
ABSTRACT
Introduction
Amyotrophic lateral sclerosis (ALS) is a fatal disorder characterized by the progressive loss of upper and lower motor neurons. ALS has traditionally been classified within the domain of neuromuscular diseases, which are a unique spectrum of disorders that predominantly affect the peripheral nervous system. However, over the past decades compounding evidence has emerged that there is extensive involvement of the central nervous system. Therefore, one can question whether it remains accurate to classify ALS as a neuromuscular disorder.
Areas covered
In this review, the authors sought to discuss current approaches toward disease classification and how we should classify ALS based on novel insights from clinical, imaging, pathophysiological, neuropathological and genetic studies.
Expert opinion
ALS exhibits the cardinal features of a neurodegenerative disease. Therefore, classifying ALS as a neuromuscular disease in the strict sense has become untenable. Diagnosing ALS however does require significant neuromuscular expertise and therefore neuromuscular specialists remain best equipped to evaluate this category of patients. Designating motor neuron diseases as a separate category in the ICD-11 is justified and adequately deals with this issue. However, to drive effective therapy development the fields of motor neuron disease and neurodegenerative disorders must come together.
1. Introduction
Through classification diseases that have more in common with one another than with others are grouped together[Citation1]. Classification is of major importance as it shapes medicine and guides its practice. The World Health Organization (WHO) states that the international classification of disease (ICD) defines the universe of diseases, disorders, injuries and other related health conditions. By doing so, it provides the diagnostic classification standard for all clinical and research purposes. This data subsequently forms the foundation for evidenced-based decision-making, permits the monitoring of the incidence and prevalence of diseases, guides the allocation of resources and reimbursement, allows for the tracking of safety and quality guidelines, facilitates the sharing and comparison of health information between hospitals, regions, settings, and countries as well as across different time periods (https://icd.who.int/en).
There needs to be a solid rationale for grouping diseases together, ensuring that there are enough similarities for items in a category to be associated, while not obfuscating differences. Therefore, effective classifications must recognize differences as well as similarities[Citation1].
Although one can classify neurological disorders in a variety of ways (e.g., etiology, symptoms or pathology), clinical anatomical reasoning formed the foundation and still is the cornerstone of modern neurology [Citation2,Citation3]. Localizing the site of the lesion is essential to making any neurological diagnosis. The first major step is to determine whether the lesion is in the central or peripheral nervous system.
Diseases of the peripheral nervous system are categorized as neuromuscular diseases. There are over 600 distinct neuromuscular disorders, each with a unique set of signs and symptoms, depending on which part of the peripheral nervous system is (predominantly) affected ()[Citation4]. Also, the heterogeneity of underlying causes at specific anatomical sites results in variability in the age of onset (ranging from neonatal to late-onset), rate of progression, and disease severity. Nevertheless, most neuromuscular diseases converge to similar downstream effects, ultimately presenting with muscle weakness and atrophy, sensory disturbances, and/or reduced tendon reflexes. Other features that have been listed in subsets of patients with neuromuscular disorders, include marked bulbar signs, involvement of cranial nerves, and respiratory or cardiac dysfunction[Citation4]. A detailed summary of all neuromuscular disorders falls outside the scope of this article.
Figure 1. Overview of the peripheral nervous system and corresponding neuromuscular disorders.
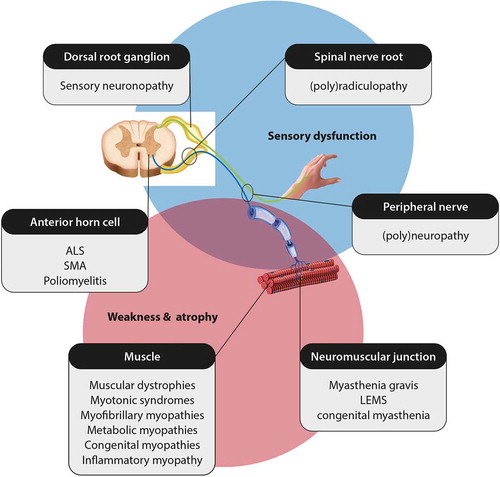
Amyotrophic lateral sclerosis (ALS) has classically been viewed as a neuromuscular disorder, because there is impairment of lower motor neurons at a different extent leading to muscle atrophy, fasciculations, severe weakness, respiratory failure, and death[Citation5]. However, ALS does not solely affect the peripheral nervous system and in fact pathological hyperreflexia (indicating involvement of the central nervous system) is even required for the diagnosis[Citation6]. Over the past 15 years, it has become evident that the involvement of the central nervous system is far more wide-spread than previously recognized. We, therefore, ask the question whether it remains accurate to classify ALS as a neuromuscular disorder.
2. Amyotrophic lateral sclerosis (ALS)
ALS is a fatal disorder characterized by the progressive loss of upper and lower motor neurons with a prevalence 2.6–3.0 cases per 100,000 people in European populations. The lifetime risk of developing ALS for women is 1:400 and is slighter higher for men at 1:350. The disease may run in families (familial ALS, FALS) or be (apparently) sporadic (SALS). On average, onset is in the early sixties, but with a wide range. Patients in their late teens and early twenties have been reported as well as cases well over the age of 90 [Citation7,Citation1602]. At present, several drugs have been approved for ALS, which include riluzol [Citation9], nuedexta [Citation10] and edaravone[Citation11]. Unfortunately, all of these drugs have limited effects and the prognosis remains grim with a median survival of 3 to 4 years.
Patients gradually develop progressive motor deficits over the course of weeks or months, which can affect any voluntary muscle. This means that the initial presentation is very heterogeneous and ranges from dysarthria to a foot drop. Disease onset is usually focal, but with subsequent spreading to other regions of the body. Spread of the disease may be within the same region (from hand to upper arm) as well as between neuro-anatomically connected regions (rostro-caudal or contra-lateral)[Citation12]. The degree of involvement of upper and lower motor neurons is also variable. Some patients predominantly show upper motor signs, with lower motor neuron involvement only becoming evident through neurophysiological studies or over the course of time. Similarly, there are patients with predominant lower motor neuron presentations[Citation13]. Whether pure upper motor neuron (primary lateral sclerosis) and pure lower motor neuron syndromes (progressive spinal muscular atrophy) actually exist, should be seen as separate diseases or are restricted phenotypes of ALS remains a topic of debate [Citation14–Citation16].
These highly heterogeneous presentations make diagnosing ALS challenging. In particular, because there is no diagnostic test that definitively demonstrates ALS (with the exception of DNA testing). ALS is therefore a diagnosis, which is made per exclusion and is based on the El Escorial [Citation6] or Awaji criteria[Citation17]. Summarized, these criteria require a history of progressive weakness that has spread within a region or to other regions (bulbar, cervical, thoracic, or lumbar), with evidence of lower motor neuron (clinical or electrophysiological) and upper motor neuron (clinical) involvement, and that no other disease processes explain the presentation [Citation6,Citation17].
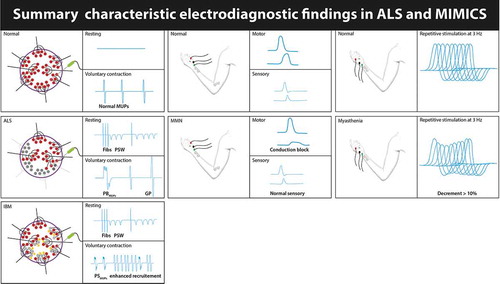
The loss of lower motor neurons in ALS leads to muscle atrophy, fasciculations, severe weakness and sometimes regional hyporeflexia. Underlying these LMN signs are degeneration and loss of peripheral axons, destruction of the neuromuscular junction, and even muscle changes. Whether these peripheral changes in muscle, NMJ, and axons are a primary event or secondary to pathological changes in the cell bodies of the LMNs is unclear. Nevertheless, given the clear involvement of the peripheral nervous system and the requirement in the diagnostic criteria to rule out other causes of LMN signs is perhaps why neuromuscular specialists are most experienced at evaluating this category of patients. Indeed, some of the most relevant alternative diagnoses that may mimic ALS are neuromuscular diseases such as Kennedy’s disease, myasthenia gravis (in particular with MuSK antibodies), distal hereditary motor neuropathies (dHMNs), multifocal motor neuropathy, pure motor CIDP, and inclusion body myositis and may require detailed neurophysiological studies ()[Citation4].
