Abstract
Carbacylamidophosphates with the general formula RC(O)NHP(O)R1R2 constitute organophosphorus compounds that are used as insecticides, pesticides and ureas inhibitors. In this work, we studied the inhibition potency of CCl3C(O)NHP(O)Cl21, CHCl2C(O)NHP(O)Cl22, CH2ClC(O)NHP(O)Cl23 and CF3C(O)NHP(O)Cl24, which are the major intermediates for carbacylamidophosphates synthesis towards human erythrocyte acetylcholinesterase (hAChe) activity using Ellman's modified kinetic method. Unexpectedly, it was observed that they were not only hydrolytically unstable but also inhibited hAChE in a similar manner to that produced by organophosphorus insecticides. Enzymatic data, bimolecular inhibition rate constants (ki) and IC50 values for inhibition of hAChE demonstrated that they are irreversible inhibitors and the inhibition potency of compound 2 (IC50 = 88 μM) was the greatest in comparison with compounds 1, 3 and 4. Also the electropositivity of the phosphorus atom and the hydrophobicity of the compounds demonstrated that these two factors play an additional effect and different role in the inhibitory activity of these compounds. Hydrolytic stability of the compounds was determined by 31P NMR monitoring of the loss of the parent molecules with D2O as a function of time. This study considers antiacetylcholinesterase activity according to the structural and the electronic aspects of compounds 1–4, according to IR, 1H, 13C and 31P NMR spectral data.
Introduction
The synthesis and biologically studies of organophosphorus compounds are of both practical relevance and academic interest [Citation1,Citation2]. The inhibitory potency of carbacylamidophosphates with the general formula RC(O)NHP(O)R1R2: which are a kind of organophosphorus derivatives, on urease activity has been studied [Citation3]. N-haloacyl phosphoramidic dichlorides are the initial molecules which have been used for synthesis of many kinds of urease inhibitors and some other carbacylamidophosphate compounds. The existences of peptide and the phosphoryl group in such molecules leads to an interesting biological activity. It is known that the phosphoramidates with a P(O) moiety have an inhibitory effect on acetylcholinesterase (AChE) activity. The inhibitory potency of some reactants (POCl3, PCl3, and PSCl3) in phosphoramidate production has been studied by Casida et. al. [Citation4,Citation5]. Their studies revealed that these compounds with a tendency for hydrolysis were AchE inhibitors. They also identified that phosphorodichloridic acid [HOP(O)Cl2] acts as the actual phosphorylating agent of AChE in POCl3. It seems that, such hydrolysis behavior may be observed in the N-haloacyl phosphoramidic dichloride. To extend and to develop, the synthesis, characterization, hydrolysis and the inhibition potency on human erythrocyte acetylcholinesterse activity of the compounds RC(O)NHP(O)Cl2 with R = CCl31, CHCl22, CH2Cl 3 and CF34, are studied. Based on spectroscopic data and hydrophobicity evaluation (πx), we considered the electronic properties of the phosphorus atom and the hydrophobicity of the surrounding substituents on the hydrolysis tendency and the inhibitory potency of the selected compounds.
Experimental
Materials and methods
All reactions in the synthesis of compounds from 1 to 4 were carried out under an argon atmosphere. Chemicals and enzymatic reagents were from the following sources: all of compounds and solvents for synthesis were from Merck, purified human plasma AChE (3.1.1.7) (50 units/785 μl) from Sigma, acetylthiocholine (ASCh), 5, 5′-dithio bis (2-nitrobenzoic acid) (DTNB) from Fluka. 1H, 13C and 31P NMR spectra were recorded on a Bruker (Avance DRS) 500 MHz spectrometer and chemical shifts were obtained in CDCl3 relative to TMS and 85% H3PO4, respectively, as external standards. IR spectra were obtained using KBr pellets on a Shimadzu IR-60 model spectrometer. Elemental analyses were performed using a Heraeus CHN-O-RAPID instrument and UV measurements were obtained with a Shimadzu UV-2100 spectrometer.
