
ABSTRACT
Introduction: Tuberous sclerosis complex (TSC) is a rare genetic disorder resulting in benign tumors in various organs. It is caused by mutations in TSC1 or TSC2 genes causing hyperactivation of the mammalian target of rapamycin (mTOR) pathway. The majority of patients with TSC develop epilepsy, and approximately two-thirds become refractory to antiepileptic drugs (AEDs). Recently, the mTOR inhibitor everolimus was approved as adjunctive therapy for TSC-associated partial seizures.
Areas covered: This article covers different characteristics of everolimus, including major clinical trials leading to its approval in TSC-associated partial seizures, safety concerns, drug pharmacokinetics/pharmacodynamics, and an overview of potential competitors and other agents used to treat TSC-associated seizures.
Expert opinion: Unlike many other therapies for treating TSC-associated seizures, everolimus addresses the underlying pathophysiology of TSC, and since it has also been shown to improve other TSC manifestations such as subependymal giant cell astrocytomas and renal angiomyolipomas, everolimus provides a potential multisystemic therapy for TSC. An important avenue for future research is exploring the possible use of everolimus as a preventative treatment for seizures as there is the potential to prevent negative developmental outcomes associated with TSC.
1. Introduction
Tuberous sclerosis complex (TSC) is an autosomal dominant, multisystem genetic disorder with a birth incidence of approximately 1:6000 [Citation1,Citation2]. It is caused by pathogenic variants in either of the TSC1 or TSC2 genes resulting in the loss of functioning protein products hamartin or tuberin, respectively, and subsequent constitutive activation of mammalian target of rapamycin complex 1 (mTORC1) [Citation2,Citation3]. The increased mTOR activation results in benign tumor growth in various organs of the body, mainly the brain, kidneys, skin, lungs, eyes, and liver [Citation2,Citation3], although there have been reports of other less common organ involvement such as bone and pancreas [Citation4,Citation5]. Neurologic involvement is the most common cause of morbidity in this condition, with epilepsy developing in up to 90% of patients with TSC, often within the first year of life [Citation6,Citation7]. In a retrospective study, approximately two-thirds of patients with TSC-associated seizures developed refractory epilepsy, which was defined as the failure to achieve seizure control with at least three first-line antiepileptic drugs (AEDs) [Citation8]. Before the implementation of mTOR inhibitors as a treatment for subependymal giant cell astrocytomas (SEGAs), lymphangioleiomyomatosis (LAM), and renal angiomyolipomas in TSC, there was no specific treatment for the disease, and all manifestations were treated symptomatically.
Consensus recommendations established in 2012 provide guidance on diagnosis and treatment of TSC [Citation9,Citation10] and advocate AEDs as first-line treatment for TSC-associated seizures [Citation10,Citation11]. Use of particular AEDs depends on age, epileptic syndrome, and seizure type [Citation11]. Typically, monotherapy is preferred to reduce the incidence of adverse events; however, combination therapy is used when monotherapy fails [Citation12]. AED combinations addressing multiple seizure mechanisms are preferable, particularly those that enhance γ-aminobutyric acid (GABA)ergic inhibition [Citation11,Citation12]. Other treatments recommended as second- and third-line for controlling TSC-associated seizures include corticosteroids or adrenocorticotropic hormone (infantile spasms), surgery, vagus nerve stimulation (VNS), the ketogenic diet and mTOR inhibitors [Citation11,Citation13]. Regarding surgery, presurgical evaluation should be promptly considered in those with drug-resistant epilepsy (those who have treatment failure with the first two AEDs), particularly in very young patients, as early surgical intervention can increase the probability of seizure-freedom [Citation11,Citation13]. Presurgical evaluation should occur even in complex cases, such as those with multiple tubers, multiple interictal foci, bilateral seizures, or epileptic spasms since a single epileptogenic tuber can be found responsible for the most impairing seizures [Citation14].
The mTOR inhibitor everolimus was recently approved in the United States and European Union as adjunctive therapy for refractory TSC-associated partial seizures based on the results of a randomized, double-blind, placebo-controlled phase 3 study [Citation15–Citation17]. As a result, more recently published recommendations advocate add-on everolimus for patients who are refractory to two AEDs [Citation13]. This drug profile summary highlights the pharmacology of everolimus, the efficacy and safety data from clinical trials, and discusses the current and future role of everolimus in the treatment of TSC-associated partial seizures.
2. Overview of the market
As previously mentioned, AEDs are currently used first-line for the treatment of TSC-associated epilepsy. In TSC, aberrant mTOR signaling associated with decreased GABAergic inhibition and increased glutamatergic excitation is believed to play a role in the neuropathology associated with TSC [Citation18]. As a result, AEDs generally used in TSC are those that target GABAergic or glutamatergic processes ().
Table 1. Overview of AEDs used in clinical studies for treatment of TSC-associated partial-onset seizures.
Among the GABAergic agents [Citation19], vigabatrin is the recommended first-line AED in treating TSC-associated seizures. In particular, it is used for management of TSC-associated infantile spasms and partial seizures [Citation10,Citation11]. Vigabatrin exerts antiepileptic effects through irreversible inhibition of GABA-T which increases the availability of GABA in the brain and reduces neuronal excitation [Citation20,Citation21]. The rate of freedom from partial seizures has been reported to be 16–46% in prospective and retrospective studies of TSC [Citation22–Citation24]. Retinal nerve fiber layer thinning and visual field deficits have been reported with vigabatrin and are a concern (boxed warning) [Citation25,Citation26]. Visual field constriction has been reported in approximately 34% of children and over 30% of adults [Citation21,Citation26,Citation27]. These figures are, however, not consistent with our clinical experience. We have not encountered a patient receiving vigabatrin for tuberous sclerosis–related epilepsy who had visual changes that adversely affected his or her activities of daily living. In our opinion, the developmental morbidity associated with uncontrolled seizures in tuberous sclerosis almost always outweighs the risk of visual field constriction, particularly in infants and children.
Benzodiazepines such as clobazam have also been used in refractory epilepsy in TSC [Citation28]. Like other benzodiazepines, clobazam exerts its anticonvulsant effects by allosteric binding to GABAA receptors, resulting in upregulation of the GABA transporters 1 and 3 [Citation28]. Approximately 69% have been reported to have ≥50% reduction in the frequency of refractory TSC seizures after initial titration (1 week to 3 months); however, only 21% were reported with this response at 12 months leading to questions of long-term efficacy [Citation28].
