Abstract
Type 3 17β-hydroxysteroid dehydrogenase (17β-HSD) catalyzes the last step in the biosynthesis of the potent androgen testosterone (T) by selectively reducing the C17 ketone of 4-androstene-3,17-dione (Δ4-dione), with NADPH as cofactor. This enzyme is thus an interesting therapeutic target for androgen-sensitive diseases. Using an efficient convergent chemical approach we synthesized a phosphorylated version of the best Δ4-dione/adenosine hybrid inhibitor of type 3 17β-HSD previously reported. An appropriately protected C2′ phosphorylated adenosine was first prepared and linked by esterification to the steroid Δ4-dione bearing an alkyl spacer. After three deprotection steps, the phosphorylated bisubstrate inhibitor was obtained. The inhibitory potency of this compound was evaluated on homogenated HEK-293 cells overexpressing type 3 17β-HSD and compared to the best non-phosphorylated bisubstrate inhibitor. Unexpectedly, the phosphorylated derivative was slightly less potent than the non-phosphorylated bisubstrate inhibitor of type 3 17β-HSD. Two hypotheses are discussed to explain this result: 1) the phosphorylated adenosine moiety does not interact optimally with the cofactor-binding site and 2) the bisubstrate inhibitors, phosphorylated or not, interact only with the substrate-binding site of type 3 17β-HSD.
Introduction
Type 3 17β-hydroxysteroid dehydrogenase (17β-HSD) is an enzyme of 310 amino acids with a molecular mass of 34 513 Da. This protein, also called androgenic 17β-HSD or testicular 17β-HSD, is a key steroidogenic enzyme in the synthesis of the most potent androgens, testosterone (T) and dihydrotestosterone (DHT) Citation1-3. It catalyzes the reduction of 4-androstene-3,17-dione (Δ4-dione) into testosterone (T) using the cofactor NADPH as illustrated in . 5α-Reductase further converts T into DHT, the most potent androgen. Contrary to other members of the 17β-HSD family that are present in both gonads and some peripheral tissues [Citation1,Citation4], type 3 17β-HSD is found mainly in the testes. Mutations in the HSD17β3 gene are responsible for male pseudohermaphrodism resulting from type 3 17β-HSD deficiency [Citation5,Citation6]. Furthermore, T and DHT possess a high affinity for the androgen receptor (AR) and thus induce androgenic activity. Thus, type 3 17β-HSD plays an important role in the development of androgen-sensitive diseases such as prostate cancer [Citation7,Citation8], benign prostatic hyperplasia [Citation9], acne [Citation10,Citation11], hirsutism [Citation12] and male-pattern baldness [Citation13,Citation14]. Selective inhibitors of this enzyme [Citation15,Citation16] are thus potential therapeutic tools for the control of active androgens.
Figure 1 Role of type 3 17β-HSD in the transformation of less potent androgen Δ4-dione into active androgens T and DHT, using the cofactor nicotinamide adenine dinucleotide phosphate (NADPH).
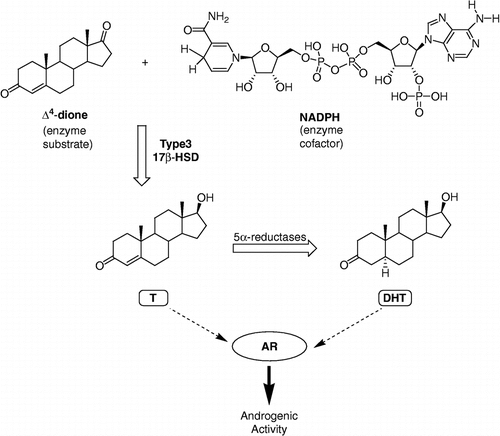
After reporting two classes of inhibitors of type 3 17β-HSD Citation17-20, our research group has recently developed a new class of inhibitors represented by Δ4dione/adenosine hybrid compounds [Citation21]. The best inhibitors of that series are those bearing an alkyl side-chain spacer of 11 or 12 methylenes between the steroid nucleus and the adenosine moiety as exemplified by compound 1 in . This class of type 3 17β-HSD inhibitors was designed by analogy to EM-1745 [Citation22], a type 1 17β-HSD inhibitor that was found to work as a bisubstrate inhibitor [Citation23]. When tested on human embryonic kidney HEK-293 cells overexpressing type 3 or type 1 17β-HSD, compound 1 proved to be a less potent inhibitor than EM-1745, for their respective enzyme. This result could be explained by the fact that different cofactors were used in the enzymatic assays: NADH for type 1 and NADPH for type 3. In fact, although both enzymes have a higher affinity for NADPH than for NADH [Citation1], type 3 has a much greater preference for NADPH, resulting in no enzyme activity at all with NADH [Citation24]. The only structural difference between NADH and NADPH is a phosphate group on C2′ of the adenosine moiety of NADPH. In order to simplify the chemical synthesis, bisubstrate inhibitors of type 1 and type 3 17βHSDs were first designed using NADH. Now, after analyzing the results, we hypothesized that a better type 3 17β-HSD inhibitor should be obtained if a phosphate group was added on C2′ of the adenosine moiety of 1 (see compound 2, ). The new bisubstrate inhibitor 2 should be more adapted than 1 to competition with the cofactor NADPH. To verify our hypothesis, we carried out the chemical synthesis of compound 2, the phosphorylated analogue of 1. Compounds 3 and 4 (), which should interact only with the cofactor-binding site, were also prepared. The potency of these compounds as inhibitors of type 3 17β-HSD will be compared and discussed.
Figure 2 (A) Bisubstrate inhibitor and phosphorylated bisubstrate inhibitor of type 3 17β-HSD, compounds 1 and 2. (B) Cofactor-like compounds 3 and 4.
Materials and methods
Chemistry
Reagents were obtained from Sigma-Aldrich Canada Co. (Oakville, ON, Canada). Usual solvents were obtained from Fisher Scientific (Montreal, QC, Canada) and VWR (Ville Mont-Royal, QC, Canada) and were used as received. Anhydrous solvents were purchased from Aldrich and VWR in SureSeal bottles, which were conserved under positive argon pressure. Tetrahydrofuran (THF) was distilled from sodium/benzophenone under argon. All anhydrous reactions were performed under positive argon pressure in oven-dried glassware. Thin-layer chromatography (TLC) was performed on 0.25-mm silica gel 60 F254 plates from Whatman (distributed by Fisher Scientific) and compounds were visualized by exposure to UV light (254 nm) and/or with a solution of ammonium molybdate/sulphuric acid/water (with heating). Flash chromatography was performed on Silicycle 60 (Québec, Canada) 230–400 mesh silica gel. Melting points were determined on an Electrothermal 9300 melting point apparatus (Cambridge, UK) and are reported uncorrected. Infrared (IR) spectra were obtained from a KBr pellet or a thin film of the solubilized compound on NaCl pellets (usually in CH2Cl2). They were recorded on a Perkin-Elmer series 1600 FT-IR spectrometer (Norwalk, CT, USA); only significant bands are reported (in cm− 1). 1H NMR and 13C NMR spectra were recorded with a Bruker AC/F 300 spectrometer (Billerica, MA, USA) at 300 and 75 MHz, respectively, and a Bruker AVANCE 400 spectrometer at 400 (1H), 100 (13C) and 162 (31P) MHz. The chemical shifts (δ) are expressed in ppm and referenced to chloroform (7.26 and 77.0 ppm), acetone (2.07 and 206.0 ppm), or methyl sulfoxide (2.51 and 39.5 ppm) for 1H and 13C respectively. For 31P NMR, the signal of H3PO4 (85%) was used as an external reference (δ = 0.00 ppm). The presence of the phosphate group increased the complexity of 13C NMR spectra and additional peaks are written between parentheses. Assignment of NMR signals was done using 1D- and 2D-NMR experiments (COSY, HSQC, HMBC, APT). Low-resolution mass spectra (LRMS) were recorded with a LCQ Finnigan apparatus (San Jose, CA, USA) equipped with an atmospheric pressure chemical ionisation (APCI) source on positive mode. High-resolution mass spectra (HRMS) were provided by the Regional Centre for Instrumental Analysis (Université de Montréal, Montréal, Canada).