Chemical synthesis
All compounds 1–4 were synthesized according to reported methods [Citation6,Citation7], by reaction of (1.04 g, 5 mmol) PCl5 in a solution of dry benzene and 2,2,2-trichloroacetamide (0.812 g, 5 mmol) for 1, 2,2-dichloroacetamide (0.64 g, 5 mmol) for 2, 2-chloroacetamide (0.47 g, 5 mmol) for 3, and 2,2,2-trifluoroacetamide (0.565 g, 5 mmol) for 4. The mixtures were refluxed for 4–18 h, then cooled to room temperature and treated with formic acid (0.23 g, 5 mmol) for 2 h at 15–20°C. The solvent was decanted leaving a white powder that was washed with dry benzene. Isolated yields for these compounds were between 90–98%. The results of spectral characterizations were as follows:
N-trichloroacetyl Phosphoramidic Dichloride [Ccl3c(o)nhp(o)cl2] 1
Anal. Calc. for C2HCl5NO2P: C, 8.58; H, 0.3, N, 5.36; found: C, 8.6; H, 0.4; N, 5.29%; 1H NMR (CDCl3), δ (ppm): 9.94(s, 1H, NH); 13C NMR(CDCl3), δ (ppm): 91.00(d., CCl3, 3JPC = 13.1 Hz), 160.3(d, C(O), 2JPC = 2.8 Hz); 31P {1H} NMR(CDCl3), δ (ppm): 8.08(s); IR(KBr), ν (cm− 1): 408(w), 521(m), 590(s), 665(vs), 817(s), 902(s), 1181(s), 1274(vs), 1426(vs), 1736(s), 2835(w), 3045(m).
N-2,2-dichloroacetyl Phosphoramidic Dichloride [Chcl2c(o)nhp(o)cl2] 2
Anal. Calc. for C2H2Cl4NO2P: C, 9.79; H, 0.81; N, 5.71; found: C, 9.81; H, 0.79; N, 5.69%; 1H NMR(CDCl3), δ (ppm): 9.68(d, 1H, NH, 2JPNH = 10.5 Hz), 5.98(1H, d, 3JP-CH = 2.3 Hz); 13C NMR(CDCl3), δ (ppm): 65.67(d, CHCl2, 3JPC = 12.0 Hz), 163.24(d, CO, 2JPC = 3.6 Hz); 31P {1H} NMR(CDCl3), δ (ppm): 8.2(s); 31P NMR(CDCl3), δ (ppm): 8.2 ((dd), 12.0 Hz); IR(KBr), ν (cm− 1): 502(w), 580(m), 663(m), 764(w), 805(w), 885(m), 952(w), 1194(m), 1280(s), 1421(vs), 1710(vs), 2830(w), 3050(m).
N-2-chloroacetyl Phosphoramidic Dichloride [Ch2clc(o)nhp(o)cl2] 3
Anal. Calc. for C2H3Cl3NO2P: C, 11.40; H, 1.42; N, 6.65; found: C, 11.18; H, 1.50; N, 6.71%; 1H NMR(CDCl3), δ (ppm): 9.16(d, NH, 2JPNH = 11.6 Hz), 4.16(d, 2H, CH2Cl, 3JPCH = 1.2 Hz); 13C NMR(CDCl3), δ (ppm): 43.65(d, CH2Cl, 3JPC = 10.5 Hz), 167.12(d, CO, 2JPC = 3.8 Hz); 31P {1H} NMR(CDCl3), δ (ppm): 6.62(s); 31P NMR(CDCl3), δ (ppm): 6.62((dd), 13.2 Hz); IR(KBr), ν (cm− 1): 531(w), 585(s), 875(m), 943(m), 1125(s), 1260(s), 1451(vs), 1721(vs), 2940(m), 32059(s).
N-2,2,2-triflouroacetyl Phosphoramidic Dichloride [Cf3c(o)nhp(o)cl2] 4
Anal. Calc. for C2HCl2F3NO2P: C, 10.43; H, 0.43; N, 6.08; found: C, 9.98; H, 0.51; N, 7.02%; 1H NMR (CDCl3), δ (ppm): 10.24(s, NH); 13C NMR (CDCl3), δ (ppm): 115.4((qd), CF3, 1JCF = 288.5 Hz, 3JCP = 16.4 Hz); 157.5((qd), CO, 2JCF = 45.8 Hz, 2JCP = 3.5 Hz); 31P {1H} NMR (CDCl3), δ (ppm): 7.42(s); IR (KBr), ν (cm− 1): 515(w), 593(m), 878(m), 1131(s), 1180(vs), 1211(m), 1279(m), 1311(w), 1466(m), 1748(m), 2900(w), 3075(m).