Figure 2. Shows the main findings on EMG that may aid to differentiate ALS from important mimics such as multifocal motor neuropathy (MMN), myasthenia gravis (MG) and inclusion body myositis (IBM).
However, in 2006 a major breakthrough was made in our understanding of ALS. Neumann and colleagues demonstrated that the presence of TDP-43-positive, ubiquitinated cytoplasmic inclusions is the main characteristic neuropathological feature of degenerating motor neurons. Importantly, they show that these TDP-43 inclusions are not only the hallmark of ALS, but also frontotemporal dementia (FTD)[Citation18]. This landmark discovery subsequently sparked a lot of research that has focused on the overlap between these two diseases[Citation19].
3. Frontotemporal dementia (FTD)
FTD is one of the most common forms of young onset dementia and is characterized by focal atrophy of the frontal and/or anterior temporal lobe. There are two major forms of FTD; behavioral variant (bvFTD) and a language variant, which is also divided into two subtypes: semantic dementia (SD) and progressive non-fluent aphasia (PNFA)[Citation20].
bvFTD is characterized by a progressive deterioration of behavior and/or cognition. Patients develop prominent changes in personality with disinhibition (socially inappropriate behavior, loss of manners or impulsive actions), apathy, loss of sympathy or empathy, stereotyped or compulsive behavior, altered food preferences, executive dysfunction and often have a profound lack of disease insight[Citation21].
The language variants of FTD are characterized by a slowly progressive, isolated language deficit that is the most prominent feature at onset and during the initial phases of the disease. The core features of semantic dementia are anomia and impaired single-word comprehension. Patients usually have quite severe naming problems, while other language domains are relatively spared. Single word comprehension is initially more problematic for low frequency words. Semantic dementia patients may not be capable of demonstrating how to use everyday items, such as a hammer or toothbrush. Despite this conceptual loss, speech remains largely fluent with intact grammar and the episodic memory is also largely spared[Citation22].
Patients with PNFA have slow, effortful speech that is due to an articulation planning deficit (apraxia of speech) or agrammatism, which results in the use of short, simple phrases and the leaving out of grammatical morphemes (functioning words that indicate tense, number, gender, or aspect). They do not suffer from conceptual loss and again episodic memory is largely spared[Citation22].
Over time patients with bvFTD develop language deficits and similarly patients with SD and PNFA develop behavioral changes.
4. The FTD-ALS continuum
Cognitive symptoms and frank dementia were reported in early descriptions of ALS. However, for a long time these changes were considered to be rare or perhaps even coincidental. Strikingly, neuropsychological studies have now clearly shown that cognitive and/or behavioral changes are an integral part of ALS. 5–15% of ALS patients also suffer from co-morbid FTD and that up to 50% have cognitive and/or behavioral deficits within the spectrum of FTD, but without meeting the formal diagnostic criteria [Citation23–Citation25].
This means that cognitive changes were frequently overlooked in the past. Perhaps because these changes may initially be subtle or were incorrectly attributed to motor symptoms. For instance, detecting language deficits in patients with severe dysarthria may be challenging and severe motor impairment may mask apathy[Citation5].
A wealth of neuropsychological research has now been conducted and has led to the characterization of the cognitive profile of ALS with deficits in fluency, language, social cognition, delayed verbal memory and executive functions[Citation26]. In ALS, loss of sympathy and apathy are the most common behavioral changes and are seen in 10–20% of patients. The language variants of FTD are understudied and probably also under recognized in ALS[Citation27]. Cognitive and/or behavioral changes are more common in certain genetic subtypes of ALS and are seen at high frequencies in particular in patients that carry repeat expansions in the C9orf72 gene[Citation23].
Only a limited number of longitudinal studies on cognition have been performed in ALS. Results from these studies, as well as, cross-sectional studies show that the frequency of patients with cognitive and/or behavioral changes increases with more advanced disease stages [Citation28,Citation29]. These findings suggest that the frequency of FTD spectrum changes in ALS is probably still underestimated.
The exact frequency of FTD patients that also develops ALS or motor involvement is unknown, as there are no population-based studies that have addressed this issue. Clinical cohort studies found that ±15% of FTD patients develop ALS, which may sometimes be many years after the onset of dementia. Approximately, 30–40% of FTD patients develop subtle clinical (mostly UMN signs) or neurophysiological evidence of motor involvement. The frequency of motor neuron involvement in FTD, therefore, seems comparable to the frequency of cognitive and behavioral changes in ALS [Citation30,Citation31].
This high frequency of cognitive/behavioral deficits in ALS and of motor involvement in FTD has led to the view that ALS and FTD form the phenotypic extremes of a clinical spectrum [Citation19,Citation23]. Screening for cognitive and behavioral changes has now become a part of the standard work-up of ALS patients and there are international consensus criteria for the ALS-FTD spectrum disorder[Citation24].
5. Imaging
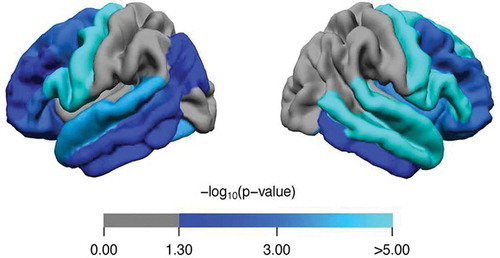
Many imaging modalities have been applied to study to brain morphologic changes in ALS, of which MRI has been used most frequently. As is to be expected, there is clear involvement of the primary motor cortex and corticospinal tract. Many studies have however also demonstrated involvement of other brain areas ()[Citation32]. Both gray and white matter involvement has been found. Widespread cerebral involvement is most prominent in patients with C9orf72 repeat expansions, but is not limited to this group[Citation33]. Intriguingly, asymptomatic C9orf72 carriers also have been found to have a thinner cerebral cortex in non-motor regions in comparison to their non-carrier family members[Citation34]. Although atrophy of posterior brain regions is not frequently reported, and a previous case report [Citation35] demonstrate that these regions might be involved and are possibly related to cognitive deficits as well. Deep gray matter has been studied less frequently than cortical gray matter, but studies have demonstrated atrophy of the deep gray matter. This includes atrophy of the hippocampi at an early stage, which is also correlated with cognitive and behavioral deficits as well as shorter survival [Citation36,Citation37]. Similarly, white matter involvement is not limited to motor connections, but also includes connections between more remote brain locations [Citation38,Citation39]. In line with this observation, it has been proposed that, although the pathogenic process might start in the motor system, it propagates to functionally linked areas through white matter connections [Citation40,Citation41].
Figure 3. Comparison of cortical thickness between ALS patients (n = 456) and healthy controls (n = 294). Analyses were corrected for age and sex. Regions with a significantly lower cortical thickness are marked in blue (p < 0.05, Bonferroni-corrected). p-values were mapped onto the surface of the entire region of interest (ROI, 34 per hemisphere) to indicate significant regional differences between ALS patients and controls.