Synthesis of 5′-O-p-methoxytrityladenosine (5) [Citation25]
Compound 5 was prepared from adenosine according to the procedure described in the literature. White solid (12.0 g, 60% yield). Mp 190–193°C, mp (Lit. [Citation25]) 189–193°C; IR (KBr) 3420, 3331, 3223 and 3072 (OH, NH2), 1643 (C = N); 1H NMR (400 MHz, DMSO-d6) 3.21 (d, J = 4.0 Hz, 5′-CH2), 3.73 (s, OCH3), 4.05 (m, 4′-CH), 4.32 (q, J = 5.4 Hz, 3′-CH), 4.70 (q, J = 5.1 Hz, 2′-CH), 5.24 (d, J = 5.9 Hz, OH), 5.58 (d, J = 5.7 Hz, OH), 5.93 (d, J = 4.5 Hz, 1′-CH), 6.85 (d, J = 8.9 Hz, 3-CH and 5-CH of p-MeOC6H4), 7.22 (d, J = 8.9 Hz, 2-CH and 6-CH of p-MeOC6H4), 7.26 and 7.36 (2m, 2 × C6H5 and NH2), 8.09 and 8.27 (2s, 2 × CH of adenine); 13C NMR (75 MHz, DMSO-d6) 55.0, 63.8, 70.3, 73.0, 82.9, 85.7, 87.9, 113.2 (2x), 119.2, 126.8 (2x), 127.8 (4x), 128.0 (4x), 130.0 (2x), 134.9, 139.6, 144.2, 144.3, 149.3, 152.6, 156.1, 158.1; LRMS calculated for C30H30N5O5 [M+H]+ 540.2, found 540.1 m/z.
Protection of 5 with a TBDMS group (Synthesis of 6 and 7)
To a solution of 5 (10.9 g, 20.3 mmol) in anhydrous DMF (100 mL) under an argon atmosphere were added imidazole (4.42 g, 64.8 mmol) and tert-butyldimethylsilyl (TBDMS) chloride (3.36 g, 22.3 mmol). The reaction was stirred for 20 h at room temperature. The mixture was extracted with EtOAc/hexanes (1:1). The organic phase was washed with water and brine, dried over MgSO4, and evaporated to dryness. The crude mixture was composed of a small amount of the bis-TBDMS product, which was not recovered, and monoTBDMS derivatives 6 and 7. The latter were purified by flash chromatography (hexanes/EtOAc, 4:6 to 2:8). This way, compound 7 was obtained first (4.9 g, 37% yield) and compound 6 was obtained next (5.4 g, 40% yield). The structures of the regioisomers 6 and 7 were confirmed by COSY-NMR experiments.
3′-O-tert-butyldimethylsilyl-5′-O-p-methoxytrityladenosine (6) [Citation26]
White solid. Mp 180–183°C, mp (Lit. [Citation26]) 178–179°C. IR (film) 3326 and 3170 (OH, NH2), 1651 (C = N); 1H NMR (400 MHz, DMSO-d6) 0.04 and 0.08 (2s, (CH3)2Si), 0.83 (s, (CH3)3CSi), 3.09 (dd, J1 = 10.5 Hz, J2 = 4.8 Hz, 1H of 5′-CH2), 3.34 (m, 1H of 5′-CH2), 3.73 (s, OCH3), 4.02 (m, 4′-CH), 4.53 (t, J = 4.7 Hz, 3′-CH), 4.86 (q, J = 5.2 Hz, 2′-CH), 5.42 (d, J = 6.1 Hz, 2′-OH), 5.89 (d, J = 5.0 Hz, 1′-CH), 6.85 (d, J = 9.0 Hz, 3-CH and 5-CH of p-MeOC6H4), 7.20 (d, J = 8.9 Hz, 2-CH and 6-CH of p-MeOC6H4), 7.26 and 7.34 (2m, 2 × C6H5, NH2), 8.08 and 8.32 (2s, 2 × CH of adenine); 13C NMR (75 MHz, DMSO-d6) − 5.1, − 4.5, 18.0, 25.8 (3x), 55.0, 63.1, 72.0, 72.1, 83.2, 85.8, 88.1, 113.1 (2x), 119.3, 126.9 (2x), 127.8 (4x), 127.9 (4x), 130.0 (2x), 134.9, 140.3, 144.17, 144.20, 149.2, 152.5, 156.1, 158.2; LRMS calculated for C36H44N5O5Si [M+H]+ 654.3, found 654.2 m/z.
2′-O-tert-butyldimethylsilyl-5′-O-p-methoxytrityladenosine (7) [Citation27]
White solid. Mp 170–174°C, mp (Lit. [Citation27]) 164–167°C. IR (film) 3402, 3313 and 3147 (OH, NH2), 1651 (C = N); 1H NMR (400 MHz, DMSO-d6) − 0.14 and − 0.04 (2s, (CH3)2Si), 0.75 (s, (CH3)3CSi), 3.27 (m, 5′-CH2), 3.74 (s, OCH3), 4.10 (m, 4′-CH), 4.26 (q, J = 5.1 Hz, 3′-CH), 4.86 (t, J = 5.0 Hz, 2′-CH), 5.14 (d, J = 5.9 Hz, 3′-OH), 5.95 (d, J = 5.0 Hz, 1′-CH), 6.87 (d, J = 8.7 Hz, 3-CH and 5-CH of p-MeOC6H4), 7.26 and 7.41 (2m, 2 × C6H5, 2-CH and 6-CH of p-MeOC6H4, NH2), 8.07 and 8.27 (2s, 2 × CH of adenine); 13C NMR (75 MHz, DMSO-d6) − 5.3, − 4.9, 17.9, 25.6 (3x), 55.0, 63.5, 70.2, 74.9, 83.3, 85.8, 87.8, 113.2 (2x), 119.2, 126.9 (2x), 127.8 (4x), 128.0 (4x), 130.1 (2x), 134.8, 139.5, 144.2, 144.3, 149.3, 152.6, 156.1, 158.2; LRMS calculated for C36H44N5O5Si [M+H]+ 654.3, found 654.2 m/z.