AChE activity assay
The activity of hAChE was determined by a modified Ellman's method [Citation8], using the level of ASCh hydrolysis by monitoring thiocholine liberation with DTNB reagents. Reaction was carried out at 25°C in 70 mM phosphate buffer (Na2HPO4/NaH2PO4, pH = 7.4, 920 μl volume) containing the enzyme (15 μl volume, diluted 100 times in phosphate buffer, pH = 7.4), DTNB (10− 4 M final concentration, 50 μl volume) and ASCh (1.35 × 10− 4 M final concentration, 15 μl volume). The absorbance change was monitored at 418 nm for 2 min, and five replicates were run in each experiment. In the absence of inhibitor, the absorbance change was directly proportional to the enzyme level.
AChE inhibition experiments
The reaction mixtures for determination of IC50 values, the median inhibitory concentration, consisted DTNB solution, 50 μl; inhibitor, x μl (5–200); acetylthiocholine (ASCh) solution, 15 μl; phosphate buffer (920 − x) μl; hAChE solution, 15 μl. The final concentrations of DTNB, ASCh, and inhibitors from 1–4 were: 10− 4, 1.35 × 10− 4, (2.16 × 10− 3 − 15.13 × 10− 3 M), (3.3 × 10− 5 − 2.3 × 10− 4 M), (9 × 10− 5 − 1.03 × 10− 3 M), (4.7 × 10− 4 − 4.1 × 10− 3 M), respectively. The enzyme concentration in the assay was 2.51 × 10− 10 M. In the four inhibitor solutions in phosphate buffer, 5 μl of dry THF was added as carrier solvent. The THF carrier (up to 5 μl) per assay did not affect the AChE activity [Citation4]. The reaction mixtures for determination of the inhibition mechanism and ki values were: DTNB and ASCh (the same as above); a solution of enzyme plus inhibitors (inhibitor concentration was adjusted to give about 50% of hAChE inhibition) 100 μl; phosphate buffer, 835 μl.
Determination of enzyme activity after incubation of inhibitors in phosphate buffer for 40 min
Enzyme activity determinations after 40 min incubation of inhibitors 1, 2, 3 and 4 in phosphate buffer (pH = 7.4) were performed for recognizing the active species that reacted with hAChE via their hydrolysis activity. It was found that at 40 min all four compounds were hydrolyzed completely and the chlorine atoms replaced by OH groups. After this time, no change in enzyme activity in the presence of the inhibitors was shown, during the enzyme activity assay (the reaction mixtures were in accordance with the section on AchE inhibition experiments).
Hydrolysis study of RC(O)NHP(O)Cl2 compounds and stability of intermediates
Hydrolytic activation and products
Hydrolytic activation of compounds 1–4 was determined by loss of the parent compounds in D2O as a function of time at 25°C. After five min. for scanning 31P NMR spectra, two major peaks were observed. The chemical shifts were as follows: Compound, 31P {1H} NMR, δ (ppm): 1, − 5.76(s), − 11.44(s); 3, − 5.59(s), − 17.53(s); 4, − 6.93(s), − 19.6(s). The intensity ratio of the two major peaks was relatively 5:1 for each compound. However, despite these results, only one single peak in the 31P NMR spectra of compound 2 in D2O was observed at δ = − 5.26 ppm which may correspond to the dihydroxylated derivative. A single peak at δ = 0–0.2 ppm with medium intensity was observed, corresponding to phosphate. The second peak in compounds 1, 3 and 4 ( − 11.44, − 17.53 and − 19.6 ppm) gradually disappeared after 30, 15 and 10 min respectively. It is proposed that the most intense peaks at δ = − 11 to − 19 ppm were related to monohydroxylated substituents, which were not stable and converted to dihydroxylated substituents at different times. Synthesis of dihydroxylated derivatives of compounds 1–4 confirmed our suggestion.
Synthesis Of Rc(o)nhp(o)(oh)2
A solution of water (6.6 mmol) in acetone (10 ml) was added dropwise to each of compounds 1–4 (3.3 mmol) in dry acetone (10 ml) while stirring at 25°C. The reaction mixtures were stirred for 20 min. 31P NMR (10% C6D6 in acetone) spectra for the synthesized compounds showed the following major peaks:
Compound, 31P {1H} NMR, δ (ppm): 1, − 5.26(s); 2, − 5.33(s); 3, − 5.48(s); 4, − 6.2(s). These major peaks corresponded to RC(O)NHP(O)(OH)2 where R = CCl3, CHCl2, CH2Cl, CF3. All of the compounds are stable in acetone.