6. Neuropathology & pathophysiology
In approximately 98% of ALS patients TDP-43-positive, ubiquitinated cytoplasmic inclusions are found in the remaining motor neurons at autopsy, making this the pathological hallmark of the disease [Citation18,Citation42]. There are a few genetic subgroups of ALS patients, in which there appears to be no or little TDP-43 pathology. In patients with SOD1 mutations, abnormal aggregation of SOD1 protein is the predominant finding and similarly there are FUS aggregates in patients with mutations in this gene[Citation20].
Neurodegenerative diseases are characterized by the predominant loss of a specific population of neurons accompanied by distinct neuropathological changes in the brain, including extracellular protein deposits, intracellular inclusions, and changes in cell morphology. In Alzheimer’s disease there is loss of neurons in the hippocampus with neurofibrillary tangles and amyloid β deposition. Parkinson’s disease is characterized by the loss of dopaminergic neurons in the substantia nigra accompanied by accumulation of α-synuclein. In FTD there is loss of cortical neurons in the frontal and/or anterior temporal lobes. In approximately half of FTD patients there is tau pathology, whereas the other half shows TDP-43 pathology highly similar to ALS. In 1–2% of FTD patients FUS pathology is found without prominent aggregation of TDP-43 or tau[Citation20]. All neurodegenerative diseases seem to start focally, but subsequently become progressive and over time affect additional regions of the brain[Citation43].
Brettschneider and colleagues elegantly demonstrated that TDP-43 pathology appears to spread throughout the brain and that this occurs in 4 stages. In stage 1 there is TDP-43 deposition in the spinal cord and brainstem or upper motor neurons. In stage 2, TDP-43 pathology progresses to the posterior frontal and anterior parietal regions, brainstem reticular formation, red nucleus and precerebellar nuclei. Stage 3 is characterized by involvement of the anterior frontal and basal forebrain, the caudate nucleus and putamen, mediodorsal nucleus, lateral thalamus and substantia nigra. In the fourth stage, TDP-43 deposition is seen in the anterior temporal lobe, including the hippocampus[Citation44].
The TDP-43 protein consists of 414 amino acid residues and can bind both DNA and RNA and has multiple functions in transcriptional repression, pre-mRNA splicing and translational regulation. Under physiological conditions TDP-43 generally resides in the nucleus. It has a nuclear export signal as well as nuclear localization signal, which means it can shuttle back and forth between nucleus and cytoplasm. The exact role of TDP-43 in the pathophysiology of ALS remains to be elucidated, but there is emerging evidence that dysregulation of TDP-43 expression plays an important role. When in nuclear excess TDP-43 binds the 3′UTR within its own pre-mRNA, which leads the use of alternative polyadenylation signals and splicing events causing these mRNA transcripts to be degraded. This feedback loop therefore auto-regulates the expression of TDP-43. Both loss of function and overexpression have been implicated in the pathogenesis.
Neuropathological studies clearly show that there is mislocalization of TDP-43 in ALS, with aggregation in the cytoplasm and depletion from the nucleus. This causes a nuclear loss-of-function leading to a widespread dysregulation of mRNA that affects hundreds of targets[Citation45]. This nuclear depletion seems to be caused by an inability (in part) of TDP-43 to reenter the nucleus, either because it has been sequestered into aggregates or by defects in nuclear transport [Citation46,Citation47].
Stress granules also seem to play a crucial in the formation of these TDP-43 aggregates. Under conditions of cell stress translation initiation is halted and stress granules are formed. These are dynamic cytoplasmic RNA–protein complexes (containing RNA-binding proteins, mRNAs, and translation initiation factors) that shield its components from degradation. The structure of TDP-43 contains aggregation-promoting prion-like domains that allow rapid self-association, which is required for the formation stress granules. Once cell stress dissipates, stress granules should dissemble allowing mRNA translation to resume.
Due to genetic mutations, chronic cell stress, protein modifications, and other mechanisms, these stress granules fail to dissemble and persist in ALS[Citation48]. The capacity to self-associate seems to lead to a self-templating process that drives the transition from stress granules into pathological aggregates. These aggregates have toxic properties and contribute to cell death[Citation49]. Moreover, TDP-43 aggregates appear to be able to spread to neighboring or functionally connected neurons and initiate this pathophysiological process[Citation50]. This process appears to be similar to the pathophysiological mechanism driving Creutzfeldt-Jakob disease and is therefore referred to as prion-like spreading[Citation51]. This mechanism of prion-like spreading also appears to play a role in the disease propagation of multiple other neurodegenerative diseases, such as Alzheimer’s disease, Parkinson’s disease and tauopathies [Citation52,Citation53].
7. Genetics
In FALS, inheritance most commonly follows an autosomal dominant pattern, but frequently with incomplete penetrance. Estimates of the frequency of FALS vary between studies and populations. In part, this may be due to differences in population genetics, but are also influenced by the fact that there is no clear definition of what constitutes familial ALS. Studies have shown that there is poor consensus amongst experts in the field. Some only consider the disease to be familial when there are one or more first degree relatives with ALS, whereas others also accept second degree or any relative. Similarly, there is no consensus on how to interpret pedigrees in which there are individuals with FTD or unspecified (young onset) dementia. Nevertheless, considering more and more studies are demonstrating that there is clustering of neurodegenerative diseases (FTD in particular) in the families of ALS patients, it seems reasonable to include FTD in the definition of FALS [Citation54–Citation57].
Most studies estimate that about 10–15% of ALS is familial and ±30 different genes have now been identified for FALS. Combined these genes explain roughly 65% of FALS cases. In the remaining cases the pathogenic mutations are yet to be identified. The most important FALS genes are C9orf72 (30–40%), SOD1 (10–20%), TARDBP (5%) and FUS (5%), while mutations in other genes are rare (±1%)[Citation58].
In the vast majority of cases (85–90%) there is no clear family history of ALS and these cases are labeled as sporadic ALS. Sporadic ALS (SALS) is thought be multifactorial in nature, with both genetic and environmental risk factors contributing to disease risk. The genetic component of SALS is nonetheless considerable and has been estimated at 40–60% [Citation59–Citation61]. Over the last 15 years studies (genome-wide association studies and others) have identified genetic risk factors for SALS at almost an exponential pace. We now know that polymorphisms and repeat expansions in genes like unc13a, NEK1, KIF5a, ATXN2 and NIPA1 are associated with ALS [Citation62–Citation65]. Interestingly, as studies in SALS are getting larger and therefore have the statistical power to analyze genetic variants with lower frequencies, overlap between FALS and SALS is emerging. For instance, common polymorphism in KIF5a appears to be a modest risk factor (SNP rs113247976 with an odds ratio of 1.38 for SALS), whereas other mutations in this gene seem to be directly pathogenic[Citation64]. Similarly, approximately 10% of SALS patients carry pathogenic mutations in FALS genes (C9orf72, SOD1, TARDBP, and FUS) and FALS patients have been found to carry mutations in more than one ALS gene [Citation58,Citation66]. A rigid distinction between FALS and SALS therefore seems artificial. ALS is perhaps best viewed as a heterogeneous disease in which genetics play a large role. Moreover, there are many reports of FALS cases that carry potentially pathogenic mutations in more than one ALS gene, suggesting the disease may be oligogenic. A recent genome-wide association study interrogated the genetic architecture of ALS and also concluded that disease risk is predominantly driven by a limited number of rare variants with large effect [Citation62,Citation66].