Synthesis of 5 from 7
Compound 7 (6.11 g, 9.35 mmol) was dissolved in dry THF (116 mL) under an argon atmosphere at 0°C. A 1M solution of tetrabutylammonium fluoride (TBAF) in THF (18.7 mL, 18.7 mmol) was added and the reaction was stirred for 2 h at 0°C. The reaction was quenched by addition of a saturated solution of NaHCO3. The product was extracted with EtOAc, washed with brine, and dried over MgSO4. Purification by flash chromatography (CH2Cl2/MeOH, 95:5 to 90:10) afforded 5 (5.03 g, 99% yield) as a white solid. The 1H NMR spectra was identical to previously obtained spectra of 5.
Synthesis of 2′-O-dibenzylphosphoryl-3′-O-tert-butyldimethylsilyl-5′-O-p-methoxytrityl-adenosine (8)
Dibenzyl-N,N-diisopropylphosphoramidite (1.43 mL, 4.25 mmol) and 1H-tetrazole (818 mg, 11.7 mmol) were added to a solution of 6 (2.32 g, 3.54 mmol) in anhydrous CH2Cl2 (120 mL) under an argon atmosphere at 0°C. The mixture was stirred at 0°C for 30 min. Then, m-chloroperbenzoic acid (m-CPBA) (2.38 g, 10.6 mmol) was added and the reaction was stirred for 30 min at 0°C. The product was extracted with CH2Cl2 and the organic phase was washed with a saturated aqueous solution of Na2S2O3, a saturated aqueous solution of NaHCO3, dried over MgSO4, and evaporated to dryness. Purification by flash chromatography (CH2Cl2/MeOH, 99:1 to 95:5) gave 8 (1.77 g, 55% yield) as a white foam. IR (film) 3327 and 3180 (NH2), 1644 (C = N), 1252 (P = O), 1000 (P-O); 1H NMR (400 MHz, DMSO-d6) − 0.01 and 0.05 (2s, (CH3)2Si), 0.76 (s, (CH3)3CSi), 3.00 and 3.43 (2m, 5′-CH2), 3.71 (s, OCH3), 4.04 (m, 4′-CH), 4.95 (m, 3′-CH, 2 × OCH2Ph), 5.74 (m, 2′-CH), 6.21 (d, J = 3.4 Hz, 1′-CH), 6.81 (d, J = 9.0 Hz, 3-CH and 5-CH of p-MeOC6H4), 7.14 (d, J = 8.9 Hz, 2-CH and 6-CH of p-MeOC6H4), 7.25 (m, 4 × C6H5), 7.42 (s, NH2), 8.13 and 8.36 (2s, 2 × CH of adenine); 13C NMR (75 MHz, DMSO-d6) − 5.3, − 4.8, 17.7, 25.6 (3x), 55.0, 62.0, 68.8 (68.9), 68.95 (69.03), 70.3, 77.0, 82.3, 85.8, 86.5, 113.1 (2x), 119.2, 126.8 (2x), 127.7 (4x), 127.77 (4x), 127.84 (4x), 127.9 (2x), 128.4 (4x), 129.9 (2x), 134.8, 135.5, 135.6, 140.7, 143.9, 144.1, 148.8, 152.4, 156.1, 158.2; 31P NMR (162 MHz, DMSO-d6) –1.21; HRMS calculated for C50H57N5O8PSi [M+H]+ 914.37085, found 914.37262 m/z.
Synthesis of 2′-O-dibenzylphosphoryl-3′-O-tert-butyldimethylsilyl-adenosine (9)
Dichloroacetic acid (4.6 mL) was added to a solution of 8 (1.69 g, 1.85 mmol) in anhydrous CH2Cl2 (60 mL) under an argon atmosphere. The reaction was stirred for 4 h at room temperature and quenched by addition of a saturated aqueous solution of NaHCO3. The crude compound was extracted with CH2Cl2 and the organic phase was dried over MgSO4, and evaporated to dryness. Purification by flash chromatography (CH2Cl2/MeOH, 97:3) afforded 9 (1.08 g, 91% yield) as a white amorphous solid. IR (film) 3327, 3182 (OH, NH2), 1651 (C = N), 1258 (P = O), 1014 (P-O); 1H NMR (400 MHz, DMSO-d6) 0.09 and 0.11 (2s, (CH3)2Si), 0.89 (s, (CH3)3CSi), 3.52 and 3.73 (2m, 5′-CH2), 4.04 (m, 4′-CH), 4.57 (dd, J1 = 4.6 Hz, J2 = 2.0 Hz, 3′-CH), 4.72 and 4.89 (2m, 2 × OCH2Ph), 5.56 (m, 2′-CH), 5.69 (m, 5′-OH), 6.18 (d, J = 6.6 Hz, 1′-CH), 7.08, 7.19 and 7.31 (3m, 2 × C6H5), 7.46 (s, NH2), 8.13 and 8.41 (2s, 2 × CH of adenine); 13C NMR (75 MHz, DMSO-d6) − 5.0, − 4.9, 17.9, 25.7 (3x), 61.0, 68.6 (68.7), 68.8 (68.9), 71.7, 76.7, 85.6 (85.7), 86.7, 119.5, 127.6 (2x), 127.7 (4x), 128.4 (4x), 135.45, 135.50, 140.2, 149.0, 152.5, 156.3; 31P NMR (162 MHz, DMSO-d6) – 1.47; HRMS calculated for C30H41N5O7PSi [M+H]+ 642.25074, found 642.25098 m/z.
Synthesis of 2′-O-dibenzylphosphoryl-3′-O-tert-butyldimethylsilyl-5′-O-nonanoyl-adenosine (10)
A mixture of nonanoic acid (590 μL, 3.38 mmol), 1-[3-(dimethylamino)propyl]-3-ethylcarbodiimide hydrochloride (EDCI) (971 mg, 5.07 mmol) and dimethylaminopyridine (DMAP) (619 mg, 5.07 mmol) in dry DMF (11 mL) under an argon atmosphere was stirred for 30 min at room temperature. Then, a solution of alcohol 9 (1.08 g, 1.69 mmol), previously dissolved in dry DMF (∼1 mL), was added and the reaction was stirred for 16 h at room temperature. The reaction was quenched by addition of water and the crude product was extracted with diethyl ether. The organic phase was washed with an aqueous solution of NaOH (10%), brine, dried over MgSO4, and evaporated to dryness. Purification by flash chromatography (CH2Cl2/MeOH, 97:3 to 96:4) yielded 10 (925 mg, 70% yield) as a white foam. IR (film) 3316, 3162 (OH, NH2), 1735 (C = O, ester), 1676 (C = N), 1253 (P = O), 1001 (P-O); 1H NMR (400 MHz, CDCl3) 0.13 and 0.14 (2s, (CH3)2Si), 0.87 (t, J = 6.9 Hz, CH2CH3), 0.93 (s, (CH3)3CSi), 1.25 (m, 5 × CH2), 1.57 (m, CH2CH2COO), 2.26 (t, J = 7.6 Hz, CH2COO), 4.21 (m, 4′-CH and 1H of 5′-CH2), 4.37 (m, 1H of 5′-CH2), 4.71 (m, 3′-CH), 4.89 and 4.95 (2d, J = 8.5 Hz, 2 × OCH2Ph), 5.34 (m, 2′-CH), 6.09 (d, J = 4.2 Hz, 1′-CH), 7.24 and 7.33 (2m, 2 × C6H5), 7.85 and 8.18 (2s, 2 × CH of adenine); 13C NMR (75 MHz, CDCl3) − 5.1, − 4.7, 14.1, 18.1, 22.6, 24.7, 25.7 (3x), 29.1 (2x), 29.2, 31.8, 34.0, 62.5, 69.75 (69.82), 69.9 (70.0), 70.9 (71.0), 77.2 (77.3), 82.4, 87.5, 119.9, 127.9 (2x), 128.0 (2x), 128.6 (2x), 128.7 (2x), 128.89 (2x), 128.94, 135.0, 141.8, 146.8, 148.8, 152.7, 173.2; 31P NMR (162 MHz, DMSO-d6) – 1.35; HRMS calculated for C39H57N5O8PSi [M+H]+ 782.37085, found 782.37219 m/z.