Among the glutamatergic agents, few have been studied in clinical studies in the TSC population. In particular, lamotrigine was evaluated in an open-label study in TSC [Citation29]. Its mechanism is believed to involve blockade of voltage-sensitive sodium channels and inhibition of release of excitatory neurotransmitters [Citation29]. In patients with TSC, it was reported to result in seizure freedom in 42%, and in a > 50% reduction in seizure frequency in 79% [Citation29].
Levetiracetam is another glutamatergic agent that has been evaluated in patients with TSC [Citation30]. While the exact mechanism is unknown, it is thought to reduce presynaptic glutamate release, and its efficacy correlates with its binding affinity to SV2A [Citation31]. In a retrospective, open-label study in patients with TSC-associated seizures, 20% became seizure free and 40% had ≥50% reduction in the frequency of seizures by 6 months of treatment [Citation30]. Other glutamatergic agents such as the sodium channel blockers valproic acid, topiramate, oxcarbazepine, and carbamazepine have been frequently used in TSC; however, they have not been evaluated in patients with TSC in clinical studies [Citation32].
While vigabatrin may have some additional effect on mTOR pathway activation [Citation33], most AED options for epilepsy provide only symptomatic management and do not treat the underlying etiology of TSC. There are other therapies available for those patients who are refractory to AED treatment [Citation8]. One of them is the ketogenic diet, whose mechanism of action likely involves inhibition of mTOR through carbohydrate restriction [Citation34]. The ketogenic diet can be utilized throughout the life span but is primarily undertaken in infancy and early childhood; it can be combined with vigabatrin and used as an alternative to surgery [Citation11,Citation13]. In a small open-label study, 4 of 12 patients with TSC were seizure-free after 3 months on the diet [Citation13]. Vagus nerve stimulation has also been used in patients with TSC refractory seizures who are not eligible for surgery [Citation13]. While significant seizure reduction has been observed, patients often do not become seizure-free, and treatment with vagus nerve stimulation is invasive and more expensive and less effective than other options [Citation13]. It should be borne in mind that, since the ketogenic diet and mTOR inhibition have similar effects, they may also have additive toxicity when used together. One advantage is that a higher degree of ketosis can be achieved with a relatively lower carbohydrate restriction. However, adverse effects such as acidosis and lipid elevation are often more pronounced, sometimes extremely so when used concurrently. When used together, accordingly, we recommend starting with a lesser degree of carbohydrate restriction and/or a decreased dosage of mTOR inhibitor when these therapies are used together.
Cannabidiol is a new agent in development that reduces seizures through a yet unknown mechanism of action, but was approved in June 2018 in the United States as adjunctive therapy to treat seizures associated either with Lennox-Gastaut syndrome or Dravet syndrome and became commercially available in late November 2018 [Citation35]. An open-label study which included patients with various epilepsy etiologies (6 of 162 had TSC) showed seizure reduction (median seizure reduction 36.5% over 12 weeks) with tolerable safety [Citation36], which led to initiation of a phase 3 placebo-controlled long-term study in patients with TSC (GWPCARE6; NCT02544763 [Citation37] and NCT02544750 which is currently ongoing). An additional expanded-access study in 56 patients with intractable epilepsy of various etiologies showed that among the 18 with TSC, median seizure frequency was reduced by 48.8% after 3 months and 57.5% after 12 months of add-on cannabidiol [Citation38].
3. Everolimus
3.1. Introduction
Everolimus, an inhibitor of mTORC1 and an analog of rapamycin, is a water-insoluble small molecule with a molecular weight of 958.2 g/mol [Citation3]. It acts by selectively binding to FKBP12 which, upon forming a complex with mTORC1, blocks the active site of mTORC1 and reduces the production of downstream effectors S6K1 and 4E-BP1 which are involved in protein synthesis [Citation3,Citation16,Citation17]. With regard to TSC-associated seizures, this is believed to reduce abnormal brain cell growth and overproliferation, reduce abnormal neuron spatial organization, and improve neuronal myelination, reduce neuronal morphology abnormalities, improve synaptic plasticity, and reduce excess inflammatory mediators in the cerebral cortex () [Citation6,Citation18,Citation39–Citation42].
Figure 1. Mechanism of action of (a) mTORC1 in the pathophysiology of TSC and (b) the mTORC1 inhibitor everolimus for TSC-associated seizures.
Akt: protein kinase B; AMPK: 5ʹ adenosine monophosphate-activated protein kinase; IGF: insulin growth factor; mTORC1: mammalian target of rapamycin complex 1; PI3K: phosphoinositide-3 kinase; PTEN: phosphatase and tensin homolog on chromosome 10; Rheb: Ras homolog enriched in brain; STRADα: STE20-related kinase adapter alpha; TSC: tuberous sclerosis complex; TSC1: tuberous sclerosis complex 1 protein; TSC2: tuberous sclerosis complex 2 protein.
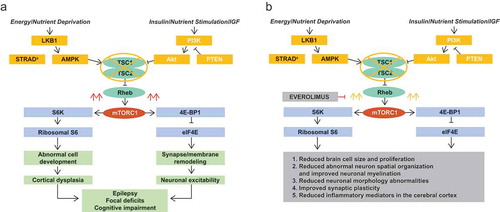
mTORC1 inhibitors are approved for adjunctive treatment of TSC-refractory seizures [Citation16,Citation17]. Everolimus, which had previously been approved for treating TSC-associated SEGA and renal angiomyolipoma, was shown to reduce seizures in phase 3 EXIST-3 study and was subsequently approved for the treatment of refractory partial seizures in patients with TSC [Citation15–Citation17]. Sirolimus is another mTORC1 inhibitor that has been evaluated to treat multiple manifestations of TSC, similarly to everolimus [Citation3]. However, in an open-label crossover study evaluating sirolimus as add-on therapy in intractable TSC seizures (patients randomly assigned to treatment with of sirolimus either during the first or second period of 6 months over a 12-month study), sirolimus did not significantly reduce seizure frequency [Citation43] (). Because of the strong phase 3 data now available for everolimus for seizure reduction in TSC, and the lack of large-scale clinical data (and regulatory approval) for sirolimus, the authors currently recommend using the mTOR inhibitor that has the approved indication, particularly if the indication has made the drug easier to obtain due to insurance issues or cost. Pharmacologically, these drugs have the same effect of inhibiting mTORC1; however, they differ in terms of lipid solubility, half-life, and metabolism [Citation3]. In clinical practice we have found that some patients can be switched from one mTOR inhibitor to the other if necessary due to adverse effects or lack of efficacy; in some cases, this may also lead to improvement.