Hydrophobic parameter evaluation
The octanol–water partition coefficient, log (Po/w), for compounds 1–4 were calculated according to Leo et al. [Citation9]. The following equation: πx = log Px − log PH was used for determining the hydrophobicity extent of substituents around the phosphorus atom, where Px is the derivative of a parent molecule, PH, and thus πx is the hydrophobicity of the function x. Because the P(O)Cl2 group was constant in all four compounds, we concentrated on the RC(O)NH moiety for calculating log (Po/w), in comparison with acetamide as the parent molecule. By this manner, the additional hydrophobicity of chlorine atoms in the terminal haloacyl group for compounds from 1–3 and fluorine atoms in compound 4 were evaluated. The πx values, shown in , indicated that molecule 1 with three chlorine atoms has the most hydrophobicity.
Table I. The spectral and enzymatic data for compounds 1–4 where R is[C(O) NHP(O)Cl2].
Relationship between the phosphorylation rate constants and phosphorus-31 NMR chemical shifts
By comparing the inhibition rate constants (ki) and the 31P NMR chemical shifts for compounds 1–4, no relationship was observed between these two factors for these molecules.
Results
Synthesis and spectral data
The synthesis of compounds 1–4 has been previously reported and characterized by IR and elemental analysis Citation3Citation6Citation7Citation10-12 and the compound 4 was additionally characterized by IR, 1H, 13C and 19F NMR spectroscopy [Citation13]. In this work, more characterization 1–4 has been achieved by IR, 1H, 13C,31P NMR spectroscopy and elemental analysis.(Scheme )
Scheme 1 Structures of compounds 1–4.
1H NMR spectroscopic data for the compounds 2 and 3 indicate two-bonds coupling between phosphorus and the NH hydrogen atom as 2JPNH = 10.5, 11.6 Hz, respectively, where these values are in agreement with other phosphoramidate compounds [Citation14]. This coupling not present in compounds 1 and 4. Also in compounds 2 and 3, the coupling between phosphorus and the hydrogen atom of the terminal haloalkyl was observed as 3JPCH = 2.3, 1.2 Hz, respectively. 13C NMR spectra showed the coupling between the phosphorus atom and the carbonyl carbon atom as 2JPC = 2.8 Hz 1, 3.6 Hz 2, 3.8 Hz 3 and 3.5 Hz 4. Three bonds coupling between phosphorus and the haloalkyl carbon atom, which were obtained by 13C NMR spectra, are 13.1 Hz 1, 12.0 Hz 2, 10.5 Hz 3. In the case of compound 4, two different couplings, as a doublet of quartet, which arise from the fluorine and phosphorus atom, were observed in the 13C NMR spectra of CF3 group, (1JFC = 288.5 Hz and 3JPC = 16.4 Hz). A similar effect was observed for the carbonyl carbon atom with 2JFC = 45.8 Hz and 2JPC = 3.5 Hz, which it leads to another doublet of quartet in 13C NMR spectra. These data were consistent with a previous report [Citation13]. Also the infrared spectral data and the elemental analys for all four compounds corresponded to other previous reports [Citation11,Citation12].
Human acetylcholinesterase inhibition
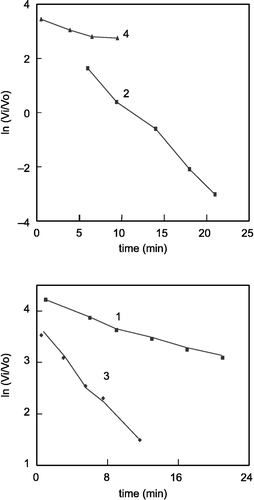
The inhibitory effect of 6 different concentrations of each of the carbacylamidophosphates 1–4 hAChE activity was determined. The IC50 values of these four compounds derived from , are given in . , shows the typical time-course for the inhibition of human erythrocyte AChE by compounds 1–4, plotted as (ln remaining enzyme activity (Vi/Vo)) vs time. The inhibition rate constants for compounds 1–4, Ki, under pseudo-first-order conditions were computed from the slopes of the plots in [Citation15,Citation16] and the values are given in . The plots in gave intercepts at Vi/Vo < 1 and the Ki values, dissociation equilibrium constants, were calculated from the equation Vi/Vo = (1+so/Km)/(1+so/Km+i/Ki) for the compounds (). Also the Km and Vmax values of hAChE under the experimental conditions used were obtained as 10.820 × 10− 4 mol L− 1 and 10.03 × 10− 6 mol L− 1 min− 1, respectively [Citation17]. The values show that, the compound 2 has the largest phosphorylation rate constant (ki = 0.31 min− 1) and the smallest dissociation equilibrium constant (ki = 0.036 mM) in comparison with the other molecules. It demonstrates that the formation of the phosphoAChE association between this molecule and the enzyme is the most effective, which leads to more inactivation of AChE activity.