Synthesis of 5′-O-nonanoyl-adenosine-2′-O-phosphate (4)
(A) Hydrogenation. A suspension of 10 (58 mg, 0.074 mmol) and Pd(OH)2 (58 mg) in cyclohexene (6 mL) and MeOH (12 mL) was refluxed for 30 min. The reaction mixture was then poured into a flask containing 50 mL of MeOH and 50 mL of THF. The suspension was filtered through a pad of celite and washed with MeOH and THF. The filtrate was evaporated to dryness to afford crude phosphate 12 (49 mg) as a white solid. (B) Hydrolysis. At room temperature, gaseous hydrogen chloride was bubbled for 30 min into a solution of 12 (44 mg, 0.074 mmol) in anhydrous CH2Cl2 (1.5 mL). The solvent was removed under vacuum and the residue was pre-adsorbed on C-18 silica gel and purified by flash chromatography using a C-18 silica gel column (reverse phase) with MeOH/H2O (5:5 to 7:3) as eluent to give 4 (10 mg, 28% yield for two steps) as a white amorphous solid. IR (KBr) 3346 and 3150 (OH, NH2), 1732 (C = O, ester), 1682 (C = N), 1076 (P-O); 1H NMR (400 MHz, DMSO-d6) 0.85 (t, J = 6.9 Hz, CH2CH3), 1.20 (m, 5 × CH2), 1.44 (m, CH2CH2COO), 2.26 (dt, J1 = 7.5 Hz, J2 = 3.1 Hz, CH2COO), 4.09 (m, 4′-CH), 4.17 and 4.37 (2m, 5′-CH2), 4.55 (t, J = 5.2 Hz, 3′-CH), 5.25 (m, 2′-CH), 6.11 (d, J = 4.4 Hz, 1′-CH), 7.41 (s, NH2), 8.14 and 8.31 (2s, 2 × CH of adenine); 13C NMR (75 MHz, DMSO-d6) 14.0, 22.1, 24.3, 28.4, 28.5, 28.6, 31.2, 33.3, 63.2, 69.3, 75.86 (75.92), 81.3, 86.6 (86.7), 119.1, 140.3, 149.1, 152.4, 155.9, 172.7; 31P-NMR (162 MHz, DMSO-d6) − 0.84; HRMS calculated for C19H31N5O8P [M+H]+ 488.19048, found 488.18969 m/z.
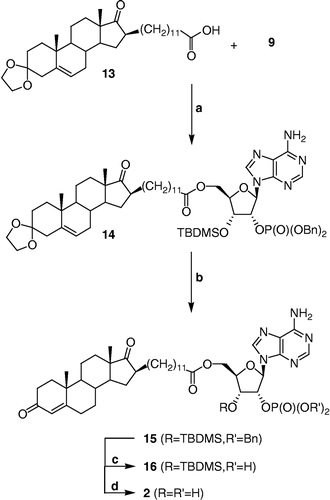
Synthesis of 2′-O-dibenzylphosphoryl-3′-O-tert-butyldimethylsilyl-5′-O-{11-[3′,3′-(ethylenedioxy)-androst-5′-en-17′-oxo-16′β-yl]-dodecanoyl}-adenosine (14)
Carboxylic acid 13 [Citation21] (50 mg, 0.095 mmol) and alcohol 9 (91 mg, 0.142 mmol) were linked by an ester using the procedure described above for the synthesis of 10 with the following modifications. Three equivalents of both EDCI and DMAP were used and the crude compound was extracted with EtOAc and the organic phase was not washed with a solution of NaOH (10%). Purification by flash chromatography (CH2Cl2/MeOH, 98:2) provided 14 (51 mg, 47% yield) as a white amorphous solid. IR (film) 3328, 3181 (NH2), 1736 (C = O, ester and ketone), 1648 (C = N), 1253 (P = O), 1010 (P-O); 1H NMR (400 MHz, acetone-d6) 0.22 (s, (CH3)2Si), 0.83 (s, 18′-CH3), 0.97 (s, (CH3)3CSi), 1.08 (s, 19′-CH3), 1.15 to 2.20 (m, 37H, CH and CH2 of steroid skeleton and alkyl chain), 2.24 (t, J = 7.5 Hz, CH2COO), 2.49 (m, 4′-CH), 3.88 (m, 2 × CH2 of ketal), 4.25 (m, 4′-CH and 5′-CH of ribose), 4.48 (m, 5′-CH of ribose), 4.98 (m, 3′-CH of ribose, 2 × OCH2Ph), 5.30 (m, 6′-CH), 5.69 (m, 2′-CH of ribose), 6.25 (d, J = 3.8 Hz, 1′-CH of ribose), 6.77 (sbr, NH2), 7.30 (m, 2 × C6H5), 8.14 and 8.22 (2s, 2 × CH of adenine); 13C NMR (100 MHz, acetone-d6) − 5.0, − 4.4, 14.0, 15.4, 19.2, 20.9, 25.4, 26.2 (3x), 28.7, 29.2 to 30.1 (6C under solvent peaks), 30.2, 30.3, 31.6, 31.81, 31.83, 32.7, 33.1, 34.3, 36.8, 37.4, 42.3, 48.3, 49.7, 50.9, 51.1, 63.0, 64.61, 64.65, 69.9 (70.0), 70.1 (70.2), 71.89 (71.94), 78.1 (78.2), 82.7, 87.98 (88.02), 109.6, 121.0, 121.8, 128.7 (2x), 128.8 (2x), 129.1 (2x), 129.19 (2x), 129.22 (2x), 141.2, 141.1, 142.1(2x), 150.3, 153.6, 157.2, 173.2, 222.6; 31P NMR (162 MHz, DMSO-d6) – 1.34; HRMS calculated for C63H91N5O11PSi [M+H]+ 1152.62165, found 1152.62085 m/z.