Table 2. Phase 1–3 clinical studies evaluating mTOR inhibitors for treatment of TSC-associated seizures.
3.2. Clinical pharmacokinetics
3.2.1. Absorption
Everolimus is available as an oral formulation (tablets or tablets for oral suspension). Peak everolimus concentration is achieved between 0.5 and 1 h in healthy patients, between 1 and 2 h in patients with advanced solid tumors, and in 1–2.2 h in stable renal transplant recipients [Citation16,Citation17,Citation44]. In healthy patients, fat content lowered area under the concentration–time curve (AUC) by 12–32% and maximum plasma concentration (Cmax) by 42–60% depending on fat content and dosage form (tablet or tablet for oral suspension) [Citation16,Citation17]. Everolimus has relatively low bioavailability (16%), with the tablet for oral suspension formulation having 20–36% lower bioavailability than the tablet formulation [Citation16,Citation17,Citation44].
3.2.2. Distribution
In both healthy subjects and patients with moderate hepatic impairment, everolimus is approximately 74% protein-bound [Citation16,Citation17]. Blood-to-plasma ratio of everolimus ranges from 17% to 73%, depending on blood concentration (5–5000 ng/ml) [Citation16,Citation17].
3.2.3. Metabolism
Everolimus is a substrate of CYP3A4 and P-glycoprotein, and to a lesser extent, CYP3A5 and CYP2C8 [Citation16,Citation17,Citation44]. At least six metabolites have been identified [Citation16,Citation17,Citation44], including three monohydroxylated metabolites, two hydrolytic ring-opened products, and a phosphatidylcholine conjugate of everolimus which have shown 100 times less activity than everolimus [Citation16,Citation17].
3.2.4. Elimination
Among renal and liver transplant recipients, terminal half-life ranged from 18 to 35 h. The mean half-life is reported to be 26–30 h [Citation3,Citation16,Citation17,Citation44]. The majority of everolimus (>80%) is eliminated in feces, with only 2–5% excreted in urine [Citation3,Citation16,Citation17]. A population pharmacokinetics model comprising data from 531 patients from the EXIST-1 (TSC-SEGA), EXIST-2 (TSC-renal angiomyolipoma), and EXIST-3 (TSC-refractory partial seizures) studies established that everolimus followed a two-compartment model with first-order absorption and elimination [Citation45].
3.3. Clinical pharmacodynamics
The magnitude of the reduction in absolute seizure frequency in patients with TSC correlates with the everolimus trough serum level achieved. Many patients nonetheless have a good clinical response at relatively low trough serum levels [Citation16,Citation17]. A logistic regression analysis using data from the core phase of the EXIST-3 study showed that a twofold increase in time-normalized minimum plasma concentration (Cmin) was associated with a 2.2-fold increase (95% CI 1.3–3.5; p = 0.0017) in the odds for a response (≥50% reduction in seizures) over the time-normalized Cmin ranges of 0.97 ng/ml to 16.40 ng/ml [Citation15,Citation17]. A significant factor in achieving response was baseline seizure frequency (odds ratio 0.978 [95% CI 0.959–0.998]) [Citation17]. Similarly, using a linear regression model, a twofold increase in time-normalized Cmin was associated with a statistically significant 28.8% (95% CI 11.7–41.8; p = 0.0019) reduction in seizure frequency [Citation15,Citation17]. Significant factors predicting the reduction in seizure frequency included baseline seizure frequency and time-normalized Cmin [Citation17]. A population PK/PD model using data from patients in EXIST-3 showed that patients with Cmin >5 ng/ml had median expected response rates (proportion with ≥50% reduction in seizures) ranging from 30% to 50% and that median response rate was positively correlated with Cmin [Citation45]. At Cmin levels <5 ng/ml, response rates became more variable and decreased more rapidly [Citation45]. The analysis thus supported using the recommended target blood concentration of 5 to 15 ng/ml when using everolimus for treating TSC-associated refractory partial seizures [Citation45]. A second analysis of PK/PD data from EXIST-3 would further recommend an initial target Cmin range of 5 to 7 ng/ml which could be expanded to 5 to 15 ng/ml in the event of inadequate response [Citation46].
3.4. Clinical efficacy
Among phase 1–3 studies in patients with TSC-associated seizures (), the first clinical study to demonstrate efficacy in partial seizures was the open-label phase 1/2 study of everolimus for the treatment of TSC-SEGA by Krueger et al. [Citation47]. In the core phase of the study, 28 patients ≥3 years of age with growing SEGA received everolimus for 6 months at an initial dose of 3 mg/m2 that was titrated to achieve blood trough concentrations of 5 to 15 ng/ml [Citation47]. While the focus of the study was treating SEGA, the effect of everolimus on seizures was assessed as a secondary end point. After 6 months of treatment, everolimus was associated with a median change of −1 seizure (p = 0.02) and, of 16 patients with available video electroencephalogram (EEG) data, 9 patients had a decrease, 6 had no change, and 1 had an increase in seizure frequency from baseline [Citation47]. Patients were eligible to continue everolimus beyond 6 months in an extension phase of the study, and results from an interim analysis (median everolimus exposure 34.2 months) showed that the proportion of patients who reported no seizures since their last visit increased from 38.5% at baseline to 65.2% at 24 months [Citation48]. In addition, only 13.0% reported at least one seizure per day at 24 months compared with 26.9% at baseline [Citation48]. In the final analysis of the study (median everolimus exposure 67.8 months), the rate was 11.1% at month 60 [Citation49]. The proportion with seizure freedom increased from 38.5% at baseline to 66.7% at 18 months and remained steady for the remainder of the study [Citation49].
Another phase 1/2 open-label study conducted by Krueger et al. focused specifically on everolimus for the treatment of TSC-associated refractory partial seizures. In the core phase of the study, 20 patients aged 2 years or older received everolimus at an initial dose of 5 mg/m2/day that was titrated to trough concentrations of 5 to 15 ng/ml over a 4-week titration interval followed by an 8-week maintenance phase for a total of 12 weeks [Citation50]. The proportion of patients with ≥50% reduction from baseline in patient-reported seizure frequency at the end of the maintenance phase, which was the primary end point of the study, was 60% (n = 12/20), with 35% (n = 7) achieving ≥90% reduction in seizure frequency [Citation50]. Three patients (15%) were considered partial responders (25% to <50% reduction in frequency of seizures) [Citation50]. Median seizure frequency decreased by 73% (p = 0.020), and four patients (20%) were seizure free [Citation50]. Eighteen continued treatment with everolimus in an extension phase with the goal of completing 4 years of treatment [Citation51]. In the final analysis of the study (everolimus duration 50.9 months), the average number of seizures per month was reduced by 72–81% throughout the extension phase [Citation51]. The proportion with ≥50% reduction in frequency of seizures was 76%, 75%, 80%, and 93% among those remaining on treatment at month 12 (n = 17), month 24 (n = 16), month 36 (n = 15), and month 48 (n = 14), respectively [Citation51]. The results of the core phase of the study led to the initiation of the large, multicenter, phase 3 EXIST-3 study.