Many of the genes that have been identified for ALS are pleiotropic. This means that a gene is associated with multiple diseases or phenotypes. For instance, repeat expansions in the C9orf72 gene are associated with ALS, FTD, parkinsonism, Huntington phenocopies and psychiatric disorders [Citation67,Citation68]. Additional examples are ANG (ALS and Parkinson’s disease)[Citation69], TARDBP (ALS, FTD and parkinsonism) [Citation70,Citation71], ATXN2 (spinocerebellar ataxia type 2, ALS and parkinsonism) [Citation72,Citation73] and FUS (ALS and essential tremor) [Citation74,Citation75].
provides an overview of ALS genes and their pleiotropic effects, which shows that the overlap is predominantly with other neurodegenerative diseases, such as FTD, spinocerebellar ataxia, parkinsonism, and hereditary spastic paraplegia. Several ALS genes (hnRNPA1, VCP, SQSTM1, MATR3 and OPTN) are also associated with a complex hereditary disease termed multisystem proteinopathy that may present as ALS, FTD, inclusion body myopathy, Paget’s disease of the bone, or a combination of these disorders[Citation76]. Only a few rare genes show overlap with myopathy, SMA or CMT phenotypes.
Table 1. Overview of ALS and pleiotropic effects.
8. Discussion
One can take many approaches toward classifying disease. Considering these classifications greatly impact the way medicine is practiced, this is not just a mere question of semantics. There is no single system of classification that is capable of accurately categorizing all neurological diseases. To overcome this, a more or less mixed approach has been implemented. Some neurological disease categories have been assigned based on etiology (vascular, trauma, infection), whereas others are based on symptoms (headache, epilepsy) or anatomy (neuromuscular, white matter, spinal cord).
Neuromuscular diseases are defined by dysfunction of the peripheral nervous system. ALS has always formed an exception within this category, given that the central nervous system (upper motor neurons) is also involved[Citation2].
Historically there has been some debate to what degree and which part of the central nervous system is involved in ALS. At autopsy, Charcot noted that in ALS patients there was an abnormal hardening of tissue on the sides of spinal cord where the corticospinal tract runs, which he described as lateral sclerosis. Some authors have argued that ALS starts in the periphery (lower motor neuron, neuromuscular junction or even muscle) and that the degeneration of the corticospinal tract is a secondary process. Data from studies in animal models of ALS has provided evidence that many of the initial pathological changes appear to occur in the peripheral motor system, which supports this dying-back hypothesis [Citation100,Citation101].
However, neuropsychological studies clearly show that cognitive and behavioral changes within the spectrum of FTD are a common feature of ALS, indicating that CNS involvement goes far beyond the corticospinal tract [Citation24,Citation25,Citation102]. Imaging studies confirm that there are indeed wide-spread brain morphologic changes outside of the motor system[Citation33]. The discovery of TDP-43 as the pathological hallmark of both ALS and FTD unequivocally cements the view that these diseases form a clinical spectrum[Citation18]. Further neuropathological studies have demonstrated that ALS exhibits the main characteristics of a neurodegenerative disease with an initial predominant loss of a specific cell population (motor neurons) accompanied by abnormal protein aggregation with progressive spread to additional regions of the brain. Moreover, the pathology (TDP-43, FUS) and disease mechanisms (stress granules, prion-like spreading, etc.) underlying ALS are shared with multiple neurodegenerative diseases [Citation18,Citation44,Citation50]. Lastly, it is clear that genetics play an important role in both FALS and SALS[Citation62]. The ever-growing list of genes that has been identified for ALS also associates with other neurodegenerative disease (FTD, SCA, parkinsonism, etc.). In short, clinical, epidemiological, imaging, neuropsychological, pathological, pathophysiological, and genetic studies show that ALS exhibits the cardinal features of a neurodegenerative disease. Moreover, there is little to no overlap with neuromuscular diseases in which pathophysiology is most commonly driven by toxic exposures, metabolic disorders, mutations in genes crucial to contractile units in muscles, synthesis of myelin and axonal transport or by inflammatory processes[Citation4].
As classifications aim to group diseases together that have more in common with one another, than with those in other categories[Citation1], it is simply untenable to classify ALS as a neuromuscular disease. Based on our current insights ALS can probably best be characterized as a neurodegenerative syndrome with extensive lower motor neuron involvement. There are indeed several other diseases within the neurodegenerative domain, which also involve the peripheral nervous system. Lower motor neuron loss and axonal neuropathy may for instance also be seen in subtypes of spinocerebellar ataxia.
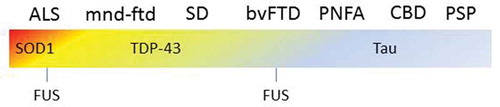
Although, it is more accurate to classify ALS within the neurodegenerative domain, it also requires the addition of an exception to address lower motor neuron degeneration. Within the ICD-11 neuromuscular diseases have been split into several categories; ‘Disorders of nerve root, plexus or peripheral nerves’ and ‘Diseases of neuromuscular junction or muscle’. Motor neuron diseases or related disorders, to which ALS belongs, forms a separate category and therefore this classification adequately deals with the issue of neuromuscular versus neurodegenerative. However, ALS is still classified separately from FTD, which falls under ‘disorders with neurocognitive impairment as a major feature’[Citation2]. This problem is not limited to the ALS-FTD spectrum. Progressive supranuclear palsy (PSP) is an atypical parkinsonism that falls into the category of movement disorders. However, in PSP the frontal and temporal lobes also degenerate, a process, which has been labeled frontotemporal lobar degeneration (FTLD). This results in cognitive and behavioral changes that overlap with FTD, which is a disease entity and not the degenerative process. These terms are however frequently used interchangeably, which leads to highly confusing literature on the topic ().
Figure 4. FTD forms a clinical with motor neuron diseases as well as with movement disorders, such as CBD and PSP. The underlying pathology and underlying genetics are equally heterogeneous. This example illustrate how challenging it can be to classify diseases with heterogeneous which form a spectrum. ALS = amyotrophic lateral sclerosis, mnd-ftd = motor neuron disease frontotemporal dementia, SD = semantic dementia, bvFTD = behavioral variant frontotemporal dementia, PNFA = progressive non-fluent aphasia, CBD = corticobasal degeneration, PSP = progressive supranuclear palsy.
Genes containing pathogenic mutations may be expressed in multiple tissues and therefore may also lead to heterogeneous phenotypes belonging to different categories. Perhaps, this is best illustrated by multisystem proteinopathy that may present as ALS (motor neuron diseases or related disorders), FTD (disorders with neurocognitive impairment as a major feature), inclusion body myopathy (diseases of neuromuscular junction or muscle), Paget’s disease of the bone (diseases of the musculoskeletal system or connective tissue), or a combination of these disorders[Citation76].
It seems we will always struggle to classify diseases that form phenotypic spectra or which are caused by mutations in genes that are expressed multiple tissue types. Perhaps we will have to accept that there are diseases that simply do not fit into predefined boxes and that in these instances, we must adopt a practical approach.
Specialized clinics for motor neuron diseases have been in place for decades and neuromuscular specialists have adapted to the insight that cognitive and behavioral changes are an integral part of the disease [Citation23,Citation102]. Neuropsychological screening has become part of the standard work-up, for which multiple ALS-specific screeners are available as well as diagnostic consensus criteria [Citation24,Citation102,Citation103]. As many neuromuscular diseases are hereditary, neuromuscular specialists are equipped to deal with genetic testing and commonly have well-established collaborations with clinical geneticists. Therefore, it seems that clinical care for patients will at present not benefit from relabeling ALS [Citation5,Citation7,Citation1602,Citation23].
In the long run, however, more extensive collaboration between the fields of motor neuron disease and neurodegeneration (FTD in particular) will be required. Understanding the penetrance of pathogenic mutations cannot be done by only studying parts of pedigrees and half of the phenotypes. Understanding the phenotype is also crucial to detecting early disease manifestations and developing biomarkers. This is of increasing importance as rapid advances have been made in developing gene-targeted therapies, in particular with antisense oligonucleotides. At present, there are multiple on-going trials for ALS patients with mutations in SOD1 (NCT02623699) and C9orf72 (NCT03626012), that have demonstrated very promising preliminary results. As other strategies to correct genetic defects are also emerging, clinicians are now even realistically considering studies with pre-symptomatic individuals with the goal of disease prevention. The identification of wet biomarkers could potentially replace clinical outcome measures in clinical trials and even facilitate the inclusion of patients with different phenotypes in the same study.
