
1. Background
Alpha-1 Antitrypsin Deficiency (AATD) is a rare disease characterized by low circulating levels of the serine protease inhibitor Alpha-1 Antitrypsin (AAT). Increased protease activity, particularly of neutrophil elastase (NE), leads to chronic obstructive pulmonary disease and bronchiectasis, and polymerization of dysfunctional AAT in the liver leads to fibrosis. It is inherited in an autosomal co-dominant fashion, and also associated with rarer manifestations including neonatal cholestatic hepatitis, panniculitis, and vasculitis. Pathogenesis of the disease is related to mutations in the SERPINA1 gene. Based on current understanding, there are multiple drug targets, involving the genetic transcription, translation, and folding of AAT in the liver; its replacement; and its inhibitory action against NE (). This editorial summarizes recent progress in lung and liver directed therapeutic trials.
Figure 1. Pathogenesis and therapeutic targets in alpha-1 antitrypsin deficiency.
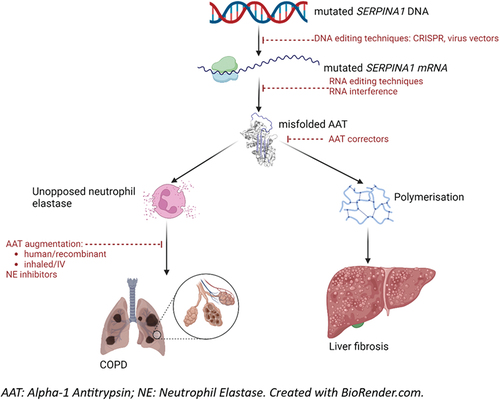
2. Existing treatment: intravenous augmentation therapy
With technological improvement, replacement of the deficient AAT became possible in the early 1980s [Citation1], and US Food and Drug Administration (FDA) approval was granted in 1987 [Citation2]. Augmentation is licensed for patients who have emphysema and the PiZZ genotype, at a dose of 60 mg/kg/week, though various regimens are used worldwide [Citation3–5]. Subsequent studies associated AAT replacement therapy with a reduction in progression of emphysema as measured by CT density, but not by FEV1 [Citation6–8]. However, CT density has been associated with disease severity and mortality in both smoking-related COPD and in AATD related COPD [Citation9,Citation10]. This indirect link is sufficient for the therapy to be recommended in guidelines in North America and in Europe, but publicly funded health agencies in many countries in these regions do not reimburse treatment, including the UK [Citation11–13]. Internationally, drug regulators also differ on which outcomes to use in trials of therapies targeting emphysema or COPD – the European Medicines Agency (EMA) accepts CT lung density as a suitable outcome, but the FDA does not.
3. Medical need
Even in countries which license AAT replacement therapy, research into alternatives is necessary. AAT augmentation relies on derivation from human plasma donations, requiring many donated units to generate one dose, which in most dosing regimens requires weekly infusions [Citation1,Citation14]. This is both time-consuming and costly, and relies on the finite resource of plasma donations which are shared with emergency, hematological, and surgical services [Citation15]. Synthetic production of AAT or NE inhibitors could reduce this limitation. Additionally, treatment so far has been intravenous, but only a minority of the AAT infused reaches the lung [Citation16]; an inhaled route may improve efficiency, whilst being less invasive to administer. Furthermore, augmentation dosing strategies vary in the literature, with equivocal evidence between regimens [Citation5,Citation14,Citation17]. Finally, augmentation therapy only addresses lung disease, such that liver directed approaches are also required if we are to manage all patients adequately. summarizes the current state of play in AATD drug development; hereafter we will focus most on those in late-stage development.
Table 1. Competitive environment: therapies currently in development.
4. Further developments in augmentation
Recently published trials of human-derived AAT (hAAT) augmentation therapy include the safety of hAAT (Prolastin-C, Grifols) in Japanese patients [Citation18]. Four patients received 60 mg/kg doses of hAAT for 4 years, with 5 serious adverse events occurring which were not thought to be related. Although the study is limited by low sample numbers, safety at this dose is consistent with clinical trials in other ethnicities [Citation17,Citation19].
Several trials continue to study safe dosing regimens of hAAT. A phase 3 trial of hAAT (GLASSIA, Takeda) posted its results in 2021 [Citation20], including 6% hAAT-antibody development, and a 2.2-fold increase in epithelial functional AAT levels on bronchoalveolar lavage. The SPARTA trial of 60 mg/kg vs 120 mg/kg hAAT doses estimates results in 2026 [Citation21]. Another phase 1/2 trial of 72 mg/kg vs 144 mg/kg hAAT expects results in 2024 [Citation22].