Before EXIST-3, the EXIST-1 study was a phase 3 study that evaluated seizure outcomes as a secondary outcome in patients with TSC treated with everolimus. EXIST-1 was a double-blind, placebo-controlled study that evaluated everolimus at a starting dose of 4.5 mg/m2 (titrated to trough concentration of 5–15 ng/ml) versus placebo for the treatment of patients of any age with growing TSC-associated SEGA for at least 6 months [Citation52]. Absolute change from baseline to 24 weeks in seizure frequency per 24 h video EEG was assessed in the core phase of the study [Citation52]. While the study consisted of 78 patients randomly assigned to everolimus and 39 to placebo, only 27 (35%) everolimus-treated patients and 13 (33%) placebo-treated patients had presence of seizure at baseline EEG [Citation52]. A sensitivity analysis of the subset of patients with ≥1 seizure at baseline showed no significant difference in the change from baseline to week 24 in seizure frequency between everolimus and placebo arms (p = 0.2988) [Citation52]. However, the study was not powered to detect differences in this outcome.
EXIST-3 was a randomized, multicenter, placebo-controlled, phase 3 study evaluating two everolimus dose levels compared with placebo as adjunctive treatment for treatment-resistant partial-onset seizures associated with TSC [Citation15]. In the core phase, patients were randomly assigned to receive everolimus high exposure (HE, 9–15 ng/ml; n = 130), everolimus low exposure (LE, 3–7 ng/ml; n = 117), or placebo (n = 119) for 18 weeks (6-week titration + 12-week maintenance) [Citation15]. The primary end points were the median percentage reduction in seizure frequency and the proportion with ≥50% reduction from baseline in seizures [Citation15]. The median percentage reduction in seizure frequency was significantly greater than placebo (14.9%) for both everolimus HE (39.6%; p < 0.0001) and everolimus LE (29.3%; p = 0.0028) [Citation15]. Similarly, response rates for everolimus HE (40.0%;p < 0.0001) and everolimus LE (28.2%; p = 0.0077) were significantly greater than placebo (15.1%) [Citation15]. A post hoc analysis of Japanese patients in the EXIST-3 study (n = 35) confirmed rates of seizure reduction and response that were consistent with the overall EXIST-3 population [Citation53]. All patients in EXIST-3 were eligible to enter into an extension phase in which they could be transitioned (including placebo patients) to everolimus at a trough range of 3 to 15 ng/ml for an additional 48 weeks or longer [Citation54]. In total, 361 of the 366 patients in EXIST-3 received everolimus in the core and/or extension phases and were included in a longer-term analysis (median duration of everolimus ~21 months) [Citation54]. Response rate increased over time from 31% at week 18 to 57.7% at 2 years. Similarly, the median percentage reduction in seizure frequency increased from 31.7% at week 18 to 56.9% at 2 years. Similar increases were observed irrespective of initial randomization group, showing that results were consistent with the primary analysis and that efficacy was sustained over time () [Citation54]. A post hoc analysis of the pediatric population (age <18 years) in the extension analysis showed that the younger children (<6 years of age) experienced relatively greater benefit than older children (6 to <18 years) with up to 1 year of treatment, although seizure reduction improved in older children with time [Citation55]. This is consistent with data showing that early, and even prophylactic, treatment with vigabatrin is particularly beneficial in TSC [Citation56]. Studies are underway to determine if even earlier treatment with mTOR inhibitors can be as or even more beneficial and vigabatrin.
Figure 2. (a) Response rate over time from the start of everolimus. Bars represent 95% confidence intervals obtained using Clopper–Pearson method. (b) Median percentage reduction in seizure frequency over time from the start of everolimus. Bars represent 95% confidence intervals based on bootstrap percentiles. Reprinted with permission from Franz DN et al. Everolimus for treatment-refractory seizures in TSC. Extension of a randomized controlled trial. Neurology Clinical Practice. Volume 8, Issue 5, Pages 412–420. https://cp.neurology.org/ [Citation54].
![Figure 2. (a) Response rate over time from the start of everolimus. Bars represent 95% confidence intervals obtained using Clopper–Pearson method. (b) Median percentage reduction in seizure frequency over time from the start of everolimus. Bars represent 95% confidence intervals based on bootstrap percentiles. Reprinted with permission from Franz DN et al. Everolimus for treatment-refractory seizures in TSC. Extension of a randomized controlled trial. Neurology Clinical Practice. Volume 8, Issue 5, Pages 412–420. https://cp.neurology.org/ [Citation54].](/cms/asset/99ca8088-a5b9-41a1-9f06-64065a5f2131/iern_a_1635457_f0002_oc.jpg)
3.5. Safety
Adverse events of everolimus for treating TSC-associated partial seizures are similar to those reported for other indications of everolimus. In the placebo-controlled EXIST-3 study, the most common all-grade adverse events of any cause reported in more than 15% of patients in either everolimus treatment group during the core phase included stomatitis, diarrhea, nasopharyngitis, pyrexia, and upper respiratory tract infection () [Citation15,Citation16]. Adverse events of potential clinical interest but that occurred in fewer than 10% of everolimus-treated patients included decreased appetite (everolimus LE: 9%; everolimus HE: 7%), pneumonia (everolimus LE: 2%; everolimus HE: 4%), aggression (everolimus LE: 2%; everolimus HE: 0.8%), proteinuria (everolimus LE: 0%; everolimus HE: 2%), menorrhagia (everolimus LE: 0.9%; everolimus HE: 0.8%), and pneumonitis (everolimus LE: 0%; everolimus HE: 0.8%) [Citation16]. In the extension analysis, the safety profile was generally consistent with previously reported data from the core phase [Citation54]. The most frequently reported adverse events (>25%) of any cause in the extension analysis included stomatitis, pyrexia, diarrhea, and mouth ulceration [Citation54]. The most frequent grade 3/4 adverse events (≥2.5%) of any cause were pneumonia (6.9%), status epilepticus (3.3%), seizures (2.8%), and stomatitis (2.5%) [Citation54]. In general, emergence of adverse events decreased after 6 months and remained stable into the second year [Citation54]. Two deaths were reported before the extension analysis cutoff including one patient who died as a result of pneumonia, which was suspected to be treatment-related, and another patient who died as a result of sudden unexplained death due to epilepsy (SUDEP) [Citation54]. An additional two deaths were reported after the cutoff, one death attributed to septic shock and the other caused by SUDEP [Citation54]. SUDEP is known to be one of the more common causes of mortality in tuberous sclerosis patients, particularly in those with refractory epilepsy [Citation57]. In the post hoc analysis of pediatric patients, long-term treatment was associated with higher incidence and severity of infections, such as pneumonia, particularly in the younger patients (<6 years); however, the incidence of pneumonia was similar to that in the general population [Citation54].