9. Conclusion
Neuromuscular diseases are a spectrum of disorders that predominantly/exclusively affect the peripheral nervous system. Current insights from clinical, imaging, neuropathological and genetic studies show that ALS exhibits the cardinal features of a neurodegenerative disease. Therefore, classifying ALS as a neuromuscular disease in the strict sense has become untenable. Diagnosing ALS however does require significant neuromuscular expertise and therefore neuromuscular specialists remain best equipped to evaluate this category of patients. Designating motor neuron diseases as a separate category in the ICD-11 is justified and adequately deals with this issue. However, to drive effective therapy development the fields of motor neuron disease and neurodegenerative disorders must come together.
10. Expert opinion
Increasingly experts are recognizing ALS as a syndrome rather than a single disease entity. There are many different subtypes of ALS, which share common features such as progressive loss of motor neurons and TDP-43 pathology. It is, however, also clear that there are considerable differences between these subtypes. Multiple genes and many different pathophysiological mechanisms are involved. Thus, it is highly unlikely that we will able to effectively treat ALS with a single drug, but rather that multiple targeted treatments will need to be developed[Citation7].
The greatest challenge in ALS research is therefore to disentangle the various subtypes, in order to facilitate the development of these targeted treatments. To this end, great progress has already been made in unraveling the genetics of FALS. Approximately 30 FALS genes have been identified to date, which account for ±65% of all familial patients[Citation58]. The identification of these genes has enabled deep phenotyping and natural history studies of genetic subgroups, such as C9orf72 and SOD1 [Citation104,Citation105]. This has also led to the identification of gene specific biomarkers. Through a process called Repeat-Associated Non-ATG translation, polymers of dipeptides (dipeptide repeats or DPRs) are derived from the repeat expansion in C9orf72, which occurs from both sense and antisense C9orf72 RNA. These DPRs can be detected in CSF and can therefore be used to assess whether experimental compounds achieve target engagement in early stage therapy development studies[Citation106]. Similarly, the degree of SOD1 knock-down in CSF has been used to evaluate target engagement for compounds targeting SOD1 [Citation105,Citation107]. A solid understanding of genotype-phenotype correlations and the natural history of the disease as well as the availability of biomarkers for target engagement, have paved the way for initiating the first trials with gene-targeted therapies in FALS.
There is great hope surrounding gene-targeted therapeutic approaches, in particular as there has been increasing success in other indications, such as SMA, Huntington, and TTR-amyloidosis [Citation108–Citation110]. It therefore only seems a matter of time before studies targeting other FALS genes will be initiated. Although these developments are highly promising, they also pose significant challenges. ALS is a relatively rare disease and further dividing the disease into multiple subgroups (>30) will complicate conducting clinical trials. Large-scale international collaboration will be required to identify and enroll patients into the appropriate studies. This infrastructure is mostly in place, as several large ALS trial consortia already exist, such as NEALS (https://www.neals.org/) and TRICALS (https://www.tricals.org/).
Alternative strategies to overcome heterogeneity in rare diseases can perhaps be copied from the field of oncology. In so-called basket trials, one targeted therapy is evaluated in multiple diseases or multiple disease subtypes[Citation111]. This means that patients with different types of cancer, but with the same molecular aberrations are enrolled into one study. For instance, BRAF mutations may be found in melanoma, non-Hodgkin’s lymphoma, colorectal cancer and papillary thyroid carcinoma and therefore BRAF inhibitors can be used as a treatment for a variety of cancers[Citation112]. Basket trials require an outcome measure that can be assessed across multiple diseases and such examples are disease free survival or tumor volume. One could envision basket trials with C9orf72 repeat expansion carriers with different clinical phenotypes (ALS, FTD, parkinsonism) in which the primary outcome would be a reduction in DPRs or neurofilaments rather than a disease specific assessment, such as the ALSFRS-R or muscle strength.
For the genetic forms of ALS, a pipeline for therapy development seems to be emerging, where after gene discovery one can move toward pre-clinical studies that aim to correct the genetic defect (antisense oligonucleotides, gene therapy, CRISPR-Cas, etc.), genotype-phenotype correlations and natural history studies, biomarker studies and finally human trials.
For SALS, the path toward effective treatment is perhaps less clear at this moment. The majority of genetic risk factors that have been identified for SALS have small odds ratios and the mechanism by which these variants contribute to disease is often also unclear. It is therefore questionable how and if targeting these variants would be useful. Nevertheless, recent studies have shown that common genetic polymorphisms may have strong effects on survival in ALS and are involved in pharmacogenetic interactions[Citation113]. A genetic post-hoc analysis of trials with lithium carbonate found evidence that patients homozygous for the C allele of rs12608932 located in the unc13a gene may have benefitted substantially from the experimental compound, whereas the overall group did not. The 12-month survival probability for unc13a carriers improved from 40.1% to 69.7% when treated with lithium carbonate[Citation114].
Obviously, there are other strategies than genetics by which patients could be sub-grouped, such as imaging characteristics, cognitive profile or biomarkers. HERV-K expression levels in blood are of potential interest, as there is mounting evidence that in (some) ALS patients reactivation of human endogenous retroviruses may play a role in the pathophysiology[Citation115]. In a recent phase 2 study HERV-K was targeted using the antiretroviral drug, Triumeq. In response to treatment with Triumeq a favorable response on HERV-K expression was seen, which was accompanied by a decline in ALSFRS-R progression rate of ±20%[Citation116].
In summary, the challenge in ALS will be to unravel disease heterogeneity and subgroup patients according to genetics, most relevant pathophysiological feature or biomarkers in order to facilitate the development of targeted treatments and introduce precision medicine.
Article highlights
ALS is disorder characterized by the loss upper and lower motor neurons. The diagnosis is made by exclusion, which means that alternative causes for lower motor neuron dysfunction (other neuromuscular diseases) need to be ruled out. Hence, ALS patients are commonly seen by neuromuscular specialist.
Frontotemporal dementia as well as cognitive and behavioral changes within the FTD spectrum are common in ALS and is seen in up to 50% of cases. Many now view ALS and FTD as the phenotypic extremes of a continuum (FTD-MND continuum).
Aggregation and mislocalization of TDP-43 is the pathological hallmark of ALS.
The genetic architecture of ALS is highly heterogeneous. Over 30 different genes have now been implicated in ALS, many of which are pleiotropic and are also involved in other neurodegenerative diseases such as FTD, parkinsonism, spinocerebellar ataxia and HSP.
Current insights from clinical, imaging, neuropathological and genetic studies show that ALS exhibits the cardinal features of a neurodegenerative disease.
Due to the highly heterogeneous nature of ALS, it seems highly unlikely that we will be able to treat the disease with a single drug. Multiple targeted therapies will need to be developed.
Declaration of interest
MA Van Es received grants from the Netherlands Organization for Health Research and Development (Veni scheme), The Thierry Latran foundation, Motor Neurone Disease Association (MNDA), FIGHT-MND, the Netherlands ALS foundation (Stichting ALS Nederland), the EU Joint Programme —Neurodegenerative Disease Research (JPND), has received travel grants from Shire (formerly Baxalta), consulted for Biogen and currently serves on the ethical review board at the UMC Utrecht, The Netherlands. LH Van den Berg reports grants from Netherlands ALS Foundation, the Netherlands Organization for Health Research and Development (Vici scheme), the Netherlands Organization for Health Research and Development (SOPHIA, STRENGTH, ALS-CarE project), funded through the EU Joint Programme—Neurodegenerative Disease Research, JPND), served on the Scientific Advisory Board of Biogen, Cytokinetics, Prinses Beatrix SpierFonds, and the Latran Foundation. The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or conflict with the subject matter or materials discussed in this manuscript apart from those disclosed.