Figure 1 The plot of Vi/Vo against Log [I] × 105 for inhibitor from 1 to 4: Vi and Vo are activity of enzyme in the presence and absence of inhibitor, respectively and [I] is inhibitor concentration (mol/lit), *means ± SE (n = 4).
![Figure 1 The plot of Vi/Vo against Log [I] × 105 for inhibitor from 1 to 4: Vi and Vo are activity of enzyme in the presence and absence of inhibitor, respectively and [I] is inhibitor concentration (mol/lit), *means ± SE (n = 4).](/cms/asset/d2e28fb7-9311-4335-95f3-00ed1bce71ae/ienz_a_145659_f0002_b.gif)
Figure 2 Plots of ln (Vi/Vo) versus time (min) for inhibitors 1–4.
Discussion
The 1H NMR spectra of the synthesized compounds present useful information about the coupling of the amidic proton with the phosphorus atom (2JPNH). As shown in the coupling constant in 2 is 2JPNH = 10.5 Hz and it for 3 is 2JPNH = 11.5 Hz. Unexspectedly, these couplings were absent in the 1 and 4. Electronwithdrawing effect of the chlorine and the fluorine atoms in 1 and 4 may be increasing the acidity of NH proton (δ = 9.94 ppm for 1 and 10.26 ppm for 4) and this may account for the absence of 2JPNH. The inhibition potency of the compounds indicates an increasing inhibitory effect on hAChE: 2>3>4>1. as obtained by IC50 values comparison. The inhibition potency of most organophosphorus compounds is dependent on the leaving group, the charge on the phosphorus atom and the phosphorus atom substitutions [Citation18]. The studies on these parameters is described as follows:
Leaving group
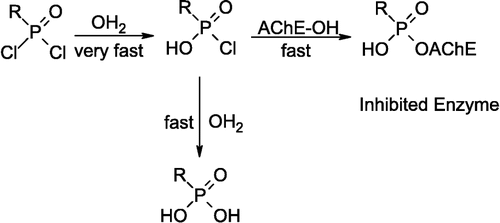
The mechanism of acetylcholinesterse inactivation by organophosphorus compounds occurs with the ejection of a leaving group, synchronous with formation of a phosphoserine linkage [Citation19,Citation20]. In all four compounds, the chlorine atom of the P–Cl bond is the best leaving group. A decreasing in bonding energy of P–Cl makes it more labile and it leads to either a better interaction with the enzyme active site and or faster displacement with an OH group of aqueous solution. As shown in , compound 2 has the smaller νP-Cl in comparison with the other compounds which leads to it being a more potent inhibitor of AchE. Also, it tends to hydrolyses faster than the others in aqueous solution. Mono- and dihydroxylated compounds from 1 to 4 are formed by replacing the chlorine atom with the OH group during the hydrolysis via bimolecular mechanism[Citation22]. The dihydroxylated compounds have no inhibitory potency on hAChE, because the OH group is a weak leaving group. This is demonstrated by determining enzyme activity in the presence of these completely hydroxylated inhibitors (after 40 min incubation in phosphate buffer). These experiments shows no change in enzyme activity and reveal that dihydroxylated compounds, RC(O)NHP(O)(OH)2, are not AChE inhibitors. Therefore, the inhibition potency and the hydrolysis tendency of compounds from 1–4, establishes that the monohydroxylated species, RC(O)NHP(O)(OH)Cl, have hAChE inhibition potency. The result compare with compound OHP(O)Cl2, which is the hydrolyzed species of POCl3, that can act as an AChE inhibitor with IC50 = 12–36 μM [Citation4]. The proposed mechanism for hAChE inhibition by 1–4 following hydrolytic activation to hydroxylated species is presented in Scheme .