Table 3. Adverse reactions reported in ≥10% of Everolimus-treated patients with TSC-associated partial-onset seizures in EXIST-3 [Citation15].
Potential drug–drug interactions with everolimus are mainly with other drugs that are inhibitors, inducers, or substrates of CYP3A4 and P-gp. Concomitant use of a strong CYP3A4 and P-gp inhibitor such as ketoconazole has resulted in a 3.9-fold increase in Cmax and 15-fold increase in AUC of everolimus [Citation16]. A moderate CYP3A4 and P-gp inhibitor such as verapamil has resulted in a 2.3-fold increase in Cmax and 3.5-fold increase in AUC of everolimus [Citation16]. A strong CYP3A4 and P-gp inducer on the other hand can result in decreased everolimus AUC by 63% and Cmax by 58% [Citation16]. With patients being treated for TSC-associated partial seizures, many AEDs they may take concomitantly are often inducers or substrates of CYP3A4 and P-gp, and everolimus concentration must be carefully monitored and dosage adjusted accordingly [Citation46]. For those patients taking CYP3A4-inducing AEDs such as carbamazepine, oxcarbazepine, phenobarbital, phenytoin, primidone, clobazam, and topiramate, a higher starting dose is recommended than in those not taking these AEDs, both in children <6 years of age (9 mg/m2 vs 6 mg/m2) and those ≥6 years of age (8 mg/m2 vs 5 mg/m2) [Citation46].
3.6. Postmarketing surveillance
While little postmarketing surveillance data are publicly available yet for the use of everolimus for TSC-associated refractory partial seizures, a long-term study (NCT02962414) is currently underway to assess safety in patients who are continuing to benefit from everolimus in EXIST-3 [Citation58]. This rollover study is expected to remain open for approximately 10 years (from first patient, first visit), or until all patients have permanently discontinued treatment, and will feature interim analyses in the fourth quarter of 2018, 2020, 2023, and 2026 [Citation58]. As of 6 August 2018, 206 patients from 18 countries have entered the study [Citation58]. Assessments will include trough pharmacokinetic levels and frequency and severity of adverse events [Citation58].
While no direct head-to-head comparisons of everolimus with competitors such as vigabatrin exist, provides an overview of differences in characteristics and potential safety concerns between these agents. Vigabatrin is regularly used in patients with TSC, particularly for infantile spasms, and prophylactic treatment in infancy. Cannabidiol has recently demonstrated efficacy in a multicenter placebo-controlled study of epilepsy in TSC, confirming the results of an earlier single-center study [Citation59]. Cannabidiol was approved for the treatment of seizures associated with Dravet’s syndrome and Lennox-Gastaut syndrome [Citation35]. A separate indication for TSC-related epilepsy may be forthcoming.
Table 4. Comparison of everolimus characteristics with potential competitors for treatment of TSC-associated partial seizures.
3.7. Regulatory affairs
Everolimus is currently approved in over 30 countries including the United States and countries of the European Union for TSC partial seizures [Citation16,Citation17,Citation60]. In the United States and European Union, the indication is as adjunctive treatment in adult and pediatric patients aged 2 years and older with TSC-associated partial-onset seizures [Citation16,Citation17]. The EU indication also further specifies that everolimus can be used for TSC-associated seizures, with or without secondary generalization [Citation17].
Everolimus is also approved in over 95 countries, including the United States and countries of the European Union, for the following indications [Citation16,Citation17,Citation60]:
Postmenopausal women with advanced hormone receptor-positive, human epidermal growth factor receptor 2-negative breast cancer in combination with exemestane after failure of treatment with letrozole or anastrozole
Adults with progressive neuroendocrine tumors of pancreatic origin (PNET) and adults with progressive, well-differentiated, nonfunctional neuroendocrine tumors (NET) of gastrointestinal (GI) or lung origin that are unresectable, locally advanced, or metastatic. Everolimus is not indicated for the treatment of patients with functional carcinoid tumors
Adults with advanced renal cell carcinoma (RCC) after failure of treatment with sunitinib or sorafenib
Adults with renal angiomyolipoma and TSC, not requiring immediate surgery
Adult and pediatric patients aged 1 year and older with TSC who have SEGA that requires therapeutic intervention but cannot be curatively resected
3.8. Conclusion
Clinical studies demonstrate that everolimus is safe and efficacious when used as adjunctive therapy for controlling TSC-associated partial seizures. Long-term safety and efficacy data are needed, particularly regarding the potential benefit of prompt seizure control leading to better developmental outcomes.
