-00552-1
- Stoll G, Nieswandt B Thrombo-inflammation in acute ischaemic stroke - implications for treatment. Nat Rev Neurol 2019;15(8):473–481. doi: 10.1038/s41582-019-0221-1