Scheme 2 Proposed mechanism for hAChE inhibition by compounds 1, 2, 3 and 4 following hydrolytic activation to RP(O)(OH)Cl, where R = CCl31, CHCl2 2, CH2Cl 3, CF3 4.
Charge on the phosphorus atom
Electropositivity of the phosphorus atom probably is the reason for better linkage of the OH-serine group with the P(O) moiety. 31P NMR spectra for these compounds showed that the decreasing order of the chemical shifts are as follows: 2 (δ = 8.2 ppm) < 1 (δ = 8.08 ppm) < 4 (δ = 7.42 ppm) < 3 (δ = 6.62 ppm)
Compound 2 has the largest amount of chemical shift as shown in . It might be expected that having the most electropositive phosphorus atom perhaps is an important factor that makes it the strongest inhibitor (IC50 = 88 μM). Surprisingly, compound 3, which has the lowest chemical shift, is a more potent inhibitor than compounds 1 and 4 due to less hydrophobicity of the substituents around the phosphorus atom.
The phosphorus atom substituents
The reaction of AChE with organophosphorus compounds displays marked stereoselectivity [Citation21], which could be utilized for an investigation of the hydrophobic and the steric interactions with the structural elements of the active center. AChE substrates or their equivalents (inhibitors such as the organophosphorus compounds) fit into a crevice which tends to exclude larger molecules [Citation25,Citation26], and also limits hydrophobic binding of large substituents, hence their absence in acetylcholine, which is the normal substrate of AChE. By calculating log (Po/w) and πx, the hydrophobicity of the compounds can be evaluated. The increasing order in hydrophobic potency according to the data in is 1>2>3>4.
By considering the fact that hydrophobic substituents hinder, rather than enhance the association of organophosphorus compounds with AChE [Citation23,Citation24], unexpectedly molecule 2 with more hydrophobicity relative to the other compounds (except 1), has the most inhibitory potency. It is proposed that this can be related to the greater electropositivity of the phosphorus atom in this molecule (δ31P NMR = 8.2 ppm), which leads to rapid nucleophilic substitution independently of the hydrophobicity of the substituents around the phosphorus atom. Structural comparison of compounds 1 and 2 shows that by replacement of a chlorine atom with hydrogen, despite a negligible difference in 31P NMR chemical shift, compound 2 is 60-fold stronger inhibitor than compound 1. Thus might be relative to the difference between the hydrophobicity of the two molecules, as the log P for compound 1 is 5 fold greater than that for compound 2.
Conclusion
This investigation establishes that the acute toxicity of the compounds on hAChE could be attributable to hydrolytic activation of RC(O)NHP(O)(OH)Cl which then phosphorylates AChE to form the enzymatically inactive phosphoAChE. The data reinforce the reported importance of the hydrophobicity of the substituents and the electropositivity on the phosphorus atom as important determinants of the inhibitory potency of organophosphorus compounds on AChE. The data reveals that the two factors mentioned above, have a different share of the inhibitory potency of each compound. It is proposed that where the 31P NMR chemical shift determining the electropositivity of specified compounds have small differences, the hydrophobic parameter of the substituents is an important factor for the differences in the inhibitory effect. Inversely, the differences in the electropositivity of the phosphorus atom might be the effective factor for the compounds with small differences in the hydrophobicity of their substituents.