4. Expert opinion
In contradistinction to all other treatments for seizures, which have efficacy irrespective of an underlying cause or specific etiology, everolimus was studied purely because of its ability to inhibit mTOR and reverse the molecular defect believed to be associated with TSC. Given that mTOR hyperactivation is believed to account for the various clinical manifestations of TSC, it is not surprising that everolimus has shown efficacy in these manifestations as well, including SEGA [Citation52], renal angiomyolipomata [Citation61–Citation63], skin lesions, such as facial angiofibromas and hypomelanotic macules [Citation62,Citation63], and cardiac rhabdomyoma [Citation64]. As such, everolimus provides a true disease-modifying multisystemic therapy for TSC. It is reasonable to expect that mTOR inhibition would be similarly beneficial for other genetic or developmental disorders characterized by mTOR hyperactivation and/or syndromes caused by mutations in genes of the AKT/PI3K/mTOR pathway. Potential therapeutic effects have already been reported in PTEN hamartoma syndrome [Citation65], Lhermitte Duclos syndrome [Citation66], Birt-Hogg-Dubé syndrome [Citation67,Citation68], and Proteus syndrome [Citation69]. An important avenue for future research exploring the possible use of everolimus as a preventative treatment for seizures, cognitive impairment, and other systemic manifestations of TSC, particularly in infants and young children [Citation70]. In addition, 25–30% of cases are identified prenatally because of the presence of cardiac rhabdomyoma on prenatal ultrasound, or postnatally but before seizure onset by the presence of hypomelanotic macules. Serial EEGs have been shown to predict the development of clinical seizures and infantile spasms in infants with tuberous sclerosis. Early (less than 1 year of age) and even prophylactic treatment of epilepsy in TSC with vigabatrin could possibly mitigate intellectual disability and autism in these children [Citation32,Citation56,Citation71]. In fact, the likelihood of not developing intellectual disability increases by 34% approximately for every month in the first 18 months of life that the onset of epilepsy is delayed [Citation70]. Use of mTOR inhibitors in infants and young children to improve or prevent epilepsy and neurodevelopmental disability is the logical next step. Important questions that need to be answered in exploring preventative treatment include identifying the best candidates for early treatment (factoring in aspects such as age, mutation status, early TSC symptoms, intracranial lesions), the timing of treatment, and whether there are any medical tests that would aid in identifying these patients.
4.1. Five-year view
Considering the success of everolimus in reducing seizures as an adjunctive therapy, future research may explore this medication as first-line treatment for TSC-associated seizures before failure with multiple AEDs and as monotherapy. Additional research may assess the efficacy and safety of everolimus in combination with specific agents/therapies such as the ketogenic diet. As mentioned previously, use of everolimus for the prevention of seizures at an early age is of great interest as this has the potential to prevent not only intellectual disability associated with early seizure onset, but also the severity of systemic manifestations such as angiomyolipomas, rhabdomyomas, SEGA, and cutaneous lesions. It is also unknown what other factors may contribute to seizure control during everolimus treatment, such as the impact of intracranial lesions and the patient’s genetic background. Future research in these areas may be beneficial.
mTOR inhibitors may be able to play an important role in treating other diseases associated with mTOR overactivation as mentioned above. It is expected that future antiepileptic drug development will focus on identification of agents with known, specific molecular mechanisms of action which target specific etiologies, genetic or otherwise. Examples include type 2b focal cortical dysplasia and other focal epilepsies associated with gain of function of mTOR such as mutations in genes like DEPDC5, MTOR, PTEN, and GATOR [Citation72–Citation76]. We expect that preemptive treatment of individuals at high risk for epilepsy will become increasingly accepted and even commonplace.
4.2. Information resources
The following websites provide support and resources for patients with TSC and their caregivers, as well as health-care providers: https://www.tsalliance.org/, http://www.tscinternational.org/, http://www.tuberous-sclerosis.org/
Article highlights
Epilepsy is prevalent among patients with TSC; approximately two-thirds of patients become refractory to treatment with AEDs
Primary therapies for TSC partial seizures (eg, AEDs) only treat symptoms without addressing underlying pathophysiology
Everolimus, which had been previously approved to treat TSC-associated SEGA and renal angiomyolipoma, was recently approved as adjunctive therapy for treating TSC-associated partial seizures based on findings from a large phase 3 study
Ongoing studies will assess long-term (~10 years) safety of everolimus for treating TSC-associated partial seizures
Future research is needed exploring everolimus for treating TSC-seizures in combination with specific therapies (such as the ketogenic diet), as monotherapy, as well as for possible prevention of TSC-seizures
Declaration of interest
D Franz has received funding to his employer, honoraria, and travel support from Novartis Pharmaceuticals Corporation. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or conflict with the subject matter or materials discussed in this manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose
Acknowledgments
Writing/editorial assistance was provided by Traci Stuve, MA, and Robert J. Schoen, PharmD, of ApotheCom (Yardley, PA, USA) and was funded by Novartis Pharmaceuticals Corporation.
Additional information
Funding
References
- Osborne JP, Fryer A, Webb D. Epidemiology of tuberous sclerosis. Ann N Y Acad Sci. 1991;615:125–127.
- Curatolo P, Bombardieri R, Jozwiak S. Tuberous sclerosis. Lancet. 2008;372(9639):657–668.
- MacKeigan JP, Krueger DA. Differentiating the mTOR inhibitors everolimus and sirolimus in the treatment of tuberous sclerosis complex. Neuro Oncol. 2015;17(12):1550–1559.
- Fraioli MF, Lecce M, Fraioli C, et al. Spinal giant cell tumor in tuberous sclerosis: case report and review of the literature. J Spinal Cord Med. 2013;36(2):157–160.
- Koc G, Sugimoto S, Kuperman R, et al. Pancreatic tumors in children and young adults with tuberous sclerosis complex. Pediatr Radiol. 2017;47(1):39–45.
- Saxena A, Sampson JR. Epilepsy in tuberous sclerosis: phenotypes, mechanisms, and treatments. Semin Neurol. 2015;35(3):269–276.
- Curatolo P, Maria BL. Tuberous sclerosis. Handb Clin Neurol. 2013;111:323–331.
- Chu-Shore CJ, Major P, Camposano S, et al. The natural history of epilepsy in tuberous sclerosis complex. Epilepsia. 2010;51(7):1236–1241.
- Northrup H, Krueger DA. Tuberous sclerosis complex diagnostic criteria update: recommendations of the 2012 international tuberous sclerosis complex consensus conference. Pediatr Neurol. 2013;49:243–254.
- Krueger DA, Northrup H. Tuberous sclerosis complex surveillance and management: recommendations of the 2012 international tuberous sclerosis complex consensus conference. Pediatr Neurol. 2013;49:255–265.
- Curatolo P, Jozwiak S, Nabbout R. Management of epilepsy associated with tuberous sclerosis complex (TSC): clinical recommendations. Eur J Paediatr Neurol. 2012;16(6):582–586.
- Moavero R, Cerminara C, Curatolo P. Epilepsy secondary to tuberous sclerosis: lessons learned and current challenges. Childs Nerv Syst. 2010;26(11):1495–1504.
- Curatolo P, Nabbout R, Lagae L, et al. Management of epilepsy associated with tuberous sclerosis complex: updated clinical recommendations. Eur J Paediatr Neurol. 2018;22(5):738–748.
- Canevini MP, Kotulska-Jozwiak K, Curatolo P, et al. Current concepts on epilepsy management in tuberous sclerosis complex. Am J Med Genet C Semin Med Genet. 2018;178(3):299–308.