Reviewer disclosures
Peer reviewers on this manuscript have no relevant financial or other relationships to disclose.
References
- Jutel A. Classification, disease, and diagnosis. Perspect Biol Med. 2011;54:189–205.
- Rajakulendran S, Dua T, Harper M, et al. The classification of neurological disorders in the 11th revision of the International Classification of Diseases (ICD-11). J Neurol Neurosurg Psychiatry. 2014;85:952–953.
- Shakir R, Rajakulendran S. The 11th revision of the International Classification of Diseases (ICD): the neurological perspective. JAMA Neurol. 2013;70:1353–1354.
- Morrison BM. Neuromuscular diseases. Semin Neurol. 2016;36:409–418.
- Turner MR, Hardiman O, Benatar M, et al. Controversies and priorities in amyotrophic lateral sclerosis. Lancet Neurol. 2013;12:310–322.
- Brooks BR, Miller RG, Swash M, et al. World federation of neurology research group on motor neuron D. El Escorial revisited: revised criteria for the diagnosis of amyotrophic lateral sclerosis. Amyotroph Lateral Scler Other Motor Neuron Disord. 2000;1:293–299.
- van Es MA, Hardiman O, Chio A, et al. Amyotrophic lateral sclerosis. Lancet. 2017;390:2084–2098.
- Brown RH Jr., Al-Chalabi A. Amyotrophic lateral sclerosis. N Engl J Med. 2017 Jul 13;377(2):162–172. DOI:10.1056/NEJMra1603471
- Bensimon G, Lacomblez L, Meininger V. A controlled trial of riluzole in amyotrophic lateral sclerosis ALS/Riluzole study group. N Engl J Med. 1994;330:585–591.
- Smith R, Pioro E, Myers K, et al. Enhanced bulbar function in amyotrophic lateral sclerosis: the nuedexta treatment trial. Neurotherapeutics. 2017;14:762–772.
- Writing G, Edaravone ALSSG. Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. Lancet Neurol. 2017;16:505–512.
- Ravits JM, La Spada AR. ALS motor phenotype heterogeneity, focality, and spread: deconstructing motor neuron degeneration. Neurology. 2009;73:805–811.
- Swinnen B, Robberecht W. The phenotypic variability of amyotrophic lateral sclerosis. Nat Rev Neurol. 2014;10:661–670.
- de Vries BS, Rustemeijer LMM, Bakker LA, et al. Cognitive and behavioural changes in PLS and PMA: challengingthe concept of restricted phenotypes. J Neurol Neurosurg Psychiatry. 2019;90:141–147.
- Turner MR, Barohn RJ, Corcia P, et al. Primary lateral sclerosis: consensus diagnostic criteria. J Neurol Neurosurg Psychiatry. 2020;91:373–377.
- Ludolph A, Drory V, Hardiman O, et al. A revision of the El Escorial criteria - 2015. Amyotroph lateral scler frontotemporal degener 2015; 16: 291–292.
- Carvalho MD, Swash M. Awaji diagnostic algorithm increases sensitivity of El Escorial criteria for ALS diagnosis. Amyotroph Lateral Scler. 2009;10:53–57.
- Neumann M, Sampathu DM, Kwong LK, et al. Ubiquitinated TDP-43 in frontotemporal lobar degeneration and amyotrophic lateral sclerosis. Science. 2006;314:130–133.
- Andersen PM. ALS and FTD: two sides of the same coin? Lancet Neurol. 2013;12:937–938.
- Seelaar H, Rohrer JD, Pijnenburg YA, et al. Clinical, genetic and pathological heterogeneity of frontotemporal dementia: a review. J Neurol Neurosurg Psychiatry. 2011;82:476–486.
- Rascovsky K, Hodges JR, Knopman D, et al. Sensitivity of revised diagnostic criteria for the behavioural variant of frontotemporal dementia. Brain. 2011;134:2456–2477.
- Gorno-Tempini ML, Hillis AE, Weintraub S, et al. Classification of primary progressive aphasia and its variants. Neurology. 2011;76:1006–1014.
- Burrell JR, Halliday GM, Kril JJ, et al. The frontotemporal dementia-motor neuron disease continuum. Lancet. 2016;388:919–931.
- Strong MJ, Abrahams S, Goldstein LH, et al. Amyotrophic lateral sclerosis - frontotemporal spectrum disorder (ALS-FTSD): revised diagnostic criteria. Amyotroph Lateral Scler Frontotemporal Degener. 2017;18:153–174.
- Woolley SC, Strong MJ. Frontotemporal dysfunction and dementia in amyotrophic lateral sclerosis. Neurol Clin. 2015;33:787–805.
- Blauw HM, Barnes CP, van Vught PW, et al. SMN1 gene duplications are associated with sporadic ALS. Neurology. 2012;78:776–780.
- Tan RH, Guennewig B, Dobson-Stone C, et al. The underacknowledged PPA-ALS: A unique clinicopathologic subtype with strong heritability. Neurology. 2019;92:e1354–e66.
- Crockford C, Newton J, Lonergan K, et al. ALS-specific cognitive and behavior changes associated with advancing disease stage in ALS. Neurology. 2018;91:e1370–e80.
- Elamin M, Bede P, Byrne S, et al. Cognitive changes predict functional decline in ALS: a population-based longitudinal study. Neurology. 2013;80:1590–1597.
- Josephs KA, Parisi JE, Knopman DS, et al. Clinically undetected motor neuron disease in pathologically proven frontotemporal lobar degeneration with motor neuron disease. Arch Neurol. 2006;63:506–512.
- Burrell JR, Kiernan MC, Vucic S, et al. Motor neuron dysfunction in frontotemporal dementia. Brain. 2011;134:2582–2594.
- Walhout R, Westeneng HJ, Verstraete E, et al. Cortical thickness in ALS: towards a marker for upper motor neuron involvement. J Neurol Neurosurg Psychiatry. 2015;86:288–294.
- Westeneng HJ, Walhout R, Straathof M, et al. Widespread structural brain involvement in ALS is not limited to the C9orf72 repeat expansion. J Neurol Neurosurg Psychiatry. 2016;87:1354–1360.
- Walhout R, Schmidt R, Westeneng HJ, et al. Brain morphologic changes in asymptomatic C9orf72 repeat expansion carriers. Neurology. 2015;85:1780–1788.
- Nijboer TCW, Nitert B, Westeneng HJ, et al. A case of ALS with posterior cortical atrophy. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:506–510.
- Westeneng HJ, Verstraete E, Walhout R, et al. Subcortical structures in amyotrophic lateral sclerosis. Neurobiol Aging. 2015;36:1075–1082.
- Machts J, Loewe K, Kaufmann J, et al. Basal ganglia pathology in ALS is associated with neuropsychological deficits. Neurology. 2015;85:1301–1309.
- Schmidt R, Verstraete E, de Reus MA, van den Heuvel MP, et al. Correlation between structural and functional connectivity impairment in amyotrophic lateral sclerosis. Hum Brain Mapp. 2014;35:4386–4395.
- Bede P, Omer T, Finegan E, et al. Connectivity-based characterisation of subcortical grey matter pathology in frontotemporal dementia and ALS: a multimodal neuroimaging study. Brain Imaging Behav. 2018;12:1696–1707.