The practicality of administration of augmentation is also an area of interest. Decisions on self-administration are currently made on a case-by-case basis by expert opinion without specific safety data [Citation3]. Recruitment is underway for a study of the self-administration of hAAT (Respreeza, CSL Behring) [Citation23].
5. Recombinant AAT (rAAT)
Addressing the limited availability of hAAT, research is underway to synthetize functional AAT, called recombinant AAT (rAAT). This also raises the potential for creating a maximally efficient AAT protein. INBRX-101 (Inhibrx) is a rAAT Fc fusion protein which has completed phase I trials showing normal AAT serum levels in 24 AATD patients, with an acceptable safety profile. A phase 2 study is currently recruiting at 2 doses vs hAAT (Zemaira, CSL Behring), with the primary outcome being change in serum AAT levels [Citation24]. A phase 1 study of another rAAT molecule (OsrhAAT, Oryzogen), at several IV doses has also begun [Citation25].
6. Inhaled AAT
Inhaled AAT offers many advantages over intravenous administration. It negates the need for IV access, and allows administration directly at the site of interest. Since a higher proportion of the treatment is distributed to lung tissue, less of the drug is needed to achieve the same lung tissue concentration, thus reducing dependence on limited plasma pools [Citation26]. An inhaled AAT product (KAMADA-AAT, Kamada), has completed phase 2 trials [Citation27]. Results included significantly higher functional AAT levels in epithelial lining fluid (ELF) at both 80 mg and 160 mg doses; increased serum plasma AAT; and increased ELF AAT-NE complexes. Phase 3 trials are now underway in centers across Europe [Citation28].
7. Neutrophil elastase inhibitors
NE inhibitors offer an alternative to AAT augmentation which does not rely on IV or nebulized routes, but still achieves NE inhibition. One such drug is alvelestat (Mereo), which has recently completed a phase 2/3 trial, known as ASTRAEUS, of reduction in desmosine and Aα-val360 biomarkers, and the St George’s Respiratory Questionnaire activity score, over 12 weeks [Citation29]. The conference abstract describes an association between circulating biomarker levels and SGRQ score but does not mention any differences between alvelestat and placebo [Citation29]. Another Phase 2 placebo-controlled trial of alvelestat, known as ATALANTa, is underway at the University of Alabama, with results expected in 2023 [Citation30]. A further NE inhibitor, PHP-303 (pH Pharma) has completed phase 1 trials, with a promising safety profile [Citation31,Citation32].
8. AAT correctors
These drugs aim to change the shape of the misfolded dysfunctional AAT protein, thereby improving its function, but also preventing the polymerization of AAT in the liver, which is the main cause of AATD liver disease. Trials of these drugs are in the very early stages [Citation33–35], but some useful information is available. A study of VX-814 (Vertex) at several doses was terminated early due to raised liver transaminases ALT and AST at all doses vs placebo [Citation36]. However, VX-864 (Vertex) showed significant improvement in functional AAT levels vs placebo (mean difference +2.2 to +2.3 µmol/L across 3 doses, p < 0.0001 in all 3 cases) in a phase 2 trial of 44 participants [Citation37].
9. Gene therapy
Gene therapy has the potential to act even earlier in the disease pathway than AAT correctors, by altering DNA or RNA to produce functional AAT instead of misfolded proteins. The ADVANCE Study is a phase 1/2 trial of ADVM-043 (Adverum), using a Virus Gene Transfer Vector to deliver a functional gene to the liver of 6 patients, to express functional human AAT [Citation38]. The safety profile at present is mixed, with 3/6 patients having severe adverse events including Clostridium difficile colitis, flu, and COPD. The results of a long-term follow-up study are expected imminently [Citation39].
The development of nucleotide editing therapies is a very active area. BEAM-302 (Beam) is described as a ‘liver-targeting lipid-nanoparticle (LNP) formulation of base editing reagents,’ and has reportedly been able to correct some SERPINA1 DNA in mouse models, with increases in serum AAT and reduced liver polymers [Citation40]. Similarly, Intellia has developed NTLA-3001, a CRISPR-mediated targeted gene editing therapy and has made a clinical trial application for a phase I study [Citation41]. Wave Life Sciences and GSK have collaborated to develop WVE-006, an RNA editing oligonucleotide which offers to correct the single nucleotide mutation at the mRNA stage, and applied for phase I trial registration [Citation42].