- French JA, Lawson JA, Yapici Z, et al. Adjunctive everolimus therapy for treatment-resistant focal-onset seizures associated with tuberous sclerosis (EXIST-3): a phase 3, randomised, double-blind, placebo-controlled study. Lancet. 2016;388(10056):2153–2163.
- Prescribing Information for Afinitor®. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022334s6lbl.pdf
- Summary of Product Characteristics for Votubia. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2010/022334s6lbl.pdf
- Curatolo P. Mechanistic target of rapamycin (mTOR) in tuberous sclerosis complex-associated epilepsy. Pediatr Neurol. 2015;52:281–289.
- Sirven JI, Noe K, Hoerth M, et al. Antiepileptic drugs 2012: recent advances and trends. Mayo Clin Proc. 2012;87(9):879–889.
- Ben-Menachem E. Mechanism of action of vigabatrin: correcting misperceptions. Acta Neurol Scand Suppl. 2011;124(192):5–15.
- Prescribing Information for Sabril®. Availbale from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2013/020427s010s011s012,022006s011s012s013lbl.pdf
- Bombardieri R, Pinci M, Moavero R, et al. Early control of seizures improves long-term outcome in children with tuberous sclerosis complex. Eur J Paediatr Neurol. 2010;14(2):146–149.
- Friedman D, Bogner M, Parker-Menzer K, et al. Vigabatrin for partial-onset seizure treatment in patients with tuberous sclerosis complex. Epilepsy Behav. 2013;27(1):118–120.
- Yum MS, Lee EH, Ko TS. Vigabatrin and mental retardation in tuberous sclerosis: infantile spasms versus focal seizures. J Child Neurol. 2013;28(3):308–313.
- Origlieri C, Geddie B, Karwoski B, et al. Optical coherence tomography to monitor vigabatrin toxicity in children. J AAPOS. 2016;20(2):136–140.
- Riikonen R, Rener-Primec Z, Carmant L, et al. Does vigabatrin treatment for infantile spasms cause visual field defects? An international multicentre study. Dev Med Child Neurol. 2015;57(1):60–67.
- Greiner HM, Lynch ER, Fordyce S, et al. Vigabatrin for childhood partial-onset epilepsies. Pediatr Neurol. 2012;46(2):83–88.
- Jennesson M, van Eeghen AM, Caruso PA, et al. Clobazam therapy of refractory epilepsy in tuberous sclerosis complex. Epilepsy Res. 2013;104(3):269–274.
- Franz DN, Tudor C, Leonard J, et al. Lamotrigine therapy of epilepsy in tuberous sclerosis. Epilepsia. 2001;42(7):935–940.
- Collins JJ, Tudor C, Leonard JM, et al. Levetiracetam as adjunctive antiepileptic therapy for patients with tuberous sclerosis complex: a retrospective open-label trial. J Child Neurol. 2006;21(1):53–57.
- Madeo M, Kovacs AD, Pearce DA. The human synaptic vesicle protein, SV2A, functions as a galactose transporter in Saccharomyces cerevisiae. J Biol Chem. 2014;289(48):33066–33071.
- Overwater IE, Bindels-de Heus K, Rietman AB, et al. Epilepsy in children with tuberous sclerosis complex: chance of remission and response to antiepileptic drugs. Epilepsia. 2015;56(8):1239–1245.
- Zhang B, McDaniel SS, Rensing NR, et al. Vigabatrin inhibits seizures and mTOR pathway activation in a mouse model of tuberous sclerosis complex. PLoS One. 2013;8(2):e57445.
- McDaniel SS, Rensing NR, Thio LL, et al. The ketogenic diet inhibits the mammalian target of rapamycin (mTOR) pathway. Epilepsia. 2011;52(3):e7–e11.
- Prescribing Information for Epidiolex®. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210365lbl.pdf
- Devinsky O, MarshE, Friedman D, et al. Cannabidiol in patients with treatment-resistant epilepsy: an open-label interventional trial. Lancet Neurol. 2016;15(3):270–278.
- Clinicaltrials.gov. A randomized controlled trial of cannabidiol (GWP42003-P, CBD) for seizures in tuberous sclerosis complex (GWPCARE6). NCT02544763. Available from: https://clinicaltrials.gov/ct2/show/NCT02544763
- Hess EJ, Moody KA, Geffrey AL, et al. Cannabidiol as a new treatment for drug-resistant epilepsy in tuberous sclerosis complex. Epilepsia. 2016;57(10):1617–1624.
- Ryther RC, Wong M. Mammalian target of rapamycin (mTOR) inhibition: potential for antiseizure, antiepileptogenic, and epileptostatic therapy. Curr Neurol Neurosci Rep. 2012;12(4):410–418.
- Wong M, Roper SN. Genetic animal models of malformations of cortical development and epilepsy. J Neurosci Methods. 2016;260:73–82.
- Meikle L, Pollizzi K, Egnor A, et al. Response of a neuronal model of tuberous sclerosis to mammalian target of rapamycin (mTOR) inhibitors: effects on mTORC1 and Akt signaling lead to improved survival and function. J Neurosci. 2008;28(21):5422–5432.
- Curatolo P, Moavero R. mTOR inhibitors as a new therapeutic option for epilepsy. Expert Rev Neurother. 2013;13(6):627–638.
- Overwater IE, Reitman AB, Bindels-de Heus K, et al. Sirolimus for epilepsy in children with tuberous sclerosis complex: a randomized controlled trial. Neurology. 2016;87(10):1011–1018.
- Kirchner GI, Meier-Wiedenbach I, Manns MP. Clinical pharmacokinetics of everolimus. Clin Pharmacokinet. 2004;43(2):83–95.
- Combes FP, Baneyx G, Coello N, et al. Population pharmacokinetics-pharmacodynamics of oral everolimus in patients with seizures associated with tuberous sclerosis complex. J Pharmacokinet Pharmacodyn. Epub 2018 July 12;45:707–719.
- Franz DN, Lawson JA, Yapici Z, et al. Everolimus dosing recommendations for tuberous sclerosis complex-associated refractory seizures. Epilepsia. 2018;59(6):1188–1197.
- Krueger DA, Care MM, Holland K, et al. Everolimus for subependymal giant-cell astrocytomas in tuberous sclerosis. N Engl J Med. 2010;363(19):1801–1811.
- Krueger D, Care M, Agricola K, et al. Everolimus long-term safety and efficacy in subependymal giant cell astrocytoma. Neurology. 2013;80(6):574–580.