- Schmidt R, de Reus MA, Scholtens LH, van den Heuvel MP, et al. Simulating disease propagation across white matter connectome reveals anatomical substrate for neuropathology staging in amyotrophic lateral sclerosis. Neuroimage. 2016;124:762–769.
- Kassubek J, Muller HP, Del Tredici K, et al. Diffusion tensor imaging analysis of sequential spreading of disease in amyotrophic lateral sclerosis confirms patterns of TDP-43 pathology. Brain. 2014;137:1733–1740.
- Brettschneider J, Arai K, Del Tredici K, et al. TDP-43 pathology and neuronal loss in amyotrophic lateral sclerosis spinal cord. Acta Neuropathol. 2014;128:423–437.
- Das S, Zhang Z, Ang LC. Clinicopathological overlap of neurodegenerative diseases: A comprehensive review. J Clin Neurosci. 2020 Aug;78:30–33. DOI:10.1016/j.jocn.2020.04.088. Epub 2020 Apr 27.
- Brettschneider J, Del Tredici K, Toledo JB, et al. Stages of pTDP-43 pathology in amyotrophic lateral sclerosis. Ann Neurol. 2013;74:20–38.
- Freibaum BD, Chitta RK, High AA, et al. Global analysis of TDP-43 interacting proteins reveals strong association with RNA splicing and translation machinery. J Proteome Res. 2010;9:1104–1120.
- Boeynaems S, Bogaert E, Van Damme P, Van Den Bosch L. Inside out: the role of nucleocytoplasmic transport in ALS and FTLD. Acta Neuropathol. 2016;132:159–173.
- Guo L, Kim HJ, Wang H, et al. Nuclear-import receptors reverse aberrant phase transitions of RNA-binding proteins with prion-like domains. Cell. 2018;173:677–92 e20.
- Molliex A, Temirov J, Lee J, et al. Phase separation by low complexity domains promotes stress granule assembly and drives pathological fibrillization. Cell. 2015;163:123–133.
- Shorter J, Taylor JP. Disease mutations in the prion-like domains of hnRNPA1 and hnRNPA2/B1 introduce potent steric zippers that drive excess RNP granule assembly. Rare Dis. 2013;1:e25200.
- McAlary L, Plotkin SS, Yerbury JJ, et al. Prion-like propagation of protein misfolding and aggregation in amyotrophic lateral sclerosis. Front Mol Neurosci. 2019;12:262.
- Ludolph AC, Brettschneider J. TDP-43 in amyotrophic lateral sclerosis - is it a prion disease? Eur J Neurol. 2015;22:753–761.
- Meisl G, Knowles TP, Klenerman D. The molecular processes underpinning prion-like spreading and seed amplification in protein aggregation. Curr Opin Neurobiol. 2020;61:58–64.
- Brunello CA, Merezhko M, Uronen RL, et al. Mechanisms of secretion and spreading of pathological tau protein. Cell Mol Life Sci. 2020;77:1721–1744.
- Byrne S, Bede P, Elamin M, et al. Proposed criteria for familial amyotrophic lateral sclerosis. Amyotroph Lateral Scler. 2011;12:157–159.
- Byrne S, Elamin M, Bede P, et al. Absence of consensus in diagnostic criteria for familial neurodegenerative diseases. J Neurol Neurosurg Psychiatry. 2012;83:365–367.
- Byrne S, Hardiman O. Familial aggregation in amyotrophic lateral sclerosis. Ann Neurol. 2010;67:554.
- Byrne S, Walsh C, Lynch C, et al. Rate of familial amyotrophic lateral sclerosis: a systematic review and meta-analysis. J Neurol Neurosurg Psychiatry. 2011;82:623–627.
- Mejzini R, Flynn LL, Pitout IL, et al. ALS genetics, mechanisms, and therapeutics: where are we now? Front Neurosci. 2019;13:1310.
- Al-Chalabi A, van den Berg LH, Veldink J. Gene discovery in amyotrophic lateral sclerosis: implications for clinical management. Nat Rev Neurol. 2017;13:96–104.
- Volk AE, Weishaupt JH, Andersen PM, et al. Current knowledge and recent insights into the genetic basis of amyotrophic lateral sclerosis. Med Genet. 2018;30:252–258.
- Al-Chalabi A, Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol. 2013;9:617–628.
- van Rheenen W, Shatunov A, Dekker AM, et al. Genome-wide association analyses identify new risk variants and the genetic architecture of amyotrophic lateral sclerosis. Nat Genet. 2016;48:1043–1048.
- Tazelaar GHP, Dekker AM, van Vugt J, et al. Association of NIPA1 repeat expansions with amyotrophic lateral sclerosis in a large international cohort. Neurobiol Aging. 2019;74:234 e9- e15.
- Nicolas A, Kenna KP, Renton AE, et al. Genome-wide analyses identify KIF5A as a novel ALS gene. Neuron. 2018;97:1268–83 e6.
- Kenna KP, van Doormaal PT, Dekker AM, et al. NEK1 variants confer susceptibility to amyotrophic lateral sclerosis. Nat Genet. 2016;48:1037–1042.
- van Blitterswijk M, van Es MA, Hennekam EA, et al. Evidence for an oligogenic basis of amyotrophic lateral sclerosis. Hum Mol Genet. 2012;21:3776–3784.
- Cooper-Knock J, Kirby J, Highley R, et al. The spectrum of C9orf72-mediated neurodegeneration and amyotrophic lateral sclerosis. Neurotherapeutics. 2015;12:326–339.
- DeJesus-Hernandez M, Mackenzie IR, Boeve BF, et al. Expanded GGGGCC hexanucleotide repeat in noncoding region of C9ORF72 causes chromosome 9p-linked FTD and ALS. Neuron. 2011;72:245–256.
- van Es MA, Schelhaas HJ, van Vught PW, et al. Angiogenin variants in Parkinson disease and amyotrophic lateral sclerosis. Ann Neurol. 2011;70:964–973.
- Sreedharan J, Blair IP, Tripathi VB, et al. TDP-43 mutations in familial and sporadic amyotrophic lateral sclerosis. Science. 2008;319:1668–1672.
- Cannas A, Borghero G, Floris GL, et al. The p.A382T TARDBP gene mutation in Sardinian patients affected by Parkinson’s disease and other degenerative parkinsonisms. Neurogenetics. 2013;14:161–166.
- Elden AC, Kim HJ, Hart MP, et al. Ataxin-2 intermediate-length polyglutamine expansions are associated with increased risk for ALS. Nature. 2010;466:1069–1075.
- Kim JM, Hong S, Kim GP, et al. Importance of low-range CAG expansion and CAA interruption in SCA2 Parkinsonism. Arch Neurol. 2007;64:1510–1518.
- Merner ND, Girard SL, Catoire H, et al. Exome sequencing identifies FUS mutations as a cause of essential tremor. Am J Hum Genet. 2012;91:313–319.
- Vance C, Rogelj B, Hortobagyi T, et al. Mutations in FUS, an RNA processing protein, cause familial amyotrophic lateral sclerosis type 6. Science. 2009;323:1208–1211.
- Taylor JP. Multisystem proteinopathy: intersecting genetics in muscle, bone, and brain degeneration. Neurology. 2015;85:658–660.
- Rosen DR, Siddique T, Patterson D, et al. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993;362:59–62.
- Yang Y, Hentati A, Deng HX, et al. The gene encoding alsin, a protein with three guanine-nucleotide exchange factor domains, is mutated in a form of recessive amyotrophic lateral sclerosis. Nat Genet. 2001;29:160–165.
- Chen YZ, Bennett CL, Huynh HM, et al. DNA/RNA helicase gene mutations in a form of juvenile amyotrophic lateral sclerosis (ALS4). Am J Hum Genet. 2004;74:1128–1135.