Fazirsiran (Takeda) is described as an RNA interference therapeutic, causing degradation of AAT RNA and therefore reduction of hepatocyte AAT production, polymerization, and consequent liver disease [Citation43]. In phase 2 trials, liver biopsies of 15 patients with PiZZ AATD liver disease were taken at baseline and at 12 weeks, with median −83.3% reduction of liver AAT, and reduced histological globule burden [Citation43]. Serum liver enzymes also reduced in all patients, and fibrosis regression was observed in the majority. The safety profile was also reassuring. Phase 3 trials in larger samples are ongoing. Another RNA interference therapeutic, belcisiran (Dicerna), is also in phase 2 trials [Citation44].
10. Conclusion
Whilst quality improvement and validatory studies continue in human-derived AAT augmentation therapy, most ongoing trials are concerned with alternatives. Although few of these new therapies have yet produced concrete results, publications of these trials are expected in the near future. Consequently, the next few years will constitute an important, and possibly transformative, period in AATD research, with benefits of interest to patients, physicians, and healthcare systems alike.
11. Expert opinion
The breadth of creativity and commitment involved in the innovation, study design, and implementation of emerging therapies for AATD give cause for optimism. Recombinant AAT has the potential to reduce financial costs and reliance on plasma donations. Nebulized AAT promises similar benefits, by way of increasing the proportion of AAT delivered to the site of interest. AAT ‘correctors’ and RNA interference therapeutics go even further, offering to change the shape of misfolded proteins to prevent liver disease, and in some cases also reduce lung function decline. Gene therapy, as it emerges from the realm of science-fiction to clinical research, even offers to cure the disease, by aiming to allow production of healthy AAT in the native liver. Ensuring that trial outcomes have clinical relevance, beyond pure biochemical correction, will be vital to ensure meaningful improvement in patients’ lives and translation into clinical practice, with reimbursement by health systems to ensure equity of access. A further key challenge is how to deliver the range of studies required when there is a limited pool of patients with such a rare disease, amongst whom only few will wish to enroll in clinical research. Reimbursement approval varies between different healthcare systems, which leads to asymmetric treatment strategies across the globe, as seen in augmentation therapy, and may hinder international research collaboration. Additionally, since disease progression occurs over many decades, long trials are necessary to demonstrate significant differences in treatment groups, which adds cost. Nonetheless, AATD therapy will certainly be an exciting field to follow over the coming years.
Declaration of interest
A M Turner has had grants and/or honoraria from CSL Behring, Grifols, Vertex, Takeda, Beam, GSK, AstraZeneca, and Chiesi.
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
Additional information
Funding
References
- Gadek JE, Fells GA, Zimmerman RL, et al. Antielastases of the human alveolar structures. Implications for the protease-antiprotease theory of emphysema. J Clin Invest. 1981 Oct;68(4):889–898.
- Wewers MD, Casolaro MA, Sellers SE, et al. Replacement therapy for alpha 1-antitrypsin deficiency associated with emphysema. N Engl J Med. 1987 Apr 23;316(17):1055–1062. doi: 10.1056/NEJM198704233161704
- CSLBehring. Respreeza summary of product characteristics. 2015.
- Soy D, de la Roza C, Lara B, et al. Alpha-1-antitrypsin deficiency: optimal therapeutic regimen based on population pharmacokinetics. Thorax. 2006 Dec;61(12):1059–1064.
- Zamora NP, Pla RV, Del Rio PG, et al. Intravenous human plasma-derived augmentation therapy in alpha 1-antitrypsin deficiency: from pharmacokinetic analysis to individualizing therapy. Ann Pharmacother. 2008 May;42(5):640–646.