- Franz DN, Agricola K, Mays M, et al. Everolimus for subependymal giant cell astrocytoma: 5-year final analysis. Ann Neurol. 2015;78(6):929–938.
- Krueger DA, Wilfong AA, Holland-Bouley K, et al. Everolimus treatment of refractory epilepsy in tuberous sclerosis complex. Ann Neurol. 2013;74(5):679–687.
- Krueger DA, Wilfong AA, Mays M, et al. Long-term treatment of epilepsy with everolimus in tuberous sclerosis. Neurology. 2016;87(23):2408–2415.
- Franz DN, Belousova E, Sparagana S, et al. Efficacy and safety of everolimus for subependymal giant cell astrocytomas associated with tuberous sclerosis complex (EXIST-1): a multicentre, randomised, placebo-controlled phase 3 trial. Lancet. 2013;381(9861):125–132.
- Mizuguchi M, Ikeda H, Kagitani-Shimono K, et al. Everolimus for epilepsy and autism spectrum disorder in tuberous sclerosis complex: EXIST-3 substudy in Japan. Brain Dev. 2018;41(1):1–10.
- Franz DN, Lawson JA, Yapici Z, et al. Everolimus for treatment-refractory seizures in TSC. Extension of a randomized controlled trial. Neurol Clin Pract. 2018;8(5):412–420.
- Curatolo P, Franz DN, Lawson JA, et al. Adjunctive everolimus for children and adolescents with treatment-refractory seizures associated with tuberous sclerosis complex: post-hoc analysis of the phase 3 EXIST-3 trial. Lancet Child Adolesc Health. 2018;2(7):495–504.
- Peters J, Prohl A, Hodgeman R et al. Time to vigabatrin”: cognitive outcome in tuberous sclerosis comples is related to timely imitiation of vigabatrin specifically. Presented at the American Epilepsy Society 69th Annual Meeting; 2015 Dec 4–8; Philadelphia, Pennsylvania. Abst. 3.193.
- Zak S, Mokhallati N, Su W, et al. Lymphangioleiomyomatosis mortality in patients with tuberous sclerosis complex. Ann Am Thorac Soc. 2019 Apr;16(4):509-512.
- Yapici Z, Fan PC, Lawson JA et al. Long-term safety, roll-over study of adjunctive everolimus in patients with tuberous sclerosis complex (TSC)-associated treatment-refractory seizures who continue to benefit from everolimus treatment after completion of EXIST-3. Poster presented at 13th European Congress of Epileptology; 2018 Aug 26–30; Vienna, Austria.
- Pharmaceuticals GW. GW pharmaceuticals reports positive phase 3 pivotal trial results for EPIDIOLEX® (cannabidiol) oral solution in patients with seizures associated with tuberous sclerosis complex. [cited 2019 May 8]. Available from: http://ir.gwpharm.com/news-releases/news-release-details/gw-pharmaceuticals-reports-positive-phase-3-pivotal-trial
- Novartis Pharmaceuticals Corporation. Novartis drug Afinitor Disperz® receives FDA approval to treat TSC-associated partial-onset seizures. [cited 2019 Feb 22]. Available from: https://www.pharma.us.novartis.com/print/15441
- Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus for angiomyolipoma associated with tuberous sclerosis complex or sporadic lymphangioleiomyomatosis (EXIST-2): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet. 2013;381(9869):817–824.
- Franz DN, Belousova E, Sparagana S, et al. Long-term use of everolimus in patients with tuberous sclerosis complex: final results from the EXIST-1 study. PLoS One. 2016;11(6):e0158476.
- Bissler JJ, Kingswood JC, Radzikowska E, et al. Everolimus long-term use in patients with tuberous sclerosis complex: four-year update of the EXIST-2 study. PLoS One. 2017;12(8):e0180939.
- Dahdah N. Everolimus for the treatment of tuberous sclerosis complex-related cardiac rhabdomyomas in pediatric patients. J Pediatr. 2017;190:21–26 e7.
- Schmid GL, Kassner F, Uhlig HH, et al. Sirolimus treatment of severe PTEN hamartoma tumor syndrome: case report and in vitro studies. Pediatr Res. 2014;75(4):527–534.
- Zak M, Ledbetter M, Maertens P. Infantile Lhermitte-Duclos disease treated successfully with rapamycin. J Child Neurol. 2017;32(3):322–326.
- Schmidt LS, Linehan WM. Clinical features, genetics and potential therapeutic approaches for Birt-Hogg-Dube syndrome. Expert Opin Orphan Drugs. 2015;3(1):15–29.
- Nakamura M, Yao M, Sano F, et al. A case of metastatic renal cell carcinoma associated with Birt-Hogg-Dube syndrome treated with molecular-targeting agents. Hinyokika Kiyo. 2013;59(8):503–506.
- Marsh DJ, Trahair TN, Martin JL, et al. Rapamycin treatment for a child with germline PTEN mutation. Nat Clin Pract Oncol. 2008;5(6):357–361.
- Capal JK, Bernardino-Cuesta B, Horn PS, et al. Influence of seizures on early development in tuberous sclerosis complex. Epilepsy Behav. 2017;70(Pt A):245–252.
- O’Callaghan FJ, Edwards SW, Alber FD, et al. Safety and effectiveness of hormonal treatment versus hormonal treatment with vigabatrin for infantile spasms (ICISS): a randomised, multicentre, open-label trial. Lancet Neurol. 2017;16(1):33–42.
- Franz DN, Capal JK. mTOR inhibitors in the pharmacologic management of tuberous sclerosis complex and their potential role in other rare neurodevelopmental disorders. Orphanet J Rare Dis. 2017;12(1):51.
- Weckhuysen S, Marsan E, Lambrecq V, et al. Involvement of GATOR complex genes in familial focal epilepsies and focal cortical dysplasia. Epilepsia. 2016;57(6):994–1003.
- Moller RS, Weckhuysen S, Chipaux M, et al. Germline and somatic mutations in the MTOR gene in focal cortical dysplasia and epilepsy. Neurol Genet. 2016;2(6):e118.
- Chassoux F, Landre E, Mellerio C, et al. Type II focal cortical dysplasia: electroclinical phenotype and surgical outcome related to imaging. Epilepsia. 2012;53(2):349–358.
- Pitkänen A, Buckmaster P, Galanopoulou AS, et al. Models of seizures and epilepsy. Cambridge (MA), USA: Academic Press; 2017.