Synthesis of 2′-O-dibenzylphosphoryl-3′-O-tert-butyldimethylsilyl-5′-O-{11-[androst-4′-en-3′,17′-dioxo-16′β-yl]-dodecanoyl}-adenosine (15)
Compound 14 (178 mg, 0.155 mmol) was dissolved in a minimum of dry CH2Cl2 (∼0.3 mL) under an argon atmosphere. After the addition of dry MeOH (3 mL) and p-TSA (88 mg, 0.464 mmol), the mixture was stirred for 1 h at room temperature. The reaction was quenched by addition of a saturated aqueous solution of NaHCO3 and the crude compound was extracted with CH2Cl2. The organic phase was washed with brine and dried over MgSO4. Purification by flash chromatography (hexanes/acetone, 7:3 to 6:4) afforded 15 (102 mg, 59% yield) as a white amorphous solid. IR (film) 3326, 3182 (NH2), 1734 (C = O, ester and ketone), 1674 (C = O, conjugated ketone and C = N), 1254 (P = O), 1010 (P-O); 1H NMR (400 MHz, CDCl3) 0.13 and 0.14 (2s, (CH3)2Si), 0.87 (s, 18′-CH3), 0.90 to 2.50 (m, 38H, CH and CH2 of steroid skeleton and alkyl chain), 0.93 (s, (CH3)3CSi), 1.21 (s, 19′-CH3), 2.22 (t, J = 7.4 Hz, CH2COO), 4.19 (m, 4′-CH and 5′-CH of ribose), 4.41 (m, 5′-CH of ribose), 4.85 (m, 3′-CH of ribose), 4.90 and 4.94 (2d, J = 8.6 Hz, 2 × OCH2Ph), 5.39 (m, 2′-CH of ribose), 5.75 (s, 4′-CH), 6.07 (d, J = 3.5 Hz, 1′-CH of ribose), 6.13 (sbr, NH2), 7.22 and 7.30 (2m, 2 × C6H5), 7.77 and 8.24 (2s, 2 × CH of adenine); 13C NMR (75 MHz, CDCl3) − 5.1, − 4.7, 13.8, 17.4, 18.1, 20.2, 24.7, 25.7 (3x), 28.1, 28.8, 29.1, 29.2, 29.5 (3x), 29.6 (2x), 29.7, 30.9, 31.6, 32.5, 32.6, 33.9, 34.8, 35.7, 38.7, 47.9, 49.3, 49.4, 53.9, 62.5, 69.7 (69.8), 70.0, 70.8, 77.2, 81.8, 87.7, 119.6, 124.1, 127.9 (2x), 128.1 (2x), 128.58 (2x), 128.63 (2x), 128.8 (2x), 135.2 (2x), 140.8, 149.2, 150.8, 154.5, 170.4, 173.2, 199.4, 222.4; 31P NMR (162 MHz, DMSO-d6) – 1.34; HRMS calculated for C61H87N5O10PSi [M+H]+1108.59543, found 1108.59642 m/z.
Synthesis of 5′-O-{11-[androst-4′-en-3′,17′-dioxo-16′β-yl]-dodecanoyl}-2′-O-phosphate-adenosine (2)
The hydrogenation and the hydrolysis procedure described above for the synthesis of 4 were used without modification for the synthesis of 2 from 15. The crude residue was pre-adsorbed on C-18 silica gel and purified by flash chromatography using a C-18 silica gel column (reverse phase) with MeOH/H2O (75:25 to 90:10) as eluent to give 2 (14 mg, 56% yield for two steps) as a white amorphous solid. IR (KBr) 3423 and 3130 (OH and NH2), 1735 (C = O, ester and ketone), 1685 (C = O, conjugated ketone and C = N), 1085 (P-O); 1H NMR (400 MHz, DMSO-d6) 0.78 (s, 18′-CH3), 0.80 to 2.50 (m, 38H, CH and CH2 of steroid skeleton and alkyl chain), 1.17 (s, 19′-CH3), 2.23 (dt, J1 = 7.3 Hz, J2 = 2.9 Hz, CH2COO), 4.09 (m, 4′-CH of ribose), 4.16 and 4.37 (2m, 5′-CH2 of ribose), 4.55 (t, J = 5.3 Hz, 3′-CH of ribose), 5.23 (m, 2′-CH of ribose), 5.66 (s, 4′-CH), 6.10 (d, J = 4.4 Hz, 1′-CH of ribose), 7.37 (s, NH2), 8.14 and 8.30 (2s, 2 × CH of adenine); 13C-NMR (75 MHz, DMSO-d6) 13.5, 16.8, 19.8, 24.3, 27.4, 28.2, 28.4, 28.6, 28.85 (2x), 28.91 (2x), 30.4, 30.6, 31.3, 31.7, 31.8, 33.2, 33.6, 34.0, 35.0, 38.2, 47.3, 48.0, 48.4, 53.3, 63.2, 69.4, 75.9, 81.3, 86.7, 119.1, 123.2, 140.2, 149.1, 152.4, 156.0, 170.7, 172.6, 198.0, 221.3; 31P NMR (162 MHz, DMSO-d6) -0.59; HRMS calculated for C41H61N5O10P [M+H]+ 814.41506, found 814.41405 m/z.
Enzymatic assay of type 3 17β-HSD inhibition
This enzymatic assay was performed as described previously [Citation21,Citation28]. Briefly, an expression vector encoding for type 3 17β-HSD was transfected into human embryonic kidney (HEK)-293 cells using the Exgen 500 procedure (Fermentas, Burlington, ON, Canada). To obtain cellular fragmentation, heat shock was performed in 50 mM sodium phosphate buffer (pH 7.4), containing 20% glycerol and 1 mM EDTA ( − 80°C to 37°C, three times, 5 min). The crude enzymes were used without further purification. The enzymatic assay was performed at 37°C for 2 h in 1 mL of a solution containing 975 μL of 50 mM sodium phosphate buffer (pH 7.4, 20% glycerol and 1 mM EDTA), 0.5 mM NADPH and 0.05 μM [4-14C]-4-androstene-3,17-dione (53.6 mCi / mmol, Perkin-Elmer Life Sciences Inc., Boston, MA, USA), 10 μL of the indicated inhibitor dissolved in ethanol (a few drops of DMSO and heating were necessary to dissolve compounds 2 and 4) and 15 μL of the diluted enzymatic source in phosphate buffer. Each inhibitor was assessed in triplicate. In order to obtain the IC50 values, 1 and Δ4-dione were assessed in triplicate at 10, 5, 1, 0.7, 0.4, 0.1, 0.05, 0.01, 0.001 μM and 2 was assessed in triplicate at 10, 5, 1, 0.5, 0.1, 0.05, 0.01, 0.005, 0.001 μM. Afterward, radiolabeled steroids were extracted from the reaction mixture with 1 mL of diethyl ether. The organic phases were pooled and evaporated to dryness with nitrogen. Residues were dissolved in 50 μL of CH2Cl2, applied on silica gel 60 F254 thin-layer chromatography plates (EMD Chemicals Inc., Gibbstown, NJ, USA) and eluted with a mixture of toluene/acetone (4:1). Substrate ([14C]-Δ4-dione) and metabolite ([14C]-T) were identified by comparison with reference steroids and quantified using the Storm 860 system (Molecular Dynamics, Sunnyvale, CA, USA). The percentage of transformation of [14C]-Δ4-dione into [14C]-T was calculated as follows: % transformation = 100 × ([14C]-T / ([14C]-T+[14C]-Δ4-dione)), and subsequently, % inhibition = 100 × ((% transformation without inhibitor - % transformation with inhibitor) / % transformation without inhibitor). From the inhibition curve, the IC50 value was calculated by computer (DE50 program, CHUL Research Center, Quebec, Canada).