- Daoud H, Zhou S, Noreau A, et al. Exome sequencing reveals SPG11 mutations causing juvenile ALS. Neurobiol Aging. 2012;33(839):e5–9.
- Nishimura AL, Mitne-Neto M, Silva HC, et al. A mutation in the vesicle-trafficking protein VAPB causes late-onset spinal muscular atrophy and amyotrophic lateral sclerosis. Am J Hum Genet. 2004;75:822–831.
- Greenway MJ, Andersen PM, Russ C, et al. ANG mutations segregate with familial and ‘sporadic’ amyotrophic lateral sclerosis. Nat Genet. 2006;38:411–413.
- Chow CY, Landers JE, Bergren SK, et al. Deleterious variants of FIG4, a phosphoinositide phosphatase, in patients with ALS. Am J Hum Genet. 2009;84:85–88.
- Hortobagyi T, Troakes C, Nishimura AL, et al. Optineurin inclusions occur in a minority of TDP-43 positive ALS and FTLD-TDP cases and are rarely observed in other neurodegenerative disorders. Acta Neuropathol. 2011;121:519–527.
- Johnson JO, Mandrioli J, Benatar M, et al. Exome sequencing reveals VCP mutations as a cause of familial ALS. Neuron. 2010;68:857–864.
- Deng HX, Chen W, Hong ST, et al. Mutations in UBQLN2 cause dominant X-linked juvenile and adult-onset ALS and ALS/dementia. Nature. 2011;477:211–215.
- Belzil VV, Daoud H, Camu W, et al. Genetic analysis of SIGMAR1 as a cause of familial ALS with dementia. Eur J Hum Genet. 2013;21:237–239.
- Parkinson N, Ince PG, Smith MO, et al. ALS phenotypes with mutations in CHMP2B (charged multivesicular body protein 2B). Neurology. 2006;67:1074–1077.
- Smith BN, Vance C, Scotter EL, et al. Novel mutations support a role for profilin 1 in the pathogenesis of ALS. Neurobiol Aging. 2015;36(1602):e17–27.
- Takahashi Y, Fukuda Y, Yoshimura J, et al. ERBB4 mutations that disrupt the neuregulin-ErbB4 pathway cause amyotrophic lateral sclerosis type 19. Am J Hum Genet. 2013;93:900–905.
- Kim HJ, Kim NC, Wang YD, et al. Mutations in prion-like domains in hnRNPA2B1 and hnRNPA1 cause multisystem proteinopathy and ALS. Nature. 2013;495:467–473.
- Johnson JO, Pioro EP, Boehringer A, et al. Mutations in the matrin 3 gene cause familial amyotrophic lateral sclerosis. Nat Neurosci. 2014;17:664–666.
- Pensato V, Tiloca C, Corrado L, et al. TUBA4A gene analysis in sporadic amyotrophic lateral sclerosis: identification of novel mutations. J Neurol. 2015;262:1376–1378.
- Smith BN, Topp SD, Fallini C, et al. Mutations in the vesicular trafficking protein annexin A11 are associated with amyotrophic lateral sclerosis. Sci Transl Med. 2017 May 3;9(388):eaad9157. DOI:10.1126/scitranslmed.aad9157
- Bannwarth S, Ait-El-Mkadem S, Chaussenot A, et al. A mitochondrial origin for frontotemporal dementia and amyotrophic lateral sclerosis through CHCHD10 involvement. Brain. 2014;137:2329–2345.
- Fecto F, Yan J, Vemula SP, et al. SQSTM1 mutations in familial and sporadic amyotrophic lateral sclerosis. Arch Neurol. 2011;68:1440–1446.
- Freischmidt A, Muller K, Ludolph AC, et al. Association of mutations in TBK1 with sporadic and familial amyotrophic lateral sclerosis and frontotemporal dementia. JAMA Neurol. 2017;74:110–113.
- Cirulli ET, Lasseigne BN, Petrovski S, et al. Exome sequencing in amyotrophic lateral sclerosis identifies risk genes and pathways. Science. 2015;347:1436–1441.
- Conforti FL, Spataro R, Sproviero W, et al. Ataxin-1 and ataxin-2 intermediate-length PolyQ expansions in amyotrophic lateral sclerosis. Neurology. 2012;79:2315–2320.
- Dadon-Nachum M, Melamed E, Offen D. The “dying-back” phenomenon of motor neurons in ALS. J Mol Neurosci. 2011;43:470–477.
- Chou SM, Norris FH. Amyotrophic lateral sclerosis: lower motor neuron disease spreading to upper motor neurons. Muscle Nerve. 1993;16:864–869.
- Abrahams S, Newton J, Niven E, et al. Screening for cognition and behaviour changes in ALS. Amyotroph Lateral Scler Frontotemporal Degener. 2014;15:9–14.
- Gosselt IK, Nijboer TCW, Van Es MA. An overview of screening instruments for cognition and behavior in patients with ALS: selecting the appropriate tool for clinical practice. Amyotroph Lateral Scler Frontotemporal Degener. 2020 Mar 11;1–13. DOI:10.1080/21678421.2020.1732424. Online ahead of print.
- Cammack AJ, Atassi N, Hyman T, et al. Prospective natural history study of C9orf72 ALS clinical characteristics and biomarkers. Neurology. 2019;93:e1605–e17.
- Bali T, Self W, Liu J, et al. Defining SOD1 ALS natural history to guide therapeutic clinical trial design. J Neurol Neurosurg Psychiatry. 2017;88:99–105.
- Todd TW, Petrucelli L. Insights into the pathogenic mechanisms of chromosome 9 open reading frame 72 (C9orf72) repeat expansions. J Neurochem. 2016;138(Suppl 1):145–162.
- Li Q, Vande Velde C, Israelson A, et al. ALS-linked mutant superoxide dismutase 1 (SOD1) alters mitochondrial protein composition and decreases protein import. Proc Natl Acad Sci U S A. 2010;107:21146–21151.
- Berk JL, Barroso FA, Coelho T. Oligonucleotide drugs for transthyretin amyloidosis. N Engl J Med. 2018;379:2085–2086.
- Bishop KM, Montes J, Finkel RS. Motor milestone assessment of infants with spinal muscular atrophy using the hammersmith infant neurological exam-part 2: experience from a nusinersen clinical study. Muscle Nerve. 2018;57:142–146.
- Tabrizi SJ, Leavitt BR, Landwehrmeyer GB, et al. Targeting huntingtin expression in patients with huntington’s disease. N Engl J Med. 2019;380:2307–2316.
- Park JJH, Hsu G, Siden EG, et al. An overview of precision oncology basket and umbrella trials for clinicians. CA Cancer J Clin. 2020;70:125–137.
- Zaman A, Wu W, Bivona TG. Targeting oncogenic BRAF: past, present, and future. Cancers (Basel). Pharmacogenomics J. 2020 Apr;20(2):220–226. DOI:10.1038/s41397-019-0111-3. Epub 2019 Oct 17.
- van Eijk RPA, Eijkemans MJC, Nikolakopoulos S, et al. Pharmacogenetic interactions in amyotrophic lateral sclerosis: a step closer to a cure? Pharmacogenomics J. 2020;20:220–226.
- van Eijk RPA, Jones AR, Sproviero W, et al. Meta-analysis of pharmacogenetic interactions in amyotrophic lateral sclerosis clinical trials. Neurology. 2017;89:1915–1922.
- Li W, Lee MH, Henderson L, et al. Human endogenous retrovirus-K contributes to motor neuron disease. Sci Transl Med. 2015;7:307ra153.
- Gold J, Rowe DB, Kiernan MC, et al. Safety and tolerability of Triumeq in amyotrophic lateral sclerosis: the lighthouse trial. Amyotroph Lateral Scler Frontotemporal Degener. 2019;20:595–604.