References
- Haux EJ, Lockridge O, Casida JE. Specificity of ethephon as a butyrylcholinesterase inhibitor and phosphorylating agent. Chem Res Toxicol 2002; 15: 1527–1533
- Lassiter TL, Marshall Renee S, Jackson leron S, Hunter DL, Vu Jenifer T, Padilla S. Automated measurements of acetylcholinesterase inhibitor and phosphorylating agenta. Toxicol 2003; 186: 241–253
- Jaroslav K, Swerdloff F. N-acyl phosphoric triamide urease inhibitors and urease inhibited, urea based fertilizer compositions. 1985, US Pat 4,517,003
- Quistad GB, Zhang N, Srarks SE, Casida JE. Phosphoacetylcholinesterase: Toxicity of phosphorus oxychloride to mammals and insects that can be attributed to selective phosphorylation of acetylcholinesterase by phosphodichloridric acid. Chem Res Toxicol 2000; 13: 652–657
- Segall Y, Quistad GB, Srarks SE, Casida JE. Major intermediates in organophosphate synthesis (PCl3, POCl3, PSCl3, and their diethyl ester) are anticholinesterase agents directly or on activation. Chem Res Toxicol 2002; 16: 350–356
- Mao TJ, Dresdner RD, Young JA. The preparation of polyfluoroalkylphosphonitrilates and N-(prefluoracyl) phosphoramidic dichlorides. J Inorg Nucl Chem 1962; 24: 53–58
- Tesslesy M, Medison NJ. Methods for the preparation of starch phosphate esters. 1974, US Pat 3,842,071
- Ellman GL, Coutney KD, Andres VR, Featherstone M. A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 1961; 7(88)95
- Leo A, Hansch C, Elkins D. Partition coefficients and their uses. Chem Rev 1971; 71: 525–616
- Rudavskii VP, Sadovskaya NP. 1991, PCT. Int.Appl.WO.9,008. p 151
- Durrell WS, Young JA, Dresdner RD. Flurocarbon nitrogen compounds IX. The reaction of nitriles with carboxylic acids. J Org Chem 1963; 28: 831
- Davidson D, Skovronek H. The acylation of amides. J Am Chem Soc 1958; 80: 376
- Narala PM, Day CS, Powers BA, Odian MA, Lachgar A, Pennington WT, Noftle RE. Characterization and crystal structures of some fluorinated imides. Polyhedron 1999; 18: 1751–1759
- Quin LD. Phosphorus-31 NMR spectra properties in compound, characterization and structural analysis. VCH, New York 1994
- Ordentlich A, Kronman C, Barak D, Stein D, Ariel N, Marcus D, Velan B, Shafferman A. Engineering resistance to ‘aging’ of phosphylated human acetylcholinesterase, Role of hydrogen bonds network in the active center. FEBS Lett 1993; 334: 215–220
- Barak D, Ordentlich A, Kaplan D, Barak R, Mizrahi D, Kornman C, Segall Y, Velan B, Shafferman A. Evidence for P-N bond scission in phosphoramidate nerve agent adducts of human acetylcholineasterase. Biochemistry 2000; 39(1156)1161
- Gholivand K, Shariatinia Z, Khajeh K, Naderimanesh H. Synthesis and spectroscopic characterization of some phosphoramidates as reversible inhibitors of human acetylcholinesterase and determination of their IC50 values. J Enz Inhib Med Chem 2003, In Press
- Thompson CM, Surez AI, Rodriguez OP. Synthesis and 31P chemical shift identification of tripeptide active site models that represent human serum acetylcholinesterase covalently modified at serine by certain organophosphorus. Chem Res Toxicol 1996; 9: 1325–1332
- Wong L, Bruggenann Roger JM, Hosea N, Berman Harvay A, Tailor P. Mechanism of oxime reactivation of acetylcholinesterase analyzed by chirality and mutagenesis. Biochemistry 2000; 39(5750)5757
- Kaplan D, Ordentlich A, Barak D, Ariel N, Kronman C, Velan B, Shafferman A. “Does Butyryllization” of acetylcholinesterase through substitution of the six divergent aromatic amino acids in the active center gorge generate an enzyme mimic of butyrylcholinesterase. Biochemistry 2001; 40: 7433–7845
- Masson P, Legrand P, Bartels CF, Froment MT, Schopfet LM, Lockridge O. Roll of aspartate 70 and tryptophan 82 in binding of succinyldithiocholine to human butyrylcholinesterase. Biochemistry 1997; 36: 2266–2277
- Hudson RF, Moss G. The mechanism of hydrolysis of phosphorochloridates and related compounds. J Chem Soc 1962; 3599–3604
- Hansch C, Deutsch EW. The use of substituent constants in the study of structure-activity relationships in cholinesterase inhibitors. Biochem Biophys 1966; 126: 117–128
- Kemp JR, Wallace KB. Molecular determinants of the species-selective inhibition of brain acetylcholinesterase. Toxicol Appl Pharmacol 1999; 104: 246–258
- Anderson RA, Auraas I, Gaare G, Fonnum F. Inhibition of acetylcholinesterase from different species by organophosphorus compounds, carbamates and methylsolphonylflouride. Gen Pharmacol 1977; 8: 331–334
- Fukuto TR, Metcalf RL, Winton M. Alkylphosphonic acid esters as insecticides. Econ Entomol 1959; 52: 1121–1127