Results
Chemical synthesis
A convergent strategy was applied to the preparation of the complex molecule 2. Thus, the steroid backbone with the appropriate alkyl side-chain spacer of 11 methylenes, acid 13, and the protected phosphorylated adenosine 9 were prepared separately. They were next joined by esterification under mild conditions. After careful removal of the three protective groups, the phosphorylated bisubstrate inhibitor 2 was obtained in only four steps starting from the two advanced synthons 9 and 13. Since the synthesis of steroid 13 had already been reported [Citation21], most of the synthetic efforts were put on the synthesis of 9.
The synthesis of protected phosphorylated adenosine 9, illustrated in Scheme , began with the selective protection of the 5′-hydroxyl of ( − )-adenosine over the 2′- and 3′-hydroxyls. This strategy using the 4-methoxytrityl protective group afforded the trityl derivative 5 as reported previously [Citation25]. The next step, the selective protection of the 3′-hydroxyl of 5, was not easy since the 2′- and 3′-hydroxyls have similar chemical properties. Noyori's research group [Citation26] has however reported a way of obtaining the 3′-O-silylated intermediate 6. Accordingly, when 5 was reacted with a slight excess of tert-butyldimethylsilyl chloride (TBDMSCl) in the presence of imidazole and DMF, a 1:1 mixture of 2′- and 3′-O-silylated compounds 7 and 6, respectively, was obtained. Noyori's group reported that these two regioisomers equilibrate rapidly in a 4:4:5:100 mixture of triethylamine, MeOH, EtOAc and diethyl ether and that the desired regioisomer 6 crystallizes from this medium more readily than 7 does. However, in our hands, this crystallization process was unsuccessful. Nonetheless, a flash chromatography using hexanes and EtOAc as eluent proved to be appropriate for separating the two regioisomers 6 and 7. The non-desired regioisomer 7 was converted back to 5 in quantitative yield by removal of the TBDMS group with TBAF in THF. COSY-NMR experiments were done to determine the exact structure of each regioisomer. Starting from the most deshielded signal corresponding to 1′-CH, NMR signals were easily assigned to their proton on the adenosine structure since the 1H-1H COSY only shows the interactions between protons from the same and from the next adjacent carbons. Furthermore, the NMR data allowed assigning the carbon bearing the OH group (C2′ or C3′) without doubt when DMSO-d6 was used, since the OH peak is apparent with this solvent.
Scheme 1 Synthesis of phosphorylated adenosine 9. Reagents, conditions and yields: (a) 4-MeOTrtCl, DMF, rt, 3 days (60%); (b) TBDMSCl, imidazole, DMF, rt, 20 h (6: 40%, 7: 37%); (c) 7, TBAF, THF, 0°C, 2 h (99%); (d) i. 6, dibenzyl diisopropylphosphoramidite, 1H-tetrazole, CH2Cl2, 0°C, 30 min; ii. m–CPBA, 0°C, 30 min (55%); (e) dichloroacetic acid, CH2Cl2, rt, 4 h (91%).
The 2′-hydroxyl of 6 was next phosphorylated using a two-step procedure [Citation29]. Thus, a phosphitylation with dibenzyl-N,N-diisopropylphosphoramidite and tetrazole followed by subsequent oxidation with m-CPBA afforded the desired phosphate 8 protected with two benzyl groups. Optimization of reaction conditions (temperature, quantity of phosphoramidite reagent) was necessary to limit the phosphitylation on the free amino group. The best yields were obtained when the phosphitylation step was carried out at 0°C with 1.2 equivalent of dibenzyl-N,N-diisopropylphosphoramidite. The benzyl protective group was chosen because it can easily be removed by hydrogenation. Thus, the phosphate derivative can be obtained simply by filtration through a pad of celite avoiding an aqueous work-up procedure, which is not appropriate when working with complex molecules like compounds 2 and 4. Finally, a selective deprotection of 5′-O-4-MeO-trityl over 3′-O-TBDMSCl of 8 was performed with dichloroacetic acid in CH2Cl2 to provide 9 in 91% yield and 12% overall yield from adenosine, without taking into account the recuperation of compound 7.
Before preparing the more complex Δ4-dione/phosphorylated adenosine hybrid 2, compound 4 was synthesized to perfect the chemical approach (Scheme ). The first step was the esterification of 9 with nonanoic acid pre-activated with EDCI and DMAP in DMF to produce 10 in 70% yield. The next step was to remove the 3′-O-silylated protective group. However, formation of compound 11 was not observed after treatment with HCl, HF, p-TSA or TBAF under various conditions well known to hydrolyse a silyl ether. It seems that the steric hindrance caused by the dibenzyl phosphate group does not allow the cleavage of the 3′-O-TBDMS group of 10. Monophosphorylation of the 2′-hydroxy of diol 5 was then attempted to overcome the 3′-O-silylated deprotection step. However, only a mixture of the starting material 5 and of a 2′,3′-diphosphorylated product was obtained, even when using only one equivalent of dibenzyl-N,N-diisopropylphosphoramidite. Another chemical approach was then envisaged. In a first step, benzyl groups of phosphate 10 were removed by hydrogenation. Classical hydrogenation procedure with hydrogen catalyzed by 10% Pd/C afforded the desired phosphate 12, but these conditions were not appropriate with a steroid backbone containing a tertiary olefin. Alternatively, a milder procedure known not to reduce olefin was used [Citation30]. Thus, removal of the benzyl groups of 10 was achieved by catalytic hydrogen transfer using palladium hydroxide as catalyst and cyclohexene as transfer reagent providing 12 in a more efficient way. Moreover, under these conditions, the benzyl groups were removed in just 30 min instead of 16 h for the classical hydrogenation. In the second step, the 3′-O-silylated protective group of 12 was removed easily by treatment with bubbling gaseous HCl in CH2Cl2 to give 4 by evaporation of the solvent. Compound 4 was purified by flash reverse-phase chromatography with C-18 silica gel and IR, 1H NMR, 13C NMR, 31P NMR, and HRMS data confirmed the expected structure.
Scheme 2 Synthesis of 4. Reagents, conditions and yields: (a) i. nonanoic acid, EDCI, DMAP, DMF, rt, 30 min; ii. 9, DMF, rt, 16 h (70%); (b) Pd(OH)2, cyclohexene, MeOH, reflux, 30 min; (c) HClg, CH2Cl2, rt, 30 min (28% for two steps).
As presented in Scheme , compound 2 was prepared using the synthetic approach developed for 4. The carboxylic acid 13 was first synthesized in 7 steps from testosterone as previously reported by us [Citation21]. Esterification of 9 with pre-activated carboxylic acid 13 as described above provided 14 in 47% yield. Hydrolysis of the 3′-ketal with p-TSA afforded the conjugated ketone 15. Since the classical hydrogenation procedure for cleaving benzyl groups (H2 with 5% or 10% Pd/C) gave the phosphate group, but unfortunately also reduced the Δ4-double bond on the steroid backbone, the milder hydrogenation treatment described above was used to afford the phosphate 16 in quantitative yield. Final deprotection of the 3′-O-silylated adenosine 16 was achieved by reaction with HCl in CH2Cl2. The phosphorylated bisubstrate compound 2 was then purified on C-18 silica gel and fully characterized using IR, 1H NMR, 13C NMR, 31P NMR and HRMS to validate its structure.
Scheme 3 Synthesis of 2. Reagents, conditions and yields: (a) i. 13, EDCI, DMAP, DMF, rt, 30 min; ii. 9, DMF, rt, 16 h (47%); (b) p-TSA, CH2Cl2, MeOH, rt, 1 h (59%); (c) Pd(OH)2, cyclohexene, MeOH, reflux, 30 min; (d) HClg, CH2Cl2, rt, 30 min (56% for two steps).