- Chapman KR, Burdon JG, Piitulainen E, et al. Intravenous augmentation treatment and lung density in severe alpha1 antitrypsin deficiency (RAPID): a randomised, double-blind, placebo-controlled trial. Lancet. 2015 Jul 25;386(9991):360–368. doi: 10.1016/S0140-6736(15)60860-1
- McElvaney NG, Burdon J, Holmes M, et al. Long-term efficacy and safety of alpha1 proteinase inhibitor treatment for emphysema caused by severe alpha1 antitrypsin deficiency: an open-label extension trial (RAPID-OLE). Lancet Respir Med. 2017 Jan;5(1):51–60. doi: 10.1016/S2213-2600(16)30430-1
- Dirksen A, Piitulainen E, Parr DG, et al. Exploring the role of CT densitometry: a randomised study of augmentation therapy in alpha1-antitrypsin deficiency. Eur Respir J. 2009 Jun;33(6):1345–1353. doi: 10.1183/09031936.00159408
- Green CE, Parr DG, Edgar RG et al. Lung density associates with survival in alpha 1 antitrypsin deficient patients. Respir med. 2016 Mar;112:81–87. doi: 10.1016/j.rmed.2016.01.007
- Crossley D, Renton M, Khan M, et al. CT densitometry in emphysema: a systematic review of its clinical utility. Int J Chron Obstruct Pulmon Dis. 2018;13:547–563. doi: 10.2147/COPD.S143066
- National Institute for Health and Care Excellence: Guidelines. Chronic obstructive pulmonary disease in over 16s: diagnosis and management. In: Health and care excellence (NICE) copyright © NICE 2018. London: National Institute for; 2019. https://www.nice.org.uk/guidanceng115evidencefull-guideline-pdf-6602767453page2
- Miravitlles M, Dirksen A, Ferrarotti I, et al. European Respiratory Society statement: diagnosis and treatment of pulmonary disease in α 1 -antitrypsin deficiency. Eur Respir J. 2017 Nov;50(5):1700610.
- Sandhaus RA, Turino G, Brantly ML, et al. The diagnosis and management of alpha-1 antitrypsin deficiency in the adult. Chronic Obstr Pulm Dis. 2016 Jun 6;3(3):668–682. doi: 10.15326/jcopdf.3.3.2015.0182
- Piitulainen E, Bernspang E, Bjorkman S, et al. Tailored pharmacokinetic dosing allows self-administration and reduces the cost of IV augmentation therapy with human alpha(1)-antitrypsin. Eur J Clin Pharmacol. 2003 Jun;59(2):151–156.
- Sieluk J, Slejko JF, Silverman H, et al. Medical costs of alpha-1 antitrypsin deficiency-associated COPD in the United States. Orphanet J Rare Dis. 2020 Sep 23;15(1):260. doi: 10.1186/s13023-020-01523-4
- Kropp J, Wencker M, Hotze A, et al. Inhalation of [123I]alpha1-protease inhibitor: toward a new therapeutic concept of alpha1-protease inhibitor deficiency? J Nucl Med. 2001 May;42(5):744–751.
- Stocks JM, Brantly ML, Wang-Smith L, et al. Pharmacokinetic comparability of Prolastin®-C to Prolastin® in alpha1-antitrypsin deficiency: a randomized study. BMC Clin Pharmacol. 2010 Sep 30;10(1):13. doi: 10.1186/1472-6904-10-13
- Seyama K, Suzuki M, Tasaka S, et al. Long-term safety of Prolastin(R)-C, an alpha1-proteinase inhibitor, in Japanese patients with alpha1-antitrypsin deficiency. Respir Investig. 2022 Nov;60(6):831–839.
- Campos MA, Kueppers F, Stocks JM, et al. Safety and pharmacokinetics of 120 mg/kg versus 60 mg/kg weekly intravenous infusions of alpha-1 proteinase inhibitor in alpha-1 antitrypsin deficiency: a multicenter, randomized, double-blind, crossover study (SPARK). COPD: J Chronic Obstructive Pulmonary Dis. 2013 Dec;10(6):687–695.