Inhibition of type 3 17β_it;-HSD
The ability of the phosphorylated bisubstrate compound 2 to inhibit the reduction of [14C]-Δ4-dione into [14C]-T by type 3 17β-HSD was evaluated and compared to that of the non-phosphorylated derivative 1. The inhibitory potency of 3 and 4, which contain only a cofactor-like moiety, was also evaluated and compared. Thus, an enzymatic assay was performed using homogenated HEK-293 cells transfected with a vector encoding for type 3 17β-HSD. The reaction was carried out for 2 h at 37°C with the cofactor NADPH [21,28]. The results obtained are illustrated in . Unlabeled Δ4-dione was used as a reference inhibitor and gave similar results to those previously reported [Citation31].
Figure 3 Inhibition of the transformation of [14C]-Δ4-dione into [14C]-T by compounds 1–4 and unlabeled Δ4-dione at different concentrations in homogenated HEK-293 cells overexpressing type 3 17β-HSD. A: First enzymatic assay. B: Second enzymatic assay. See Experimental Section for more details on the experimental conditions used.
![Figure 3 Inhibition of the transformation of [14C]-Δ4-dione into [14C]-T by compounds 1–4 and unlabeled Δ4-dione at different concentrations in homogenated HEK-293 cells overexpressing type 3 17β-HSD. A: First enzymatic assay. B: Second enzymatic assay. See Experimental Section for more details on the experimental conditions used.](/cms/asset/68057ffc-a621-455a-95ba-4da77463853f/ienz_a_205046_f0006_b.gif)
In a first enzymatic assay (), the bisubstrate phosphorylated compound 2 inhibited 46 and 21% of the enzymatic transformation compared to 65 and 19% for the non-phosphorylated derivative 1 at 1 and 0.1 μM, respectively. A second enzymatic assay was performed, in order to confirm the results of the first enzymatic assay, and similar results were obtained () with 94, 48 and 25% for compound 2 and 83, 58 and 24% for compound 1 at 10, 1 and 0.1 μM, respectively. Taken together, these data indicate only a slight difference in inhibitory potency between the bisubstrate inhibitors 2 and 1 (phosphorylated or not). As expected, no inhibition of type 3 17β-HSD was observed with compounds 3 and 4 at 1 and 0.1 μM, although a weak effect appears at the higher concentration of 10 μM (, 31 and 43% for 3 and 4, respectively).
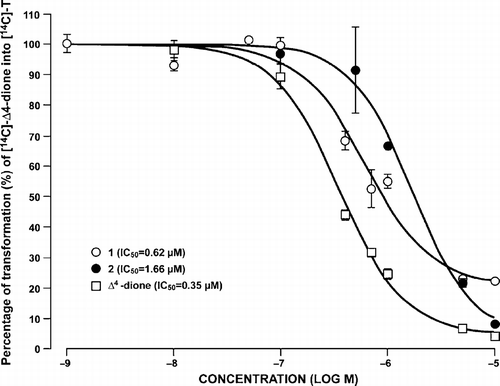
Since similar inhibitions were obtained for 1 and 2, IC50 values were calculated and presented in . In this enzymatic assay, the IC50 value represents the concentration of inhibitor at which type 3 17β-HSD activity was reduced by half. The non-phosphorylated compound 1 gave a lower IC50 value than the phosphorylated compound 2 with 0.62 and 1.66 μM, respectively.
Figure 4 Inhibition of type 3 17β-HSD in transfected homogenated HEK-293 cells by compounds 1, 2 and unlabeled Δ4-dione, at various concentrations and determination of IC50 values. See Experimental Section for more details on the conditions used in the enzymatic assay.
Discussion
The inhibitory potency of the non-phosphorylated compound 1 and the phosphorylated compound 2 were compared. Unfortunately and contrary to our working hypothesis, compound 2 proved to be a slightly less potent inhibitor of type 3 17β-HSD than 1 with IC50 values of 1.66 μM and 0.62 μM, respectively. First, it occurred to us that these unexpected results might be explained by a degradation of the inhibitor during storage or the enzymatic assay. Thus, to eliminate the possibility that 2 could have undergone phosphate hydrolysis during storage, its stability in ethanol was assessed. For this purpose, three weeks-old ethanol solutions of 1 and 2 were applied on TLC and eluted. Comparison of eluted spots with freshly prepared solutions of 1 and 2 revealed that the phosphate group of 2 was not hydrolysed in ethanol, even after three weeks. The stability of 2 during the enzymatic process was also evaluated using a similar procedure. After the assay, the medium was extracted with diethyl ether, applied on TLC and eluted. If the phosphate on compound 2 had been hydrolysed, compound 1 should have been formed. However, no spot corresponding to 1 was observed on the TLC. Clearly, the slightly lower inhibition of 2 when compared to 1 cannot be attributed to a transformation or a degradation of compound 2.
One hypothesis to explain our results is that the spacer length (n = 11) between the steroid and the C2′-phosphate adenosine is not optimal. In fact, the first Δ4-dione/adenosine hybrid inhibitors, which were used for length optimization, were developed without the phosphate group. Furthermore, type 3 17β-HSD is inactive when NADH (non-phosphorylated cofactor) is used, as is the case in these first inhibitors. The preference of type 3 17β-HSD for the cofactor NADPH is explained by the interaction between the C2′ phosphate group of adenosine and Arg80 [Citation24]. In fact, the guanidinium group found on the side chain of this aminoacid interacts with the C2′-phosphate in a bi-dentate fashion. For compound 2, it may be that this interaction does not occur efficiently because of an inadequate spacer length, thus resulting in lower inhibition than first expected.
Another hypothesis to explain the results is that inhibitors 1 and 2 might interact only with the substrate-binding site. This hypothesis is based on our knowledge of the mechanism of action of the enzyme and from the study of interactions between bisubstrate inhibitor EM-1745 and type 1 17β-HSD, an enzyme belonging to the same family as type 3. Thus, when EM-1745 was tested with the cofactor NADPH on homogenated HEK-293 cells overexpressing type 1 17β-HSD, weak inhibition was observed with 19% at 1 μM, compared to 91% at 1 μM with the cofactor NADH [Citation21]. These results are in agreement with those obtained for type 3 17β-HSD since NADPH was used for the enzymatic assay. Few studies have been performed to elucidate the exact mechanism of action of type 1 and type 3 17β-HSDs. Nevertheless, it was proposed that type 1 and type 3 17β-HSDs display a random mechanism for binding [Citation32,Citation33]. This means that either the substrate or the cofactor binds first to the enzyme, at random. However, in both enzymatic assays (type 1 and type 3 17β-HSDs), the concentration of NADPH and labeled substrate (E1 or Δ4-dione) used was 500 μM and 0.05 μM, respectively. Since the concentration of cofactor used is 10 000 times higher than that of the labeled substrate and 500 times higher than that of the inhibitor (1 μM), it is very probable that the cofactor binds first to the enzyme. Thus, when inhibitors 1, 2 or EM-1745 attempt to enter into the catalytic site of the enzyme (type 1 or type 3), the cofactor-binding site is already occupied by NADPH, which is known to possess a greater affinity for the cofactor-binding site than NADH [Citation1]. Consequently, 1, 2 or EM-1745 might not truly act as bisubstrate inhibitors when NADPH is the cofactor, and only interact efficiently with the substrate-binding site, resulting in lower inhibition than would be the case with an actual bisubstrate inhibitor. Moreover, the better inhibition of type 1 17β-HSD by EM-1745 when NADH is used as cofactor instead of NADPH could be explained this way. Because the concentration of NADH used (100 μM) is higher than the concentrations of the labeled substrate and EM-1745, the enzyme should also bind first with the cofactor NADH. But, since NADH possesses a weak affinity for the cofactor-binding site compared to NADPH, EM-1745 can remove the cofactor from its binding site and then interact with both the substrate and the cofactor-binding sites. Finally, even if compound 2 has a phosphate group like NADPH, this inhibitor of type 3 17β-HSD is not strong enough to remove the cofactor NADPH from its binding site. Based on this hypothesis and the results reported in and , it appears that compounds 1 and 2 do not act as bisubstrate inhibitors of type 3 17β-HSD, but probably only interact with the substrate- (Δ4-dione) binding site.
Conclusions
A phosphorylated bisubstrate inhibitor of type 3 17β-HSD, compound 2, has been synthesized using a convergent strategy. This complex molecule was obtained in four steps from the coupling of two advanced synthons: the carboxylic acid steroid 13 and the appropriately protected phosphorylated adenosine 9. This latter key intermediate was prepared using an appropriate protective group strategy. This could also be applied in the synthesis of phosphorylated bisubstrate inhibitors of other enzymes that use the NADPH cofactor. The inhibitory potency of the phosphorylated Δ4-dione/adenosine hybrid compound 2 was evaluated on homogenated HEK-293 cells overexpressing type 3 17β-HSD and compared to its non-phosphorylated analogue 1. However, unexpectedly, the phosphorylated inhibitor 2 is a slightly less potent inhibitor than 1. Hydrolysis of the phosphate group of 2 during the enzymatic assay was tested, but it appears that 2 is stable. Two hypothesis were discussed to explain the lower than expected inhibition of compound 2: 1) the adenosine part of 2 might not form optimum interactions with the cofactor-binding site of the enzyme, and 2) the cofactor NADPH could bind first to the enzyme, preventing compound 2 from removing the cofactor from its binding site, with the result that compound 2 would only interact with the substrate-binding site.
Acknowledgements
This work was supported by the Canadian Institutes of Health Research (CIHR) (financial support and M.B., scholarship). We are grateful to Yannick Laplante and Dr Van Luu-The for providing HEK-293 cells overexpressing type 3 17β-HSD. We also thank Ms Sylvie Méthot for careful reading of the manuscript.
References
- Luu-The V. J Steroid Biochem Mol Biol 2001; 76: 143–151
- Labrie F, Luu-The V, Lin SX, Simard J, Labrie C, El-Alfy M, Pelletier G, Bélanger A. J Mol Endocrinol 2000; 25: 1–16
- Labrie F, Luu-The V, Lin SX, Simard J, Labrie C. Trends Endocrinol Metab 2000; 11: 421–427
- Mindnich R, Möller G, Adamski J. Mol Cell Endocrinol 2004; 218: 7–20
- Geissler WM, Davis DL, Wu L, Bradshaw KD, Patel S, Mendonc BB, Elliston KO, Wilson JD, Russell DW, Andersson S. Nat Genet 1994; 7: 34–39
- Boehmer ALM, Brinkmann AO, Sandkuijl LA, Halley DJJ, Niermeijer MF, Andersson S, Jong FH, Kayserili H, Vroede MA, Otten BJ, Rouwé CW, Mendonça BB, Rodrigues C, Bode HH, Ruiter PE, Delemarre-Van de Wall HA, Drop SLS. J Clin Endocrinol Metab 1999; 84: 4713–4721
- Labrie F, Bélanger A, Cusan L, Labrie C, Simard J, Luu-The V, Diamond P, Gomez JL, Candas B. Endocr-Relat Cancer 1996; 3: 243–278
- Denmeade SR, Isaacs JT. Prostate 2004; 58: 211–224
- Andriole G, Bruchovsky N, Chung LWK, Matsumoto AM, Rittmaster R, Roehrborn C, Russell D, Tindall D. J Urol 2004; 172: 1399–1403
- Sansone GL, Reisner RM. J Invest Dermatol 1971; 56: 366–372
- Thiboutot D, Martin P, Volikos L, Gilliland K. J Invest Dermatol 1998; 111: 390–395
- Kuttenn F, Mowszowicz I, Schaison G, Mauvais-Jarvis P. J Endocrinol 1977; 75: 83–91
- Bingham KD, Shaw DA. J Endocrinol 1973; 57: 111–121
- Sato T, Tadokoro T, Sonoda T, Asada Y, Itami S, Takayasu S. J Dermatol Sci 1999; 19: 123–125
- Poirier D. Curr Med Chem 2003; 10: 453–477
- Smith HJ, Nicholls PJ, Simons C, Le Lain R. Exp Opin Ther Patents 2001; 11: 789–824
- Ngatcha BT, Luu-The V, Labrie F, Poirier D. J Med Chem 2005; 48: 5257–5268
- Maltais R, Luu-The V, Poirier D. J Med Chem 2002; 45: 640–653
- Maltais R, Luu-The V, Poirier D. Bioorg Med Chem 2001; 9: 3101–3111
- Ngatcha BT, Laplante Y, Labrie F, Luu-The V, Poirier D. Mol Cell Endocrinol 2006; 248: 225–232
- Bérubé M, Laplante Y, Poirier D. Med Chem 2006; 2: 329–347
- Poirier D, Boivin RP, Tremblay MR, Bérubé M, Qiu W, Lin SX. J Med Chem 2005; 48: 8134–8147
- Qiu W, Campbell RL, Gangloff A, Dupuis P, Boivin RP, Tremblay MR, Poirier D, Lin SX. FASEB J 2002; 16: 1829–1831
- McKeever BM, Hawkins BK, Geissler WM, Wu L, Sheridan RP, Mosley RT, Andersson S. Biochim Biophys Acta 2002; 1601: 29–37
- Lohrmann R, Khorana HG. J Am Chem Soc 1964; 86: 4188–4194
- Noyori R, Uchiyama M, Nobori T, Hirose M, Hayakawa Y. Aust J Chem 1992; 45: 205–225
- Ogilvie KK, Beaucage SL, Schifman AL, Theriault NY, Sadana KL. Can J Chem 1978; 56: 2768–2780
- Luu-The V, Zhang Y, Poirier D, Labrie F. J Steroid Biochem Mol Biol 1995; 55: 581–587
- Lindberg J, Ekeroth J, Konradsson P. J Org Chem 2002; 67: 194–199
- Shuto S, Horne G, Marwood RD, Potter BVL. Chem Eur J 2001; 7: 4937–4946
- Ngatcha BT, Luu-The V, Poirier D. J Enz Inhib Med Chem 2002; 17: 155–165
- Penning TM. Endocr Relat Cancer 1996; 3: 41–56
- Penning TM, Ricigliano JW. J Enz Inhib 1991; 5: 165–198