- Bnpo S. Takeda. GLASSIA safety, immunogenicity, and bronchoalveolar lavage study. 2016. Available from: https://classic.clinicaltrials.gov/show/NCT02525861
- LLC GT. Efficacy and safety of Alpha1-proteinase inhibitor (human), modified process (alpha-1 MP) in subjects with pulmonary emphysema due to Alpha1 antitrypsin deficiency (AATD). 2013. Available from: https://classic.clinicaltrials.gov/show/NCT01983241
- LLC GT. A study to evaluate safety, tolerability and pharmacokinetics of two different doses of Alpha1-proteinase inhibitor subcutaneous (human) 15% in participants with Alpha1-antitrypsin deficiency. 2021. Available from: https://classic.clinicaltrials.gov/show/NCT04722887
- Behring C. Respreeza self-administration and learning program (AmAretti study). 2019. Available from: https://classic.clinicaltrials.gov/show/NCT04262284
- Inhibrx I. Study of INBRX-101 compared to plasma-derived A1PI therapy in adults with AATD emphysema. 2023. Available from: https://classic.clinicaltrials.gov/show/NCT05856331
- Corp HB. Study of OsrhAAT or Placebo in Healthy Volunteers. 2022. Available from: https://classic.clinicaltrials.gov/show/NCT05315921
- Petrache I, Hajjar J, Campos M. Safety and efficacy of alpha-1-antitrypsin augmentation therapy in the treatment of patients with alpha-1-antitrypsin deficiency. Biologics. 2009;3:193–204. doi: 10.2147/BTT.S4414
- Kamada L. Phase II, safety and efficacy study of Kamada-alpha-1-antitrypsin (AAT) for inhalation. 2014. Available from: https://classic.clinicaltrials.gov/show/NCT02001688;
- Kamada L, Health S. Evaluate efficacy and safety of “Kamada-AAT for inhalation” in patients with AATD. 2019. Available from: https://classic.clinicaltrials.gov/show/NCT04204252
- Rabbie S, Black I, Inshaw J. Analysis from phase II clinical trial, alvelestat, NE (Neutrophil Elastase) inhibitor in AATD-LD: correlation between biomarker response (desmosine and Aa-val360) and clinical outcome (SGRQ). B22. New treatments and novel targets for COPD and ALPHA-1. 2023. A2844–A2844.
- UoAa B, NIo H, BioPharma M. Alvelestat (MPH966) for the treatment of ALpha-1 ANTitrypsin deficiency. 2019. Available from: https://classic.clinicaltrials.gov/show/NCT03679598
- Watz H, Nagelschmitz J, Kirsten A, et al. Safety and efficacy of the human neutrophil elastase inhibitor BAY 85-8501 for the treatment of non-cystic fibrosis bronchiectasis: a randomized controlled trial. Pulm Pharmacol Ther. 2019 Jun 01;56:86–93.
- A study of safety, tolerability, and pharmacokinetics of single doses of PHP-303 in healthy subjects. Available from: https://classic.clinicaltrials.gov/show/NCT03627845
- Incorporated VP. A phase 1, first-in-human study of VX-634. 2022. Available from: https://classic.clinicaltrials.gov/show/NCT05579431
- Limited ZF, Research HM, Plc CP. A single ascending and repeated dose study of oral ZF874 in healthy volunteers and PiXZ subjects. 2020. Available from: https://classic.clinicaltrials.gov/show/NCT04443192
- Incorporated VP. A study to evaluate efficacy and safety of VX-864 in participants with the PiZZ genotype. 2023. Available from: https://classic.clinicaltrials.gov/show/NCT05643495
- Incorporated VP. Evaluation of the efficacy and safety of VX-814 in subjects with the PiZZ genotype. 2020. Available from: https://classic.clinicaltrials.gov/show/NCT04167345
- Incorporated VP. Evaluation of the efficacy and safety of VX-864 in subjects with the PiZZ genotype. 2020. Available from: https://classic.clinicaltrials.gov/show/NCT04474197;
- Adverum Biotechnologies I. Safety dose finding study of ADVM-043 Gene therapy to treat alpha-1 antitrypsin (A1AT) deficiency. 2017. Available from: https://classic.clinicaltrials.gov/show/NCT02168686
- Adverum Biotechnologies I. Long-term follow-up study of ADVM-043. 2018. Available from: https://classic.clinicaltrials.gov/show/NCT03804021
- Beam therapeutics presents preclinical data highlighting utility of BEAM-302 to correct an alpha-1 antitrypsin (AAT) deficiency disease-causing mutation [Internet]. 2023. Available from: https://investors.beamtx.com/node/8731/pdf
- Intellia. Intellia therapeutics announces Second Quarter 2023 financial results and highlights recent company progress. 2023.
- Wave life sciences announces submission of first clinical trial application for WVE-006, the first-ever RNA editing clinical Candidate, and plans for upcoming virtual “R&D day” [Internet]. 2023. Available from: https://ir.wavelifesciences.com/news-releases/news-release-details/wave-life-sciences-announces-submission-first-clinical-trial
- Strnad P, Mandorfer M, Choudhury G, et al. Fazirsiran for liver disease associated with Alpha(1)-antitrypsin deficiency. N Engl J Med. 2022 Aug 11;387(6):514–524. doi: 10.1056/NEJMoa2205416
- Dicerna Pharmaceuticals I, a Novo Nordisk Company. A study of Belcesiran in patients with AATLD. 2021. Available from: https://classic.clinicaltrials.gov/show/NCT04764448